DOI:
10.1039/D2RA07164A
(Paper)
RSC Adv., 2023,
13, 7503-7513
Triterpenoid saponins and C21 steroidal glycosides from Gymnema tingens and their glucose uptake activities†
Received
11th November 2022
, Accepted 21st February 2023
First published on 8th March 2023
Abstract
Four new triterpenoid saponins, tigensides A–D (1–4), and one new C21 steroid, tipregnane A(9), together with six known compounds were isolated from the EtOAc fraction of the roots and stems of Gymnema tingens. The chemical structures of the new compounds were determined based on their spectroscopic data, including IR, UV, NMR, and mass spectrometric analysis. All compounds were isolated for the first time. Compounds 1–11 promoted glucose uptake in the range of 1.12 to 2.52 fold, respectively. Compound 2 showed the most potent glucose uptake, with 2.52 fold enhancement. Additionally, compound 2 showed a medium effect on the GLUT4 translocation activity in L6 cells in further study.
Introduction
Diabetes mellitus (DM) is a metabolic chronic disease characterized by hyperglycemia and insulin resistance (IR), which is caused by a lack of insulin secretion or insulin response.1 In 2017, approximately, 425 million people (1 in 11 adults) were affected with diabetes and this may rise to 693 million in 2045.2 Type 2 DM (T2DM) accounts for 90–95% of global diabetes.3 The treatment of diabetes accounts for around 12% of overall global health-care expenditures with respect to socioeconomic burden.4 Current synthetic agents and insulin used effectively for the treatment of diabetes are scarce and expensive with prominent adverse effects. Natural products and their active ingredients have advantages such as minimal side effects, being rich in resources, and low price. Therefore, the complementary and alternative approach to screen new effective hypoglycemic drugs through isolation of phytochemicals from natural products is imperative5,6
The genus Gymnema of the family Asclepiadaceae comprises 25 species, which are distributed in tropical region of Asia, and subtropical regions of southern Africa and Oceania.7 Previous phytochemical studies on Gymnemas have led to the isolation and identification of triterpenoids, triterpenoid saponins, C21 steroids, C21 steroidal glycosides, flavonoids, peptides, polysaccharides and several other components.8–14 All of the components described above play an important role related to several bioactivities, such as anti-diabetic, anti-inflammatory, anti-bacterial, anti-arthritic, anti-hyperlipidemic, cytotoxic, and immunostimulatory activities.15–21 Some triterpenoid saponins from the genus Gymnema were found to be able to attenuate hyperglycemia induced by pituitary growth hormone or adrenocorticotropic hormone and to inhibit intestinal glucose absorption in diabetic rats.22–27 In traditional herbal medicine, the Gymnema species has been well known for the anti-diabetic potential.28
Gymnema tingens Spreng. is a plant of the genus Gymnema. In China, it is distributed in the provinces of Guangdong, Guangxi, Guizhou and Yunnan, and grows in hillsides, forests or shrubs. It is a folklore medicine in China used for the treatment of rheumatism and polio.29 Previous phytochemical studies on Gymnema tingens have led to the isolation and identification of triterpenoids, steroids, phenolic glycosides, and several other components.30–32 However, few studies have been reported on its bioactive components with respect to possible antidiabetic activity. In our previous investigation, we found that the EtOAc fraction of G. tingens showed a good activity on glucose uptake. As part of an ongoing search for bioactive compounds from Gymnema plants, we performed purification of the EtOAc fraction of G. tingens by chromatography column and semipreparative phase HPLC, and obtained four new triterpenoid saponins, tigensides A–D (1–4), and one new C21 steroid, tipregnane A (9), together with six known compounds. The structures were identified by spectroscopic analysis and comparison of observed and reported spectroscopic data.
In addition, we tested the glucose uptake activity of compounds 1–11 and the GLUT4 translocation activity of compound 2 in L6 cells in the further study. As the important protein for preserving whole-body glucose homeostasis, GLUT4 is considered to be a potential therapeutic target for the treatment of T2DM.33 Herein, we report the isolation and structure elucidation of these isolated compounds, as well as their biological activities. These results suggest that the G. tingens have the potential to become a source for new antidiabetic drug discovery.
Results and discussion
Structural elucidation of the isolated compounds
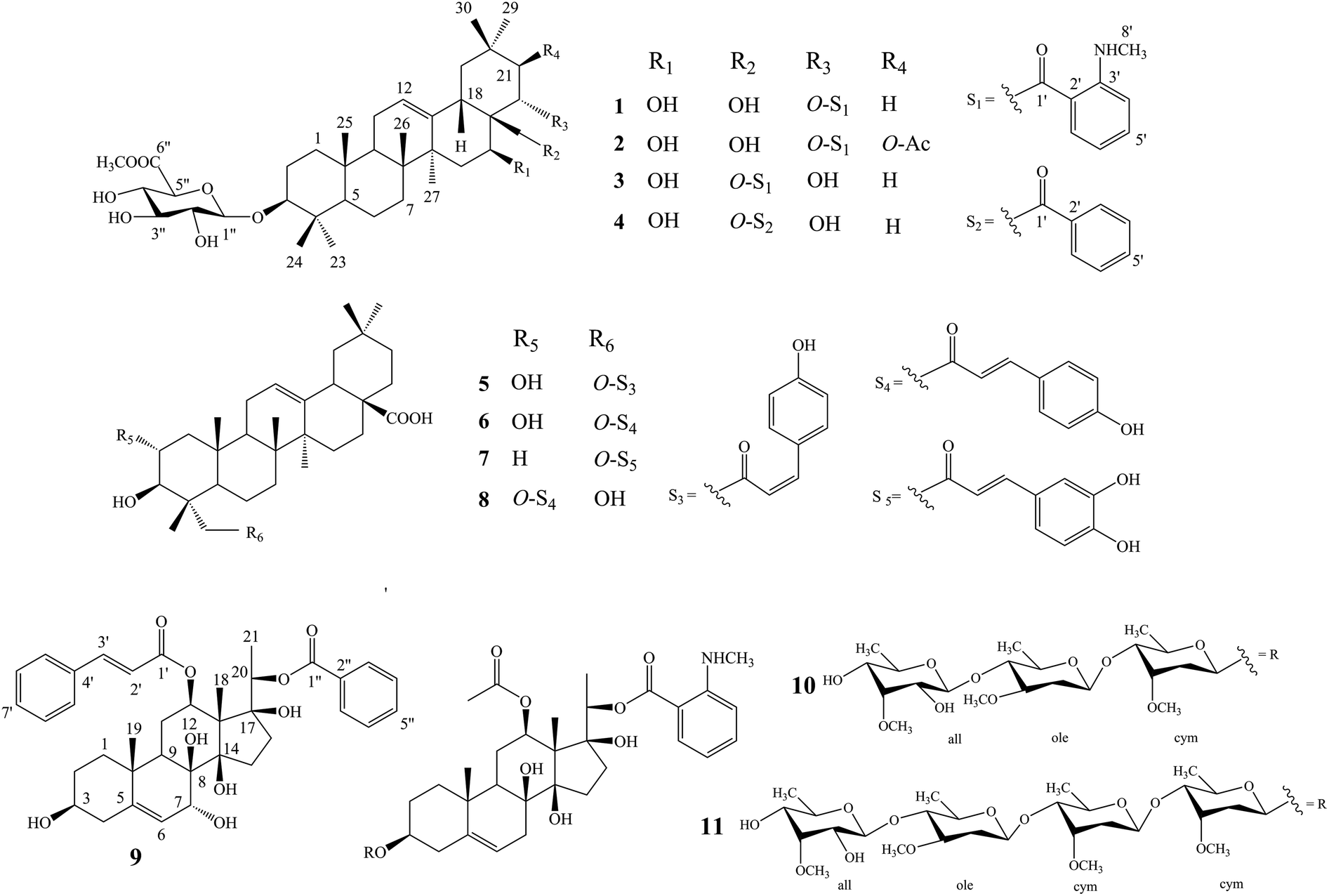
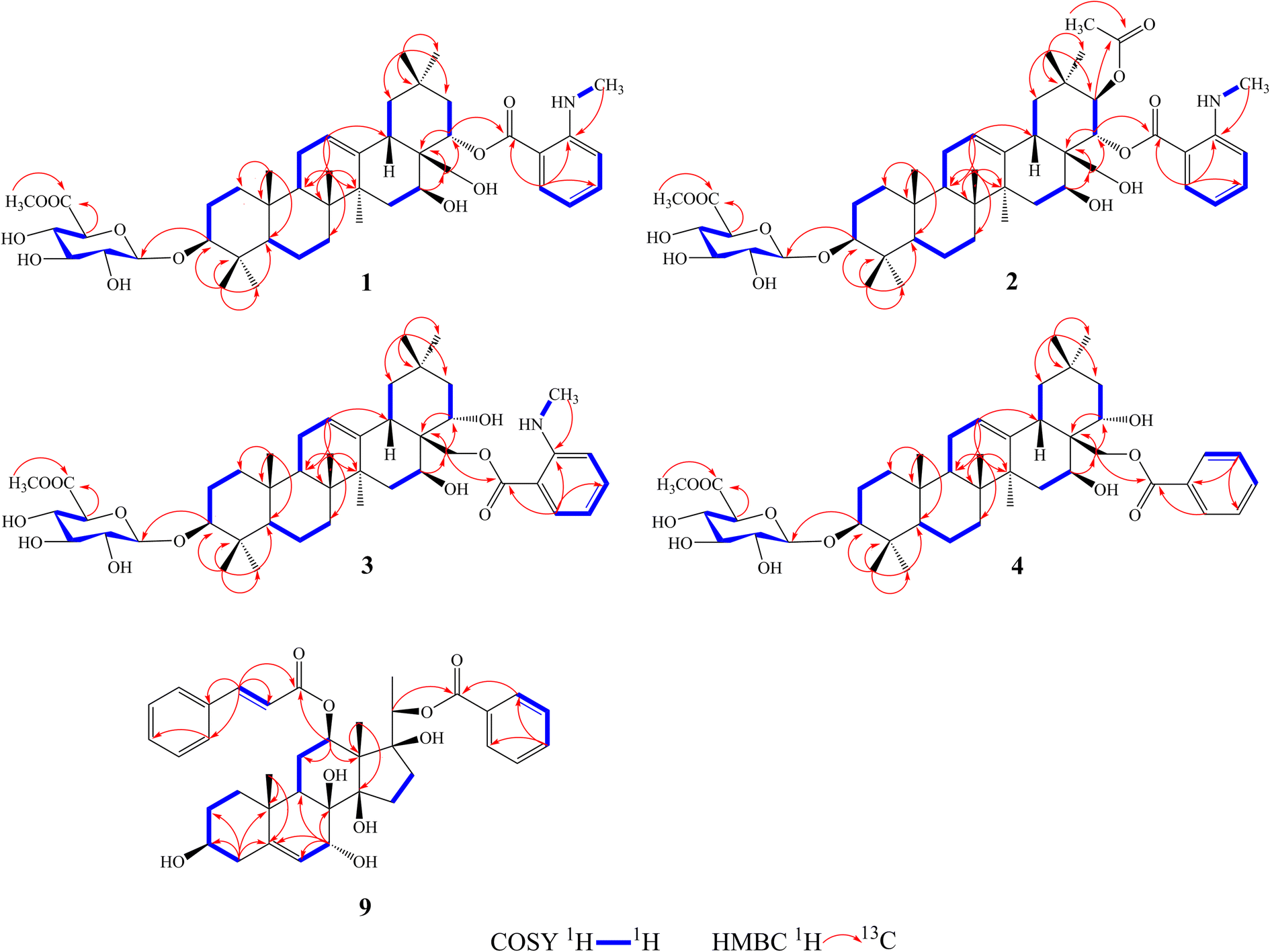
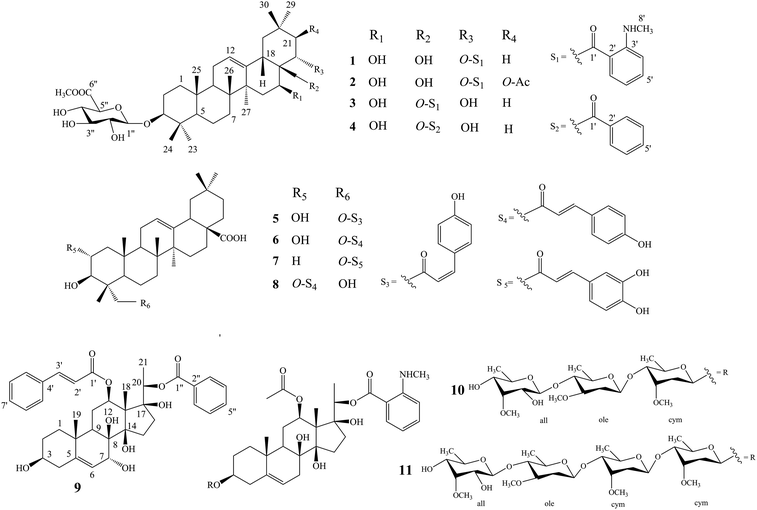
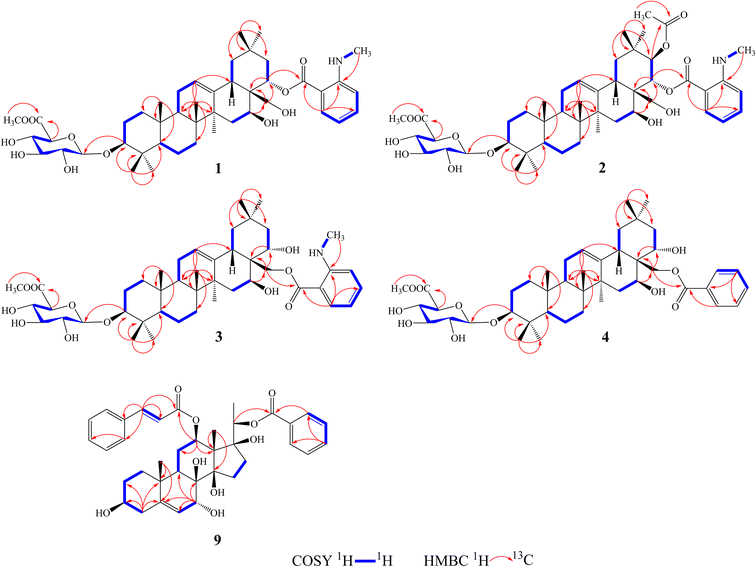
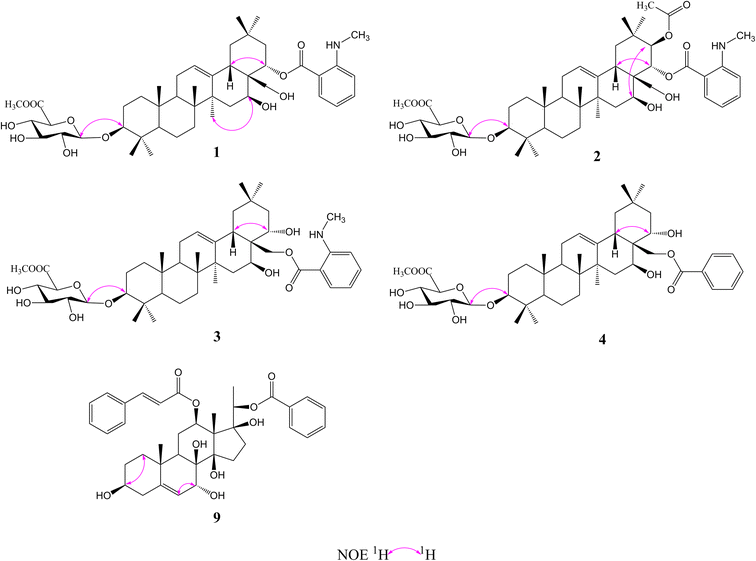
Compound 1 was isolated as a yellow, amorphous powder with an [α]25D −0.9 (c 0.5, MeOH). Its molecular formula of C45H67NO11 was suggested from a deprotonated high-resolution electrospray ionization mass spectrometric (HR-ESIMS) ion peak at m/z 798.4796 [M + H]+ (calcd for C45H68NO11, 798.4787), indicating 12 degrees of unsaturation. Absorptions for NH stretch (3390 cm−1), olefinic (1608 cm−1), and NH bend (1519 cm−1) groups were observed in its IR spectrum. The UV absorption maxima at 220 nm. The 1H NMR spectrum (Table 1) of 1 showed signals for one olefinic proton at δH 5.37 (1H, brs), three oxygenated methine protons at δH 3.38 (1H, dd, J = 11.8, 4.4 Hz), 5.11 (1H, dd, J = 11.5, 5.2 Hz), and 6.37 (1H, dd, J = 10.9, 5.1 Hz), a pair of oxygenated methylene protons at δH 4.51 (1H, m) and 4.09 (1H, d, J = 10.6 Hz), seven methyl groups at δH 1.45, 1.33, 1.30, 1.01, 1.00, 0.95 and 0.82 (each 3H, s), four aromatic protons at δH 8.48 (1H, dd, J = 8.4, 1.8 Hz), 7.39 (1H, ddd, J = 7.8, 7.2, 1.8 Hz), 6.67 (1H, dd, J = 7.8, 1.2 Hz), and 6.66 (1H, ddd, J = 8.4, 7.2, 1.2 Hz), and one nitrogenated methyl group at δH 2.80 (3H, overlapped). The 13C NMR spectrum (Table 2) exhibited resonances for one carbonyl carbon at δC 168.8, two olefinic carbons at δC 143.0 and 124.0, and six aromatic carbons at δC 152.2, 135.1, 133.4, 115.1, 112.7 and 111.9. 1H–1H COSY (Fig. 2) correlations of H-1/H-2/H-3, H-5/H-6/H-7, H-9/H-11/H-12, H-15/H-16, H-18/H-19, H-21/H-22, H-4′/H-5′/H-6′/H-7′ and H-8′/H-9′ were observed in its structure. An observation of 1H–13C long-range correlations (Fig. 2) of H-24 (δH 1.00) to C-3 (δC 89.5), C-4 (δC 40.0), C-5 (δC 56.0) and C-23 (δC 28.6), H-25 (δH 0.82) to C-1 (δC 39.2), C-5 (δC 56.0) and C-10 (δC 37.2), H-26 (δH 0.95) to C-7 (δC 33.3), C-8 (δC 40.7), C-9 (δC 47.5) and C-14 (δC 43.5), H-12 (δH 5.37) to C-9 (δC 47.5), C-14 (δC 43.5) and C-18 (δC 44.5), H-30 (δH 1.30) to C-19 (δC 46.7), C-20 (δC 32.8), C-21 (δC 39.8) and C-29 (δC 33.7), and H-22 (δH 6.37) to C-17 (δC 46.4) enabled the establishment of pentacyclic triterpene skeleton.34 Further HMBC correlations of H-22 (δH 6.37) to C-1′ (δC 168.8), H-7′ (δH 8.48) to C-1′ (δC 168.8), C-3′ (δC 152.2) and C-5′ (δC 135.1), and H-8′ (δH 2.80) to C-3′ (δC 152.2) revealed that an anthranilate group was connected to C-22. Comparison of the NMR data with those of (3β,16β,22α)-22-(N-methylanthraniloxy)-16,28-dihydroxyolean-12-en-3-yl-3-O-β-D-glucopyra nosyl-β-D-glucopyranosidur-onic acid isolated from Gymnema inodorum,35 suggested that 1 had the same aglycone moiety. In addition, a group of sugar-proton signals [δH 5.03 (d, J = 7.8 Hz, H-1′′), 4.14 (dd, J = 9.0, 7.8 Hz, H-2′′), 4.31 (dd, J = 9.6, 9.0 Hz, H-3′′), 4.52 (dd, J = 9.7, 9.6 Hz, H-4′′), 4.64 (d, J = 9.7 Hz, H-5′′)] were observed in the 1H NMR spectra, of which one anomeric proton signal at δH 5.03 (d, J = 7.8 Hz) suggested a β-D-linkage of the sugar moiety in 1. The HMBC correlations between the carbomethoxyl protons at δH 3.75 and the carboxyl group carbon of the glucuronic acid at δC 171.4 indicated that glucuronic acid had been methyl esterified. Further analysis of the 13C NMR data of the glycone [δC 107.8 (C-1′′), 75.9 (C-2′′), 78.4 (C-3′′), 73.7 (C-4′′), 77.7 (C-5′′) 171.4 (C-6′′), and 52.5 (–OCH3)] indicated that the glucuronic acid was the same as the sugar moiety of caraganoside C isolated from Caragana microphylla.36 The sugar chain at the C-3 position was indicated by the long-range coupling in the HMBC spectrum of H-3 (δH 3.38) to C-1′′ (δC 107.8). Moreover, the NOESY correlations (Fig. 3) between H-16 (δH 5.11) and H-27 (δH 1.45) implied that H-16 was α-orientated. The attachment of an anthranilate group at C-22 was deduced as α-oriented based on the NOESY correlation between H-22 (δH 6.37) and H-18 (δH 3.05). Thus, compound 1 was established as (3β,16β,22α)-22-(N-methylanthraniloxy)-16,28-dihydroxyolean-12-en-3-yl-3-O-β-D glucuronopyranosyl methyl ester, and named tigensides A.
Table 1 1H-NMR (600 MHz) data for compounds 1–4 and 9 (J in Hz)
| No. |
δH (J in Hz) |
| 1a |
2a |
3a |
4a |
9b |
| Pyridine-d5 as solvent. CDCl3 as solvent. |
| 1 |
1.44 (m)/0.87 (m) |
1.42 (m)/0.86 (m) |
1.40 (m)/0.86 (m) |
1.39 (m)/0.85 (m) |
1.15 (m)/1.90 (m) |
| 2 |
2.18 (m)/1.88 (m) |
2.17 (m)/1.87 (m) |
2.15 (m)/1.86 (m) |
2.15 (m)/1.85 (m) |
1.55 (m)/1.82 (m) |
| 3 |
3.38 (dd, 11.8, 4.4) |
3.39 (dd, 12.1, 4.5) |
3.36 (dd, 11.8, 4.4) |
3.36 (dd, 11.8, 4.0) |
3.60 (m) |
| 4 |
|
|
|
|
2.38 (m)/2.41 (m) |
| 5 |
0.79 (m) |
0.78 (m) |
0.79 (m) |
0.78 (m) |
|
| 6 |
1.49 (m)/1.31 (m) |
1.49 (m)/1.30 (m) |
1.48 (m)/1.30 (m) |
1.47 (m)/1.29 (m) |
5.67 (d, 4.7) |
| 7 |
1.57 (m)/1.34 (m) |
1.50 (m)/1.26 (m) |
1.56 (m)/1.32 (m) |
1.56 (m)/1.33 (m) |
4.06 (d, 5.2) |
| 8 |
|
|
|
|
|
| 9 |
1.59 (m) |
1.56 (m) |
1.59 (m) |
1.58 (m) |
1.82 (m) |
| 10 |
|
|
|
|
|
| 11 |
1.83 (m) |
1.84 (m)/1.80 (m) |
1.80 (m) |
1.80 (m) |
1.84 (m)/2.02 (m) |
| 12 |
5.37 (brs) |
5.42 (brs) |
5.35 (brs) |
5.35 (brs) |
4.86 (overlapped) |
| 13 |
|
|
|
|
|
| 14 |
|
|
|
|
|
| 15 |
2.20 (d, 12.2)/1.26 (m) |
2.11 (m)/1.67 (m) |
2.16 (m)/1.72 (m) |
2.15 (m)/1.73 (m) |
2.32 (m) |
| 16 |
5.11 (dd, 11.5, 5.2) |
5.15 (dd, 11.1, 5.2) |
5.17 (dd, 11.5, 5.3) |
5.19 (overlapped) |
2.07 (m) |
| 17 |
| 18 |
3.05 (d, 13.5) |
3.16 (dd, 11.1, 4.3) |
2.95 (dd, 13.9, 4.6) |
2.96 (dd, 14.0, 3.5) |
1.64 (s) |
| 19 |
2.00 (m)/1.26 (m) |
2.28 (m)/1.39 (m) |
2.05 (m)/1.23 (m) |
2.05 (m)/1.23 (m) |
1.14 (s) |
| 20 |
4.91 (d, 6.2) |
| 21 |
2.00 (m) |
5.89 (d, 11.3) |
2.11 (m)/1.96 (m) |
2.10 (m)/1.97 (m) |
1.36 (d, 6.2) |
| 22 |
6.37 (dd, 10.9, 5.1) |
6.51 (d, 11.2) |
4.86 (overlapped) |
4.87 (dd, 11.6, 2.3) |
|
| 23 |
1.33 (s) |
1.34 (s) |
1.32 (s) |
1.32 (s) |
|
| 24 |
1.00 (s) |
1.00 (s) |
0.97 (s) |
0.98 (s) |
|
| 25 |
0.82 (s) |
0.81 (s) |
0.82 (s) |
0.82 (s) |
|
| 26 |
0.95 (s) |
0.90 (s) |
1.13 (s) |
1.11 (s) |
|
| 27 |
1.45 (s) |
1.40 (s) |
1.43 (s) |
1.43 (s) |
|
| 28 |
4.51 (m)/4.09 (d, 10.6) |
4.36 (d, 10.8)/4.09 (d, 10.9) |
5.35 (m)/4.89 (d, 10.9) |
5.43 (d, 10.8)/4.95 (d, 11.1) |
|
| 29 |
1.01 (s) |
1.07 (s) |
1.04 (s) |
1.05 (s) |
|
| 30 |
1.30 (s) |
1.34 (s) |
1.17 (s) |
1.17 (s) |
|
| |
Anth |
Anth |
Anth |
Bz |
Cin |
| 1′ |
|
|
|
|
|
| 2′ |
|
|
|
|
6.11 (d, 16.0) |
| 3′ |
|
|
|
8.26 (dd, 7.8, 2.4) |
7.45 (d, 16.0) |
| 4′ |
6.67 (dd, 7.8, 1.2) |
6.64 (dd, 8.4, 1.8) |
6.66 (dd, 8.4, 1.2) |
7.45 (t, 7.8) |
|
| 5′ |
7.39 (ddd, 7.8, 7.2, 1.8) |
7.36 (ddd, 8.4, 8.1, 1.2) |
7.42 (ddd, 8.4, 7.8, 1.5) |
7.53 (t, 7.8) |
7.28 (d, 7.2) |
| 6′ |
6.66 (ddd, 8.4, 7.2, 1.2) |
6.60 (ddd, 8.1, 7.8, 1.8) |
6.71 (ddd, 8.0, 7.8, 1.2) |
7.45 (t, 7.8) |
7.33 (m) |
| 7′ |
8.48 (dd, 8.4, 1.8) |
8.46 (dd, 7.8, 1.2) |
8.26 (dd, 8.0, 1.5) |
8.26 (dd, 7.8, 2.4) |
7.37 (m) |
| 8′ |
2.80 (overlapped) |
2.75 (overlapped) |
2.64 (overlapped) |
|
7.33 (m) |
| 9′ |
8.40 (m) |
8.17 (m) |
7.99 (m) |
|
7.28 (d, 7.2) |
| |
Glucuronopyranosyl methyl ester |
Glucuronopyranosyl methyl ester |
Glucuronopyranosyl methyl ester |
Glucuronopyranosyl methyl ester |
BZ |
| 1′′ |
5.03 (d, 7.8) |
5.04 (d, 7.8) |
5.01 d (7.8) |
5.01 d (7.8) |
|
| 2′′ |
4.14 (dd, 9.0, 7.8) |
4.14 (dd, 9.0, 7.8) |
4.12 (dd, 9.0, 7.8) |
4.12 (dd, 9.0, 7.8) |
|
| 3′′ |
4.31 (dd, 9.6, 9.0) |
4.31 (dd, 9.6, 9.0) |
4.30 (dd, 9.6, 9.0) |
4.30 (dd, 9.6, 9.0) |
7.92 (dd, 7.2, 1.5) |
| 4′′ |
4.52 (dd, 9.7, 9.6) |
4.52 (dd, 9.7, 9.6) |
4.51 (dd, 9.7, 9.6) |
4.51 (dd, 9.7, 9.6) |
7.36 (m) |
| 5′′ |
4.64 (d, 9.7) |
4.65 (d, 9.7) |
4.63 (d, 9.7) |
4.63 (d, 9.7) |
7.56 (t, 7.2) |
| 6′′ |
|
|
|
|
7.36 (m) |
| 7′′ |
3.75, s |
3.75, s |
3.75, s |
|
7.92 (dd, 7.2, 1.5) |
Table 2 13C-NMR (150 MHz) data for compounds 1–4 and 9
| No. |
δC |
| 1a |
2a |
3a |
4a |
9b |
| Pyridine-d5 as solvent. CDCl3 as solvent. |
| 1 |
39.2 (t) |
39.1 (t) |
39.2 (t) |
39.2 (t) |
38.6 (t) |
| 2 |
27.1 (t) |
27.1 (t) |
27.1 (t) |
27.1 (t) |
30.8 (t) |
| 3 |
89.5 (d) |
89.5 (d) |
89.5 (d) |
89.5 (d) |
71.8 (d) |
| 4 |
40.0 (s) |
40.0 (s) |
40.0 (s) |
40.0 (s) |
42.0 (t) |
| 5 |
56.0 (d) |
56.0 (d) |
56.0 (d) |
56.0 (d) |
144.2 (s) |
| 6 |
18.8 (t) |
18.8 (t) |
18.8 (t) |
18.8 (t) |
122.7 (d) |
| 7 |
33.3 (t) |
33.2 (t) |
33.3 (t) |
33.3 (t) |
68.3 (d) |
| 8 |
40.7 (s) |
40.6 (s) |
40.7 (s) |
40.7 (s) |
76.2 (s) |
| 9 |
47.5 (d) |
47.4 (d) |
47.5 (d) |
47.5 (d) |
38.8 (d) |
| 10 |
37.2 (s) |
37.1 (s) |
37.1 (s) |
37.1 (s) |
37.8 (s) |
| 11 |
24.3 (t) |
24.3 (t) |
24.3 (t) |
24.3 (t) |
25.2 (t) |
| 12 |
124.0 (d) |
124.6 (d) |
124.4 (d) |
124.5 (d) |
73.2 (d) |
| 13 |
143.0 (s) |
142.0 (s) |
142.5 (s) |
142.5 (s) |
58.0 (s) |
| 14 |
43.5 (s) |
43.2 (s) |
43.0 (s) |
43.0 (s) |
87.7 (s) |
| 15 |
37.6 (t) |
37.8 (t) |
36.6 (t) |
36.6 (t) |
33.6 (t) |
| 16 |
66.0 (d) |
67.0 (d) |
66.7 (d) |
66.7 (d) |
33.4 (t) |
| 17 |
46.4 (s) |
48.6 (s) |
44.9 (s) |
44.9 (s) |
86.8 (s) |
| 18 |
44.5 (d) |
43.4 (d) |
44.3 (d) |
44.3 (d) |
10.8 (q) |
| 19 |
46.7 (t) |
46.3 (t) |
46.5 (t) |
46.5 (t) |
19.8 (q) |
| 20 |
32.8 (s) |
37.2 (s) |
32.6 (s) |
32.6 (s) |
75.6 (d) |
| 21 |
39.8 (t) |
76.9 (d) |
44.5 (t) |
44.5 (t) |
15.2 (q) |
| 22 |
74.0 (d) |
74.0 (d) |
70.1 (d) |
70.0 (d) |
|
| 23 |
28.6 (q) |
28.6 (q) |
28.6 (q) |
28.6 (q) |
|
| 24 |
17.4 (q) |
17.4 (q) |
17.3 (q) |
17.4 (q) |
|
| 25 |
16.1 (q) |
16.0 (q) |
16.1 (q) |
16.1 (q) |
|
| 26 |
17.4 (q) |
17.3 (q) |
17.6 (q) |
17.6 (q) |
|
| 27 |
28.1 (q) |
28.0 (q) |
28.1 (q) |
28.1 (q) |
|
| 28 |
60.1 (t) |
60.6 (t) |
63.0 (t) |
63.7 (t) |
|
| 29 |
33.7 (q) |
29.7 (q) |
33.8 (q) |
33.8 (q) |
|
| 30 |
25.3 (q) |
20.3 (q) |
25.3 (q) |
25.4 (q) |
|
| |
Anth |
Anth |
Anth |
Bz |
Cin |
| 1′ |
168.8 (s) |
168.9 (s) |
169.4 (s) |
167.2 (s) |
166.6 (s) |
| 2′ |
112.7 (s) |
111.6 (s) |
111.7 (s) |
131.6 (s) |
118.9 (d) |
| 3′ |
152.2 (s) |
152.5 (s) |
152.9 (s) |
130.1 (d) |
144.5 (d) |
| 4′ |
111.9 (d) |
111.8 (d) |
111.9 (d) |
129.5 (d) |
134.4 (s) |
| 5′ |
135.1 (d) |
135.4 (d) |
135.5 (d) |
133.8 (d) |
128.3(d) |
| 6′ |
115.1 (d) |
115.2 (d) |
115.2 (d) |
129.5 (d) |
128.8 (d) |
| 7′ |
133.4 (d) |
133.3 (d) |
131.8 (d) |
130.1 (d) |
130.3 (d) |
| 8′ |
30.1 (q) |
30.0 (q) |
29.8 (q) |
|
128.8 (d) |
| 9′ |
|
|
|
|
128.3 (d) |
| |
Glucuronopyranosyl methyl ester |
Glucuronopyranosyl methyl ester |
Glucuronopyranosyl methyl ester |
Glucuronopyranosyl methyl ester |
Bz |
| 1′′ |
107.8 (d) |
107.8 (d) |
107.8 (d) |
107.8 (d) |
164.8 (s) |
| 2′′ |
75.9 (d) |
75.9 (d) |
75.9 (d) |
75.9 (d) |
130.2 (s) |
| 3′′ |
78.4 (d) |
78.4 (d) |
78.4 (d) |
78.4 (d) |
129.8 (d) |
| 4′′ |
73.7 (d) |
73.7 (d) |
73.7 (d) |
73.7 (d) |
128.6 (d) |
| 5′′ |
77.7 (d) |
77.7 (d) |
77.7 (d) |
77.7 (d) |
133.2 (d) |
| 6′′ |
171.4 (s) |
171.4 (s) |
171.4 (s) |
171.4 (s) |
128.6 (d) |
| 7′′ |
52.5 (q) |
52.5 (q) |
52.5 (q) |
52.5 (q) |
129.8 (d) |
| |
|
Ac |
|
|
|
| 1′′′ |
|
171.2 (s) |
|
|
|
| 2′′′ |
|
21.1 (q) |
|
|
|
 |
| | Fig. 1 Structures of compounds 1–11. | |
 |
| | Fig. 2 1H–1H COSY and key HMBC correlations and for compounds 1–4 and 9. | |
 |
| | Fig. 3 Key NOE correlations and for compounds 1–4 and 9. | |
Compound 2, a yellow amorphous powder with an [α]25D +2.4 (c 0.5, MeOH), was found to possess a molecular formula of C47H69NO13 based on its HRESIMS peak at m/z 856.4807 [M + H]+ (calcd for C47H70NO13, 856.4842), which indicated 13 degrees of unsaturation. The IR spectrum indicated the presence of NH stretch (3433 cm−1) and ester (1743 cm−1) groups. The UV chromatogram of 2 showed maxima absorption at 220 nm. The 1H and 13C NMR data (Tables 1 and 2) of 2 were very similar to those of 1. The only difference was the presence of signals for an acetyl group at C-21. This was substantiated by means of the observed COSY and HMBC (Fig. 2) correlations of H-21 (δH 5.89)/H-22 (δH 6.51), H-30 (δH 1.34) to C-21 (δC 76.9), H-21 (δH 5.89) to C-1′′′ (δC 171.2), and H-2′′′ (δH 1.89) to C-1′′′ (δC 171.2). Furthermore, the NOE cross-peaks (Fig. 3) were observed between H-21 (δH 5.89) and H-16 (δH 5.15), and between H-22 (δH 6.51) and H-18 (δH 3.16), confirming α-orientation of H-21 and β-orientation of H-22, respectively Thus, compound 2 was assigned as (3β,16β,21β,22α)-22-(N-methylanthraniloxy)-21-acetyl- 16,28-dihydroxyolean-12-en-3-yl-3-O-β-D glucuronopyranosyl methyl ester, and named tigensides B.
Compound 3 was obtained as a yellow amorphous powder with an [α]25D +40.9 (c 0.5, MeOH). The molecular formula C45H67NO11 was determined from the HRESIMS ion peak at m/z 798.4797 [M + H]+ (calcd for C45H68NO11, 798.4787), consistent with 12 degrees of unsaturation. The IR bands at 3390, 1681, and 1519 cm−1 were indicative of NH stretch, olefinic, and NH bend groups. The UV chromatogram of 3 indicated the presence of maxima absorption at 220 nm. The 1H and 13C NMR data (Tables 1 and 2) obtained for 3 were very similar to those of 1. The only observed difference was that the position of an anthranilate group at C-22 in 1 was exchanged to C-28 in 3. The HMBC correlations (Fig. 2) of H-16 (δH 5.17) to C-28 (δC 63.0), H-22 (δH 4.87) to C-17 (δC 44.9), H-28 (δH 5.35) to C-17 (δC 44.9), C-22 (δC 70.1) and C-1′ (δC 169.4), and H-30 (δH 1.17) to C-21 (δC 44.5) indicated that a hydroxyl group was attached to C-22, and an anthranilate group was connected to C-28, which was further confirmed by its 1H–1H COSY correlation (Fig. 2) of H-21 (δH 2.11)/H-22 (δH 4.86). In addition, the proton on H-22 was determined as β-orientation based on a NOESY correlation (Fig. 3) between H-22 (δH 4.86) and H-18 (δH 2.95). Thus, the structure of 3 was identified as (3β,16β,22α)-28-(N-methylanthraniloxy)-16,22-dihydroxyolean-12-en-3-yl-3-O-β-D glucuronopy -ranosyl methyl ester, and named tigensides C.
Compound 4 was obtained as a yellow amorphous powder with an [α]25D +4.0 (c 0.5, MeOH). The molecular formula C44H64O11 was determined from the HRESIMS ion peak at m/z 769.4540 [M + H]+ (calcd for C44H65O11, 769.4521), indicating 13 degrees of unsaturation. The IR bands at 3421, 1720, and 1458 cm−1 were indicative of the occurrence of hydroxy, ester and olefinic groups. Maxima absorption at 230 nm was observed in the UV chromatogram of 4. The 1H and 13C-NMR data (Tables 1 and 2) indicated that the structure of 4 was very similar to 3 except the absence of the signals of the methyl amine group at C-3′ of the aromatic ring. The NMR spectra illustrated typical signals of A2B2X system for one benzoyl moiety [δH 8.26 (dd, J = 7.8, 2.4 Hz, H-3′/H-7′), δC 130.1; δH 7.53 (t, J = 7.8 Hz, H-5′), δC 133.8; δH 7.45 (t, J = 7.8 Hz, H-4′/H-6′), δC 129.5]. Furthermore and importantly, the HMBC correlation (Fig. 2) form H-28 (δH 5.43) to C-1′ (δC 167.2) confirmed the position of the benzoyl group at C-28. Moreover, the configuration of H-22 was characterized as β-orientation based on a NOESY correlation (Fig. 3) between H-22 (δH 4.89) and H-18 (δH 2.96). Consequently, the structure of compound 4 was established as (3β,16β,22α)-28-O-benzoyl-16,22-dihydroxyo -lean-12-en-3-yl-3-O-β-D glucuronopyranosyl methyl ester, and named tigensides D.
Compound 9 was obtained as a yellow amorphous powder with an [α]25D +154.6 (c 0.5, MeOH). The molecular formula C37H44O9 was determined from the HRESIMS ion peak at m/z 633.3062 [M + H]+ (calcd for C37H45O9, 633.3075), consistent with 16 degrees of unsaturation. The IR bands at 3421, 1708 and 1635 cm−1 were indicative of the presence of hydroxyl, ester, and olefinic groups. The maxima absorption of 9 was at 220 in the UV chromatogram. The 1H NMR spectrum (Table 1) demonstrated signals for one olefinic proton δH 5.67 (1H, d, J = 4.7 Hz), three methyl groups [δH 1.64 (3H, s), 1.36 (3H, d, J = 6.2 Hz), and 1.14 (3H, s)], and four oxygenated methine protons [δH 3.60 (1H, m), 4.06 (1H, d, J = 5.2 Hz), 4.86 (1H, overlapped), and 4.91 (1H, d, J = 6.2 Hz)]. Furthermore, the presence of two olefinic carbons (δC 144.2 and 122.7), four oxygenated tertiary carbons (δC 75.6, 73.2, 71.8, and 68.3), and three oxygenated quaternary carbons (δC 87.7, 86.8, and 76.2) in its 13C NMR spectrum suggested the presence of 3,7,8,12,14,17,20-pentahydroxypregnane skeleton.37 In further support of the skeleton, the following key HMBC correlations (Fig. 2) of δH 2.38 (H-4) to 30.8 (C-2), 71.8 (C-3), 144.2 (C-5) and 37.8 (C-10), δH 1.14 (H-19) to 144.2 (C-5) and 37.8 (C-10), δH 4.06 (H-7) to δC 144.2 (C-5), 122.7 (C-6), 76.2 (C-8) and 38.8 (C-9), δH 4.86 (H-12) to δC 25.2 (C-11) and 58.0 (C-13), and δH 1.64 (H-18) to δC 58.0 (C-13) and 87.7 (C-14) were observed. The fragments H-1/H-2/H-3/H-4, H-6/H-7, H-9/H-11/H-12, and H-15/H-16 were observed in the 1H–1H COSY spectrum (Fig. 2). In addition, the 1H and 13C NMR data of 9 displayed signals for a benzoyl group [20-Bz: δH 7.92 (2H, dd, J = 7.2, 1.5 Hz, H3′′/H-7′′), 7.56 (1H, t, J = 7.2 Hz, H-5′′), and 7.36 (2H, m, H-4′′/6′′); δC 164.8, 133.2, 130.2, 129.8, and 128.6] and a cinnamoyl group [12-Cin: δH 7.45 (1H, d, J = 16.0 Hz, H-3′), 7.37 (1H, m, H-7′), 7.33 (2H, m, H-6′/8′), 7.28 (2H, d, J = 7.2 Hz, H-5′/9′), and 6.11 (1H, d, J = 16.0 Hz, H-2′); δC 166.6, 144.5, 134.4, 130.3, 128.8, 128.3, and 118.9]. The benzoyl and cinnamoyl groups were located at C-20 and C-12, respectively, by interpretation of the HMBC correlations of H-20 (δH 4.91) to C-1′′ (δC 164.8), and H-12 (δH 4.86) to C-1′ (δC 166.6). The NMR data of 9 were very similar to those of dibenzoylgagaimol isolated form Metaplexis japonica MAKINO.38 Thus, the structure of 9 was elucidated as 12-O-cinnamoyl-20-O-benzoyl gagaimol, and named tipregnane A.
The other isolated compounds (Fig. 1) were identified as 23-cis-p-coumaroyloxy-2α,3β-dihydroxyolean-12-en-28-oic acid (5),39 23-trans-p-coumaroyloxy-2α,3β-dihydroxyolean-12-en-28-oic acid (6),40 3β,23-dihydroxy-olean-12-en-28-oic acid 23-caffeate (7),41 tomentoid A (8),42 stephanoside B (10),43 tinctoroside B (11),44 by comparison of their NMR spectra with those of the known compounds.
Glucose uptake assay
Glucose uptake of the tissues and cells is an important link to maintain the stability of blood glucose concentration, and the increase in glucose uptake can improve insulin resistance of T2DM.
In order to test the glucose uptake activity of the isolated compounds, all compounds were selected to evaluate their glucose uptake activity in L6 cells. We treated insulin as the positive control group. Compared with the control group, Compounds 1–11 presented the effects of glucose uptake in L6 cells at 1.12- to 2.52-fold, respectively. Compounds 1, 3 and 6 exerted weak activity, and showed glucose uptake activity with the enhancement by 1.75-, 1.90-, and 1.72-fold at 30 μg mL−1 (Fig. 4). Compounds 4 and 7 possessed a moderate effect on promoting glucose uptake with the enhancement by 2.08- and 1.62-fold at 30 μg mL−1. In addition, compounds 5 and 8 were toxic to L6 cells. Compound 2 was the most active compound, and exhibited glucose uptake activity with 2.52-fold enhancement at 30 μg mL−1.
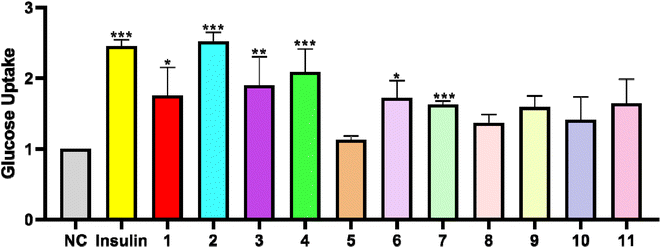 |
| | Fig. 4 Effects of compounds 1–11 (30 μg mL−1) on glucose uptake in L6 cells. (*P < 0.05, **P < 0.01, ***P < 0.001, compared with the non-treated group). | |
GLUT-4 translocation assay
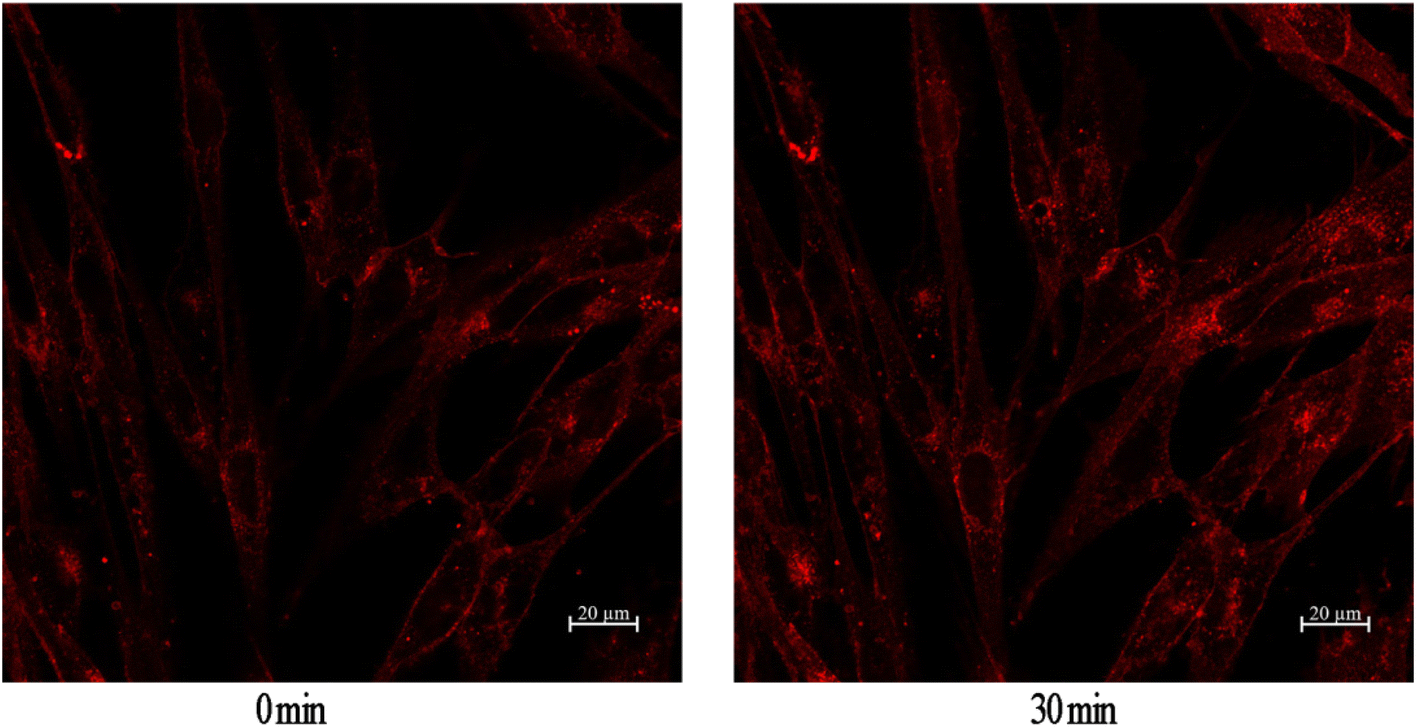
GLUT4 is a glucose transporter expressed primarily in adipose and muscle tissues, and is a key regulator of wholebody glucose homeostasis,45 as its reduced translocation from the intracellular pool to the plasma membranes is reportedly a possible cause of insulin resistance.46 Therefore there has been considerable interest in targeting GLUT4 for the treatment of T2DM. Based on this target, a cell-based GLUT4 translocation assay using stable L6 cells expressing IRAP-mOrange was established to identify isolated compounds that may have potential antidiabetic effects, by evaluating their effects on the translocation of GLUT4 to the plasma membrane. The confocal microscopy was used to detect IRAP-mOrange trafficking to characterize GLUT4 localization. IRAP, as insulin-regulated aminopeptidase, has been identified as the major protein of GLUT4 storage vesicles (GSVs), which shows strong co-localization with GLUT4 stored in GSVs and may indirectly reflect GLUT4 translocation. The effect of compound 2 on GLUT4 translocation was assessed in L6 cells which stably expressed IRAP-mOrange. As shown in Fig. 5, compound 2 treatment increased fluorescence intensity on L6 cell membranes. The fluorescence reached the greatest intensity (1.36-fold) at 30 minutes after addition of compound 2. Based on our established GLUT4 translocation assay system, we found that compound 2 displayed a medium effect in promoting GLUT4 translocation and stimulating glucose uptake in L6 cells, indicating that compound 2 possessed potential antidiabetic effects.
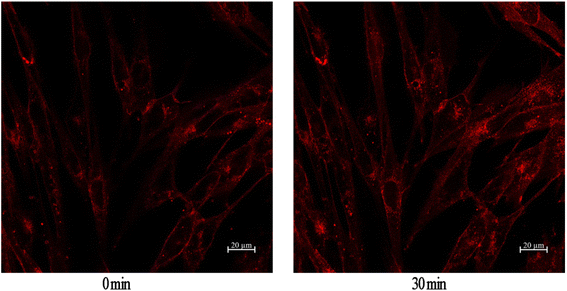 |
| | Fig. 5 Effect of compound 2 on GLUT4 translocation in L6 cells. | |
Conclusions
In summary, four new triterpenoid saponins (1–4) and one new C21 steroid (5) have been isolated from G. tingens, together with six known compounds (6–11) in this work. Compounds 1–11 presented the effects of glucose uptake in L6 cells at 1.12- to 2.52-fold, respectively. Specifically, compound 2 exhibited the most potent glucose uptake, with 2.52 – fold enhancement, which was even more potent than the positive control group of insulin. The fluorescence reached the greatest intensity (1.36-fold) at 30 minutes after addition of compound 2, showing that have a medium effect on the GLUT4 translocation activity in L6 cells in the further study. In addition, the C21 steroids show weak activity of glucose uptake. Therefore, triterpenoid saponins are thought to be the major antidiabetic compounds of G. tingens. The study indicates that the separation and characterization of these compounds plays an important role in the research and development of new anti-diabetic phytoconstituents, and the Gymnema tingens Spreng. has potential to become a natural resource for developing diabetes drugs.
Experimental section
General experimental procedures
Semi-preparative HPLC purification was performed on a Waters 2535 HPLC connected with a 2998 PDA Detector and a 2707 Autosampler (Waters, Milford, MA, USA). Separations were performed on a COSMOSIL C18 column (5 μm, 10 × 250 mm) (Nacalai Tesque, Kyoto, Japan), a COSMOSIL C8 column (5 μm, 10 × 250 mm) (Nacalai Tesque, Kyoto, Japan) and a YMC-pack diol column (5 μm, 10 × 50 mm; 5 μm, 20 × 150 mm) (Yamamura Chemical Research, Kyoto, Japan). Direct injection high resolution ESIMS and LC-DAD-ESIMS analyses were recorded on an ultra-performance liquid chromatography-quadrupole/electrostatic field orbitrap high resolution mass spectrometry (Thermo Fisher Scientific, Waltham, MA, USA). The NMR spectra were recorded on an AVANCE III 600 MHz spectrometer (Bruker BioSpin, Ettlingen, Germany). Optical rotations were recorded on an Autopol IV Automatic Polarimeter (Rudolph Research Analytical, Hackettstown, NJ, USA). Chromatography grade solvents were used for HPLC, and all other chemical reagents were analytical grade. Sephadex LH-20 dextran gel was purchased from Amersham Pharmacia Biotech Co. (Piscataway, NJ, USA).
Plant materials
The roots and stems of Gymnema tingens Spreng. (20191011a) were collected from Guizhou Province, China in October 2019. The roots and stems were identified by Professor Songji Wei of College of Pharmacy, Guangxi University of Chinese Medicine, Nanning, China.
Extraction and isolation
The roots and stems of G. tingens (18.2 kg) were exhaustively extracted with 80% aqueous EtOH (2 times × 200 L, each 3 h) at reflux, and the EtOH extract was concentrated to dryness under reduced pressure. The obtained residue (2521.4 g) was suspended in H2O and partitioned with petroleum ether (PE) (8 × 3.5 L), ethyl acetate (EtOAc) (10 × 3.5 L), and n-butyl alcohol (n-BuOH) (15 × 3.5 L) to afford 12.2 g of PE fraction, 292.2 g of EtOAc fraction, and 1104.5 g of n-BuOH fraction after evaporation, respectively. The EtOAc fraction (292.2 g) was separated on an HP-20 macroporous resin column eluted with EtOH–H2O (from 0 to 95%, v/v) to give seven fractions Fr.1–Fr.7. Fr.5 (85.1 g) was subjected to silica-gel chromatography column (CC), eluted with a gradient of CH2Cl2–CH3OH (from 200![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 to 1:1, v/v) to afford eight fractions Fr.5.1–Fr.5.8. Fr.5.4 (35.0 g) was separated into two fractions (Fr.5.4.1, Fr.5.4.2) over silica gel CC with CH2Cl2–CH3OH (from 200:1 to 50:1, v/v) as the eluent. Fr.5.4.2 (14.8 g) was subjected to an RP-18 column that was eluted with MeOH–H2O (from 40% to 100%, v/v) to obtain two subfractions Fr.5.4.2.1–Fr.5.4.2.2. Fr.5.4.2.2 (9.0 g) was chromatographed via a Sephadex LH-20 column with MeOH as solvent to give two subfractions Fr.5.4.2.2.1 and Fr.5.4.2.2.2. Compounds 10 (13.7 mg) and 11 (17.9 mg) were isolated from Fr.5.4.2.2.1 using semipreparative normal-phase HPLC [n-hexane-(n-hexaneiso:propyl alcohol, 7:3), 15:85 to 0:100, 9.0 mL min−1, 0 to 30 min] and semipreparative reversed-phase HPLC (MeCN–H2O, 55:45 to 85:15, 4.0 mL min−1, 0 to 50 min). Fr.5.5 (7.2 g) was separated by Sephadex LH-20 column (MeOH), and then semipreparative normal-phase HPLC [n-hexane-(n-hexaneiso:propyl alcohol, 7:3), 25:75 to 0:100, 9.0 mL min−1, 0 to 30 min] to yield seven subfractions Fr.5.5.2.1–Fr.5.5.2.7. Compounds 5 (3.0 mg) and 7 (7.0 mg) were isolated from Fr.5.5.2.3 by semipreparative reversed-phase HPLC (MeCN–H2O, 40:60 to 100:0, 4.0 mL min−1, 0 to 20 min). Fr.5.5.2.4 was purified by semipreparative reversed-phase HPLC (MeCN–H2O, 40:60 to 100:0, 4.0 mL min−1, 0 to 30 min) to give compounds 6 (76.9 mg) and 8 (10.0 mg). Compounds 1 (23.6 mg), 2 (1.8 mg), 3 (13.8 mg), 4 (26.3 mg), and 9 (7.0 mg) were isolated from Fr.5.5.2.7 by using semipreparative reversed-phase HPLC (MeCN–H2O, 45:55 to 80:20, 4.0 mL min−1, 0 to 30 min).
:1 to 1:1, v/v) to afford eight fractions Fr.5.1–Fr.5.8. Fr.5.4 (35.0 g) was separated into two fractions (Fr.5.4.1, Fr.5.4.2) over silica gel CC with CH2Cl2–CH3OH (from 200:1 to 50:1, v/v) as the eluent. Fr.5.4.2 (14.8 g) was subjected to an RP-18 column that was eluted with MeOH–H2O (from 40% to 100%, v/v) to obtain two subfractions Fr.5.4.2.1–Fr.5.4.2.2. Fr.5.4.2.2 (9.0 g) was chromatographed via a Sephadex LH-20 column with MeOH as solvent to give two subfractions Fr.5.4.2.2.1 and Fr.5.4.2.2.2. Compounds 10 (13.7 mg) and 11 (17.9 mg) were isolated from Fr.5.4.2.2.1 using semipreparative normal-phase HPLC [n-hexane-(n-hexaneiso:propyl alcohol, 7:3), 15:85 to 0:100, 9.0 mL min−1, 0 to 30 min] and semipreparative reversed-phase HPLC (MeCN–H2O, 55:45 to 85:15, 4.0 mL min−1, 0 to 50 min). Fr.5.5 (7.2 g) was separated by Sephadex LH-20 column (MeOH), and then semipreparative normal-phase HPLC [n-hexane-(n-hexaneiso:propyl alcohol, 7:3), 25:75 to 0:100, 9.0 mL min−1, 0 to 30 min] to yield seven subfractions Fr.5.5.2.1–Fr.5.5.2.7. Compounds 5 (3.0 mg) and 7 (7.0 mg) were isolated from Fr.5.5.2.3 by semipreparative reversed-phase HPLC (MeCN–H2O, 40:60 to 100:0, 4.0 mL min−1, 0 to 20 min). Fr.5.5.2.4 was purified by semipreparative reversed-phase HPLC (MeCN–H2O, 40:60 to 100:0, 4.0 mL min−1, 0 to 30 min) to give compounds 6 (76.9 mg) and 8 (10.0 mg). Compounds 1 (23.6 mg), 2 (1.8 mg), 3 (13.8 mg), 4 (26.3 mg), and 9 (7.0 mg) were isolated from Fr.5.5.2.7 by using semipreparative reversed-phase HPLC (MeCN–H2O, 45:55 to 80:20, 4.0 mL min−1, 0 to 30 min).
Compound 1. Yellow colour, amorphous powder; [α]25D −0.9 (c 0.5, MeOH); UV (MeOH) λmax (logε) 220 (1.14) nm; IR (KBr)vmax3390, 2924, 1743, 1681, 1608, 1577, 1519, 1458, 1242, 1172, 1087, 1053, 752, 702 cm−1; HR-ESI-MS (positive): m/z 798.4796 [M + H]+ (calcd for C45H68NO11, 798.4787); NMR data (pyridine-d5), see Tables 1 and 2.
Compound 2. Yellow colour, amorphous powder; [α]25D +2.4 (c 0.5, MeOH); UV (MeOH) λmax (logε) 220 (0.21) nm; IR (KBr)vmax3433, 2924, 2357, 1743, 1246, 1172, 1056, 752 cm−1; HR-ESI-MS (positive): m/z 856.4807 [M + H]+ (calcd for C47H70NO13, 856.4842); NMR data (pyridine-d5), see Tables 1 and 2.
Compound 3. Yellow colour, amorphous powder; [α]25D +40.9 (c 0.5, MeOH); UV (MeOH) λmax (logε) 220 (1.79) nm; IR (KBr)vmax3390, 2951, 2924, 1743, 1681, 1581, 1519, 1458, 1330, 1242, 1172, 1130, 1087, 1049, 748, 702 cm−1; HR-ESI-MS (positive): m/z 798.4797 [M + H]+ (calcd for C45H68NO11, 798.4787); NMR data (pyridine-d5), see Tables 1 and 2.
Compound 4. Yellow colour, amorphous powder; [α]25D + 4.0 (c 0.5, MeOH); UV (MeOH) λmax (logε) 230 (0.55) nm; IR (KBr)vmax3421, 2951, 1720, 1458, 1377, 1276, 1172, 1049, 713 cm−1; HR-ESI-MS (positive): m/z 769.4540 [M + H]+ (calcd for C44H65O11, 769.4521); NMR data (pyridine-d5), see Tables 1 and 2.
Compound 9. Yellow colour, amorphous powder; [α]25D +154.6 (c 0.5, MeOH); UV (MeOH) λmax (logε) 220 (1.87) nm; IR (KBr)vmax 3421, 2931, 1708, 1635, 1450, 1280, 1176, 1072, 987, 771, 713 cm−1; HR-ESI-MS (positive): m/z 633.3062 [M + H]+ (calcd for C37H45O9, 633.3075); NMR data (CDCl3), see Tables 1 and 2.
Glucose uptake and GLUT-4 translocation
Cell culture. L6 cells were added with the prepared whole medium after they were resuscitated and then cultured in a humidified incubator with 37 °C, ambient oxygen, and 5% CO2. When the cells grew to about 80%, they were replaced with a differentiation medium and continued to differentiate into myotubes at 37 °C and 5% CO2 for one week. The medium was changed every 2 days, and the medium was still a differentiation medium. The whole medium was MEM-α (Hyclone, USA) medium containing 10% fetal bovine serum (FBS, Hyclone, USA) and 1% antibiotics (100 U mL−1 penicillin and 100 μg mL−1 streptomycin), and the differentiation medium was MEM-α medium containing 2% fetal bovine serum and 1% antibiotics.47,48
Glucose uptake assay. We used a cell-based fluorescently-labeled deoxyglucose analog kit (2-NBDG kit, Cayman Chemical, USA) to evaluate whether compounds 1–11 increased glucose uptake in L6 cells.49,50 Before the experiment, the differentiated L6 myotube was inoculated into a 96-well plate at a density of 1 × 104–4 × 104 cells/well. After 2 h of starvation in serum-free α-MEM medium, we add 100 μL MEM-α medium, which contained different drugs, to each well, and cells were incubated for 12 h to 100% fusion. After overnight incubation, cells were treated with insulin (100 nM), compounds 1–11 (30 μg mL−1) or normal control (0.1% DMSO) in 100 μL glucose-free α-MEM containing 150 μg mL−1 2-NBDG. At the end of the treatment, plates were centrifuged for five minutes at 400 g at room temperature. The supernatant was aspirated, and 200 μL of cell-based assay buffer was added to each well. Plates were centrifuged for five minutes at 400 g at room temperature. Then the supernatant was aspirated, and 100 μL of cell-based assay buffer was added to each well. The 2-NBDG taken up by cells was detected with fluorescent filters (excitation/emission = 485/535 nm).
GLUT-4 translocation assay. Construction of myc-GLUT4-mOrange plasmid and cell line were according to the previous report.51,52 Myc-GLUT4-mOrange-L6 cells were cultured on glass coverslips for 12 h, and then L6 myoblasts are differentiated to L6 myotubes. Cells were starved in a PSS solution for 2 h. After starvation, mOrange fluorescence was detected by laser-scanning confocal microscopy at an excitation wavelength of 555 nm. Cells were treated with 10 μg mL−1 tested samples and images were taken every 5 min over a period of 30 min. Zen 2010 Software (Carl Zeiss, Jena, Germany) was used to analyze the fluorescence intensity of mOrange. The detailed method of GLUT4 fusion with the plasma membrane was described as previous reports.
Statistical analysis
The data were shown by means ± standard error of the mean (SEM). One-way analysis of variance (ANOVA) was used to analyse differences between groups, followed by Tukey's post hoc test using Graphpad prime software. A p value of < 0.05 was considered as the levels of statistical significance.
Conflicts of interest
The authors declare that they have no conflict of interest.
Acknowledgements
We are grateful to Prof. Song-Ji Wei of Guangxi University of Chinese medicine, for the identification of the plant. This work was supported by the National Natural Science Foundation of China (No. 81860750) and Inheritance and innovation team of Guangxi Traditional Chinese Medicine (2022B005).
Notes and references
- F. M. Ashcroft and P. Rorsman, Cell, 2012, 148, 1160–1171 CrossRef CAS PubMed.
- A. Ranjan, R. K. Singh, S. Khare, R. Tripathi, K. Pandey, A. K. Singh, V. Gautam, J. S. Tripathi and S. K. Singh, Sci. Rep., 2019, 9, 1–13 CrossRef CAS PubMed.
- Y. Li, Y. Liu, J. Liang, T. Wang, M. Sun and Z. Zhang, J. Agric. Food Chem., 2019, 67, 13051–13060 CrossRef CAS PubMed.
- C. Bommer, V. Sagalova, E. Heesemann, J. M. Goehler, R. Atun, T. Bärnighausen, J. Davies and S. Vollmer, Diabetes care, 2018, 41, 963–970 CrossRef PubMed.
- O. O. Elekofehinti, Pathophysiology, 2015, 22, 95–103 CrossRef CAS PubMed.
- M. Y. Liu, T. X. Zhou, J. Y. Zhang, G. F. Liao, R. M. Lu and X. Z. Yang, Molecules, 2021, 26, 6549 CrossRef CAS PubMed.
- Y. Jiang, B. T. Li, Flora of China, Science Press, Beijing, 1977, vol 63, p. 415 Search PubMed.
- G. D. Fabio, V. Romanucci, A. D. Marco and A. Zarrelli, Molecules, 2014, 19, 10956–10981 CrossRef PubMed.
- N. P. Sahu, S. B. Mahato, S. K. Sarkar and G. Poddar, Phytochemistry, 1996, 41, 1181–1185 CrossRef CAS PubMed.
- K. Yoshikawa, K. Matsuchika, K. Takahashi, M. Tanaka, S. Arihara, H. C. Chang and J. D. Wang, Chem. Pharm. Bull., 1999, 47, 798–804 CrossRef CAS PubMed.
- X. Liu, W. C. Ye, B. Yu, S. X. Zhao, H. M. Wu and C. T. Che, Carbohydr. Res., 2004, 339, 891–895 CrossRef CAS PubMed.
- K. Kamei, R. Takano, A. Miyasaka, T. Imoto and S. Hara, J. Biochem., 1992, 111, 109–112 CrossRef CAS PubMed.
- X. Y. Wu, G. H. Mao, Q. Y. Fan, T. Zhao, J. L. Zhao, F. Li and L. Q. Yang, Food Res. J., 2012, 48, 935–939 CrossRef CAS.
- S. Vats and R. Kamal, Pak. J. Biol. Sci., 2013, 16, 1771–1775 CrossRef PubMed.
- P. Nagella, I. M. Chung and H. N. Murthy, Methods Mol. Biol., 2011, 11, 537–540 CAS.
- P. V. Diwan, I. Margaret and S. Ramakrishna, Inflammopharmacology, 1995, 3, 271–277 CrossRef.
- S. N. Sinha, G. C. Saha and M. Biswas, Adv. Biores., 2010, 125, 28 Search PubMed.
- J. K. Malik, F. V. Manvi, B. K. Nanjwade and D. K. Dwivedi, Der Pharm. Lett., 2010, 2, 336–341 CAS.
- P. Daisy, J. Eliza and K. A. M. M. Farook, J. Ethnopharmacol., 2019, 126, 339–344 CrossRef PubMed.
- K. M. Ramkuma, C. Manjula, B. Elango, K. Krishnamurthi, S. S. Devi and P. Rajaguru, Cell Proliferation, 2013, 46, 263–271 CrossRef PubMed.
- S. Kumar, P. Gupta, S. Sharma and D. Kumar, Chin. J. Integr. Med., 2011, 9, 117–128 CrossRef PubMed.
- S. S. Gupta, C. B. Seth and M. C. Variyar, J. Med. Res., 1962, 50, 73–81 CAS.
- B. Anupam and C. H. Malay, Phytother. Res., 1994, 8, 118–120 CrossRef.
- M. Yoshikawa, T. Murakami, M. Kadoya, Y. H. Li, N. Murakami, J. Yamahara and H. Matsuda, Chem. Pharm. Bull., 1997, 45, 1671–1676 CrossRef CAS PubMed.
- K. Shimizu, A. Iino, J. Nakajima, K. Tanaka, S. Nakajyo, N. Urakawa, M. Atsuchi, T. Wada and C. Yamashita, J. Vet. Med. Sci., 1997, 59, 245–251 CrossRef CAS PubMed.
- S. Yoshioka, T. Imoto, M. Miyoshi, T. Kasagi, R. Kawahara and Y. Hiji, WakanIyaku Zasshi, 1996, 13, 300–303 CAS.
- M. Yoshikawa, T. Murakami and H. Matsuda, Chem. Pharm. Bull., 1997, 45, 2034–2038 CrossRef CAS PubMed.
- H. J. H. Kahksha, O. Alam, S. Naaz, V. Sharma, A. Manaithiya, J. Khan and A. Alam, J. Ethnopharmacol., 2020, 286, 114908 CrossRef PubMed.
- Y. Jiang and B. T. Li, Flora of China, Science Press, Beijing, 1977, vol 63, p. 422 Search PubMed.
- D. P. Khare, A. Khare and M. P. Khare, Carbohydr. Res., 1980, 81, 285–294 CrossRef CAS.
- J. C. He, M. Y. Wang and Z. C. Dong, Nat. Prod. Res. Dev., 2013, 25, 1673–1675 CAS.
- J. Tian, Studies on Chemical Constituents of Gymnema tingens, Peking Union Medical College, 2013 Search PubMed.
- Y. Huang, J. Hao, D. Tian, Y. Z. Wen, P. Zhao, H. Chen, Y. B. Lv and X. Z. Yang, Front. Pharmacol., 2018, 9, 760 CrossRef PubMed.
- J. L. Li, J. Zhou, Z. H. Chen, S. Y. Guo, C. Q. Li and W. M. Zhao, J. Nat. Prod., 2015, 78, 1548–1555 CrossRef CAS PubMed.
- J. P. An, E. J. Park, B. Ryu, B. W. Lee, H. M. Cho, T. P. Doan, T. T. Pham and W. K. Oh, J. Nat. Prod., 2020, 83, 1265–1274 CrossRef CAS PubMed.
- G. L. Jin, C. J. Zheng, W. B. Xin, Z. J. Mao, P. X. Sun, Z. X. Zeng and L. P. Qin, Arch. Pharmacal Res., 2011, 34, 869–873 CrossRef CAS PubMed.
- M. A. Raees, H. Hussain, A. A. Rawahi, R. Csuk, A. A. Ghafri, N. U. Rehman, A. Elyassi, I. R. Green, T. Mahmood and A. A. Harrasi, Phytochem. Lett., 2016, 16, 230–235 CrossRef CAS.
- T. Nomura, T. Fukai and T. Kuramochi, Planta Med., 1981, 41, 206–207 CrossRef CAS PubMed.
- J. Xiong, Y. Huang, Y. Tang, M. You and J. F. Hu, Chinese J. Org. Chem., 2013, 33, 1304–1308 CrossRef CAS.
- J. Q. Gu, E. J. Park, L. Luyengi, M. E. Hawthorne, R. G. Mehta, N. R. Farnsworth, J. M. Pezzuto and A. D. Kinghorn, Phytochemistry, 2001, 58, 121–127 CrossRef CAS PubMed.
- H. Pan, L. N. Lundgren and R. Andersson, Phytochemistry, 1994, 37, 795–799 CrossRef CAS.
- Y. B. Zhang, W. Li, Z. M. Zhang, N. H. Chen, X. Q. Zhang, J. W. Jiang, G. C. Wang and Y. L. Li, Chem. Lett., 2016, 45, 368–370 CrossRef CAS.
- K. Yoshikawa, N. Okada, Y. Kann and S. Arihara, Chem. Pharm. Bull., 1996, 44, 1790–1796 CrossRef CAS.
- Z. L. Gao, H. P. He, Y. T. Di, X. Fang, C. S. Li, Q. Zhang, P. J. Zhao, S. L. Li and X. J. Hao, Helv. Chim. Acta, 2009, 92, 1775–1781 CrossRef CAS.
- S. Yonemitsu, H. Nishimura, M. Shintani, R. Inoue, Y. Yamamoto, H. Masuzaki, Y. Ogawa, K. Hosoda, G. Inoue, T. Hayashi and K. Nakao, Diabetes, 2001, 50, 1093–1101 CrossRef CAS PubMed.
- D. Leto and A. R. Saltiel, Nat. Rev. Mol. Cell Biol., 2012, 13, 383–396 CrossRef CAS PubMed.
- R. J. Koenig and R. J. Smith, J. Clin. Invest., 1985, 76, 878–881 CrossRef CAS PubMed.
- Q. Zhou, X. Yang, M. Xiong, X. Xu, L. Zhen, W. Chen, Y. Wang, J. Shen, P. Zhao and Q. H. Liu, Cell. Physiol. Biochem., 2016, 38, 2030–2040 CrossRef CAS PubMed.
- Y. Huang, T. Zhou, Y. Zhang, H. Huang, Y. Ma, C. Wu, C. Wang, Q. Lin, X. Yang and K. Pang, J. Funct. Foods, 2021, 77, 104338 CrossRef CAS.
- G. Song, Y. Huang, M. Xiong, Z. Yang, Q. Liu, J. Shen, P. Zhao and X. Yang, Front. Pharmacol., 2021, 11, 561956 CrossRef PubMed.
- Y. Lv, P. Zhao, K. Pang, Y. Ma, H. Huang, T. Zhou and X. Yang, J. Ethnopharmacol., 2021, 268, 113654 CrossRef CAS PubMed.
- Y. Ma, T. Zhou, P. Zhao, H. Y. Choi, J. Hao, H. Huang, C. Wu, X. Yang and K. Pang, Bioorg. Chem., 2021, 106, 104500 CrossRef CAS PubMed.
|
| This journal is © The Royal Society of Chemistry 2023 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence *ab
*ab