
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceInconsistent hydrogen bond-mediated vibrational coupling of amide I†
Suranjana Chakrabarty and
Anup Ghosh *
*
a, Department of Condensed Matter of Physics and Materials Sciences, S. N. Bose National Centre for Basic Sciences, JD Block, Sector-III, Salt Lake City, Kolkata – 700 106, India. E-mail: anupg86@gmail.com; anup.ghosh@bose.res.in
First published on 5th January 2023
Abstract
Using infrared spectroscopy and density functional theory (DFT) calculations, we scrutinized an amide (dimethylformamide) as a “model” compound to interpret the interactions of amide 1 with different phenol derivatives (para-chlorophenol (PCP) and para-cresol (CP)) as “model guest molecules”. We established the involvement of amide I in vibrational coupling with symmetric and asymmetric C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C modes of different phenolic derivatives and how their coupling was dependent upon different guest aromatic phenolic compounds. Interestingly, substitution of phenol perturbed the pattern of vibrational coupling with amide I. The symmetric and asymmetric CC modes of PC were coupled significantly with amide 1. For PCP, the symmetric CC mode coupled significantly, but the asymmetric mode coupled negligibly, with amide I. Here, we reveal the nature of vibrational coupling based on the structure of a guest molecule hydrogen-bonded with amide I. Our conclusions could be valuable for depiction of the unusual dynamics of coupled amide-I modes as well as the dependency of vibrational coupling on altered factors.
C modes of different phenolic derivatives and how their coupling was dependent upon different guest aromatic phenolic compounds. Interestingly, substitution of phenol perturbed the pattern of vibrational coupling with amide I. The symmetric and asymmetric CC modes of PC were coupled significantly with amide 1. For PCP, the symmetric CC mode coupled significantly, but the asymmetric mode coupled negligibly, with amide I. Here, we reveal the nature of vibrational coupling based on the structure of a guest molecule hydrogen-bonded with amide I. Our conclusions could be valuable for depiction of the unusual dynamics of coupled amide-I modes as well as the dependency of vibrational coupling on altered factors.
Introduction
Hydrogen bonds are pervasive in protein molecules, and are involved in biological processes such as molecular association, catalysis, and signal transmission.1–15 How biologically active, small organic molecules interact with other molecules to elicit different biological effects is an important research area.16–20 However, identifying the vibrational modes of biologically active molecules (e.g., proteins) is difficult. To overcome such difficulties, the structural and environmental properties of biomolecules have been investigated using “vibrational probes”.21–26Many biomolecules contain “amide I”, “amide II”, “amide III”, and “amide A” modes of vibration. However, the amide-I mode is studied widely as a vibrational probe.27,28 The amide bond is present in many organic molecules and biomolecules.29–32 Most importantly, as an infrared (IR) probe, amide I is employed extensively because of its sensitivity to the native electric field, solvation, and large molar extinction coefficient.33–38 In particular, vibrational spectroscopic measurements of the amide-I band are used to monitor shifts in the transition frequency, which is sensitive to the local electric fields as well as interactions with specific “guest molecules”.24–27 Many studies have focused on the relationship between vibrational couplings and conformational dynamics of proteins/peptides.34–41 Various theoretical methodologies and multidimensional IR-spectroscopy methods have been employed to investigate the vibrational coupling and structural details of biological systems.42–49 Vibrational coupling and the interactions between different vibrational modes have been investigated.50 The vibrational coupling between hydrogen bonds associated with amide-A and amide-I/II modes within the same amide component for several dipeptides has been studied using two-dimensional IR spectroscopy.51 The hydrogen bonding between amide I and phenol derivatives, dimethylformamide (DMF), and dimethyl acetamide has been considered.52–54
Investigation of the amide-I vibrational mode is very complex because it is delocalized in biomolecules. However, to study the molecular perceptions and sensitivity of the amide-I mode in the presence of intermolecular hydrogen bonds, we used DMF as a “model” molecule. We measured the IR absorbance of the CC mode involved in vibrational coupling during intermolecular hydrogen bonding with amide I. Correlations between the hydrogen bond-induced vibrational coupling of CC and CO transitions with different factors were investigated by employing linear IR spectroscopy. We revealed how pervasive formation of hydrogen bonds in the presence of phenolic compounds (hydrogen-bond contributors) could disturb the amide-I transition and symmetric/asymmetric CC transition of guest molecules. Hydrogen-bond formation as well as the dependency of vibrational coupling upon different orientations between coupled modes were also investigated in our work.
We employed linear IR spectroscopy and density functional theory (DFT) calculations as theoretical approaches. The frequency gap between symmetric and asymmetric CC stretching of phenol derivatives and the CO vibrational mode of DMF, as well as the enhancement factor in IR absorption during vibrational coupling, were monitored in the presence of different donor molecules. Vibrational coupling in biomolecules is important to understand the many biological interactions and processes at the microscopic level, so the coupling of amide I must be investigated. Overall, this structural evidence of vibrational coupling can be used to elucidate many biological and chemical effects.
Experimental section
Para-chlorophenol (PCP; 99.9% purity), para-cresol (PC; 99.9% purity), and DMF (99.9% purity) were purchased from MilliporeSigma and used without additional purification. The chemical structure of PCP and PC are drawn in Scheme 1. A solution of DMF (0.1 M) in carbon tetrachloride (CCl4) was used for linear IR spectroscopy. The sample was placed in a homemade Fourier transform infrared (FTIR) sample cell with CaF2 windows and a Teflon™ spacer (60 μm). Linear IR absorption spectroscopy was undertaken using an FTIR spectrometer (JASCO-FTIR-6300). The background of the solvent (CCl4) was measured and subtracted from all spectra of interactions between DMF and phenol derivatives. The Beer–Lambert law was validated by plotting the area of IR absorbance for the CC mode vs. concentration (Fig. S1, ESI†).
 | ||
| Scheme 1 Chemical structure of the phenol derivatives para-cresol (PC) and para-chlorophenol (PCP). | ||
Theoretical section
We wished to gain detailed knowledge about the IR absorption spectra of the CO mode in DMF and CC mode of different phenol substitutions, so we undertook DFT calculations employing Gaussian 09. In a preliminary manner, all the initial geometries of DMF and different phenolic complexes were optimized by the B3LYP/6-311G+ (D, P) level of theory. Then, calculations to determine the frequency of IR absorption were done for all DMF–phenol hydrogen-bonded complexes.
Results and discussion
A series of linear IR spectra of DMF solution (0.1 M) in CCl4 were taken with increasing concentrations of PC and PCP from 0 M to 0.05 M (Fig. 1). Preliminarily, the IR absorption frequency of the CO mode was shown to be 1686 cm−1 (black single peak) in the absence of phenolic compounds (0.00 M). With gradual addition of PC or PCP, the IR absorbance of amide 1 decreased progressively and the frequency shifted towards a lower-wavenumber region (Fig. 1a and b, respectively). Fig. 1 reveals that increasing the concentration of PC and PCP led to hydrogen-bond formation of CO and gradual shifting of the peak position of IR absorption. However, in PC (0.1 M) and PCP (0.1 M), with a gradual increase in the DMF concentration, the IR absorption spectra for symmetric and asymmetric CC modes showed anomalous behaviours (Fig. 2). For PC, the IR absorption peaks for symmetric and asymmetric CC modes were at 1597.5 cm−1 and 1618.5 cm−1 (Fig. 2a) whereas, for PCP they were at 1594.0 cm−1 and 1606.5 cm−1, respectively (Fig. 2b). For PC and PCP, with an increasing concentration of DMF (0.000–0.10 M), a significant difference in IR absorbance was observed between symmetric and asymmetric CC modes (Fig. 2). For PC, with a gradual increase in the DMF concentration from 0.000 M to 0.10 M, IR absorbance for symmetric and asymmetric CC modes was enhanced significantly. However, in contrast with PCP, though IR absorbance for symmetric CC stretching was enhanced, IR absorbance for asymmetric CC stretching was altered negligibly throughout the experiment (Fig. 2). The IR-absorbance enhancement ratio for the symmetric and asymmetric CC modes of PC was 1.35 and 1.40 whereas, for PCP, it was 5.97 and 1.00, respectively (Table 1), as calculated from Fig. 2. The transition dipole moment of the symmetric and asymmetric modes of PC and PCP changed accordingly (Table 1). These unusual phenomena focused our attention on the intermolecular interactions between amide (CO) and phenolic compounds (PC and PCP).
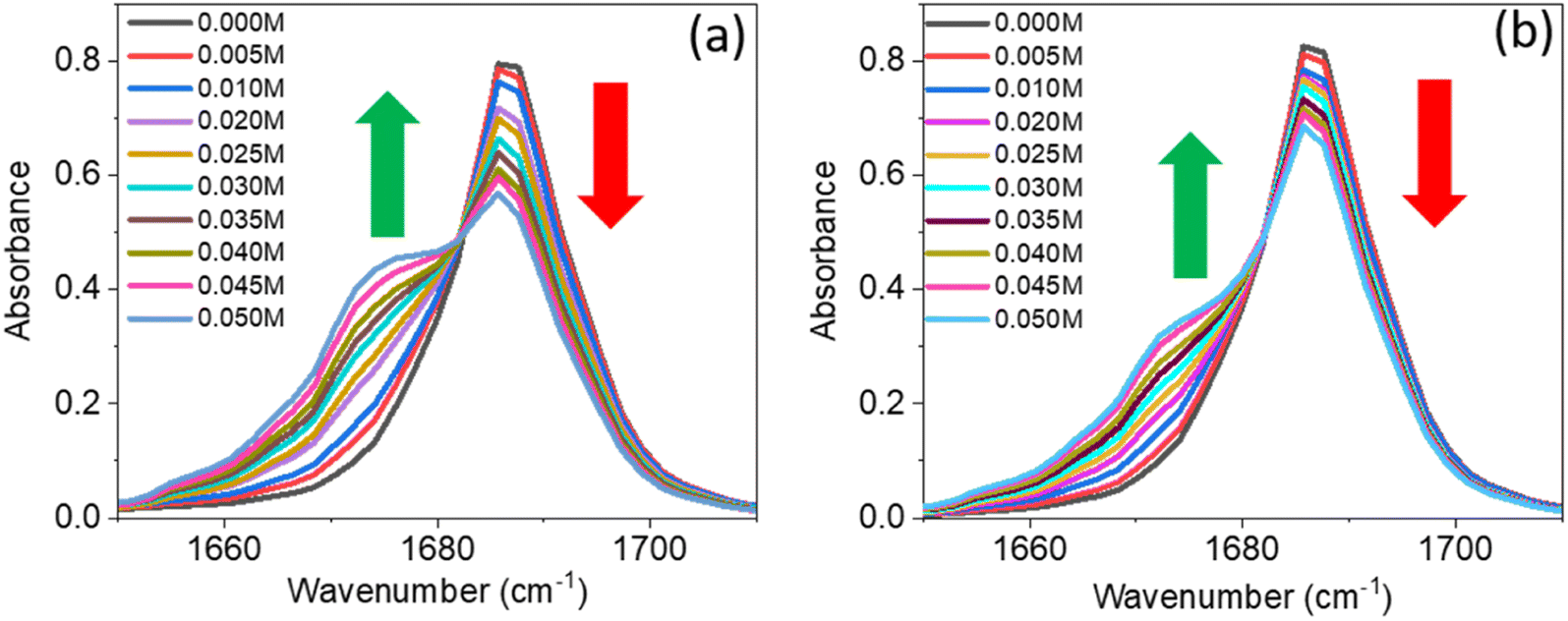 | ||
| Fig. 1 Linear IR spectra of amide I (dimethylformamide) in the presence of different concentrations of (a) para-cresol and (b) para-chlorophenol. The colour of the spectra is described in the inset with different concentrations of para-cresol and para-chlorophenol. The green (up) and red arrow (down) represent enhancement and suppression of absorbance, respectively. | ||
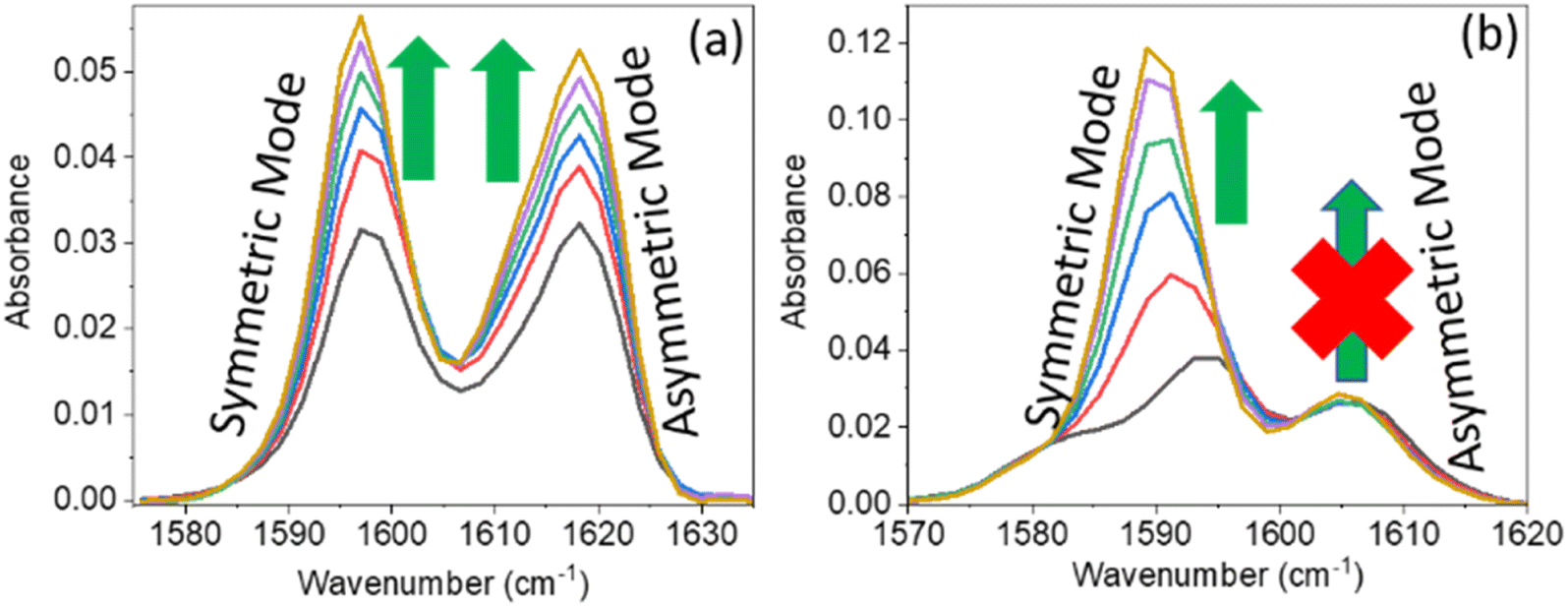 | ||
| Fig. 2 Linear IR absorption of the CC mode of (a) para-cresol (0.1 M) and (b) para-chlorophenol (0.1 M) in the presence of dimethylformamide at 0.00 M (black), 0.02 M (red), 0.04 M (blue), 0.06 M (green), 0.08 M (violet), and 0.10 M (dark-yellow). The green arrow represents enhancement of absorbance. The red cross represents no enhancement of absorbance. | ||
C mode and the ratio of IR absorption area for the hydrogen-bonded CC mode and free CC mode of phenol derivatives. The enhancement factor and transition dipole moment ratio were calculated from the experimental data shown in Fig. 2
| Phenol derivatives | Stretching mode | Frequency (cm−1) | IR enhancement ratio (R) | Transition dipole moment ratio |
|---|---|---|---|---|
| PC | Symmetric | 1597.5 | 1.35 | 1.16 |
| Asymmetric | 1618.5 | 1.44 | 1.20 | |
| PCP | Symmetric | 1694.0 | 5.97 | 2.44 |
| Asymmetric | 1606.5 | 1 | 1 |
The increase in IR absorbance of the CC mode could have been due to the altered hydrogen bonding with vibrational modes or n–π* bonding of PC and the PCP ring with the CO mode of DMF (Fig. 2a and b). To elucidate the precise reason underpinning the enhancement, we undertook IR spectroscopy of a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 DMF:anisole mixture. The unaltered enhancement of IR absorbance for the CC mode of the DMF:anisole (1:1) mixture signified no interaction of the non-bonded electron of the oxygen atom of DMF with the π electron cloud of anisole. We did not know whether the enhancement was due to the altered electron density of the phenolic ring (CC mode) after hydrogen bonding with DMF, so we undertook IR spectroscopy of a mixture of acetonitrile (ACN) and phenol at a ratio of 1:1 (Fig. 3b). The shifting to a higher IR absorbance frequency of CN signified formation of a hydrogen bond between ACN with phenolic OH (Fig. 3b). An absence of enrichment of IR absorbance of CC explained the non-involvement of hydrogen bonds. Hence, these results suggested that the enhancement in IR absorbance of the CC mode was not due to n–π* bonding or hydrogen bonding. Therefore, the enhancement was probably due to vibrational coupling between the CC and CO modes of amide (DMF) and phenol derivatives (PC and PCP).
:1 DMF:anisole mixture. The unaltered enhancement of IR absorbance for the CC mode of the DMF:anisole (1:1) mixture signified no interaction of the non-bonded electron of the oxygen atom of DMF with the π electron cloud of anisole. We did not know whether the enhancement was due to the altered electron density of the phenolic ring (CC mode) after hydrogen bonding with DMF, so we undertook IR spectroscopy of a mixture of acetonitrile (ACN) and phenol at a ratio of 1:1 (Fig. 3b). The shifting to a higher IR absorbance frequency of CN signified formation of a hydrogen bond between ACN with phenolic OH (Fig. 3b). An absence of enrichment of IR absorbance of CC explained the non-involvement of hydrogen bonds. Hence, these results suggested that the enhancement in IR absorbance of the CC mode was not due to n–π* bonding or hydrogen bonding. Therefore, the enhancement was probably due to vibrational coupling between the CC and CO modes of amide (DMF) and phenol derivatives (PC and PCP).
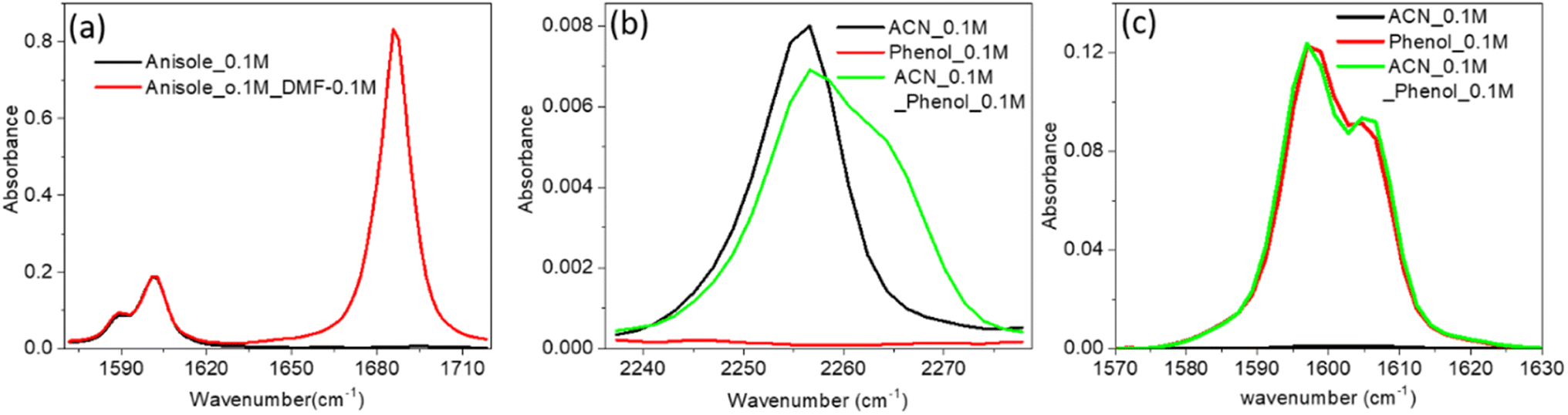 | ||
| Fig. 3 (a) Linear IR absorbance spectra of the CC mode of anisole (0.1 M) in the absence (black) and presence (red) of dimethylformamide (0.1 M). (b) IR spectra of the CN mode of ACN (0.1 M) in the absence (black) and presence (green) of phenol (0.1 M) in CCl4. (c) IR spectra of the CC mode of phenol (0.1 M) in the absence (black) and presence (red) of ACN (0.1 M). | ||
To check that our hypotheses on vibrational coupling were robust, we carried out DFT calculations (b3lyp, 6311 G (d, p)) for PC and PCP hydrogen-bonded with DMF, and the videos are provided in ESI† (AV1, AV2, AV3, AV4). Vibrational couplings were visualized between the CC of phenol derivatives and CO of DMF. Interestingly, our experimental observations aligned with the videos for DFT calculations. In AV1 and AV2, the symmetric and asymmetric CC modes of PC were coupled significantly with amide 1 of DMF; for PCP, the symmetric CC mode was coupled significantly (AV3) but the asymmetric mode was not (AV4). PC and PCP are phenol derivatives and form hydrogen bonds with amide 1 of DMF, but they showed different vibrational coupling. Hence, phenolic substitution altered the pattern of vibrational coupling.
Vibrational coupling is dependent upon the frequency gap of IR absorption, the distance between two vibration modes, and orientation. The frequency gap between the asymmetric CC mode of PC and CO stretching mode of DMF was smaller (56 cm−1) than that of the symmetric CC and CO (76 cm−1) modes of PC (Fig. 4a). However, the intensity of asymmetric and symmetric CC modes was enhanced simultaneously. The frequency gap was smaller for the asymmetric CC mode (67 cm−1) than symmetric CC mode (79.5 cm−1) of PCP with amide 1 of DMF. Significant enhancement in IR absorbance was observed for the symmetric CC mode, but negligible enhancement was observed for the asymmetric CC mode (Fig. 4b). The distance between the CC and CO modes of phenol derivatives and DMF according to DFT calculations are shown in Fig. 4c and d. An identical distance (5.2 Å) between CC and CO could not explain the disparity in vibrational coupling. Hence, we assumed that a different transition dipole angle between the CC mode and amide-I mode was the cause of this difference in vibrational coupling. To ascertain the reason for this anomalous coupling behaviour, we exposed different orientations between vibration modes (CC and CO) (Fig. 5). The sign of the coupling constant (βij) was dependent upon the geometry of the hydrogen-bonded DMF and phenol derivatives (eqn (1)).55 If CC and CO modes are parallel (Fig. 5a) and βij is positive, the intensity of the CO vibrational mode will be enhanced, with a transition dipole moment μi + μj. In contrast, the intensity of the CC vibrational mode will be weaker, with a transition dipole moment μi − μj. In the case of antiparallel CC and CO modes (Fig. 5b) and negative coupling constant βij, the CC mode will be weaker and correspondingly the CO mode will be stronger, with a transition dipole moment of μi + μj and μi − μj, respectively. In the head-to-tail (Fig. 5c) or head-to-head orientation (Fig. 5d) of CC and CO, the lower frequency mode of CC will carry more oscillator strength compared with the CO mode. An unperturbed intensity will result if the CC mode and CO mode are perpendicular to each other (Fig. 5e). The geometries of the DMF–PC complex and DMF–PCP complex are in-between the limits we modelled in Fig. 5. The symmetric and asymmetric CC modes of PC and symmetric CC mode of PCP are coupled in the manner shown in Fig. 5c and d. Probably, the asymmetric CC mode of PCP was coupled with the CO mode of DMF as shown in Fig. 5e or they were coupled very weakly according to the model shown in Fig. 5a–d.
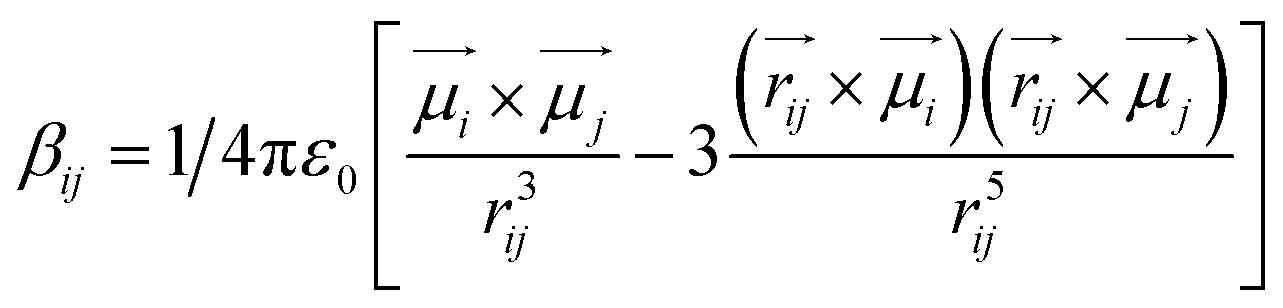 | (1) |
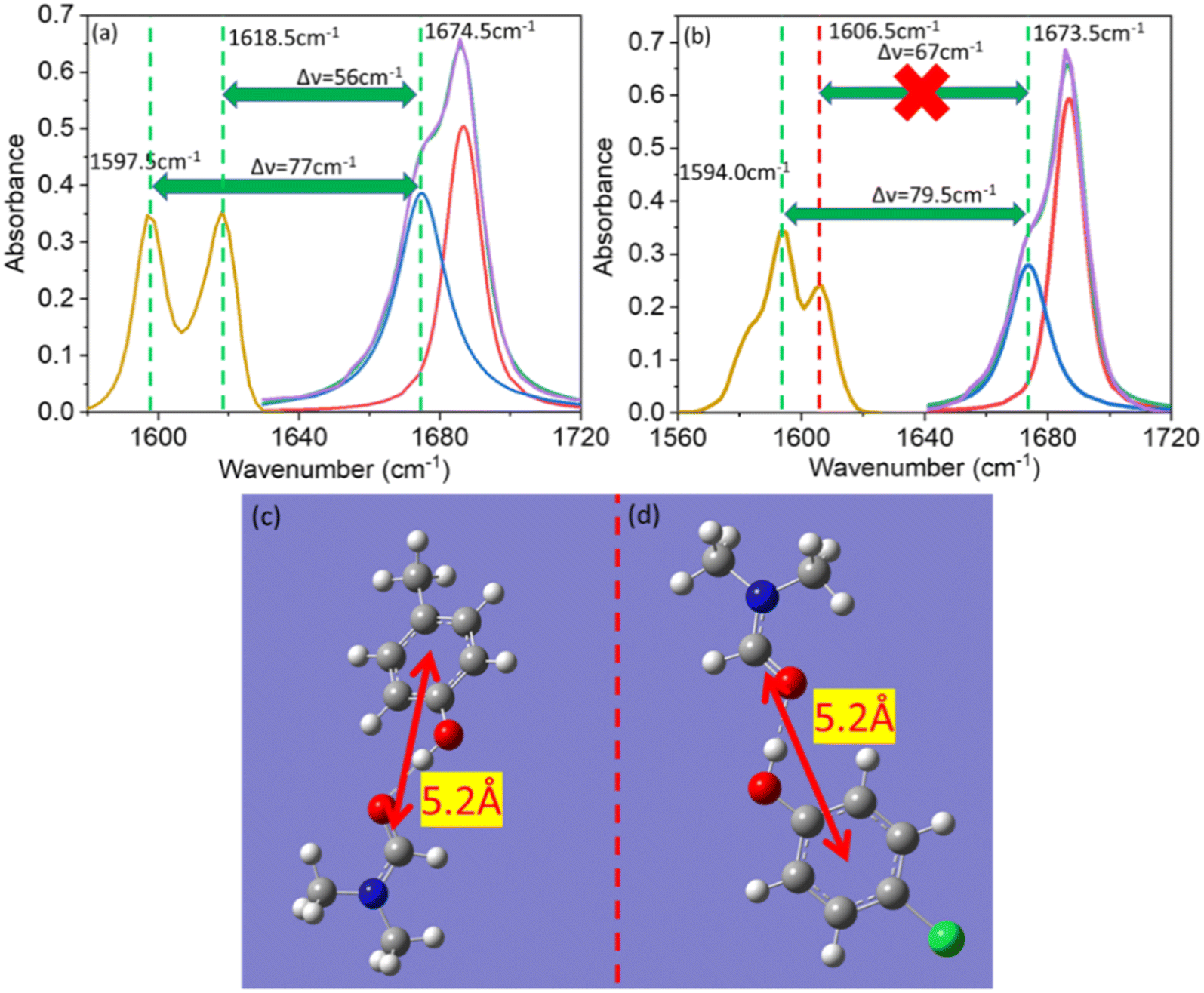 | ||
| Fig. 4 Linear IR spectra of amide I (dimethylformamide) and the CC mode of (a) para-cresol and (b) para-chlorophenol. The IR spectra of amide I (violet), free amide I (red), hydrogen-bonded amide I (blue), cumulative spectra (green), and CC (gold) are shown. (c) Distance between CO and CC of dimethylformamide and para-cresol. (d) Distance between CO and CC of dimethylformamide and para-chlorophenol. A distance of 5.2 Å was calculated (by DFT) for para-cresol and para-chlorophenol, respectively, with amide I of dimethylformamide. | ||
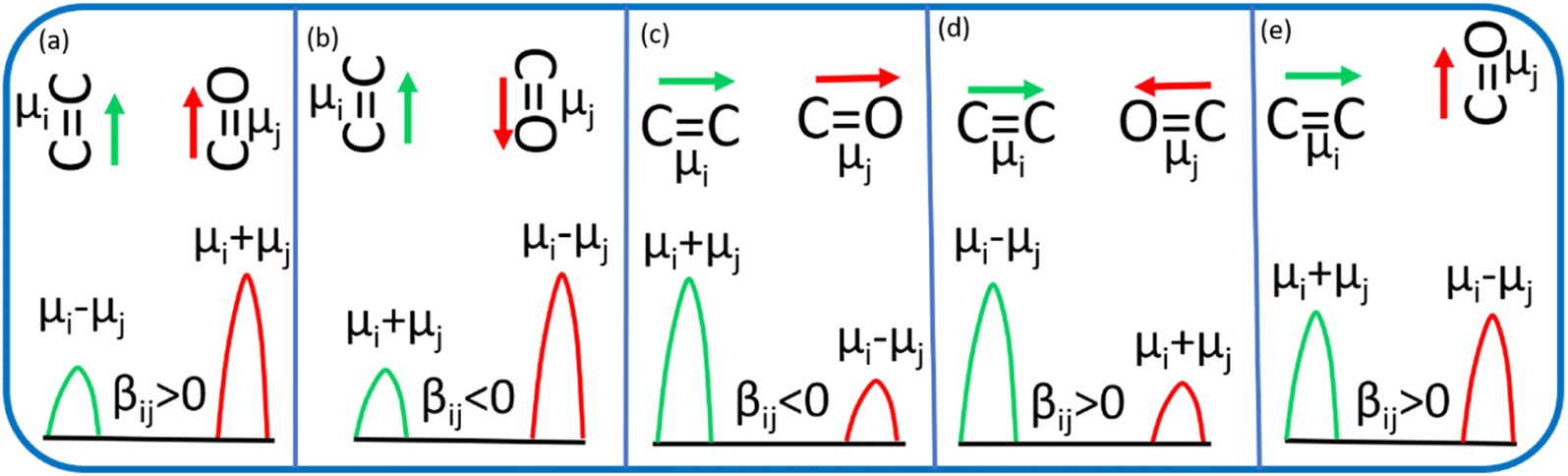 | ||
| Fig. 5 Red and green denote the linear vibration modes of CO and CC, respectively. βij is the coupling constant, and μi, and μj are the transition dipole moments of the CC mode and CO mode, respectively. Orientation of CO and CC modes- (a) parallel (b) antiparallel (c) linearly parallel (d) oppositely parallel and (e) perpendicular. | ||
We wished to validate our hypotheses of coupling of the symmetric and asymmetric CC modes of PC and PCP with amide 1. Hence, we carried out DFT calculations of para-ethylphenol and para-nitrophenol. Surprisingly, we observed the same results as PC for para-ethylphenol (AV5, AV6) and as PCP for para-nitrophenol (AV7, AV8). We calculated the vibrational coupling for meta-chlorophenol and ortho-chlorophenol complexes with DMF. The symmetric and asymmetric CC modes of meta-chlorophenol were coupled with amide I (AV9, AV10). The symmetric CC mode of ortho-chlorophenol was coupled with amide I, but the asymmetric CC mode was not (AV11, AV12). Hence, the CC symmetric and asymmetric modes coupled with amide 1 for para-electron-promoting phenolic compounds. Only the CC symmetric mode coupled with amide 1 for para- and ortho-electron-withdrawing-substituted phenolic compounds. The symmetric and asymmetric CC modes of electron-withdrawing meta-substituted phenolic compounds were coupled like para-electron-promoting phenolic compounds.
Conclusions
Employment of FTIR spectroscopy and DFT calculations revealed the anomalous vibrational coupling between amide 1 and the CC mode of phenol derivatives. For PC, IR absorbance was enhanced markedly for symmetric and asymmetric CC modes upon gradual addition of DMF. For PCP, the symmetric CC mode was enhanced significantly, whereas the asymmetric CC mode was not. Even though the frequency gap was less for the asymmetric CC transition compared with that for the CO transition, and the distance between CC and CO was constant for symmetric and asymmetric CC modes for PC and PCP, distinctive behaviour was observed for different phenol derivatives in the presence of DMF. Theoretical and experimental observations revealed that, with alteration of phenol substituents, the coupling pattern changed. The area of IR absorbance was enhanced ≈5.97 times for the CC symmetric mode, whereas it was altered negligibly for the asymmetric mode, in the case of PCP. For PC, the area of IR absorbance for symmetric and asymmetric modes was enhanced significantly (≈1.4 times). Thus, we revealed the nature of vibrational coupling based on the structure of a guest molecule hydrogen-bonded with amide I. Our conclusions could be valuable for depiction of the unusual dynamics of coupled amide-I modes as well as the dependency of vibrational coupling on altered factors.
Conflicts of interest
The authors declare no competing financial interest.Acknowledgements
AG thanks SNBNCBS, Kolkata, India, for instrumental facilities and financial support from the DST-India. SC thanks DST India for a fellowship.References
- R. E. Dickerson and I. Geis, The Structure and Action of Proteins, Harper and Row, New York, Evanston, London, 1969 Search PubMed.
- D. L. Nelson and M. M. Cox, Lehninger Principles of Biochemistry, W. H. Freeman and Company, New York, 2005 Search PubMed.
- J. M. Berg, J. L. Tymoczko and L. Stryer, Biochemistry, W. H. Freeman and Company, New York, 2007 Search PubMed.
- S. Krimm and J. Bandekar, Adv. Protein Chem., 1986, 38, 181–364 CrossRef CAS.
- A. Barth and C. Zscherp, Q. Rev. Biophys., 2002, 35, 369–430 CrossRef CAS.
- S. Woutersen and P. Hamm, J. Phys.: Condens. Matter, 2002, 14, R1035–R1062 CrossRef CAS.
- P. Hamm, M. Lim and R. M. Hochstrasser, J. Phys. Chem. B, 1998, 102, 6123–6138 CrossRef CAS.
- J. Ma, I. M. Pazos, W. Zhang, R. M. Culik and F. Gai, Annu. Rev. Phys. Chem., 2015, 66, 357–377 CrossRef CAS.
- R. Adhikary, J. Zimmermann and F. E. Romesberg, Chem. Rev., 2017, 117, 1927–1969 CrossRef CAS.
- S. D. Fried and S. G. Boxer, Acc. Chem. Res., 2015, 48, 998–1006 CrossRef CAS PubMed.
- H. Kim and M. Cho, Chem. Rev., 2013, 113, 5817–5847 CrossRef CAS PubMed.
- W. Huan, Z. Yanfei, Z. Fengtao, K. Zhengang, H. Buxing, H. Junfeng, W. Zhenpeng and L. Zhimin, Sci. Adv., 2021, 7(22), 1–9 Search PubMed.
- A. Ghosh, B. Cohn, A. K. Prasad and L. Chuntonov, J. Chem. Phys., 2018, 149(18), 184501 CrossRef.
- S. Chakrabarty, A. Barman and A. Ghosh, J. Phys. Chem. B, 2022, 126, 5490–5496 CrossRef CAS.
- S. Chakrabarty, S. H. Deshmukh, A. Barman, S. Bagchi and A. Ghosh, J. Phys. Chem. B, 2022, 126, 4501–4508 CrossRef CAS PubMed.
- M. K. Gilson and H. X. Zhou, Annu. Rev. Biophys. Biomol. Struct., 2007, 36, 21–42 CrossRef CAS.
- R. U. Lemieux, Acc. Chem. Res., 1996, 29, 373–380 CrossRef CAS.
- Y. Levy and J. N. Onuchic, Annu. Rev. Biophys. Biomol. Struct., 2006, 35, 389–415 CrossRef CAS.
- D. L. Mobley, E. Dumont, J. D. Chodera and K. A. Dill, J. Phys. Chem. B, 2007, 111, 2242–2254 CrossRef CAS.
- T. H. Plumridge and R. D. Waigh, J. Pharm. Pharmacol., 2002, 54, 1155–1179 CrossRef CAS.
- N. S. Myshakina, Z. Ahmed and S. A. Asher, J. Phys. Chem. B, 2008, 112(38), 11873–11877 CrossRef CAS.
- J. Ma, I. M. Pazos, W. Zhang, R. M. Culik and F. Gai, Annu. Rev. Phys. Chem., 2015, 66, 357–377 CrossRef CAS.
- R. Adhikary, J. Zimmermann and F. E. Romesberg, Chem. Rev., 2017, 117, 1927–1969 CrossRef CAS PubMed.
- S. D. Fried and S. G. Boxer, Acc. Chem. Res., 2015, 48, 998–1006 CrossRef CAS PubMed.
- H. Kim and M. Cho, Chem. Rev., 2013, 113, 5817–5847 CrossRef CAS.
- A. I. Ahmed and F. Gai, Protein Sci., 2017, 26(2), 375–381 CrossRef.
- S. Chakrabarty, S. Maity, D. Yazhini and A. Ghosh, Langmuir, 2020, 36, 11255–11261 CrossRef CAS.
- A. Ghosh, A. K. Prasad and L. Chuntonov, J. Phys. Chem. Lett., 2019, 10, 2481–2486 CrossRef CAS.
- D. G. Brown and J. Bostrom, J. Med. Chem., 2016, 59, 4443–4458 CrossRef CAS.
- V. R. Pattabiraman and J. W. Bode, Nature, 2011, 480, 471–479 CrossRef CAS.
- A. B. Hughes, Origins and Synthesis of Amino Acids, Amino Acids, Peptides and Proteins in Organic Chemistry, Wiley-VCH, Weinheim, Germany, 2009, vol. 1 Search PubMed.
- A. A. Kaspar and J. M. Reichert, Drug Discovery Today, 2013, 18, 807–817 CrossRef CAS PubMed.
- R. B. Dyer, F. Gai, W. H. Woodruff, R. Gilmanshin and R. H. Callender, Acc. Chem. Res., 1998, 31, 709–716 CrossRef CAS.
- S. Woutersen, R. Pfister, P. Hamm, Y. Mu, D. S. Kosov and G. Stock, J. Chem. Phys., 2002, 117, 6833–6840 CrossRef CAS.
- N. Demirdöven, C. M. Cheatum, H. S. Chung, M. Khalil, J. Knoester and A. Tokmakoff, J. Am. Chem. Soc., 2004, 126, 7981–7990 CrossRef.
- M. M. Waegele, R. M. Culik and F. Gai, J. Phys. Chem. Lett., 2011, 2, 2598–2609 CrossRef CAS.
- A. L. Serrano, M. M. Waegele and F. Gai, Protein Sci., 2012, 21, 157–170 CrossRef CAS PubMed.
- H. Kim and M. Cho, Chem. Rev., 2013, 113(8), 5817–5847 CrossRef CAS PubMed.
- P. Hamm, and R. M. Hochstrasser, Ultrafast Infrared, and Raman Spectroscopy, 2001 Search PubMed.
- A. M. Cunha, E. Salamatova, R. Bloem, S. J. Roeters, S. Woutersen, M. S. Pshenichnikov and T. L. C. Jansen, J. Phys. Chem. Lett., 2017, 8, 2438–2444 CrossRef CAS.
- H. Li, R. Lantz and D. Du, Molecules, 2019, 24, 186 CrossRef.
- W. M. Zhang, V. Chernyak and S. Mukamel, J. Chem. Phys., 1999, 110, 5011–5028 CrossRef CAS.
- C. Scheurer, A. Piryatinski and S. Mukamel, J. Am. Chem. Soc., 2001, 123, 3114–3124 CrossRef CAS PubMed.
- A. Moran and S. Mukamel, Proc. Natl. Acad. Sci. U. S. A., 2004, 101, 506–510 CrossRef CAS.
- E. G. Buchanan, W. H. James, S. H. Choi, L. Guo, S. H. Gellman, C. W. Müller and T. S. Zwier, J. Chem. Phys., 2012, 137, 094301 CrossRef.
- M. Lima, R. Chelli, V. V. Volkov and R. Righini, J. Chem. Phys., 2009, 130, 204518 CrossRef.
- S. Krimm and Y. Abe, Proc. Natl. Acad. Sci. U. S. A., 1972, 69, 2788–2792 CrossRef CAS PubMed.
- S. Roy, T. L. C. Jansen and J. Knoester, Phys. Chem. Chem. Phys., 2010, 12, 9347–9357 RSC.
- C. Liang, M. Louhivuori, S. J. Marrink, T. L. C. Jansen and J. Knoester, J. Phys. Chem. Lett., 2013, 4, 448–452 CrossRef CAS PubMed.
- J. Schaefer, H. G. Ellen, Y. N. Backus and M. Bonn, J. Phys. Chem. Lett., 2016, 7, 4591–4595 CrossRef CAS PubMed.
- I. V. Rubtsov, J. Wang and R. M. Hochstrasser, J. Phys. Chem. A, 2003, 107, 3384–3396 CrossRef CAS.
- J. V. Hatton and R. E. Richards, Mol. Phys., 1960, 3, 253–263 CrossRef CAS.
- M. Malathi, R. Sabesan and S. Krishnan, Curr. Sci., 2002, 86, 838–842 Search PubMed.
- A. Ghosh, J. Phys. Chem. B, 2019, 123, 7771–7776 CrossRef CAS.
- P. Hamm and M. Zanni, Concepts and Methods of 2D Infrared Spectroscopy, Cambridge University Press, New York, 2011 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: We have provided videos showing coupling of the: symmetric CC and CO modes of PC and DMF (AV1); asymmetric CC and CO modes of PC and DMF (AV2); symmetric CC and CO modes of PCP and DMF (AV3); asymmetric CC and CO modes of PCP and DMF (AV4); symmetric CC and CO modes of para-ethylphenol and DMF (AV5); asymmetric CC and CO modes of para-ethylphenol and DMF (AV6); symmetric CC and CO modes of para-nitrophenol and DMF (AV7); asymmetric CC and CO modes of para-nitrophenol and DMF (AV8); symmetric CC and CO modes of meta-chlorophenol and DMF (AV9); asymmetric CC and CO modes of meta-chlorophenol and DMF (AV10); symmetric CC and CO modes of ortho-chlorophenol and DMF (AV11); asymmetric CC and CO modes of ortho-chlorophenol and DMF (AV12). See DOI: https://doi.org/10.1039/d2ra07177k |
| This journal is © The Royal Society of Chemistry 2023 |