DOI:
10.1039/D2RA07400A
(Paper)
RSC Adv., 2023,
13, 6191-6198
Rhodium-catalyzed selenylation and sulfenylation of quinoxalinones ‘on water’†
Received
21st November 2022
, Accepted 8th February 2023
First published on 20th February 2023
Abstract
A rhodium-catalysed, regioselective synthetic methodology for selenylation and sulfenylation of 3-phenyl quinoxolinones has been developed through N-directed C–H activation in the presence of silver triflimide, and silver carbonate using dichalcogenides ‘on water’. The methodology has been proven to be efficient, regioselective and green. Using this method, a range of selenylations and sulfenylations of the substrates has been carried out in good to excellent yields. Further, late-stage functionalisation produced potential anti-tumour, anti-fungal and anti-bacterial agents making these compounds potential drug candidates.
Introduction
Green chemistry, also known as sustainable chemistry, is composed of twelve principles that provide a scientific blueprint for researchers and scientists to reduce the negative impact of chemicals and chemical processes on the environment and to advance new technologies to improve economic, environmental and societal outcomes.1 These twelve principles emphasize avoiding or eliminating the consumption and the production of hazardous chemicals in a chemical process. In the quest of developing a sustainable synthetic protocol, the choice of solvent is a constant source of worry as it is wasted in bulk quantities after the completion of a reaction and causes significant impact on health and the environment, if disposed of untreated.2 From the perspective of green chemistry, water proved to be a sustainable solvent due to its benign nature, high availability, cost-effective, non-toxic and non-flammable properties.3 Moreover, water exhibits exceptional reactivity and selectivity by stabilizing the transition state through hydrogen bonding and by exhibiting a hydrophobic effect.4 Depending upon the solubility of reactants and products in water, the water-mediated reactions are categorized as “in water” and “on water” reactions. “In water” conditions refer to a system that utilizes additives to enhance the solubility of less soluble catalysts, adducts and other components to make the system homogeneous. On the other hand, the “on water” is a comparatively new approach and was developed by Sharpless and his group in 2005 to demonstrate the enhancement of reaction rate in an aqueous medium despite the insolubility of reactants and other components.5 Moreover, heterogeneity of the “on water” system reduces tedious workups and introduces further sustainability to the protocol, unlike the “in water” system. The high dielectric constant of water, high heat capacity, high redox stability and ease of workup make the “on water” system highly desirable for the development of green and sustainable protocols for chemical synthesis.6
Organosulphur and organoselenium compounds have gained a prominent place in modern organic synthesis owing to their widespread applications in the field of pharmaceuticals,7 agrochemicals8 and polymer materials.9 The pharmaceutical evaluation of FDA-approved drugs revealed that 25% of the USA's top two hundred prescribed drugs contain organosulphur compounds.10 Some of the important biological activities of organosulphur and organoselenium compounds include human breast cancer cell growth inhibitors,11 antitumor agents,12 RAR agonists,13 vortioxetine,14 nicotine acetylcholine receptor ligands,15 antioxidants,16 albendazole,17 antifolates,18 etc. (Fig. 1).
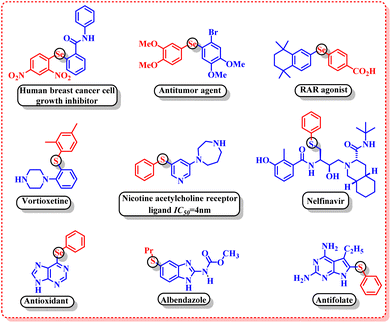 |
| | Fig. 1 Biologically active organosulphur and organoselenium compounds. | |
Moreover, the introduction of selenium or sulphur into organic molecules framework dramatically affects compound's photo-physical and electronic properties.19 Consequently, these organochalcogenides have significant applications as effective organic materials and as fluorescent probes.20 Considering the immense importance of organosulphur and organoselenium compounds, the development of new synthetic protocols for the formation of C–Se and C–S linkages has attracted considerable attention in recent years.21
Over the past few decades, transition metal-catalysed cross-coupling reactions have emerged as a powerful synthetic tool for the construction of carbon–carbon and carbon–heteroatom bonds.22 Several metal-mediated coupling reactions have been recently developed with selenium and sulphur nucleophiles for the preparation of biologically active compounds.23 However, these cross-coupling reactions required several steps to prepare pre-functionalized substrates, limiting the reaction's atom economy and making the methodology expensive and laborious.24 Conversely, direct C–H selenylation and sulfenylation provide a straightforward method for the installation of sulphur and selenium groups into the organic framework as it offers shorter routes with higher atom economy, requires less time and utilizes the readily available starting materials.25 In this direction, palladium, nickel and copper complexes have been extensively explored for the direct sp2 C–H chalcogenylation and have served the purpose well. Despite the vast progress, these catalytic systems suffer from less functional group tolerance, narrow substrate scope and high catalytic loading. Moreover, metal additives and stabilizing ligands are also required to prevent the decomposition of metal complexes which further restricts the practicality of these reactions on a large or industrial scale.26
In the field of C–H bond functionalization, rhodium-based catalysts outperform the catalytic ability of Pd, Ni and Cu by offering exceptional functional group tolerance, high efficiency, low catalytic loading, and broad synthetic applications. Rhodium(III), especially in the form of [Cp*RhCl2]2 performs exceptionally well due to the more polarised Rh(III)–C(aryl) bond and the bulkier Cp* ligand promotes enough steric hindrance during the C–H activation process and make the catalyst thermally stable.27 Owing to the inherent advantages of rhodium catalysis, remarkable progress has been witnessed in the area of rhodium(III) catalyzed direct C–H chalcogenylation, over the past few years. In 2014, Li and his co-workers carried out directed sulfenylation of arene C–H bonds using [Cp*RhCl2]2.28 Subsequently, rhodium-catalyzed direct C–H selenylation was reported by Xingwei Li and coworkers.29
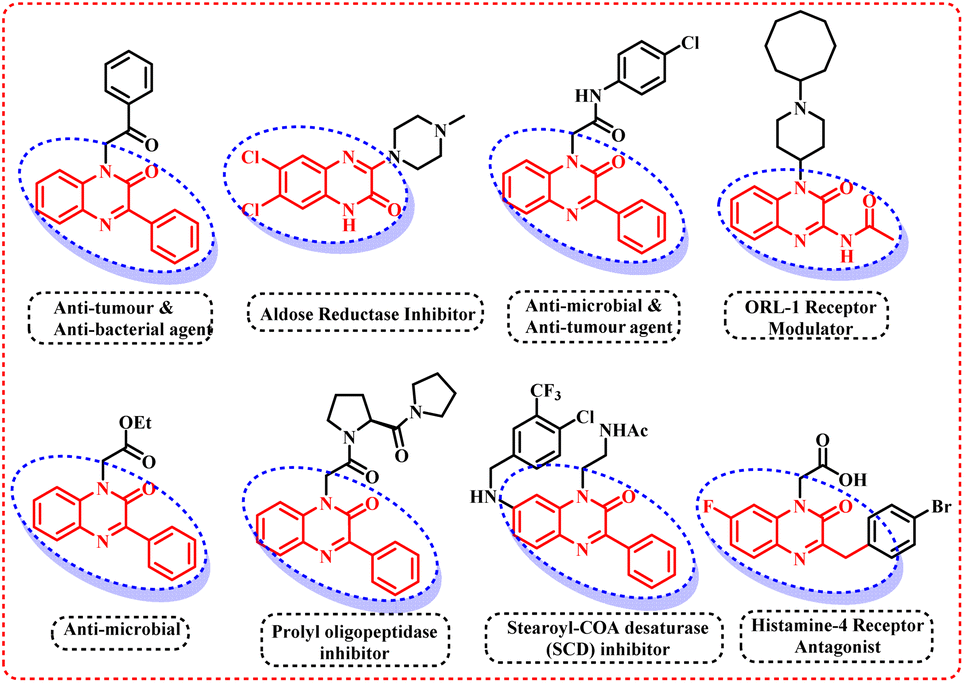
Afterwards, many groups have explored the rhodium catalyst for direct C–H selenylation and sulfenylation30 and have utilized a wide variety of organic solvents such as DMF, DMSO, 1,4-dioxane, etc. However, till date, direct selenylation and sulfenylation of heterocyclic compounds have not been reported ‘on water’. Thus, in continuation of our efforts to develop the sustainable and greener protocols for the C–H functionalization of various biologically active heterocycles,31 we herein are reporting the regioselective selenisation and sulfenylation of pharmaceutically important 3-phenyl quinoxalinones (Fig. 2) using rhodium(III) catalyst ‘on water’.
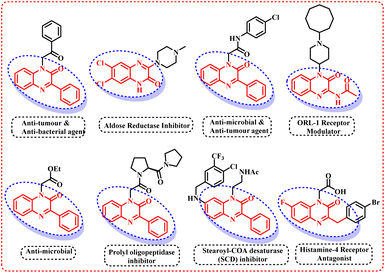 |
| | Fig. 2 Pharmaceutically active quinoxalinones. | |
Results and discussion
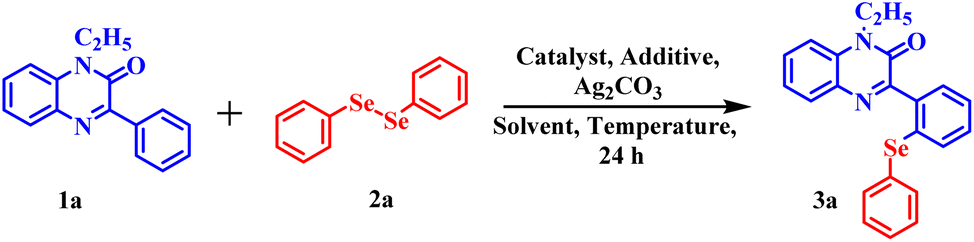
For our preliminary examination, 1-ethyl-3-phenylquinoxalin-2(1H)-one 1a and commercially available 1,2-diphenyl diselenide 2a were chosen as model substrates to carry out regioselective selenylation. The reaction was allowed to run at a ceiling temperature of 110 °C with 5 mol% of [Cp*RhCl2]2 as a catalyst, 60 mol% of silver triflimide as an additive, 1.0 equivalent of silver carbonate as an oxidant and 1,4-dioxane as a solvent. The reaction completed in 24 hours and the regioselective, mono-selenylated product 3a was obtained in 35% yield (Table 1, entry 1). In order to improve the product yield, a series of solvents were screened (Table 1, entries 2–6). Solvents such as DMSO and DMA were found completely ineffective for the reaction (Table 1, entries 3 and 4). However, in DMF, product 3a was obtained in 25% yield (Table 1, entry 2). A dramatic increase in the product yield was observed when the reaction was performed in water (Table 1, entry 6). In water, 94% of desired product 3a was isolated. Further, the effect of catalysts on the performance of the reaction was evaluated using water as a solvent. Inferior product yield was obtained with Rh(COD)Cl2 and [RuCl2(p-cymene)]2 catalysts (Table 1, entries 7 and 8). To improve the product yield further, we next screened different additives (Table 1, entries 9 and 10). No improvement in product yield was observed on changing the additive from silver triflimide to silver triflate and silver hexafluoroantimonate. Similarly, inferior product yield was obtained on replacing Ag2CO3 with Ag2O and AgNO3 (Table 1, entries 11 and 12). After a brief examination of the reaction temperature, 110 °C was found as the optimum temperature for this reaction. Incomplete conversion of the reactant was observed when the reaction was carried out at lower temperatures (Table 1, entries 13 and 14). Nevertheless, a slight decrease in product yield was observed when the reaction was carried out at higher temperatures (Table 1, entries 15 and 16). Further, neither the change in additive concentration nor the change in substrate concentration provided any improvement in the product yield (Table 1, entries 17–20). However, slightly inferior product yield was noted with 3 mol% and approximately the same yield was observed with 7 mol% of the catalyst respectively (Table 1, entries 21 and 22). Change of oxidant from silver carbonate to Cu(OAc)2 did not have any fruitful outcome (Table 1, entry 23). Finally, we established the optimised condition as 1-ethyl-3-phenylquinoxalin-2(1H)-one (1a), 2 equiv. of commercially available 1,2-diphenyl diselenide (2a), 5 mol% of [Cp*RhCl2]2 catalyst, 60 mol% of silver triflimide, 1 equiv. of silver carbonate and 1.5 mL of water.
Table 1 Optimization of reaction conditionsa
Substrate scope
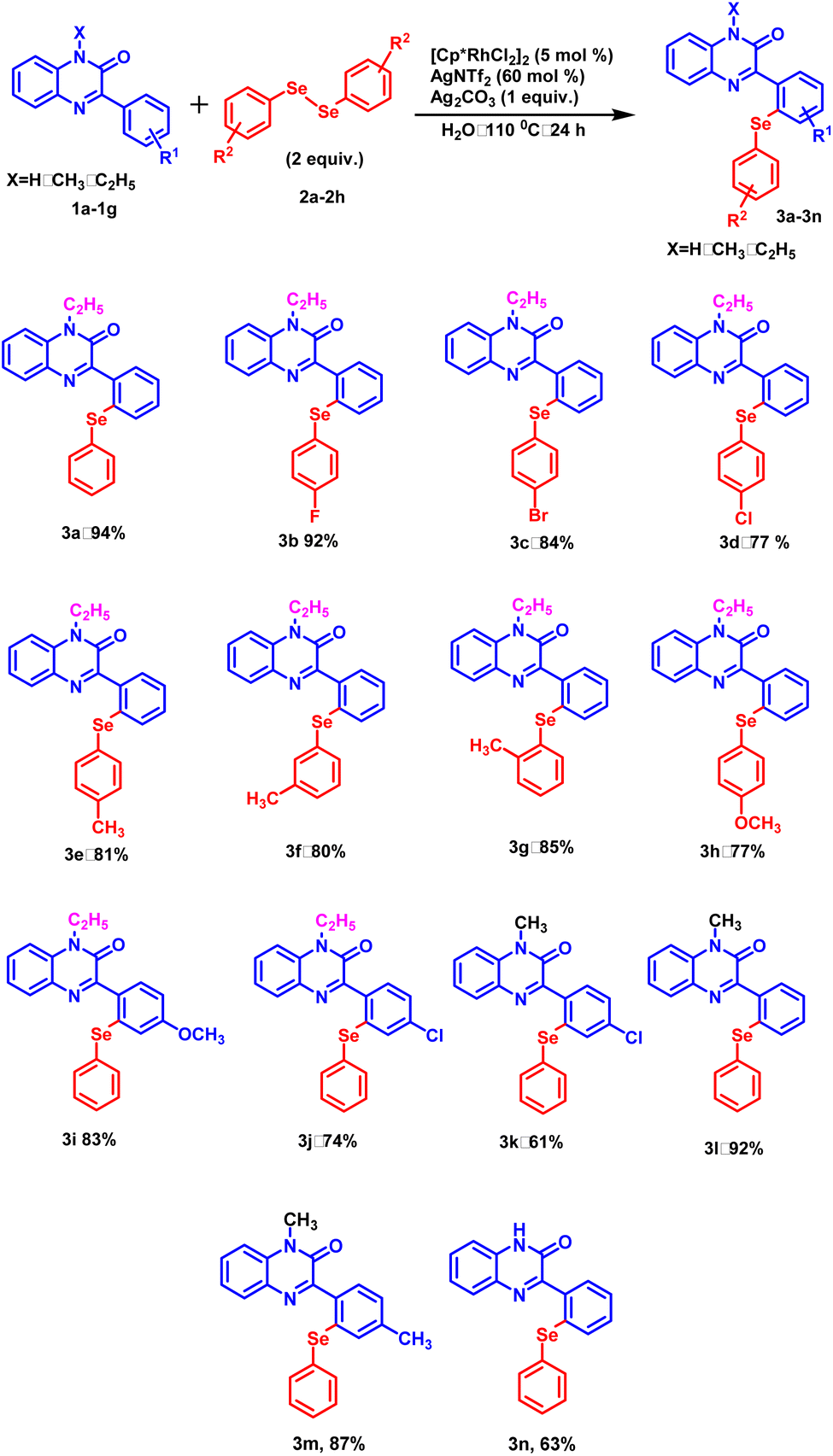
With this optimized protocol, we examined the scope and generality by employing a variety of 3-phenylquinoxalin-2(1H)-ones bearing both electron-donating and electron-withdrawing groups (Scheme 1). All the 3-phenylquinoxalin-2(1H)-one derivatives afforded the desired products in good to excellent yields. Electron-withdrawing group such as –Cl exhibited a significant adverse effect on product yield in comparison to the electron-donating group such as –CH3 (Scheme 1, 3k, 3m). A similar trend was observed with N-substituted 3-phenylquinoxalin-2(1H)-ones. Stronger electron-donating groups such as –C2H5 provided better product yield as compared to milder electron-donating groups such as –CH3 and –H (Scheme 1, 3a, 3l and 3n). Electron-withdrawing groups present on the para-position of the benzene ring exhibited a disadvantageous effect on product yields as compared to electron donating group –H (Scheme 1, 3a and 3j). Subsequently, the electronic effect of substituent present on diphenyl diselenide was studied. Among the halogens substituted at the para-position of the diselenide, fluoro-diselenide afforded a better product yield than bromo and chloro counterparts (Scheme 1, 3b, 3c and 3d). We also carried out the reaction with diselenides with methyl group at ortho, meta and para positions and interestingly all of them produced the excellent yields (Scheme 1, 3e, 3f and 3g).
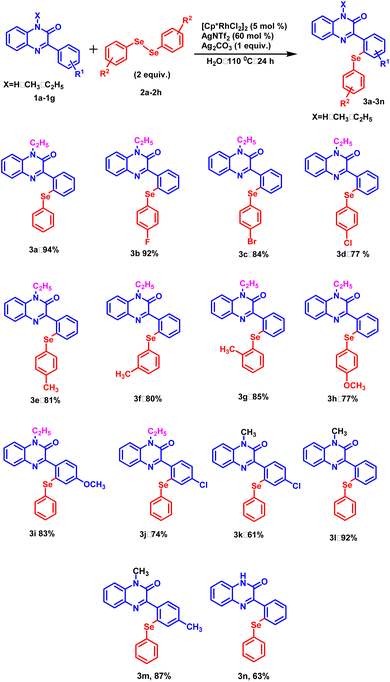 |
| | Scheme 1 Substrate scope of 3-phenylquinoxalin-2(1H)-ones and diphenyl diselenides. | |
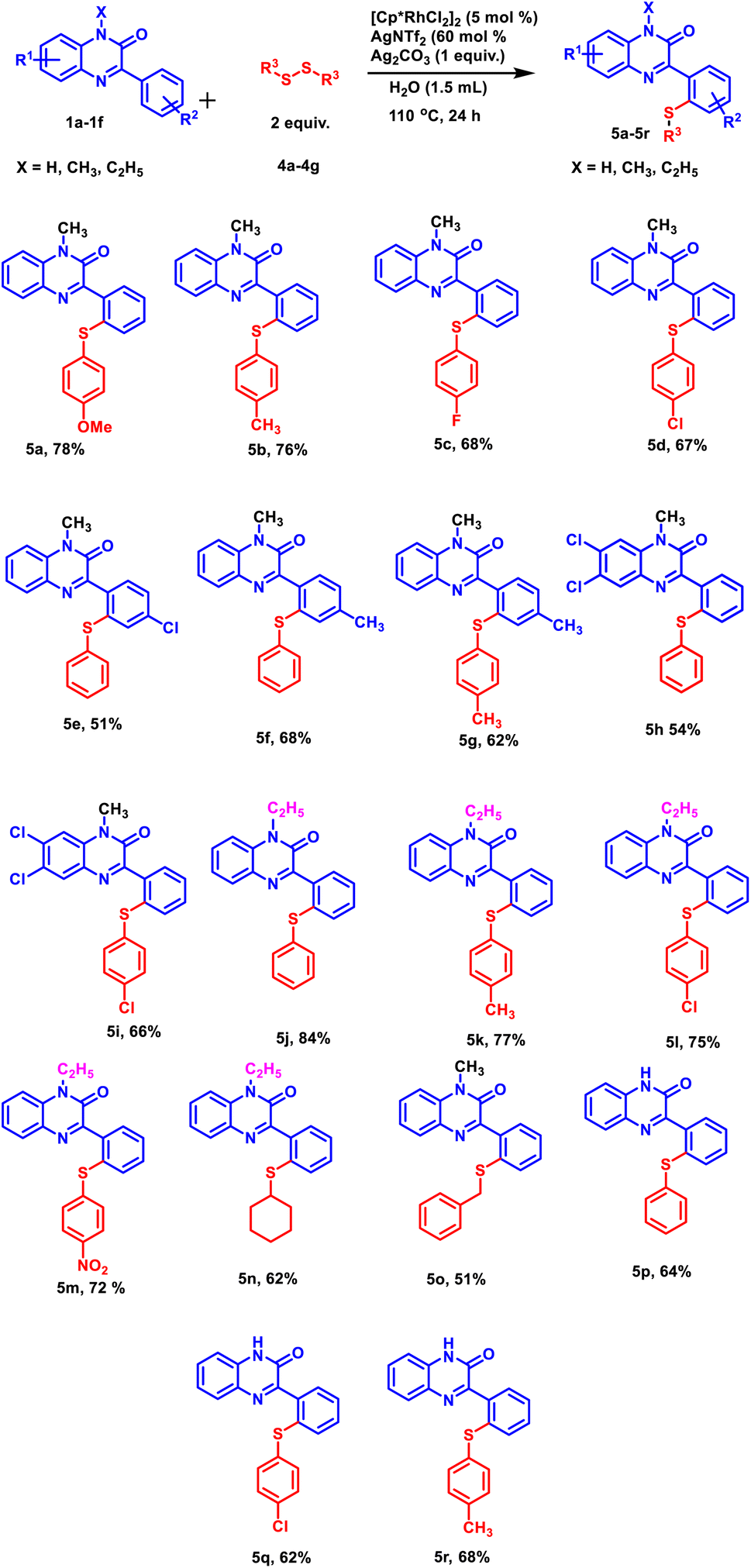
Further, to explore the substrate scope, a range of differently substituted diphenyl disulphides were allowed to react with 3-phenylquinoxalin-2(1H)-ones under the optimized reaction condition (Scheme 2, 5a–5d, 5m, 5n, and 5o). The results demonstrated that all the diphenyl disulphide derivatives afforded the corresponding products in good to moderate yields. It was observed that diphenyl disulphide having electron-donating groups (–OCH3 and –CH3) (Scheme 2, 5a and 5b) furnished the desired products in higher yields as compared to electron-withdrawing groups such as (–F and –Cl) (Scheme 2, 5c and 5d). After studying the electronic effect of diphenyl disulphide, the electronic effect of substituents present on 3-phenylquinoxalin-2(1H)-one was examined. For this purpose, –CH3 and –Cl groups were taken into the consideration at the para-position of the phenyl ring. It was observed that the methyl group afforded better product yield than the chloro group (Scheme 2, 5e and 5f). We were pleased to find that N-substituted as well as N-unsubstituted 3-phenylquinoxalin-2(1H)-ones delivered the corresponding products in good to moderate yields. However, stronger electron-donating groups afforded higher product yields as compared to less electron-donating groups (Scheme 2, 5j and 5p). Additionally, the established protocol was also found compatible with dialkyl disulfides (Scheme 2, 5n and 5o), suggesting a wide substrate scope of the established protocol.
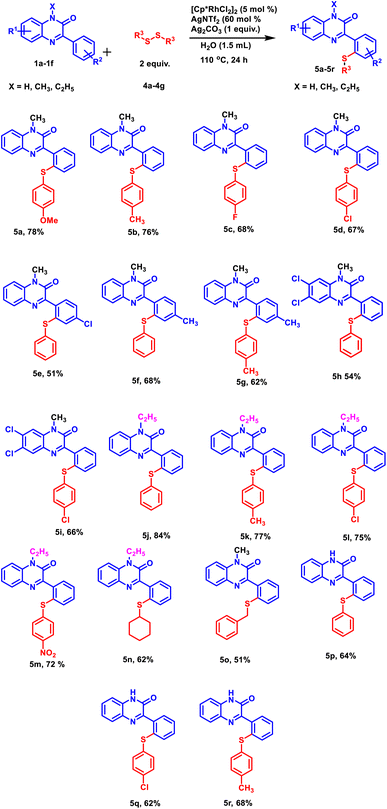 |
| | Scheme 2 Substrate scope of 3-phenylquinoxalin-2(1H)-ones and diphenyl(dialkyl) disulfides. | |
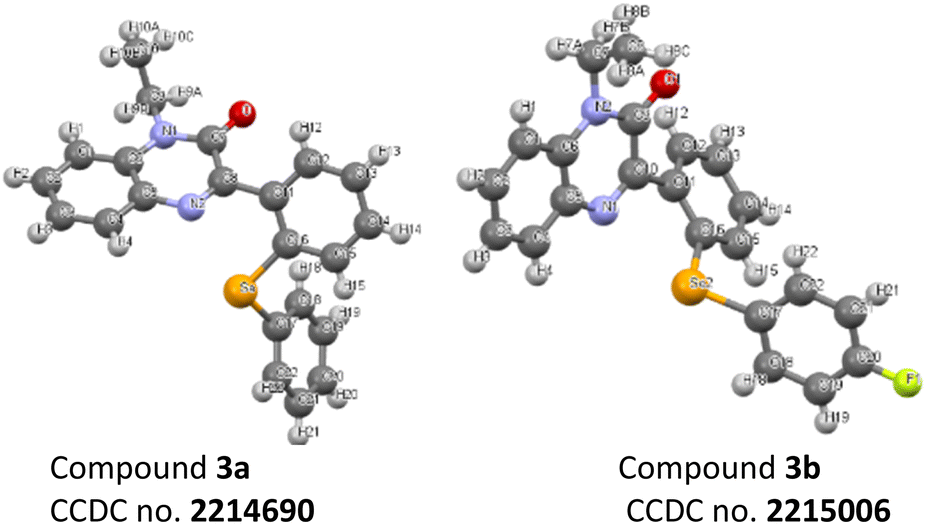
A substantial improvement in the product yields was recorded with diphenyl diselenides as a coupling partner as compared to diphenyl disulphides (Schemes 1 and 2). The higher reactivity of diphenyl diselenide is attributed to the higher nucleophilicity of the Se atom and the low bond dissociation energy of the Se–Se bond.32 Although diphenyl disulphides smoothly delivered the targeted products in good to moderate yields, the chemical reactivity of diphenyl disulphide is lower due to its strong coordinating and absorptive affinity towards the catalysts.33 The structure of compounds 3a and 3b are also confirmed by crystallography (Fig. 3).
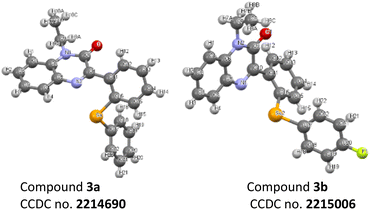 |
| | Fig. 3 Single crystal X-ray structure of the compounds 3a and 3b. | |
Synthetic application
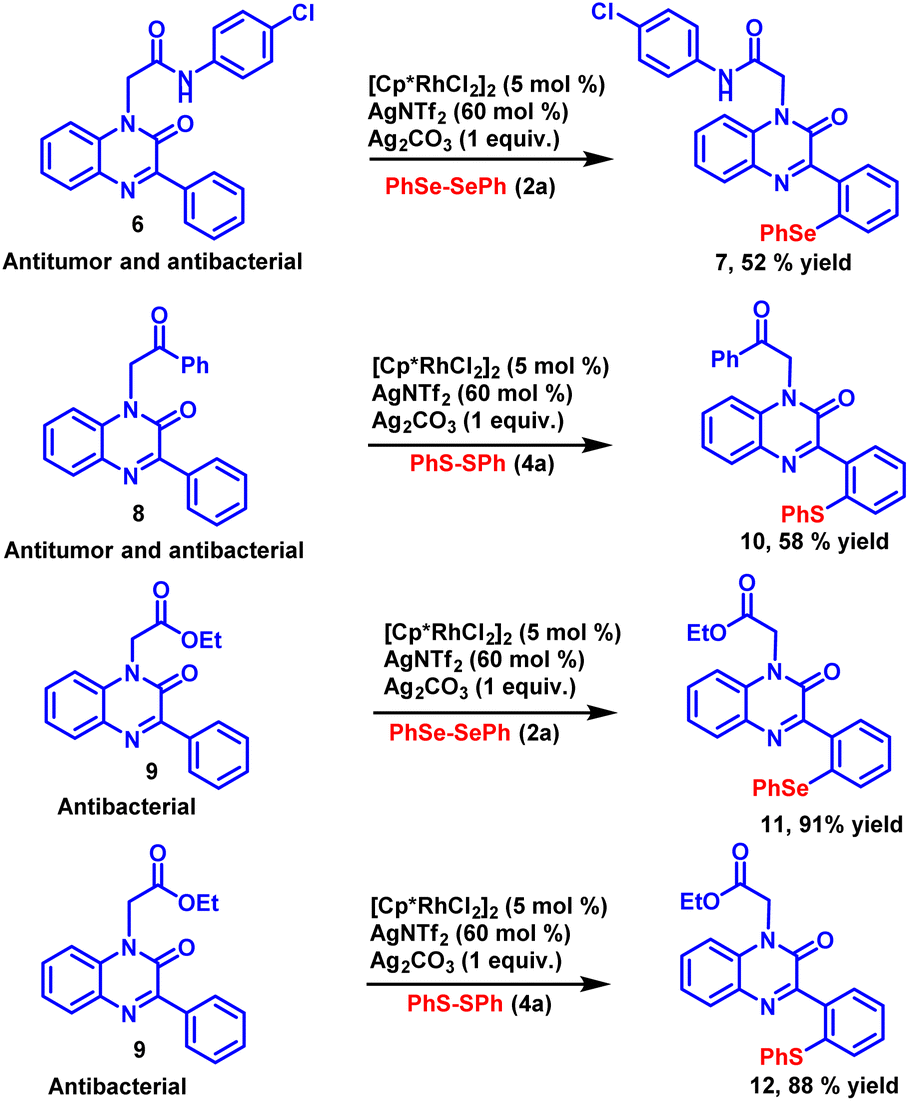
Modern drug discovery relies heavily on the identification of lead compounds and the synthesis of their related analogues. The late-stage functionalization of drug candidates offers a practical and viable alternative to access a large number of related analogous for structure–activity relationship (SAR) studies. To investigate the synthetic potential of the established methodology, late-stage functionalization of potential antimicrobial and antitumor compounds34 was carried out (Scheme 3). N-(4-Chlorophenyl)-2-(2-oxo-3-phenylquinoxalin-1(2H)-yl)acetamide (6) was allowed to react with diphenyl diselenide 2a under the standard reaction conditions. The reaction proceeded smoothly and selenylated product 7 was obtained with good yield. Late-stage functionalization of another potential antimicrobial and antitumor compound 1-(2-oxo-2-phenylethyl)-3-phenylquinoxalin-2(1H)-one (8) was also carried out with diphenyl disulfide 4a as coupling partner with the newly developed reaction condition, the sulfenylated product 10 was obtained in 58% yield. Similarly, 1-(2-oxobutyl)-3-phenylquinoxalin-2(1H)-one (9), a synthon having anti-bacterial activity was subjected to sulfenylation and selenylation under the standard reaction condition, the respective selenylated and sulfenylated products 11 and 12 were obtained in excellent yields. The successful implementation of C–H chalcogenation on pharmacologically active compounds paves a rapid way to access drug-related analogues that finds a huge application in the process of drug discovery.
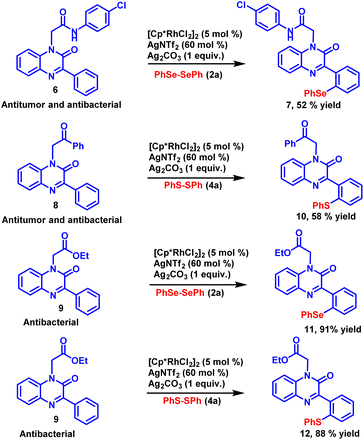 |
| | Scheme 3 Late-stage functionalization of potential anti-tumour and anti-microbial agents. | |
Control experiments
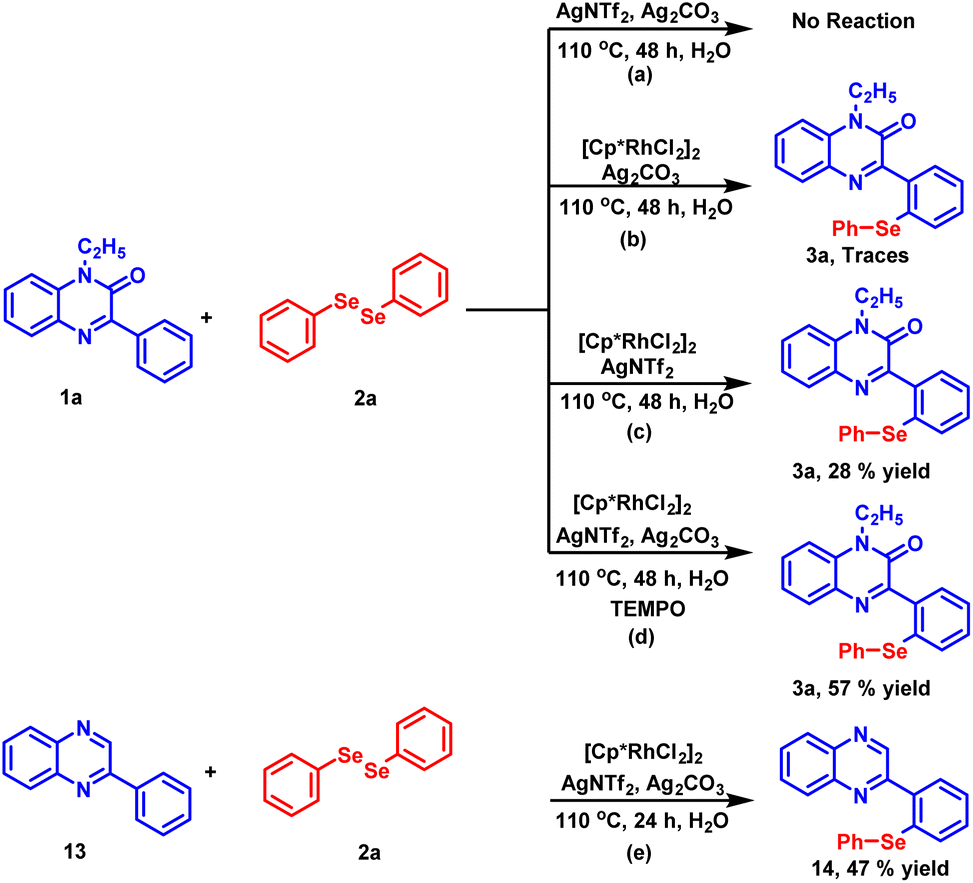
To gain an insight into the reaction mechanism, several control experiments were carried out (Scheme 4). In the first experiment, the coupling reaction between 1a and 2a was performed in the absence of the catalyst (Scheme 4a). As anticipated, no reaction took place and the starting material was recovered even after 48 hours. Next, the reaction was conducted by excluding silver triflimide from the standard reaction conditions (Scheme 4b) and the product was detected in traces. The absence of product formation indicates that the catalyst [Cp*RhCl2]2 alone is unable to drive the reaction. In the next experiment, the reaction was performed in the absence of silver carbonate (Scheme 4c). The reaction did not proceed to completion and 28% of the desired product 3a was isolated after 48 hours. The observation demonstrates the important role of silver carbonate as a co-oxidant. Further, when the radical scavenger experiment was carried out using TEMPO (Scheme 4d), product 3a was obtained in 57% yield, which suggests that the radical process may not be involved in this reaction. To check whether the carbonyl group present at the C-2 position of the substrate has any directing influence on the reaction instead of nitrogen, we found that the reaction proceeds in the absence of the carbonyl group and compound 13 provided the synthesized product 14 in 47% yield.
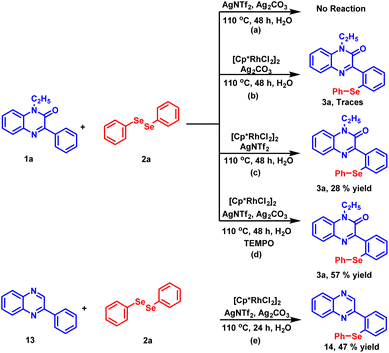 |
| | Scheme 4 Preliminary mechanistic experiments. | |
H/D exchange and kinetic studies
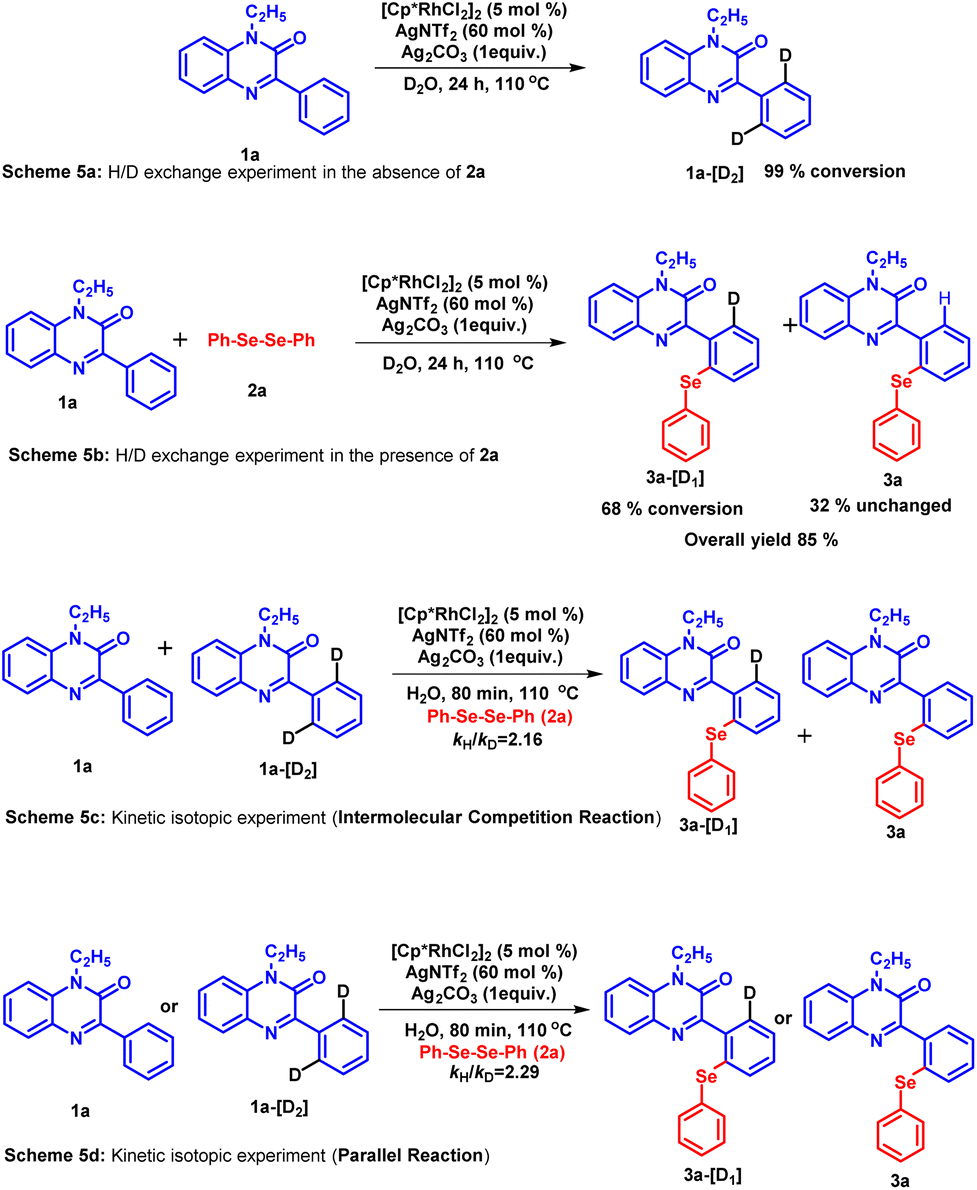
To assess the reversibility of the C–H activation step, a hydrogen–deuterium (H/D) exchange experiment was carried out using D2O as a solvent (Scheme 5a and b).35 These experiments were performed in the absence as well as in the presence of coupling partner 2a under the standard condition. The 1H NMR analysis of the recovered compound revealed a 99% conversion of substrate 1a into 1a-[D2] (Scheme 5a). In the presence of coupling partner 2a, the mixture of the product obtained contains 68% of 3a-[D1] and 32% of product 3a (Scheme 5b). A significant amount of deuterium incorporation in both these experiments infers the reversibility of the C–H activation step. Furthermore, the kinetic isotopic experiment (KIE) was performed to investigate whether the C–H activation step is involved in the rate-determining step or not (Scheme 5c). The parallel experiment using 1a and 1a-[D2] was subjected to react with diphenyl diselenide 2a under the standard reaction conditions for 80 minutes. The 1H NMR studies of the isolated product mixture revealed the kinetic isotopic effect value (kH/kD) as 2.29. While the intermolecular competition KIE experiment between the same coupling partners for 80 minutes yielded the kH/kD value as 2.16. The kinetic isotopic effect suggests the possible involvement of the C–H activations step in the rate-limiting step.36
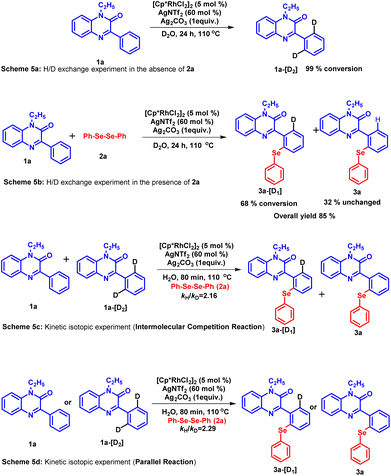 |
| | Scheme 5 H/D exchange and deuterium kinetic isotope studies. | |
Mechanism
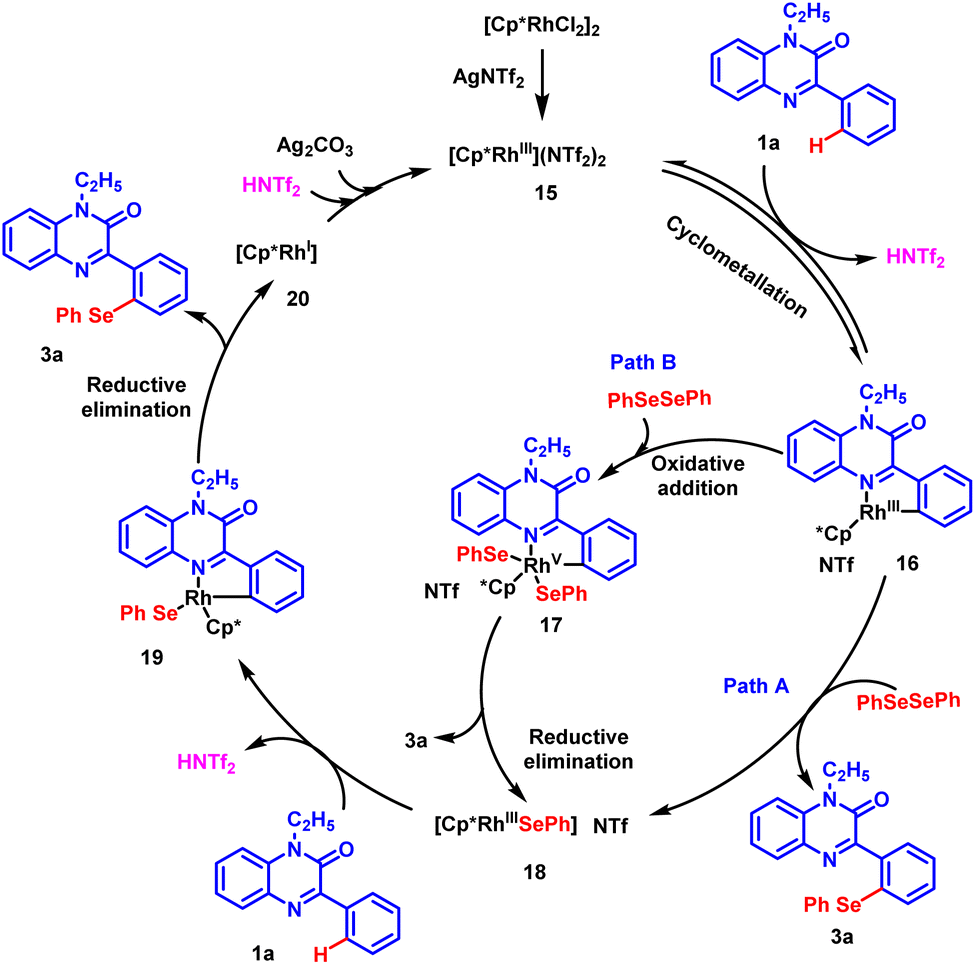
Based on the experimental results and the literature reports, a rational catalytic mechanism for Rh-catalyzed, regioselective chalcogenation of 1-ethyl-3-phenylquinoxalin-2(1H)-one is depicted in Scheme 6. It is believed that [Cp*RhCl2]2 precursor reacts with silver triflimide (AgNTf2) in situ to generate an active rhodium catalyst (15). The catalyst reacts with substrate 1a and a cyclo-metallated rhodium complex 16 is generated possibly through a C–H activation step. Intermediate 16 further reacts with diphenyl diselenide 2a through a nucleophilic substitution reaction and delivers the desired product 3a and intermediate 18. Alternatively, intermediate 16 may prefer path B, where oxidative addition with diphenyl diselenide produces a rhodium(V) complex 17. Following this, the reductive elimination of complex 17 affords the desired product 3a and Rh(III) intermediate 18 back.37,38 Subsequently, another molecule of substrate 1a participates in C–H activation with intermediate 18 to generate cyclo-metalled rhodium intermediate 19. Afterwards, the reductive elimination of intermediate 19 results in the formation of Rh(I) complex 20 and the desired product 3a. The oxidation of Rh(I) species by the action of silver carbonate and triflimide regenerates the active rhodium(III) catalyst 15 to complete the catalytic cycle.
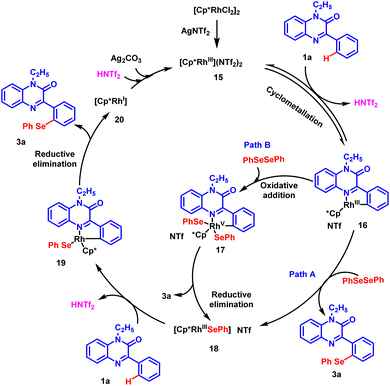 |
| | Scheme 6 Plausible reaction mechanism for the regioselective chalcogenation of phenylquinoxalinones. | |
Conclusion
In conclusion, we have successfully developed a green and sustainable method in an aqueous medium for the regioselective direct C–H chalcogenation of phenylquinoxalinones using [Cp*RhCl2]2 as a catalyst, ‘on water’. The metal-directing property of the nitrogen atom is utilized to afford the chalcogenated products in good to excellent yields. A wide variety of diphenyl disulphides, diphenyl diselenides and phenylquinoxalinones were found to be compatible with the established protocol thus offering remarkable substrate scope and excellent functional group tolerance. The tedious work-up step has been avoided. With this ‘on water’ method, the straightforward synthesis of the four bioactive compounds has been afforded via the late-stage functionalization of the potential antitumor, antifungal and the antimicrobial candidates and this has further widened the scope of the established protocol in the field of organic synthesis and drug discovery.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
This work has been supported financially by Institution of Eminence (IoE), University of Delhi. We also thank University Science Instrumentation Centre (USIC), University of Delhi for instrumental facilities. Prince acknowledges CSIR, New Delhi, India for providing Senior Research Fellowship and SK acknowledges University Grant Commission for Senior Research Fellowship.
References
-
(a) R. A. Sheldon, Green Chem., 2005, 7, 267 RSC;
(b) R. Ballini, L. Barboni, F. Fringuelli, A. Palmieri, F. Pizzo and L. Vaccaro, Green Chem., 2007, 9, 823 RSC;
(c) C. J. Li and B. M. Trost, Proc. Natl. Acad. Sci. U. S. A., 2008, 105, 13197 CrossRef CAS PubMed;
(d) I. T. Horváth and P. T. Anastas, Chem. Rev., 2007, 107, 2169 CrossRef PubMed;
(e) R. Breslow, The Principles and Reasons for Using Water as a Solvent for Green Chemistry, Handbook of Green Chemistry, 2010, pp. 1–29 Search PubMed;
(f) A. R. Katritzky, Comprehensive Heterocyclic Chemistry III, Elsevier, Amsterdam, NY, 2008 Search PubMed;
(g) G. Brahmachari, Green Synthetic Approaches for Biologically Relevant Heterocycles, Elsevier, Amsterdam, 2015, pp. 185–208 Search PubMed;
(h) K. L. Ameta and A. Dandia, Green Chemistry: Synthesis of Bioactive Heterocycles, Springer, 2014 Search PubMed.
-
(a) M. O. Simon and C. J. Li, Chem. Soc. Rev., 2012, 41, 1415 RSC;
(b) D. J. C. Constable, A. D. Curzons and V. L. Cunningham, Green Chem., 2002, 4, 521 RSC.
-
(a) C. J. Li and L. Chen, Chem. Soc. Rev., 2006, 35, 68 RSC;
(b) C. J. Li and T. H. Chan, Comprehensive Organic Reactions in Aqueous Media, John Wiley and Sons, New York, 2007 CrossRef;
(c) P. Dixneuf and V. Cadierno, Metal-Catalyzed Reactions in Water, John Wiley & Sons, 2013 CrossRef;
(d) S. Minakata and M. Komastsu, Chem. Rev., 2009, 109, 711 CrossRef CAS PubMed;
(e) A. Chanda and V. V. Fokin, Chem. Rev., 2009, 109, 725 CrossRef CAS PubMed.
-
(a) D. C. Rideout and R. Breslow, J. Am. Chem. Soc., 1980, 102, 7816 CrossRef CAS;
(b) F. M. Moghaddam, L. Hojabri, S. Taheri and P. Pirani, J. Iran. Chem. Soc., 2010, 7, 781 CrossRef CAS;
(c) S. Otto, G. Boccaletti and J. B. F. N. Engberts, J. Am. Chem. Soc., 1998, 120, 4238 CrossRef CAS;
(d) X. Y. Liu and C. M. Che, Angew. Chem., Int. Ed., 2008, 47, 3850 Search PubMed;
(e) C. I. Herrerias, X. Yao, Z. Li and C. J. Li, Chem. Rev., 2007, 107, 2546 CrossRef CAS PubMed;
(f) U. M. Lindstrom and G. Andersson, Angew. Chem., Int. Ed., 2006, 45, 548 CrossRef PubMed;
(g) F. Zhou and C. J. Li, Nat. Commun., 2014, 5, 4254 CrossRef CAS PubMed.
- S. Narayan, J. Muldoon, M. G. Finn, V. V. Fokin, H. C. Kolb and K. B. Sharpless, Angew. Chem., Int. Ed., 2005, 44, 3275 CrossRef CAS PubMed.
- Y. Cheng, F. Xue, S. Yu, S. Du and Y. Yang, Molecules, 2021, 26, 4004 CrossRef CAS PubMed.
-
(a) S. Patai, The Chemistry of the Thiol Group, Wiley, 1974 Search PubMed;
(b) L. A. Damani, Sulfur Containing Drugs and Related Organic Compounds: Chemistry, Biochemistry, and Toxicology, Part B: Metabolism of Sulfur Functional Groups, Ellis Horwood Ltd, Chichester, UK, 1989, vol. 1 Search PubMed;
(c) R. J. Cremlyn, An Introduction to Organosulfur Chemistry, Wiley & Sons, New York, 1996 Search PubMed;
(d) M. D. McReynolds, J. M. Doughtery and P. R. Hanson, Chem. Rev., 2004, 104, 2239 CrossRef CAS PubMed;
(e) J. Zhao and X. Jiang, Chin. Chem. Lett., 2018, 29, 1079 CrossRef CAS;
(f) P. Chauhan, S. Mahajan and D. Enders, Chem. Rev., 2014, 114, 8807 CrossRef CAS PubMed.
-
(a) C. B. Mishra, S. Kumari and M. Tiwari, Eur. J. Med. Chem., 2015, 92, 1 CrossRef CAS PubMed;
(b) A. Ayati, S. Emami, A. Asadipour, A. Shabee and A. Foroumadi, Eur. J. Med. Chem., 2015, 97, 699 CrossRef CAS PubMed;
(c) P. Sun, D. Yang, W. Wei, L. Jiang, Y. Wang, T. Dai and H. Wang, Org. Chem. Front., 2017, 4, 1367 RSC;
(d) M. Klečka, R. Pohl, J. Čejka and M. Hocek, Org. Biomol. Chem., 2013, 11, 5189 RSC.
-
(a) F. Bernardi, I. G. Csizmadia and A. Mangini, Organic Sulfur Chemistry, Elsevier Science Pub. Co., 1985 Search PubMed;
(b) K. C. Nicolaou, C. R. H. Hale, C. Nilewski and H. A. Ioannidou, Chem. Soc. Rev., 2012, 41, 5185 RSC;
(c) M. Feng, B. Tang, S. H. Liang and X. Jiang, Curr. Top. Med. Chem., 2016, 16, 1200 CrossRef CAS PubMed.
- E. A. Ilardi, E. Vitaku and J. T. Njardarson, J. Med. Chem., 2014, 57, 2832 CrossRef CAS PubMed.
- L. Engman, I. Cotgreave, M. Angulo, C. W. Taylor, G. D. P. Murrieta and G. Powis, Anticancer Res., 1997, 17, 4599 CAS.
- J. A. Woods, J. A. Hadfield, A. T. McGown and B. W. Fox, Bioorg. Med. Chem., 1993, 5, 333 CrossRef PubMed.
- P. Diaz, F. Gendre and J. M. Bernardon, Tetrahedron Lett., 1998, 39, 9003 CrossRef CAS.
- C. Sanchez, K. E. Asin and F. Artigas, Pharmacol. Ther., 2015, 145, 43 CrossRef CAS PubMed.
- S. F. Nielsen, E. O. Nielsen, G. M. Olsen, T. Liljefors and D. Peters, J. Med. Chem., 2000, 43, 2217 CrossRef CAS PubMed.
- L. F. B. Duarte, R. L. Oliveira, K. C. Rodrigues, G. T. Voss, B. Godoi, R. C. Schumacher, G. Perin, E. A. Wilhelm, C. Luchese and D. Alves, Bioorg. Med. Chem., 2017, 25, 6718 CrossRef CAS PubMed.
- R. J. Horton, Acta Trop., 1997, 64, 79 CrossRef CAS PubMed.
- A. Gangjee, Y. Zeng, T. Talreja, J. J. McGuire, R. L. Kisliuk and S. F. Queener, J. Med. Chem., 2007, 50(13), 3046 CrossRef CAS PubMed.
-
(a) E. J. Lenardão, C. Santi and L. Sancineto, New Frontiers in Organoselenium Compounds, Springer, Cham, 2018, pp. 157–183 CrossRef;
(b) E. Regis, L. D. O. Aguiar, P. Tuzimoto, E. Girott, T. E. Frizon, A. G. D. Bo, E. Zapp, R. Marra, H. Gallardo and A. A. Vieira, Dyes Pigm., 2018, 157, 109 CrossRef CAS;
(c) G. C. Hoover and D. S. Seferos, Chem. Sci., 2019, 1, 9182 RSC;
(d) A. E. Baumann, D. A. Burns, B. Liu and V. S. Thoi, Commun. Chem., 2019, 2, 86 CrossRef.
-
(a) B. Wang, P. Li, F. Yu, J. Chen, Z. Qu and K. A. Han, Chem. Commun., 2013, 49, 5790 RSC;
(b) B. Wang, P. Li, F. Yu, P. Song, X. Sun, S. Yang, Z. Lou and K. A. Han, Chem. Commun., 2013, 49, 1014 RSC;
(c) M. R. Detty, P. B. Merkel, S. L. Gibson and R. Hilf, Oncol. Res., 1992, 4, 367 CAS;
(d) D. A. Bellnier, D. N. Young, M. R. Detty, S. Camacho and A. R. Oseroff, Photochem. Photobiol., 1999, 70, 630 CrossRef CAS PubMed;
(e) K. A. Leonard, J. P. Hall, M. I. Nelen, S. R. Davies, S. O. Gollnick, S. Comacho, A. R. Oseroff, S. L. Gibson, R. Hilf and M. R. Detty, J. Med. Chem., 2000, 43, 4488 CrossRef CAS PubMed;
(f) S. W. Kaldor, V. J. Kalish, J. F. Davies, B. V. Shetty, J. E. Fritz, K. Appelt, J. A. Burgess, K. M. Campanale, N. Y. Chirgadze, D. K. Clawson and B. A. Dressman, J. Med. Chem., 1997, 40, 3979 CrossRef CAS PubMed.
-
(a) D. A. Boyd, Angew. Chem., Int. Ed., 2016, 55, 15486 CrossRef CAS PubMed;
(b) D. K. Dang, C. Sundaram, Y. L. T. Ngo, W. M. Choi, J. S. Chung, E. J. Kim and S. H. Hur, Dyes Pigm., 2019, 165, 327 CrossRef CAS;
(c) P. Gaur, A. Kumar, G. Dey, R. Kumar, S. Bhattacharyya and S. Ghosh, ACS Appl. Mater. Interfaces, 2016, 8, 10690 CrossRef CAS PubMed;
(d) G. Wang, Q. Guo, D. Chen, Z. Liu, X. Zheng, A. Xu, S. Yang and G. Ding, ACS Appl. Mater. Interfaces, 2018, 10, 5750 CrossRef CAS PubMed;
(e) S. Yang, J. Sun, P. He, X. Deng, Z. Wang, C. Hu, G. Ding and X. Xie, Chem. Mater., 2015, 27, 2004 CrossRef CAS.
-
(a) Metal-Catalyzed Cross-Coupling Reactions, ed. A. D. Meijere and F. Diederich, Wiley-VCH, Weinheim, 2004 Search PubMed;
(b) Handbook of Organopalladium Chemistry for Organic Synthesis, ed. E. I. Negishi and A. De Meijere, Wiley-Interscience, New York, 2002 Search PubMed.
-
(a) J. F. Hartwig, Acc. Chem. Res., 2008, 41, 1534 CrossRef CAS PubMed;
(b) T. Kondo and T. Mitsudo, Chem. Rev., 2000, 100, 3205 CrossRef CAS PubMed;
(c) S. V. Ley and A. W. Thomas, Angew. Chem., Int. Ed., 2003, 42, 5400 CrossRef CAS PubMed.
- Y. Yang, W. Hou, L. Qin, J. Du, H. Feng, B. Zhou and Y. Li, Chem.–Eur. J., 2014, 20, 416 CrossRef CAS PubMed.
-
(a) S. Guin, A. Deb, P. Dolui, S. Chakraborty, V. K. Singh and D. Maiti, ACS Catal., 2018, 8, 2664 CrossRef CAS;
(b) S. Yu, B. Wan and X. Li, Org. Lett., 2015, 17, 58 CrossRef CAS PubMed;
(c) S. Jana, P. Aseeva and R. M. Koenigs, Chem. Commun., 2019, 55, 12825 RSC;
(d) M. Arisawa, R. Suzuki, K. Ohashi and M. Yamaguchi, Asian J. Org. Chem., 2020, 9, 553 CrossRef CAS;
(e) X. Zhao, Z. Yu, S. Yan, S. Wu, R. Liu, W. He and L. Wang, J. Org. Chem., 2005, 70, 7338 CrossRef CAS PubMed;
(f) Q. Chen, P. Wang, T. Yan and M. Cai, J. Organomet. Chem., 2017, 840, 38 CrossRef CAS;
(g) P. Franzmann, S. B. Beil, D. Schollmeyer and S. R. Waldvogel, Chem.–Eur. J., 2019, 25, 1936 CrossRef CAS PubMed;
(h) M. Wang and L. Wang, Adv. Synth. Catal., 2009, 351, 1586 CrossRef CAS;
(i) R. Chatterjee, A. Mukherjee, S. Santra, K. R. Zyryanov and G. V. Majee, Tetrahedron, 2019, 43, 130624 CrossRef;
(j) F. H. Cui, J. Chen, S. X. Su, Y. L. Xu, H. S. Wang and Y. Pan, Adv. Synth. Catal., 2017, 359, 3950 CrossRef CAS;
(k) E. Q. Luz, D. Seckler, J. S. Araujo, L. Angst, D. B. Lima, E. A. M. Rios, R. R. Ribeiro and D. S. Rampon, Tetrahedron, 2019, 75, 1258 CrossRef CAS;
(l) H. Qiao, B. Sun, Q. Yu, Y. Y. Huang, Y. Zhou and F. L. Zhang, Org. Lett., 2019, 21, 6914 CrossRef CAS PubMed.
- G. Song, F. Wang and X. Li, Chem. Soc. Rev., 2012, 41, 3651 RSC.
- W. Ma, N. Kaplaneris, X. Fang, L. Gu, R. Mei and L. Ackermann, Org. Chem. Front., 2020, 7, 1022 RSC.
- Y. Yang, W. Hou, L. Qin, J. Du, H. Feng, B. Zhou and Y. Li, Chem.–Eur. J., 2014, 20, 416 CrossRef CAS PubMed.
- S. Yu, B. Wan and X. Li, Org. Lett., 2015, 17, 58 CrossRef CAS PubMed.
-
(a) S. Yang, B. Feng and Y. Yang, J. Org. Chem., 2017, 82, 12430 CrossRef CAS PubMed;
(b) W. Xie, B. Li and B. Wang, J. Org. Chem., 2016, 81, 396 CrossRef CAS PubMed;
(c) S. Maity, U. Karmakar and R. Samanta, Chem. Commun., 2017, 53, 12197 RSC;
(d) F. Wang, X. Yu, Z. Qi and X. Li, Chem.–Eur. J., 2016, 22, 511 CrossRef CAS PubMed;
(e) D. Wang, K. Zhou, J. Zhang and Y. Zhao, Org. Chem. Front., 2020, 7, 3229 RSC;
(f) T. K. Vats, A. Mishra and I. Deb, Adv. Synth. Catal., 2018, 360, 2291 CrossRef CAS;
(g) Y. R. Jian, Z. C. Yan, Z. Xiang, Y. S. Xiong and X. M. Duan, Org. Biomol. Chem., 2021, 19, 2901 RSC.
-
(a) M. Gupta, P. Kumar, V. Bahadur, K. Kumar, V. S. Parmar and B. K. Singh, Eur. J. Org. Chem., 2018, 896 CrossRef CAS;
(b) P. Kumar, M. Gupta, V. Bahadur, V. S. Parmar and B. K. Singh, Eur. J. Org. Chem., 2018, 1552 CrossRef CAS;
(c) M. Gupta, S. Kumar, P. Kumar, A. K. Singh, V. Bahadur and B. K. Singh, ChemistrySelect, 2019, 4, 13992 CrossRef CAS;
(d) R. S. K. Lalji, P. Kumar, M. Gupta, V. S. Parmar and B. K. Singh, Adv. Synth. Catal., 2020, 362, 552 CrossRef CAS;
(e) P. Kumar, S. Dutta, S. Kumar, V. Bahadur, E. V. V. Eycken, K. S. Vimaleswaran, V. S. Parmar and B. K. Singh, Org. Biomol. Chem., 2020, 18, 7987 RSC;
(f) S. Kumar, R. S. K. Lalji, M. Gupta, P. Kumar, R. Kumar and B. K. Singh, Org. Biomol. Chem., 2022, 20, 8944 RSC;
(g) S. Kumar, M. Gupta, R. S. K. Lalji and B. K. Singh, RSC Adv., 2023, 13, 2365 RSC.
- H. J. Reich and R. J. Hondal, ACS Chem. Biol., 2016, 11, 821 CrossRef CAS PubMed.
-
(a) L. L. Hegedus and R. W. McCabe, Catalyst Poisoning, Marcel Dekker, New York, 1984 Search PubMed;
(b) C. H. Bartholomew, Appl. Catal., A, 2001, 212, 17 CrossRef CAS.
- S. A. El-Hawash, N. S. Habib and M. A. Kassem, Arch. Pharm., 2006, 339, 564 CrossRef CAS PubMed.
- J. H. Chu, S. T. Chen, M. F. Chiang and M. J. Wu, Organometallics, 2015, 34, 953 CrossRef CAS.
-
(a) A. Mandal, S. Dana, H. Sahoo, G. S. Grandhi and M. Baidya, Org. Lett., 2017, 19, 2430 CrossRef CAS PubMed;
(b) E. M. Simmons and J. F. Hartwig, Angew. Chem., Int. Ed., 2012, 51, 3066 CrossRef CAS PubMed.
-
(a) Y. Yang, W. Hou, L. Qin, J. Du, H. Feng, B. Zhou and Y. Li, Chem.–Eur. J., 2014, 20, 416 CrossRef CAS PubMed;
(b) L. Yu, J. Meng, L. Xia and R. Guo, J. Org. Chem., 2009, 74, 5087 CrossRef CAS PubMed;
(c) A. M. S. Recchi, D. F. Back and G. Zeni, Tetrahedron, 2021, 90, 132188 CrossRef CAS.
-
(a) S. H. Park, J. Kwak, K. Shin, J. Ryu, Y. Park and S. Chang, J. Am. Chem. Soc., 2014, 136, 2492 CrossRef CAS PubMed;
(b) J. Wang, D. Qin, J. Lan, Y. Cheng, S. Zhang, Q. Guo, J. Wu, D. Wu and J. You, Chem. Commun., 2015, 51, 6337 RSC;
(c) X. Huang, Y. Wang, J. Lan and J. You, Angew. Chem., Int. Ed., 2015, 54, 9404 CrossRef CAS PubMed;
(d) J. Q. Wu, S. S. Zhang, H. Gao, Z. Qi, C. J. Zhou, W. W. Ji, Y. Liu, Y. Chen, Q. Li, X. Li and H. Wang, J. Am. Chem. Soc., 2017, 139, 3537 CrossRef CAS PubMed;
(e) X. Wang, T. Gensch, A. Lerchen, C. G. Daniliuc and F. Glorius, J. Am. Chem. Soc., 2017, 139, 6506 CrossRef CAS PubMed.
|
| This journal is © The Royal Society of Chemistry 2023 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence ab,
Princea,
Mohit Gupta
ab,
Princea,
Mohit Gupta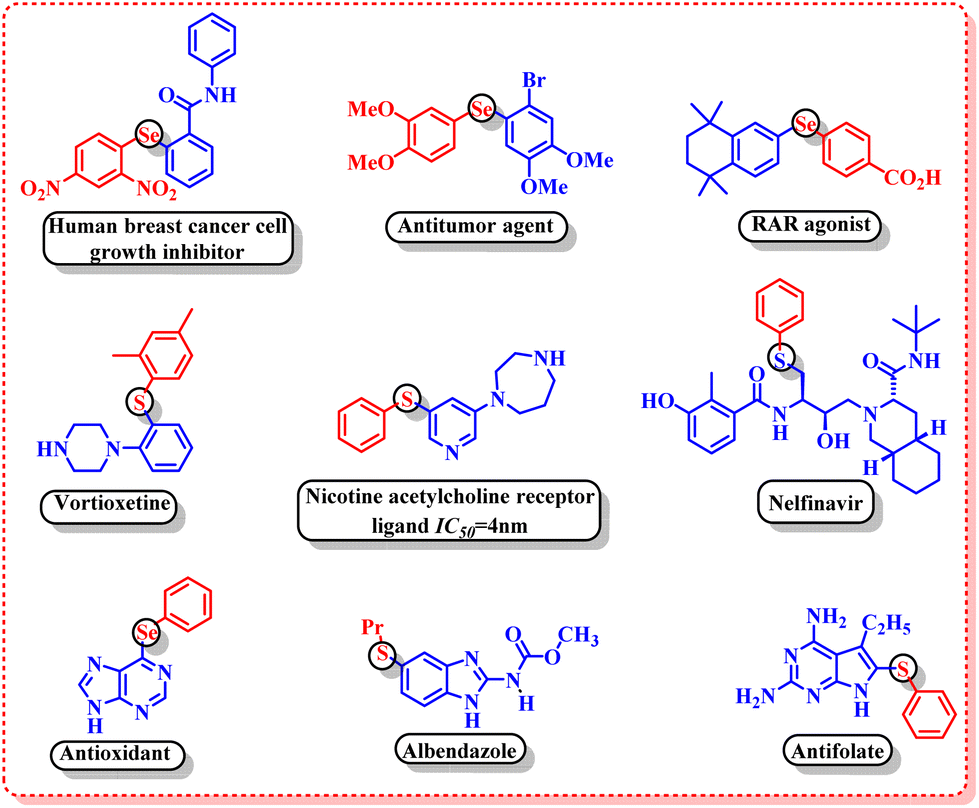
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :
: