
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceBiodegradable porous polymeric drug as a drug delivery system: alleviation of doxorubicin-induced cardiotoxicity via passive targeted release†
Qiuhong Jiao‡
a,
Baoting Liu‡a,
Xiufeng Xu‡b,
Tao Huang‡a,
Bufan Caoa,
Lide Wanga,
Qingguo Wanga,
Ailing Dua,
Jingtian Li*a,
Baolong Zhou *c and
Tao Wang*a
*c and
Tao Wang*a
aDepartment of Cardiology, Affiliated Hospital of Weifang Medical University, Weifang 261031, Shandong, China. E-mail: wfkljt@126.com; wangtao575.happy@163.com
bDepartment of Geriatrics, Affiliated Hospital of Weifang Medical University, Weifang 261031, Shandong, China
cSchool of Pharmacy, Weifang Medical University, Weifang 261031, Shandong, China. E-mail: zhoubaolong@wfmc.edu.cn
First published on 13th February 2023
Abstract
Doxorubicin (DOX) is an effective chemotherapeutic drug developed against a broad range of cancers, and its clinical applications are greatly restricted by the side effects of severe cardiotoxicity during tumour treatment. Herein, the DOX-loaded biodegradable porous polymeric drug, namely, Fc-Ma-DOX, which was stable in the circulation, but easy to compose in the acidic medium, was used as the drug delivery system avoiding the indiscriminate release of DOX. Fc-Ma was constructed via the copolymerization of 1,1′-ferrocenecarbaldehyde with D-mannitol (Ma) through the pH-sensitive acetal bonds. Echocardiography, biochemical parameters, pathological examination, and western blot results showed that DOX treatment caused increased myocardial injury and oxidative stress damage. In contrast, treatment with Fc-Ma-DOX significantly reduced myocardial injury and oxidative stress by DOX treatment. Notably, in the Fc-Ma-DOX treatment group, we observed a significant decrease in the uptake of DOX by H9C2 cells and a significant decrease in reactive oxygen species (ROS) production.
1. Introduction
Doxorubicin (DOX) is a powerful antitumour drug against a broad range of cancers.1 However, the high risk of complications posed during anticancer therapy, such as cardiotoxicity, anaphylaxis, bone marrow suppression, rash, hair loss, mouth inflammation and vomiting, particularly cardiotoxicity, severely limits the clinical applications of DOX.2–4 DOX has been widely reported to induce dose-dependent, progressive, and potentially lethal myocardial damage. Despite decades of research, the molecular mechanisms of DOX-induced cardiotoxicity remain controversial5 There are several mechanisms that have been proposed for doxorubicin-induced cardiotoxicity; free radical generation and apoptosis are the most widely reported. Apart from this, other mechanisms are also involved in DOX-induced cardiotoxicity such as impaired mitochondrial function, a perturbation in iron regulatory protein, disruption of Ca2+ homeostasis, autophagy, the release of nitric oxide and inflammatory mediators and altered gene and protein expression that involved apoptosis.6 Therefore, for the real application of DOX in the clinic, development of a new drug delivery method is urgently needed to protect against the cardiotoxicity induced by DOX.In recent years, various methods have been developed to reduce the cardiotoxic potency of DOX, including the concurrent administration of cardioprotective agents (e.g., dexrazoxane, beta-blockers, angiotensin-converting enzyme inhibitors, and angiotensin receptor blockers),7–9 the use of anthracycline analogues and the employment of novel drug delivery systems (polymeric, lipid and inorganic nanoparticle-based nanoformulations, nanogels, nanotubes and nanocrystals).10 However, there are still many issues that need to be addressed. For example, as the only FDA-approved cardioprotective agent for anthracycline-induced cardiotoxicity, the simultaneous administration of dexrazoxane with DOX might increase the incidence of second primary malignancies (myelodysplastic syndrome and acute myeloid leukaemia) in paediatric patients.11–13 Cardiotoxicity can be minimized with the use of a carrier-based DOX delivery system, which could alter the pharmacological distribution of DOX, ultimately reducing its accumulation in the heart.14 Lipophilic lipid formulations with high affinity for skin commonly cause palmar-plantar erythrodysesthesia, known as hand foot syndrome (HFS).15,16 Meanwhile, inorganic carriers, mainly metal-based particles, face the difficulty of biodegradation.17 Additionally, traditional drug delivery systems suffer from instability, complexity of synthesis, and low drug efficacy, which greatly limit their treatment efficiency in tumours.18,19 Hence, it is necessary to develop new drug delivery systems that not only overcome the issue of cardiotoxicity but also retain or even enhance the therapeutic efficacy of anticancer drugs.
Porous organic polymers (POPs) represent an emerging class of lightweight multifunctional network materials constructed by covalently linking organic building blocks.20 The pure organic structure endows POPs with unique physicochemical characteristics, such as good biocompatibility, large specific surface area, high porosity, and excellent stability, as well as customizable structure and functions.21,22 Over decades of development, thousands of POPs with numerous structures and functions have been developed for various applications in fields ranging from the environment to biomedicine, involving gas storage and separation, heterogeneous catalysis, biomedicine, sensing, optoelectronics, and energy storage and conversion.23–27 The intrinsic organic and porous structure of POPs not only facilitates drug loading but is also favourable for the desired postmodification, further enhancing their application properties.28,29
In this work, a biocompatible ferrocenyl-based (Fc-Ma) porous drug delivery system (Fc-Ma-DOX) connected by pH-sensitive acetal bonds was designed for the controllable release of DOX to tumour sites. The drug delivery system could ultimately alter the pharmacological distribution of DOX, thereby reducing the myocardial injury caused by it.30 For the first time, we concurrently studied the potential of a POP to enhance cancer therapy and minimize DOX-induced cardiotoxicity. Both the biochemical parameters and pathological examination results demonstrated that Fc-Ma could serve as a smart carrier with a specific acidic tumour microenvironment (TME)-induced release behaviour, which could effectively avoid the early release of DOX in normal tissues, significantly reducing the aggregation of DOX in normal tissues. Therefore, the drug delivery system could reduce myocardial oxidative stress and apoptosis by reducing the DOX-induced overproduction of oxygen free radicals, significantly lowering the cardiotoxicity induced by DOX (Fig. 1).
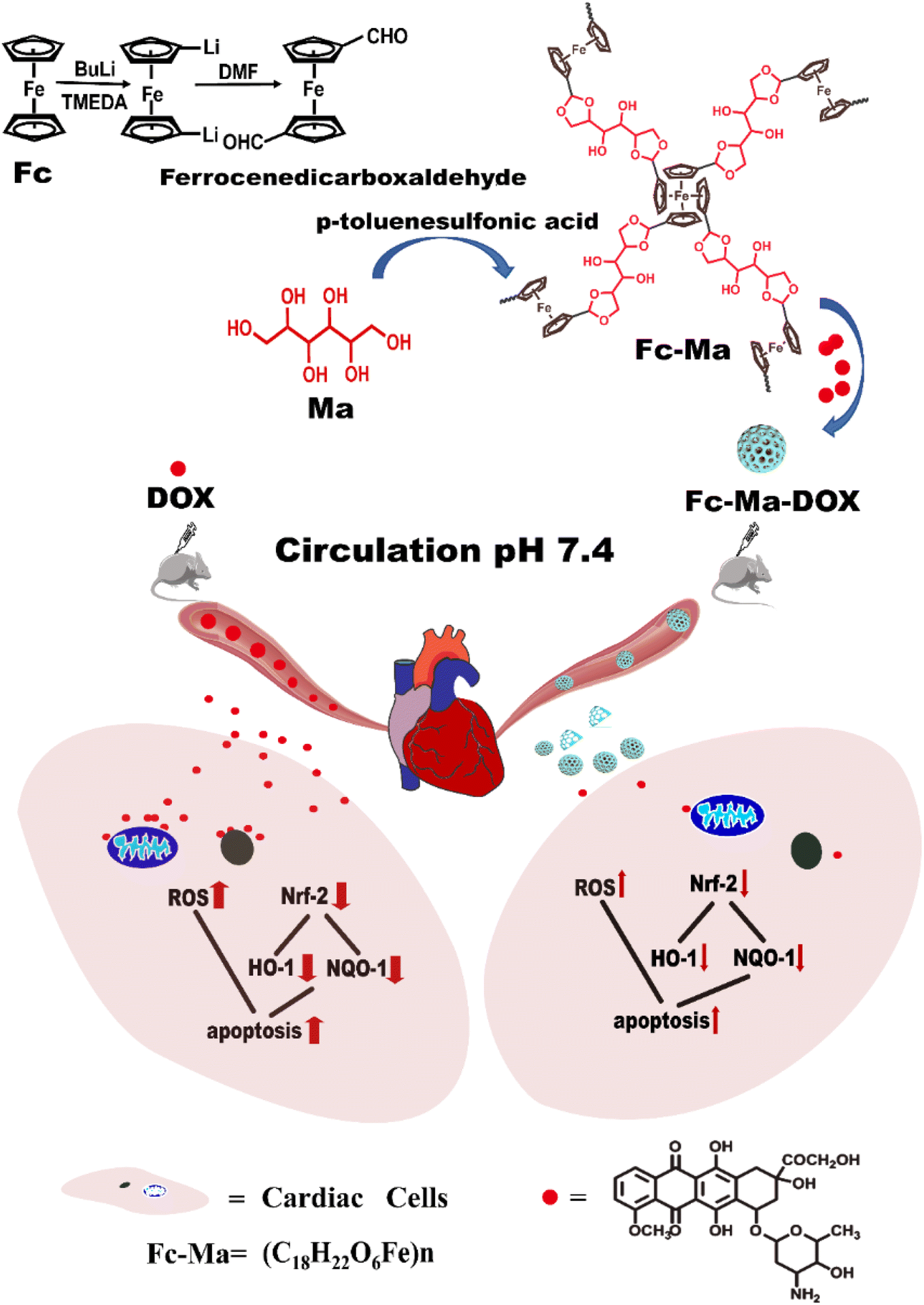 | ||
| Fig. 1 Schematic illustrating the formation of the Fc-Ma-DOX and mechanism to reduce cardiotoxicity. | ||
2. Materials and methods
2.1. Synthesis of Fc-Ma
The biocompatible ferrocenyl-based (Fc-Ma) porous drug delivery system was formed according to our previous studies.31 Briefly, ferrocene (Fc) pre-dissolved in n-hexane, n-butyllithium solution and tetramethyl ethylenediamine was added to a round bottom flask with a magnetic stirrer. Under continuous stirring, anhydrous dimethylformamide (DMF) was slowly injected into the reaction system. 1,1′-Ferrocenecarbaldehyde was obtained as a red solid after the full reaction.Under ultrasonication, 1,1′-ferrocenecarbaldehyde (0.50 g, 2.07 mmol), D-mannitol (Ma) (0.38 g, 2.07 mmol), and p-methylbenzene sulfonic acid (7.64 g, 40.16 mmol) were added to the reaction tube. The reaction system was heated to 180 °C. After 72 h, a black solid was obtained and named Fc-Ma. Fc-Ma and DOX were dissolved in deionized water, and the prepared Fc-Ma-DOX was obtained after continuous stirring.
2.2. Characterization of Fc-Ma and Fc-Ma-DOX
Fourier transform infrared spectroscopy (FTIR) was performed on KBr pellets in the range from 4000 to 400 cm−1 using a Spectrum Spotlight 400 system. Transmission electron microscopy (TEM, Tecnai G2 20 TWIN) images were obtained by dipping the prepared samples on a Cu-net. The morphology was further confirmed by employing field-emission scanning electron microscopy (FESEM, Ultra 55).2.3. In vitro DOX release
DOX release from Fc-Ma-DOX was measured under different pH conditions (pH 5.5, 6.5 and 7.4) by the dialysis method. Briefly, Fc-Ma-DOX was transferred into a dialysis bag (MWCO: 8000–14000) and immersed in 50 mL of phosphate-buffered saline (PBS) with different pH conditions at 37 °C. At fixed time intervals, 1 mL of the solution outside the dialysis bag was removed for fluorescent measurements, and an equal volume of fresh buffer was added. The amount of DOX released from the POPs was determined using an automatic enzyme immunoassay instrument (Multiskan Go, America) at 481 nm by comparison with DOX standard curves.2.4. Animals and treatments
All animal experiments were carried out with the approval of the Institutional Animal Care and Use Committee of Weifang Medical University. Male C57BL/6 J mice (8–10 weeks old) were purchased from the Institute of Medical Laboratory Animal Center, Weifang Medical University (Shandong, China) and kept in a specific pathogen-free barrier system with free access to the standard diet and water. After acclimation for one week, mice were randomly divided into 4 groups: (I) control group; (II) Fc-Ma group; (III) DOX group; and (IV) Fc-Ma-DOX group. To mimic chronic DOX exposure, the mice in the DOX groups (n = 10 for each group) were injected intraperitoneally with free DOX (5 mg kg−1) every week for 4 weeks.32 The mice in the Fc-Ma and Fc-Ma-DOX groups (n = 10 for each group) were injected intraperitoneally with Fc-Ma and Fc-Ma-DOX (Fc-Ma-DOX was administered at a dose with an equivalent amount of DOX to that of free DOX, and the DOX concentration in Fc-Ma-DOX was calculated based on drug loading31) every week for 4 weeks. The control mice received saline, and the animals were weighed once a week. One week after the last injection, cardiac functions in the mice were examined, all animals were sacrificed, and blood samples were harvested. The hearts were rapidly removed and weighed. Part of the heart tissue was fixed, and the remaining samples were stored at −80 °C for further assays. To observe the survival rate in mice exposed to DOX, the mice (n = 5–15 for each group) were injected intraperitoneally with free DOX (5 mg kg−1, every three days), Fc-Ma, Fc-Ma-DOX (at a DOX equivalent dose of 5 mg kg−1) and saline 7 times. After the first injection of DOX, the mice were observed daily for 30 days.2.5. Echocardiography
After mice were anaesthetized with 1.5% isoflurane, echocardiography was conducted using a high-resolution Vevo 3100LT (Visual Sonics, Toronto, Canada) imaging system to analyze cardiac functions. M-mode images of the left ventricle were recorded at the papillary muscle level. Ejection fraction (EF), fractional shortening (FS), left ventricular end-diastolic dimension (LVEDD) and left ventricular end-systolic dimension (LVESD) were measured.33 At least three separate cardiac cycles were averaged for the echocardiographic measurements.2.6. Histopathology
Myocardial tissues were fixed in 4% paraformaldehyde for 24 hours at room temperature. The fixed tissues were embedded in conventional paraffin wax and cut into 5 μm sections, and then the sections were stained with haematoxylin and eosin (H&E). Horizontal heart sections were stained with Masson's trichrome blue (Sigma Aldrich, USA) for the analysis of fibrosis. Specimens were observed under a light microscope (400 amplification: Nikon Tokyo, Japan).2.7. Biochemical analysis
The blood was centrifuged at 3000 rpm for 10 min, the supernatant was then taken, and the serum levels of lactate dehydrogenase (LDH), cardiac isoform of troponin T (cTnT) and the creatine kinase isoenzyme (CK-MB) were detected by performing enzyme-linked immunosorbent assay (ELISA) in strict accordance with the manufacturer's instructions (JianCheng, Nanjing, China). Oxidative stress indices, such as malonyl dialdehyde (MDA), superoxide dismutase (SOD), and glutathione peroxidase (GSH-Px), were analyzed with commercial kits (Beyotime, Shanghai, China). The experiment was performed in triplicate.2.8. Cell culture
H9C2 cells were maintained in Dulbecco's modified Eagle's medium (DMEM) (Gibco, NY, USA) supplemented with 10% fetal bovine serum (FBS, Life Technologies, UK) and antibiotics (100 IU mL−1 penicillin and 100 mg mL−1 streptomycin) in a humidified atmosphere of 5% CO2 and 95% O2 at 37 °C.2.9. Intracellular ROS detection
Superoxide production in the myocardium of mice and H9C2 cells was detected by dihydroethidium (DHE) staining and 2′,7′-dichlorofluorescin diacetate (DCFH-DA) staining according to the manufacturer's instructions, respectively (Beyotime, Shanghai, China). Briefly, frozen heart sections were stained with DHE (10 μmol L−1), while H9C2 cells were incubated with DCFH-DA (10 μmol L−1) in a dark chamber at 37 °C for 30 min, and then fluorescence microscopy (Olympus; Tokyo, Japan) was used to examine the sections in a blinded manner.2.10. DOX-induced cell injury
A Cell Counting Kit-8 (CCK-8) assay kit (Dojindo Laboratories, Tokyo, Japan) was used to determine cell viability following the manufacturer's instructions. H9C2 cells were seeded into 96-well plates at a density of 1 × 105 cells per well, incubated for 48 hours, and then pretreated for 24 hours with the indicated concentrations of free DOX, Fc-Ma and Fc-Ma-DOX. The cells in the control groups were treated with 0.1% dimethyl sulfoxide (DMSO, Sigma–Aldrich Chemical Co., USA) using an automatic enzyme immunoassay instrument (Multiskan Go, America) to measure the absorbance at a wavelength of 450 nm. The experiments were repeated 3 times.2.11. DOX uptake in H9C2 cells
H9C2 cells were seeded into 6-well plates at a density of 6 × 104 cells per well in 1 mL DMEM and incubated for 48 hours. Free DOX and Fc-Ma-Dox were added to each well. After 6 hours of further incubation, the culture medium was removed, and the cells were washed three times with PBS buffer. The fluorescence was observed under an inverted fluorescence microscope (Olympus; Tokyo, Japan).2.12. TUNEL staining
H9C2 cells were fixed with 4% paraformaldehyde, and a terminal deoxynucleotidyl transferase-mediated nick-end labelling (TUNEL) assay was performed according to the manufacturer's protocol to detect apoptosis (Promega Corporation, Madison, WI, USA). The nucleus was labelled with DAPI.2.13. Western blotting assay
Immunoblotting was performed as previously described.34 In brief, protein lysates were extracted from frozen heart samples or H9C2 cells. A BCA Protein Assay Kit (Thermo Fisher Scientific, Waltham, MA, USA) was used to determine the protein concentrations of the samples. Equal amounts of protein were separated by 8–12% SDS–PAGE and then transferred onto PVDF membranes (Merck Millipore, MA, USA). The membranes were then blocked with 5% bovine serum albumin for 2 hours at room temperature and incubated at 4 °C overnight with primary antibodies (anti-HO-1, 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1000, Abcam; anti-Nrf2, 1:1000, Abcam; anti-NQO-1, 1:1000, Abcam; and anti-GAPDH, 1:1000, Abcam). After incubation with horseradish peroxidase-conjugated secondary antibodies (1:2000, Abcam, Cambridge, MA) for 60 min, immune reactive protein bands were detected by a BioSpectrum Gel Imaging System (UVP, California, USA) and quantified by Odyssey Image Studio 5.1. GAPDH was used as a loading control for whole cellular protein.
:1000, Abcam; anti-Nrf2, 1:1000, Abcam; anti-NQO-1, 1:1000, Abcam; and anti-GAPDH, 1:1000, Abcam). After incubation with horseradish peroxidase-conjugated secondary antibodies (1:2000, Abcam, Cambridge, MA) for 60 min, immune reactive protein bands were detected by a BioSpectrum Gel Imaging System (UVP, California, USA) and quantified by Odyssey Image Studio 5.1. GAPDH was used as a loading control for whole cellular protein.
2.14. Statistical analysis
GraphPad Prism 8.0 (GraphPad, USA) statistical software was used for data analysis, and one-way analysis of variance (one-way ANOVA) was used for comparisons among multiple groups. A t test was used to analyze differences between two groups. A repeated-measures ANOVA was used to examine the alteration in body weight. Data are expressed as the mean ± standard deviation (x ± s), and statistical significance was accepted at values of p < 0.05.3. Results and discussions
3.1. Morphology and characterization of Fc-Ma and Fc-Ma-DOX
The route for the preparation of Fc-Ma-DOX is shown in Fig. S1.† Fc-Ma was obtained via the copolymerization of ferrocenedicarboxaldehyde and D-mannitol (Ma) in 1,2-dichlorobenzene with catalytic p-toluene sulfonic acid, which was further used as the carrier for the noncovalent loading of DOX through the formation of H-bonds, π–π conjugation, and porous adsorption.35,36The successful preparation of the porous networks was concurrently verified by the absence of a characteristic vibration band of the aldehyde group in Fc at 1690 cm−1, and the appearance of new absorption peaks ascribed to the tensional vibration of the cyclic acetal appeared at 1704 cm−1 (Fig. S2a†), corresponding to the acetal bonds. Notably, the characteristic absorption peaks of DOX located at 1621, 1407, and 1007 cm−1 were also observed in Fc-Ma-DOX (Fig. S2b†), indicating the successful loading of DOX into the carrier. In addition, according to our previous study, the change in the porosity also confirmed the loading of DOX into Fc-Ma.31 The morphology of Fc-Ma and Fc-Ma-DOX was confirmed by scanning electron microscopy (SEM) and transmission electron microscopy (TEM), respectively. As shown in Fig. S3,† tiny near-spherical particles with numerous macropores were observed by SEM of Fc-Ma, and they were well retained after the entry of DOX (Fig. S3a†). Furthermore, widely distributed pores were also observed in the TEM images (Fig. S3b†). Due to the presence of a pH-labile acetal bond in Fc-Ma-DOX, it should show pH-responsive DOX release behavior, with DOX release increasing with decreasing pH. As exhibited in Fig. S4,† the cumulative release amount of DOX (72 h) reached 80.3% and 63.9% in the buffers at pH 5.5 and 6.5 (pH of tumour tissues), respectively, which was much higher than that in the neutral media (pH 7.4, equal to blood plasma). Overall, the evaluation results indicated that the pH-triggered drug delivery system can evade normal tissues, which would be critically important to reduce the toxicity and side effects of anticancer drugs in normal tissues.
3.2. Verification of DOX-induced cardiotoxicity in a mouse model
One of the manifestations of chronic DOX-induced cardiotoxicity is congestive heart failure.37 Accordingly, we conducted echocardiography to investigate cardiac dysfunction in mice induced by DOX (Fig. 2a). Compared with the control group, echocardiography showed that EF and FS were markedly decreased, while LVESD and LVEDD were increased in DOX-treated mice (Fig. 2b–e), suggesting that a marked decrease in left ventricular contractile function was caused by DOX. Pathological changes in the myocardial tissue of mice were observed by H&E staining, which showed that the myocardial tissue cells from the mice treated with DOX were disordered with vacuolar degeneration and severe cell necrosis, while the myocardial tissue cells of the control groups appeared grossly normal (Fig. 2f). Masson's trichrome staining (Fig. 2g) revealed that the cardiac collagen fraction was significantly increased in DOX-treated mice. As shown in Fig. 2h–j, compared with those in the control group, the levels of serum cTnT, CK-MB, and LDH were obviously increased in mice injected with DOX, demonstrating severe damage to the cardiac membrane integrity. These results indicated the successful establishment of DOX-induced cardiotoxicity in a mouse model.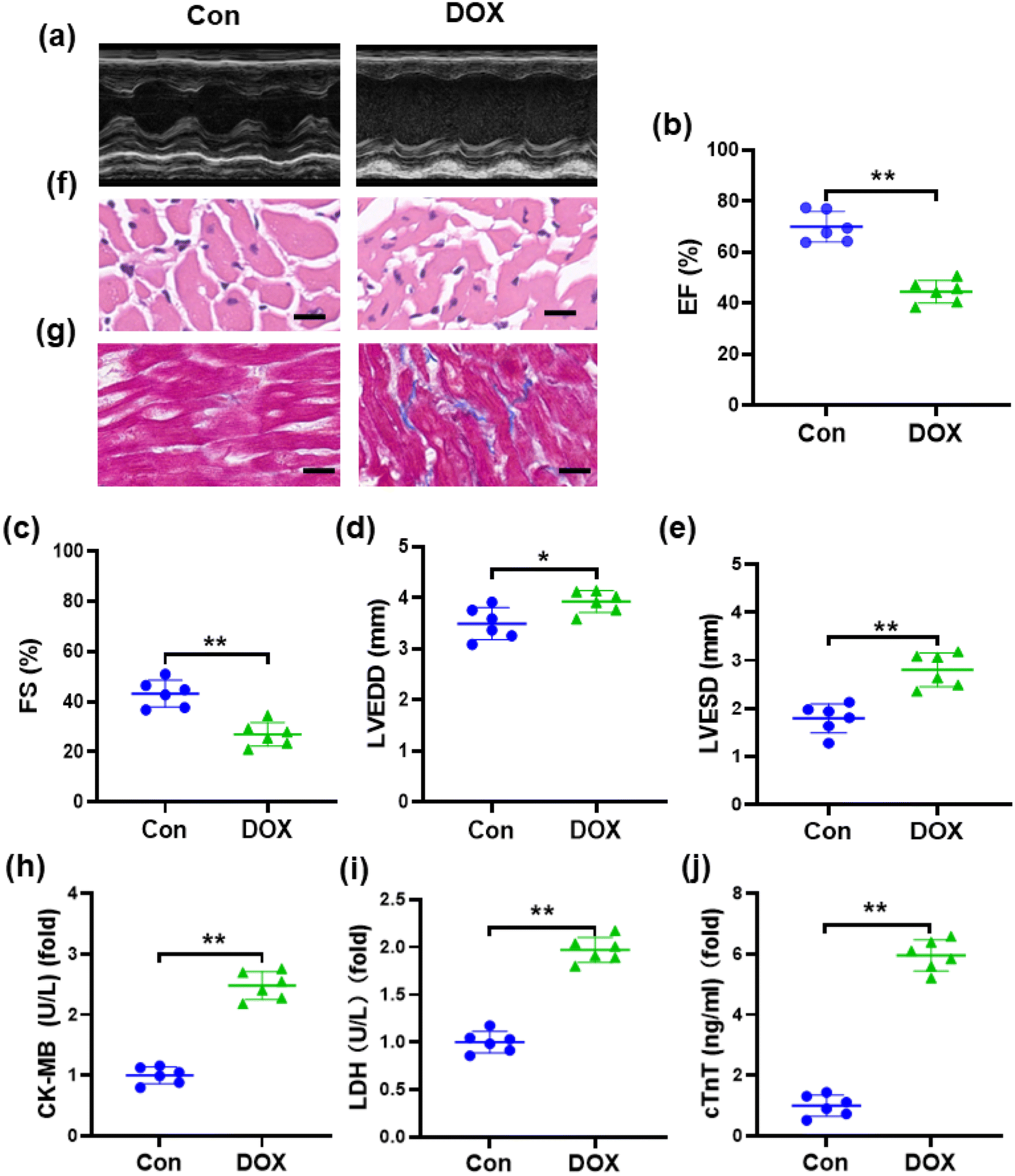 | ||
| Fig. 2 Verification of the DOX induced cardiotoxicity in mice model: (a) representative M-mode echocardiography of left ventricular chamber change from mice (n = 6); (b)–(e) ejection fraction (EF%), fraction shortness (FS%), left ventricular end-diastolic dimension (LVEDD) and left ventricular end-systolic dimension (LVESD) of mice as determined via echocardiography (n = 6); (f) representative images of H&E staining (n = 6, scale bar 20μm); (g) representative images of Masson's trichrome staining (n = 6, scale bar, 20μm); (h)–(j) biochemical determination of CK-MB, LDH and cTnT serum levels (n = 6). Values represent the mean ± SD. *p < 0.05, **p < 0.01 vs. control group. | ||
3.3. DOX aggravates oxidative damage in a mouse model
Oxidative stress and reduced antioxidant status are thought to be primarily responsible for DOX-induced cardiotoxicity because myocardial tissues lack sufficient antioxidant mechanisms.38 Therefore, the reactive oxygen species (ROS) level in the heart was detected by DHE staining, which demonstrated that the injection of DOX resulted in increased ROS production in the heart of mice (Fig. 3a and b). To assess the effects of DOX on oxidative stress, typical indicators of oxidative stress, such as SOD, MDA and GSH-Px, were also examined. Compared with the control group, MDA was significantly increased, while SOD and GSH-Px were decreased in the hearts of mice treated with DOX (Fig. 3c–e). Furthermore, nuclear factor erythroid 2-related factor 2 (Nrf-2), which is an emerging regulator of cellular resistance to oxidants controlling the basal and induced expression of an array of antioxidant response element-dependent genes, was also examined.39,40 As shown in Fig. 3f and g, compared with the control group, the expression of Nrf-2 was downregulated after DOX treatment. Accordingly, the expression levels of the downstream molecules haem oxygenase-1 (HO-1) and NADPH quinone oxidoreductase-1 (NQO-1) were also reduced.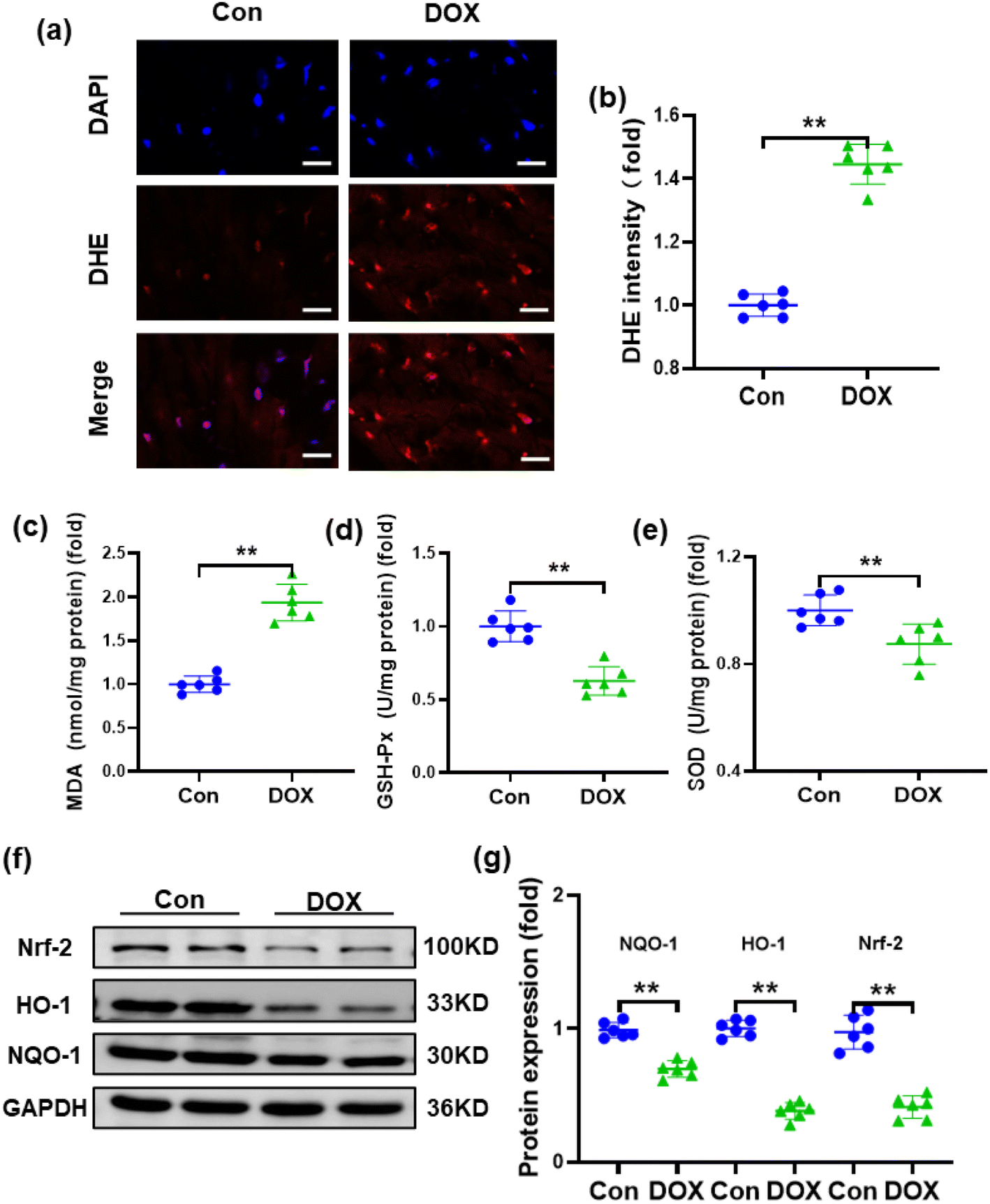 | ||
| Fig. 3 DOX aggravates oxidative damage in mice model: (a) and (b) representative DHE staining images and the quantitative results, from top to bottom, the images show cell nuclei stained by DAPI (blue), ROS fluorescence (red) and the merge of the two images (n = 6, scale bar 20μm); (c)–(e) the levels of GSH-Px, MDA and SOD in heart samples (n = 6); (f) and (g) Western blots and the statistical results of Nrf-2, HO-1 and NQO-1 proteins (n = 6). Values represent the mean ± SD. **p < 0.01 vs. control group. | ||
3.4. Fc-Ma-Dox alleviates doxorubicin-induced cardiomyopathy in vivo
To determine whether Fc-Ma-DOX treatment could effectively prevent DOX-induced heart dysfunction through the pH-responsive effect, we examined the effect of Fc-Ma-DOX treatment by echocardiography (Fig. 4a). Ejection fraction (EF) and fractional shortening (FS) were significantly increased and LVESD and LVEDD were significantly decreased in the mice treated with Fc-Ma-DOX compared with those given free DOX. Compared with the control group, none of these conditions were markedly varied in the Fc-Ma-treated group (Fig. 4b–e). In the histopathological examinations, the H&E staining results showed no differences between the Fc-Ma-DOX and Fc-Ma-treated groups and the control group, revealing normal heart histology, myocardial degeneration, and necrosis; occasional vacuolization was observed in the DOX-treated group (Fig. 4f). Myocardial fibrosis was significantly increased in the DOX-treated group compared to the control group and was decreased in the Fc-Ma-DOX-treated group compared to the DOX-treated group (Fig. 4g). Furthermore, typical biochemical markers, such as LDH, CK-MB and cTnT, were detected to evaluate the effect of Fc-Ma-DOX treatment on cardiac membrane integrity. As expected, the levels of these markers were significantly increased in the mice treated with DOX and were much higher than those in the mice treated with Fc-Ma-DOX. Meanwhile, as seen from Fig. 4h–j, no significant differences were detected in the myocardial levels of cTnT, LDH, or CK-MB for the mice treated with Fc-Ma compared with the control group. Additionally, we found that Fc-Ma-DOX treatment significantly attenuated DOX-induced body weight loss in mice, which raised the possibility for further clinical use (Fig. 4k). As shown in Fig. 4l, compared with the control group, the survival rate of mice treated with DOX was much lower, which was effectively alleviated by Fc-Ma-DOX treatment, and the survival rate of mice treated with Fc-Ma was not significantly different.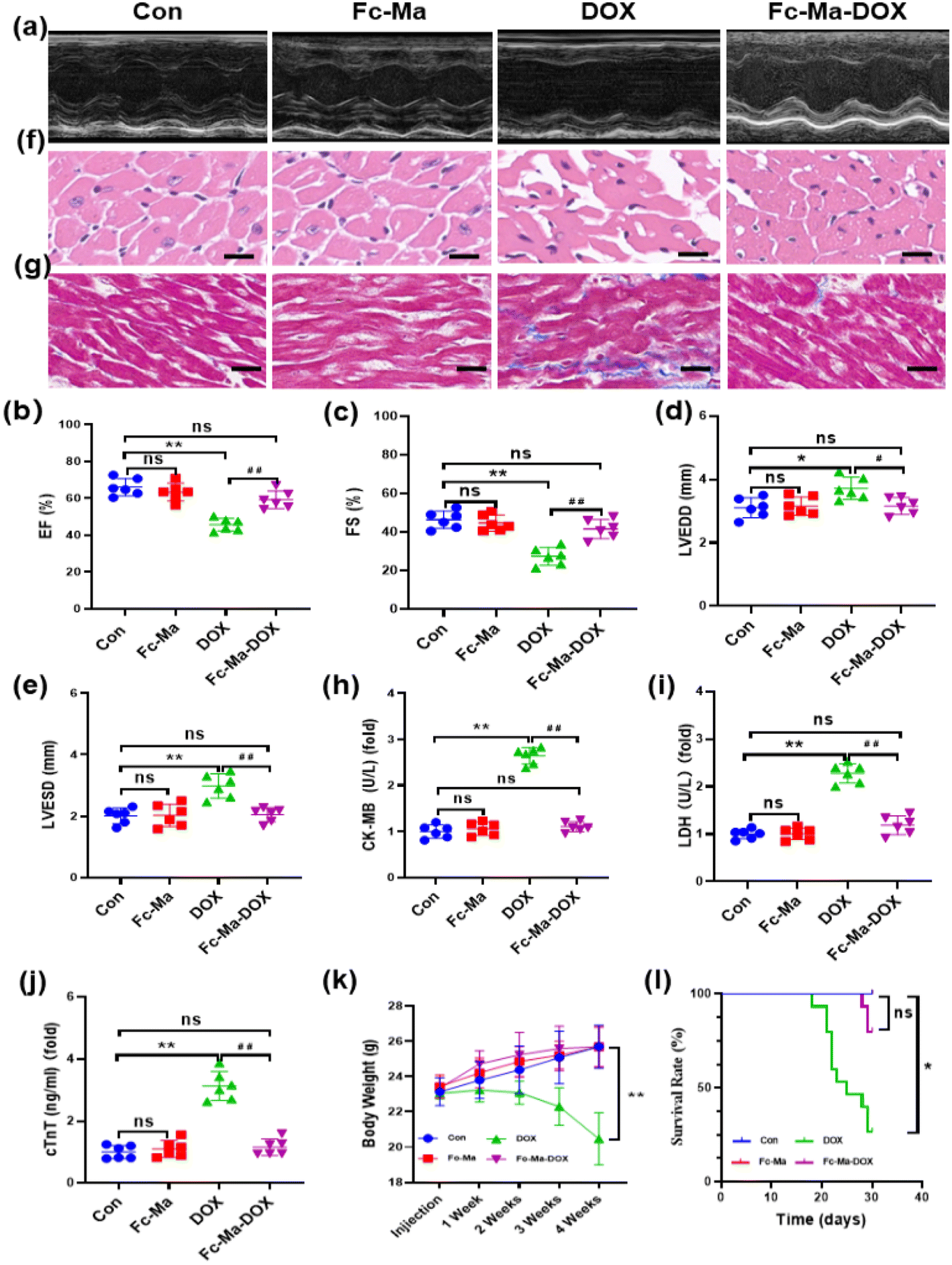 | ||
| Fig. 4 Fc-Ma-DOX alleviates Doxorubicin induced cardiomyopathy in vivo: (a) representative M-mode echocardiography of left ventricular chamber change from mice (n = 6); (b)–(e) ejection fraction (EF%), fraction shortness (FS%), left ventricular end-diastolic dimension (LVEDD) and left ventricular end-systolic dimension (LVESD) of mice as determined via echocardiography (n = 6); (f) H&E staining (n = 6, scale bar 50μm); (g) Masson's trichrome staining (n = 6, scale bar 50μm); (h)–(j) the levels of CK-MB, LDH and cTnT in serum (n = 6); (k) body weight change (n = 6); (l) the survival curves up to 30 days after the first DOX injection in mice (n = 5–15). Values represent the mean ± SD. *p < 0.05, **p < 0.01 vs. control group, #p < 0.05, ##p < 0.01 vs. DOX group, ns indicates no significance. | ||
3.5. Fc-Ma-DOX attenuated DOX-induced oxidative stress in vivo
Oxidative stress, as a chief component of the molecular and cellular determinants of doxorubicin cardiotoxicity, contributes to the development of DOX-related cardiac injury.6 As displayed in Fig. 5a and b, compared with the control group, DHE staining demonstrated that the mice treated with Fc-Ma displayed no significant differences in ROS generation. Cardiac ROS generation in mice was notably increased after DOX treatment; conversely, it was decreased by Fc-Ma-DOX treatment. In addition, the levels of important antioxidant enzymes, including SOD and GSH-Px, were significantly decreased in the mice treated with DOX. However, this downregulation was remarkably blocked by Fc-Ma-DOX treatment. Moreover, MDA, a biomarker related to lipid peroxidation, was significantly increased in the mice treated with DOX, but the abnormal accumulation of MDA was inhibited by Fc-Ma-DOX treatment. Compared with the control group, none of the antioxidant enzyme levels markedly varied in the Fc-Ma-treated group (Fig. 5c–e). Nrf-2 acts as a critical transcription factor to maintain redox homeostasis.41 Interestingly, Fig. 5f and g clearly shows that Fc-Ma-DOX treatment significantly attenuated DOX-induced downregulation of Nrf-2, HO-1 and NQO-1 in the heart, and the above protein levels were not significantly different in the Fc-Ma-treated group compared with the control group.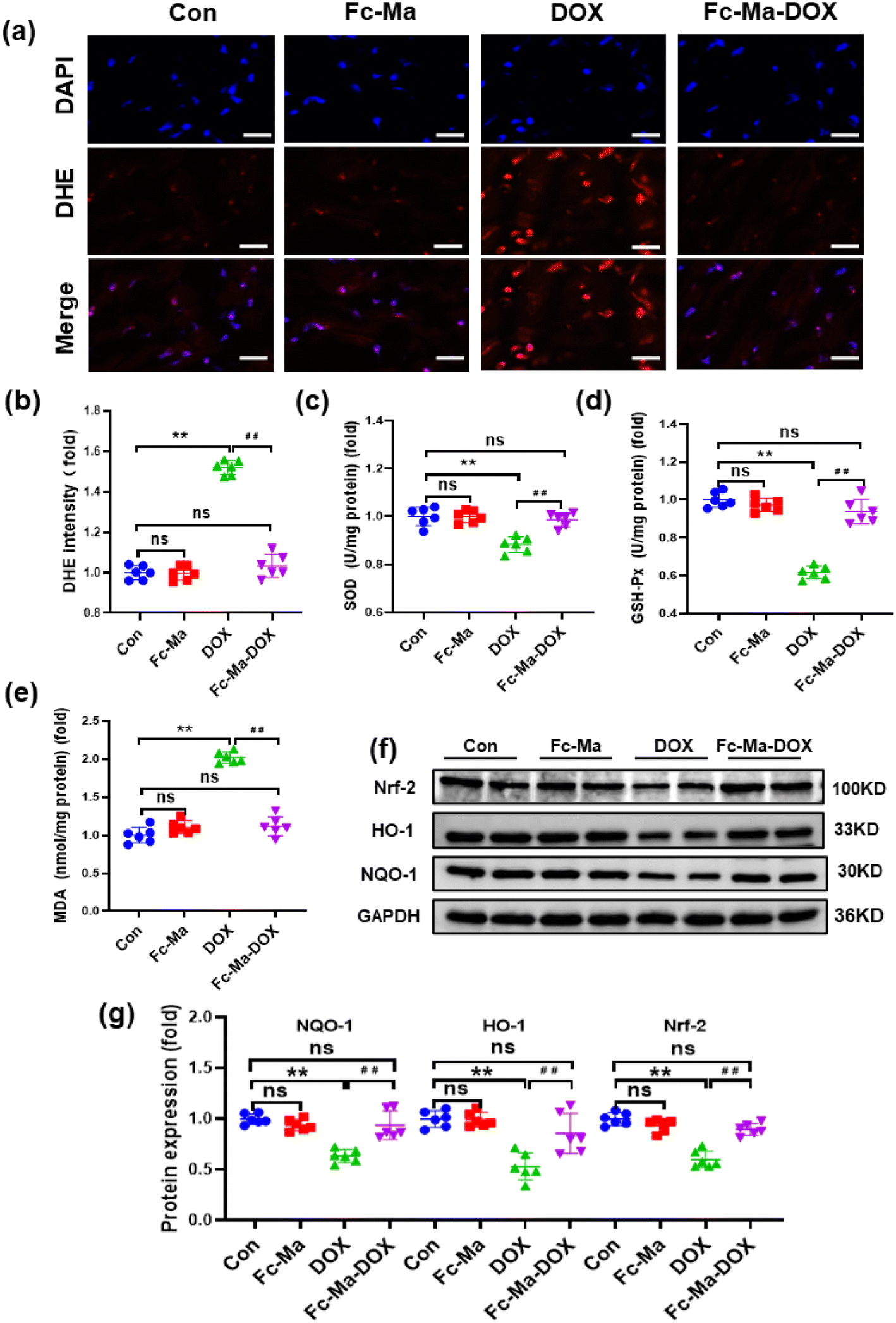 | ||
| Fig. 5 Fc-Ma-DOX attenuated DOX-induced oxidative stress in vivo: (a) and (b) representative DHE staining images and the quantitative results, from top to bottom, the images show cell nuclei stained by DAPI (blue), ROS fluorescence (red) and the merge of the two images (n = 6, scale bar 20μm); (c)–(e) the levels of MDA, SOD and GSH-PX in heart samples (n = 6); (f) and (g) Western blots and the statistical results of Nrf-2, HO-1 and NQO-1 proteins (n = 6). Values represent the mean ± SD. **p < 0.01 vs. control group, ##p < 0.01 vs. DOX group, ns indicates no significance. | ||
3.6. DOX uptake in H9C2 cells
We exposed H9C2 cells to various concentrations of free DOX (0.1, 0.625, 1.25, 2.5, 5.0, 10.0, 20.0, and 40.0 μg mL−1), Fc-Ma-DOX (Fc-Ma-DOX was administered at doses with equivalent amounts of DOX to those of the various concentrations of free DOX) and Fc-Ma (25, 50, 75, 100, 125, 150, 175, and 200 μg mL−1) for 24 hours to assess the cell viability of H9C2 cells by CCK-8 assay. As shown in Fig. 6a, the viability of H9C2 cells administered 5 μg mL−1 DOX was significantly reduced; therefore, we chose this condition for the subsequent in vitro studies. We found that the viability of H9C2 cells did not decrease significantly even with large doses of Fc-Ma, indicating that Fc-Ma has good biosafety (Fig. 6b). To evaluate the endocytosis capacity of DOX in cardiomyocytes, H9C2 cells were exposed to Fc-Ma-DOX at different pH values (6.5 and 7.4) for 6 hours. Inverted fluorescence microscopy images showed strong DOX fluorescence in H9C2 cells administered Fc-Ma-DOX at a pH value of 6.5, whereas only a weak fluorescence (Fig. 6c) was found at a pH of 7.4. Moreover, as presented in Fig. 6d, compared to the strong fluorescence of H9C2 cells incubated with free DOX, the cells incubated with Fc-Ma-DOX only showed low fluorescence in neutral media (pH = 7.4). Notably, the data in our previous study also suggested that the accumulation of DOX in the heart of mice treated with Fc-Ma-DOX was significantly decreased compared with that in mice treated with free DOX.31 All these results suggested that the degradation of Fc-Ma-DOX was markedly decreased under neutral conditions, which could effectively reduce the accumulation of DOX in cardiomyocytes, thus effectively reducing DOX cardiotoxicity. | ||
| Fig. 6 DOX uptake in H9C2 cells: (a) and (b) cell viability was assessed by the Cell Counting Kit-8 method after 24 hours of exposure to the various concentrations of free DOX, Fc-Ma, Fc-Ma-DOX; (c) inverted fluorescent microscopy images of H9C2 cells treated with Fc-Ma-DOX at different pH values (pH 6.5 and 7.4) for 6 hours, from top to bottom, the images show cell nuclei stained by DAPI (blue), DOX fluorescence (red), and the merge of the two images (scale bar 20μm); (d) inverted fluorescent microscopy images of H9C2 cells treated with Fc-Ma-DOX and DOX at the pH value was 7.4 for 6 hours, from top to bottom, the images show cell nuclei stained by DAPI (blue), DOX fluorescence (red), and the merge of the two images (scale bar 20μm). | ||
3.7. Fc-Ma-DOX inhibits oxidative stress and apoptosis in vitro
Previous studies showed that ROS generated by DOX caused cardiac injury and cardiomyocyte apoptosis.42,43 We detected ROS levels in H9C2 cells by DCFH-DA staining to further investigate the effect of Fc-Ma-DOX treatment on DOX-induced cardiotoxicity in vitro. The H9C2 cells were plated in 6-well culture plates at a density of 6 × 104 cells per mL and then treated with free DOX (5 μg mL−1), Fc-Ma (5 μg mL−1) and Fc-Ma-DOX (at a DOX equivalent dose of 5 μg mL−1) for 24 hours, as shown in Fig. 7a. DOX treatment resulted in increased oxidative stress in the H9C2 cells, and Fc-Ma-DOX treatment almost completely inhibited ROS production. Moreover, the production of ROS was hardly increased in the H9C2 cells treated with Fc-Ma. As expected, western blotting revealed that the Nrf-2 protein level was downregulated in the H9C2 cells treated with DOX and that the downstream HO-1 and NQO-1 protein levels were also decreased, while the above protein levels were preserved in the H9C2 cells treated with Fc-Ma-DOX (Fig. 7b and c). TUNEL staining results suggested that DOX incubation caused massive apoptosis in H9C2 cells, which was notably reversed by Fc-Ma-DOX treatment (Fig. 7d).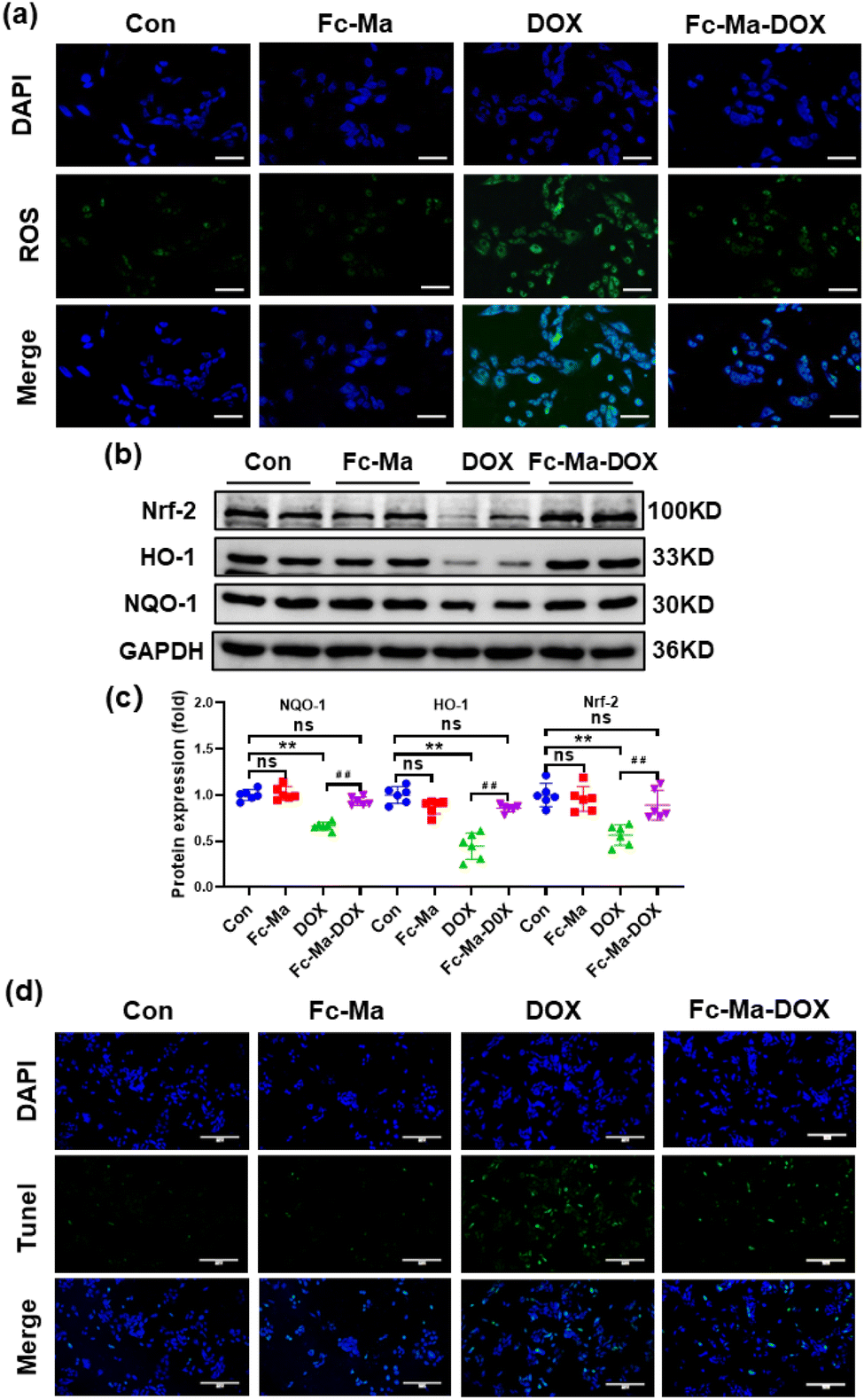 | ||
| Fig. 7 Fc-Ma-DOX inhibits oxidative stress and apoptosis in vitro: (a) representative images of DCFH-DA staining, from top to bottom, the images show cell nuclei stained by DAPI (blue), ROS fluorescence (green) and the merge of the two images (scale bar 20μm); (b) and (c) Western blots and statistical results in cultured H9C2 cells (n = 6); (d) representative images of TUNEL staining, from top to bottom, the images show cell nuclei stained by DAPI (blue), tunel fluorescence (green) and the merge of the two images (scale bar 200μm). Values represent the mean ± SD. **p < 0.01 vs. control group, ##p < 0.01 vs. DOX group, ns indicates no significance. | ||
3.8. Discussion
Oxidative stress and the increased generation of intracellular ROS are widely considered to be primarily responsible for DOX cardiotoxicity.41,44 Due to the strong affinity of DOX for cardiolipin, it is especially retained in the mitochondrial inner membrane in the form of DOX-cardiolipin complexes, which can inhibit mitochondrial enzymes involved in oxidative phosphorylation, interfering with the normal electron transport chain to increase free radical reactive oxygen species (ROS) production. Moreover, DOX can be converted to the unstable intermediate metabolite semiquinone in the presence of molecular oxygen, which in turn accelerates superoxide anion generation during the redox cycling process.38,45,46 Excessive free radicals can directly trigger oxidative damage to cardiomyocytes characterized by lipid peroxidation, leading to cell membrane damage, DNA damage and finally the initiation of apoptosis.6,47 Because myocardial tissues lack sufficient antioxidant mechanisms, ROS accumulation in DOX-induced cardiac injury is accompanied by a disturbed antioxidant defence system in the heart. Nuclear factor erythroid 2-related factor 2 (Nrf-2) is a master regulator of multiple antioxidant enzymes and is known as a cytoprotective transcription factor. It has a key role in modulating cell redox balance and sensing the status of cellular oxidative stress by stimulating the activity of components of antioxidant defence, such as antioxidant enzymes (superoxide dismutase (SOD), glutathione peroxidase (GSH-Px), and haem oxygenase-1 (HO-1)) and the major detoxifying enzyme quinone oxidoreductase 1 (NQO-1).48–50 Consistently, in our study, cardiac Nrf-2 expression was found to be decreased by DOX treatment, and the expression of downstream HO-1 and NQO-1 was also reduced.Porous organic polymers (POPs), due to their unique chemical and physical properties, are frequently used for the design of drug delivery vehicles.21,22,28 Stimuli-responsive drug delivery vehicles may assist in the controlled release of a drug because they can respond to changes in their environmental parameters. Both internal and external stimuli can trigger the release of drugs by evoking a change in the drug carrier. Internal stimuli include various changes in target tissues, such as pH, redox status, ionic strength, and protein and enzyme concentrations, and external (physical) stimuli include temperature, light, ultrasound, magnetic force, and electric fields.51–53 In fact, there are pH differences between many tissues and cellular compartments of the human body; moreover, tumour tissue has a lower pH than surrounding normal tissue due to metabolic glycolysis and lactic acid production. Due to the widely varying pH conditions in diverse biological systems and tissues, it has been possible to design pH-sensitive polymers for drug delivery.54–56 The use of pH-sensitive polymers may reduce nonspecific exposure to chemotherapeutic drugs to not only contribute to targeted drug release but also reduce drug leakage and reduce side effects.14,57–59
In this study, we constructed a biodegradable porous organic polymer (POP) composed of ferrocene (Fc) and D-mannitol (Ma) through pH-responsive acetal bonding, which was then used as the host for the loading of DOX. The acid-unstable acetal linkage was stable in a physiological environment but degraded at a lower pH, drug release mainly occurred due to hydrolysis of the linker bonds in response to the decrease in pH. The DOX released from Fc-Ma-DOX under normal physiological conditions was significantly lower than that in the acidic environment.60,61 Compared with free DOX treatment, the accumulation of DOX in normal and neutral myocardium with Fc-Ma-DOX treatment was significantly reduced. Accordingly, the excessive generation of ROS, oxidative stress and apoptosis induced by DOX were significantly reduced in hearts treated with Fc-Ma-DOX. Fc-Ma-DOX reduced doxorubicin-induced myocardial damage by reducing DOX accumulation in cardiomyocytes.
4. Conclusions
In summary, we have successfully developed a biocompatible ferrocenyl-based pH-responsive porous drug delivery system combined with DOX, which can effectively overcome DOX-induced cardiotoxicity. These results demonstrate the potential of this drug delivery system as a rational tool for further studies of doxorubicin-induced cardiomyopathy.Author contributions
Q. J. (Qiuhong Jiao), B. L. (Baoting Liu), X. X. (Xiufeng Xu), and T. H. (Tao Huang) designed the experiment and wrote the paper; B. C. (Bufan Cao), L. W. (Lide Wang) completed the experiment and data analysis together; A. D. (Ailing Du) and Q. W. (Qingguo Wang) revised the paper; J. L. (Jingtian Li), B. Z. (Baolong Zhou), and T. W. (Tao Wang) guided the experiment and revised the paper. All authors have read and agreed to the published version of the manuscript.Institutional Review Board statement
The animal study protocol was approved by the Institutional Animal Care and Use Committee of Weifang Medical University (protocol code 2021SDL388).Conflicts of interest
The authors declare no conflict of interest.Acknowledgements
This project was supported by the National Natural Science Foundation of China (No. 82000441), Shandong Provincial Natural Science Foundation, China (No. ZR2020QH011, ZR2020QB067). Medicine and Health Science Technology Development Program of Shandong Province (No. 202203010959). Traditional Chinese Medicine Science and Technology Development Plan of Shandong Province (M-2022241). The author would like to express sincere gratitude to all the participants in this study.References
- F. M. Muggia and M. D. Green, New anthracycline antitumor antibiotics, Crit. Rev. Oncol./Hematol., 1991, 11(1), 43–64 CrossRef CAS.
- S. Gorini, et al., Chemotherapeutic Drugs and Mitochondrial Dysfunction: Focus on Doxorubicin, Trastuzumab, and Sunitinib, Oxid. Med. Cell. Longevity, 2018, 2018, 7582730 Search PubMed.
- L. Rochette, et al., Anthracyclines/trastuzumab: new aspects of cardiotoxicity and molecular mechanisms, Trends Pharmacol. Sci., 2015, 36(6), 326–348 CrossRef CAS PubMed.
- F. S. Carvalho, et al., Doxorubicin-induced cardiotoxicity: from bioenergetic failure and cell death to cardiomyopathy, Med. Res. Rev., 2014, 34(1), 106–135 CrossRef CAS PubMed.
- B. B. Wu, K. T. Leung and E. N. Poon, Mitochondrial-Targeted Therapy for Doxorubicin-Induced Cardiotoxicity, Int. J. Mol. Sci., 2022, 23(3), 1912 CrossRef CAS PubMed.
- P. S. Rawat, et al., Doxorubicin-induced cardiotoxicity: an update on the molecular mechanism and novel therapeutic strategies for effective management, Biomed. Pharmacother., 2021, 139, 111708 CrossRef CAS PubMed.
- M. Sterba, et al., Oxidative stress, redox signaling, and metal chelation in anthracycline cardiotoxicity and pharmacological cardioprotection, Antioxid. Redox Signaling, 2013, 18(8), 899–929 CrossRef CAS.
- H. Liu, et al., Pharmaceutical Measures to Prevent Doxorubicin-Induced Cardiotoxicity, Mini-Rev. Med. Chem., 2017, 17(1), 44–50 CrossRef CAS PubMed.
- V. B. Pai and M. C. Nahata, Cardiotoxicity of chemotherapeutic agents: incidence, treatment and prevention, Drug Saf., 2000, 22(4), 263–302 CrossRef CAS.
- S. Marchal, et al., Anticancer Drug Delivery: An Update on Clinically Applied Nanotherapeutics, Drugs, 2015, 75(14), 1601–1611 CrossRef CAS.
- S. W. Langer, Dexrazoxane for the treatment of chemotherapy-related side effects, Cancer Manage. Res., 2014, 6, 357–363 CrossRef.
- C. K. Tebbi, et al., Dexrazoxane-associated risk for acute myeloid leukemia/myelodysplastic syndrome and other secondary malignancies in pediatric Hodgkin's disease, J. Clin. Oncol., 2007, 25(5), 493–500 CrossRef CAS PubMed.
- R. T. Dorr, Cytoprotective agents for anthracyclines, Semin. Oncol., 1996, 23(4 suppl. 8), 23–34 CAS.
- M. Fojtu, et al., Reduction of Doxorubicin-Induced Cardiotoxicity Using Nanocarriers: A Review, Curr. Drug Metab., 2017, 18(3), 237–263 CrossRef CAS PubMed.
- A. Gabizon, H. Shmeeda and Y. Barenholz, Pharmacokinetics of pegylated liposomal Doxorubicin: review of animal and human studies, Clin. Pharmacokinet., 2003, 42(5), 419–436 CrossRef CAS PubMed.
- D. N. Waterhouse, et al., A comparison of liposomal formulations of doxorubicin with drug administered in free form: changing toxicity profiles, Drug Saf., 2001, 24(12), 903–920 CrossRef CAS PubMed.
- U. Kanwal, et al., Advances in nano-delivery systems for doxorubicin: an updated insight, J. Drug Targeting, 2018, 26(4), 296–310 CrossRef CAS PubMed.
- M. Cagel, et al., Doxorubicin: nanotechnological overviews from bench to bedside, Drug Discovery Today, 2017, 22(2), 270–281 CrossRef CAS PubMed.
- S. Tran, et al., Cancer nanomedicine: a review of recent success in drug delivery, Clin. Transl. Med., 2017, 6(1), 44 Search PubMed.
- D. H. Yang, et al., Porous organic polymers for electrocatalysis, Chem. Soc. Rev., 2022, 51(2), 761–791 RSC.
- Y. Zhu, et al., Emerging porous organic polymers for biomedical applications, Chem. Soc. Rev., 2022, 51(4), 1377–1414 RSC.
- N. Singh, et al., Nanoscale porous organic polymers for drug delivery and advanced cancer theranostics, Chem. Soc. Rev., 2021, 50(23), 12883–12896 RSC.
- J. Wu, et al., Porous Polymers as Multifunctional Material Platforms toward Task-Specific Applications, Adv. Mater., 2019, 31(4), e1802922 CrossRef PubMed.
- W. Chen, et al., Macrocycle-derived hierarchical porous organic polymers: synthesis and applications, Chem. Soc. Rev., 2021, 50(20), 11684–11714 RSC.
- S. Wang, et al., Porous organic polymers as a platform for sensing applications, Chem. Soc. Rev., 2022, 51(6), 2031–2080 RSC.
- S. B. Yu, et al., Water-soluble and dispersible porous organic polymers: preparation, functions and applications, Chem. Soc. Rev., 2022, 51(2), 434–449 RSC.
- Z. Li and Y. W. Yang, Macrocycle-Based Porous Organic Polymers for Separation, Sensing, and Catalysis, Adv. Mater., 2022, 34(6), e2107401 CrossRef PubMed.
- J. H. Kim, et al., Post-synthetic modifications in porous organic polymers for biomedical and related applications, Chem. Soc. Rev., 2022, 51(1), 43–56 RSC.
- Y. Tang, et al., Porous organic polymers for drug delivery: hierarchical pore structures, variable morphologies, and biological properties, Biomater. Sci., 2022, 10(19), 5369–5390 RSC.
- L. J. C. Albuquerque, et al., pH-responsive polymersome-mediated delivery of doxorubicin into tumor sites enhances the therapeutic efficacy and reduces cardiotoxic effects, J. Controlled Release, 2021, 332, 529–538 CrossRef CAS PubMed.
- H. Lou, et al., Biodegradable Porous Polymeric Drug with pH-Stimuli-Responsive Delivery Capacity for Combined Cancer Therapy, ACS Appl. Polym. Mater., 2021, 4(1), 714–724 CrossRef.
- E. Y. Podyacheva, et al., Analysis of Models of Doxorubicin-Induced Cardiomyopathy in Rats and Mice. A Modern View from the Perspective of the Pathophysiologist and the Clinician, Front. Pharmacol., 2021, 12, 670479 CrossRef CAS PubMed.
- A. Schlitt, et al., Cardiotoxicity and oncological treatments, Deutsches Ärzteblatt international, 2014, 111(10), 161–168 Search PubMed.
- T. Wang, et al., The MEK inhibitor U0126 ameliorates diabetic cardiomyopathy by restricting XBP1's phosphorylation dependent SUMOylation, Int. J. Biol. Sci., 2021, 17(12), 2984–2999 CrossRef CAS PubMed.
- S. N. Dada, et al., Covalent and Noncovalent Loading of Doxorubicin by Folic Acid-Carbon Dot Nanoparticles for Cancer Theranostics, ACS Omega, 2022, 7(27), 23322–23331 CrossRef CAS PubMed.
- K. R. Karnati and Y. Wang, Understanding the co-loading and releasing of doxorubicin and paclitaxel using chitosan functionalized single-walled carbon nanotubes by molecular dynamics simulations, Phys. Chem. Chem. Phys., 2018, 20(14), 9389–9400 RSC.
- E. Oikonomou, et al., Cancer Therapeutics-Related Cardiovascular Complications. Mechanisms, Diagnosis and Treatment, Curr. Pharm. Des., 2018, 24(37), 4424–4435 CrossRef CAS PubMed.
- C. Y. Kong, et al., Underlying the Mechanisms of Doxorubicin-Induced Acute Cardiotoxicity: Oxidative Stress and Cell Death, Int. J. Biol. Sci., 2022, 18(2), 760–770 CrossRef CAS PubMed.
- P. D. Ray, B. W. Huang and Y. Tsuji, Reactive oxygen species (ROS) homeostasis and redox regulation in cellular signaling, Cell. Signalling, 2012, 24(5), 981–990 CrossRef CAS PubMed.
- C. Yu and J. H. Xiao, The Keap1-Nrf2 System: A Mediator between Oxidative Stress and Aging, Oxid. Med. Cell. Longevity, 2021, 2021, 6635460 Search PubMed.
- B. Kalyanaraman, Teaching the basics of the mechanism of doxorubicin-induced cardiotoxicity: have we been barking up the wrong tree?, Redox Biol., 2020, 29, 101394 CrossRef CAS PubMed.
- L. F. Hu, et al., A Systematic Review of the Potential Chemoprotective Effects of Resveratrol on Doxorubicin-Induced Cardiotoxicity: Focus on the Antioxidant, Antiapoptotic, and Anti-Inflammatory Activities, Oxid. Med. Cell. Longevity, 2021, 2021, 2951697 Search PubMed.
- L. Jiang, et al., Peroxiredoxin-1 Overexpression Attenuates Doxorubicin-Induced Cardiotoxicity by Inhibiting Oxidative Stress and Cardiomyocyte Apoptosis, Oxid. Med. Cell. Longevity, 2020, 2020, 2405135 Search PubMed.
- M. Songbo, et al., Oxidative stress injury in doxorubicin-induced cardiotoxicity, Toxicol. Lett., 2019, 307, 41–48 CrossRef CAS PubMed.
- D. Cappetta, et al., Oxidative Stress and Cellular Response to Doxorubicin: A Common Factor in the Complex Milieu of Anthracycline Cardiotoxicity, Oxid. Med. Cell. Longevity, 2017, 2017, 1521020 Search PubMed.
- N. F. Sangweni, et al., Molecular insights into the pathophysiology of doxorubicin-induced cardiotoxicity: a graphical representation, Arch. Toxicol., 2022, 96(6), 1541–1550 CrossRef CAS PubMed.
- P. Angsutararux, S. Luanpitpong and S. Issaragrisil, Chemotherapy-Induced Cardiotoxicity: Overview of the Roles of Oxidative Stress, Oxid. Med. Cell. Longevity, 2015, 2015, 795602 Search PubMed.
- R. L. Maute, et al., tRNA-derived microRNA modulates proliferation and the DNA damage response and is down-regulated in B cell lymphoma, Proc. Natl. Acad. Sci. U. S. A., 2013, 110(4), 1404–1409 CrossRef CAS PubMed.
- A. Loboda, et al., Role of Nrf2/HO-1 system in development, oxidative stress response and diseases: an evolutionarily conserved mechanism, Cell. Mol. Life Sci., 2016, 73(17), 3221–3247 CrossRef CAS PubMed.
- P. Shaw and A. Chattopadhyay, Nrf2-ARE signaling in cellular protection: mechanism of action and the regulatory mechanisms, J. Cell. Physiol., 2020, 235(4), 3119–3130 CrossRef CAS PubMed.
- S. Mura, J. Nicolas and P. Couvreur, Stimuli-responsive nanocarriers for drug delivery, Nat. Mater., 2013, 12(11), 991–1003 CrossRef CAS PubMed.
- X. Zhao, J. Bai and W. Yang, Stimuli-responsive nanocarriers for therapeutic applications in cancer, Cancer Biol. Med., 2021, 18(2), 319–335 Search PubMed.
- A. Xie, et al., Stimuli-responsive prodrug-based cancer nanomedicine, EBioMedicine, 2020, 56, 102821 CrossRef PubMed.
- M. Karimi, et al., pH-Sensitive stimulus-responsive nanocarriers for targeted delivery of therapeutic agents, Wiley Interdiscip. Rev.: Nanomed. Nanobiotechnol., 2016, 8(5), 696–716 CAS.
- T. Yoshida, et al., pH- and ion-sensitive polymers for drug delivery, Expert Opin. Drug Delivery, 2013, 10(11), 1497–1513 CrossRef CAS PubMed.
- Z. Li, J. Huang and J. Wu, pH-Sensitive nanogels for drug delivery in cancer therapy, Biomater. Sci., 2021, 9(3), 574–589 RSC.
- F. Meng, et al., pH-sensitive polymeric nanoparticles for tumor-targeting doxorubicin delivery: concept and recent advances, Nanomedicine, 2014, 9(3), 487–499 CrossRef CAS PubMed.
- R. Gannimani, et al., Acetal containing polymers as pH-responsive nano-drug delivery systems, J. Controlled Release, 2020, 328, 736–761 CrossRef CAS PubMed.
- N. M. AlSawaftah, et al., pH-Responsive Nanocarriers in Cancer Therapy, Polymers, 2022, 14(5), 936 CrossRef CAS PubMed.
- P. Mi, Stimuli-responsive nanocarriers for drug delivery, tumor imaging, therapy and theranostics, Theranostics, 2020, 10(10), 4557–4588 CrossRef CAS PubMed.
- M. Karimi, et al., Smart micro/nanoparticles in stimulus-responsive drug/gene delivery systems, Chem. Soc. Rev., 2016, 45(5), 1457–1501 RSC.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d2ra07410a |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2023 |