
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceVisible light-induced oxidative α-hydroxylation of β-dicarbonyl compounds catalyzed by ethylenediamine–copper(II)†
Yujie Hea,
Hao Yinb,
Yifeng Wang *b,
Mingming Chu*a and
Yiming Li*a
*b,
Mingming Chu*a and
Yiming Li*a
aCollege of Biological, Chemical Sciences and Engineering, Jiaxing University, Jiaxing, 314001, China. E-mail: chumingming@zjxu.edu.cn; liyiming@zjxu.edu.cn
bDepartment of Green Chemistry and Technology, Zhejiang University of Technology, Hangzhou 310014, P. R. China. E-mail: wangyifeng@zjut.edu.cn
First published on 9th March 2023
Abstract
We have developed an efficient oxidative α-hydroxylation of β-keto esters with firstly using the structurally simple ethylenediamine–copper(II) as a catalyst for β-keto esters activation and using visible light as the driving force for generating more active singlet oxygen (1O2) from triplet state oxygen (3O2) in the air, providing a series of α-hydroxy β-keto esters in excellent yields (up to 99%) under extremely low photosensitizer loading (0.01 mol%) and catalyst loading (1 mol%) within a short time. Moreover, the gram-scale synthesis showed the practical utility of this protocol.
Introduction
The α-hydroxy β-dicarbonyl moiety is a functional and structural motif in numerous natural products and pharmaceuticals such as hamigeran A,1 vindoline analogue,2 Y224A,3 kjellmanianone,4 and pramanicin.5 Furthermore, the intact α-hydroxy β-dicarbonyl compound is the key intermediate in synthetic transformations, as represented by the manufacture of the insecticide indoxacarb.6 The oxidative α-hydroxylation of dicarbonyl compounds, providing a direct access to such a skeleton, have therefore been extensively studied over the past years. In this respect, the extensive investigation revealed that most of these works often rely on using strong oxidants such as organic peroxide,7 oxaziridine,8 dimethyldioxirane9 or nitrosobenzene.10 From economic as well as environmental viewpoints, visible light has been recognized as a type of environmentally-friendly sustainable energy and molecular oxygen is considered a green oxidant, so visible light-induced aerobic oxidation is undoubtedly a more appealing strategy for the α-hydroxylation of β-dicarbonyl compounds. At present, considerable achievements have been made in this promising project by Meng's groups using the salan-copper(II) and cinchona alkaloid derivatives as a catalyst for activating the β-keto esters and tetraphenyl-porphyrin (TPP) or phthalocyanine (PC) as the optimal photosensitizer.11 Meanwhile, the combination of bis(oxazoline)–Ni(II) complexes and a visible light photosensitizer in grafting or physically mixed way, reported by Xiao's group12 and our group,13 respectively, providing alternative catalysis systems to realize this transformation. Despite these important advances, when we consider the long-standing challenges that lowing catalyst and photosensitizer loading, using structurally simple catalyst and achieving excellent catalyst performance and practicable reaction scale-up in such light-mediated oxidative α-hydroxylation of β-dicarbonyl compounds, developing new competent catalytic system to be compatible with photooxidation remain highly desirable (Scheme 1 and Fig. 1). | ||
| Scheme 1 The visible-light induced oxidative α-hydroxylation of β-keto esters catalyzed by structurally simple ligand/metal complexes. | ||
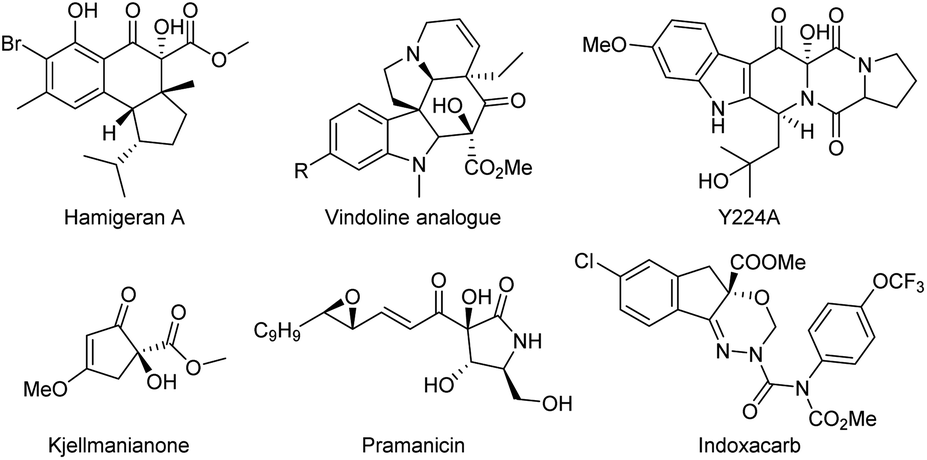 | ||
| Fig. 1 The natural products and pharmaceuticals containing α-hydroxy β-dicarbonyl moiety. | ||
In our previous works, the chiral bisoxazolines have been demonstrated as a type of competent N-donor ligands in the asymmetric α-functionalization of dicarbonyl compounds, such as the electrophilic fluorination and Michael addition reactions of β-keto esters and the brominative dearomatization of naphthol derivatives.14 Basing on the recognition of the activating nature of bisoxazoline–metal complexes, we turn our attention to developing structurally simple N-donor ligands, particularly some commercial organic molecule, to coordinate with metal salts and realize the α-functionalization of dicarbonyl compounds in enantioselective or nonenantioselective manner.
Results and discussion
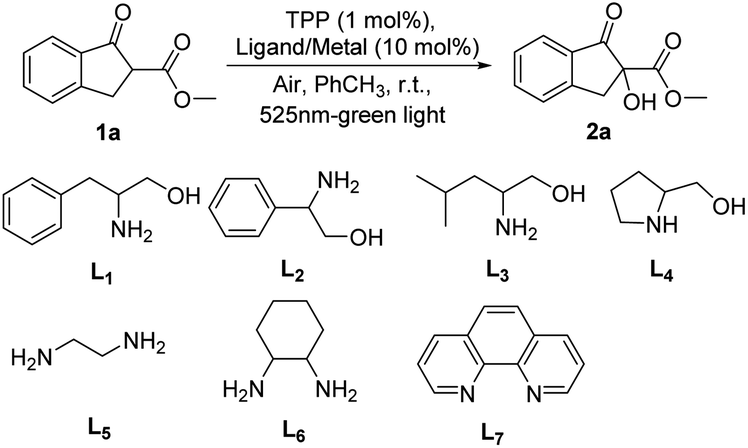
The β-amino alcohol, being the starting material of bisoxazoline ligands to provide nitrogen coordination center to metal ion, was chosen as the ligand in our initial study. We employed 1-methyl indanone carboxylate 1a as a model substrate and O2 in the air as an oxidant for the α-hydroxylation reaction under the green light of 525 nm, the desired product 2a was obtained in 86% yield in one hour with using benzylethanolamine L1 as ligand and Cu(OAc)2 as metal salt (Table 1, entry 1). Encouraged by this result, the other β-amino alcohols L2–L4 were investigated, the α-hydroxylation product would be provided in an excellent yield of 99% within one hour when using phenylglycinol L2 as ligand (entry 2 vs. entry 1), and the ligands L3 and L4 led to comparable results in longer time (entries 3 and 4 vs. entry 1). The no-substituted ethylenediamine L5 and cyclohexane diamine L6 which were generally used to construct salen ligands were then tested, it was shown that better catalyst performance was observed with using ethylenediamine L5 as ligand, affording the α-hydroxylation product in 99% yield (entry 5 vs. entry 6). We continued the investigation by using 1,10-phenanthroline L7 as the ligand, providing an eroded result (entry 7). From the viewpoint of economy and accessibility, the ethylenediamine L5 was identified as the optimal ligand. Next, various metal salts complexing with ethylenediamine L5 were investigated. When using Zn(OAc)2·2H2O, Ni(OAc)2·4H2O instead of Cu(OAc)2, the yield of reaction would be slightly decreased (entries 8 and 9 vs. entry 5). Considering the influence of counterions, different copper(II) salts were then assessed, and the results showed that the coordination of L5 with Cu(OAc)2 gave better yield (entries 10–12). Subsequently, other photosensitizers such as rose Bengal and thioxanthone were tested, while the isolated yield of desire products were obviously decreased (entries 13 and 14 vs. entry 5). The indispensability of the ligand, metal salts and visible light in this reaction was demonstrated by single factor test (entries 15–17).
|
||||
|---|---|---|---|---|
| Entry | Ligand | Lewis acid | Time/h | Yieldb/% |
| a Reactions were performed with 1a (0.1 mmol), TPP (1 mol%) and 10 mol% of ligand and Lewis acid in PhMe (2.0 mL) under an air atmosphere.b Isolated yields.c Thioxanthone was used as photosensitizer.d Rose Bengal was used as photosensitizer.e No photosensitizer. | ||||
| 1 | L1 | Cu(OAc)2 | 1 | 86 |
| 2 | L2 | Cu(OAc)2 | 1 | 99 |
| 3 | L3 | Cu(OAc)2 | 2 | 95 |
| 4 | L4 | Cu(OAc)2 | 2 | 83 |
| 5 | L5 | Cu(OAc)2 | 1 | 99 |
| 6 | L6 | Cu(OAc)2 | 1 | 95 |
| 7 | L7 | Cu(OAc)2 | 2 | 50 |
| 8 | L5 | Zn(OAc)2·2H2O | 1 | 95 |
| 9 | L5 | Ni(OAc)2·4H2O | 1 | 89 |
| 10 | L5 | CuSO4 | 1 | 91 |
| 11 | L5 | CuCl2 | 1 | 83 |
| 12 | L5 | Cu(OTf)2 | 1 | 86 |
| 13c | L5 | Cu(OAc)2 | 1 | 40 |
| 14d | L5 | Cu(OAc)2 | 1 | 82 |
| 15 | — | Cu(OAc)2 | 1 | 28 |
| 16 | L5 | — | 1 | Trace |
| 17e | L5 | Cu(OAc)2 | 1 | Trace |
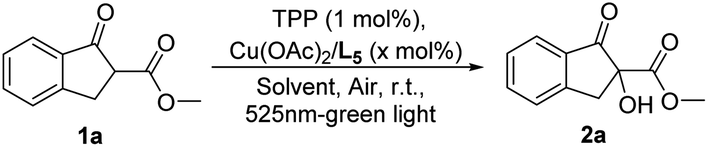
With the optimum catalyst and photosensitizer identified, other parameters such as solvent and catalyst loading were investigated. As shown in Table 2, the screening of organic solvent revealed that an excellent yield could be obtained when using toluene, MTBE or THF as the solvent (entries 1–7). To further explore the efficiency of this catalytic system, the lower catalyst loadings were tested (entries 8–10). In detail, the decrease to 1 mol% metallic catalyst loading still enabled the transformation to work well when using THF solvent, providing the product 2a in the maintained yield (99% yield) within two hours (entries 8 and 9 vs. entry 10). With further decrease in catalyst loading, the much-diminished yield would be observed albeit in longer reaction time (entry 11). The next study shown that the TPP loading could be reduced to as low as 0.01 mol%, providing same perfect reaction outcome (99% yield) via prolonging the reaction time to 7 h (entry 13), and the lower TPP loading of 0.005 mol% would result in observably eroded catalytic performance (entry 14).
|
||||
|---|---|---|---|---|
| Entry | Solvent | Cu(OAc)2/L5 (mol%) | Time/h | Yieldb/% |
| a Reactions were performed with 1a (0.1 mmol), TPP (1 mol%) in solvent (2.0 mL) under an air atmosphere.b Isolated yields.c With 0.1 mol% TPP loading.d With 0.01 mol% TPP loading.e With 0.005 mol% TPP loading. | ||||
| 1 | Toluene | 10 | 1 | 99 |
| 2 | DCE | 10 | 1 | 58 |
| 3 | MeOH | 10 | 1 | 91 |
| 4 | DMF | 10 | 1 | 41 |
| 5 | EtOAc | 10 | 1 | 94 |
| 6 | MTBE | 10 | 1 | 99 |
| 7 | THF | 10 | 1 | 99 |
| 8 | Toluene | 1 | 12 | 82 |
| 9 | MTBE | 1 | 12 | 67 |
| 10 | THF | 1 | 2 | 99 |
| 11 | THF | 0.5 | 12 | 35 |
| 12c | THF | 1 | 4 | 99 |
| 13d | THF | 1 | 7 | 99 |
| 14e | THF | 1 | 12 | 42 |
Under the above optimized conditions, we initially assessed the generality of the α-hydroxylation reaction for different β-keto esters. As shown in Scheme 2, in most cases, the α-hydroxylation reaction of various β-keto esters proceeded efficiently with high yields. In detail, the ester functionality with different steric hindrance (1a–1f) was well tolerated and the corresponding products were afforded with excellent yields (94–99% yields). The β-keto esters with the aromatic ring bearing electron-withdrawing halogen groups at different positions (1g–1l) underwent the reaction smoothly, give the desired products in comparable yields (up to 84–99% yields). Besides, when the electron-donating groups such as MeO and Me were tethered to the aromatic ring, a maintained reaction outcome could be obtained (1m–1o). Next, benzofuranone, tetralone and cyclohexanone-derived β-keto esters (1p–1r) were demonstrated to be amenable to the reaction protocol, furnishing the desired products in satisfactory yields (76–93% yields). In order to investigate the possible extension of this reaction, the β-keto amide 1s were chose as substrates, allowing the reaction to provide α-hydroxylation products in still high yields (96% yield). Meanwhile, the substrates with no α-substituent such as pyrazolin-5-ones, oxindole, acetoacetic ether, acetophenone and 3-phenylpropionic acid esters have been tested under the optimized reaction conditions, while these transformations did not proceed smoothly and no desired products were isolated, presumedly because the C–H bond enthalpy or dissociation energy of primary carbon and secondary carbon was higher than tertiary carbon, making it hard for above substrates to be oxidized in present catalytic system.
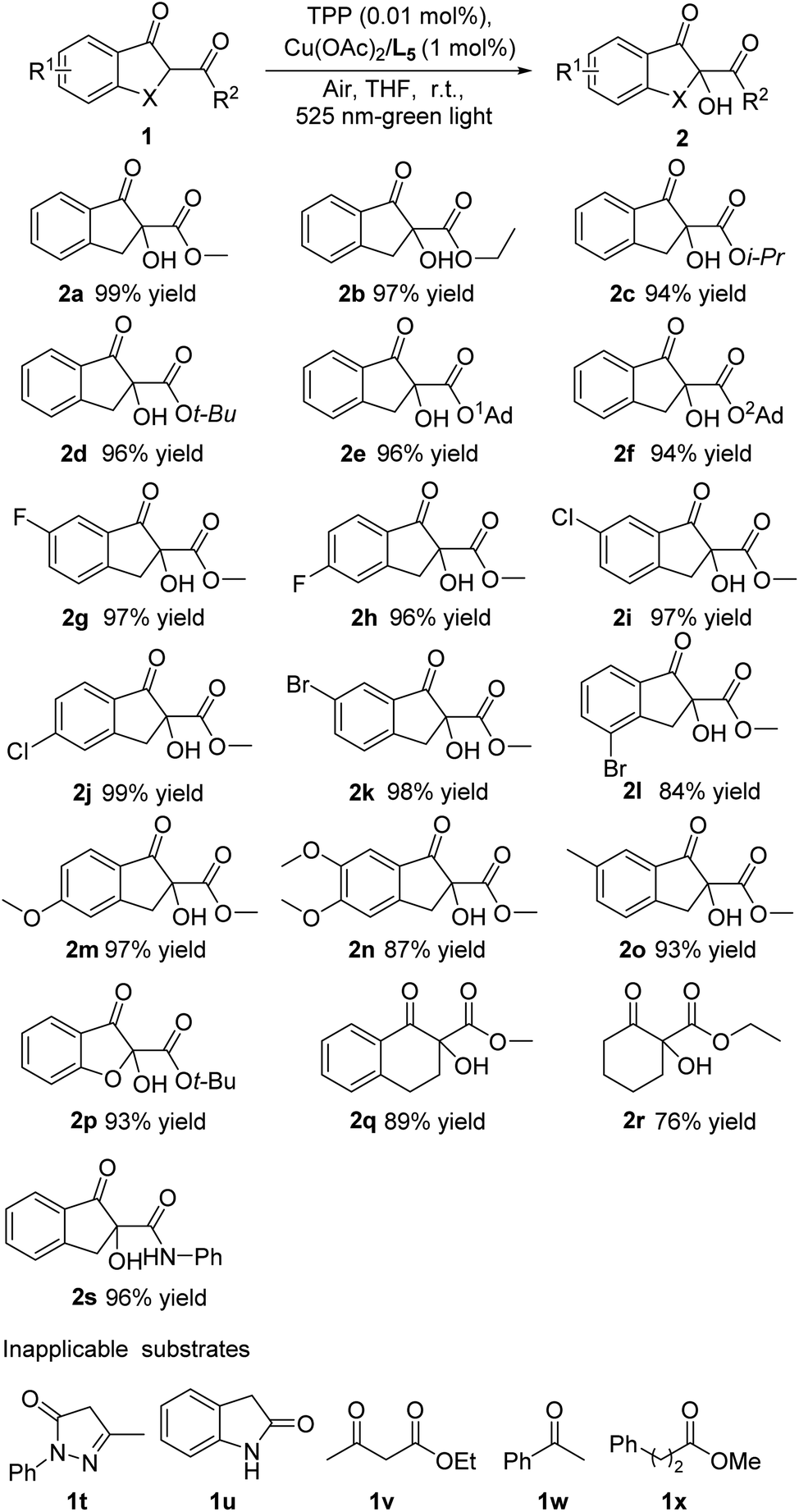 | ||
| Scheme 2 The evaluation of substrate scope. All yields are of isolated products. Unless otherwise noted, the reactions were performed with 1a (0.1 mmol), TPP (0.01 mol%), 1 mol% of Cu(AcO)2 and L5 in THF (2.0 mL) under air atmosphere. | ||
To demonstrate the synthetic potential of this aerobic hydroxylation of β-keto esters, the scale-up of this reaction was performed. The result shown that the reaction of β-keto esters 1a at 5.5 mmol proceeded well under the standard conditions to generate the corresponding adduct 2a in 97% yield, suggesting that this aerobic α-hydroxylation has the potential for a large-scale production without a decrease in chemical yield (Scheme 3).
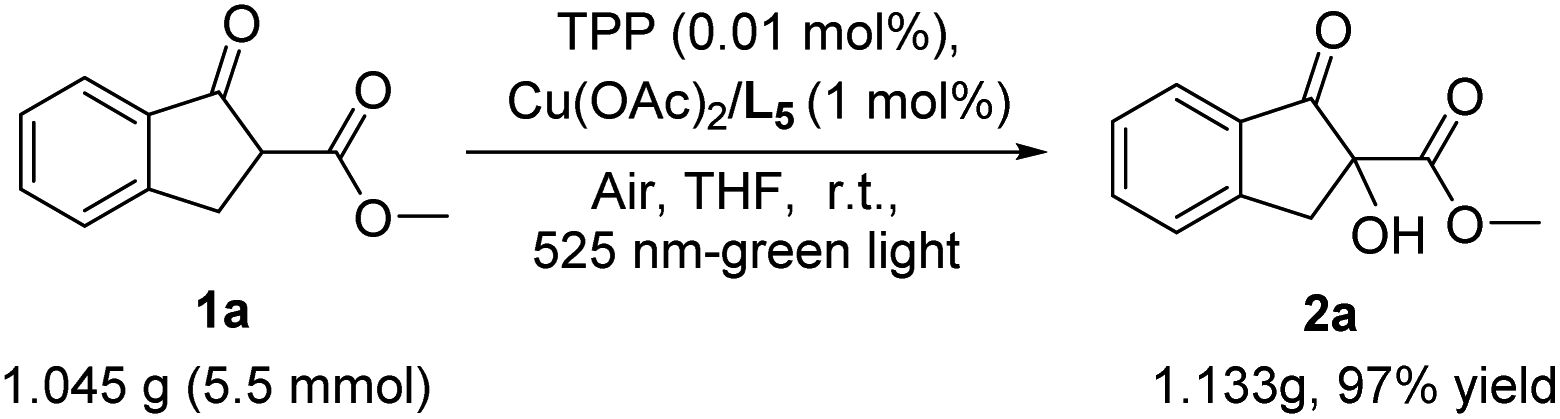 | ||
| Scheme 3 Synthetic potential. | ||
Some control experiments have also been conducted to better understand the reaction mechanism. The hydroperoxide 2A was prepared in situ at lower temperature of 0 °C, and the high-resolution mass spectrum of the reaction mixture could certify the generation of intermediate 2A.15 While because of the high reactivity and the instability of 2A, we could not isolate such intermediate product. Moreover, tertiary butyl indanone carboxylate 1d was added to above reaction solution (Scheme 4a), the α-hydroxylation product 2d could be obtained with no light irradiation, revealing that the hydroperoxide 2A may also as oxidizing agent in this photooxygenation. Comparing with the result of the entry 11 in Table 2, the control experiment using 0.5 mol% of Cu/ethylenediamine 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2 mixture as catalyst shown almost same catalysis performance (Scheme 4b). That is, only increasing the amount of ligand didn't improve the transformation efficiency, indicating that the molar ratio of Cu(II) ion and ethylenediamine ligand in the transition state of this reaction might be 1:1, even though the bis(ethylenediamine)copper complex could also be formed in literature precedents.16
:2 mixture as catalyst shown almost same catalysis performance (Scheme 4b). That is, only increasing the amount of ligand didn't improve the transformation efficiency, indicating that the molar ratio of Cu(II) ion and ethylenediamine ligand in the transition state of this reaction might be 1:1, even though the bis(ethylenediamine)copper complex could also be formed in literature precedents.16
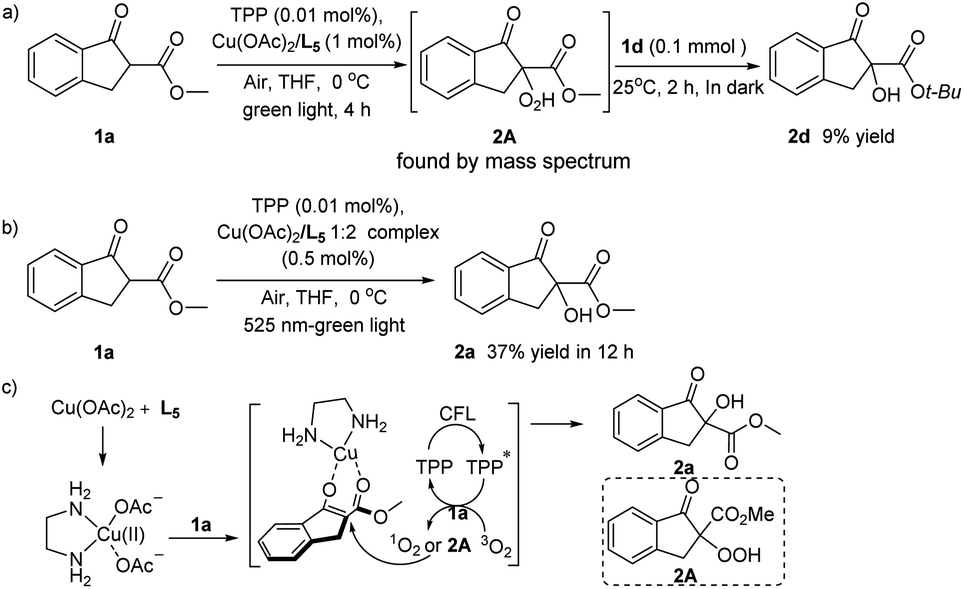 | ||
| Scheme 4 The control experiments (a and b) and the proposed mechanism (c). | ||
Based on the experimental results and the previous studies,11–14,17 a plausible reaction pathway was suggested. As shown in Scheme 4c, the pre-synthetic ethylenediamine–copper(II) complex acting as a Lewis acid catalyst to form the copper enolate species bearing increased nucleophilicity. Under the visible light, molecular oxygen was activated from the triplet state (3O2) to the singlet state (1O2) with the photosensitizer by energy transfer, and the hydroperoxide intermediate 2A would been simultaneously yielded in this transformation. The subsequent attack of 1O2 or hydroperoxide intermediate 2A to the activated 1a afforded the α-hydroperoxide product 2a.
Conclusions
In summary, with firstly using structurally simple ethylenediamine–copper(II) catalyst for β-keto esters activation and TPP photosensitizer for generating highly active singlet oxygen (1O2) in the air atmosphere, an oxidative α-hydroxylation reaction of β-keto esters was developed under visible light, allowing an high-efficient access to various α-hydroxy β-keto esters with a wide substrate scope. Moreover, the low catalyst and photosensitizer loading, structurally simple ligand, easy scalability also demonstrated the fascination of this reaction protocol.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the Program for Science and Technology of Zhejiang Province (LGG21B030003), the Program for Science and Technology of Jiaxing (2020AY10016), and the National Natural Science Foundation of China (21603085).Notes and references
- G. Olack and H. J. Morrison, J. Org. Chem., 1991, 56, 4969 CrossRef CAS.
- K. D. Wellington, R. C. Cambie, P. S. Rutledge and P. Bergquist, J. Nat. Prod., 2000, 63, 79 CrossRef CAS PubMed.
- H. Xue, C. Lu, L. Liang and Y. Shen, Rec. Nat. Prod., 2012, 6, 28 CAS.
- (a) M. Nakayama, Y. Fukuoka, H. Nozaki, A. Matsuo and S. Hayashi, Chem. Lett., 1980, 9, 1243 CrossRef; (b) J. Christoffers, T. Werner, W. Frey and A. Baro, Chem.–Eur. J., 2004, 10, 1042 CrossRef CAS PubMed.
- S.-Y. Aoki, T. Tsukude, Y. Miyazaki, K.-I. Takao and K.-I. Tadano, Heterocycles, 2006, 69, 49 CrossRef CAS PubMed.
- S. F. McCann, G. D. Annis, R. Shapiro, D. W. Piotrowski, G. P. Lahm, J. K. Long, K. C. Lee, M. M. Hughes, B. J. Myers, S. M. Griswold, B. M. Reeves, R. W. March, P. L. Sharpe, P. Lowder, W. E. Barnette and K. D. Wing, Pest Manage. Sci., 2001, 57, 153 CrossRef CAS.
- (a) M. R. Acocella, O. G. Mancheño, M. Bella and K. A. Jørgensen, J. Org. Chem., 2004, 69, 8165 CrossRef CAS PubMed; (b) H.-J. Yao, M.-M. Lian, Z. Li, Y.-K. Wang and Q.-W. Meng, J. Org. Chem., 2012, 77, 9601 CrossRef CAS PubMed; (c) C.-K. Yin, W.-D. Cao, L.-L. Lin, X.-H. Liu and X.-M. Feng, Adv. Synth. Catal., 2013, 355, 1924 CrossRef CAS; (d) Z. Li, M.-M. Lian, F. Yang, Q.-W. Meng and Z.-X. Gao, Eur. J. Org. Chem., 2014, 2014, 3491 CrossRef CAS; (e) Y.-K. Wang, Z. Li, T. Xiong, J.-N. Zhao and Q.-W. Meng, Synlett, 2014, 25, 2155 CrossRef CAS; (f) H. Qing, Y.-K. Wang, Z.-H. Zheng, S. Chen and Q.-W. Meng, Tetrahedron: Asymmetry, 2016, 27, 834 CrossRef CAS; (g) Y.-K. Wang, H. Yin, H. Qing, J.-N. Zhao, Y.-F. Wu and Q.-W. Meng, Adv. Synth. Catal., 2016, 358, 737 CrossRef CAS; (h) F. Yang, J.-N. Zhao, X.-F. Tang, G.-L. Zhou, W.-Z. Song and Q.-W. Meng, Org. Lett., 2017, 19, 448 CrossRef CAS PubMed; (i) J. Chen, H.-Y. Gu, X.-Y. Zhu, W.-W. Nam and B. Wang, Adv. Synth. Catal., 2020, 362, 2976 CrossRef CAS.
- (a) T. Ishimaru, N. Shibata, J. Nagai, S. Nakamura, T. Toru and S. Kanemasa, J. Am. Chem. Soc., 2006, 128, 16488 CrossRef CAS PubMed; (b) L.-W. Zou, B.-M. Wang, H.-F. Mu, H.-R. Zhang, Y.-M. Song and J.-P. Qu, Org. Lett., 2013, 15, 3106 CrossRef CAS PubMed; (c) J.-J. Jiang, J. Huang, D. Wang, M.-X. Zhao, F.-J. Wang and M. Shi, Tetrahedron: Asymmetry, 2010, 21, 794 CrossRef CAS; (d) J. Li, G. Chen, Z. Wang, R.-Z. Zhang, X.-M. Zhang and K.-L. Ding, Chem. Sci., 2011, 2, 1141 RSC; (e) X. Gu, Y. Zhang, Z.-J. Xu and C.-M. Che, Chem. Commun., 2014, 50, 7870 RSC; (f) X.-B. Lin, S. Ruan, Q. Yao, C.-K. Yin, L.-L. Lin, X.-M. Feng and X.-H. Liu, Org. Lett., 2016, 18, 3602 CrossRef CAS PubMed; (g) J. Novacek, J. A. Izzo, M. J. Vetticatt and M. Waser, Chem.–Eur. J., 2016, 22, 17339 CrossRef CAS PubMed.
- A. M. R. Smith, D. Billen and K. K. Hii, Chem. Commun., 2009, 26, 3925 RSC.
- M. Lu, D. Zhu, Y.-P. Lu, X.-F. Zeng, B. Tan, Z.-J. Xu and G.-F. Zhong, J. Am. Chem. Soc., 2009, 131, 4 CrossRef PubMed.
- (a) Y.-K. Wang, Z.-H. Zheng, M.-M. Lian, H. Yin, J.-N. Zhao, Q.-W. Meng and Z.-X. Gao, Green Chem., 2016, 18, 5493 RSC; (b) Y.-K. Wang, H. Yin, X.-F. Tang, Y.-F. Wu, Q.-W. Meng and Z.-X. Gao, J. Org. Chem., 2016, 81, 7042 CrossRef CAS PubMed; (c) F. Yang, J.-N. Zhao, X.-F. Tang, Y.-F. Wu, Z.-Y. Yu and Q.-W. Meng, Adv. Synth. Catal., 2019, 361, 1673 CrossRef CAS; (d) J.-N. Zhao, F. Yang, Z.-Y. Yu, X.-F. Tang, Y.-F. Wu, C.-F. Ma and Q.-W. Meng, Chem. Commun., 2019, 55, 13008 RSC.
- W. Ding, L.-Q. Lu, Q.-Q. Zhou, Y. Wei, J.-R. Chen and W.-J. Xiao, J. Am. Chem. Soc., 2017, 139, 63 CrossRef CAS PubMed.
- H. Yin, C.-J. Wang, Y.-G. Zhao, Z.-Y. He, M.-M. Chu, Y.-F. Wang and D.-Q. Xu, Org. Biomol. Chem., 2021, 19, 6588 RSC.
- (a) Y.-F. Wang, H.-J. Wang, Y.-D. Jiang, C. Zhang, J.-J. Shao and D.-Q. Xu, Fast, Green Chem., 2017, 19, 1674 RSC; (b) B. Wang, Y.-F. Wang, Y.-D. Jiang, M.-M. Chu, S.-S. Qi, W.-Z. Ju and D.-Q. Xu, Org. Biomol. Chem., 2018, 16, 7702 RSC; (c) Y.-F. Wang, J.-J. Shao, B. Wang, M.-M. Chu, S.-S. Qi, X.-H. Du and D.-Q. Xu, Adv. Synth. Catal., 2018, 360, 2285 CrossRef CAS; (d) Y.-F. Wang, Z.-H. Jiang, M.-M. Chu, S.-S. Qi, H. Yin, H.-T. Han and D.-Q. Xu, Org. Biomol. Chem., 2020, 18, 4927 RSC; (e) Y.-F. Wang, C.-J. Wang, Q.-Z. Feng, J.-J. Zhai, S.-S. Qi, A.-G. Zhong, M.-M. Chu and D.-Q. Xu, Chem. Commun., 2022, 58, 6653 RSC.
- The crude organic solution was immediately detected by HR-ESI-MS, and the spectrogram of the mass spectrum could be seen in the ESI†.
- L. J. Kirschenbaum and K. Kustin, J. Chem. Soc. A, 1970, 684 RSC.
- A. Keivanloo, M. Bakherad, M. Khosrojerdi and A. H. Amin, Res. Chem. Intermed., 2018, 44, 2571 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d2ra07411g |
| This journal is © The Royal Society of Chemistry 2023 |