
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceDFT insights into Nb-based 211 MAX phase carbides: Nb2AC (A = Ga, Ge, Tl, Zn, P, In, and Cd)†
Prima Das,
N. Jahan * and
M. A. Ali*
* and
M. A. Ali*
Department of Physics, Chittagong University of Engineering and Technology (CUET), Chattogram-4349, Bangladesh. E-mail: nusrat83@cuet.ac.bdashrafphy31@cuet.ac.bd
First published on 14th February 2023
Abstract
In this study, we performed the first-principles calculations to study the 211 MAX phase carbides: Nb2AC (A = Ga, Ge, Tl, Zn, P, In, Cd, and Al). The structural characteristics are in good agreement with those of the prior studies. The mechanical behavior has been explored by calculating the stiffness constants, elastic moduli, and Vickers hardness. The stiffness constants and phonon dispersion curves were used to check the structural stability of the selected compounds. 2D and 3D plotting of elastic moduli and calculated anisotropy indices disclosed the anisotropy of the elastic properties. We utilized the Mulliken atomic and bond overlap population to explain the mixture of ionic and covalent bonding among these carbides. The metallic behavior has been confirmed by calculating the band structure and density of states (DOS). Partial DOS was also used to discuss the bonding nature and strength among the different states. The optical properties of these phases have also been computed and analyzed to reveal possible relevance in diverse fields. The Debye temperature (ΘD), Grüneisen parameter (γ), melting temperature (Tm), and minimum thermal conductivity (Kmin) were studied to bring out their possible relevance in high-temperature technology. The outcomes of this research indicate that the titled carbides are suitable for use as solar radiation-protecting coating and thermal barrier coating (TBC) materials.
1 Introduction
Many ternary compounds such as carbides, nitrides, and borides have been included in the MAX phase materials family and can be expressed as Mn+1AXn, where M belongs to the early transition metal group, A is the element that exists within the groups 12–16, and X can only be C, N, and B, and n is a positive integer with a value of 1–3.1–4 The MAX phase was first revealed in the 1960s by Nowotny et al.1–4 In the 1990s, Barsoum et al.5,6 renewed the interest by revealing their remarkable characteristics. They have remarkable mechanical strength, machinability, electronic conductivity, and thermal conductivity. They are also machinable, similar to metals, have better mechanical properties at high temperatures, and have superior corrosion and oxidation resistance including ceramics.7 The MAX phase family has become a significant materials group from both research and application points of view. Owing to the magnificent fusing of metallic and ceramic properties, the number of published articles is increasing day by day.8Use of C and N as X elements was confined for a long time9–11 and has been extended recently, where B has been used as an X element. The extension has opened a new platform for the MAX phase materials, owing to the interesting properties and potential applications of B and B-containing compounds.12,13 To date, only a few of the MAX phase borides have been synthesized so far in spite of a large number of predicted phases.14 On the contrary, a significant number of MAX phase carbides have already been synthesized and characterized, revealing their prospective applications. In parallel to the experimental study, a large number of computational approaches15,16 have also already been performed throughout the last decades. Cover et al.17 performed the first-principles calculation of the elastic properties of 240 elemental combinations, revealing the role of A elements and their interaction with M elements. Keast et al.18 computed the total energies of competing phases to check the stability of five different schemes (Crn+1-Al-Cn, Tin+1-Al-Cn, Tin+1-Si-Nn, Tin+1-Al-Nn, and Tin+1-Si-Cn, where n is a positive integer with a value of 1 ∼ 4). Aryal et al.19 studied 792 MAX phases, and 665 phases were found to be thermodynamically and elastically stable. 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 314 solid solutions and 216 possible M2AX phases were revealed by Ashton et al.20 Khaledialidusti et al.21 explored a large group of MAX phases, where M is taken as Sc, Ti, Y, Zr, V, Cr, Nb, Hf, Ta, Mo, and W and A is taken as P, S, Al, Si, Zn, Cu, Ga, As, Ge, Sn, Cd, In, Bi, Ir, Tl, Au, and Pb, and revealed their possibility of exfoliation to produce 2D systems. More than 80 MAX phases have been synthesized, most of which are 211 phases (58 prominent members).22 These studies have inspired us to think about Nb-based 211 MAX phases.
314 solid solutions and 216 possible M2AX phases were revealed by Ashton et al.20 Khaledialidusti et al.21 explored a large group of MAX phases, where M is taken as Sc, Ti, Y, Zr, V, Cr, Nb, Hf, Ta, Mo, and W and A is taken as P, S, Al, Si, Zn, Cu, Ga, As, Ge, Sn, Cd, In, Bi, Ir, Tl, Au, and Pb, and revealed their possibility of exfoliation to produce 2D systems. More than 80 MAX phases have been synthesized, most of which are 211 phases (58 prominent members).22 These studies have inspired us to think about Nb-based 211 MAX phases.
Nb-based 211 MAX phases have also attracted attention in recent years. For example, Nb2AC (A = Al, Ge, Ga, Sn, In, As, P, S, and Cu) MAX phases were studied by Hadi et al.23 to explore the elastic behavior and radiation tolerant behavior. Superconducting nature has been found in the Nb2SnC, Nb2InC, Nb2AsC, and Nb2SC phases23 with the lowest superconducting temperature for Nb2AsC.22 Bouhemadou et al.24,25 performed first-principles calculations to study the structural and elastic properties of Nb2InC and Nb2GeC. The Nb2InC phase was first synthesized by Jeitschko et al.26 Nb2AlC has attracted much attention as a viable material because of its better mechanical and thermal properties.27,28 When compared with several MAX phases, it has been seen that Nb2PC has higher elastic constants.17 The exfoliation possibility of Nb2GaC and Nb2InC into 2D MXene systems has been reported.29 The electrochemical properties of Nb2SnC have been investigated for use in a Li-ion electrolyte.30 The noble transition metal Cu has also been selected as an A element in Nb2CuC.31 Shein et al.32 explored the structural, electrical, and elastic properties of M2GaC (M = Mo, V, and Nb). Cover et al.17 studied only the elastic and structural properties of Nb2SiC, whereas some fundamental properties need to be investigated. Bouhemadou et al.33 carried out a theoretical study to calculate the structural, elastic, electronic, and thermal properties of Nb2SiC.
Moreover, so far, we know only the electronic and mechanical characteristics of Nb2AC (A = Ga, Ge, P, and In) phases have been investigated, whereas Nb2AC (A = Tl, Zn, and Cd) phases have been predicted to be stable via the calculation of formation energy,21 and the physical properties are not disclosed yet. Exploration of the physical properties of new materials carries the same significance as prediction of new materials: it is impossible to take any advantage of new materials unless their physical properties are brought out. Several essential physical aspects, important optical properties, mechanical anisotropy, Vickers hardness including Mulliken populations, and thermal properties of Nb2AC (A = Ga, Ge, P, and In) are still unexplored. Mechanical anisotropy is critical for structural materials since it is linked to important mechanisms such as crack formation (and propagation), plastic deformation, and elastic instability, all of which limit their utility. The Vickers hardness describes the total strength properties of a solid's particular bonds. Mulliken population analysis is important to prove the existence of the combination of the ceramic and metallic nature. Understanding the thermal properties is important to forecast their appropriateness for use in extreme conditions. The optical characteristics of standard MAX phase compounds are necessary to predict their possible applications, for instance, as coating materials for protection from solar heating. These are the motivations behind this study, which demonstrates the significance of in-depth research on MAX phase carbides Nb2AC (A = Ga, Ge, Tl, Zn, P, In, and Cd).
Therefore, we aimed to provide a theoretical insight into the MAX phase carbides Nb2AC (A = Ga, Ge, Tl, Zn, P, In, and Cd), in which we will consider the first-time investigation of Nb2AC (A = Tl, Zn, and Cd) and some important unexplored properties of Nb2AC (A = Ga, Ge, P, and In) phases. Last of all, the calculated parameters of the titled phases are compared with those of the most known Nb-based phase, Nb2AlC, to make this research a systematic one.
2 Computational methodology
The Nb2AC (A = Ga, Ge, Tl, Zn, P, In, and Cd) carbides' physical properties have been calculated using the pseudopotential-based DFT, implemented in CASTEP (CAmbridge Serial Total Energy Package).34,35 For the term of exchange-correlation, the GGA (generalized gradient approximation) of the PBE (Perdew–Burke–Ernzerhof)36 and PBEsol (Perdew–Burke–Ernzerhof for solids)37 were used. The PBE has been widely used for DFT calculations, whereas PBEsol gives a more accurate lattice constant for solids.38 Recently, the effect of different functionals on the calculated values of the parameters has been reported, in which it is seen that the PBEsol gives more accurate results of the lattice parameters compared with other functionals for solids. The more accurate values of the lattice parameters give more accurate results, especially the mechanical behavior characterizing parameters. Thus, we have selected PBEsol in association with the mostly used GGA-PBE functional for this study.39,40 The electronic structure was optimized by density mixing, and the atomic configuration was relaxed using BFGS (Broyden Fletcher Goldfarb Shanno).41 The electronic orbitals of Nb- 4d4 5s1, C- 2s2 2p2, Ga- 4s2 4p1, Ge- 3d10 4s2 4p2, Tl- 5d10 6s2 6p1, Zn- 3d10 4s2, P- 3s2 3p3, In- 4d10 5s2 5p1 and Cd- 4d10 5s2 were accomplished for pseudo-atomic calculations. The cutoff energy was set to 500 eV, and the convergence was assured using a k-point mesh of 9 × 9 × 2.42 The total energy self-consistent convergence was carried out using 5 × 10−6 eV per atom, with 0.01 eV Å as the maximum force on the atom. Again, 5 × 10−4 Å is used for an ionic displacement, which is the maximum range, with a maximum stress of 0.02 GPa. The phonon dispersion curves were calculated using the Density Functional Perturbation Theory (DFPT) linear-response method.43 Most of our data have been compared to those of ref. 23 (and 25), where the calculations were performed using the following inputs: exchange-correlation – GGA-PBE (LDA-CA), cut-off energy- 550 (350) eV, k-points-10 × 10 × 2 (9 × 9 × 2).3 Results and discussion
3.1 Structural properties and phase stability
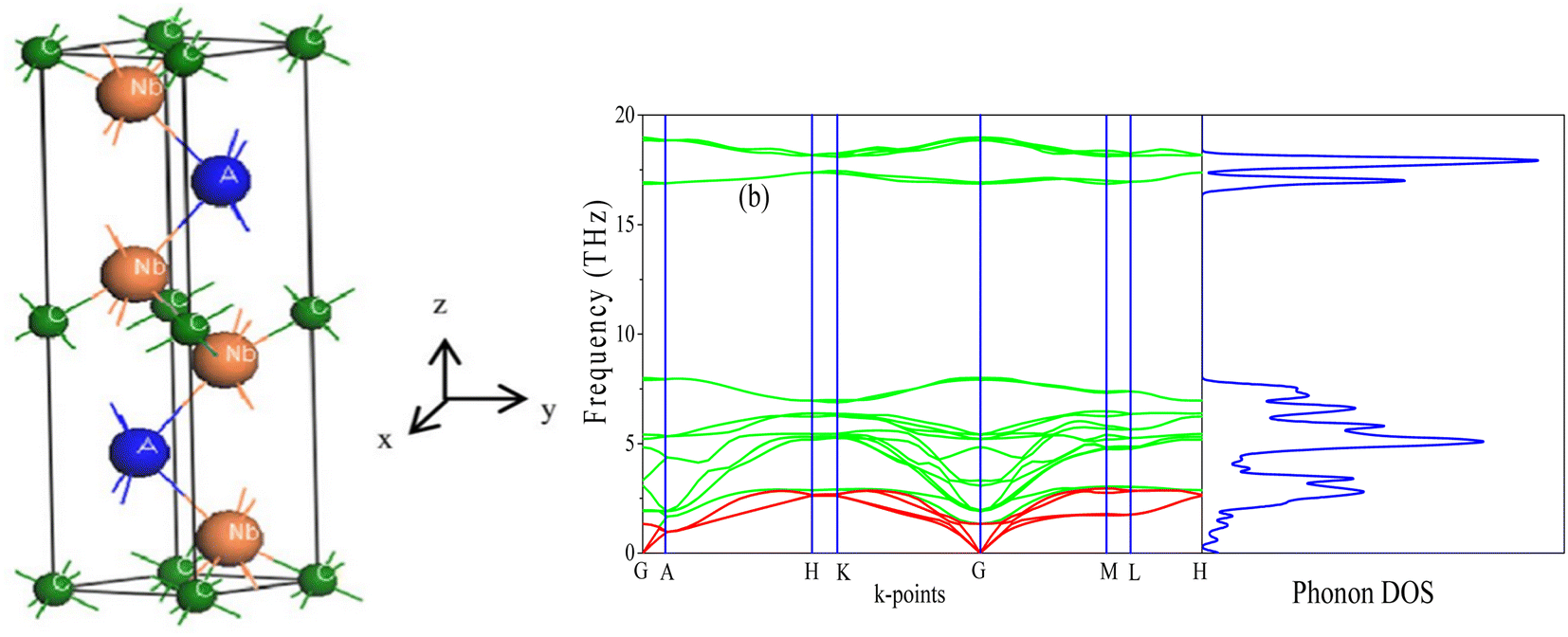 | ||
| Fig. 1 (a) The unit cell of Nb2AC (A = Ga, Ge, Tl, Zn, P, In, Cd, and Al); (b) phonon dispersion curve and DOS of Nb2GaC calculated using GGA PBEsol. | ||
| Phases | a (Å) | c (Å) | Zm | c/a | Density (ρ) | Volume (Å3) | Od | Pd | Ref. |
|---|---|---|---|---|---|---|---|---|---|
| a Calculated values using GGA PBE.36b Calculated values using GGAPBEsol37 and *calculated values using reported data. | |||||||||
| Nb2GaC | 3.145 | 13.73 | 0.0896 | 4.366 | 7.59 | a116.97 | 1.039 | 0.689 | aThis |
| 3.115 | 13.51 | 4.337 | b113.53 | 1.044 | 0.834 | bThis | |||
| 3.143 | 13.64 | 4.340 | 116.66 | *1.044 | *0.834 | 23 | |||
| 3.13 | 13.56 | 4.332 | 115.05 | *1.046 | *0.836 | 6 | |||
| Nb2GeC | 3.244 | 12.69 | 0.095 | 3.914 | 7.76 | 115.75 | 1.086 | 0.904 | aThis |
| 3.194 | 12.77 | 3.998 | 112.83 | 1.066 | 0.890 | bThis | |||
| 3.255 | 12.59 | 3.868 | *1.097 | *0.912 | 32 | ||||
| 3.237 | 12.35 | 3.815 | *1.109 | *0.959 | 25 | ||||
| 3.228 | 12.76 | 3.953 | 115.10 | *1.076 | *0.898 | 23 | |||
| Nb2TlC | 3.229 | 12.74 | 0.0795 | 3.944 | 10.18 | 131.11 | 1.254 | 0.891 | aThis |
| 3.199 | 14.27 | 4.461 | 126.46 | 1.131 | 0.781 | bThis | |||
| Nb2ZnC | 3.145 | 13.73 | 0.0875 | 4.366 | 7.43 | 117.58 | 1.060 | 0.829 | aThis |
| 3.117 | 13.53 | 4.341 | 113.83 | 1.066 | 0.833 | bThis | |||
| Nb2PC | 3.299 | 11.59 | 0.0999 | 3.515 | 8.28 | 109.33 | 1.149 | 0.978 | aThis |
| 3.267 | 11.52 | 3.526 | 106.48 | 1.146 | 0.976 | bThis | |||
| 3.292 | 11.578 | 3.517 | 108.68 | *1.139 | *0.978 | 23 | |||
| 3.28 | 11.5 | 3.506 | 107.15 | *1.152 | *0.979 | 6 | |||
| Nb2InC | 3.185 | 14.54 | 0.0821 | 4.563 | 11.49 | 127.69 | 1.103 | 0.799 | aThis |
| 3.152 | 14.37 | 4.559 | 123.67 | 1.079 | 0.799 | bThis | |||
| 3.186 | 14.528 | 4.560 | 127.72 | *1.079 | *0.799 | 23 | |||
| 3.172 | 14.37 | 4.530 | 125.21 | *1.085 | *0.804 | 47 | |||
| Nb2CdC | 3.172 | 14.68 | 0.0830 | 4.628 | 8.06 | 127.87 | 1.055 | 0.790 | aThis |
| 3.140 | 14.45 | 4.602 | 123.37 | 1.060 | 0.794 | bThis | |||
| Nb2AlC | 3.245 | 12.69 | 0.0897 | 3.911 | 6.34 | 115.74 | 1.142 | 0.902 | aThis |
| 3.102 | 13.79 | 4.446 | 114.99 | 1.021 | 0.819 | bThis | |||
| 3.12 | 13.93 | 4.463 | 117.41 | *1.017 | *0.817 | 23 | |||
Table 1 shows the lattice constants (a, c) for an optimized cell, internal parameters, and the hexagonal ratio (c/a) ratio of Nb2AC (A = Ga, Ge, Tl, Zn, P, In, Cd, and Al). The fundamental polyhedrons of Nb2AC (A = Ga, Ge, Tl, Zn, P, In, Cd, and Al) are studied using Hug's distortion indexes (DIs).44,45 Two parameters- the distortion of octahedral (Od) and trigonal prism (Pd) of the M6X octahedrons and M6A trigonal prisms, respectively, are used to describe the distortions in the structure of the 211 MAX phases using the following equations:46
| Od = √3/2{4zM2(c/a)2 + 1/12}1/2 |
| Pd = 1/{1/3 + (1/4 − zM)2(c/a)2}1/2 |
The polyhedron would be ideal when both the octahedron and the trigonal parameters are equal to 1.46 The distortion of the polyhedron is measured by the deviation from 1, where a low distortion value indicates a more stable structure.46 Table 1 shows the value of the Od and Pd of Nb2AC (A = Ga, Ge, Tl, Zn, P, In, Cd, and Al). Table 1 also contains the Od and Pd of Nb2AC (A = Ga, Ge, P, In, and Al) as calculated from the reported lattice parameters. A very good consistency is observed for the previously studied phases, indicating the reliability of our present calculations. The comparison is not possible for Nb2AC (A = Tl, Zn, and Cd) phases because of their first-time calculation. The accuracy of the present calculations is also revealed by the close agreement of the values of a and c [Table 1] of Nb2AC (A = Ga, Ge, P, In, and Al) phases with previously reported values.23,25,32
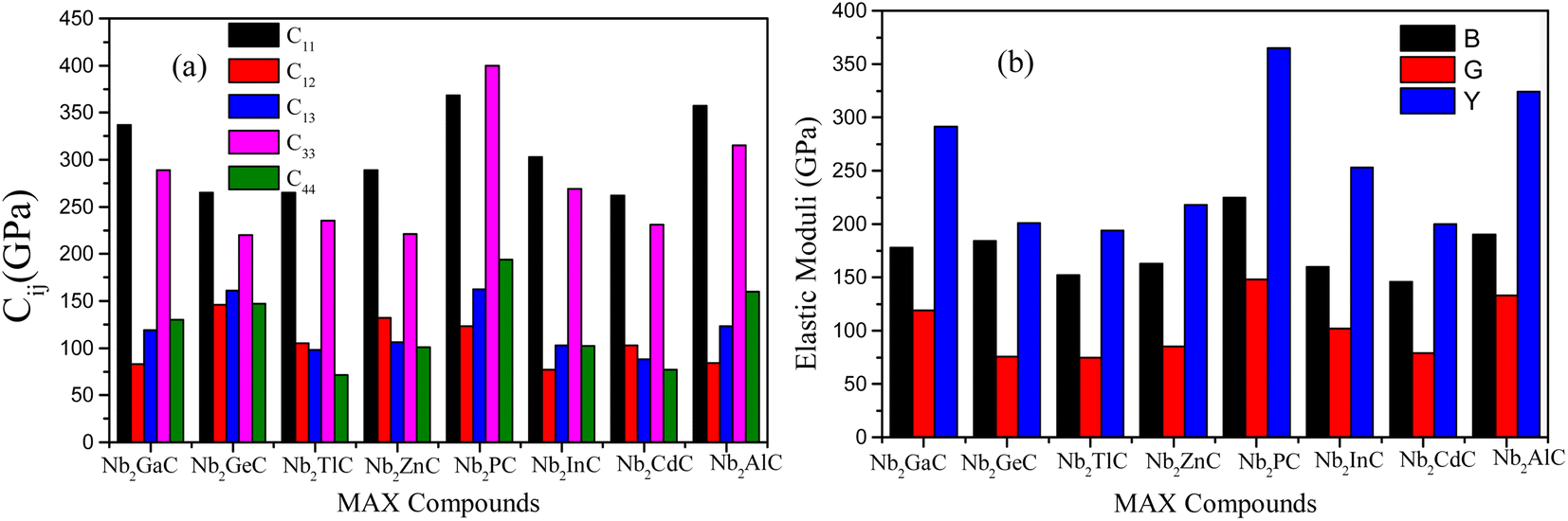 | ||
| Fig. 2 Comparison of (a) stiffness constants and (b) elastic moduli of Nb2AC (A = Ga, Ge, Tl, Zn, P, In, Cd, and Al) MAX phases calculated using GGA PBEsol. | ||
3.2 Mechanical properties
| Parameters | Nb2GaC | Nb2GeC | Nb2TlC | Nb2ZnC | Nb2PC | Nb2InC | Nb2CdC | Nb2AlC | Ref. |
|---|---|---|---|---|---|---|---|---|---|
| a Calculated values using GGA PBE.36b Calculated values using GGA PBEsol37 and *calculated values using reported data. | |||||||||
| C11 (GPa) | 337 | 265 | 265 | 289 | 368 | 303 | 262 | 357 | aThis |
| 363 | 306 | 283 | 294 | 394 | 331 | 282 | 368 | bThis | |
| 323 | 284 | 373 | 280 | 333 | 23 | ||||
| 308 | 25 | ||||||||
| C33 (GPa) | 289 | 220 | 235 | 221 | 400 | 269 | 231 | 315 | aThis |
| 313 | 295 | 258 | 225 | 423 | 295 | 258 | 314 | bThis | |
| 281 | 275 | 407 | 266 | 284 | 23 | ||||
| 306 | 25 | ||||||||
| C44 (GPa) | 130 | 147 | 71 | 101 | 194 | 102 | 77 | 160 | aThis |
| 141 | 151 | 86 | 113 | 212 | 112 | 89 | 164 | bThis | |
| 126 | 152 | 193 | 104 | 138 | 23 | ||||
| 177 | 25 | ||||||||
| C12 (GPa) | 83 | 146 | 105 | 132 | 123 | 77 | 103 | 98 | aThis |
| 92 | 137 | 106 | 137 | 132 | 85 | 123 | 84 | bThis | |
| 77 | 136 | 114 | 80 | 84 | 23 | ||||
| 133 | 25 | ||||||||
| C13 (GPa) | 119 | 161 | 98 | 106 | 162 | 103 | 88 | 123 | aThis |
| 131 | 161 | 118 | 122 | 179 | 112 | 102 | 119 | bThis | |
| 130 | 161 | 172 | 113 | 117 | 23 | ||||
| 168 | 25 | ||||||||
| CP (GPa) | −47 | −1 | 34 | 31 | −71 | −25 | 26 | −62 | aThis |
| −49 | −14 | 20 | 24 | −80 | −27 | 34 | −80 | bThis | |
| *−49 | *−16 | *−79 | *−24 | *−54 | 23 | ||||
| *−44 | 25 | ||||||||
| B (GPa) | 178 | 184 | 152 | 163 | 225 | 160 | 146 | 190 | aThis |
| 194 | 203 | 168 | 173 | 242 | 175 | 163 | 188 | bThis | |
| 178 | 195 | 230 | 160 | 176 | 23 | ||||
| 206 | 25 | ||||||||
| G (GPa) | 119 | 76 | 75 | 85 | 148 | 102 | 79 | 133 | aThis |
| 128 | 101 | 84 | 88 | 153 | 112 | 85 | 140 | bThis | |
| 114 | 101 | 150 | 96 | 122 | 23 | ||||
| 109 | 25 | ||||||||
| Y (GPa) | 291 | 201 | 194 | 218 | 365 | 253 | 200 | 324 | aThis |
| 314 | 261 | 215 | 225 | 379 | 277 | 216 | 337 | bThis | |
| 282 | 258 | 369 | 240 | 298 | 23 | ||||
| 279 | 25 | ||||||||
| ν | 0.23 | 0.32 | 0.29 | 0.28 | 0.23 | 0.24 | 0.27 | 0.22 | aThis |
| 0.23 | 0.29 | 0.29 | 0.28 | 0.24 | 0.23 | 0.28 | 0.20 | bThis | |
| *0.24 | *0.28 | *0.23 | *0.25 | *0.22 | 23 | ||||
| *0.28 | 25 | ||||||||
| G/B | 0.67 | 0.41 | 0.49 | 0.52 | 0.65 | 0.63 | 0.54 | 0.70 | aThis |
| 0.66 | 0.49 | 0.50 | 0.51 | 0.63 | 0.64 | 0.52 | 0.74 | bThis | |
| *0.64 | *0.52 | *0.65 | *0.60 | *0.69 | 23 | ||||
| *0.53 | 25 | ||||||||
| B/C44 | 1.36 | 1.25 | 2.14 | 1.61 | 1.15 | 1.56 | 1.89 | 1.18 | aThis |
| 1.38 | 1.34 | 1.95 | 1.53 | 1.14 | 1.56 | 1.83 | 1.15 | bThis | |
| *1.41 | *1.28 | *1.19 | *1.54 | *1.28 | 23 | ||||
| *1.16 | 25 | ||||||||
Furthermore, the stiffness constants are used to predict the ductile/brittle behavior of Nb2AC (A = Ga, Ge, Tl, P, Zn, In, Cd, and Al) phases by computing the Cauchy pressure (CP). The difference between C11 and C44 is defined as the CP;52 a negative and positive value represents the brittle and ductile nature, respectively. Negative and positive values also indicate the directional covalent and ionic bonds, respectively. As evident from Table 2, Nb2GaC, Nb2GeC, Nb2PC, Nb2InC, and Nb2AlC have directional covalent bonds and behave as brittle solids. On the contrary, Nb2TlC, Nb2ZnC, and Nb2CdC phases behave as ductile solids with a positive CP. Though most MAX phases are brittle, a few of them, such as Zr3CdB4, Ti2CdC, and Ti2ZnX (X = C and N), have already been reported to be ductile.53–55
Finally, the stiffness constants were used to calculate the polycrystalline elastic moduli. Hill's approximation56 was used to compute the bulk modulus (B) and shear modulus (G), which is the average of the Voight57 and the Reuss58 models as follows: [B = (BV + BR)/2]; here, BV = [2 (C11 + C12) + C33 + 4C13]/9 and BR = C2/M, where C2 = C11 + C12C33 − 2C132; M = C11 + C12 + 2C33 − 4C13. BV and BR are expressed as the upper (Voight) and lower limit (Reuss) of B, respectively. Again, [G = (GV + GR)/2]; here, GV = [12C44 + 12C66]/30 and GR = (5/2) [C2C44C66]/[3BVC44C66 + C2(C44 + C66)], where C66 = (C11 − C12)/2. Here, like B, GV and GR are also expressed as the upper (Voight) and lower limit (Reuss) of G, respectively. The Poisson's ratio (υ) and Young's modulus (Y) are also computed from B and G by using these relations: Y = 9BG/(3B + G) and υ = (3B − Y)/(6B).59,60
As known, the pure deformations (volume and shape) are studied by the bulk modulus (mostly known for the study of elastic response against pressure) and the shear modulus (mostly known for the study of rigidity of solids against pressure). It is seen from Table 2 that Nb2PC (Nb2CdC) has the highest (lowest) resistance against hydrostatic pressure, whereas the lowest resistance to plastic deformation is noted for Nb2TlC among the studied compounds. However, the compounds can be ranked based on the values of B (calculated using GGA-PBEsol) as follows: Nb2PC > Nb2GeC > Nb2GaC > Nb2AlC > Nb2InC > Nb2ZnC > Nb2TlC > Nb2CdC, whereas the ranking for G will be as follows: Nb2PC > Nb2AlC > Nb2GaC > Nb2InC > Nb2GeC > Nb2ZnC > Nb2CdC > Nb2TlC. For Young's modulus, Y is the measure of the stiffness of solids that relates the stiffness with the thermal shock resistance (inverse relationship) of solids. Therefore, a solid with a high Y value indicates high stiffness and low thermal shock resistance (TSR).61 The Y-based ranking of the phases also follows the G-based ranking. Table 2 implies that Nb2TlC exhibits high TSR, while Nb2PC has the lowest TSR, followed by the Y-based reverse ranking among the herein-studied phases. Although these moduli do not indicate hardness, they are usually higher for harder materials.62 Compared to Y, B and G have a close relationship with the material's hardness. In some cases, these parameters are used to predict the hardness of solids using the following formulae: 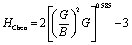 ,63 and
,63 and 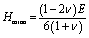 ,64 Table 2 also includes the previously reported values.23 As evident from Table 2, the obtained values are in good accord with reported values, ensuring the accuracy of the present calculation that helps other researchers to consider our calculated values as a reference for both application and research purposes. A comparison of the elastic moduli for the studied compounds is shown in Fig. 2(b).
,64 Table 2 also includes the previously reported values.23 As evident from Table 2, the obtained values are in good accord with reported values, ensuring the accuracy of the present calculation that helps other researchers to consider our calculated values as a reference for both application and research purposes. A comparison of the elastic moduli for the studied compounds is shown in Fig. 2(b).
The Machinability Index (MI) is commonly used in the tribological sector to forecast a solid's performance and is defined as the B/C44 ratio. The MI index is presented in Table 2. As evident, the MI of Nb2TlC, Nb2ZnC, and Nb2CdC is higher than that of other studied phases owing to their ductile nature. Though Nb2GeC is ductile, its C44 is comparatively higher, which results in a lower MI value. Based on the values [Table 2], the titled phases are expected to be more machinable than the widely known Nb2AlC phase except for Nb2PC. The lowest MI is found for Nb2PC, as expected, due to its highest C44. In addition, a good relationship between machinability and ductility is observed as expected. The MI values are also different for PBE and PBEsol functions because of the different values of B and C44. However, the obtained values of MI are comparable with those of some other 211 MAX phases, like Ti2AlC, whose machinability index is 1.23.69 Though some 211 phases exhibit a very high value of MI, such as W2SnC (MI = 33.3) and Mo2PbC (MI = 15.8),17,70 it should be noted that their C44 values are much lower (W2SnC, 6 GPa, and Mo2PbC, 10 GPa).17
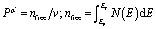 , Ep indicates the energy of the pseudogap and EF indicates the energy of the Fermi level. The volume of the μ-type bond is denoted by vμb, which is calculated in the following way:
, Ep indicates the energy of the pseudogap and EF indicates the energy of the Fermi level. The volume of the μ-type bond is denoted by vμb, which is calculated in the following way: 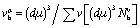 . At last, the equation for Vickers hardness is as follows:
. At last, the equation for Vickers hardness is as follows: 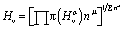 ; nμ = μ-type bond number. Table 3 shows the computed Vickers hardness of Nb2AC (A = Ga, Ge, Tl, Zn, P, In, Cd, and Al). As shown in Table 3, Nb2PC has higher Vickers hardness than other studied compounds, wherein Nb2TlC possesses the lowest value of Hv, in agreement with mechanical parameters such as C44 and G, which are assumed to be more related to the hardness of solids. The Hv values of Nb2AC (A = Ga, Ge, Tl, Zn, In, Cd, and Al) are comparable with the exception of Nb2PC, which has a much higher value of Hv. This can be explained on the basis of the bond overlap population (Pμ). As seen in Table 3, the Pμ of Nb2PC is 1.01 and 0.98 for C–Nb and P–Nb bonds, respectively, which indicates that strong covalent bonding is found between both Nb–C and Nb–P atoms, whereas for Nb2AC (A = Ga, Ge, Tl, Zn, In, Cd, and Al), Pμ is found only for Nb–C atoms, and no other significant covalent bond is noticed for these phases. Thus, both significant covalent bonding among Nb–C and Nb–P atoms is assumed to be responsible for such higher hardness. However, the variation in Hv of Nb2AC (A = Ga, Ge, Tl, Zn, In, Cd, and Al) phases is due to the combined effect of the variation in both bond population Pμ and bond length dμ.
; nμ = μ-type bond number. Table 3 shows the computed Vickers hardness of Nb2AC (A = Ga, Ge, Tl, Zn, P, In, Cd, and Al). As shown in Table 3, Nb2PC has higher Vickers hardness than other studied compounds, wherein Nb2TlC possesses the lowest value of Hv, in agreement with mechanical parameters such as C44 and G, which are assumed to be more related to the hardness of solids. The Hv values of Nb2AC (A = Ga, Ge, Tl, Zn, In, Cd, and Al) are comparable with the exception of Nb2PC, which has a much higher value of Hv. This can be explained on the basis of the bond overlap population (Pμ). As seen in Table 3, the Pμ of Nb2PC is 1.01 and 0.98 for C–Nb and P–Nb bonds, respectively, which indicates that strong covalent bonding is found between both Nb–C and Nb–P atoms, whereas for Nb2AC (A = Ga, Ge, Tl, Zn, In, Cd, and Al), Pμ is found only for Nb–C atoms, and no other significant covalent bond is noticed for these phases. Thus, both significant covalent bonding among Nb–C and Nb–P atoms is assumed to be responsible for such higher hardness. However, the variation in Hv of Nb2AC (A = Ga, Ge, Tl, Zn, In, Cd, and Al) phases is due to the combined effect of the variation in both bond population Pμ and bond length dμ.
| Compounds | Bond | nμ | dμ (Å) | Pμ | Pμ′ | vμb (Å3) | Hμv (GPa) | Hv (GPa) | Ref. |
|---|---|---|---|---|---|---|---|---|---|
| a Calculated values using GGA PBE.36b Calculated values using GGA PBEsol.37 | |||||||||
| Nb2GaC | C–Nb | 4 | 2.177 | 0.93 | 0.0223 | 0.0036 | 2.418 | 2.418 | aThis |
| 2.167 | 0.93 | 0.0209 | 0.0037 | 2.55 | 2.55 | bThis | |||
| Nb2GeC | C–Nb | 4 | 2.208 | 0.99 | 0.0443 | 0.0037 | 1.498 | 1.498 | aThis |
| 2.183 | 1.02 | 0.0100 | 0.0038 | 2.86 | 2.86 | bThis | |||
| Nb2TlC | C–Nb | 4 | 2.194 | 0.94 | 0.0358 | 0.0029 | 1.994 | 1.994 | aThis |
| 2.176 | 0.96 | 0.0207 | 0.0031 | 2.19 | 2.19 | bThis | |||
| Nb2ZnC | C–Nb | 4 | 2.177 | 0.92 | 0.0504 | 0.0036 | 2.297 | 2.297 | aThis |
| 2.160 | 0.91 | 0.0741 | 0.0037 | 2.33 | 2.33 | bThis | |||
| Nb2PC | C–Nb | 4 | 2.229 | 1.01 | 0.0195 | 0.0187 | 13.706 | 9.312 | aThis |
| P–Nb | 4 | 2.580 | 0.98 | 0.0089 | 6.326 | aThis | |||
| C–Nb | 4 | 2.211 | 1.00 | 0.0166 | 0.0200 | 14.56 | 10.02 | bThis | |
| P–Nb | 4 | 2.557 | 0.98 | 0.0096 | 6.89 | bThis | |||
| Nb2InC | C–Nb | 4 | 2.193 | 0.96 | 0.0184 | 0.0031 | 2.160 | 2.160 | aThis |
| 2.174 | 0.95 | 0.0105 | 0.0032 | 2.82 | 2.82 | bThis | |||
| Nb2CdC | C–Nb | 4 | 2.177 | 0.99 | 0.0206 | 0.0031 | 2.224 | 2.224 | aThis |
| 2.160 | 0.93 | 0.0648 | 0.0032 | 2.11 | 2.11 | bThis | |||
| Nb2AlC | C–Nb | 4 | 2.165 | 0.98 | 0.0179 | 0.0037 | 2.63 | 2.63 | aThis |
| 2.166 | 1.00 | 0.0112 | 0.0037 | 2.71 | 2.71 | bThis | |||
3.3 Electronic properties, and Mulliken atomic and bond population analysis
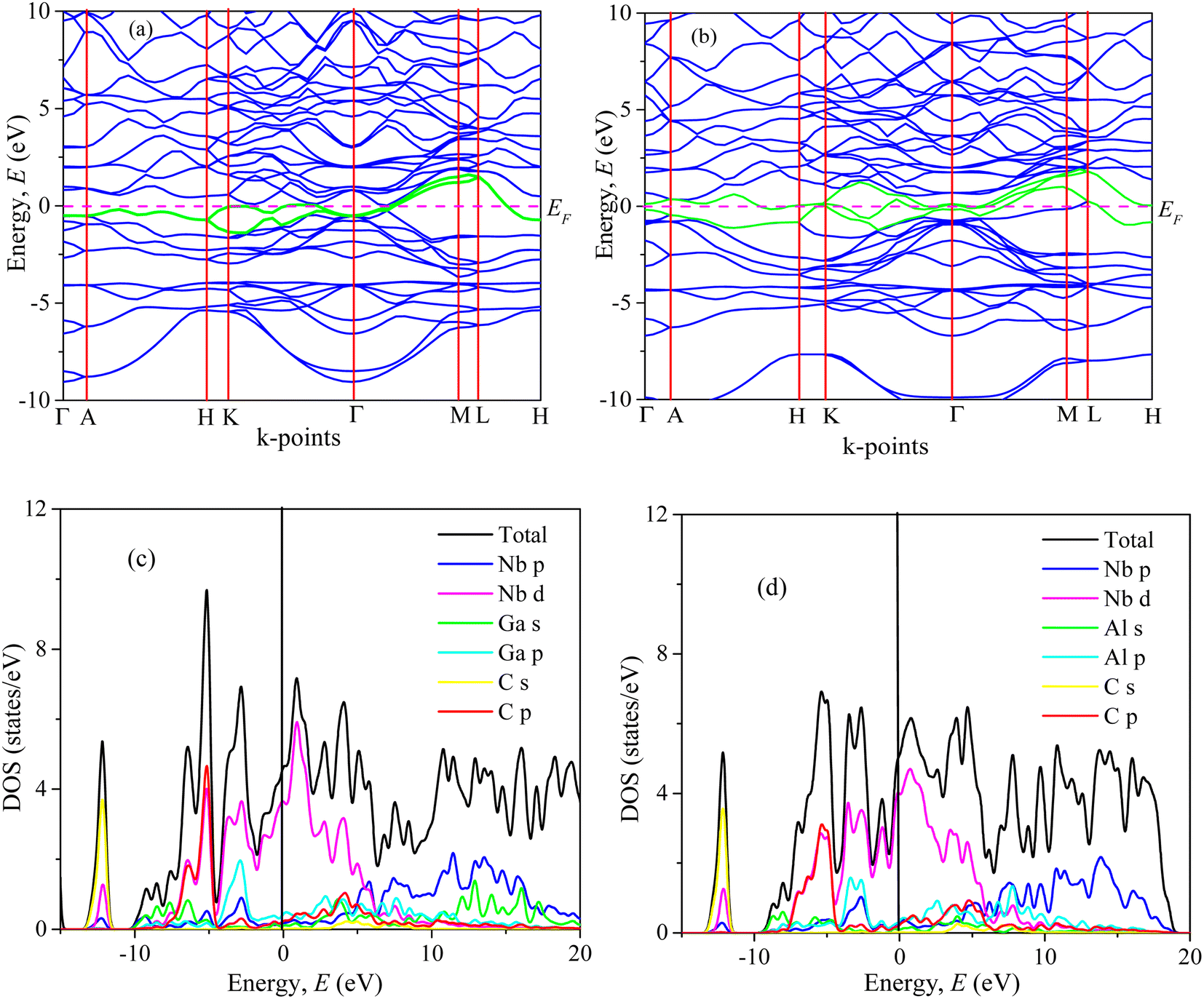 | ||
| Fig. 3 Band structure, and total and partial DOS of (a, c) Nb2GaC and (b, d) Nb2AlC calculated using GGA PBEsol. | ||
We have also computed the total and partial density of states (DOS) of Nb2AC (A = Ga, Ge, Tl, Zn, P, In, Cd, and Al), which are also shown in Fig. 3(c), (d) and S4(a)–(f)† [calculated using PBEsol]. Here, EF is the Fermi level, where the DOS values are 3.2, 3.3, 4.0, 3.5, 3.0, 3.1, 2.8, and 3.0 for Nb2GaC, Nb2GeC, Nb2TlC, Nb2ZnC, Nb2PC, Nb2InC, Nb2CdC, and Nb2AlC, respectively. The computed compound's DOSs of the studied phases are found to be similar to those of reported MAX phases25,72 and Nb2AlC, which is presented here for comparison.
We have also computed the partial density of states (PDOS) to understand better the chemical bonding of Nb2AC (A = Ga, Ge, Tl, Zn, P, In, Cd, and Al). Fig. 3(c), (d), and S4(a)–(f)† [calculated using PBEsol] show the PDOS for Nb2AC. As seen, C-2s is not involved in the DOS at EF. As a result, the conduction properties are not attributed to carbon. On the other hand, at the Fermi level, Nb-d electrons contribute significantly to the DOS; therefore, the conduction properties ought to be involved in Nb. The A-p (A = Ga, Ge, Tl, Zn, P, In, and Cd) electrons are also involved in the conduction mechanism, with a much lower level of contribution. C-p states also slightly contribute to the conduction properties. This outcome is in line with an earlier MAX phase reported.68 The degenerate states concerning both lattice sites and angular momentum indicate that a covalent interaction exists between the atoms of the compounds. Hybridized states include C-p and Nb-d and A (A = Ga, Ge, Tl, Zn, P, In, Cd, and Al) p and Nb d states. Moreover, some ionic characteristics can be anticipated because of the disparity in electro-negativity between the constituent atoms. There is a covalent–ionic combination in the bonding character, which has been explained in Section 3.3.2. When compared to Nb-d and A-p states, the hybridization peak of Nb-d and C-p lies in the lower energy side, as seen in the PDOS; consequently, covalent bonding due to hybridization between Nd-d and C-p states is stronger than that of Nb-d and A p states (A = Ga, Ge, Tl, Zn, P, In, and Cd). The peak position of hybridization between Nb-X and Nb and A states is also responsible for the variation in the hardness of the studied phases. For example, the hybridization among Nb-d, P-p and C-p states is observed in the lowest energy side (below – 5 eV), which results in strong hybridization among them, and a higher bond overlap population is found, which results in the hardest phase of Nb2PC among the considered phases. The same hybridization peak for other phases is found to appear at an energy scale of above −5 eV. Similar results are also reported for the MAX phases.25,72 Additionally, we demonstrated the PDOS of Nb2AlC, which is similar to those of Nb2AC (A = Ga, Ge, Tl, Zn, P, In, Cd, and Al).
| Phases | Atoms | s | p | d | Total | Charge (e) | Bond | Bond number nμ | Bond overlap population Pμ |
|---|---|---|---|---|---|---|---|---|---|
| Nb2GaC | C | 1.43 | 3.22 | 0.00 | 4.65 | −0.65 | C–Nb | 4 | 0.93 |
| Ga | 0.88 | 1.84 | 9.99 | 12.71 | 0.29 | ||||
| Nb | 2.27 | 6.62 | 3.94 | 12.82 | 0.18 | ||||
| Nb2GeC | C | 1.45 | 3.23 | 0.00 | 4.67 | −0.67 | C–Nb | 4 | 1.02 |
| Ge | 0.99 | 2.53 | 0.00 | 3.52 | 0.48 | ||||
| Nb | 2.35 | 6.56 | 3.99 | 12.90 | 0.10 | ||||
| Nb2TlC | C | 1.43 | 3.22 | 0.00 | 4.65 | −0.65 | C–Nb | 4 | 0.96 |
| Tl | 3.11 | 7.84 | 9.92 | 20.87 | 0.13 | ||||
| Nb | 2.25 | 6.58 | 3.92 | 12.74 | 0.26 | ||||
| Nb2ZnC | C | 1.43 | 3.23 | 0.00 | 4.66 | −0.66 | C–Nb | 4 | 0.91 |
| Zn | 0.53 | 1.32 | 9.93 | 11.78 | 0.22 | ||||
| Nb | 2.28 | 6.63 | 3.87 | 12.78 | 0.22 | ||||
| Nb2PC | C | 1.44 | 3.20 | 0.00 | 4.63 | −0.63 | C–Nb | 4 | 1.00 |
| P | 1.57 | 3.45 | 0.00 | 5.02 | 0.04 | P–Nb | 4 | 0.98 | |
| Nb | 2.24 | 6.45 | 3.98 | 12.67 | 0.33 | ||||
| Nb2InC | C | 1.43 | 3.22 | 0.00 | 4.65 | −0.65 | C–Nb | 4 | 0.95 |
| In | 0.99 | 1.81 | 9.97 | 12.77 | 0.23 | ||||
| Nb | 2.22 | 6.63 | 3.94 | 12.79 | 0.21 | ||||
| Nb2CdC | C | 1.43 | 3.23 | 0.00 | 4.66 | −0.66 | C–Nb | 4 | 0.93 |
| Cd | 0.55 | 1.28 | 9.91 | 11.73 | 0.27 | ||||
| Nb | 2.29 | 6.65 | 3.87 | 12.81 | 0.19 | ||||
| Nb2AlC | C | 1.45 | 3.22 | 0.00 | 4.67 | −0.67 | C–Nb | 4 | 1.00 |
| Al | 0.97 | 1.83 | 0.00 | 2.80 | 0.20 | ||||
| Nb | 2.22 | 6.57 | 3.98 | 12.76 | 0.24 |
3.4 The elastic anisotropy
The study of the elastic anisotropy of the MAX phases is essential because of their potential use in practical applications. Some important physical processes, such as plastic deformation, unusual phonon modes, dislocation dynamics, crack behavior, etc., are caused by mechanical anisotropy in solids.76,77 Since the values of C11 and C33 are unequal [Table 2], other elastic moduli are calculated using these elastic constants. Thus anisotropic nature of the elastic properties is expected for these compounds. These facts encourage us to study the mechanical anisotropy of the titled carbides in the 211 MAX phases. It is possible to demonstrate the level of anisotropy by plotting the elastic moduli in different directions. In this manner, we used the ELATE code78 to compute the values of Young's modulus, compressibility, shear modulus, and Poisson's ratio, which are presented in Fig. 4(a–d) for Nb2GaC, 5(a–d) for Nb2AlC, S5(a–d) for Nb2GeC, S6(a–d) for Nb2TlC, S7(a–d) for Nb2ZnC, S8(a–d) for Nb2PC, S9(a–d) for Nb2InC and S10(a–d)† for Nb2CdC. The 3D and 2D plots will help to explain the anisotropic nature. The isotropic nature of solids is represented by the sphere in 3D plots and the circle in 2D plots. In contrast, the anisotropy is indicated by a departure from a perfect circle or sphere, and the degree depends on the departure level. Young's modulus (Y) is anisotropic in the xz and yz planes but isotropic in the xy planes, as seen in Fig. 4(a), 5(a), S5(a), S6(a), S7(a), S8(a), S9(a), and S10(a).† Y has minimum values at the vertical axis of the xz and yz planes and a maximum value at an intermediate angle of 45° of those axes. Fig. 4(b), 5(b), S5(b), S6(b), S7(b), S8(b), S9(b), and S10(b)† depict the compressibility (K), which exhibits a similar anisotropic character to Y. The compressibility (K) is isotropic in the xy plane but anisotropic in the xz and yz planes, where K has maximum values on the axes of the xz and yz planes and a minimum value at an angle of 45° to those axes. For the considered compounds presented in Fig. 4(c), 5(c), S5(c), S6(c), S7(c), S8(c), S9(c), and S10(c),† the shear modulus (G) displays two surfaces for both 2D and 3D representations. The green line shows the minimum values for a 45° angle, while the blue line shows the maximum values for the same angle. In the xy and yz planes, G is maximum along both axes, with the minimum value found at an angle of 45° between the axes. In the xy plane, it is seen to be isotropic. In Fig. 4(d), 5(d), S5(d), S6(d), S7(d), S8(d), S9(d), and S10(d),† a different anisotropic characteristic is seen for Poisson's ratio. Like G, there are two surfaces for both 2D and 3D representations, except Nb2GeC. The blue line indicates maximum values at a 45° angle, whereas the green line indicates minimum values at the same angle for all compounds except Nb2GeC. For Nb2GeC, the green line indicates positive values, where the values are maximum at an angle of 45°, and the red line indicates maximum negative values for the same angle. Poisson's ratio is also shown to be anisotropic, with the minimum values found within the vertical axes in the xz and yz planes. In contrast, the maximum values are found within the horizontal axes for both compounds. In the xy plane, Poisson's ratio is found to be isotropic.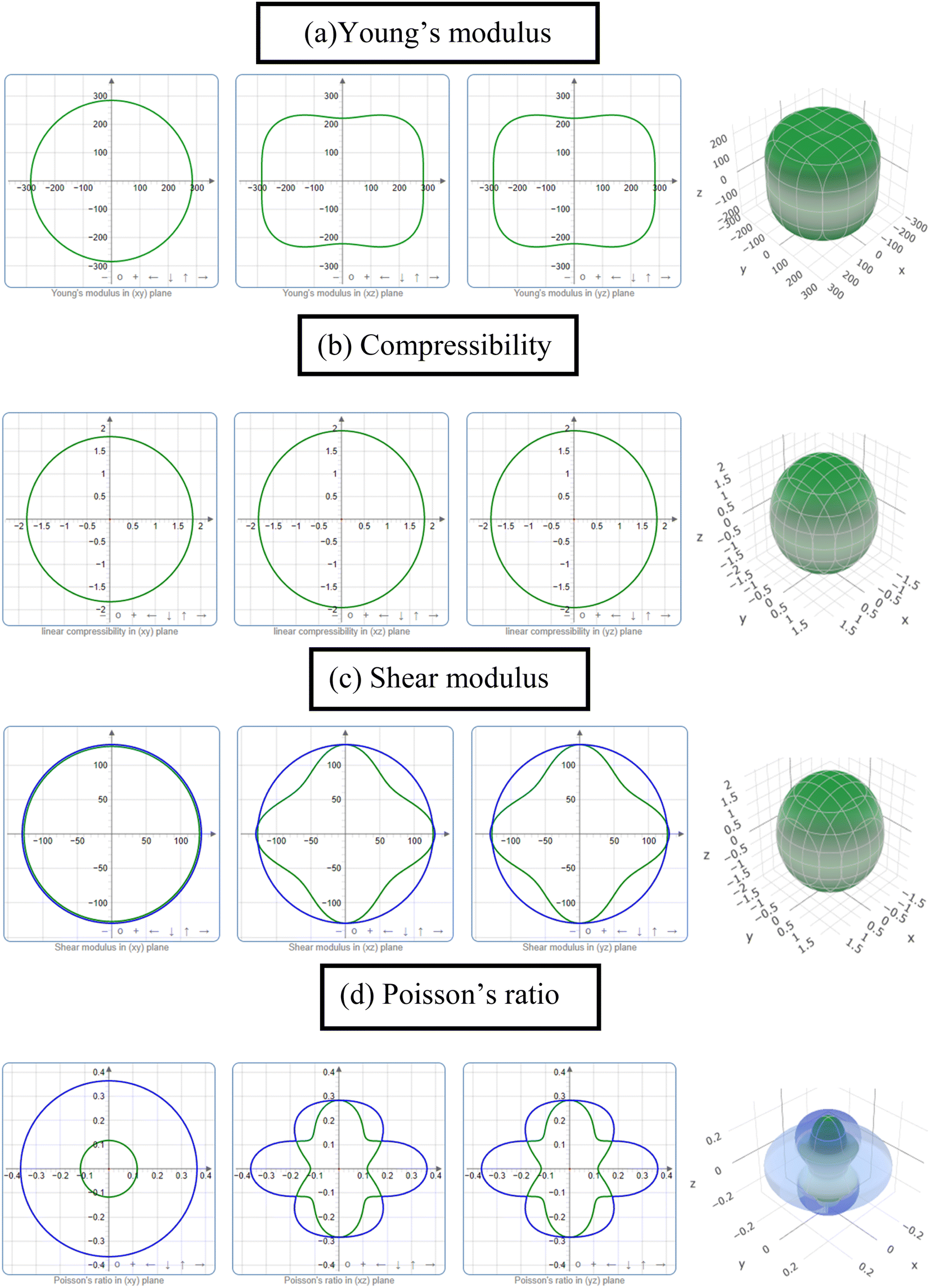 | ||
| Fig. 4 The 2D and 3D plots of (a) Y, (b) K, (c) G and (d) υ of Nb2GaC for GGA PBEsol. | ||
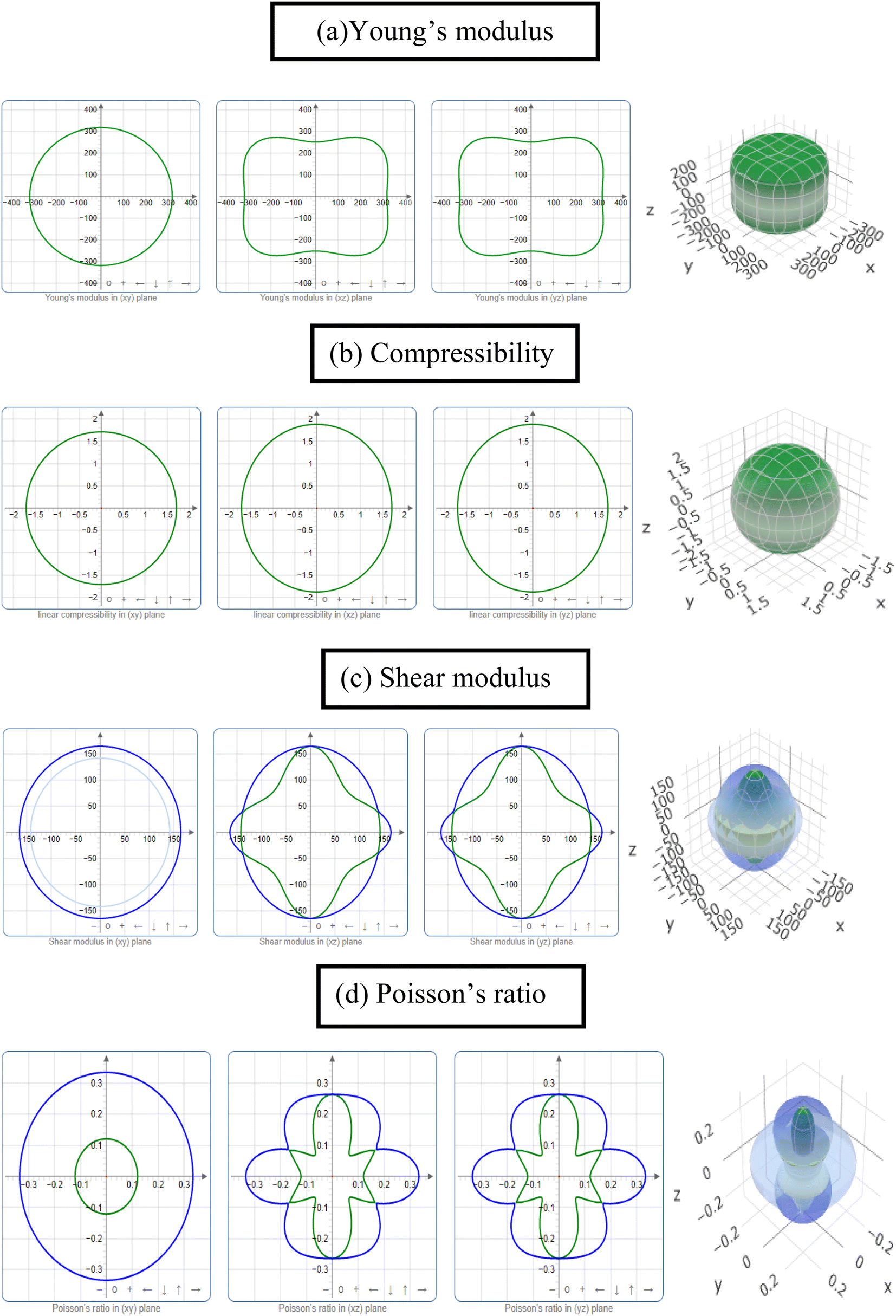 | ||
| Fig. 5 The 2D and 3D plots of (a) Y, (b) K, (c) G and (d) υ of Nb2AlC for GGA PBEsol. | ||
Other important anisotropic indices have also been calculated. Using the following relationships, the three shear anisotropic factors Ai (i = 1, 2, and 3) are computed: 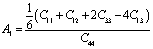 ,
, 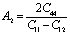 , and A3 = A1.
, and A3 = A1. 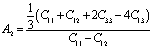 ,79 for the {100}, {011}, and {001}, respectively. Using the following relations, the elastic anisotropy for the bulk modulus (Ba and Bc) across both the a and c-axes is computed:80
,79 for the {100}, {011}, and {001}, respectively. Using the following relations, the elastic anisotropy for the bulk modulus (Ba and Bc) across both the a and c-axes is computed:80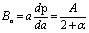 and
and 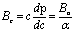 , where A = 2(C11 + C12) + 4C13α + C33α2, where
, where A = 2(C11 + C12) + 4C13α + C33α2, where 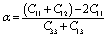 . Additionally, the elastic anisotropy for the ratio of the linear compressibility coefficients (kc/ka) [ka for a and kc for c-directions] is computed using the following relation:81
. Additionally, the elastic anisotropy for the ratio of the linear compressibility coefficients (kc/ka) [ka for a and kc for c-directions] is computed using the following relation:81 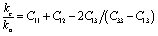 . Table 5 and S2† show the value of the obtained anisotropy factors. The value of Ai = 1 implies isotropy, otherwise anisotropic nature; thus, the Nb2AC (A = Ga, Ge, Tl, Zn, P, In, Cd, and Al) compounds are anisotropic owing to their non-unit (1) value. The equality of kc and ka, and Ba and Bc also implies the isotropic nature. As evident, these parameters also suggest the anisotropic nature of the studied compounds. Furthermore, the percentage anisotropies of compressibility and shear modulus were computed as follows:82
. Table 5 and S2† show the value of the obtained anisotropy factors. The value of Ai = 1 implies isotropy, otherwise anisotropic nature; thus, the Nb2AC (A = Ga, Ge, Tl, Zn, P, In, Cd, and Al) compounds are anisotropic owing to their non-unit (1) value. The equality of kc and ka, and Ba and Bc also implies the isotropic nature. As evident, these parameters also suggest the anisotropic nature of the studied compounds. Furthermore, the percentage anisotropies of compressibility and shear modulus were computed as follows:82 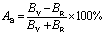 and
and 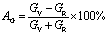 , also certifying the anisotropic nature.
, also certifying the anisotropic nature.
| Phase | A1 | A2 | A3 | Ba | Bc | Kc/ka | AB | AG | Au | Ref. |
|---|---|---|---|---|---|---|---|---|---|---|
| a *Calculated values using reported data. | ||||||||||
| Nb2GaC | 0.658 | 1.041 | 0.685 | 491.61 | 1130.96 | 1.06 | 0.001 | 0.013 | 0.130 | This |
| *0.585 | *1.024 | *0.599 | *505.19 | *1483.25 | 0.93 | *0.001 | *0.022 | *0.224 | 23 | |
| Nb2GeC | 0.429 | 1.787 | 0.767 | 475.71 | 1792.77 | 0.90 | 0.003 | 0.057 | 0.607 | This |
| *0.358 | *2.054 | *0.734 | *448.88 | *1317.91 | 0.86 | *0.004 | *0.085 | *0.932 | 23 | |
| *0.359 | *2.023 | *0.723 | *470.79 | *2125.46 | *0.76 | *0.001 | *0.084 | *0.922 | *25 | |
| Nb2TlC | 0.839 | 0.972 | 0.816 | 420.78 | 1034.08 | 1.09 | 0.002 | 0.003 | 0.031 | This |
| Nb2ZnC | 0.579 | 1.439 | 0.833 | 468.84 | 869.98 | 1.82 | 0.012 | 0.027 | 0.296 | This |
| Nb2PC | 0.515 | 1.618 | 0.833 | 563.72 | 2019.99 | 0.69 | 0.004 | 0.038 | 0.407 | This |
| *0.529 | *1.490 | *0.789 | *520.15 | *2105.85 | 0.61 | *0.007 | *0.033 | *0.358 | *23 | |
| Nb2InC | 0.830 | 0.911 | 0.756 | 448.67 | 951.08 | 1.05 | 0.006 | 0.005 | 0.051 | This |
| *0.705 | *1.037 | *0.731 | *387.65 | *1095.07 | 0.88 | *0.005 | *0.008 | *0.095 | 25 | |
| Nb2CdC | 0.960 | 1.119 | 1.074 | 437.09 | 782.85 | 1.29 | 0.003 | 0.002 | 0.023 | This |
| Nb2AlC | 0.614 | 1.155 | 0.709 | 487.51 | 986.40 | 1.09 | 0.003 | 0.016 | 0.165 | This |
| 0.656 | 1.074 | 0.704 | *438.87 | *1042.45 | 0.95 | 0.006 | 0.012 | 0.129 | 23 | |
Finally, we have calculated the universal anisotropy index AU based on the Voight, V (upper limit), and Reuss, R (lower limit), models using the following relation:83 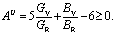 If the value of AU is zero, it implies isotropic behavior, whereas a non-zero value reveals anisotropic behavior; the non-zero values of AU reveal the anisotropic behavior of the studied compounds. In summary, we have found the anisotropic nature of Nb2AC (A = Ga, Ge, Tl, Zn, P, In, Cd, and Al) compounds.
If the value of AU is zero, it implies isotropic behavior, whereas a non-zero value reveals anisotropic behavior; the non-zero values of AU reveal the anisotropic behavior of the studied compounds. In summary, we have found the anisotropic nature of Nb2AC (A = Ga, Ge, Tl, Zn, P, In, Cd, and Al) compounds.
3.5 Optical properties
The MAX phase materials have already been identified as prospective candidates for use as a coating layer to lessen solar heating.84 They have also been used in other sectors, such as optical systems.84 Therefore, it is also hoped that the studied carbides will also be appropriate for the above mentioned ones. We have computed a variety of optical constants in an approach to reveal the optical response of these carbides when electromagnetic radiation is incident upon them.To estimate the optical properties, it is essential to use the equation ε(ω) = ε1(ω)+iε2(ω). Based on the electronic states of each momentum matrix element's occupied and unoccupied states, it is possible to state that ε2(ω) is the imaginary portion of the related dielectric function and fully calculated by CASTEP using the formula below:
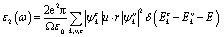
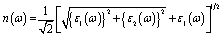
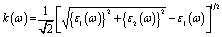
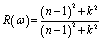
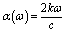
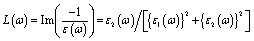
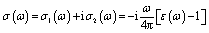
A Drude correction must be made for the study of the dielectric function of metallic materials, which is usually done by adding the plasma frequency and a broadening factor during first-principles calculations.88,89 Because of the metallic nature of the studied carbides, a damping of 0.05 eV and plasma frequency of 3 eV were used to enhance the computed spectra lower energy side. Moreover, a Gaussian smearing value of 0.5 eV was also used to smear out the k-points around the Fermi level. The calculated optical constants of the titled phases are presented in Fig. 6, along with those of Nb2AlC for comparison. The real part ε2(ω) of the dielectric function, where the low energy peaks are attributed to electron intra-band transitions,90 is shown in Fig. 6(a). Because of the electron intra-band transitions, the assigned value for each peak is less than 1 eV. The materials exhibit Drude-like behavior, as indicated by the massive negative values of ε1(ω), whereas inter-band transitions occur at higher energies. Fig. 6(b) shows the imaginary part of the dielectric function ε2(ω). At around 16 eV, it has been seen that the values of ε2(ω) pass through zero from above. This is another example of the compound's metallic nature. A similar nature of the real and imaginary parts of the dielectric function was reported for the most studied MAX phase Ti3SiC2 (ref. 84) and widely used 211 MAX phase Ti2AlC.91 The refractive index, n(ω), of Nb2AC (A = Ga, Ge, Tl, Zn, P, In, Cd, and Al) is depicted in Fig. 6(c). This significant optical constant contributes to the design of optical systems like photonic crystals and wave guides. As shown in Fig. 6(c), the static value of n (0) for Nb2AC (A = Ga, Ge, Tl, Zn, P, In, Cd, and Al) is 7.0, 8.9, 9.4, 11, 8.3, 10.9, 9.5 and 8.9, respectively. Fig. 6(d) shows the extinction coefficient, k(ω), for the Nb2AC (A = Ga, Ge, Tl, Zn, P, In, Cd, and Al) MAX phases. The extinction coefficient, k(ω), is used for measuring the loss of electromagnetic radiation due to absorption and is found to vary similarly to ε2(ω), like other MAX phases.89,90 In Fig. 6(e), the absorption coefficient of Nb2AC (A = Ga, Ge, Tl, Zn, P, In, Cd, and Al) MAX phases is illustrated, where the spectra are shown to increase from zero photon energy due to the metallic behavior of the researched compounds. The spectra are seen to increase as incident energy increases. It showed the strongest absorption region in the spectral range of 7–10 eV; it decreases with a further increase in photon energy. Because of the high absorption coefficients in the high energy range (7–10 eV), Nb2AC (A = Ga, Ge, Tl, Zn, P, In, Cd, and Al) MAX phases can be considered potential absorbing materials in this energy range. The photoconductivity of Nb2AC (A = Ga, Ge, Tl, Zn, P, In, Cd, and Al) is shown in Fig. 6(f), which is also found to start at the beginning from zero photon energy because of the metallic behavior of the selected phases. The band structure and electronic DOS results are well consistent with the absorption coefficient and photoconductivity results.
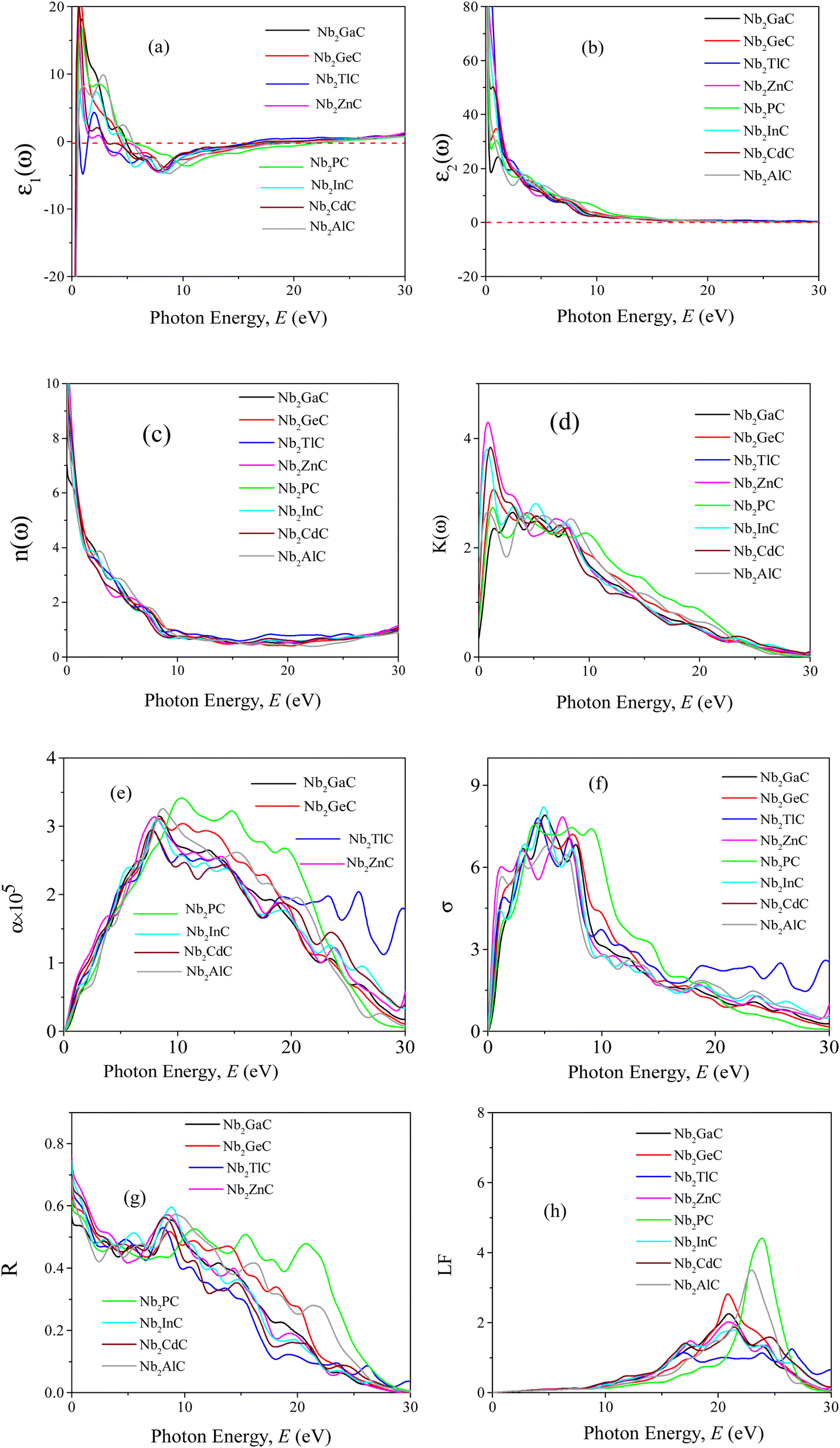 | ||
| Fig. 6 (a) Real part (ε1) and (b) imaginary part (ε2) of dielectric function (ε), (c) refractive index (n), (d) extinction coefficient (k), (e) absorption coefficient (α), (f) photoconductivity (σ), (g) reflectivity (R), and (h) loss function (LF) of Nb2AC (A = Ga, Ge, Tl, Zn, P, In, Cd, and Al) MAX phases as a function of photon energy calculated using GGA PBEsol. | ||
MAX phases are used as coating materials to reduce solar heating, which is one of their most significant applications. The reflectivity of the target materials has been investigated to disclose this possibility, shown in Fig. 6(g). It was reported by Li et al.84,92 that if a MAX compound has a reflectivity of 44% in the visible range, it will be able to reduce solar heating. The reflectivity spectrum for Nb2GaC begins with a value of 0.569 (56.9%), the minimum value among the studied phases. Nb2TlC has the highest value, which is 0.982 (98.2%). For Ti3SiC2, the spectrum has an initial value of ∼0.75 (75%), going down at around 1 eV, and then remaining almost constant up to 6 eV, whereas for Nb2AlC, it starts with an initial value of 0.68 (68%), which is down to below 44% at around 2.1 eV. Though Nb2AC (A = Ga, Ge, and P) has a lower initial value of R than Nb2AlC, neither of their spectra are down to less than 44% up to the visible light range. However, each spectrum of the titled compounds exhibits average higher values (also higher than 44% up to the visible light range) than that of Nb2AlC. Thus, in comparison with Ti3SiC2 and Nb2AlC, it can be concluded that Nb2AC (A = Ga, Ge, Tl, Zn, P, In, and Cd) compounds are candidates for use as cover materials to lessen solar heating. In the energy range of 5.5 to 11 eV, there are a few sharp peaks in the reflectivity spectra, and around 30 eV, reflectivity finally approaches zero.
When electrons move through materials, they lose their energy. An optical constant called the loss function is used to evaluate this type of energy loss. Fig. 6(h) displays the calculated loss functions for the aforementioned MAX phase compounds. The loss function's peak frequency is referred to as the plasma frequency (ωp), which is observed at 20.89, 20.92, 16.38, 17.50, 23.91, 20.54, 17.21 and 23.11 for Nb2GaC, Nb2GeC, Nb2TlC, Nb2ZnC, Nb2PC, Nb2InC, Nb2CdC, and Nb2AlC, respectively. In a loss function, this energy is determined by its characteristic frequency when ε1(ω) and ε2(ω) both pass through zero from below and above, respectively. Reflectivity also identifies the falling edges. This is the critical value that is established by the plasma frequency when the materials are transformed into transparent dielectrics from the metallic system. We have also calculated the optical constant using GGA PBE, but not been shown due to their similar nature.
3.6 Thermal properties
MAX phases are excellent candidates for applications at high-temperature because of their excellent mechanical properties at high temperatures. Therefore, the study of the basic parameters required to predict their application carries significant interest. The Debye temperature (ΘD), minimum thermal conductivity (Kmin), Grüneisen parameter (γ), melting temperature (Tm), etc. of the researched compounds have been calculated for predicting their high temperature applications.The Debye temperature (ΘD), one of the key characteristic parameters of solids, is closely related to the material's bonding strength, melting temperature, thermal expansion, thermal conductivity, etc. The ΘD of studied phases has been calculated using sound velocity following Anderson's method.93 The relevant formulae are as follows:
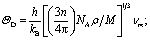
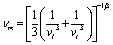 ; vl and vt can be obtained from their relationships with the polycrystalline bulk modulus (B) and shear modulus (G): vl = [(3B + 4G)/3ρ]1/2 and vt = [G/ρ]1/2. The calculated ΘD of Nb2AC (A = Ga, Ge, Tl, Zn, P, In, Cd, and Al) is presented in Table 6 for GGA PBEsol and Table S3† for GGA PBE.
; vl and vt can be obtained from their relationships with the polycrystalline bulk modulus (B) and shear modulus (G): vl = [(3B + 4G)/3ρ]1/2 and vt = [G/ρ]1/2. The calculated ΘD of Nb2AC (A = Ga, Ge, Tl, Zn, P, In, Cd, and Al) is presented in Table 6 for GGA PBEsol and Table S3† for GGA PBE.
| Phases | ρ (g cm−3) | vl (m s−1) | vt (m s−1) | vm (m s−1) | ΘD (K) | Kmin (W mK−1) | Γ | Tm (K) | Ref. |
|---|---|---|---|---|---|---|---|---|---|
| a Calculated values using GGA PBESOL37 and *calculated values using reported data. | |||||||||
| Nb2GaC | 7.59 | 6931 | 4106 | 4547 | 548 | 1.05 | 1.41 | 1913 | This |
| Nb2GeC | 7.76 | 6596 | 3607 | 4022 | 485 | 0.93 | 1.71 | 1715 | This |
| 6632 | 3695 | 4114 | 508 | *0.95 | *1.66 | *1737 | 25 | ||
| Nb2TlC | 10.18 | 5244 | 2872 | 3202 | 372 | 0.68 | 1.71 | 1590 | This |
| Nb2ZnC | 7.43 | 6248 | 3440 | 3834 | 461 | 0.88 | 1.66 | 1574 | This |
| Nb2PC | 8.28 | 7337 | 4297 | 4764 | 640 | 1.29 | 1.45 | 2171 | This |
| Nb2InC | 11.49 | 5311 | 3121 | 3459 | 483 | 0.95 | 1.41 | 1790 | This |
| Nb2CdC | 8.06 | 5855 | 3247 | 3616 | 424 | 0.79 | 1.65 | 1587 | This |
| Nb2AlC | 6.34 | 7315 | 4405 | 4871 | 592 | 1.46 | *1.37 | 1800 | 94 |
As shown in Table 6, ΘD is highest for Nb2PC and lowest for Nb2TlC. The ranking of the compounds roughly followed the hardness-based ranking, which fairly agrees with the hardness and Debye temperature relationship.95 The ΘD of the titled compounds is lower than that of Nb2AlC, except Nb2PC. Recently, Hadi et al.96 reported a MAX (V2SnC) phase as a TBC material with a ΘD value of 472 K. Among the studied compounds, a much lower ΘD is found for only Nb2TlC (372 K) and Nb2CdC (424 K); others have ΘD either comparable to or higher than that of V2SnC. In addition, the ΘD of Y4Al2O9, a well-known TBC material, is 564 K.97 Thus, ΘD values of Nb2AC (A = P, Al, Ga, and Ge) phases [Table 6] are comparable with that of Y4Al2O9.97
The minimum thermal conductivity (Kmin) is defined as the constant value of thermal conductivity at high temperature. As its name suggests, this conductivity is minimum owing to the breaking of the pairing of phonons at high temperature. Calculation of minimum thermal conductivity is essential for predicting the use of solids at high temperature. It has already been established that the MAX phases are suitable for use in high temperature technology as a coating layer (TBC). Thus, calculation of Kmin is also required for the titled phases. We have calculated the Kmin of Nb2AC (A = Ga, Ge, Tl, Zn, P, In, Cd, and Al) compounds using the following equation:98 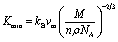 , where kB is the Boltzmann constant, vm is the average phonon velocity, NA is Avogadro's number, and ρ is the crystal's density, respectively, as listed in Table 6. The order of the Kmin value is expected to be as follows: Nb2AlC > Nb2PC > Nb2GaC > Nb2InC > Nb2GeC > Nb2ZnC > Nb2CdC > Nb2TlC; that is, the Kmin values of the studied compounds are lower than that of Nb2AlC, indicating more suitability of the phases as the smaller the Kmin is, the more suitable as TBC materials. It should be noted that the Kmin of V2SnC is 1.20 Wm−1 K−1 and Y4Al2O9 is 1.13 Wm−1 K−1.96,98 Thus, it is expected that the Kmin value suggests the studied compounds as suitable TBC materials.
, where kB is the Boltzmann constant, vm is the average phonon velocity, NA is Avogadro's number, and ρ is the crystal's density, respectively, as listed in Table 6. The order of the Kmin value is expected to be as follows: Nb2AlC > Nb2PC > Nb2GaC > Nb2InC > Nb2GeC > Nb2ZnC > Nb2CdC > Nb2TlC; that is, the Kmin values of the studied compounds are lower than that of Nb2AlC, indicating more suitability of the phases as the smaller the Kmin is, the more suitable as TBC materials. It should be noted that the Kmin of V2SnC is 1.20 Wm−1 K−1 and Y4Al2O9 is 1.13 Wm−1 K−1.96,98 Thus, it is expected that the Kmin value suggests the studied compounds as suitable TBC materials.
An essential thermal parameter that helps to explain the anharmonic effects of lattice dynamics is the Grüneisen parameter (γ); lower anharmonic effects are expected for the solids used at high temperature. Therefore, we have calculated γ of Nb2AC (A = Ga, Ge, Tl, Zn, P, In, Cd, and Al) compounds using the following equation:99 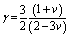 . According to Table 6, the obtained values of γ are lying in between 0.85 and 3.53, which is in line with what is predicted for the polycrystalline materials with ν values in the range of 0.05–0.46.100 Besides, the low values of γ confirm the lower anharmonic effects in the selected compounds, like other MAX phase materials.99
. According to Table 6, the obtained values of γ are lying in between 0.85 and 3.53, which is in line with what is predicted for the polycrystalline materials with ν values in the range of 0.05–0.46.100 Besides, the low values of γ confirm the lower anharmonic effects in the selected compounds, like other MAX phase materials.99
Finally, we have calculated the melting temperature (Tm) of the studied compounds using the following equation: 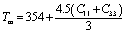 ,101 and listed it in Table 6. The melting temperature of the solids mainly depends on atomic bonding strength; the stronger the atomic bonding, the higher the Tm. Thus, a close relationship between Tm and Y is expected,94,97 and the order of Tm for the titled phases is found to be following the Y-based ranking of the phases. It is seen from Table 6 that Tm of Nb2AC (A = Ge, Tl, Zn, In, and Cd) is lower than that of the Nb2AlC, but still higher than that of the V2SnC (1533 K) MAX phase, a known TBC material.96 In addition, the Tm of Nb2AC (A = Ga and P) is not only higher than that of Nb2AlC94 but also comparable to that of Y4Al2O9 (2000 K). Tm values of Nb2AC (A = Ge, Tl, Zn, In, and Cd) are lower than that of Y4Al2O9 (2000 K) but still reasonably high. Thus, based on the values of ΘD, kmin, and Tm of the studied phases, in comparison with those of Y4Al2O9 and some other MAX phases that have already been reported as TBC materials, we conclude that the titled phases can be considered as potential TBC materials.
,101 and listed it in Table 6. The melting temperature of the solids mainly depends on atomic bonding strength; the stronger the atomic bonding, the higher the Tm. Thus, a close relationship between Tm and Y is expected,94,97 and the order of Tm for the titled phases is found to be following the Y-based ranking of the phases. It is seen from Table 6 that Tm of Nb2AC (A = Ge, Tl, Zn, In, and Cd) is lower than that of the Nb2AlC, but still higher than that of the V2SnC (1533 K) MAX phase, a known TBC material.96 In addition, the Tm of Nb2AC (A = Ga and P) is not only higher than that of Nb2AlC94 but also comparable to that of Y4Al2O9 (2000 K). Tm values of Nb2AC (A = Ge, Tl, Zn, In, and Cd) are lower than that of Y4Al2O9 (2000 K) but still reasonably high. Thus, based on the values of ΘD, kmin, and Tm of the studied phases, in comparison with those of Y4Al2O9 and some other MAX phases that have already been reported as TBC materials, we conclude that the titled phases can be considered as potential TBC materials.
4 Conclusion
A DFT investigation of 211 Nb2AC (A = Ga, Ge, Tl, Zn, P, In, and Cd) carbides has been carried out in this research. The studied phases are dynamically and mechanically stable. Among the studied phases, Nb2PC exhibits the best combination of mechanical properties, while Nb2TlC exhibits the lowest. The Nb2GaC, Nb2PC, and Nb2InC are brittle, whereas Nb2GeC, Nb2TlC, Nb2ZnC, and Nb2CdC are ductile. The Vickers hardness of Nb2PC is also higher than that of others considered here, while the lowest Vickers hardness is found for Nb2TlC, in good agreement with elastic moduli. The calculated direction-dependent (2D and 3D) elastic moduli and anisotropic indices confirm the anisotropic character of the studied phases. The electronic band structure and DOS confirm the metallic nature with a dominating contribution from Nb-3d states. Partial DOS discloses strong hybridization between Nb-d and C-2p states. Mulliken's population analysis reveals the existence of both ionic bonds and covalent bonds within the studied compounds. The optical constants, such as real and imaginary parts of the dielectric function, absorption coefficient, and photoconductivity spectra, are in good accord with band structure results. The reflectivity spectra reveal the possibility of their use as coating materials to diminish solar heating. The obtained values of the Grüneisen parameter (γ) show a lower anharmonic effect within the title carbides. The low value of Kmin and comparatively higher melting temperature with reasonable Debye temperature suggest the studied compounds as TBC materials. The results found in this study are encouraging and hoped to attract attention from the scientific community for further investigation of new MAX phase materials.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors are grateful to the Ministry of Science and Technology (MOST), Bangladesh, for providing the financial support to complete this work (Physical Science-626, 2021–2022 Special Research Grant Project). Prima Das is also grateful to the Chittagong University of Engineering and Technology (CUET) for the financial support.References
- W. Jeitschko, H. Nowotny and F. Benesovsky, Carbides of formula T2MC, J. Less-Common Met., 1964, 7, 133–138 CrossRef CAS.
- H. Wolfsgruber, H. Nowotny and F. Benesovsky, Die kristallstruktur von Ti3GeC2, Monatsh. Chem., 1967, 98, 2403–2405 CrossRef CAS.
- H. Nowotny, Struktur chemieeiniger verbindungen der ubergangsmetalemit den elementen C, Si, Ge, Sn, Prog. Solid State Chem., 1970, 2, 27–70 CrossRef.
- H. Nowotny, J. C. Schuster and P. Rogl, Structural chemistry of complex carbides and related compounds, J. Solid State Chem., 1982, 44, 126–133 CrossRef CAS.
- M. W. Barsoum and T. El-Raghy, Synthesis and characterization of a remarkable ceramic: Ti3SiC2, J. Am. Ceram. Soc., 1996, 79, 1953–1956 CrossRef CAS.
- M. W. Barsoum, The Mn+1AXn phases: a new class of solids: thermodynamically stable nanolaminates, Prog. Solid State Chem., 2000, 28, 201–281 CrossRef CAS.
- M. W. Barsoum, MAX phases: properties of machinable ternary carbides and nitrides, Wiley VCH Verlag GmbH & Co. KGaA, 2013 Search PubMed.
- P. Chakraborty, A. Chakrabarty, A. Dutta and T. Saha-Das gupta, Soft MAX phases with boron substitution: a computational prediction, Phys. Rev. Mater., 2018, 2, 103605 CrossRef.
- M. Sokol, V. Natu, S. Kota and M. W. Barsoum, On the chemical diversity of the MAX phases, Trends Chem., 2019, 1, 210–223 CrossRef CAS.
- M. S. Ali, M. A. Rayhan, M. A. Ali, R. Parvin and A. K. M. A. Islam, New MAX phase compound Mo2TiAlC2: first-principles study, J. Sci. Res., 2016, 8, 109–117 CrossRef CAS.
- M. A. Ali, M. M. Hossain, M. A. Hossain, M. T. Nasir, M. M. Uddin, M. Z. Hasan, A. K. M. A. Islam and S. H. Naqib, Recently synthesized (Zr1-xTix)2AlC (0 ≤ x ≤ 1) solid solutions: theoretical study of the effects of M mixing on physical properties, J. Alloys Compd., 2018, 743, 146–154 CrossRef CAS.
- P. Li, R. Zhou and X. C. Zeng, Computational analysis of stable hard structures in the Ti-B system, ACS Appl. Mater. Interface., 2015, 7, 15607–15617 CrossRef CAS PubMed.
- B. Feng, J. Zhang, Q. Zhong, W. Li, S. Li, H. Li, P. Cheng, S. Meng, L. Chen and K. Wu, Experimental realization of two-dimensional boron sheets, Nat. Chem., 2016, 8, 563–568 CrossRef CAS PubMed.
- M. S. Hossain, N. Jahan, M. M. Hossain, M. M. Uddin and M. A. Ali, High pressure mediated physical properties of Hf2AB (A = Pb, Bi) via DFT calculations, Mater. Today Commun., 2023, 34, 105147 CrossRef CAS.
- A. Jain, G. Hautier, C. J. Moore, S. P. Ong, C. C. Fischer, T. Mueller, K. A. Persson and G. Ceder, A high-throughput infrastructure for density functional theory calculations, Comput. Mater. Sci., 2011, 50, 2295–2310 CrossRef CAS.
- S. Curtarolo, G. Hart, M. B. Nardelli, N. Mingo, S. Sanvito and O. Levy, The high-throughput highway to computational materials design, Nat. Mater., 2013, 12, 191–201 CrossRef CAS PubMed.
- M. F. Cover, O. Warschkow, M. M. M. Bilek and D. R. McKenzie, A comprehensive survey of M2AX phase elastic properties, J. Phys.: Condens. Matter, 2009, 21, 305403 CrossRef CAS PubMed.
- V. J. Keast, S. Harris and D. K. Smith, Prediction of the stability of the Mn+1AXn phases from first principles, Phys. Rev. B: Condens. Matter Mater. Phys., 2009, 80, 214113 CrossRef.
- S. Aryal, R. Sakidja, M. W. Barsoum and W. Y. Ching, A genomic approach to the stability, elastic, and electronic properties of the MAX phases, Phys. Status Solidi B, 2014, 251, 1480–1497 CrossRef CAS.
- M. Ashton, R. G. Hennig, S. R. Broderick, K. Rajan and S. B. Sinnott, Computational discovery of stable M2AX phases, Phys. Rev. B, 2016, 94, 054116 CrossRef.
- R. Khaledialidusti, M. Khazaei, S. Khazaei and K. Ohno, High-throughput computational discovery of ternary-layered MAX phases and prediction of their exfoliation for formation of 2D MXenes, Nanoscale, 2021, 13, 7294–7307 RSC.
- M. A. Hadi, Superconducting phases in a remarkable class of metallic ceramics, J. Phys. Chem. Solids, 2020, 138, 109275 CrossRef CAS.
- M. A. Hadi, S. R. G. Christopoulos, A. Chroneos, S. H. Naqib and A. K. M. A. Islam, Elastic behaviour and radiation tolerance in Nb-based 211 MAX phases, Mater. Today Commun., 2020, 25, 101499 CrossRef CAS.
- A. Bouhemadou, Calculated structural and elastic properties of M2InC (M = Sc, Ti, V, Zr, Nb, Hf, Ta), Mod. Phys. Lett. B, 2008, 22, 2063–2076 CrossRef CAS.
- A. Bouhemadou, Calculated structural, electronic and elastic properties of M2GeC (M = Ti, V, Cr, Zr, Nb, Mo, Hf, Ta and W), Appl. Phys. A, 2009, 96, 959–967 CrossRef CAS.
- W. Jeitschko, H. Nowotny and F. Benesovsky, Die H-Phasen Ti2TiC, Ti2PbC, Nb2InC, Nb2SnC and Ta2GaC, Monatsh. fur Chem., 1964, 95, 431–435 CrossRef CAS.
- I. Salama, T. El-Raghy and M. W. Barsoum, Synthesis and mechanical properties of Nb2AlC and (Ti, Nb)2AlC, J. Alloys Compd., 2002, 347, 271–278 CrossRef.
- W. Zhang, N. Travitzky, C. F. Hu, Y. C. Zhou and P. Greil, Reactive hot pressing and properties of Nb2AlC, J. Am. Ceram. Soc., 2009, 92, 2396–2399 CrossRef CAS.
- M. Khazaei, A. Ranjbar, K. Esfarjani, D. Bogdanovski, R. Dronskowski and S. Yunoki, Insights into exfoliation possibility of MAX phases to MXenes, Phys. Chem. Chem. Phys., 2018, 20, 8579–8592 RSC.
- S. Zhao, Y. Dall'Agnese, X. Chu, X. Zhao, Y. Gogotsi and Y. Gao, Electrochemical interaction of Sn-containing MAX phase (Nb2SnC) with Li-ions, ACS Energy Lett., 2019, 4, 2452–2457 CrossRef CAS.
- H. Dinga, Y. Li, J. Lu, K. Luo, K. Chen, M. Li, P. O. A. Persson, L. Hultman, P. Eklund, S. Du, Z. Huang, Z. Chai, H. Wang, P. Huang and Q. Huang, Synthesis of MAX phases Nb2CuC and Ti2(Al0.1Cu0.9) N by A-site replacement reaction in molten salts, Mater. Res. Lett., 2019, 7, 510–516 CrossRef.
- I. R. Shein and A. L. Ivanovskii, Structural, elastic, electronic properties and Fermi surface for superconducting Mo2GaC in comparison with V2GaC and Nb2GaC from first principles, Physica C, 2010, 470, 533–537 CrossRef CAS.
- M. A. Ghebouli, B. Ghebouli, A. Bouhemadou and M. Fatmi, Theoretical study of the structural, elastic, electronic and thermal properties of the MAX phase Nb2SiC, Solid State Commun., 2011, 151, 382–387 CrossRef CAS.
- M. D. Segall, P. J. D. Lindan, M. J. Probert, C. J. Pickard, P. J. Hasnip, S. J. Clark and M. C. Payne, First principles simulation: ideas, illustrations and the CASTEP code, J. Phys.: Condens. Matter, 2002, 14, 2717–2744 CrossRef CAS.
- S. J. Clark, M. D. Segall, C. J. Pickard, P. J. Hasnip, M. I. J. Probert, K. Refson and M. C. Payne, First principles methods using CASTEP, ZeitschriftFürKrist. – Cryst. Mater., 2005, 220, 567–570 CrossRef CAS.
- J. P. Perdew, K. Burke and M. Ernzerhof, Generalized gradient approximation made simple, Phys. Rev. Lett., 1996, 77, 3865 CrossRef CAS PubMed.
- J. P. Perdew, A. Ruzsinszky, G. I. Csonka, O. A. Vydrov, G. E. Scuseria, L. A. Constantin, X. Zhou and K. Burke, Restoring the density-gradient expansion for exchange in solids and surfaces, Phys. Rev. Lett., 2008, 100, 136406 CrossRef PubMed.
- O. Beckstein, J. E. Klepeis, G. L. W. Hart and O. Pankratov, First-principles elastic constants and electronic structure of α-Pt2Si and PtSi, Phys. Rev. B: Condens. Matter Mater. Phys., 2001, 63, 134112 CrossRef.
- J. Islam, S. K. Mitro, M. M. Hossain, M. M. Uddin, N. Jahan, A. K. M. A. Islam, S. H. Naqib and M. A. Ali, Exploration of the physical properties of the newly synthesized kagome superconductor LaIr3Ga2 using different exchange–correlation functionals, Phys. Chem. Chem. Phys., 2022, 24, 29640 RSC.
- S. Masys and V. Jonausaks, A first-principles study of structural and elastic properties of bulk SrRuO3, J. Chem. Phys., 2013, 139, 224705 CrossRef CAS PubMed.
- T. H. Fischer and J. Almlof, General methods for geometry and wave function optimizatin, J. Phys. Chem., 1992, 96, 9768 CrossRef CAS.
- H. J. Monkhorst and J. D. Pack, Special points for Brillouin-zone integrations, Phys. Rev. B: Condens. Matter Mater. Phys., 1976, 13, 5188 CrossRef.
- E. I. Isaev, QHA project, https://qe-forge.org/qha, accessed May 25, 2013 Search PubMed.
- G. Hug, M. Jaouen and M. W. Barsoum, X-ray absorption spectroscopy, EELS, and full-potential augmented plane wave study of the electronic structure of Ti2Al3, Ti2AlN, Nb2AlC and (Ti0.5Nb0.5)2AlC, Phys. Rev. B: Condens. Matter, 2005, 71, 024105 CrossRef.
- G. Hug, Electronic structures of and composition gaps among the ternary carbides Ti2MC, Phys. Rev. B: Condens. Matter, 2006, 74, 184113 CrossRef.
- M. B. Kanoun, S. Gourmi-said and M. Jaouen, Steric effect on the M site of nanolaminate compounds M2SnC (M = Ti, Zr, Hf and Nb), Phys. Condens. Matter, 2009, 21, 045404 CrossRef CAS PubMed.
- A. D. Bortolozo, Z. Fisk, O. H. Sant' Anna, C. A. M. dos Santos and A. J. S. Machado, Superconductivity in Nb2InC, Physica C, 2009, 469, 57 CrossRef.
- M. A. Ali and A. K. M. A. Islam, Sn1-xBixO2 and Sn1-xTaxO2 (0≤x≤0.75): A first-principles study, Phys. Rev. B: Condens. Matter Mater. Phys., 2012, 407, 1020–1026 CrossRef CAS.
- M. Born, On the stability of crystal lattices. I, Math. Proc. Cambridge Philos. Soc., 1940, 36, 160–172 CrossRef CAS.
- Z. Sun, D. Music, R. Ahuja and J. M. Schneider, Theoretical investigation of the bonding and elastic properties of nanolayered ternary nitrides, Phys. Rev. B: Condens. Matter Mater. Phys., 2005, 71, 193402 CrossRef.
- S. H. jhi, J. Ihm, S. G. Louie and M. L. Cohen, Electronic mechanism of hardness enhancement in transition-metal carbonitrides, Nature, 1999, 399, 132–134 CrossRef CAS.
- D. G. Pettifor, Theoretical predictions of structure and related properties of intermetallics, J. Mater. Sci. Tech., 1992, 8, 345–349 CrossRef CAS.
- M. W. Qureshi, M. A. Ali and X. Ma, Screen the thermomechanical and optical properties of the new ductile 314 MAX phase boride Zr3CdB4: A DFT insight, J. Alloys Compd., 2021, 877, 160248 CrossRef CAS.
- M. Roknuzzaman, M. A. Hadi, M. J. Abden, M. T. Nasir, A. K. M. A. Islam, M. S. Ali, K. Ostrikov and S. H. Naqib, Physical properties of predicted Ti2CdN versus existing Ti2CdC MAX phase: An ab initio study, Comput. Mater. Sci., 2016, 113, 148–153 CrossRef CAS.
- M. W. Qureshi, M. A. Ali, X. Ma, G. Tang, M. U. Javed and D. Paudyal, Verification of stability and unraveling the electronic and physical properties of bulk and (001)-surfaces of newly synthesized Ti2ZnX (X = C, N) MAX phases, Surf. Interfaces, 2022, 31, 102032 CrossRef CAS.
- R. Hill, The elastic behaviour of a crystalline aggregate, Proc. Phys. Soc., London, Sect. A, 1952, 65, 349–354 CrossRef.
- W. Voigt, Lehrbuch der Kristallphysik, Teubner, Leipzig, 1928 Search PubMed.
- A. Reuss, Berechnung der fließgrenze von mischkristallen auf grund der plastizitätsbedingungfüreinkristalle, J. Appl. Math. Mech., 1929, 9, 49–58 CAS.
- M. A. Ali, M. Roknuzzaman, M. T. Nasir, A. K. M. A. Islam and S. H. Naqib, Structural, elastic, electronic and optical properties of Cu3MTe4 (M = Nb, Ta) sulvanites —An ab initio study, Int. J. Mod. Phys. B, 2016, 30, 1650089 CrossRef CAS.
- M. A. Ali, M. A. Hossain, M. A. Rayhan, M. M. Hossain, M. M. Uddin, M. Roknuzzaman, K. Ostrikov, A. K. M. A. Islam and S. H. Naqib, First-principles study of elastic, electronic, optical and thermoelectric properties of newly synthesized K2Cu2GeS4chalcogenide, J. Alloys Compd., 2018, 781, 37–46 CrossRef.
- X. Wang, H. Xiang, X. Sun, J. Liu, F. Hou and Y. Zhou, Mechanical properties and damage tolerance of bulk Yb3Al5O12 ceramic, J. Mater. Sci. Technol., 2015, 31, 369–374 CrossRef CAS.
- M. A. Ali, A. K. M. A. Islam, N. Jahan and S. Karimunnesa, First-principles study of SnO under high pressure, Int. J. Mod. Phys. B, 2016, 30, 1650228 CrossRef CAS.
- X. Q. Chen, H. Niu, D. Li and Y. Li, Modeling hardness of polycrystalline materials and bulk metallic glasses, Intermetallics, 2011, 19, 1275–1281 CrossRef CAS.
- N. Miao, B. Sa, J. Zhou and Z. Sun, Theoretical investigation on the transition-metal borides with Ta3B4-type structure: A class of hard and refractory materials, Comput. Mater. Sci., 2011, 50, 1559–1566 CrossRef CAS.
- M. Radovic and M. W. Barsoum, MAX phases: Bridging the gap between metals and ceramics, Am. Ceram. Soc. Bull., 2013, 92, 20–27 CAS.
- M. Mebrek, A. Mokaddem, B. Doumi, A. Yakoubi and A. Mir, A novel theoretical study of elastic and electronic properties of M2CdC (M = Zr, Hf, and Ta) MAX phases, Acta Phys. Pol. A, 2018, 133, 76–81 CrossRef CAS.
- S. F. Pugh, Relations between the elastic moduli and the plastic properties of polycrystalline pure metals, Philos. Mag. J. Sci., 1954, 45, 823–843 CrossRef CAS.
- M. Roknuzzaman, M. A. Hadi, M. A. Ali, M. M. Hossain, N. Jahan, M. M. Uddin, J. A. Alarco and K. Ostrikov, First hafnium-based MAX phase in the 312 family, Hf3AlC2: A first-principles study, J. Alloys Compd., 2017, 727, 616–626 CrossRef CAS.
- I. R. Shein and A. L. Ivanovskii, Graphene-like titanium carbides and nitrides Tin+1Cn, Tin+1Nn (n = 1, 2, and 3) from de-intercalated MAX phases: First-principles probing of their structural, electronic properties and relative stability, Comput. Mater. Sci., 2012, 65, 104 CrossRef CAS.
- I. R. Shein and A. L. Ivanovskii, Elastic properties of superconducting MAX phases from first-principles calculations, Phys. Status Solidi B, 2011, 248, 228–232 CrossRef CAS.
- H. Gou, L. Hou, J. Zhang and F. Gao, Pressure-induced incompressibility of ReC and effect of metallic bonding on its hardness, Appl. Phys. Lett., 2008, 92, 2419 CrossRef.
- P. Barua, M. M. Hossain, M. A. Ali, M. M. Uddin, S. H. Naqib and A. K. M. A. Islam, Effects of transition metals on physical properties of M2BC (M = V, Nb, Mo and Ta): a DFT calculation, J. Alloy. Compd., 2018, 770, 523–534 CrossRef.
- Y. Zhou and Z. Sun, Electronic structure and bonding properties of layered machinable and ceramics, Phys. Rev. B: Condens. Matter Mater. Phys., 2000, 61, 12570–12573 CrossRef CAS.
- M. A. Ali and S. H. Naqib, Recently synthesized (Ti1-xMox)2AlC (0 ≤ x ≤ 0.20) solid solutions: deciphering the structural, electronic, mechanical and thermodynamic properties via ab initio simulations, RSC Adv., 2020, 10, 31535–31546 RSC.
- A. Chowdhury, M. A. Ali, M. M. Hossain, S. H. Naqib and A. K. M. A. Islam, Predicted MAX phase Sc2InC: dynamical stability, vibrational and optical properties, Phys. Status Solidi B, 2018, 255, 1700235 CrossRef.
- H. M. Ledbetter and A. Migliori, A general elastic-anisotropy measure, J. Appl. Phys., 2006, 100, 063516 CrossRef.
- J. Chang, G. P. Zhao, X. L. Zhou, K. Liu and L. Y. Lu, Structure and mechanical properties of tantalum mononitride under high pressure: a first-principles study, J. Appl. Phys., 2012, 112, 083519 CrossRef PubMed.
- R. Gaillac, P. Pullumbi and F. X. Coudert, ELATE: an open-source online application for analysis and visualization of elastic tensors, J. Phys.: Condens. Matter, 2016, 28, 275201 CrossRef PubMed.
- H. M. Ledbetter, Elastic properties of zinc: a compilation and a review, J. Phys. Chem. Ref. Data, 1977, 6, 1181–1203 CrossRef CAS.
- A. K. M. A. Islam, A. S. Sikder and F. N. Islam, NbB2: a density functional study, Phys. Lett. A, 2006, 350, 288–292 CrossRef CAS.
- J. Wang, Y. Zhou, T. Liao and Z. Lin, First-principles prediction of low shear-strain resistance of Al3BC3: a metal borocarbide containing short linear BC2 units, Appl. Phys. Lett., 2006, 89, 021917 CrossRef.
- Anisotropy in single crystal refractory compound, ed. D. H. Chung, W. R. Buessem, F. W. Vahldiek and S. A. Mersol, Plenum Press, New York, 1968, vol. 2 Search PubMed.
- S. I. Ranganathan and M. Ostoja-Starzewski, Universal elastic anisotropy index, Phys. Rev. Lett., 2008, 101, 055504 CrossRef PubMed.
- S. Li, R. Ahuja, M. W. Barsoum, P. Jena and B. Johansson, Optical properties of Ti3SiC2 and Ti4AlN3, Appl. Phys. Lett., 2008, 92, 221907 CrossRef.
- M. F. Li, Physics of Semiconductor, Science Press, Beijing, 1991 Search PubMed.
- R. C. Fang, Solid Spectroscopy, Chinese Science Technology University Press, Hefei, 2003 Search PubMed.
- Y. Zhang and W. M. Shen, Basic of Solid Electronics, Zhe Jiang University Press, Hangzhou, 2005 Search PubMed.
- M. T. Nasir, M. A. Hadi, M. A. Rayhan, M. A. Ali, M. M. Hossain and M. Roknuzzaman, et al., First-principles study of superconducting ScRhP and ScIrP pnictides, Phys. Status Solidi B, 2017, 254, 1700336 CrossRef.
- F. Sultana, M. M. Uddin, M. A. Ali, M. M. Hossain, S. H. Naqib and A. K. M. A. Islam, First principles study of M2InC (M = Zr, Hf and Ta) MAX phases: the effect of M atomic species, Results Phys, 2018, 11, 869–876 CrossRef.
- K. Akter, F. Parvin, M. A. Hadi and A. K. M. A. Islam, Insights into the predicted Hf2SN in comparison with the synthesized MAX phase Hf2SC: a comprehensive study, Comput. Condens. Matter., 2020, 24, e00485 CrossRef.
- N. Haddad, E. G. Caurel, L. Hultman, M. W. Barsoum and G. Hug, Dielectric Properties of Ti2AlC and Ti2AlN MAX Phases: The Conductivity Anisotropy, J. Appl. Phys., 2008, 104, 023531 CrossRef.
- M. A. Ali and M. W. Qureshi, Newly synthesized MAX phase Zr2SeC: DFT insights into physical properties towards possible applications, RSC Adv., 2021, 11, 16892 RSC.
- O. L. Anderson, A simplified method for calculating in the Debye temperature from elastic constants, J. Phys. Chem. Solids, 1963, 24, 909–917 CrossRef CAS.
- M. A. Hadi, N. Kelaidis, S. H. Naqib, A. K. M. A. Islam, A. Chroneos and R. V. Vovk, Insights into the physical properties of a new 211 MAX phase Nb2CuC, J. Phys. Chem. Solids, 2020, 149, 109759 CrossRef.
- M. A. Ali, M. M. Hossain, M. M. Uddin, M. A. Hossain, A. K. M. A. Islam and S. H. Naqib, Physical properties of new MAX phase borides M2SB (M = Zr, Hf and Nb) in comparison with conventional MAX phase carbides M2SC (M = Zr, Hf and Nb): Comprehensive insights, J. Mater. Res. Tech., 2021, 11, 1000–1018 CrossRef CAS.
- M. A. Hadi, M. Dahlqvist, S. R. G. Christopoulos, S. H. Naqib, A. Chroneos and A. K. M. A. Islam, Chemically stable new MAX phase V2SnC: a damage and radiation tolerant TBC material, RSC Adv., 2020, 10, 43783–43798 RSC.
- Y. Zhou, X. Lu, H. Xiang, Z. Feng and Z. Li, Theoretical prediction on mechanical and thermal properties of a promising thermal barrier material: Y4Al2O9, J. Adv. Ceram., 2015, 4, 83–93 CrossRef CAS.
- G. A. Slack, The thermal conductivity of nonmetallic crystals, Solid State Phys., 1979, 34, 1–71 CAS.
- V. N. Belomestnykh and E. P. Tesleva, Interrelation between anharmonicity and lateral strain in quasi-isotropic polycrystalline solids, Tech. Phys., 2004, 49, 1098–1100 CrossRef CAS.
- S. I. Mikitishin, Interrelationship of Poisson's ratio with other characteristics of pure metals, Sov. Mater. Sci., 1982, 18, 262–265 CrossRef.
- M. E. Fine, L. D. Brown and H. L. Marcus, Elastic constants versus melting temperature in metals, Scr. Metall., 1984, 18, 951–956 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d2ra07468k |
| This journal is © The Royal Society of Chemistry 2023 |