
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceContemporary progress in the green synthesis of spiro-thiazolidines and their medicinal significance: a review
Surbhi Dhadda
 a,
Shaily Sharma
b,
Prakash Jakhar
b and
Himanshu Sharma
*b
a,
Shaily Sharma
b,
Prakash Jakhar
b and
Himanshu Sharma
*b
aDepartment of Chemistry, Faculty of Basic and Applied Sciences, Vivekananda Global University, Jagatpura, Jaipur, Rajasthan 303012, India
bMicrowave Chemistry Lab, Department of Chemistry, UCOS, Mohanlal Sukhadia University, Udaipur, Rajasthan 313001, India. E-mail: himanshu@mlsu.ac.in
First published on 25th January 2023
Abstract
The development of new strategies for the production of nitrogen and sulfur-containing heterocycles remains an extremely alluring but challenging proposition. Among these heterocyclic compounds, spiro-thiazolidines are a distinct class of heterocyclic motifs with an all-encompassing range of pharmaceutical activities such as anti-histaminic, anti-proliferative, anesthetic, hypnotic, anti-fungal, anti-inflammatory, anti-HIV, anthelmintic, CNS stimulant, and anti-viral potentials. Consequently, investigators have produced these heterocycles through diversified intricate pathways as object structures for medicinal studies. Notwithstanding their innumerable manmade solicitations, there is yet no special periodical on MCRs concerning spiro-thiazolidine via green synthesis. Thus, this in-depth review encompasses the excursion of MCRs to spiro-thiazolidines, including the environment-friendly synthetic approaches, reaction situations, rationale behind the optimal selection of catalyst, scope, anticipated mechanism, and biological activities. In this review, we have focussed on the furthermost current developments in spiro-thiazolidine creation under different conditions, such as ionic liquid-assisted, microwave-assisted, on-water, solid-supported acid-catalyzed, asymmetric, and nanocatalyst-assisted syntheses, developed over the last 8 years. This study details works regarding the total amalgamation of spiro-thiazolidines under N- and S-containing heterocycles. Furthermore, this article summarizes the developments of artificially and pharmaceutically important spiro-thiazolidine candidates.
 Surbhi Dhadda | Dr Surbhi Dhadda obtained her PhD in Synthetic Organic Chemistry from Department of Chemistry, University of Rajasthan, Jaipur, India in 2021. She started her career in 2011 from Vedic Kanya P.G. College, Rajapark, Jaipur as Assistant Professor in Department of Chemistry. She is currently working in Faculty of Basic and Applied Sciences, Vivekananda Global University, Jagatpura, Jaipur. She has published many research articles in various national and international reputed journals in the field of synthetic organic chemistry, green chemistry, and medicinal chemistry. Her current research interests include the design, synthesis and pharmacological evaluation of synthesized heterocyclic compounds, and nanotechnology. |
 Shaily Sharma | Shaily Sharma has done her post-graduation from JECRC University, Jaipur in 2020 and received gold medal in M.Sc. Chemistry in 2020. Presently, she is pursuing her PhD under the supervision of Dr Himanshu Sharma. Her research interests are photochemistry and nanotechnology. |
 Prakash Jakhar | Prakash Jakhar received his M.Sc. from JNV University, Jodhpur in 2014. He is NET qualified and recipient of CSIR- UGC JRF fellowship. Presently, he is pursuing his PhD under the supervision of Dr Himanshu Sharma. His research interests are photochemistry and polymer chemistry. |
 Himanshu Sharma | Dr Himanshu Sharma is presently working as Assistant Professor in Department of Chemistry, Mohanlal Sukhadia University, Udaipur, Rajasthan, India. She is NET and GATE qualified and is a recipient of Junior and Senior research fellowship from CSIR, New Delhi. She was awarded PhD degree from University of Rajasthan, Jaipur. She has been co-principal investigator of one research project funded by RUSA-India. She has published many research articles in reputed national and international journals of high visibility including conference proceedings and also contributed several books chapters in esteemed chemistry books. Her research interests are in synthetic organic chemistry, green chemistry, photochemistry, nanotechnology, and polymer chemistry. |
Introduction
N- and S-containing heterocyclic motifs are universal structural units that extensively exist in naturally occurring molecules and medicinal agents.1,2 Thiazolidines embrace an advantageous position in both organic synthesis and medicinal chemistry. Thiazolidine derivatives, which are found as fundamental structural components in a variety of medically active compounds, have been used to represent a major subdivision of heterocycles. Thiazolidinone are thiazolidine derivatives that include carbonyl groups at positions 2, 4, or 5. With a carbonyl group on the fourth carbon, thiazolidinone is a saturated version of thiazole and is known as the "mystic moiety" since it contains practically all known biological functions. A cyclic structure was reported based on the hydrolysis of 3-phenyl-2-phenyl-amino-4-thiazolidinone with mercaptoacetic acid being the main byproduct. The structures of thiazolidinone are given in Fig. 1.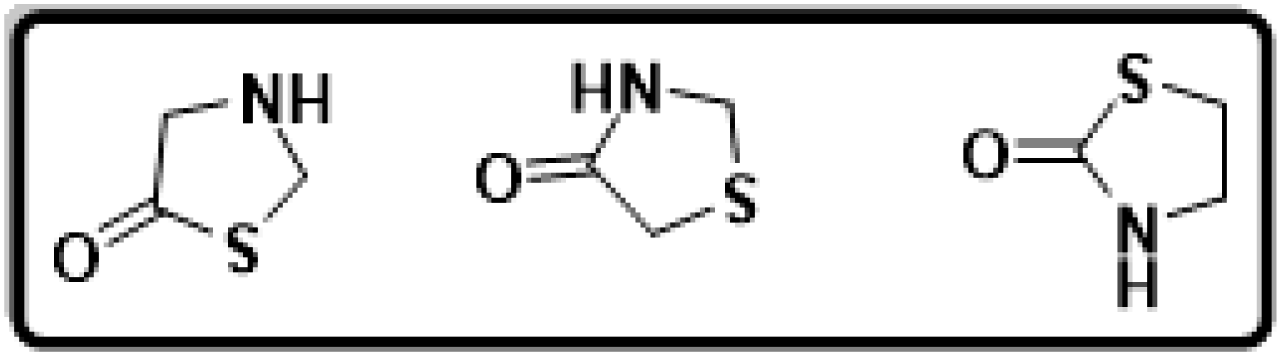 | ||
| Fig. 1 The cyclic structures of various thiazolidinones. | ||
Although substitutions can occur on the 2, 3, and 5 positions of the thiazolidinone ring, the second position carbon atom of the ring has the maximum impact on the structure and characteristics of thiazolidinones. The structure–activity connection of thiazolidinone, as an important family of N- and S-containing heterocycles, makes it a particularly alluring target for combinatorial library synthesis. Thiazolidinone is widely employed as the main building block in the areas of pharmaceuticals and pharmacological agents.3 Thiazolidinone scaffold has been linked to a variety of biological processes, including those that are anti-bacterial, anti-fungal, anti-tubercular, anti-convulsant, anti-thyroid, and anti-histaminic.4,5
For more than 40 years, the synthesis of thiazolidines has been made possible by the reaction of 3-mercaptoalkylamines with aldehydes and ketones.6 Numerous investigations have been published to create innovative lead or hit compounds that include thiazolidines.7–13 Thiazolidine-2,4-dione heterocyclic ring system has several uses, such as in an acidic solution, where it prevents mild steel from corroding. These can also be utilized as very sensitive reagents for heavy metals in analytical chemistry14 and as a brightener in the electroplating industry.15
Spiro compounds generally have cyclic structures bearing one mutual sp3 hybridized carbon atom surrounded by two rings and fascinating synthetic encounters because of their significant structural inflexibility and complexity.16,17 Spiro heterocyclic compounds encompass nitrogen, sulfur, and oxygen atoms, and have unique features of several natural and artificial products with notable roles in biological processes and have exhibited significant pharmacological activities.18 Additionally, they have various usages as photochromic resources19 and asymmetric catalysts.20 Diverse synthetic methodologies concerning spiro compounds have been offered, in which the quaternary core was constructed by rearrangements, ring expansions/contractions, alkylations, transition metal-promoted reactions, cycloadditions, etc.21–23
Spiro-thiazolidines are versatile frameworks and a mesmerizing class of heterocyclic compounds that have advanced ominously in recent years owing to their influential nature of establishment in the grounds of material science, catalysis, drug discovery, pharmacological, and combinatorial chemistry. Because of their extensive significance, spirocyclic derivatives have been synthesized by both chemists and biologists in recent decades. Simultaneously, many heterocycles that comprise a thiazolidine and its analogues are also connected with various pharmaceutical effects, like anti-HIV, antiviral, antifungal, antibacterial, diuretic, antihistaminic, anticonvulsant, anti-inflammatory, tuberculostatic, anticancer, and analgesic actions.24–27 Here, some bioactive spiro-thiazolidine scaffolds are shown in Fig. 2.28,29
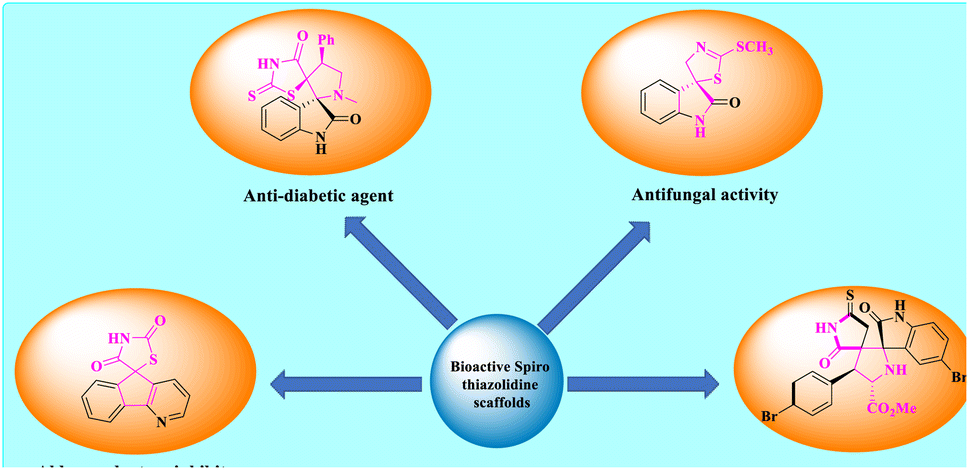 | ||
| Fig. 2 Bioactive spiro-thiazolidine scaffolds. | ||
Regardless of their innumerable synthetic solicitations, there is yet no special review on the MCRs regarding the green amalgamation of spiro-thiazolidine-based derivatives. This critical review purposes the greatest current developments in spiro-thiazolidine production under various conditions like ionic liquid-assisted synthesis, microwave-assisted synthesis, solid-supported acid-catalyzed synthesis, on water synthesis, nanocatalyst-assisted synthesis, asymmetric synthesis, miscellaneous, and so forth developed over the last 8 years. Hence, this review insurances recent advances in the passage of MCRs to spiro-thiazolidines containing the artificial tactics, possibilities, reaction situations, rationale behind the catalyst selection, and the anticipated mechanism. Moreover, this article will undeniably be underwritten in the scientific domain for emerging artificially and medicinally imperative spiro-thiazolidine analogs. We have also termed a reasonable study of numerous synthetic strategies of spiro-thiazolidines with gains (+) and drawbacks (−) (Fig. 3).
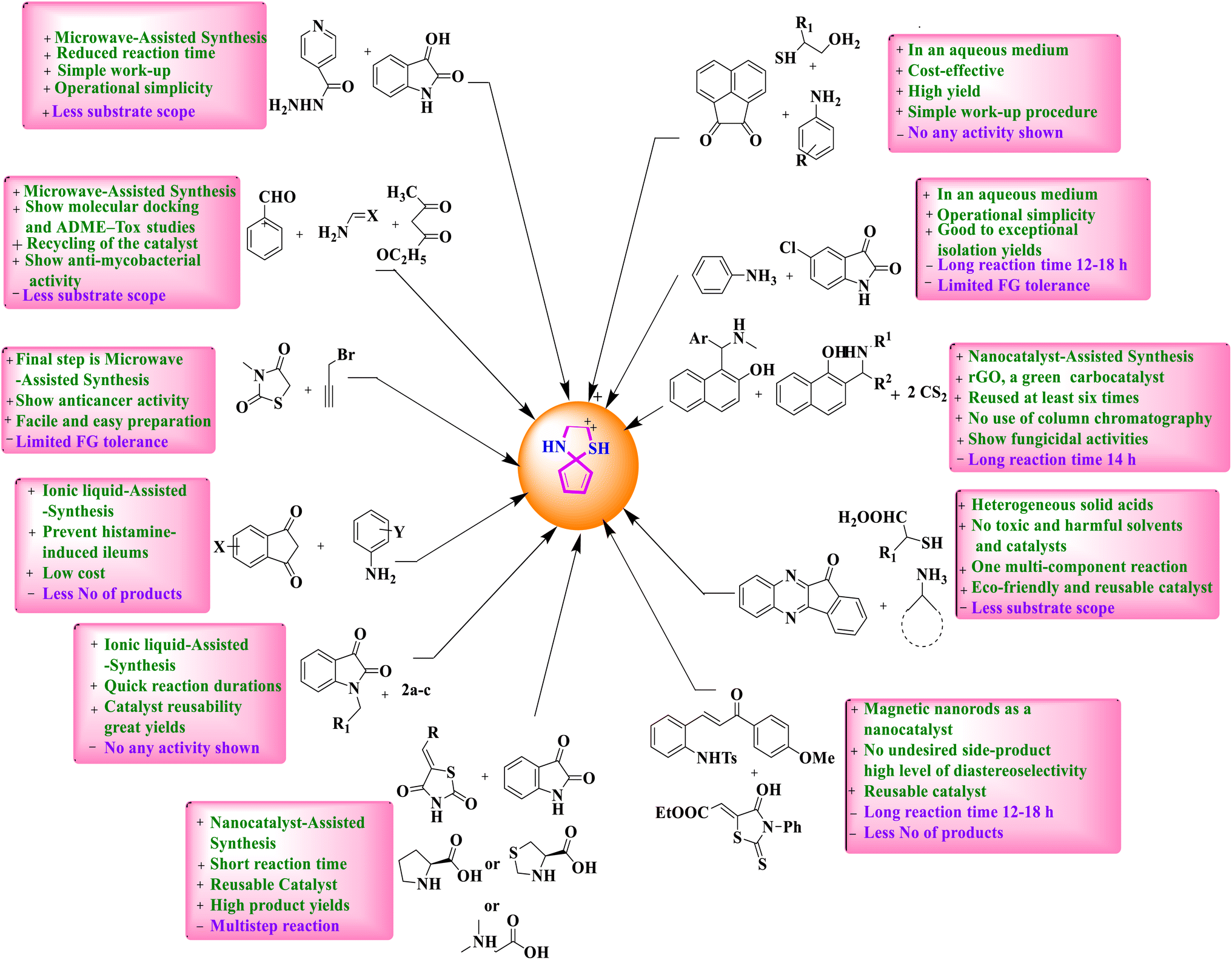 | ||
| Fig. 3 A comparative study of various synthetic strategies. | ||
Recent advances in the synthesis of spiro-thiazolidines under green conditions
A comprehensive account of topical progressions in the green production of spiro-thiazolidine derivatives with their systematic instincts is enclosed in the subsequent sections. A comparative study of various techniques of spiro-thiazolidine synthesis is given in Table 1.| S. no. | Green approach | Reaction condition | Yield | Advantages | Reference |
|---|---|---|---|---|---|
| 1 | Microwave-assisted synthesis | Scheme 1 | 40–90% | ✓Operational simplicity | P. N. Shinde et al.31 |
| Steps: | |||||
| EtOH, glacial CH3COOH, Mw, 3 min | ✓Reduced reaction time | ||||
| HSCH2COOH, ZnCl2/DMF, Mw, 10 min | ✓Simple work-up | ||||
| EtOH, ArCHO/CH3COOH, Mw, 17 min | ✓Show antimicrobial potency | ||||
| Scheme 2 | 80.7–85.6% | ✓Effective workup | M. A. Borad et al.32 | ||
| Steps: | |||||
| Aluminosilica (10 mol%)/C2H5OH, reflux, 2 h | ✓Great product yields | ||||
| NH2NH2H2O/C2H5OH, conc. H2SO4, reflux, 3 h | ✓Use of recyclable catalyst | ||||
| CNBr, C2H5OH, NaHCO3 | ✓Recycling of the catalyst | ||||
| 5-Substituted isatin, CH3OH/glacial CH3COOH, reflux, 45 min | ✓Show molecular docking and ADME–Tox studies | ||||
| Thioglycolic acid, ZrSiO2 (10 mol%), DMF, 120 °C, 9–12 min, Mw | ✓Show anti-mycobacterial activity | ||||
| Scheme 3 | 80% | ✓The final step is microwave-assisted synthesis | S. Kotha et al.33 | ||
| Steps: | |||||
| K2CO3, DMF, rt, 1–14 h | ✓Apoptosis inducers | ||||
| Propargyl bromide, Mo(CO)6 (5 mol%), MeCN, Mw, 10 min, 90 °C | |||||
| 2 | Ionic liquid-assisted synthesis | Scheme 4 | 90–97% | ✓Use of green reaction media | K. Arya et al.41 |
| ✓The ease of production, separation, and recycling of the catalyst | |||||
| [MIM] + BF4−, Mw, 2–4 min | ✓Minimal cost and environmentally beneficial aspect | ||||
| ✓Prevent histamine-induced ileum | |||||
| 3 | Solid supported acid-catalysed synthesis | Scheme 5 | 62–98% | ✓Quick reaction durations | E. M. Hussein et al.42 |
| MCM-SO3H, EtOH, 20–90 min | ✓Catalyst reusability | ||||
| ✓Great yield | |||||
| 4 | On water synthesis | Scheme 7 | 85–91% | ✓In green solvent | R. Singh et al.48 |
| ✓Easy handling of the catalyst | |||||
| ✓Cost-effective | |||||
| Thiamine hydrochloride, 80 °C, stirring | ✓High yield | ||||
| ✓Simple work-up procedure | |||||
| Scheme 9 | 81% | ✓In an aqueous medium | M. Nath et al.49 | ||
| Steps: | ✓Cost-effective | ||||
| DBSA, H2O, 18 h, 25 °C | ✓High yield | ||||
| Thioglycolic acid, 12 h, 25 °C | ✓Simple work-up procedure | ||||
| 5 | Nanocatalyst-assisted synthesis | Scheme 10 | 55–98% | ✓Nanocatalyst-assisted synthesis | N. Ma et al.73 |
| ✓rGO as a green carbocatalyst | |||||
| Na2CO3, CH3OH, rt, 14 h | ✓Energy efficiency | ||||
| ✓Show fungicidal activities | |||||
| Scheme 12 | 70–70% | ✓Heterogeneous solid acids | R. Singh et al.74 | ||
| ✓No toxic and harmful solvents and catalysts | |||||
| DES, 80 °C, solid C–SO3H, 4–4.5 h | ✓One multi-component reaction | ||||
| ✓Eco-friendly and reusable catalyst | |||||
| Scheme 14 | 78–90% | ✓High yield | A. Bekhradnia et al.75 | ||
| ✓High level of diastereoselectivity | |||||
| [MnCoCuFe2O4@L-proline], EtOH, 100 °C, 9 h | ✓The prevention of undesired side-product formation | ||||
| ✓Easy recovery of catalyst | |||||
| 6 | Asymmetric synthesis | Scheme 15 | 99% | ✓Magnetic nanorods as a nanocatalyst | D. M. Du et al.76 |
| ✓No undesired side-product | |||||
| Rhodanine derivatives, 2-tosylaminochalcone, squaramide catalyst, CHCl3, 35 °C, 48 h | ✓High level of diastereoselectivity | ||||
| ✓Reusable catalyst | |||||
| 7 | Miscellaneous synthesis | Scheme 16 | 61–67% | ✓Highly regioselective | R. Raghavachary et al.77 |
| ✓Gives a single product only | |||||
| Toluene, reflux, 8 h | ✓More functional group tolerance | ||||
| Scheme 17 | 54% | ✓Easy formation steps | T. Sakata et al.78 | ||
| Steps: | |||||
| CS2, DCC, in C5H5N | ✓Less reaction times | ||||
| CS2, in C5H5N | ✓No side products | ||||
| NH2NH2 | ✓Functional group tolerance | ||||
| Scheme 18 | 60–70% | ✓Show antimicrobial activity | B. S. Kumar et al.79 | ||
| Steps: | |||||
| NaNO2, HCl, CCl3COCH2COOC2H5 | |||||
| DMF, Al2O3, MW, 2 min | |||||
| ClCH2COOC2H5, DMF, K2CO3, stirring, rt, 8 h | |||||
| SHCH2COOH, 120–125 °C, 12 h | |||||
| N2H4 H2O, EtOH, reflux, 5 h | |||||
| Benzaldehyde, acetic acid, reflux, 1–4 h | |||||
| 8 | Scheme 19 | 82–96% | ✓Green media | J. M. Khurana et al.80 | |
| CuSO4·5H2O | ✓Easy handling of the catalyst | ||||
| Sodium ascorbate (20 mol%) | ✓One-pot, four-component procedure | ||||
| 9 | Scheme 21 | 87–98% | ✓High yields | E. M. Hussein et al.81 | |
| Steps: | |||||
| Dry toluene | ✓Ease of work-up | ||||
| Ar-CHO, DBSNa, 20 mol% | ✓Quick reaction times | ||||
| 10 | Scheme 22 | 44–70% | ✓Biologically imperative units | R. R. Kumar et al.82 | |
| MeOH, reflux | ✓Wide substrate scope |
Microwave-assisted synthesis
Microwave-assisted synthesis is a region of emergent concern, which is not only used in engineering laboratories but also in the educational arena. Microwave (MW) irradiation is a solitary tool in modern organic chemistry to produce an inclusive array of products in a small reaction time with a high level of yields, fewer or insignificant formation of by-products, as well as stereoselectivity.30P. N. Shinde et al.31 reported the synthesis of spiro-[indole-thiazolidine] derivatives (5) in the reaction of isoniazid (1) and isatin (2) in the manifestation of the catalytic amount of glacial acetic acid provided N-(2-oxo-1,2-dihydro-3′H-indol-3-ylidene)pyridine-4-carbohydrazide (3), which was further cyclo-condensed with mercaptoacetic acid in the occurrence of anhydrous ZnCl2 to produce spiro-[indole-thiazolidine] derivatives (4). Further, in the last step, compound (4) was abridged with aromatic aldehydes to provide arylidene derivatives (5) (Scheme 1). The synthetic procedure applied in this research had several advantages: operational simplicity, reduced reaction time, and simple work-up. The authors studied the antimicrobial potency of the synthesized compounds against Gram-negative and Gram-positive microorganisms and found that some derivatives were significantly active.
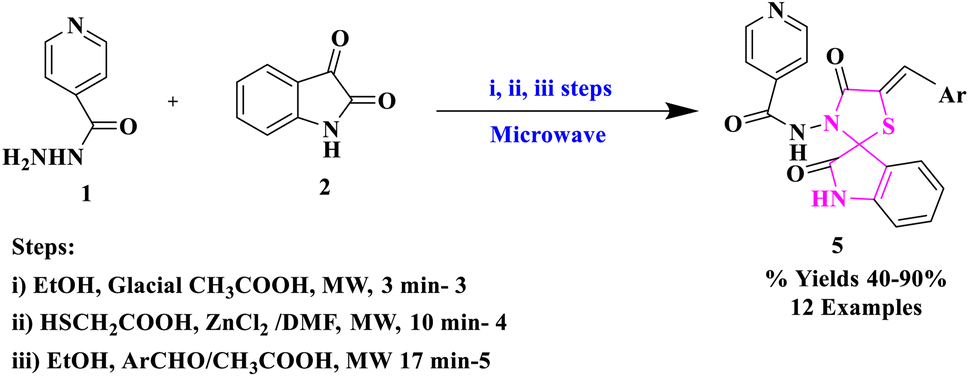 | ||
| Scheme 1 Synthesis of spiro-arylidene derivatives. | ||
M. A. Borad and co-authors32 synthesized precursors by the previously reported methods namely 6-methyl-4-phenyl-2-oxo/thioxo-1,2,3,4-tetrahydropyrimidine-5-carbohydrazide (9), 5-(5-amino-1,3,4-oxadiazol-2-yl)-6-methyl-4-phenyl-3,4-dihydropyrimidin-2(1H)-one/thione (10) and 5-substituted-3-[{5-(6-methyl-2-oxo/thioxo-4-phenyl-1,2,3,4-tetrahydropyrimidin-5-yl)-1,3,4-oxadiazol-2-yl}imino]-1,3-dihydro-2H-indol-2-one (11). Further, they produced several novel derivatives of spiro-[indole-thiazolidines] (12) from substituted isatins and thioglycolic acid (TGA) catalyzed by ZrSiO2 under microwave influence (Scheme 2). This approach has certain key advantages, such as ease of use, a simple, efficient workup, good product yields, and the use of recyclable catalysts. The anti-mycobacterial potential of all the spiro scaffolds was tested in vitro in contrast to the Mycobacterium tuberculosis (H37Rv) bacteria. The synthesized molecules were also subjected to a molecular docking study and the ADME–Tox characteristics of produced drugs were also estimated theoretically.
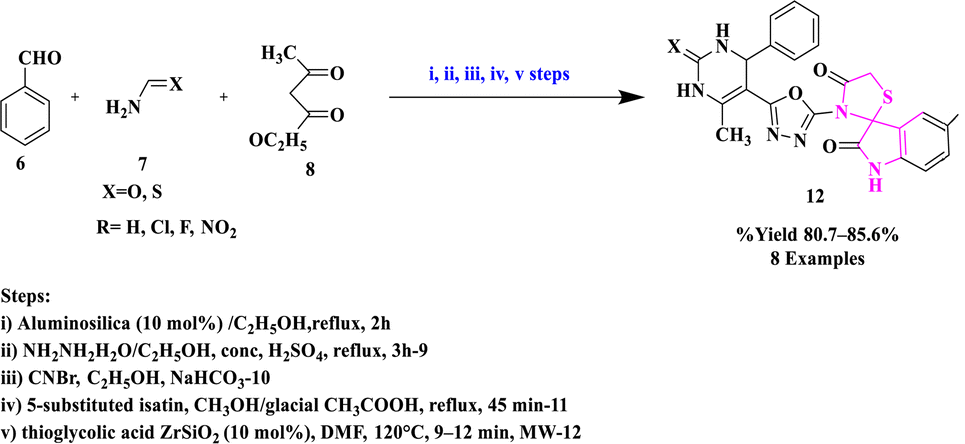 | ||
| Scheme 2 Synthesis of spiro-[indole-thiazolidines]. | ||
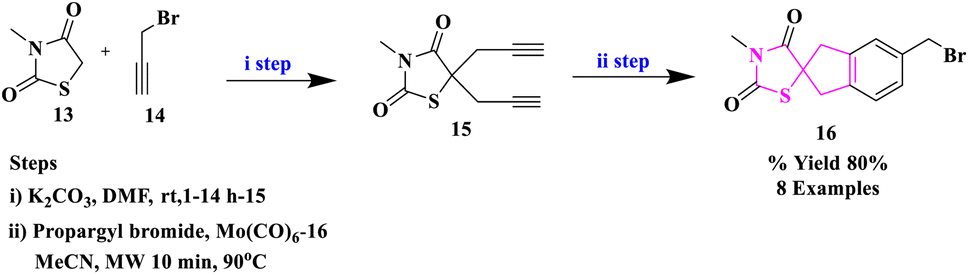
S. Kotha and colleagues33 described a new synthetic method for assembling spiro-thiazolidinediones using a [2 + 2 + 2] cyclotrimerization, followed by functionalization with DA chemistry and the click reaction (Scheme 3). The novel benzyl alcohol derivatives of thiazolidine-2,4-dione were prepared and they were proved to be effective apoptosis inducers in the Hek293, HeLa, Jurkat, U937, and K562 (cancer) cell lines for the first time using flow cytometry.
 | ||
| Scheme 3 Synthesis of diverse spirocyclic thiazolidinediones. | ||
In this instance, the necessary diyne precursor of thiazolidinedione was synthesized from N-methylthiazolidine-2,4-dione (13) and propargyl bromide (14) in DMF in the presence of K2CO3 to obtain the dipropargylated intermediate thiazolidinedione (15) in 85% yield. The co-trimerized spiro derivative (16) was obtained by reacting diyne (15) with propargyl bromide (14) in acetonitrile in the manifestation of Mo(CO)6 at 90 °C under the influence of microwave irradiation (MWI).
Ionic liquid-assisted synthesis
Ionic liquids (ILs) have fascinated widespread research interest as environment-friendly solvents owing to their favorable characteristics for instance insignificant vapor pressure, non-inflammability, high thermal stability, and reusability.34–37 They have also been called “designer solvents” because their physical and chemical characteristics may be tweaked by carefully selecting the right cation and anion. Ionic liquids are developing as a ‘green reaction medium’ (catalyst as well as solvent) by combining these unique features. Ionic liquids as a reaction medium offer a practical solution to both the catalytic recycling issues and solvent emission.38–40K. Arya and researchers41 reported Brønsted acidic Ils, including N-based organic cations 1-butyl-3-methylimidazolium and 1-methylimidazolium with inorganic anions like PF6, BF4, and PTSA, as catalysts and reaction media for the production of fluorinated spiro[3H-indole-3,2′-tetrahydro-1,3-thiazine]. Fluorinated spiro[3H-indole-3,2′-thiazolidine]-2,4′(1H)-diones-2,4′(1H)-diones (19) were produced using a one-pot environment-friendly microwave-generated process (Scheme 4). Significant substrate transformation and artifact selectivity were produced by the amalgamation of physiologically potent fluorinated spiro-indole[thiazine/thiazolidinone] with the use of catalytic quantities of ionic liquids. The creation of spiro derivatives using Ils as the reaction medium went well, and the products were easily decanted from the ionic liquid. The study concluded that such a reaction medium was abundant in manufacture, separation, and reutilization as the catalyst, which was inexpensive and advantageous to the environment. The aforementioned substances also shielded guinea pig ileum against histamine induction. The assessment of pA2 levels confirmed the existence of H1-antagonism.
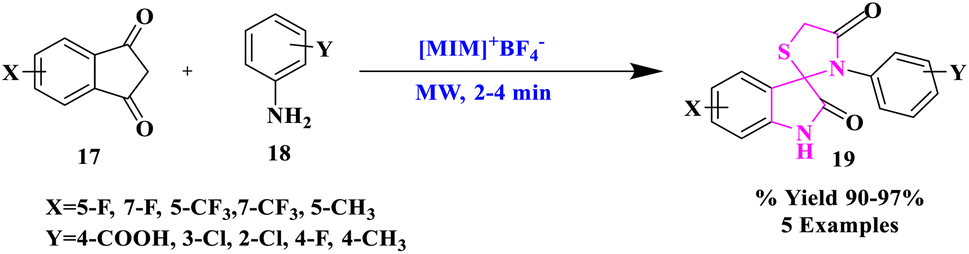 | ||
| Scheme 4 Synthesis of fluorinated spiro[indole-thiazolidiones]. | ||
Solid supported acid-catalysed synthesis
E. M. Hussein and colleagues42 used sulfonated mesoporous silica (MCM-SO3H) as a heterogeneous and reusable acidic catalyst to establish a simple and effective one-pot production of polyfunctionalized spirothiazolidin-4-ones (23) (Scheme 5). This approach avoided the use of harmful solvents, resulting in greater yields under benign conditions as compared to the previously described synthetic procedures for spirothiazolidin-4-one molecules. Product (23) was produced under optimal conditions by reacting indoline-2,3-dione derivatives (20) with aromatic amines (21) to produce the proper imine derivative, followed by a reaction with thioglycolic acid (22). This approach improved yields while employing reasonably non-hazardous solvents. The primary features of this protocol included environment-friendly conditions, ease of reaction, quick reaction times, high yields, ease of work-up, as well as catalyst reusability.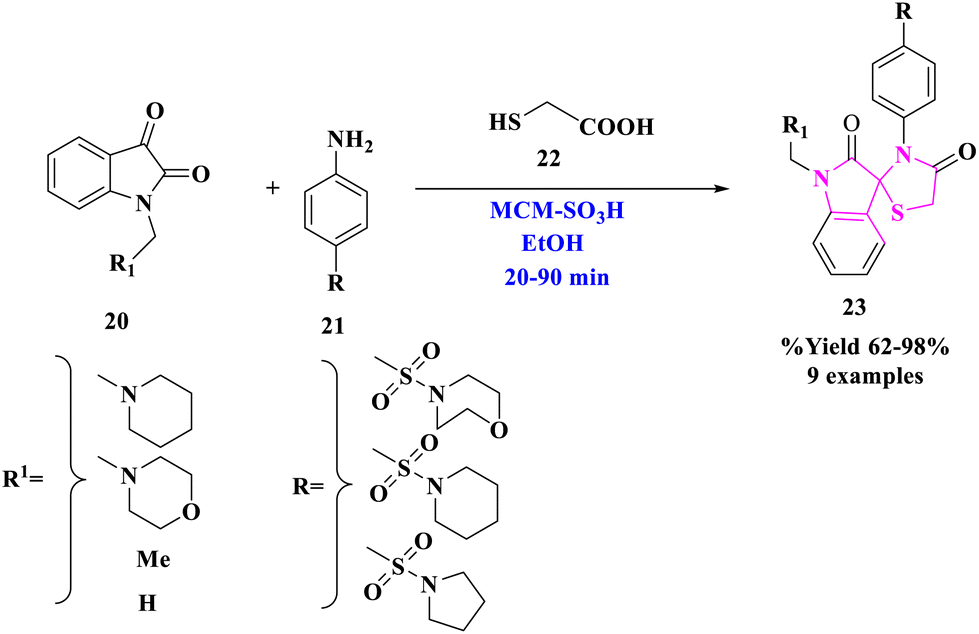 | ||
| Scheme 5 The production of new spiro[indoline-3,2′-thiazolidine]-2,4′-diones. | ||
The putative mechanism proposed a viable process for the manufacturing of spirothiazolidin-4-ones (23) by applying MCM-SO3H as a catalyst. MCM-SO3H worked as a Brønsted acid catalyst, activating the cyclic ketone's carbonyl group, followed by a nucleophilic attack by the aromatic amine's –NH2 group. Imine intermediate I was formed after further dehydration. Following that, a nucleophilic attack of the thiol group of thioglycolic acid at the imino group of intermediate I (activated by MCM-SO3H) resulted in the construction of intermediate II, which then underwent an intramolecular nucleophilic attack of the –NH group at the carboxyl group, yielding intermediate III, and the final product was made by the elimination of an H2O molecule (Scheme 6).
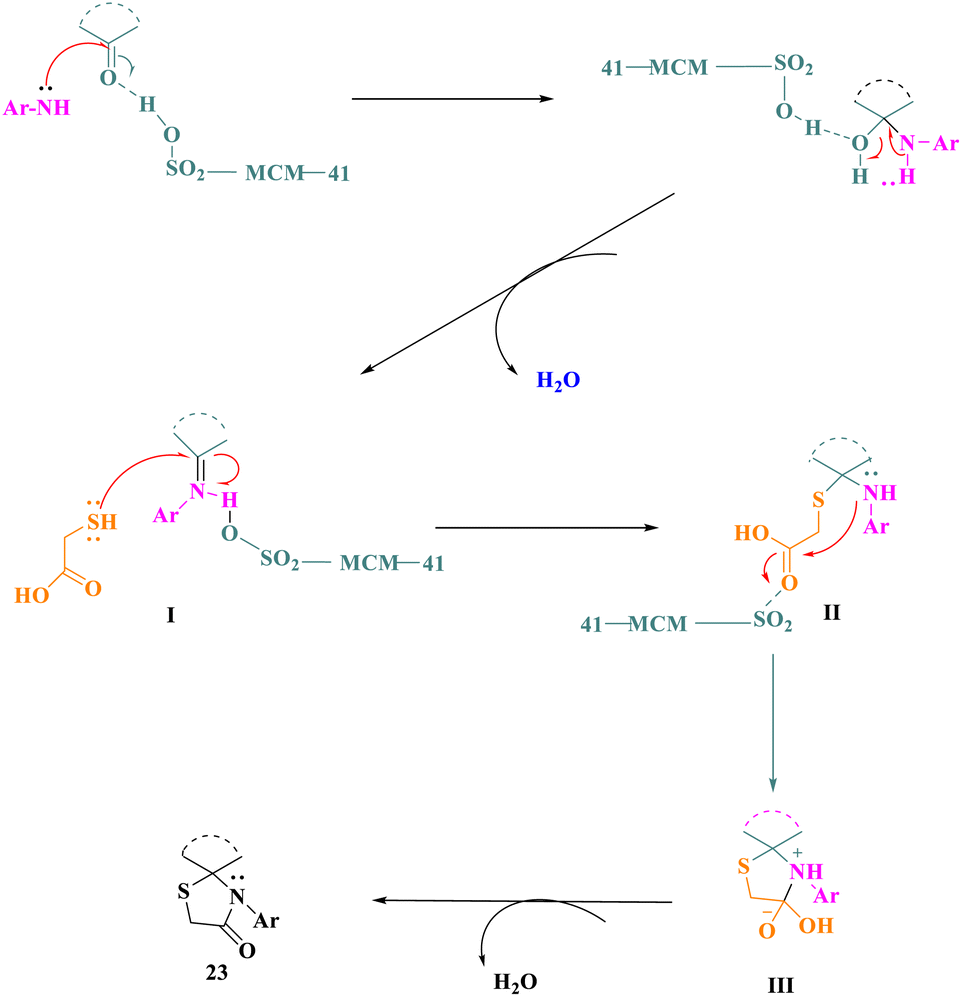 | ||
| Scheme 6 The feasible mechanism for the production of spiro-thiazolidin-4-ones by MCM-SO3H. | ||
On-water synthesis
Water is regarded as the ideal solvent for carrying out chemical reactions in green chemistry since it is inexpensive, non-toxic, safe, and provides no environmental harm.43–47 Furthermore, because typical organic molecules are poorly soluble in water, using water as a solvent creates product purification by simple filtration or extraction quite straightforward.R. Singh and colleagues48 described a water-facilitated and thiamine hydrochloride promoted efficient and eco-compatible process for the production of spiro[acenaphthylene-1,2'[1,3]-thiazolidine]-2,4′(1H)-diones (27) via MCR of substituted anilines, α-mercaptocarboxylic acid, and acenaphthylene-1,2-dione at 80 °C (Scheme 7). One C–S bond and two C–N bonds were formed during this conversion, which resulted in the one-pot assembly of a five-membered ring. The process was very enticing, sustainable, and economical because the catalyst was easily accessible and recoverable, the excellent product yield, a rapid work-up process, simple catalyst handling, and no toxic or organic solvents were used.
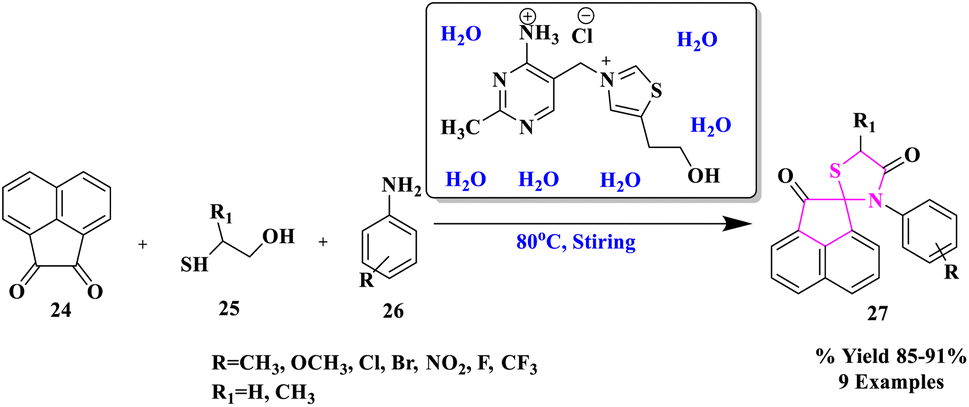 | ||
| Scheme 7 Production of spiro[acenaphthylene-1,2′[1,3]-thiazolidine]-2,4′(1H)-dione derivatives. | ||
The suggested reaction mechanism showed that the removal of a water molecule caused condensation between acenapthoquinone and aniline that resulted in the creation of the imine derivative A. Intermediate B was formed by the nucleophilic addition of the thiol group of α-mercaptocarboxylic acid (25) to intermediate A. Finally, the intermediate B was cyclized and dehydrated intramolecularly to produce the spiro[acenaphthylene-1,2′[1,3]-thiazolidine]-2,4′(1H)-dione derivatives (27) (Scheme 8).
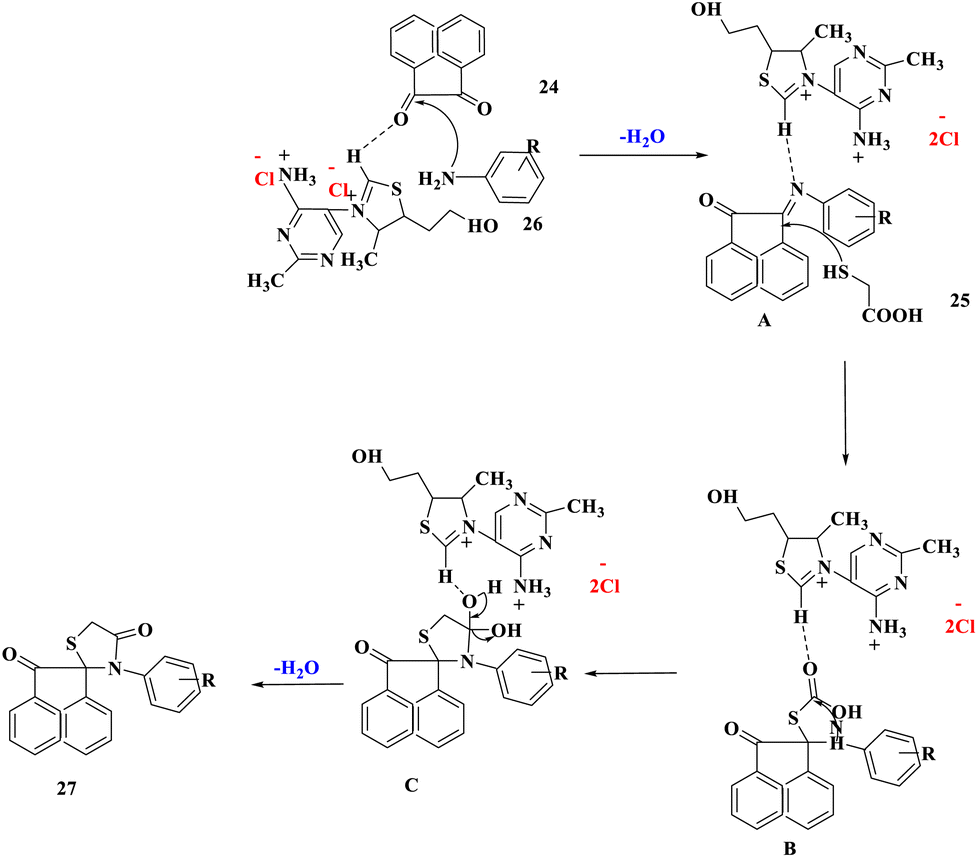 | ||
| Scheme 8 Plausible mechanism for the creation of spiro[acenaphthylene-1,2′[1,3] thiazolidine]-2,4′(1H)-dione derivatives. | ||
M. Nath and group49 developed an energy-efficient, environmentally acceptable synthetic approach for the production of a range of pharmaceutically potent spiro[indoline-3,2′-thiazolidinone] derivatives (30) (Scheme 9). These derivatives were prepared by reacting primary amines (28) with different isatins (29) and thioglycolic acid in the occurrence of p-dodecyl benzenesulfonic acid (DBSA) as an effective Brønsted acid surfactant joined catalyst in an aqueous medium at 25 °C. This synthetic technique had the advantages of operational simplicity, energy efficiency, high to remarkable isolation yields, and utilization of a favored green solvent system for product manufacturing.
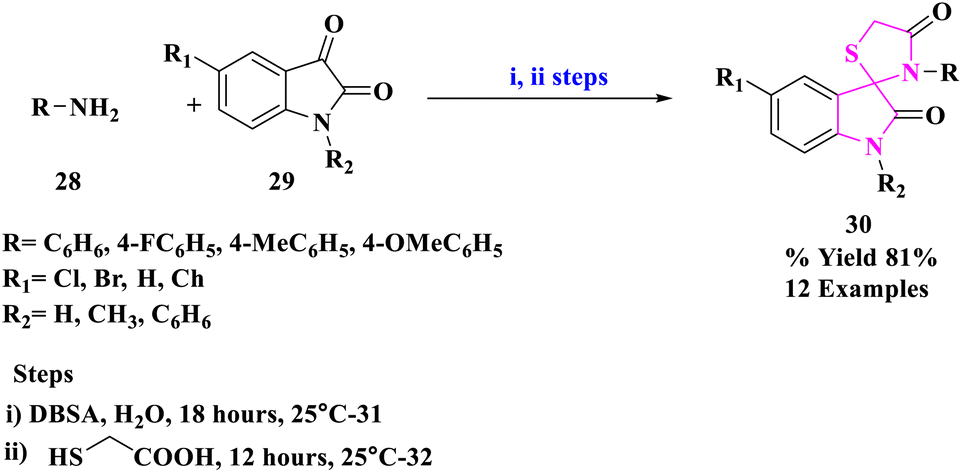 | ||
| Scheme 9 Synthesis of 5-chlorospiro[indoline-3,2′-thiazolidine]-2,4′-dione. | ||
Nanocatalyst-assisted synthesis
Research fields in physics,50–52 chemistry,53–55 biology,56–58 medicine,59–62 and particularly catalysis63–71 have taken incredible advantage of the hasty developments in nanotechnology and nanoscience. The morphologies of nanoparticles (NPs) include nanospheres, nanosheets, nanoclusters, nanograins, and nanofibers. They are substances that have a cross-section of less than 100 nm and are shaped as spherical dots, rods, thin plates, or any irregular form.72N. Ma et al.73 developed a new and effective one-pot access to spiro-thiazolidinethione derivatives (35 or 35′) readily from naphthol Mannich bases and CS2 promoted by rGO, an eco-friendly and reusable carbocatalyst under mild conditions (Scheme 10). Under moderate circumstances, reduced graphene oxide (rGO) worked as a green catalyst for dearomatizing cyclization of naphthol Mannich bases (33 or 33′) with carbon disulfide (34) with ambient oxygen as a clean oxidant. Without losing considerable catalytic activity, rGO was reused at least six times. Furthermore, the synthesized products were tested for fungicidal activities. Some of the target compounds showed inhibition against Thanatephorus cucumeris, Botrytis cinerea, Sclerotinia sclerotiorum, and other fungi.
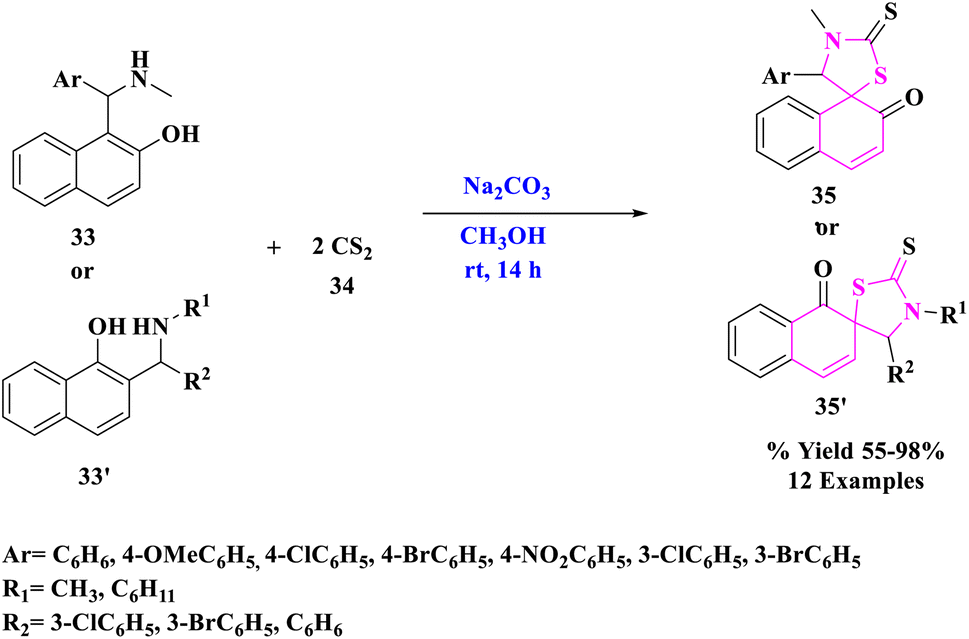 | ||
| Scheme 10 Synthesis of spiro-thiazolidinethione from naphthol Mannich bases. | ||
Scheme 11 depicts a possible mechanism for the synthesis of compounds (35 or 35′). 1-[(Methylamino)phenylmethyl]-2-naphthol interacted with carbon disulfide and a base to generate intermediate I and H-base. Then, by releasing H2O2, O2 could take hydrogen atoms from the hydroxy group in intermediate I and a proton from the H-base to produce resonance structures II, III, and IV. The product was formed when III loses an electron in the intramolecular cyclization process. At the same time, one electron was accepted by the rGO cation to restore its original structure.
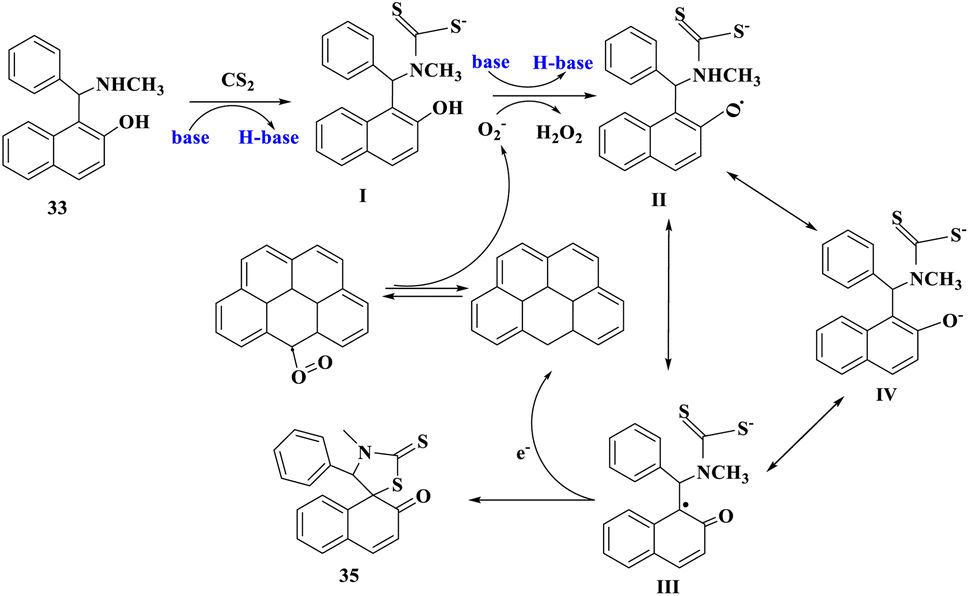 | ||
| Scheme 11 The plausible mechanism for synthesis of spiro-thiazolidinethiones. | ||
Due to high volatility, toxicity, corrosive character, and lack of recovery and reuse, homogeneous catalysts pose certain challenges in their use. As a result, following the principles of green chemistry, it is strongly advised that eco-friendly and reusable heterogeneous solid acids can be used as a replacement for traditional, poisonous, and polluting homogeneous acid catalysts. R. Singh and his co-workers74 designated a convenient and environmentally-friendly method for the synthesis of new hybrid spiro[indeno[1,2-b]quinoxaline[11, 2']-thiazolidine]-4′-ones (39) via a multi-component reaction involving indeno[1,2-b]quinoxalinone, mercaptocarboxylic acids (37) and various types of amines (38) using urea-choline chloride as a green deep eutectic solvent and carbon-SO3H as a solid acid catalyst (Scheme 12). This procedure had the benefits of avoiding hazardous solvents and catalysts as well as high to outstanding product yields. Furthermore, both catalyst and DES were quantitatively recovered from the reaction mixture and utilized many times.
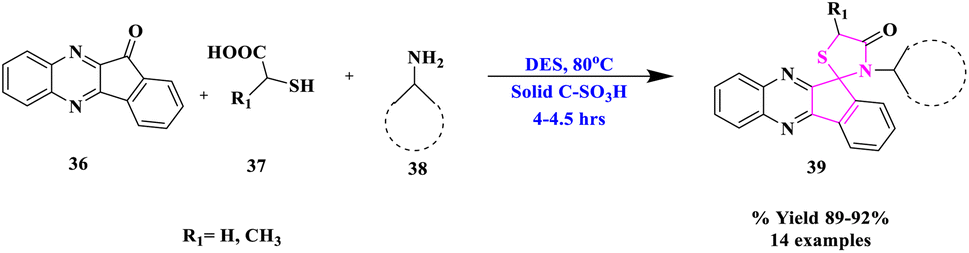 | ||
| Scheme 12 Synthesis of spiro[indeno[1,2-b]quinoxaline-[11,2′]-thiazolidine]-4′-ones. | ||
A plausible mechanism was proposed for the production of spiro[indeno[1,2-b]quinoxaline-[11,2']-thiazolidine]-4′-ones (39). The carbon-SO3H increased the electrophilic nature of carbonyl carbon (36), which was attacked at the same time by the –NH2 group of aniline (38) to create intermediate imine A. The nucleophilic addition of the thiol group of acid (37) to intermediate A initiated by C–SO3H led to the development of intermediate B, which then underwent intramolecular cyclization and dehydration to yield spiro[indeno[1,2-b]quinoxaline-[11,2']-thiazolidine]-4′-ones (39) (Scheme 13).
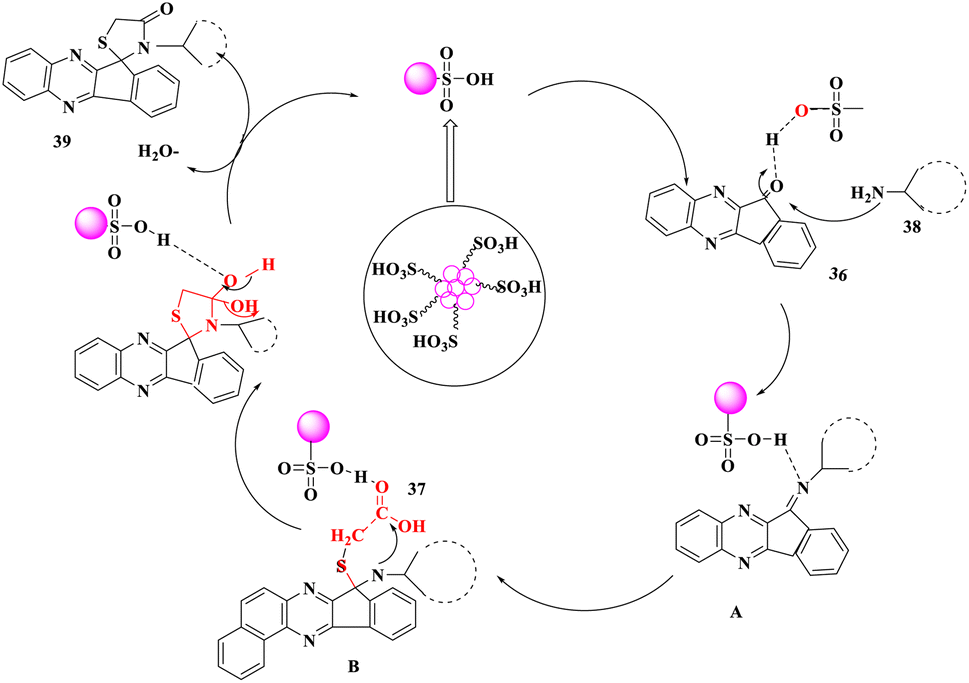 | ||
| Scheme 13 Proposed mechanism for the formation of thiazolidine-4'ones derivatives. | ||
L-Proline MNR-supported catalyzed, effectual, one-pot, green, and three-component approach was investigated by A. Bekhradnia and his group75 for the stereoselective construction of a novel class of spirothiazolidine derivatives. The interaction of 5-arylidene thiazolidine-2,4-diones (43), isatin (40), and secondary amino acids (41a–c) with MCCFe2O4@L-proline (MnCoCuFe2O4@L-proline) magnetic nanorods as a new nanocatalyst yielded a series of spiro-heterocycle derivatives (44a–c) stereoselectively and in high yields (Scheme 14). Thermal stability, magnetic characteristics, and other physicochemical features of the produced catalyst were all thoroughly investigated using a variety of methodologies. This catalyst was demonstrated to be an effective and reusable catalyst when it was used to produce endo-isomers of spirocyclic pyrrolidine/pyrrolizidine/pyrrolothiazolidine derivatives. The presented method's primary appealing features included its high yield, high degree of diastereoselectivity, avoidance of the production of undesirable side products, and simplicity of catalyst recovery without suffering a significant loss of catalytic activity.
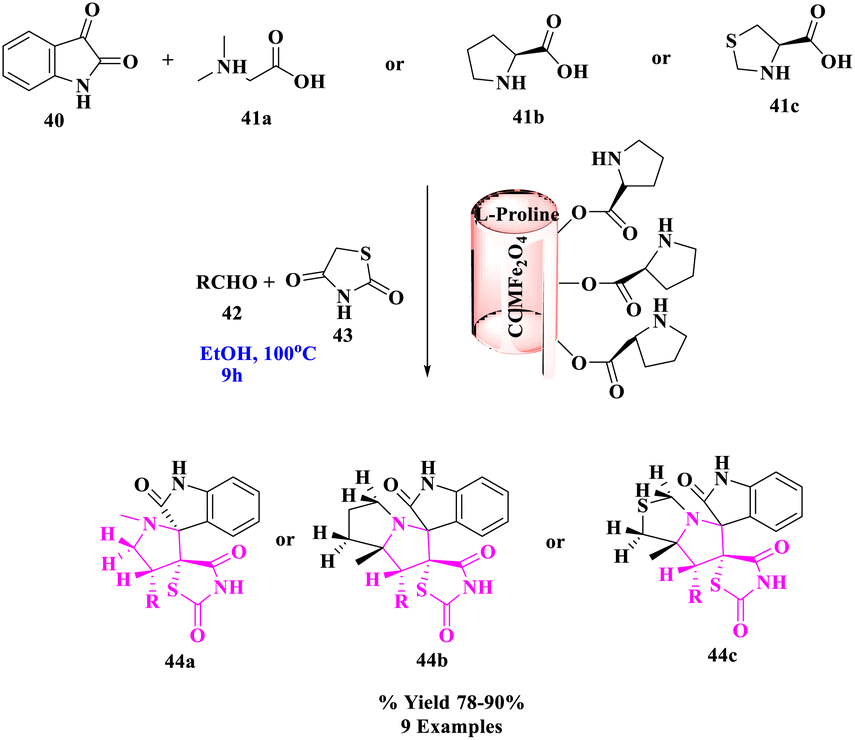 | ||
| Scheme 14 Green one-pot, three-component asymmetric 1,3-dipolar cycloaddition catalyzed by the CCMFe2O4@L-proline MNRs catalyst. | ||
Asymmetric synthesis
D. M. Du et al.76 developed a bifunctional squaramide-catalyzed asymmetric cascade aza-Michael/Michael addition process to synthesize chiral spirothiazolidinone tetrahydroquinolines with three contiguous stereocenters. To generate spirothiazolidinone tetrahydroquinolines (47), numerous functionalized rhodanine derivatives (45) and 2-tosylaminochalcone (46) with squaramide catalyst were reacted in CHCl3 solvent and stirred at 35 °C for 48 h (Scheme 15). Under moderate circumstances, this cascade reaction yielded the required products in good to exceptional yields (up to >99% yield) with outstanding diastereoselectivity (>25![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 dr) and high enantioselectivity (up to 96 percent ee). More significantly, the stereoselectivity was unaffected by the amplification and derivation procedures.
:1 dr) and high enantioselectivity (up to 96 percent ee). More significantly, the stereoselectivity was unaffected by the amplification and derivation procedures.
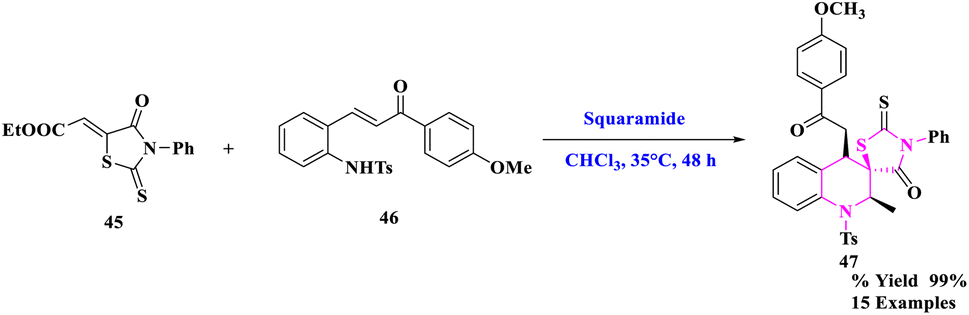 | ||
| Scheme 15 Asymmetric synthesis of highly functionalized spirothiazolidinone tetrahydroquinolines. | ||
Miscellaneous synthesis
R. Raghavachary et al.77 synthesized a diverse sugar-fused chromanono thiolizidine derivatives (51) by the reaction of several 3-arylidenechroman-4-ones as dipolarophiles (49) with thiazolidine-4-carboxylic acid (50), and sugar aldehyde (48) under the toluene, reflux for 8 h in good yields (Scheme 16). The cycloaddition was found to be extremely regioselective and the o-benzyl group was found to be removed. Highly regioselective, majorly a single product formation and more functional group tolerance were significant to this reaction.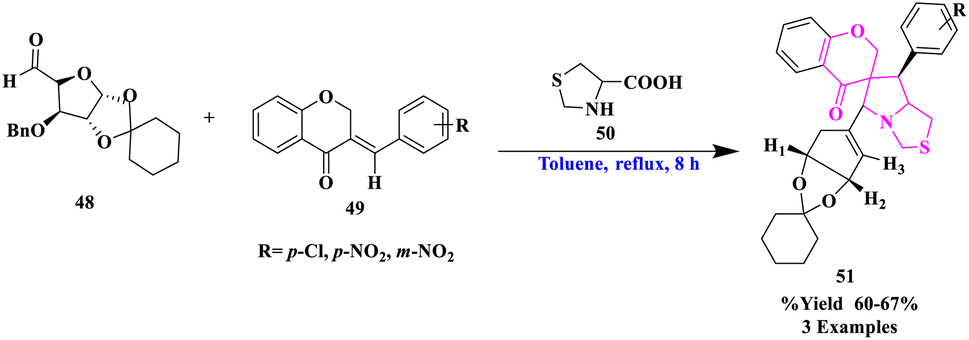 | ||
| Scheme 16 Synthesis of sugar-fused spiro-chromanono thiolizidines. | ||
The production of 3′-aminofluorene-9-spiro-5′-imidazolidine-2′,4′-dithione (54) via the reaction of fluorene-9-spiro-4′-thiazolidine-2′,5′-dithione (53) with hydrazine was described by T. Sakata and his group78 (Scheme 17). The main advantages of this reaction were easy formation steps, less reaction time, no side products, and functional group tolerance.
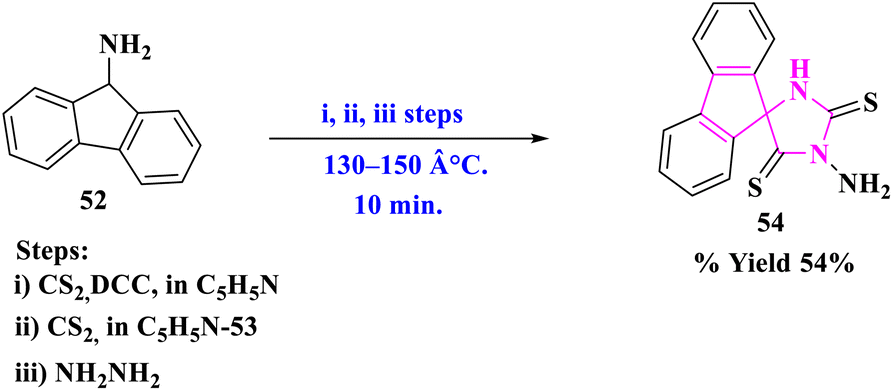 | ||
| Scheme 17 Synthesis of 3′-aminofluorene-9-spiro-5′-imidazolidine-2′,4′-dithione. | ||
B. S. Kumar and his colleagues79 designed the multistep synthetic strategy of some new spiro compounds comprising thiazolidinone (56) and pertaining antimicrobial evaluation (Scheme 18). The antimicrobial activity of the recently produced compounds was assessed by the cup plate method. The spiro derivatives with –OCH3, –Cl, and –Br substituents displayed promising in vitro antimicrobial activity.
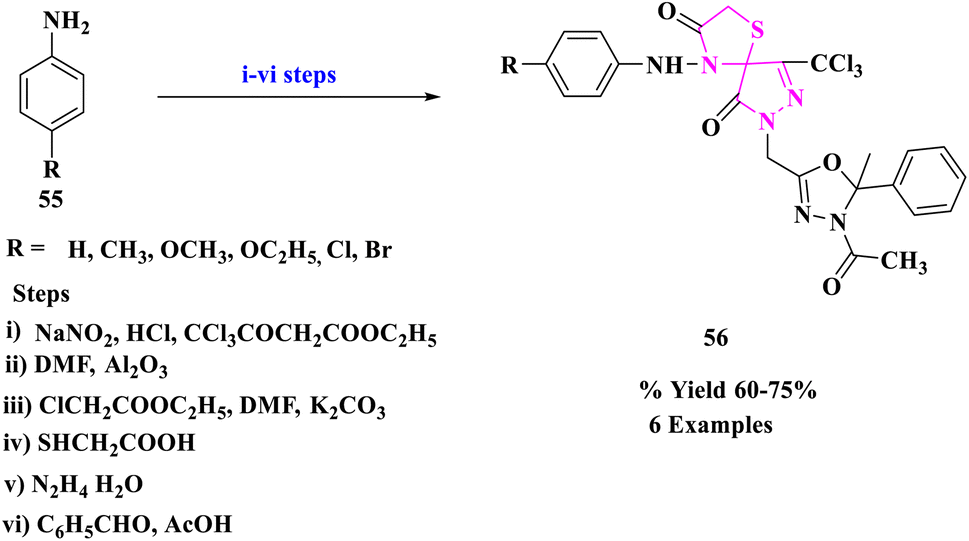 | ||
| Scheme 18 Synthesis of [5-oxo-4-(4-substituted aryl hydrazono)-3-trichloro methyl-4,5-dihydro-pyrazol-1-yl]-acetic acid ethyl ester. | ||
A range of novel dispiro-thiazolidine-2,4-dione associated 1,2,3- triazole derivatives (61) were synthesized by J. M. Khurana et al.80 via a one-pot, four-component procedure that used 5-arylidene-3-(prop-2-ynyl)thiazolidine-2,4-dione (57), isatin (58), sarcosine (60), and substituted azides (59) using Cu(I) produced in situ as a catalyst in PEG-400 as extremely effective and green media (Scheme 19). That was the first time a four-component reaction using a traditional Huisgen reaction was reported, in which the two dipolar moieties (substituted azides and in situ produced azomethine ylides) reacted with acetylenic and olefinic dipolarophiles, respectively.
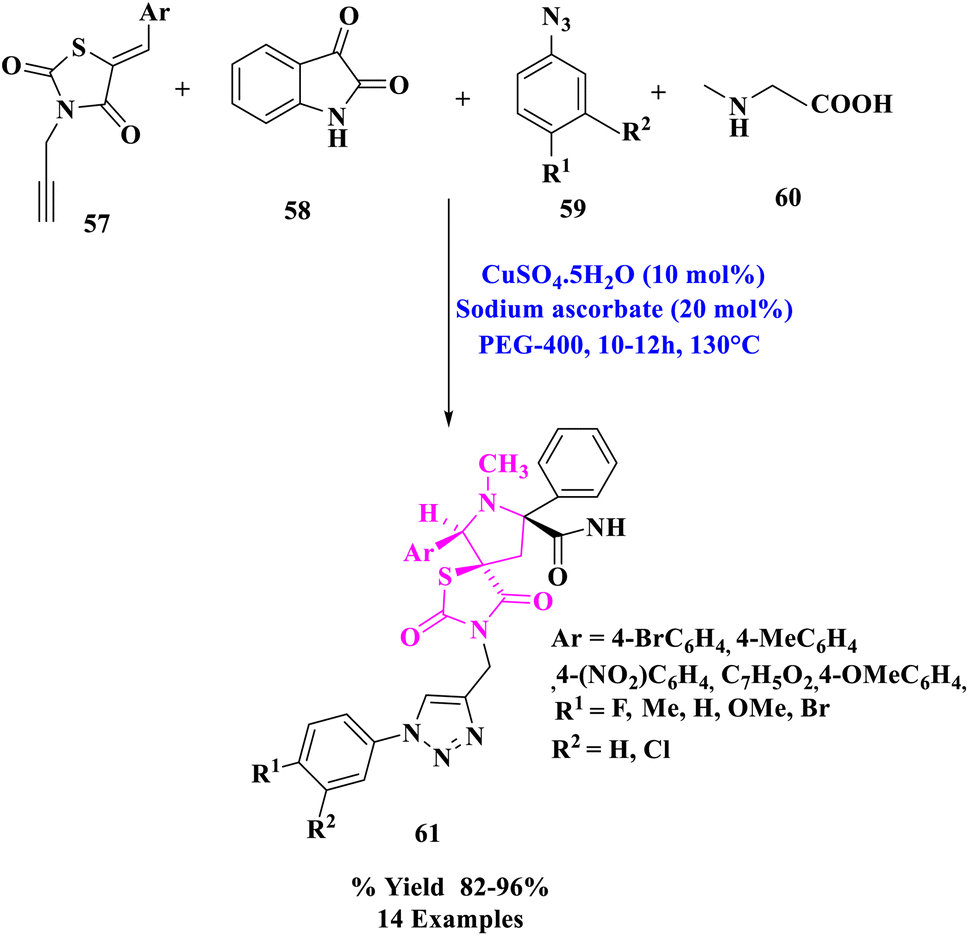 | ||
| Scheme 19 Construction of thiazolidine-2,4-dione linked 1,2,3- triazole derivatives. | ||
The hypothesized mechanism for the production of the final product 61 was depicted (Scheme 20). The planned route was divided into two sections, the first stage comprised a Cu(I) catalyzed [3 + 2] azide–alkyne cycloaddition to produce intermediate A. Cu(I) was created in situ by sodium ascorbate's well-known reduction of Cu(II) to Cu(I).32 In the second portion, intermediate A was subjected to a [3 + 2] cycloaddition reaction with azomethine ylide produced in situ to yield the desired products 61 (Path b). CO-TLC analysis of a genuine sample of A confirmed the production of A during the four-component condensation. The intermediate of A was further validated by an independent reaction of A with isatin and sarcosine in PEG-400, which produced 61 and thus the intermedite of A in the four-component condensation in general. An independent reaction of A with isatin and sarcosine corroborated the involvement of Cu(I) in catalyzing just the first phase of the route.
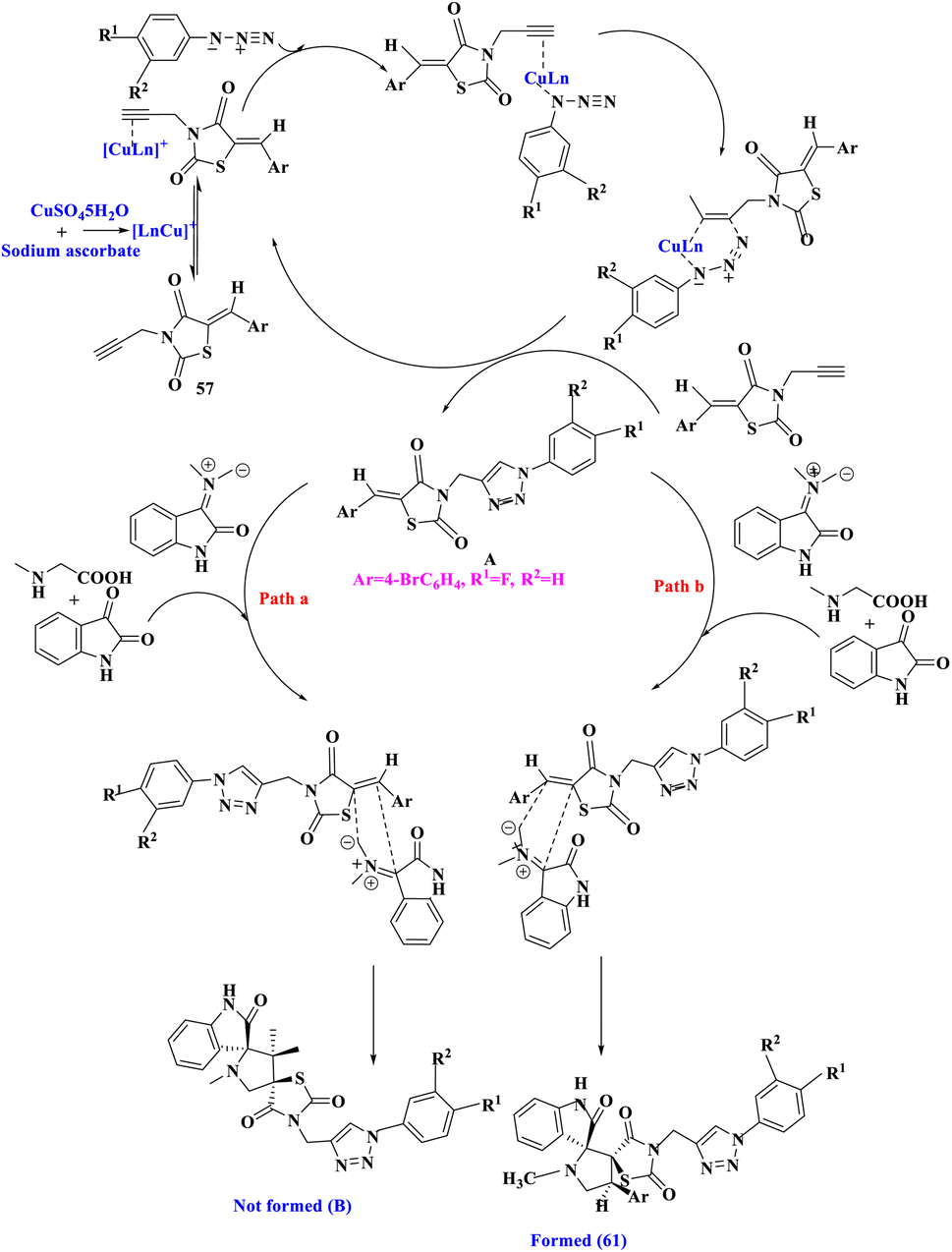 | ||
| Scheme 20 The plausible mechanism for the regio- and stereo-selective formation of thiazolidine-2,4-dione linked 1,2,3- triazole derivatives. | ||
E. M. Hussein and coworkers81 designed a novel, rapid and efficient pathway to synthesize spiro-thiazolidinones (67) for the production of diversified significant drugs essential for the treatment of bacterial infections, inflammations, and hypertension. The production of certain 2-arylidine-1-thia-4-azaspiro[4.5]decan-3-ones (67) by condensation of 1-thia-4-azaspiro[4.5]decan-3-one (65) with aromatic aldehydes (66) in acetic acid at room temperature using sodium dodecylbenzene sulfonate (DBSNa) (20 mol%) (Scheme 21). High yields, ease of work-up, and quick reaction times were the key benefits of this process.
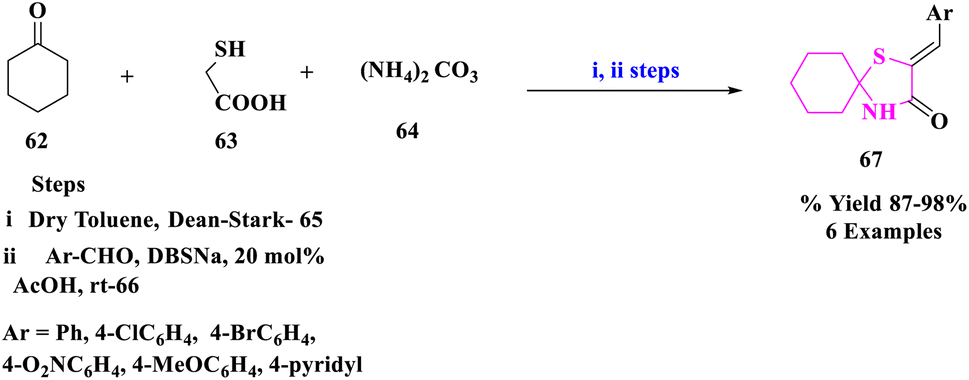 | ||
| Scheme 21 Synthesis of 2-arylidine-1-thia-4-azaspiro[4.5]decan-3-ones. | ||
R. R. Kumar et al.82 introduced the three-component 1,3-dipolar cycloaddition reaction of isatin (68), proline (70), and (Z)-5-arylidene-3-(2-cyclopropyl-2-oxo-1-phenylethyl)thiazolidine-2,4-diones (69) gave new dispiro oxindole–pyrrolizine–thiazolidine-2,4-dione derivatives (71) in opposing to the frequently detected regiochemistry (Scheme 22). These structurally fascinating hybrid heterocycles, including biologically imperative units, served as noteworthy leading compounds.
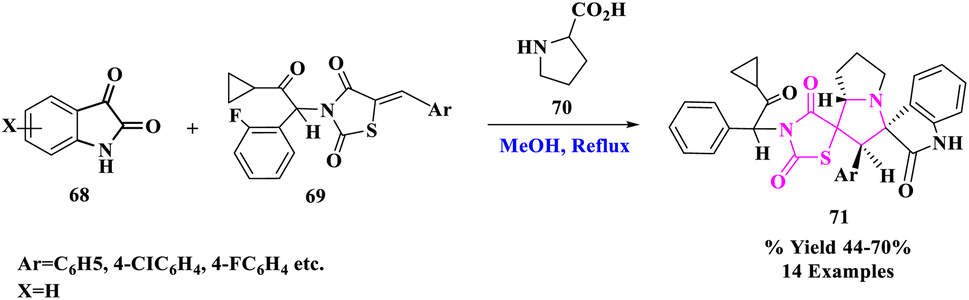 | ||
| Scheme 22 Synthesis of dispiro oxindole–pyrrolizine–thiazolidine-2,4-dione hybrids. | ||
Biological activity of spiro-thiazolidine derivatives
A broad outlook of pharmaceutical activities such as antimicrobial, anticancer, antidiabetic, antioxidant, and antitubercular, displayed by spiro-thiazolidine derivatives with the best candidates reported in the past eight years are mentioned in this review, explaining the importance of spiro-thiazolidines in medicinal chemistry (Fig. 4 and Table 2).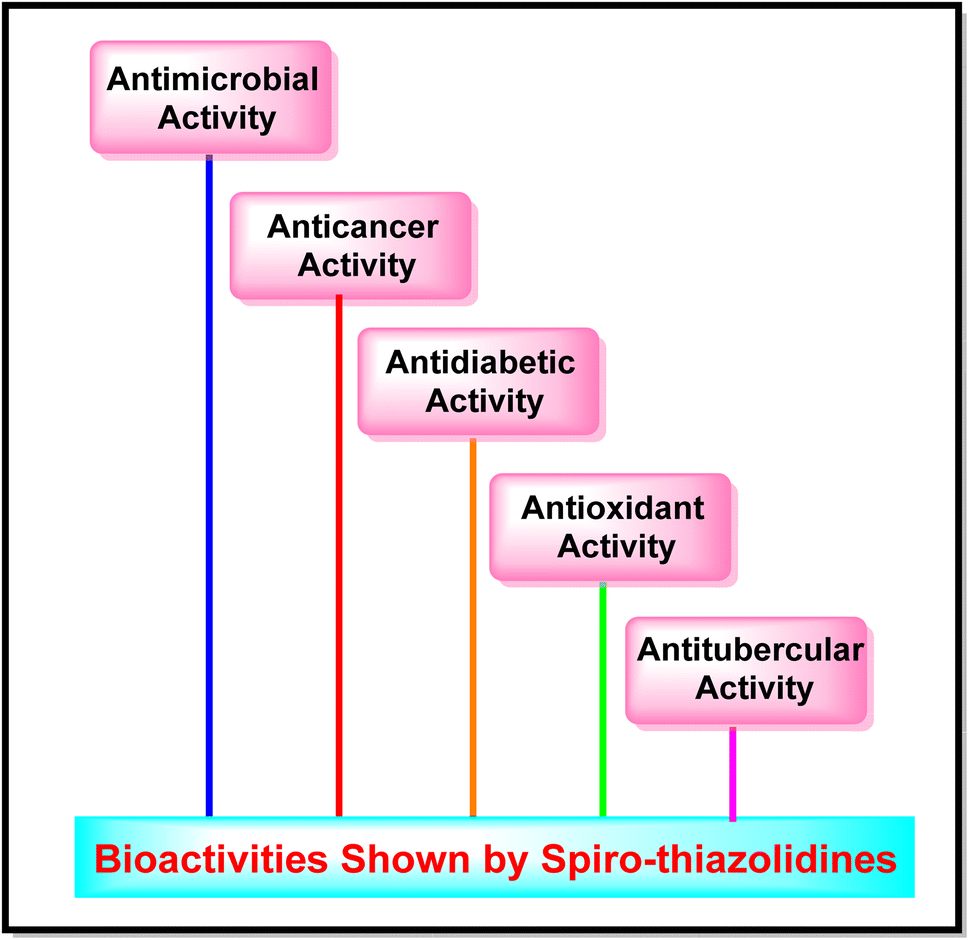 | ||
| Fig. 4 Bioactivities induced by spiro-thiazolidine derivatives. | ||
| S. no. | Activity | Bacteria/fungus/human cell lines | Methods | Standard drug molecules | Ref. |
|---|---|---|---|---|---|
| 1 | Antimicrobial activity | Bacterial strains | Disc diffusion, microdilution/turbidometric | Antibacterial:streptomycin | Y. S. Patel et al.83 |
| Bacillus subtilis | |||||
| Bacillus sphaericus | |||||
| Staphylococcus aureus | |||||
| Pseudomonas aeruginosa | |||||
| Klebsiella aerogenes | |||||
| Chromobacterium violaceum | |||||
| Fungal strains | Agar diffusion and broth dilution | Antifungal:amphotericin B | |||
| Candida albicans | |||||
| Aspergillus fumigatus | |||||
| Trichophyton rubrum | |||||
| Trichophyton mentagrophytes | |||||
| Bacterial strains | Broth dilution | Antibacterial:sulfamethoxazole | G. N. Kandile et a.84 | ||
| Staphylococcus aureus | |||||
| Staphylococcus epidermidis | |||||
| Escherichia coli | |||||
| Klebsiella pneumonia | Antifungal:fluconazole | ||||
| Fungal strains | |||||
| Aspergillus fumigatu | |||||
| Candida albicans | |||||
| Bacterial strains | Nil | Antibacterial:benzylpenicillin | A. N. Al-Romaizan et al.85 | ||
| Escherichia coli | |||||
| Bacillus subtilis | |||||
| Fungal strains | Antifungal:imidil | ||||
| Aspergillus flavus | |||||
| Aspergillus niger | |||||
| Bacterial strains | Disc diffusion | Antibacterial:cefoxitin | S. S. Shaban et al.86 | ||
| Staphylococcus aureus | |||||
| Escherichia coli | |||||
| Bacterial strains | Agar diffusion | Antibacterial:ampicillin | P. N. Shinde et al.31 | ||
| Staphylococcus aureus | |||||
| Escherichia coli | |||||
| Fungal strains | Nil | Nil | N. Ma et al.73 | ||
| Sclerotinia sclerotiorum | |||||
| Botrytis cinerea | |||||
| Thanatephorus cucumeris | |||||
| Phytophthora infestans | |||||
| Cercospora rachidicola | |||||
| Alternaria solani | |||||
| Gibberella zeae | |||||
| Macrophoma kuwatsukai | |||||
| Rhizoctonia cerealis | |||||
| Bacterial strains | Cup plate method | Antibacterial:ciprofloxacin | B. S. Kumar et al.79 | ||
| Staphylococcus aureus | |||||
| Bacillus cereus | |||||
| Escherichia coli | |||||
| Pseudomonas aeruginosa | |||||
| Fungal strains | Antifungal:clotrimazole | ||||
| Aspergillus niger | |||||
| Candida albicans | |||||
| Bacterial strains | Cup plate method | Antibacterial:ciprofloxacin | E. M. Hussein et al.81 | ||
| Staphylococcus aureus | |||||
| Escherichia coli | |||||
| Pseudomonas aeruginosa | |||||
| Fungal strains | Antifungal:nystatin | ||||
| Aspergillus niger | |||||
| Candida albicans | |||||
| Fusarium oxysporium | |||||
| Bacterial strains | Agar diffusion well | Antibacterial:ampicillin, gentamicin | A. Barakat et al.87 | ||
| Staphylococcus pneumonia | |||||
| Bacillus subtilis | |||||
| Pseudomonas aeruginosa | |||||
| Escherichia coli | |||||
| Fungal strains | Antifungal:amphotricin A, fluconazole | ||||
| Aspergillus fumigatus | |||||
| Syncephalastrum racemosum | |||||
| Geotricum candidum | |||||
| Candida albicans | |||||
| 2 | Anticancer and antidiabetic activities | Human liver cancer (HepG-2) cell lines | Lactate dehydrogenase (LDH) assay | Doxorubicin (HepG-2) | M. El-Shahat et al.88 |
| Human normal Retina pigmented epithelium (RPE-1) cell lines | |||||
| Human breast carcinoma (MCF-7) | Cytotoxicity assay | Anticancer:doxorubicin | M. El-Shahat et al.89 | ||
| Human liver carcinoma (HepG-2) | |||||
| Alpha-amylase inhibitor | Alpha-amylase inhibitory assay | Antidiabetic:acarbose | |||
| Alpha-glucosidase inhibitor | Alpha-glucosidase inhibitory assay | ||||
| Cervical cancer cell line (HeLa) | MTT assay | Etoposide | S. Kotha et al.33 | ||
| Human embryonic kidney cell line (Hek293) | Cytotoxicity assay | ||||
| Human histiocytic lymphoma cell line (U937) | |||||
| T-cell leukemia cell line (Jurkat) | Annexin V-FITC/PI assay | Camptothecin | |||
| Myelogenous leukemia cell line (K562) | |||||
| Human liver carcinoma (HepG-2) | SRB assay | LSD | E. M. Flefel et al.90 | ||
| Human breast carcinoma (MCF-7) | |||||
| 3 | Antioxidant activity | NA | 2,2-Diphenyl-1-picrylhydrazyl technique | Ascorbic acid | M. N. Meeran et al.91 |
| Butylated hydroxytoluene | |||||
| 1,1-Diphenyl-2-picrylhydrazyl (DPPH) method | Ascorbic acid | T. E. Ali et al.92 | |||
| DPPH scavenging test | BHA | E. M. Flefel et al.90 | |||
| 4 | Antitubercular activity | Mycobacterium tuberculosis (MTB) H37RV | Lowenstein–Jensen (L-J) MIC technique | Isoniazid | M. A. Borad et al.32 |
Antimicrobial activity
P. N. Patel and Y. S. Patel83 synthesized various spiro-thiazolinone heterocyclic compounds and tested them for antibacterial (MIC/MZI) and antifungal (MIC/MZI) activities against Gram-positive bacteria such as Bacillus subtilis, Bacillus sphaericus, Staphylococcus aureus, and Gram-negative bacteria such as Pseudomonas aeruginosa, Klebsiella aerogenes, Chromobacterium violaceum by disc diffusion, microdilution/turbidometric methods. Agar diffusion and broth dilution methods were used to test the antifungal activity against Candida albicans, Aspergillus fumigatus, Trichophyton rubrum, and Trichophyton mentagrophytes in DMSO.Antimicrobial screening results validated that practically all the candidates were active and had moderate to good antibacterial activity compared to standard drugs. At the studied doses, compounds 7′-(4-chlorophenyl)-3′-(4-((6,7-dihydrothieno[3,2-c]pyridin-5(4H)-yl)sulfonyl)phenyl)-6′,7′-dihydro-3′H-spiro[cyclohexane-1,2′-thiazolo[4,5-d]pyrimidine]-5′(4′H)-thione (72), 3′-(4-chlorophenyl)-6′-(4-((6,7-dihydrothieno[3,2-c]pyridin-5(4H)-yl)sulfonyl)phenyl)-2′-phenyl-1′,2′,3′,6′-tetrahydrospiro[cyclohexane-1,5′-pyrazolo[3,4-d]thiazole] (73), 5′-amino-7′-(4-chlorophenyl)-3′-(4-((6,7-dihydrothieno[3,2-c]pyridin-5(4H)-yl)sulfonyl)phenyl)-3′H-spiro[cyclohexane-1,2′-thiazolo[4,5-b]pyridine]-6′-carbonitrile (74), and 7′-(4-chlorophenyl)-3′-(4-((6,7-dihydrothieno[3,2-c]pyridin-5(4H)-yl)sulfonyl)phenyl)-5′-oxo-4′,5′-dihydro-3′H-spiro[cyclohexane-1,2′-thiazolo[4,5-b]pyridine]-6′-carbonitrile (75) having a huge heterocyclic system on the thiazolidine-4-one ring exhibited a robust inhibitory effect compared to the reference medication (antibacterial-streptomycin and antifungal-amphotericin B), whereas, other compounds showed considerable antibacterial activity. According to the findings, an increase in the combination of heterocyclic rings with the thiazolidine-4-one ring might be the cause of an increase in considerable inhibitory action. The inclusion of chlorophenyl in the molecules also boosted the inhibitory effect (Fig. 5).
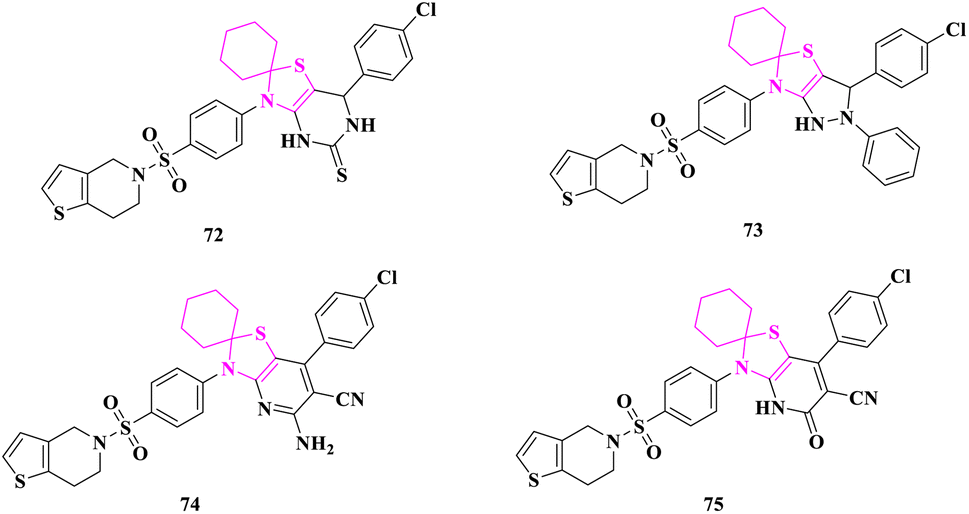 | ||
| Fig. 5 3′H-spiro[cyclohexane-1,2′-thiazolo[4,5-b]pyridine]-6′-carbonitrile derivatives as antimicrobial agents. | ||
G. N. Kandile and co-researchers84 synthesized new Schiff bases containing thiazolidine motifs and further checked them for in vitro antimicrobial activity using broth dilution procedure against two Gram-positive bacterial strains (Staphylococcus aureus and Staphylococcus epidermidis), two Gram-negative bacterial strains (Escherichia coli and Klebsiella pneumonia) and two fungal strains (Aspergillus fumigatu and Candidaalbicans) in minimal inhibitory concentration (MIC), minimal bactericidal concentration (MBC), and minimum fungicidal concentration (MFC) terms. The standard drugs used for antibacterial activity were sulfamethoxazole and antifungal activity was fluconazole. Bisspirothiazolodine 3′,3′′′-(3,3′-dimethoxy-[1,1′-biphenyl]-4,4′-diyl)bis(spiro[indoline-3,2′-thiazolidine]-2,4′-dione) (76) displayed the best antimicrobial activity among thiazolidine derivatives (Fig. 6).
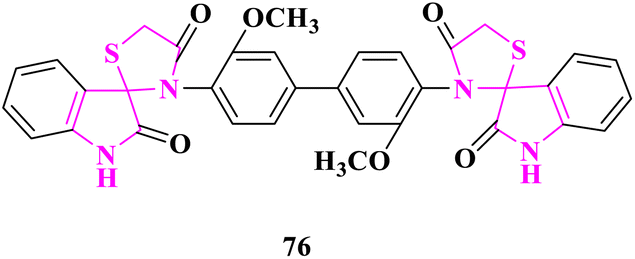 | ||
| Fig. 6 Bis(spiro[indoline-3,2′-thiazolidine]-2,4′-dione) derivative as the antimicrobial agent. | ||
A. N. Al-Romaizan85 reported the synthesis of novel spiro[indol-thiazolidine-2,4-diones] and bis(5-fluorospiro[indoline-3,2′-thiazolidine]-2,4′-dione) derivatives. The in vitro antimicrobial activity of the synthesized derivatives was tested against Escherichia coli and Bacillus subtilis bacterial strains and Aspergillus flavus and Aspergillus niger fungi. Benzylpenicillin (antibacterial) and imidil (antifungal) were used as the standard antibiotics in DMSO at a 100 μg mL−1 control concentration. 3′,3′′′-(1,4-phenylene)bis(5-fluoro-5′-(2,2,2-trifluoroacetyl)spiro[indoline-3,2′-thiazolidine]-2,4′-dione) (77) was found to display most prominent activity because it had the electron-withdrawing groups –C–F, –CF3, indole, and thiazolidinone motifs that exhibited high activity against microbes (Fig. 7).
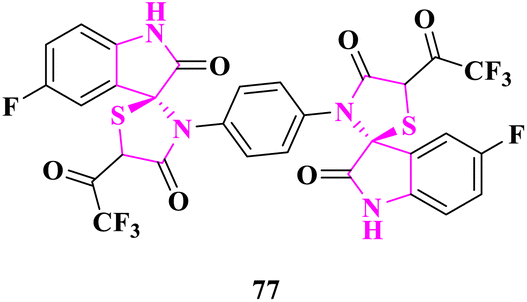 | ||
| Fig. 7 Bis(5-fluoro-5′-(2,2,2-trifluoroacetyl)spiro[indoline-3,2′-thiazolidine]-2,4′-dione) derivative as an antimicrobial drug. | ||
S. S. Shaban and co-researchers86 synthesized spiro derivatives and some of them were checked against Gram-negative bacteria (Escherichia Coli) and Gram-positive bacteria (Staphylococcus aureus) via the disc diffusion method at 1 mg mL−1 disc concentration. Altogether, these derivatives displayed moderate to good antibacterial activity. Molecule 6-(4-bromophenyl)-4-(4-methoxyphenyl)-2-((3-oxo-1-thia-4-azaspiro[4.4]nonan-4-yl)amino) nicotinamide (78) possesses the highest activity concerning Gram-negative and Gram-positive bacterias. Cefoxitin was used as an antibacterial reference drug in DMSO as a solvent (Fig. 8).
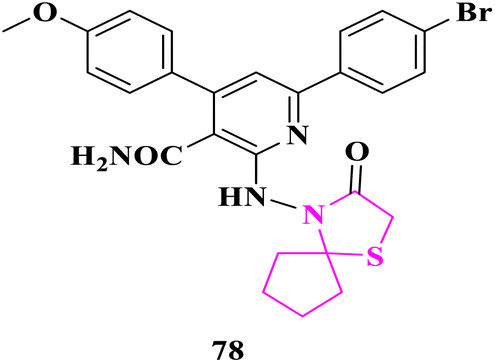 | ||
| Fig. 8 Aza spiro derivative as an anti-bacterial molecule. | ||
P. N. Shinde and co-authors31 studied the antimicrobial activity of newly synthesized spiro(indole-thiazolidine) derivatives using the agar diffusion method at 25 μg, 50 μg, and 100 μg concentrations in DMF. The standard drug ampicillin was utilized as a reference for antimicrobial activity. Compounds (Z)-N-(5′-(3-chlorobenzylidene)-2,4′-dioxospiro[indoline-3,2′-thiazolidin]-3′-yl)isonicotinamide (79) bearing electron-withdrawing group exhibited excellent activity against S. aureus and (Z)-N-(5'-(4-isopropylbenzylidene)-2,4′-dioxospiro[indoline-3,2′-thiazolidin]-3′-yl)isonicotinamide (80) bearing electron-donating group displayed high activity against E. coli according to the zone of inhibition (Fig. 9).
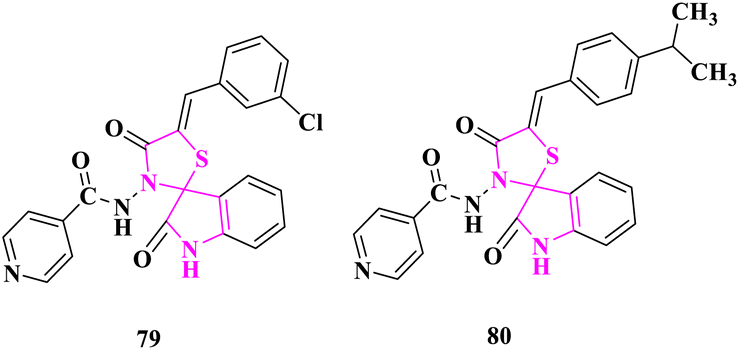 | ||
| Fig. 9 Dioxospiro[indoline-3,2′-thiazolidin] derivatives as antibacterial agents. | ||
Spiro-thiazolidinethione derivatives were synthesized and some of them were evaluated for in vitro antifungal activity against Sclerotinia sclerotiorum, Botrytis cinerea, Thanatephorus cucumeris, Phytophthora infestans, Cercospora rachidicola, Alternaria solani, Gibberella zeae, Macrophoma kuwatsukai, and Rhizoctonia cerealis fungal strains at 50 μg mL−1 concentration by N. Ma and co-researchers.73 3′-Methyl-4′-phenyl-2′-thioxo-1H-spiro[naphthalene-2,5′-thiazolidin]-1-one (81) displayed enhanced activity than other derivatives (Fig. 10).
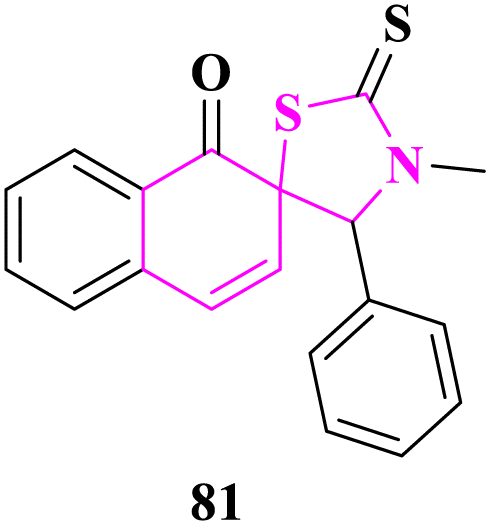 | ||
| Fig. 10 Spiro[naphthalene-2,5′-thiazolidin] as an antifungal agent. | ||
B. S. Kumar and researchers66 produced novel spiro-thiazolidinone derivatives and evaluated antimicrobial activity by cup plate technique. Staphylococcus aureus, Bacillus cereus, Escherichia coli, Pseudomonas aeruginosa were bacterial strains, and Aspergillus niger, Candida albicans were fungal strains for which reference drugs were ciprofloxacin for antibacterial and clotrimazole for antifungal evaluation. According to the zone of inhibition (ZOI), compounds 7-((4-acetyl-5-methyl-5-phenyl-4,5-dihydro-1,3,4-oxadiazol-2-yl)methyl)-4-((4-methoxyphenyl)amino)-9-(trichloromethyl)-1-thia-4,7,8-triazaspiro[4.4]non-8-ene-3,6-dione (82), 7-((4-acetyl-5-methyl-5-phenyl-4,5-dihydro-1,3,4-oxadiazol-2-yl)methyl)-4-((4-chlorophenyl)amino)-9-(trichloromethyl)-1-thia-4,7,8-triazaspiro[4.4]non-8-ene-3,6-dione (83), and 7-((4-acetyl-5-methyl-5-phenyl-4,5-dihydro-1,3,4-oxadiazol-2-yl)methyl)-4-((4-bromophenyl)amino)-9-(trichloromethyl)-1-thia-4,7,8-triazaspiro[4.4]non-8-ene-3,6-dione (84) having substituents –OCH3, –Cl and –Br, respectively, displayed excellent activity (Fig. 11).
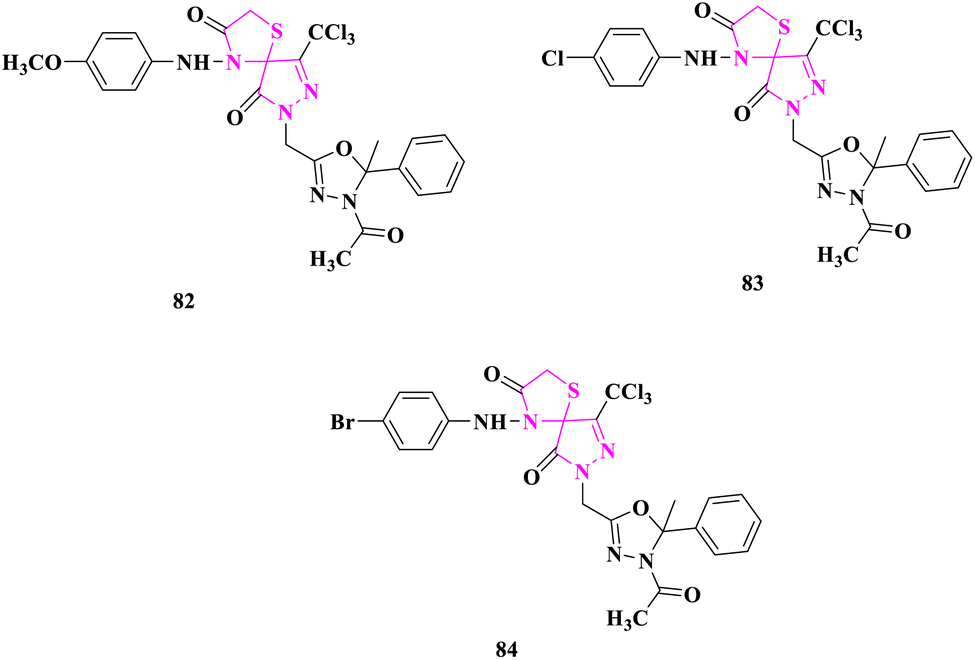 | ||
| Fig. 11 Spiro derivatives with antimicrobial potential. | ||
E. M. Hussein and team81 reported thiazolidine moiety-based spiro heterocycles synthesis and their in vitro antimicrobial activity was studied using the cup plate procedure. All the synthesized derivatives were assessed for antibacterial activity at 50 mg mL−1 concentration against Gram-positive (Staphylococcus aureus) bacteria and Gram-negative (Escherichia Coli, Pseudomonas aeroginosa) bacteria using standard antibacterial reference ciprofloxacin. Compound 2-(4-methoxybenzylidene)-4-(morpholinomethyl)-1-thia-4-azaspiro[4.5]decan-3-one (85) showed excellent activity against both Gram-positive and Gram-negative bacterial strains. The results specified that the aromatic and aliphatic substituents type was the governing factor for the activity of synthesized compounds. Based on the structure–activity relationships (SAR), the phenyl ring attached with an electron-donating group (MeO-) as demonstrated in compound (85) but the phenyl ring with electron-withdrawing groups (–Cl, –NO2) showed less antibacterial potential.
The produced compounds were checked for in vitro antifungal activity against fungal strains Aspergillus niger, Candida albicans, and Fusarium oxysporium by Nystatin as a standard antifungal drug at 50 mg mL−1 concentration. Here also the results indicated that the type of substituent was the controlling factor for antifungal properties. Compound 2-(4-nitrobenzylidene)-4-(pyrrolidin-1-ylmethyl)-1-thia-4-azaspiro[4.5]decan-3-one (86) exhibited excellent activities against all fungal strains. SAR showed that the phenyl ring attached with electron-withdrawing groups (–NO2, –Cl) had more antifungal behavior (Fig. 12).
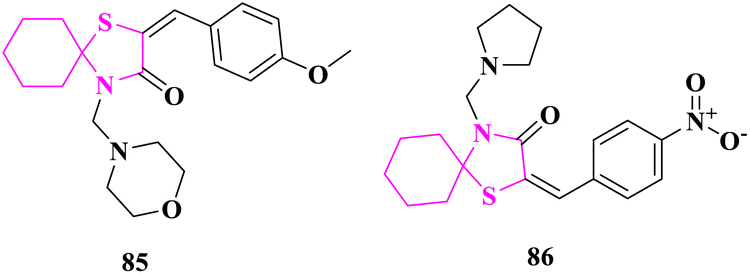 | ||
| Fig. 12 Azaspiro derivatives as antimicrobial agents. | ||
Polycyclic heterocycles containing spiro thioxothiazolidin-4-one were produced and tested for antimicrobial activity by A. Barakat and co-authors87 via the agar diffusion well process against bacterial strains (Staphylococcus pneumonia, Bacillus subtilis, Pseudomonas aeruginosa, and Escherichia coli) and fungal strains (Aspergillus fumigatus, Syncephalastrum racemosum, Geotricum candidum, and Candida albicans). In terms of antibacterial activity, (1′R, 7a′R)-2′′-Thioxo-2'-(p-tolyl)-5′,6′,7′,7a′-tetrahydro-2′H-dispiro[indoline-3,3′-pyrrolizine-1′,5′′-thiazolidine]-2,4′′-dione (87) and (1′R, 7a′R)-2′-phenyl-2′′-thioxo-5′,6′,7′,7a′-tetrahydro-2′H-dispiro[indoline-3,3′-pyrrolizine-1′,5′′-thiazolidine]-2,4′′-dione (88) compounds outperformed chosen benchmarks ampicillin and gentamicin, and in terms of antifungal activity, amphotericin A and fluconazole. The phenyl group played an essential role in determining drug interaction inside the receptors, according to a molecular docking investigation of the produced compounds (Fig. 13).
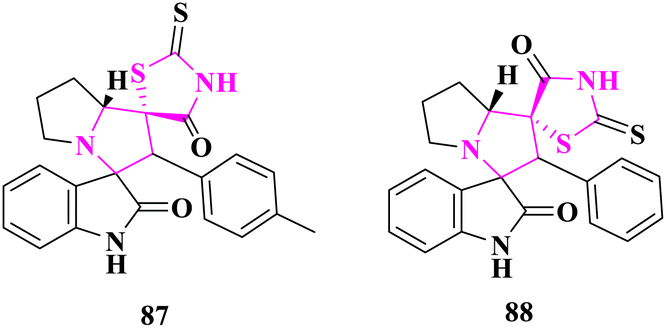 | ||
| Fig. 13 Spiro derivatives with antimicrobial potential. | ||
Anticancer and antidiabetic activities
The arylidene end products of the new bis spirothiazolidine ring system were produced and their in vitro anticancer activity was considered studied by M. El-Shahat and associates.88 The newly synthesized chemicals were tested in vitro against HepG-2 (human liver cancer) and RPE-1 (human normal Retina pigmented epithelium) cell lines, utilizing the lactate dehydrogenase (LDH) assay to detect cellular membrane permeabilization (rupture) and severe irreversible cell damage. The results indicated that four compounds 4,4′-(1,4-phenylene)bis(2-(4-fluorobenzylidene)-1-thia-4-azaspiro [4.5]decan-3-one) (89), 4,4′-(1,4-phenylene)bis(2-(4-nitrobenzylidene)-1-thia-4-azaspiro [4.5]decan-3-one) (90), 4,4′-(1,4-phenylene)bis(2-(4(dimethylamino)benzylidene)-1-thia-4-azaspiro[4.5]decan-3-one) (91), and 4,4′-(1,4-phenylene)bis(2-(4(dimethylamino)benzylidene)-1-thia-4-azaspiro [4.5]decan-3-one) (92) had considerably higher anticancer efficacy than the positive control doxorubicin in the case of HepG-2 cancer (Fig. 14).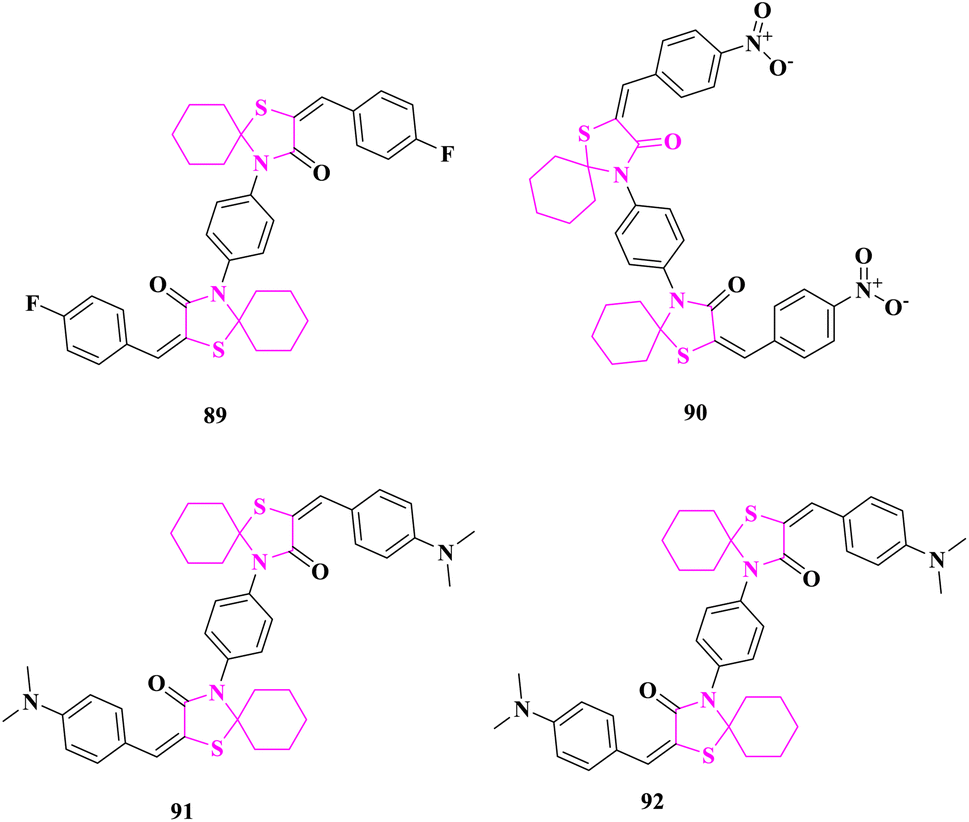 | ||
| Fig. 14 Bis spirothiazolidines with anticancer activity. | ||
Compound 96 exhibited 100 times greater noxiousness on cancer cells than on normal cells, respectively, when the findings of both liver cancer and normal cells were compared. The positive control, on the other hand, revealed just 2.6 times greater toxicity on tumor cells than on normal cells. These findings suggested that these derivatives might be employed as anticancer medication candidates since they were less harmful to normal cells than the positive control.
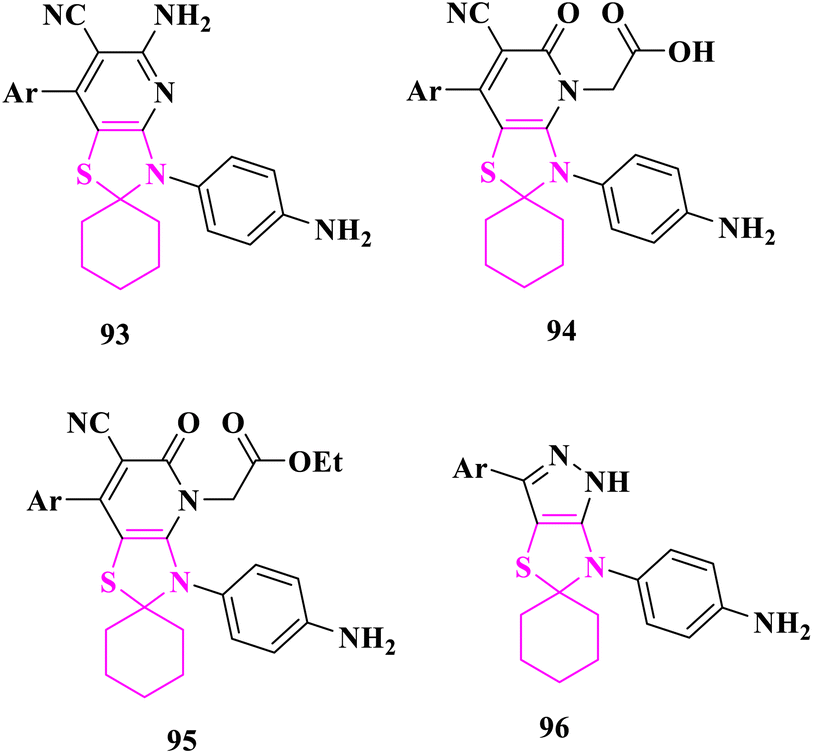
M. El-Shahat and co-researchers89 synthesized new spiro thiazolidene compounds and their fused analogues and tested them for anticancer and antidiabetic efficacy. Three compounds 5′-amino-3′-(4-aminophenyl)-7′-argio-3′H-spiro[cyclohexane-1,2′-thiazolo[4,5-b]pyridine]-6′-carbonitrile (92), 2-(3′-(4-aminophenyl)-7′-argio-6′-cyano-5′-oxo-3′,5′-dihydro-4′H-spiro[cyclohexane-1,2′-thiazolo[4,5-b]pyridin]-4′-yl)acetic acid (93), and ethyl 2-(3'-(4-aminophenyl)-7′-argio-6′-cyano-5′-oxo-3′,5′-dihydro-4′H-spiro[cyclohexane-1,2′-thiazolo[4,5-b]pyridin]-4′-yl)acetate (94), demonstrated considerable anticancer activity against human breast carcinoma (MCF-7) and human liver carcinoma (HepG-2) cell lines when compared to doxorubicin as positive control. At all the concentrations (3.90, 7.8, 15.6, 31.25, 62.5, 125, 250, and 500 μg mL−1), spiro thiazolopyridine–carbonitrile derivatives with amino, acetic acid, or propanoic acid groups demonstrated remarkable anticancer activity. When compared to the antidiabetic acarbose, compounds (92) and 4-(3′-argiospiro[cyclohexane-1,5′-pyrazolo[3,4-d]thiazol]-6′(1′H)-yl)aniline (96) at different concentrations (7.81, 15.63, 31.25, 62.5, 125, 250, 500, and 1000 μg mL−1) demonstrated superior therapeutic indices for both alpha-amylase inhibitor and alpha-glucosidase inhibitor as the positive control. Furthermore, compounds (92) and (96), which contain amino spiro thiazolopyridine-carbonitrile and pyrazolo spirothiazolidine groups, had strong activity against alpha-amylase and alpha-glucosidase enzymes at all doses (Fig. 15).
 | ||
| Fig. 15 Spiro-thiazolo derivatives with anti-tumor activity. | ||
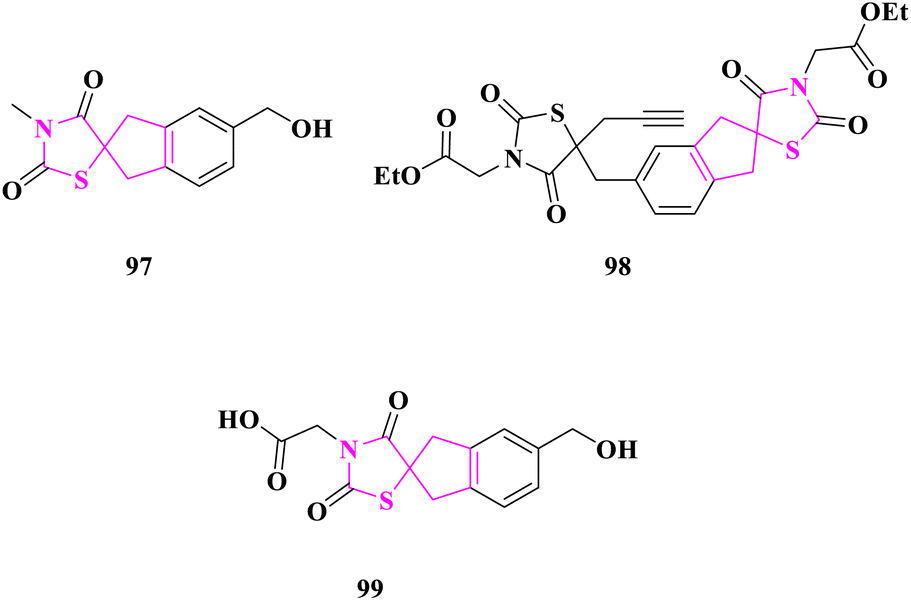
Spirothiazolidinediones were assembled by S. Kotha and researchers33 and were evaluated for anticancer efficacy as an efficient apoptosis inducer in the cervical cancer cell line (HeLa), human embryonic kidney cell line (Hek293), human histiocytic lymphoma cell line (U937), T-cell leukemia cell line (Jurkat), and myelogenous leukemia cell line (K562). On Jurkat, K562, HEK293, HELA, A549, and U937 cell lines, anticancer activity was compared to camptothecin and etoposide. Ethyl 2-(5-((3′-(2-ethoxy-2-oxoethyl)-2′,4′-dioxo-1,3-dihydrospiro[indene-2,5′-thiazolidin]-5-yl)methyl)-2,4-dioxo-5-(prop-2-yn-1-yl)thiazolidin-3-yl)acetate (98) and 5-(hydroxymethyl)-3′-methyl-1,3-dihydrospiro[indene-2,5′-thiazolidine]-2′,4′-dione (97) had the greatest activity (IC50 = 0.29 and 0.36 nM, respectively) against leukemic monocytic lymphoma cells (U937), whereas 2-(5-(hydroxymethyl)-2′,4′-dioxo-1,3-dihydrospiro[indene-2,5′-thiazolidin]-3′-yl)acetic acid (99) had an IC value of 0.34 nM against T-cell leukaemia (Jurkat). When compound 99 was introduced to the culture of Jurkat cells at a dose of 0.75 nM, the greatest percentage of apoptosis was recorded, which was 68.65%. Compound 98 exhibited around 95% late apoptosis in K562 cells under comparable circumstances, whereas compound 97 showed about 30.28% for cell line U937. Flow cytometry data revealed that 24 hours following the action of compounds 99, 98, and 97, a hypodiploidal DNA peak occurred in all three cell lines, Jurkat, K562, and U937, indicating a failure to stop the fission cycle at the checkpoints and eventually leading to cell death (Fig. 16).
 | ||
| Fig. 16 Dihydrospiro[indene-2,5′-thiazolidin] derivatives with anticancer efficacy. | ||
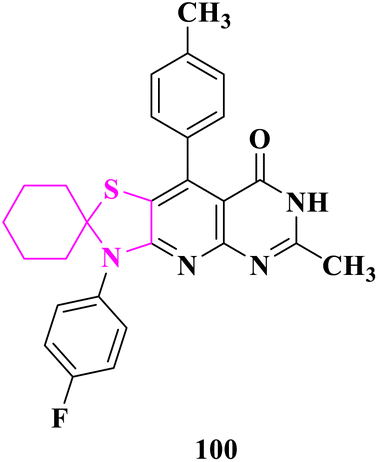
Novel spiro(cyclohexane-1,2′-thiazolidine) and spiro(cyclohexane-1,2′-thiazole) derivatives were prepared by E. M. Flefel and co-authors90 and evaluated for anticancer activity against two tumor cell lines HepG2 and MCF-7. The crude extract demonstrated adequate efficacy against HepG2 and MCF-7 cell lines, with potencies ranging from 15 to 84 percent at 100 μg mL−1. The HepG2 cell lines, on the other hand, showed stronger resistance to the synthetic molecules, with resistance ranging from 15 to 78 percent at 100 μg mL−1. In addition, 3′-(4-fluorophenyl)-6′-methyl-9′-(p-tolyl)-3′H-spiro[cyclohexane-1,2′-thiazolo[5′,4′:5,6]pyrido[2,3-d]pyrimidin]-8′(7′H)-one (100) had the strongest anticancer effect against both the cell lines (MCF-7 and HepG2). The significance of antioxidant substances in cancer cell growth suppression might be explained as hypothesized methods, such as the participation of heteroatom lone pairs in the chelation process, which increases ROS (reactive oxygenated species) generation and radical creation, causing DNA damage in cancer cells (Fig. 17).
 | ||
| Fig. 17 Spiro derivatives with antitumor potential. | ||
Antioxidant activity
M. N. Meeran and coworkers91 created spirothiozolidin-4-one and 5′-methyl-spiro-4-thiazolidione derivatives, which were tested for antioxidant activity using the 2,2-diphenyl-1-picrylhydrazyl technique. In comparison to the standards, ascorbic acid and butylated hydroxytoluene, (5′S)-3′-(2-benzoyl-4-chlorophenyl)-5′-methylspiro[indoline-3,2′-thiazolidine]-2,4′-dione (101) was shown to have high antioxidant activity (Fig. 18).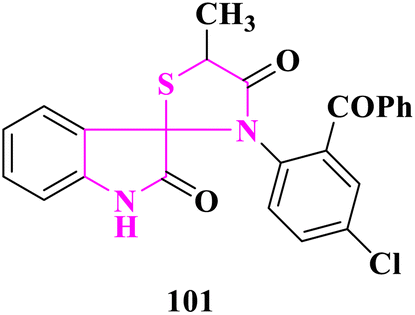 | ||
| Fig. 18 Spiro[indoline-3,2′-thiazolidine] derivative as an antioxidant. | ||
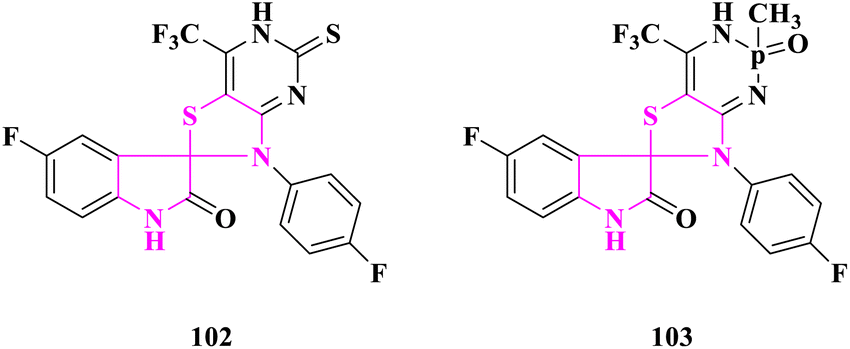
Novel sulfur and phosphorous fused fluorinated spiro[oxindole-thiazolidinone] derivatives were produced by T. E. Ali et al.92 and screened for antioxidant activity using the 1,1-diphenyl-2-picrylhydrazyl (DPPH) method. Ascorbic acid's radical scavenging activity was employed as a benchmark. At 150, 300, and 450 μmol L−1, all of the produced compounds were scavenged between 49 and 78 percent of the DPPH radicals. The interaction between the compounds under investigation and DPPH radicals caused the rise in percent DPPH inhibition. Furthermore, 5-fluoro-3′(4-fluorophenyl)-5′-thioxo-7′-(trifluoromethyl)-spiro{indole-3,2′-thiazolo[4′,5′-d]pyrimidin}-2-one (102) and 5-fluoro-7′-(4-fluorophenyl)-4′-(trifluoromethyl)-2′-methyl-2′-oxido-1′,2′-dihydro-spiro{indole-3,6′-thiazolo[4′,5′-d][1,3,2]diazaphosphinin}-2-one (103) was revealed to have antioxidative activity in the 70–78 percent range. The antioxidant activity of thiazolopyrimidinethione or thiazolodiazaphosphorine was improved by the inclusion of a trifluoromethyl group and an oxindole moiety (Fig. 19).
 | ||
| Fig. 19 Spiro-thiazolidine moiety as an antioxidant. | ||
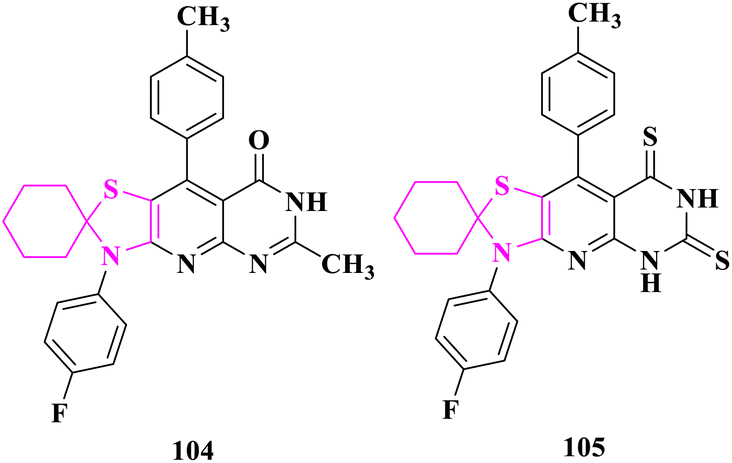
The antioxidant activity of the synthesized compounds was determined using the DPPH scavenging test by Flefel and co-researchers.90 At 50 and 100 μg mL−1, 3′-(4-fluorophenyl)-6′-methyl-9′-(p-tolyl)-3′H-spiro[cyclohexane-1,2′-thiazolo[5′,4′:5,6]pyrido[2,3-d]pyrimidin]-8′(7′H)-one (104) had the greatest antioxidant activity, ranging from 65 to 92.52 percent. At 100 μg mL−1, the activity of 3′-(4-fluorophenyl)-9′-(p-tolyl)-3′H-spiro[cyclohexane-1,2′-thiazolo[5′,4′:5,6]pyrido[2,3-d]pyrimidine]-6′,8′(5′H,7′H)-dithione (105) was more or less equal with 84.61%. The antioxidant activity of the studied substances, however, was lower than that of BHA, a synthetic antioxidant standard (93.6 percent at 100 μg mL−1), according to the data. The antioxidant activity of the promising compound (104) was related to the production of the pyrmidinone moiety's enol form, which was accountable for its antioxidant action. Under the impact of the prolonged conjugation, the resultant oxygen-centered radical from the DPPH radical quenching process was more stable in this enol form. Additionally, the inclusion of electron-donating groups like methyl, amino, and thialkyl groups contributes to compound 104′s higher antioxidant activity than the other compounds studied. Similarly, compound 105's antioxidant activity could be attributed to its thiol form; though, the less effective interaction between the resultant sulfur-centered radical and the p-cloud of aromatic rings elucidates compound 105's lesser antioxidant activity compared to compound 104 (Fig. 20).
 | ||
| Fig. 20 Spiro-thiazolidine derivatives as an antioxidant. | ||
Antitubercular activity
Newly synthesized 5-substituted-3-(5-(6-methyl-2-oxo/thioxo-4-phenyl-1,2,3,4-tetrahydropyrimidin-5-yl)-1,3,4-oxadiazol-2-yl)spiro[indoline-3,2′-thiazolidine]-2,4′-diones derivatives were selected for in vitro anti-mycobacterial activity against Mycobacterium tuberculosis (MTB) H37RV strain by the Lowenstein–Jensen (L–J) MIC technique by M. A. Borad and the team.32 Isoniazid was employed as a control medication with a MIC of 0.20 μg mL−1. In comparison to other compounds, compound 3′-(5-(6-methyl-2-oxo-4-phenyl-1,2,3,4-tetrahydropyrimidin-5-yl)-1,3,4-oxadiazol-2-yl)-5-nitrospiro [indoline-3,2′-thiazolidine]-2,40-dione (106) showed greater activity against the H37RV strain (12.5 μg mL−1), which was close to the MIC value of isoniazid. All derivatives were docked into the same pocket to study their binding energy to an inhibitor of enoyl-acyl carrier protein reductase (InhA). Despite displaying one breach of Lipinski's criterion, compound 106 bonds with the maximum energy. The calculated ADMET parameters showed that the pharmacokinetic qualities were good. According to the results of molecular docking, compound 106 might be employed as a template for the development of novel anti-tuberculosis drugs (Fig. 21).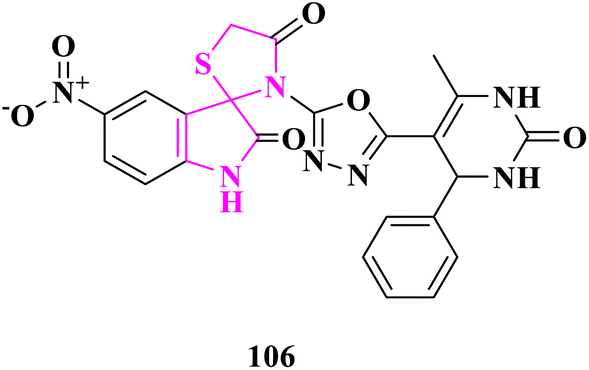 | ||
| Fig. 21 Spiro-thiazolidine derivative as an antimycobacterial agent. | ||
Conclusions
The current review has highlighted the tremendous impact of the green, effective and eco-benign synthetic strategies for the generation of bioactive spiro-thiazolidines via several steps, different reactants, catalysts, and various conditions. Spiro-thiazolidines have been and will continue to be the cherished synthetic targets of heterocycles due to their high potential importance as pharmaceuticals and materials. The protocols for their synthesis are highly demanding and will remain to be an important endeavor in the years to come. In this review, we have concentrated on the most recent advancements in spiro-thiazolidine synthesis under varied conditions, such as microwave-assisted synthesis, ionic liquid-assisted synthesis, solid-supported acid-catalyzed synthesis, on-water synthesis, nanocatalyst-assisted synthesis, and asymmetric synthesis, developed over the last 8 years.The antiviral, antifungal, antibacterial, diuretic, antihistaminic, tuberculostatic, anticancer, anticonvulsant, analgesic, and anti-inflammatory actions were significantly influenced by the heterocyclic framework of these compounds. As a result, we hope that this review will assist researchers in developing further into the many aspects of this subject matter to uncover hidden prospects and serve as a roadmap for the development of many more unique, innovative, and environmentally friendly synthetic techniques. These synthetic techniques will continue to gain in popularity, with a wide range of applications in chemical synthesis, pharmacology, and medicine. The goal of this review is to emphasize the importance of spiro-thiazolidines in synthetic, pharmaceutical, and other disciplines, making them a useful topic in organic chemistry.
Author contributions
Himanshu Sharma: conceptualization, reviewing, evaluation and supervision; Surbhi Dhadda: conceptualization, reviewing, and evaluation; Shaily Sharma, Prakash Jakhar: literature collection, evaluation, and manuscript preparation.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors are grateful to the Department of Chemistry, M.L.S.U. Udaipur (Raj.) and Department of Chemistry, F.B.A.S., Vivekananda Global University, Jagatpura, Jaipur, (Raj.) for providing necessary laboratory and library facilities. Prakash is grateful to CSIR for providing necessary funding.References
- E. Vitaku, D. T. Smith and J. T. Njardarson, J. Med. Chem., 2014, 57, 10257–10274 CrossRef CAS PubMed.
- R. D. Taylor, M. MacCoss and A. D. G. Lawson, J. Med. Chem., 2014, 57, 5845–5859 CrossRef CAS PubMed.
- J. E. Downer and P. Synke, J. Chem. Soc., 1960, 12, 963–965 RSC.
- A. A. Zagade and G. P. Senthilkumar, Der Pharma Chem., 2011, 3, 523–537 CAS.
- Y. S. Prabhakar, V. R. Solomon, M. K. Gupta and S. B. Katti, QSAR Top. Heterocycl. Chem., 2006, 4, 161–249 CAS.
- H. Soloway, F. Kipnis, J. Ornfelt and P. E. Spoerri, J. Am. Chem. Soc., 1948, 70, 1667–1668 CrossRef CAS.
- M. A. Gouda and A. A. Abu-Hashem, Arch. Pharm., 2011, 344, 170–177 CrossRef CAS PubMed.
- S. Nishida, H. Maruoka, Y. Yoshimura, T. Goto, R. Tomita, E. Masumoto, F. Okabe, K. Yamagata and T. Fujioka, J. Heterocycl. Chem., 2012, 49, 303–309 CrossRef CAS.
- Q. Zhang, H. Zhou, S. Zhai and B. Yan, Curr. Pharm. Des., 2010, 16, 1826–1842 CrossRef CAS PubMed.
- D. Havrylyuk, B. Zimenkovsky, O. Vasylenko and R. Lesyk, J. Heterocycl. Chem., 2013, 50, E55–E62 CrossRef CAS.
- D. Osmaniye, S. Levent, C. M. Ardıç, Ö. Atlı, Y. Özkay and Z. A. Kaplancıklı, Sulfur Silicon Relat. Elem., 2018, 193, 249–256 CrossRef CAS.
- M. S. A. El-Gaby, Z. H. Ismail, S. M. Abdel-Gawad, H. M. Aly and M. M. Ghorab, Sulfur Silicon Relat. Elem., 2009, 184, 2645–2654 CrossRef CAS.
- R. P. Singh, M. N. Aziz, D. Gout, W. Fayad, M. A. El-Manawaty and C. J. Lovely, Bioorg. Med. Chem., 2019, 27, 115047, DOI:10.1016/j.bmc.2019.115047.
- E. S. Raper, Coord. Chem. Rev., 1985, 61, 115–184 CrossRef CAS.
- G. R. Form, E. S. Raper and T. C. Downie, Acta Crystallogr., Sect. B: Struct. Crystallogr. Cryst. Chem., 1975, B31, 2181, DOI:10.1107/S0567740875007212.
- X. N. Zhang, X. Dong, M. Wei and M. Shi, Tetrahedron, 2014, 70, 2838–2846 CrossRef CAS.
- V. Padmavati, K. Sudheer, A. Muralikrishna and A. Padmaja, Indian J. Chem., 2015, 54B, 283–289 Search PubMed.
- P. Saraswat, G. Jeyabalan, M. Z. Hassan, M. U. Rahman and N. K. Nyola, Synth. Commun., 2016, 46, 1643–1664 CrossRef CAS.
- R. Pradhan, M. Patra, A. K. Behera, B. K. Mishra and R. K. Behera, Tetrahedron, 2006, 62, 779–828 CrossRef CAS.
- K. Ding, Z. Han and Z. Wang, Chem. - Asian J., 2009, 4, 32–41 CrossRef CAS PubMed.
- M. Sannigrahi, Tetrahedron, 1999, 55, 9007–9071 CrossRef CAS.
- S. Kotha, A. C. Deb, K. Lahiri and E. Manivannan, Synthesis, 2009, 2, 165–193 CrossRef.
- R. Rios, Chem. Soc. Rev., 2012, 41, 1060–1074 RSC.
- S. C. Jain, J. Sinha, S. Bhagat, W. Errington and C. E. Olsen, Synth. Commun., 2003, 33, 563–577 CrossRef CAS.
- R. Ottanà, R. Maccari, M. L. Barreca, G. Bruno, A. Rotondo, A. Rossi, G. Chiricosta, R. D. Paola, L. Sautebin, S. Cuzzocrea and M. G. Vigorita, Bioorg. Med. Chem., 2005, 13, 4243–4252 CrossRef PubMed.
- C. V. Kavitha, S. Basappa, N. Swamy, K. Mantelingu, S. Doreswamy, M. A. Sridhar, J. S. Prasad and S. Rangappa, Bioorg. Med. Chem., 2006, 14, 2290–2299 CrossRef CAS PubMed.
- G. Küçükgüzel, A. Kocatepe, E. De Clercq, F. Şahin and M. Güllüce, Eur. J. Med. Chem., 2006, 41, 353–359 CrossRef PubMed.
- Y. X. Song and D. M. Du, Org. Biomol. Chem., 2020, 18, 6018–6041 RSC.
- M. Suchý, P. Kutschy, K. Monde, H. Goto, N. Harada, M. Takasugi, M. Dzurilla and E. Balentová, J. Org. Chem., 2001, 66, 3940–3947 CrossRef PubMed.
- M. A. Borad, M. N. Bhoi, J. A. Parmar and J. A. Patel, Int. Lett. Chem., Phys. Astron., 2015, 53, 122–129 Search PubMed.
- P. N. Shinde and M. A. Raskar, Int. J. Curr. Pharm. Res., 2019, 11, 71–74 CrossRef CAS.
- M. A. Borad, M. N. Bhoi, S. K. Rathwa, M. S. Vasava, H. D. Patel, C. N. Patel, H. A. Pandya, E. A. Pithawala and J. J. Georrge, Interdiscip. Sci.: Comput. Life Sci., 2018, 10, 411–418 CrossRef CAS PubMed.
- S. Kotha, G. Sreevani, L. U. Dzhemileva, M. M. Yunusbaeva, U. M. Dzhemilev and V. A. D’yakonov, Beilstein J. Org. Chem., 2019, 15, 2774–2781 CrossRef CAS PubMed.
- P. Wasserscheid and T. Welton, Ionic Liquids in Synthesis, Wiley-VCH Verlag, 2008 Search PubMed.
- J. Ranke, S. Stolte, R. Störmann, J. Arning and B. Jastorff, Chem. Rev., 2007, 107, 2183–2206 CrossRef CAS PubMed.
- N. V. Plechkova and K. R. Seddon, Chem. Soc. Rev., 2008, 37, 123–150 RSC.
- T. L. Greaves and C. J. Drummond, Chem. Rev., 2008, 108, 206–237 CrossRef CAS PubMed.
- M. J. Earle, P. B. McCormac and K. R. Seddon, Chem. Commun., 1998, 20, 2245–2246 RSC.
- F. Liu, M. B. Abrams, R. T. Baker and W. Tumas, Chem. Commun., 2001, 5, 433–434 RSC.
- E. D. Bates, R. D. Mayton, I. Ntai and J. H. Davis, J. Am. Chem. Soc., 2002, 124, 926–927 CrossRef CAS PubMed.
- K. Arya, D. S. Rawat, A. Dandia and H. Sasai, J. Fluorine Chem., 2012, 137, 117–122 CrossRef CAS.
- E. M. Hussein and S. A. Ahmed, Chem. Heterocycl. Compd., 2017, 53, 1148–1155 CrossRef CAS.
- C. J. Li and L. Chen, Chem. Soc. Rev., 2006, 35, 68–82 RSC.
- S. Chitra, N. Paul, S. Muthusubramanian and P. A. Manisankar, Green Chem., 2011, 13, 2777–2785 RSC.
- P. Klumphu and B. H. Lipshutz, J. Org. Chem., 2014, 79, 888–900 CrossRef CAS PubMed.
- J. H. Clark, Nat. Chem., 2009, 1, 12–13 CrossRef CAS PubMed.
- K. Eskandari, B. Karami and S. Khodabakhshi, J. Chem. Res., 2014, 38, 600–603 CrossRef CAS.
- R. Singh and S. A. Ganaie, Res. Chem. Intermed., 2017, 43, 45–55 CrossRef CAS.
- A. Preetam and M. Nath, Tetrahedron Lett., 2016, 57, 1502–1506 CrossRef CAS.
- W. C. Chan, A. Khademhosseini, H. Möhwald, W. J. Parak, J. F. Miller, A. Ozcan and P. S. Weiss, ACS Nano, 2017, 11, 3423–3424 CrossRef CAS PubMed.
- A. K. Hussein, Renewable Sustainable Energy Rev., 2016, 62, 767–792 CrossRef.
- J. E. Contreras, E. A. Rodriguez and J. Taha-Tijerinac, Electr. Power Syst. Res., 2017, 143, 573–584 CrossRef.
- R. Sadeghi, R. J. Rodriguez, Y. Yao and J. L. Kokini, Annu. Rev. Food Sci. Technol., 2017, 8, 467–492 CrossRef PubMed.
- T. A. Saleh and I. M. Abdullahi, Nanotechnology in Oil and Gas Industries, DOI:10.1007/978-3-319-60630-9_3.
- A. M. Abdalla, S. Hossain, A. T. Azad, P. M. Petra, F. Begum, S. G. Eriksson and A. K. Azad, Renewable Sustainable Energy Rev., 2018, 82, 353–368 CrossRef CAS.
- N. C. Seeman and H. F. Sleiman, Nat. Rev. Mater., 2017, 3, 1–23 Search PubMed.
- K. E. Bujold, A. Lacroix and H. F. Sleiman, Chem, 2018, 4, 495–521 CAS.
- P. Chidchob and H. F. Sleima, Curr. Opin. Biotechnol., 2018, 46, 63–70 CrossRef CAS PubMed.
- A. P. Ramos, M. A. Cruz, C. B. Tovani and P. Ciancaglini, Biophys. Rev., 2017, 9, 79–89 CrossRef CAS PubMed.
- J. H. Myung, K. A. Tam, S. J. Park, A. Cha and S. Hong, Interdiscip. Rev.: Nanomed. Nanobiotechnol., 2016, 8, 223–239 Search PubMed.
- A. Chen and S. Chatterjee, Chem. Soc. Rev., 2013, 42, 5425–5438 RSC.
- Q. Zhang, Z. Wu, N. Li, Y. Pu, B. Wang, T. Zhang and J. Tao, Mater. Sci. Eng., C, 2017, 77, 1363–1375 CrossRef CAS PubMed.
- O. Y. Podyacheva and Z. R. Ismagilov, Catal. Today, 2015, 249, 12–22 CrossRef CAS.
- M. B. Gawande, A. Goswami, T. Asefa, H. Guo, A. V. Biradar, D. L. Peng, R. Zboril and R. S. Varma, Chem. Soc. Rev., 2015, 44, 7540–7590 RSC.
- N. Sharma, H. Ojha, A. Bharadwaj, D. P. Pathak and R. K. Sharma, RSC Adv., 2015, 5, 53381–53403 RSC.
- A. Chen and C. Ostrom, Chem. Rev., 2015, 115, 11999–12044 CrossRef CAS PubMed.
- K. Shen, X. Chen, J. Chen and Y. Li, ACS Catal., 2016, 6, 5887–5903 CrossRef CAS.
- M. B. Gawande, A. Goswami, F. X. Felpin, T. Asefa, X. Huang, R. Silva, X. Zou, R. Zboril and R. S. Varma, Chem. Rev., 2016, 116, 3722–3811 CrossRef CAS PubMed.
- S. Navalón and H. García, Nanomaterials, 2016, 6, 123–125 CrossRef PubMed.
- A. Zuliani, F. Ivars and R. Luque, ChemCatChem, 2018, 10, 1968–1981 CrossRef CAS.
- Z. Tang and H. Zhao, Chin. J. Catal., 2017, 38, 949–950 CrossRef CAS.
- V. Polshettiwar and R. S. Varma, Green Chem., 2010, 12, 743–754 RSC.
- J. Li, M. Wang, Y. Zhang, Z. Fan, W. Zhang, F. Sun and N. Ma, ACS Sustainable Chem. Eng., 2016, 4, 3189–3195 CrossRef CAS.
- R. Singh, S. A. Ganaie, A. Singh and A. Chaudhary, Synth. Commun., 2019, 49, 80–93 CrossRef CAS.
- M. Akhavan and A. Bekhradnia, RSC Adv., 2021, 11, 14755–14768 RSC.
- Y. X. Song and D. M. Du, Org. Biomol. Chem., 2018, 16, 9390–9401 RSC.
- S. Nallamala, S. Mannem and R. Raghavachary, SynOpen, 2017, 1, 0063–0067 CrossRef CAS.
- Y. Takazawa, T. Yamamoto, M. Suzuki and T. Sakata, Heterocycles, 2018, 96, 2087–2095 CrossRef CAS.
- K. R. Raju, G. P. Raghavendra, K. B. Santosh and L. K. Ravindranath, J. Chem. Technol. Metall., 2014, 49, 238–246 CAS.
- J. Sindhu, H. Singh and J. M. Khurana, Mol. Diversity, 2014, 18, 345–355 CrossRef CAS.
- E. M. Hussein, G. S. Masaret and K. S. Khairou, Chem. Cent. J., 2015, 9, 1–10 CrossRef CAS PubMed.
- S. Ponnuchamy, R. V. Sumesh and R. R. Kumar, Tetrahedron Lett., 2015, 56, 4374–4376 CrossRef CAS.
- P. N. Patel and Y. S. Patel, Cogent Chem., 2015, 1, 1048558 CrossRef.
- N. G. Kandile, M. I. Mohamed and H. M. Ismaeel, J. Enzyme Inhib. Med. Chem., 2017, 32, 119–129 CrossRef CAS PubMed.
- A. N. Al-Romaizan, Int. J. Org. Chem., 2020, 10, 77–87 CrossRef CAS.
- M. A. EL-Hashash, S. S. Shaban and R. S. Ali, J. Heterocycl. Chem., 2021, 58, 329–339 CrossRef CAS.
- A. Barakat, S. M. Soliman, A. M. Al-Majid, M. Ali, M. S. Islam, Y. A. Elshaier and H. A. Ghabbour, J. Mol. Struct., 2018, 1152, 101–114 CrossRef CAS.
- E. M. Flefel, W. I. El-Sofany, H. M. Awad and M. El-Shahat, Med. Chem., 2020, 20, 152–160 CAS.
- E. M. Flefel, W. I. El-Sofany, R. A. Al-Harbi and M. El-Shahat, Molecules, 2019, 24, 2511–2529 CrossRef PubMed.
- E. M. Flefel, H. H. Sayed, A. I. Hashem, E. A. Shalaby, W. El-Sofany and F. M. Abdel-Megeid, Med. Chem. Res., 2014, 23, 2515–2527 CrossRef CAS.
- M. N. Meeran and A. Z. Hussain, Indian J. Pharm. Sci., 2017, 79, 641–645 CAS.
- T. E. Ali and R. M. Abdel-Rahman, J. Sulfur Chem., 2014, 35, 399–411 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2023 |