
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceBiodegradable smart materials with self-healing and shape memory function for wound healing†
Siqin Suna,
Chaoxian Chen*ab,
Jianghong Zhanga and
Jianshe Hu *a
*a
aDepartment of Chemistry, College of Science, Northeastern University, Shenyang 110819, P. R. China. E-mail: hujs@mail.neu.edu.cn
bDepartment of Material Science and Engineering, Key Laboratory of Polymer Chemistry and Physics of Ministry of Education, Peking University, Beijing 100871, P. R. China. E-mail: Chenchaoxian1001818@pku.edu.cn
First published on 23rd January 2023
Abstract
Notwithstanding the rapid development of suture elastomers to meet the needs of practical surgery, utilizing the elastomers' self-healing function as a surgical suture to facilitate the healing of wounds has not been addressed. Here, a biodegradable aliphatic polycarbonate smart elastomer, mPEG113-b-PMBCn, was synthesized from aliphatic polycarbonate monomer with methoxy polyethylene glycol (mPEG113, 5.0 kDa) as initiator, which exhibited excellent mechanical properties, highly efficient self-repairing, and remarkable shape memory behavior. The polymers possess outstanding self-healing ability for 150 min. Meanwhile, after 46.33 ± 1.18 s, the temporary shape of the obtained polymer had been recovered. The results of biocompatibility tests reveal that the polymers have excellent biocompatibility and can be regarded as good biomedical materials. Then, in vivo experiments were used to prove the self-healing knotting ability of the polymers and quickly close a wound surface using a programmed shape at physiological temperature. The results demonstrated that the injury wound can be effectively healed compared with traditional sutures, which will offer new study suggestions for subsequent healing areas.
1. Introduction
In surgery, healing behavior can be promoted by exact wound closure, which can reduce the suffering of patients.1,2 Surgical sutures can facilitate wound healing. However, the skill of knotting presents particular challenges for doctors. Especially, the necrosis of surrounding tissues occurs when the knotting force is too large, which leads to the injury wound being susceptible to risk of secondary infection.3,4 Suture polymers have been used to facilitate the needs of practical surgery; however, exploiting the polymers' self-healing function, appropriate mechanical strength, knot security, and little inflammatory response as a surgical suture to enhance wound healing efficiency has not been reported.Shape-memory polymers (SMPs) have excellent potential because they can respond to heat, electric fields, light, magnetic fields, etc., promoting the possible applications in many fields such as intelligent sutures, actuators, and so on.5–13 In other words, the network's movement can promote the restoration of permanent shapes of SMPs, and thus they are regarded as smart materials.14–17 Undoubtedly, SMPs have been manufactured as surgical sutures to facilitate wound healing.13,16,17
Furthermore, polymeric materials' self-healing ability has been used in wound healing, body sensors,18–21 and other fields.21–25 With the advancement of ever-increasing stimuli-responsive actuation elastomers, actuating polymers have been embraced in many fields.21–23,26 Intrinsic self-healing elastomers are manufactured via non-covalent bonds. These bonds, including hydrogen bonding,22,27–29 hydrophobic interactions,28–31 polymer entanglements,27,32 and others,10,33 can be facilitated to synthesize self-healing elastomers with good mechanical properties. The variation of such non-covalent interactions can be used to adjust the self-healing behavior of materials.10,34–36 Many researchers are devoted to designing smart materials, promoting their excellent properties for use in biomedical applications.5,22,24,26,37,38 For instance, Biswas et al.37 prepared polyurethane nanohybrid based on in situ polymerization. The polymers have a 95.0% shape recovery ability, and the healing time of wounds is 9 days, but the technology of self-healing knotting was not reported. Zeng et al.38 synthesized shape-memory polymeric materials that indicated a shape recovery ratio of ∼95.0% and tensile fracture stress of 27.2 MPa. After being processed into a spiral-like stent through the shape memory function of the materials, the blended polymers showed an efficient self-expansion within 20 s at 37 °C.
However, if the polymers were used in biomedical applications, their mechanical strength, self-healing, and shape memory function are widely challenged and contradictory.5,14,22–24,26,37,38 Until now, none of the literature reports have addressed the above issues when polymers are applied to wound healing.
Here, we report a suggestion for self-healing knotting for wound healing to synthesize and fabricate surgical sutures. The polymers were formed from 2-methyl-2-benzyloxycarbonyl propylene carbonate (MBC)39,40 with Sn(Oct)2 and methoxy polyethylene glycol (mPEG113, 5.0 kDa) as initiators based on ring-opening polymerization. The obtained surgical sutures can properly close a wound edge under temperature induction and have good self-healing knotting ability at room temperature for self-tightening sutures in key-hole surgery. The mechanical properties, self-repair efficiency, shape memory, biocompatibility, and degradation properties of the surgical sutures were investigated thoroughly. The experimental results demonstrated that the prepared novel sutures have potential as excellent biomaterials for wound healing.
2. Results and discussion
2.1 Design of the elastomer and mechanical and self-repairing properties
In our work, the aliphatic polycarbonate network is synthesized by the bulk ring-opening polymerization of MBC using mPEG113 as an initiator (Fig. 1(B)). The resultant elastomers were named P1–P3. Synergistic interactions between hydrophobic interactions and hydrogen bonds of the network will occur, which lead to excellent properties, including appropriate mechanical properties, self-repairing, and shape memory of the network structure (see Experimental section, ESI† for more details). As shown in Fig. 1(E), in the NMR spectrum of monomer MBC, the signal at δ = 7.45–7.28 corresponds to unsaturated hydrogen in the benzene ring; the methylene group directly connected to the benzene ring has a single peak at δ = 5.20 because there is no hydrogen on its adjacent carbon; signals at δ = 4.66 and δ = 4.21 correspond to the methylene on the carbonate ring, corresponding to the vertical bond and the equatorial bond, respectively, thus appearing in the NMR spectrum in two positions and will affect each other and split into double peaks. Furthermore, δ = 1.28 corresponds to the methyl signal, which presents a single peak because there is no hydrogen on the adjacent carbon. The 1H NMR spectrum indicated the typical signal peak of the initiator mPEG113 at 3.64 ppm. After polymerization, the hydrophobic association was formed between the benzene ring groups on the side chain of the polymer. The signal of unsaturated hydrogen on the benzene ring and the signal of the methylene group directly connected to the benzene ring of MBC disappeared, becoming a broadband peak, which may be due to the formation of intermolecular/intramolecular hydrophobic associative interactions formed by the benzene ring groups after cross-linking, thereby shielding their chemical signals. In addition, both the methylene signal and the methyl group signal of MBC were weakened, which was due to the presence of terminal hydrogen bonding interactions in the cross-linked network. Under the action of the electrostatic field, the density of the electron cloud around the proton decreases, so that the precession of the extranuclear electrons of the hydrogen atom is further bound by the hydrogen bond, and the paramagnetic shielding and deshielding effects increase, resulting in a deshielding effect. | ||
| Fig. 1 (A) SMP suture with self-healing property for the interface of a wound. (B) Schematic process of the obtained SMPs. (C) Schematic of the construction of the SMPs based on aliphatic polycarbonate. (D) The network structure of the obtained SMPs. (E) The 1H NMR spectrum of the monomer MBC, initiator, and elastomer P2. | ||
The FTIR spectra of the obtained SMPs are shown in Fig. 2(a). After polymerization, the hydroxyl associations have been established at 3400–3000 cm−1. Meanwhile, the peaks of the carbon–carbon double bond, methyl, and methylene C–H stretching vibration are reinforced. Accordingly, the monomer unit has been successfully ring-opened. Furthermore, the thermal transitions of the obtained SMPs were investigated (Fig. 2(b)). The results demonstrated that the glass transition temperature (Tg) of the SMPs based on aliphatic polycarbonate was increased with a decrease of the catalyst, and the Tg values of P1, P2, and P3 are 17.30 °C, 17.46 °C, and 17.55 °C. The hard and soft domains of the obtained SMPs were enhanced to reinforce the physical cross-linked density of the elastomer network,8 indicating that Tg of the obtained SMPs can be slightly enhanced. Meanwhile, the thermogravimetric analysis of the SMPs shows that they have good stability up to temperatures as high as 300 °C. Importantly, compared with those for wound healing reported previously,38,41 the mechanical strength of the obtained SMPs is comparable (Fig. 2(d)).
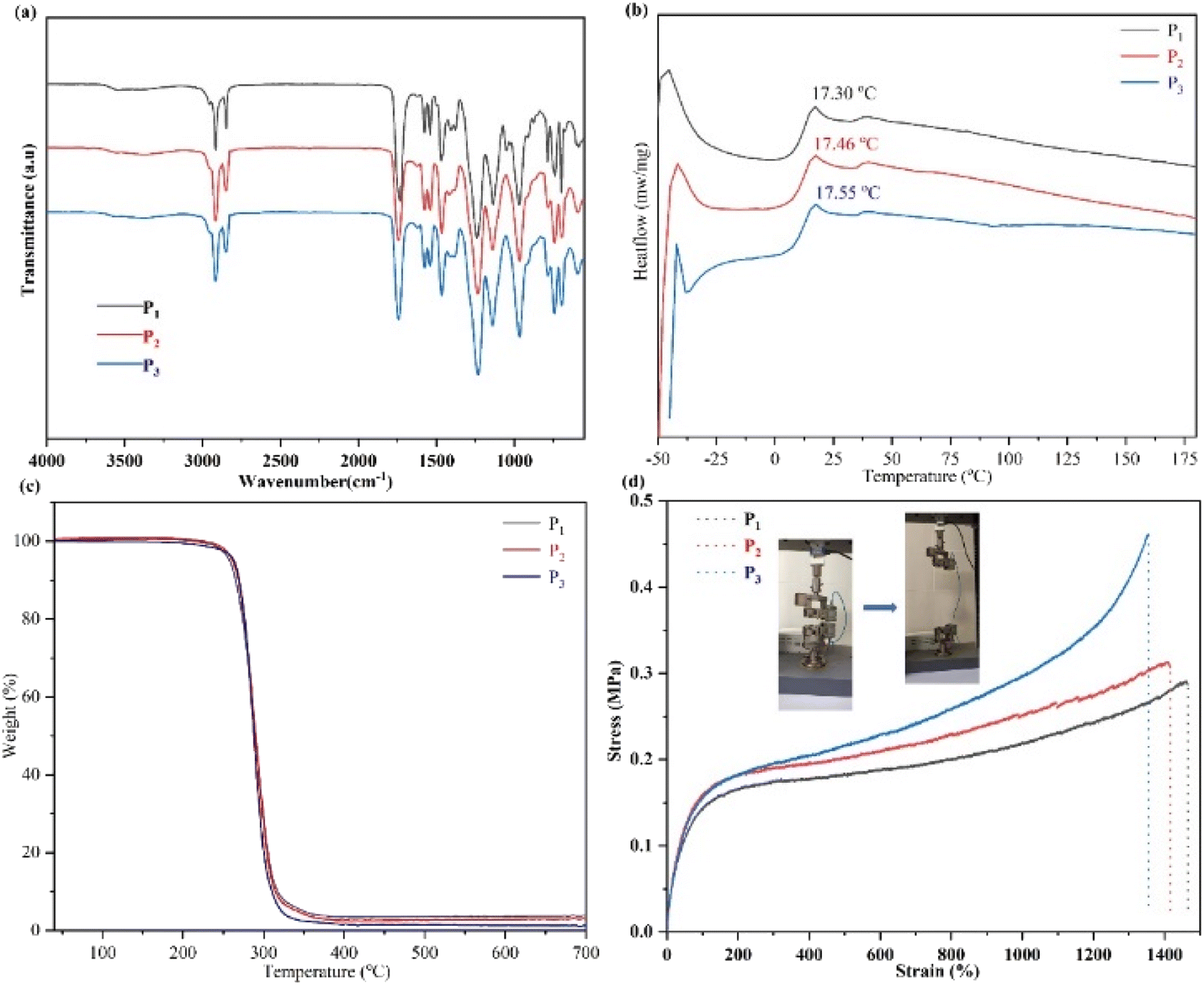 | ||
| Fig. 2 (a) FTIR spectra of the SMPs. Note: the chemical structure of the monomer is reported in our previous work.8 (b and c) The thermal behavior of the SMPs: (b) DSC curves of the obtained SMPs; (c) TGA curves of the obtained SMPs. (d) Curves of mechanical properties of the SMPs. | ||
The mechanical properties of SMP P3 were determined. Owing to the physical cross-linking interactions of the network structure, the mechanical properties of P3 are dependent on frequency (Fig. S1(a and b)†). As the temperature increases, the moduli (G′ and G′′) of P3 decreased (Fig. S1(b)†), demonstrating that physical cross-linked interactions were established, and the polymer has potentially self-healing ability.
Fig. 3(a) shows curves of the tensile strength of P2. The results indicated that the mechanical properties of polymer P2 were notably increased with enhanced speeds, indicating that the network was cross-linked by physical interactions.8,22,23 Then, the mechanical strength curves of the self-healing behavior of P3 are shown in Fig. 3(b). After 120 min, the self-healing efficiency of P2 is 98.88%, suggesting that the polymers possess excellent self-healing ability duo to the physical cross-linked interactions in the network. In addition, Fig. 3(c) shows images of the self-healing of P1 and P3. After 60 min, almost complete regeneration of the surface is achieved, revealing that the broken non-covalent bonds in the network can be quickly reconnected. Furthermore, the self-healing study was completed at 25 °C, higher than Tg of the elastomer, which will promote the movement capability of the polymer segments to further facilitate the self-healing of the polymers. Importantly, the synthesized polymers' self-healing behavior can be greatly reinforced by the catalyst content, and the self-healing time of P3 is 120 min. Comparing with the self-healing time of P1, with a decrease of the catalyst content, the self-healing ability of the SMPs increase, which is due to the increase of cross-linking density.8 Therefore, the self-repairing ability of the obtained SMPs had been significantly enhanced, promoting the high efficiency of self-repairing of the SMPs.
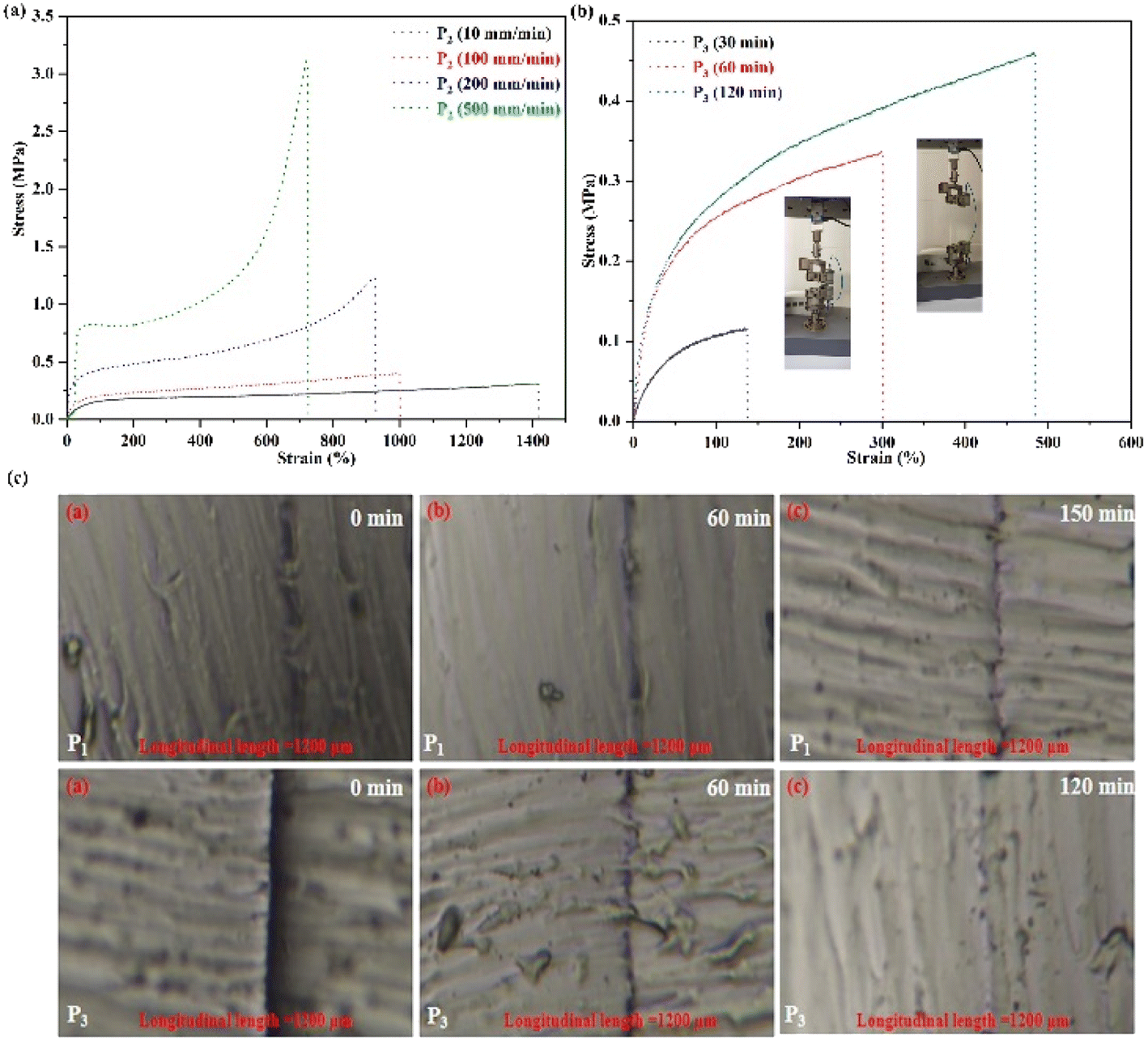 | ||
| Fig. 3 (a) Curves of mechanical strength of P2. (b) Mechanical strength curves of self-healing behavior of P3. (c) Images of self-healing of the SMPs. | ||
To further investigate the relationship between healing efficiency and temperature for the SMPs, the self-healing abilities of the SMPs were explored at high temperature (Fig. S2†). After 130 min and 110 min at 37 °C, P1 and P3 can be fully healed, respectively. Then, with increased temperature, the self-healing ability of the SMPs can be notably enhanced, and the full healing time is only 20 min and 10 min at 50 °C for P1 and P3, respectively. Therefore, the self-healing efficiency of the obtained SMPs can be notably increased by temperature. Meanwhile, the polymer movement of the SMPs can be driven, which will promote the re-formation of the hydrogen bonds and hydrophobic associative interactions of the network with variation of the temperature.
2.2 Shape memory behaviors
As demonstrated above, the obtained SMPs possess controllable mechanical properties and high-efficiency self-healing ability. Next, the function of shape memory needs to be investigated for the obtained SMPs. As shown in Fig. 4, the stored deformation energy can be fixed when the synthesized polymer is immersed in 7 °C water, which will limit the mobility of the polymer's chain (Video S1†) and afford programmed temporary shapes. Then, the mobility of the polymer's chain and fixed stored deformation energy can be stimulated by increased temperature. This is attributed to the view that enough toughness of the polymers when the polymers were input below Tg may restore the deformation energy and fix any deformed state of the polymers. Meanwhile, the temporary shape of the polymer can be recovered to its original shape under the driving force of the synergistic effect. Furthermore, after 46.33 ± 1.18 s, the temporary shape of the elastomer can be restored (Fig. 4(a), Video S1 and S2†). Fig. 4(d) shows the shape changes of P2 as a response to body temperature, demonstrating that the structural changes of the polymer had been induced by enhanced temperature, and the subsequent biomedical applications can be realized by thermal responsiveness of the polymer. | ||
| Fig. 4 (a) The shape recovery ratio of elastomer P2. (b) The shape recovery ratio of the obtained sample P2 within three cycles. (c) Rr and Rf' curves of polymers P1–P3. (d) The shape changes of P2 as a response to body temperature. | ||
2.3 In Vitro enzymatic degradation tests
The degradation behavior of an elastomer can be examined in terms of the mass loss of the polymer.37,42,43 As shown in Fig. 5, the polymer's degradation behavior was investigated by degradation tests in vitro. The results revealed that a good linear relationship of mass loss of the polymer can be obtained, shown in Fig. 5(a). This is because the aliphatic polycarbonate has good biocompatibility and no acidic degradation products resulting from its degradation.43 Therefore, the degradation behavior of the polymer was studied, indicating that the pH value of the polymer can remain at a value of ∼7.0 and does not widely reduce during the degradation time (Fig. 5(b)), which enables the materials to be applied in biomedical fields. Furthermore, Fig. 5(c–e) shows SEM images (×5.0 K, ×10.0 K) of the polymer before and after degradation. The degradation holes of the polymer can be obviously observed, and the interface of the polymer became wrinkled and rough after degradation.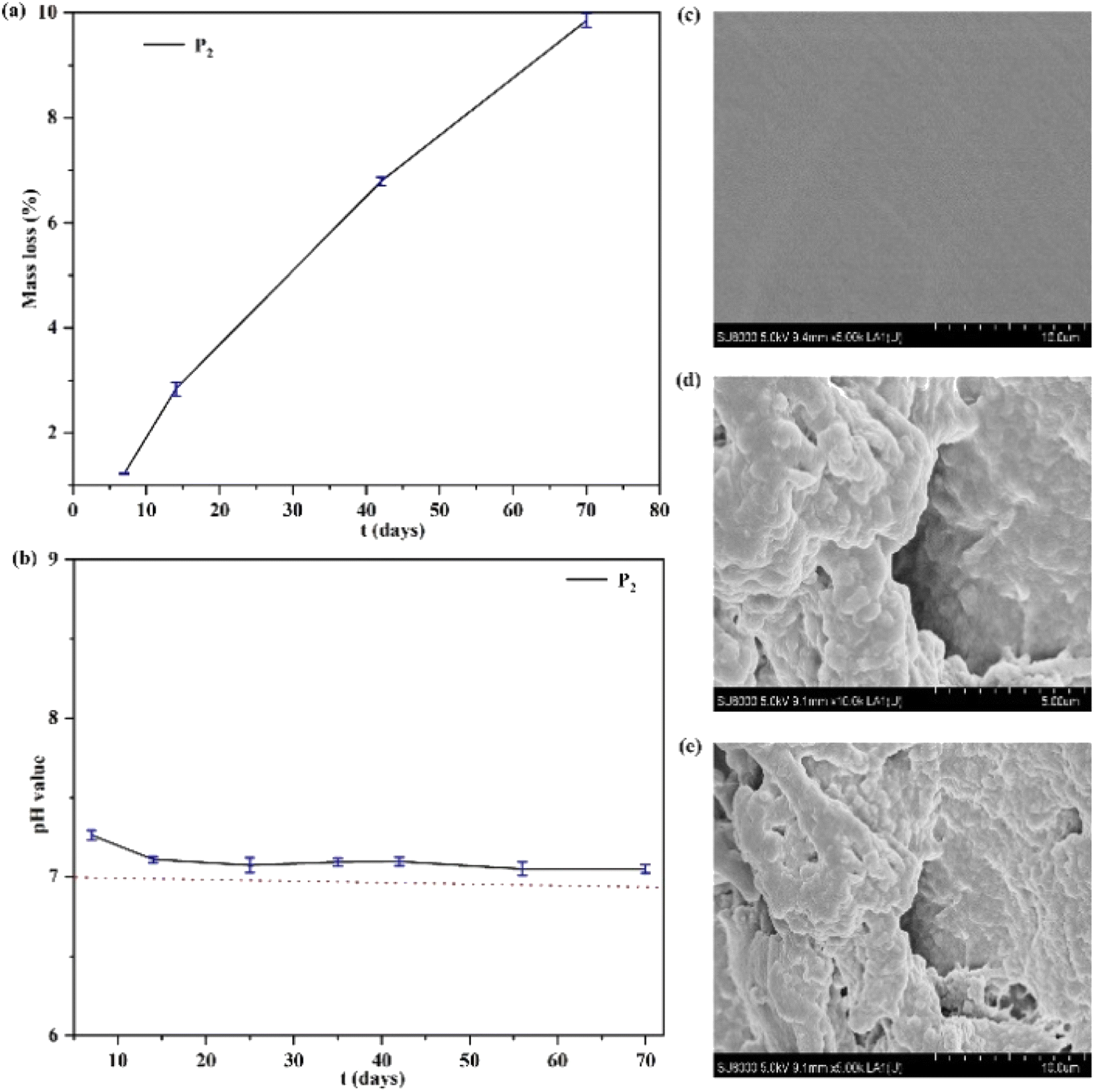 | ||
| Fig. 5 (a) The mass loss curve of polymer P2 with increasing number of days. (b) The pH curve of polymer P2 with increasing number of days. (c, d and e) SEM images (×5.0 K, ×10.0 K) of polymer P2 before and after degradation at 37 °C. | ||
The mechanical strengths of the obtained P3 changed after 8 days of degradation as shown in Fig. S3,† demonstrating that the mechanical strengths of the obtained P3 were reduced with an increase of the degradation time. The stress value of P3 was 0.41 MPa and 0.33 MPa after 2 and 8 days of degradation, respectively. This is attributed to the view that after degradation, the surface of SMPs becomes wrinkled and rough, and the physical cross-linked interactions of SMPs are weakened, which will lead to a decrease in mechanical strength.
2.4 Biocompatibility behavior
The biocompatibility tests of the elastomer can be conducted using HUVECs in vitro. As shown in Fig. 6(a), after culturing for 3 days in vitro, compared with the optical density (OD) values reported by Vancso,42 the OD values of the obtained SMPs were approximate, showing that the obtained SMPs exhibit excellent cell proliferation in our work. Then, the blood compatibility of the polymer can be tested in terms of the hemolysis ratio obtained between RBCs and the polymer. As shown in Fig. 6(b), the appearance of hemolysis was not observed after the tests, and the hemolysis ratio was 0.28%, far below the ASTM standard, proving that the polymer possesses good RBC compatibility.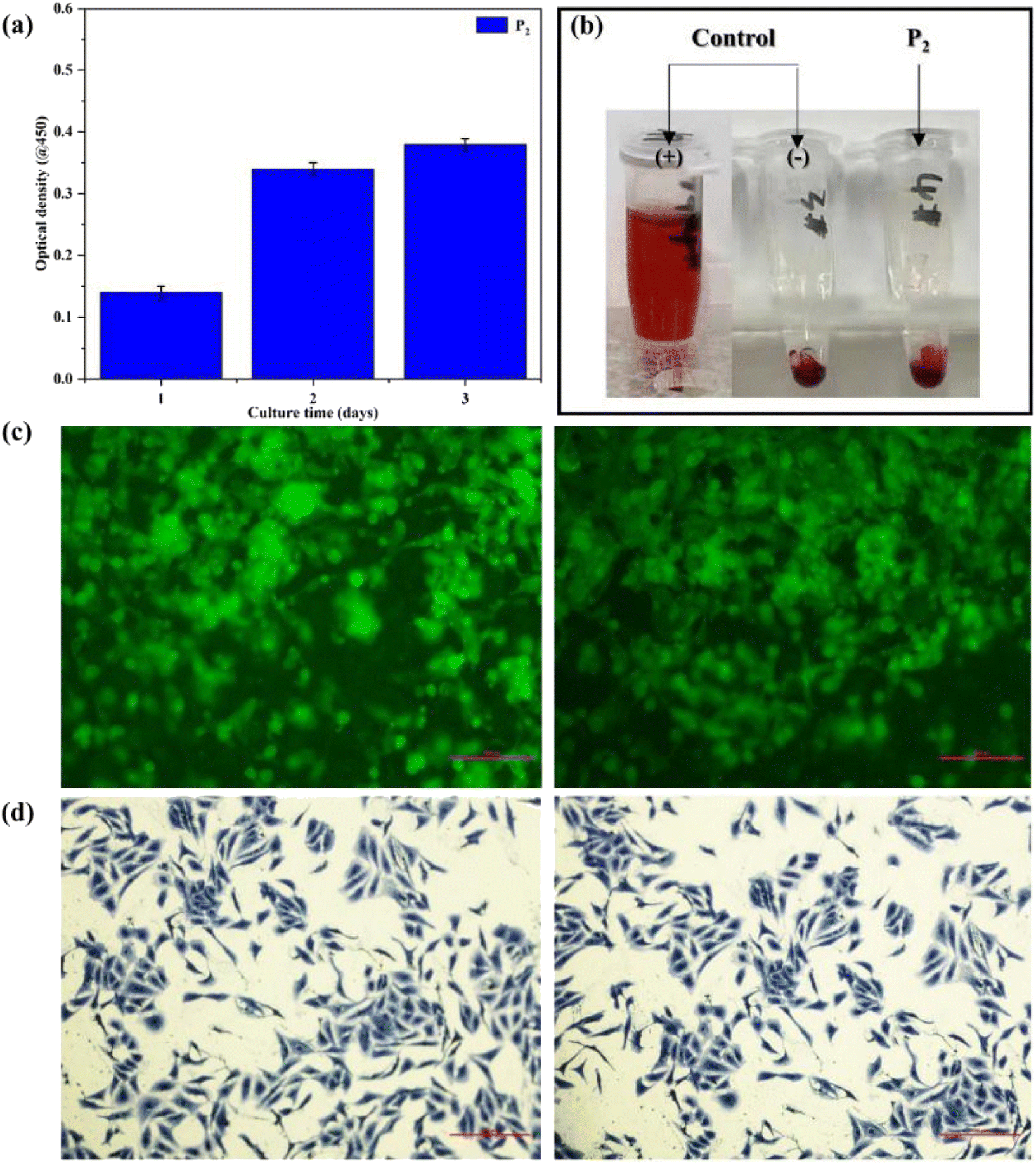 | ||
| Fig. 6 (a) The OD values of the polymer. (b) Results of hemolysis tests of the polymer. (c) Results of confocal tests of the polymer. (d) Clone-forming of HUVECs. | ||
Furthermore, the cell viability of the polymer was examined by confocal and clone-forming tests, the results being shown in Fig. 6(c and d). The tests revealed that HUVECs can induce the formation of an elliptical structure and cellular clusters (Fig. 6(c)). Meanwhile, the cell viability of the sample with HUVEC lines was enhanced notably after clone-forming, indicating the reinforced proliferation behavior of cells (Fig. 6(d)). Consequently, the polymer exhibits good cell affinity to afford opportunities in smart biomedical applications.
2.5 SMPs for wound healing applications
In vivo, a test of self-tightening of sutures was used to evaluate the wound healing of the elastomers with self-healing and shape memory function. The SMPs' suture and the technique of self-healing knotting were used to investigate the conditions of wound healing based on a mouse skin suture-wound model. As shown in Fig. 7(A), the shape of the polymer can be fixed below Tg, resulting in the deformation energy of SMPs being stored to promote wound healing when the mice used in the experiment are injured. Meanwhile, the postoperative knotting technique was executed by the self-repairing function of the polymer for the first time (Fig. 7A(f–h)). A standard surgical needle was used to suture at 2 mm away from the wound edge (Fig. 7A(e) and 7B(a)). The process of wound healing is shown in Fig. 7B. The wounds (length 2 cm and width 2 mm) can be healed by heat treatment with increased time, and the wounds were completely closed after 5 days due to the excellent shape recovery function of the polymer (Fig. 7B and Video S3†) compared with a traditional suture. Importantly, the knotting problem is still not solved by all SMP sutures reported.3,4,35,37 However, the need for wound healing can be satisfied by only one knot after suturing. This is attributed to the view that the polymer's self-repairing behavior can be induced automatically by the physical cross-linking of the network in the knotting process.8 Furthermore, after complete healing, scar formation was not produced, suggesting that the self-healing function of the obtained SMPs is effective for closing the wound, which is most important for wound healing. The histology of the suture neighboring tissue was studied by H&E staining examination, seen in Fig. S4.† The results reveal that an inflammatory phenomenon was not observed obviously after 5 days of wound healing compared with the traditional suture, showing the beneficial anti-inflammatory ability of the polymer. | ||
| Fig. 7 (A) The process diagrams of SMPs surgical sutures in vivo animal experiment; (B) heat irradiation suture for wound closure: images showing the wound healing process after tightening of the wound lips as a function of time in days at room temperature. | ||
3. Conclusions
In summary, we have successfully designed a smart elastomer. The elastomer with good biocompatibility embraces controllable mechanical, high-efficiency self-repairing, and excellent shape memory properties. The elastomer possesses an excellent wound-closing ability, and the injury wound can be efficiently healed by thermal treatment. Significantly, compared with previously reported studies, the knotting problem of sutures was improved by the polymer's self-repairing function for the first time, and the wound-healing efficiency can be reinforced notably. The results of this study are expected to promote the development of the wound healing field.Ethical statement
All animal procedures were performed in accordance with the Guidelines for Care and Use of Laboratory Animals of Northeastern University and experiments were approved by the Guidelines of the Liaoning Administrative Committee on Animal Research. The volunteer declares that she knows and agrees to participate in the work of this paper.Conflicts of interest
The authors declare no competing financial interest.Acknowledgements
This work was supported by the Fundamental Research Funds for the Central Universities (N2105005).References
- J. Huang, L. Chen and Q. Yuan, J. Biomed. Nanotechnol., 2019, 15, 1371–1383 CrossRef CAS PubMed.
- F. E. Muysoms, S. A. Antoniou and K. Bury, Hernia, 2015, 19, 1–24 CrossRef CAS PubMed.
- A. Lendlein, A. M. Schmidt and R. Langer, Proc. Natl. Acad. Sci. U. S. A., 2001, 98, 842–847 CAS.
- A. Lendlein and R. Langer, Science, 2002, 296, 1673 CrossRef PubMed.
- J. G. Hardy, M. Palma, S. J. Wind and M. J. Biggs, Adv. Mater., 2016, 28, 5717–5724 CrossRef CAS PubMed.
- Q. Zhao, H. J. Qi and T. Xie, Prog. Polym. Sci., 2015, 79, 49–50 Search PubMed.
- M. D. Hager, S. Bode, C. Weber and U. S. Schubert, Prog. Polym. Sci., 2015, 3, 49–50 Search PubMed.
- C. X. Chen, S. W. Chen, Z. H. Guo, W. R. Hu, Z. P. Chen, J. W. Wang, J. S. Hu, J. Guo and L. Q. Yang, J. Mater. Chem. A., 2020, 8, 16203–16211 RSC.
- T. Xie, Nature, 2010, 464, 267–270 CrossRef CAS PubMed.
- J. Liu, C. S. Tan, Z. Yu, N. Li, C. Abell and O. A. Scherman, Adv. Mater., 2017, 29, 1605325 CrossRef PubMed.
- H. Yang, W. R. Leow, T. Wang, J. Wang, J. Yu, K. He, D. Qi, C. Wan and X. Chen, Adv. Mater., 2017, 29, 1701627 CrossRef PubMed.
- C. Löwenberg, M. Balk, C. Wischke, M. Behl and A. Lendlein, Acc. Chem. Res., 2017, 50, 723–732 CrossRef PubMed.
- C. X. Chen, Z. Hou, S. Chen, J. Guo, Z. Chen, J. S. Hu and L. Q. Yang, Composites, Part B, 2022, 240, 109985 CrossRef CAS.
- A. Lendlein and S. Kelch, Angew. Chem., Int. Ed., 2002, 41, 2034–2057 CrossRef CAS.
- W. Wang, D. Shen, X. Li, Y. Yao, J. Lin, A. Wang, J. Yu, Z. L. Wang, S. W. Hong, Z. Lin and S. Lin, Angew. Chem., Int. Ed., 2018, 57, 2139–2143 CrossRef CAS PubMed.
- C. Liu, H. Qin and P. Mather, J. Mater. Chem. A., 2007, 17, 1543–1558 RSC.
- K. Hearon, M. A. Wierzbicki, L. D. Nash, T. L. Landsman, C. Laramy, A. T. Lonnecker, M. C. Gibbons, S. Ur, K. O. Cardinal, T. S. Wilson, K. L. Wooley and D. J. Maitland, Adv. Healthcare Mater., 2015, 4, 1386–1398 CrossRef CAS PubMed.
- Z. Chen, F. Fu, Y. Yu, H. Wang, Y. Shang and Y. Zhao, Adv. Mater., 2019, 31, 1805431 CrossRef PubMed.
- C. X. Chen, N. Duan, S. W. Chen, Z. H. Guo, J. S. Hu, J. Guo, Z. P. Chen and L. Q. Yang, J. Mol. Liq., 2020, 319, 114134 CrossRef CAS.
- J. Wu, L. H. Cai and D. A. Weitz, Adv. Mater., 2017, 29, 1702616 CrossRef PubMed.
- H. U. Rehman, Y. Chen, M. S. Hedenqvist, H. Li, W. Xue, Y. Guo, Y. Guo, H. Duan and H. Liu, Adv. Funct. Mater., 2018, 28, 1704109 CrossRef.
- Z. Wei, J. H. Yang, J. Zhou, F. Xu, M. Zrinyi, P. H. Dussault, Y. Osada and Y. M. Chen, Chem. Soc. Rev., 2014, 43, 8114–8131 RSC.
- R. Liang, H. Yu, L. Wang, L. Lin, N. Wang and K. U. R. Naveed, ACS Appl. Mater. Interfaces, 2019, 11, 43563–43572 CrossRef CAS PubMed.
- Y. Zheng, Y. Li, X. Hu, J. Shen and S. Guo, ACS Appl. Mater. Interfaces, 2017, 9, 13988–13998 CrossRef CAS PubMed.
- Y. Yanagisawa, Y. Nan, K. Okuro and T. Aida, Science, 2018, 359, 72–76 CrossRef CAS PubMed.
- J. Yang, J. Zhou, M. Zrinyi, Y. Osada and Y. M. Chen, Chem. Soc. Rev., 2014, 43, 8114–8131 RSC.
- S.-M. Kim, H. Jeon, S.-H. Shin, S.-A. Park, J. Jegal, S. Y. Hwang, D. X. Oh and J. Park, Adv. Mater., 2018, 30, 1705145 CrossRef PubMed.
- J. Hao and R. A. Weiss, Macromolecules, 2011, 44, 9390–9398 CrossRef CAS.
- H. Zhang, H. Xia and Y. Zhao, ACS Macro Lett., 2012, 1, 1233–1236 CrossRef CAS PubMed.
- Y. Yang, X. Wang, F. Yang, H. Shen and D. Wu, Adv. Mater., 2016, 28, 7178–7184 CrossRef CAS PubMed.
- Q. Chen, L. Zhu, H. Chen, H. Yan, L. Huang, J. Yang and J. Zheng, Adv. Funct. Mater., 2015, 25, 1598–1607 CrossRef CAS.
- D. Y. Wu, S. Meure and D. Solomon, Prog. Polym. Sci., 2008, 33, 479–522 CrossRef CAS.
- X. Chen, M. A. Dam, K. Ono, A. Mal, H. Shen, S. R. Nutt, K. Sheran and F. Wudl, Science, 2002, 295, 1698–1702 CrossRef CAS PubMed.
- J. Kang, D. Son, G. J. N. Wang, Y. Liu, J. Lopez, Y. Kim, J. Y. Oh, T. Katsumata, J. Mun, Y. Lee, L. H. Jin, J. B. Tok and Z. N. Bao, Adv. Mater., 2018, 30, 1706846 CrossRef PubMed.
- S. Yoshida, H. Ejima and N. Yoshie, Adv. Funct. Mater., 2017, 27, 1701670 CrossRef.
- L. Zhang, Z. Liu, X. Wu, Q. Guan, S. Chen, L. Sun, Y. F. Guo, S. L. Wang, J. C. Song, E. M. Jeffries, C. He, F. L. Qing, X. G. Bao and Z. W. You, Adv. Mater., 2019, 31, 1901402 CrossRef PubMed.
- A. Biswas, A. P. Singh, D. Rana, V. K. Aswal and P. Maiti, Nanoscale, 2018, 10, 9917 RSC.
- B. B. Zeng, Y. Li, L. S. Wang, Y. Zheng, J. B. Shen and S. Y. Guo, ACS Sustainable Chem. Eng., 2020, 8, 1538–1547 CrossRef CAS.
- C. X. Chen, Z. C. Li, S. W. Chen, J. S. Hu, Z. P. Chen and L. Q. Yang, J. Mol. Liq., 2021, 329, 115581 CrossRef CAS.
- Y. Yu, P. Li, C. Zhu, N. Ning, S. Zhang and G. J. Vancso, Adv. Funct. Mater., 2019, 29, 1904402 CrossRef.
- Y. Haramiishi, N. Chanthaset and K. Kan, Polym. Degrad. Stab., 2016, 130, 78–82 CrossRef CAS.
- Z. P. Hou, W. Zhang, J. Guo, Z. P. Chen, J. S. Hu and L. Q. Yang, Eur. Polym. J., 2019, 112, 51–59 CrossRef CAS.
- Positive control material introduced to ASTM Hemolysis Standard, Stand News., 2000, 28, pp. 9–10 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: The synthesis of the polymer; the structure, thermal behavior, mechanical properties, shape memory performance, enzymatic degradation, and biological recognition characterization of the obtained elastomers; in vivo studies; H&E stain testing of the self-healing of the SMPs (PDF). See DOI: https://doi.org/10.1039/d2ra07493a |
| This journal is © The Royal Society of Chemistry 2023 |