
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceStructural and photoelectron spectroscopic study on the heterotrinuclear nickel–titanium dioxide carbonyl complexes Ni2TiO2(CO)n− (n = 2–4)†
Shihu Duad,
Haiyan Han *a,
Yongliang Yanb,
Yantao Lvb,
Zhihui Fana,
Xiuhong Liua,
Xiaoqing Liangc,
Hua Xied,
Zhi Zhao*a and
Ruili Shi*a
*a,
Yongliang Yanb,
Yantao Lvb,
Zhihui Fana,
Xiuhong Liua,
Xiaoqing Liangc,
Hua Xied,
Zhi Zhao*a and
Ruili Shi*a
aSchool of Mathematics and Physics Science and Engineering, Hebei University of Engineering, Handan 056038, China. E-mail: hanhy0226@163.com; zhaozhi@hebeu.edu.cn; shiruili@hebeu.edu.cn
bSchool of Information and Electrical Engineering, Hebei University of Engineering, Handan 056038, China
cDepartment of Physics, Taizhou University, Taizhou 318000, China
dState Key Laboratory of Molecular Reaction Dynamics, Dalian Institute of Chemical Physics, Chinese Academy of Sciences, Dalian 116023, China
First published on 20th January 2023
Abstract
Herein, the configurations and intrinsic electronic properties of heteronuclear transition metal dioxide carbonyl anions Ni2TiO2(CO)n− (n = 2–4) in the gas phase were investigated using mass spectrometry coupled anionic photoelectron spectroscopy, ab initio calculations, and simulated density-of-state (DOS) spectra. The results clearly show that the binding of electrons is enhanced by the addition of CO. The ground state structures of Ni2TiO2(CO)n− (n = 2–4) are characterized to show that three transition metal atoms (one Ti atom and two Ni atoms) forming a quasi-line is favored. The interaction between Ni and C becomes weaker as the cluster size increases. The natural electron configuration shows that the extra electron is enriched on O atoms attached to Ti, and there is strong interaction between Ti and O atoms. This work gives significant insight into the configuration and electronic structures of nickel-titanium dioxide carbonyl anions, which has potential application in adsorption of carbon monoxide on the surfaces/interfaces of alloys.
1. Introduction
The interaction between carbon monoxide (CO) and transition metal (TM) containing metal compounds, and oxide complexes attracts lots of attention due to its great significance in catalytic chemistry, coordination chemistry, medicine, and material science.1–4 Transition metal carbonyls or transition metal oxide carbonyls could provide well-defined models of CO adsorption on the surfaces/interfaces of alloys or the binding at active sites of catalysts.5,6 For muti adsorption of carbon monoxide on a transition metal (oxide), great effort has been made to search the ground state structures both in experiment and theory.7–9 The CO molecules usually adsorb to the transition metal atoms to form side-on-bonded, bridging, or terminal modes.10,11 Meanwhile, the maximum carbonyl-coordination number of mononuclear metal has been extensively studied.12–16 The multinuclear transition metal carbonyl compounds have also gained increasing interest in the last few years.17–30Subsequently, the study on the reaction of metal oxide clusters with CO also gradually enriched. For the metal oxide carbonyls, the MO(CO)5+ (M = Sc, Y, La and Ce) is similar to a pentagonal pyramidal structure with C5V symmetry.31 The maximum carbonyl-coordination number of ScO(CO)n+, YO(CO)n+ and LaO(CO)n+ is 6, 7 and 9, respectively.32–34 On the side of heterobinuclear transition metal oxide carbonyls, the sequential adsorption of CO in NbNiO(CO)5–8− and TaNiO(CO)4–8− clusters can promote the competitive binding with oxygen atom to the transition metal centers.35,36 Many researches focus on the oxidation of CO when studying the interaction between CO and transition metal oxide (TMO) since CO oxidation is one of the most important prototypical reactions.35–39 The TM atom acts as a preferred trapping site for CO adsorption and accepts the electron from CO. Then the ligand CO could be oxidized by the oxygen species on the TMO cluster. Moreover, the CO could directly react with the oxygen atom in TMO.36 For heteronuclear TMO, experimental observations combined with quantum chemistry calculations confirm that there is oxidation of CO in ScOFe(CO)5−, YOFe(CO)5−,40 NbNiO(CO)7–8−,35 Ni2VO4,5CO−,41 and TaNiO(CO)8− complexes.36 While there is no oxidation of CO in LaOFe(CO)5− anion.40 Therefore, different ratios and types of TMO will affect the results of CO oxidation reaction.
The above-mentioned researches rarely involve the interaction between CO and heteronuclear transition metal dioxide. The geometry, electronic structures, and the containing of CO oxidation for the heteronuclear transition metal dioxide carbonyl complexes are unclear. In this work, using experimental PES and theoretical investigation, we report the structures and electronic properties of the heteronuclear transition metal dioxide carbonyl anions Ni2TiO2(CO)n− (n = 2–4) in gas phase. The anions are produced via a laser vaporization supersonic cluster source, selected by a dual channel mass spectrometer (D-TOFMS), and then analyzed using a velocity-map imaging spectrometer. Meanwhile, the structures, energetic and electronic properties are performed by ab initio calculations to further understand the experimental observation. The consistency of experimental and simulated results reveals that the three transition metal atoms form a quasi-line and there is no oxidation of CO in Ni2TiO2(CO)n− (n = 2–4) clusters.
2. Experimental and theoretical methods
2.1. Experiment
The photoelectron detachment experiments were carried out with a homemade device which included a laser vaporization source, a D-TOFMS and a velocity-map imaging spectrometer. A detailed description of the instrument has already been provided elsewhere,42 so what follows is merely an outline. Ni2TiO2(CO)n− (n = 2–4) clusters were fabricated by ablating a small Ti/Ni target (mole ratio, Ti/Ni = 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1) with a laser vaporization (532 nm) beam in the presence of helium carrier gas seeded with 5% CO. Stagnation of the carrier gas was approximately 4 atm. Cluster anions expanded into the source chamber after being cooled. Wiley–McLaren time-of-flight mass spectrometer was used to mass select these anionic clusters. For the detachment of these anionic clusters, photon energy of 355 nm (3.496 eV) was employed. The photoelectrons were mapped onto a detector consisting of a micro-channel plate and a phosphor screen. The two-dimensional (2D) images on the phosphor screen were recorded by a charge-coupled-device (CCD) camera. Each image was obtained by accumulating 10000–50000 laser shots at 10 Hz repetition rate. All of the raw images were reconstructed using the basis set expansion inverse Abel transform method (BASEX). The photoelectron spectra were calibrated using the known spectrum of Au−.43–45 The PES were plotted against electron binding energy eBE = hν − eKE, where hν is the photon energy. The typical energy resolution was about 50 meV full width at half maximum (FWHM) at electron kinetic energy (eKE) of 1 eV.
:1) with a laser vaporization (532 nm) beam in the presence of helium carrier gas seeded with 5% CO. Stagnation of the carrier gas was approximately 4 atm. Cluster anions expanded into the source chamber after being cooled. Wiley–McLaren time-of-flight mass spectrometer was used to mass select these anionic clusters. For the detachment of these anionic clusters, photon energy of 355 nm (3.496 eV) was employed. The photoelectrons were mapped onto a detector consisting of a micro-channel plate and a phosphor screen. The two-dimensional (2D) images on the phosphor screen were recorded by a charge-coupled-device (CCD) camera. Each image was obtained by accumulating 10000–50000 laser shots at 10 Hz repetition rate. All of the raw images were reconstructed using the basis set expansion inverse Abel transform method (BASEX). The photoelectron spectra were calibrated using the known spectrum of Au−.43–45 The PES were plotted against electron binding energy eBE = hν − eKE, where hν is the photon energy. The typical energy resolution was about 50 meV full width at half maximum (FWHM) at electron kinetic energy (eKE) of 1 eV.
2.2. Theory
We used BP86 functional, which has been verified to be outstanding for describing the transition metal carbonyl compounds in the previous works,35,46 and ma-TZVP basis set to elucidate the geometrical, energetic, and electronic properties of Ni2TiO2(CO)n− (n = 2–4) clusters. The BP86 functional combines Becke's 1988 exchange functional (B)47 with Perdew's 1986 gradient corrected correlation functional method (P86).48 The ma-TZVP basis set is a modification of the standard def2-TZVP basis set49 with diffuse functions for atoms heavier than He by dividing the smallest s and p exponential parameters already present by a factor of 3.50,51 Meanwhile, many possible spin multiplicities were considered, including doublets, quartet and sextet for Ni2TiO2(CO)n−, and singlet, triplet and quintet for neutral Ni2TiO2(CO)n, respectively. The results are shown in Tables S1 and S2 of the ESI.† The lowest spin state, i.e. doublets for Ni2TiO2(CO)n− and singlets for Ni2TiO2(CO)n are most stable. The structures were fully optimized without any symmetry constraint. Frequency analyses at the same level of theory with geometry optimizations were performed to confirm that each optimized structure is a true minimum on potential energy surface without imaginary frequency. The zero-point-energy (ZPE) corrections were considered in the total energy of each cluster isomer for relative energy and adiabatic detachment energy (ADE) calculations.The simulated photoelectron spectrum for a given isomer is based on the generalized Koopmans' theorem.52–55 The vertical detachment energy (VDE) was calculated as the difference in energy between the neutral and anionic species based on the optimized anionic geometry. The ADE was calculated as the difference in energy between the neutral and the anion both at the related optimized configurations. The calculated density of states was then globally shifted in order to align the binding energies of the HOMO with the theoretical VDE value.56,57 In order to better understand the electronic structure of the Ni2TiO2(CO)n− clusters, we calculated the natural electron configuration using natural bond orbital (NBO) analyses. All of the calculations were carried out with the Gaussian09 package.58
3. Results and discussion
3.1. Photoelectron spectra in experiment
The photoelectron images and the corresponding photoelectron spectra of Ni2TiO2(CO)n− (n = 2–4) recorded at 355 nm are shown in Fig. 1. Note that the experimental error bars are determined by our instrumental resolution. The main band in the spectrum of Ni2TiO2(CO)2− is obviously split into three peaks located at 2.44 ± 0.05, 2.65 ± 0.04 and 2.94 ± 0.03 eV, with a higher intensity in the third peak. The dominant peaks marked with the letter X of all three photoelectron spectra have a maximum band corresponding to the vertical detachment energy (VDE) values. The VDE values of the ground states for the Ni2TiO2(CO)n− (n = 2–4) complexes were directly determined to be 2.44 ± 0.05 eV, 2.76 ± 0.04 eV, and 2.86 ± 0.03 eV, respectively. The bands from the spectra without vibrational features prevent us from directly measuring the ground-state ADEs, which can be alternatively estimated by the intersection of a line drawn along the rising edge of the main band with the binding energy axis. The ADE values of the ground states for the Ni2TiO2(CO)n− (n = 2–4) complexes were evaluated to be 2.17 ± 0.07 eV, 2.43 ± 0.05 eV, and 2.60 ± 0.05 eV, respectively. With the increasing of cluster size, the VDE and ADE values increase, revealing that the electron is stabilized upon the bonding of CO molecules. Meanwhile, several peaks corresponding to the binding energies of non-highest occupied electron in the spectra could be distinguished, such as the major peaks centered at 2.65 ± 0.04, 2.94 ± 0.03 eV for Ni2TiO2(CO)2− and at 2.86 ± 0.03, 3.02 ± 0.02 eV for Ni2TiO2(CO)3−.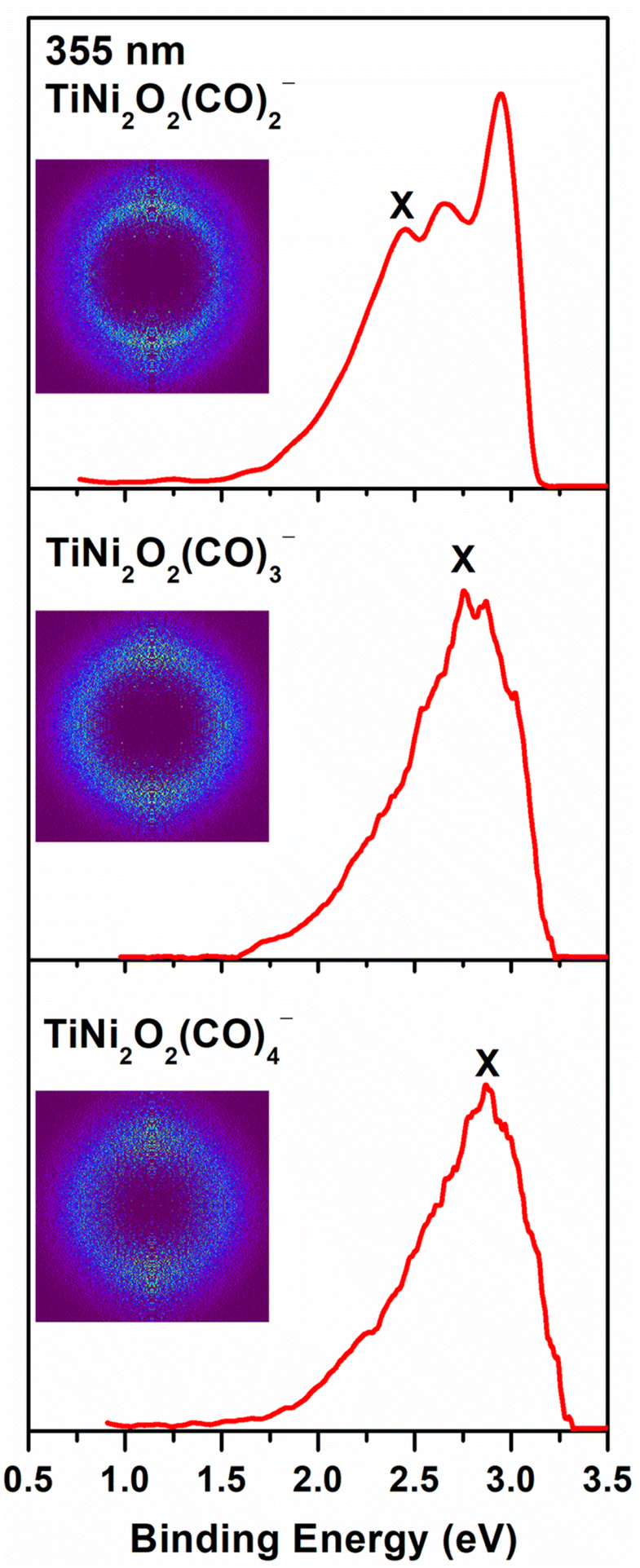 | ||
| Fig. 1 Photoelectron images of Ni2TiO2(CO)n− (n = 2–4) at 355 nm. Photoelectron images after inverse Abel transformation are embedded in the photoelectron spectra. | ||
3.2. Comparison between experimental and theoretical results
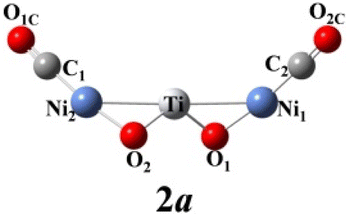
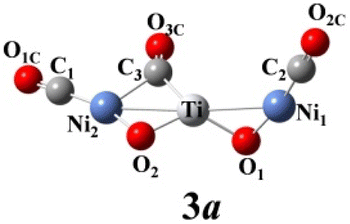
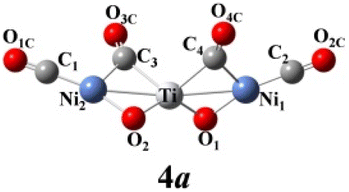
We use ab initio simulations to gain a clear insight into the geometric and electronic properties of Ni2TiO2(CO)n− (n = 2–4) clusters by comparing with the results of experiment. The structures, symmetries, and relative energies of the three lowest-energy structures for each size of Ni2TiO2(CO)n− (n = 2–4) clusters obtained from ab initio calculations are given in Fig. 2. The other isomers with higher energies of Ni2TiO2(CO)n− (n = 3, 4) clusters are listed in Fig. S1 of the ESI.† The lowest-energy structures are designated as na, and the metastable structures are designated as nb, nc, and so on. In all the structures, the three transition metal atoms (one Ti atom and two Ni atoms) form a quasi-line or triangular structure. The two oxygen atoms bond to the Ti atom to form a stoichiometric TiO2 core, which is different from the results of NbNiO(CO)5–8− and TaNiO(CO)4–8− with the competitive binding with oxygen atom to the transition metal centers.35,36 The oxidation of CO is not favored due to the strong interaction between Ti and O atoms. Unlike NbNiO(CO)7–8− or TaNiO(CO)8−,35,36 the oxidation of CO in Ni2TiO2(CO)n− (n = 2–4) clusters only occurs in some higher-energy structures such as 3e, 3f, 3g, 4i, 4j, and 4k with one of the oxygen atoms on Ti binding to the C. The comparison between the experimental photoelectron spectra of Ni2TiO2(CO)n− (n = 2–4) clusters recorded at 355 nm photons and the BP86 ones of the three lower-energy structures for each size are shown in Fig. 3–5. Table 1 lists the experimental and theoretical VDE and ADE values as well as the theoretical relative energies of the three lower-energy structures for each size of Ni2TiO2(CO)n− (n = 2–4) clusters. The experimental and theoretical VDE and ADE values as well as the theoretical relative energies of the higher energy structures of Ni2TiO2(CO)n− (n = 3, 4) clusters are given in Table S3 of the ESI.†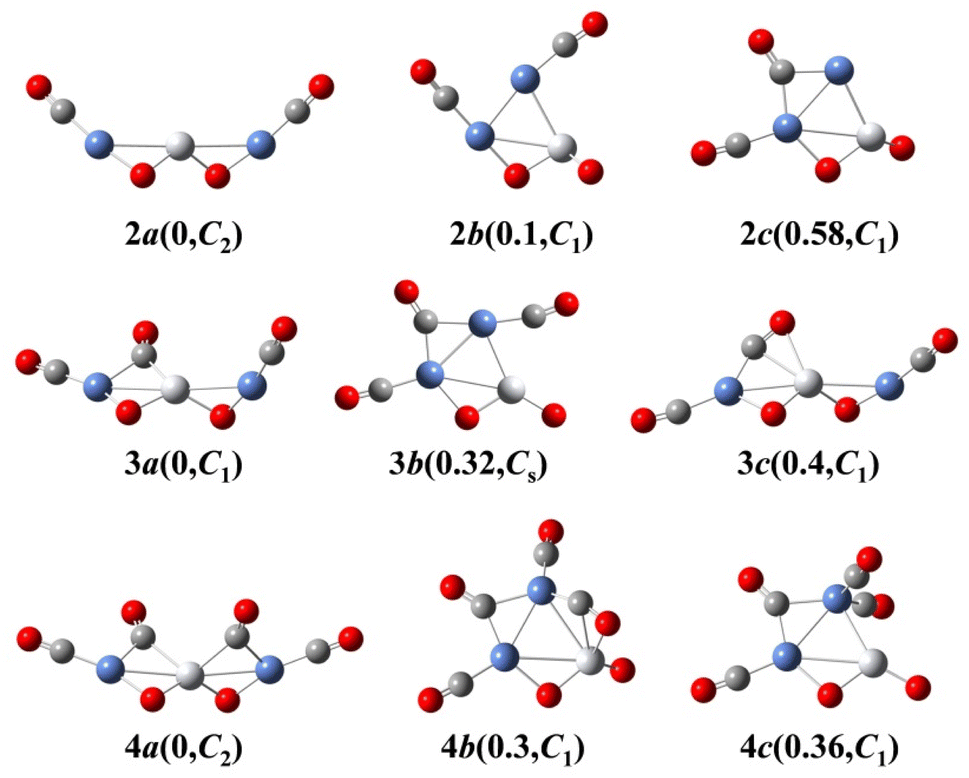 | ||
| Fig. 2 Schematic diagrams of the three lowest-energy structures for each size of Ni2TiO2(CO)n− (n = 2–4) clusters (Ti, white; Ni, blue; C, gray; O, red). The relative energy (in units of eV) and symmetry of each cluster are in parentheses. | ||
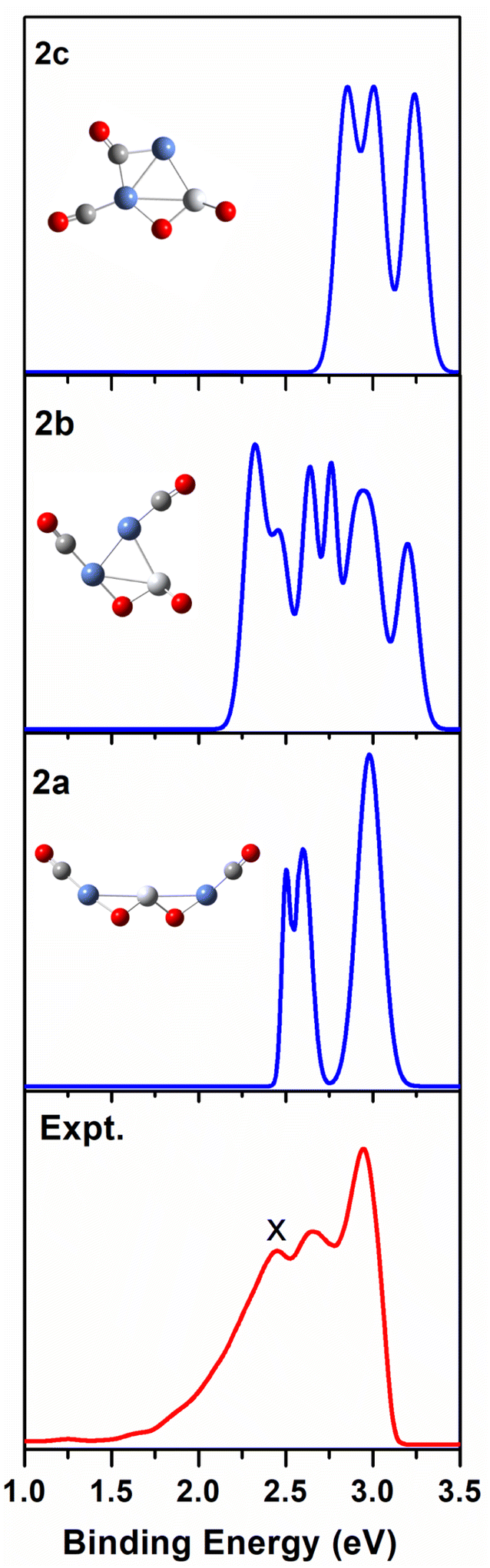 | ||
| Fig. 3 Comparison between the simulated photoelectron spectra of the three Ni2TiO2(CO)2− isomers 2a, 2b, 2c (blue line) and the experimental one at 355 nm (red line). | ||
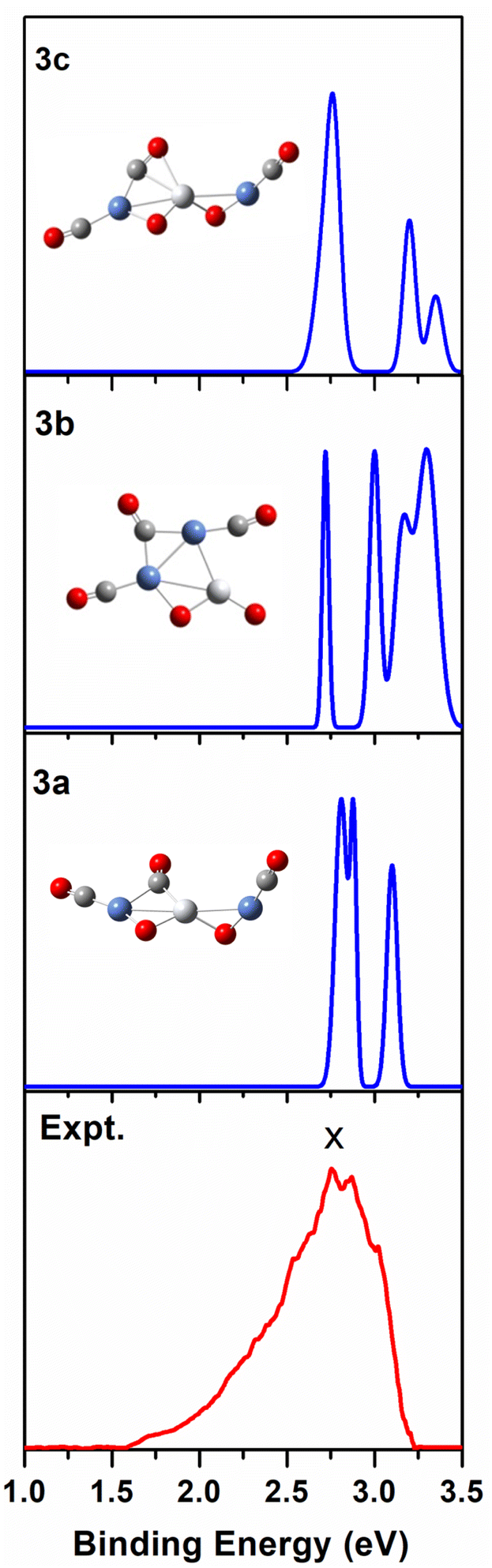 | ||
| Fig. 4 Comparison between the simulated photoelectron spectra of the three Ni2TiO2(CO)3− isomers 3a, 3b, 3c (blue line) and the experimental one at 355 nm (red line). | ||
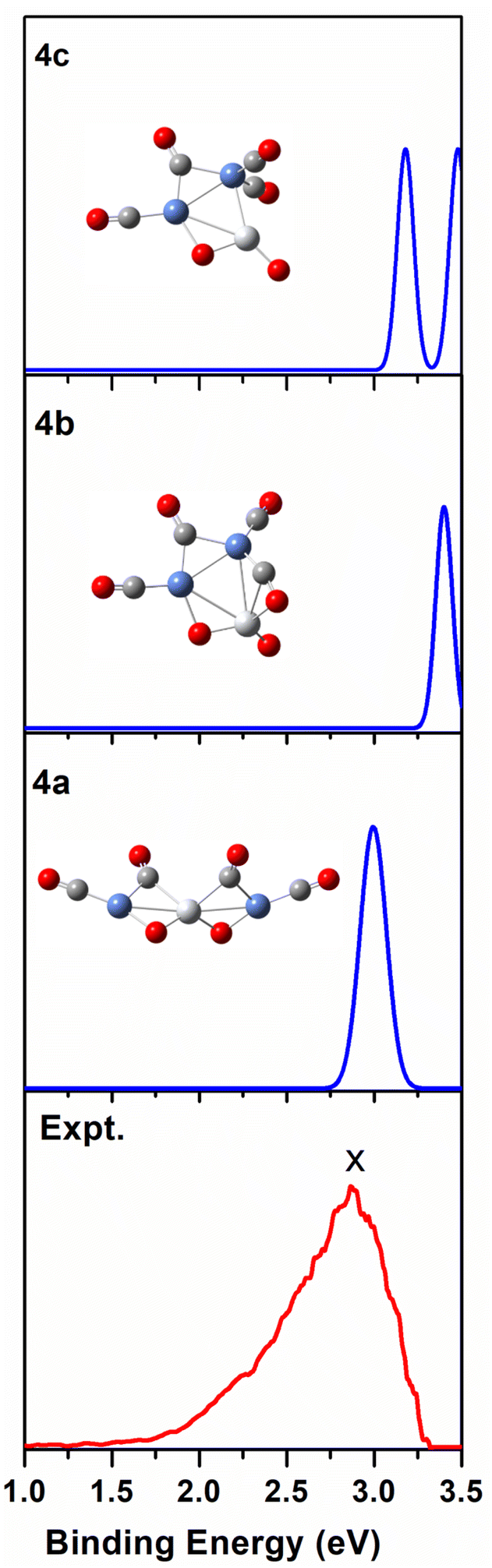 | ||
| Fig. 5 Comparison between the simulated photoelectron spectra of the three Ni2TiO2(CO)4− isomers 4a, 4b, 4c (blue line) and the experimental one at 355 nm (red line). | ||
| n | Isomers | ΔE (eV) | VDE (eV) | ADE (eV) | ||
|---|---|---|---|---|---|---|
| Theo. | Exp. | Theo. | Exp. | |||
| 2 | 2a | 0.00 | 2.52 | 2.44(5) | 2.27 | 2.17(7) |
| 2b | 0.10 | 2.32 | 2.13 | |||
| 2c | 0.58 | 2.85 | 1.81 | |||
| 3 | 3a | 0.00 | 2.81 | 2.76(4) | 2.54 | 2.43(5) |
| 3b | 0.32 | 2.71 | 2.38 | |||
| 3c | 0.40 | 2.73 | 2.40 | |||
| 4 | 4a | 0.00 | 2.98 | 2.86(3) | 2.72 | 2.60(5) |
| 4b | 0.30 | 3.40 | 2.89 | |||
| 4c | 0.36 | 3.18 | 2.86 | |||
As shown in Table 1, the calculated VDEs of both 2a (2.52 eV) and 2b (2.32 eV) are in reasonable agreement with the experimental one (2.44 ± 0.05 eV). The ADEs of both 2a (2.27 eV) and 2b (2.13 eV) are consistent with the experimental result (2.17 ± 0.07 eV). The simulated VDE of 2c is 2.85 eV, which is larger than the experimental result (2.44 ± 0.05 eV). While the ADE of 2c (1.81 eV) is smaller than the experimental one (2.17 ± 0.07 eV). In Fig. 3, the simulated spectrum of 2a has three discrete peaks at 2.52, 2.60, and 2.95 eV, what agrees very well with the experimental photoelectron spectrum (2.44 ± 0.05, 2.65 ± 0.04, 2.94 ± 0.04 eV). The peaks in the simulated spectrum of 2b are apparently more than the experimental result, leading to the conclusion that 2b should be excluded. Therefore, 2a cluster exists in the cluster beam in the experiment.
According to Table 1 and S3 of the ESI,† among the seven Ni2TiO2(CO)3− isomers, 3a, 3b, and 3c have both comparable VDE and ADE values to experimental ones. From Fig. 4 we can see that there are three peaks at 2.81, 2.88, and 3.1 eV in the simulated PES of 3a, which coincides with the experimental values (2.76 ± 0.04, 2.86 ± 0.03, 3.02 ± 0.02 eV) well. However, the number of the peaks (four peaks centered at 2.71, 3, 3.16, and 3.3 eV) in the simulated PES of 3b is larger than that in experiment. For 3c, except for the first peak at 2.73 eV in the simulated PES is comparable to the experimental result of 2.76 eV, the other two peaks at 3.19 and 3.32 eV cannot match the experimental values of 2.86 ± 0.03 and 3.02 ± 0.02 eV, respectively. This suggests that the 3a isomer should be responsible for the n = 3 spectrum.
It can be seen from Tables 1 and S3 of the ESI† that the VDE (2.98 eV) and ADE (2.72 eV) of 4a match the experimental VDE 2.86 ± 0.03 eV and ADE 2.60 ± 0.05 eV well, respectively. The VDEs of the meta-stable structures of Ni2TiO2(CO)4− clusters are larger than the experimental ones. In Fig. 5, there is only one main peak at 2.98 eV in the simulated PES of 4a, which is identical to the experimental feature. The peak position in the simulated spectrum of 4b deviated from the experimental one. Meanwhile, the peaks in the simulated spectrum of 4c are more than the experimental result. Therefore, 4a is the most probable structure detected in experiment.
3.3. Geometric and electronic properties of the lowest-energy structures
The above-mentioned comparison between experimental and theoretical results reveals that the lowest-energy structures of Ni2TiO2(CO)n− (n = 2–4) clusters are the ones with the three transition metal atoms forming a quasi-line. 2a has C2 symmetry. The two CO molecules added to the two Ni atoms form terminal ones. 3a is a structure with the third CO molecule located in the middle of Ti and Ni forming bridge coordination. The fourth CO molecule in 4a locates at the middle of Ti and Ni on the other side and 4a also has C2 symmetry. Table 2 lists several characteristic bond lengths and bond angles of the ground state structures of Ni2TiO2(CO)n− (n = 2–4) clusters. The average bond lengths of Ti–O, Ti–Ni, Ni–O, and the distance between Ni and Ni (5.295, 5.252, and 5.292 Å for 2a, 3a, and 4a, respectively) remain constant as the cluster size increases, meaning that the framework of Ni2TiO2 is stable in the lowest-energy structure of Ni2TiO2(CO)n− (n = 2–4) clusters. The average distances between Ni and C become longer as the cluster size increases. The angle of Ni–Ti–Ni decreases as the cluster size increasing. Thus, the interaction between Ni and C becomes weaker with the cluster size increasing.| n | 2 | 3 | 4 |
|---|---|---|---|
| Ti–O | 1.760 | 1.778 | 1.78 |
| Ni–C | 1.694 | 1.79 | 1.835 |
| Ti–Ni | 2.648 | 2.628 | 2.654 |
| Ni–O | 1.822 | 1.83 | 1.84 |
| ∠Ni–Ti–Ni | 178.01 | 175.6 | 171.1 |
The natural electron configuration of the lowest-energy structures of Ni2TiO2(CO)n− (n = 2–4) clusters is shown in Table 3. The valence electron configuration of the element Ti is 3d24s2, while its 4s orbital lose electrons as well as 3d, 4p and 4d orbitals gain extra electrons in the lowest-energy structures of Ni2TiO2(CO)n− (n = 2–4) clusters. The Ti atom in 2a, 3a, and 4a totally loses 1, 0.7, and 0.4 electrons, respectively. In the two Ni atoms of all the ground state structures, though the 4s orbital lose electrons and 3d and 4p orbitals gain extra electrons, the total electrons remain unchanged. The two O atoms bonded to Ti totally gain 1.6 electrons since the 2s orbital loses 0.2 electrons and the 2p orbital gains 1 electron for each O atom. The number of electrons gained by the oxygen atom is almost equal to the number of electrons lost by the C atom in one CO molecule. Thus, the CO molecules do not gain or lose electrons. In conclusion, the extra electron is enriched on O atoms attached to Ti. The Ti atom loses electrons and the O atoms attached to Ti gain electrons, indicating strong interaction between Ti and O. Meanwhile, the interaction between CO molecules and Ni2TiO2 unit is weak. All those lead to no CO oxidation in the lowest-energy structures of Ni2TiO2(CO)n− (n = 2–4) clusters.
| Valence electron | Natural electron configuration | |||
|---|---|---|---|---|
|
|
|
||
| Ti | 3d24s2 | 4s0.43d2.34p0.3 | 4s0.23d2.64p0.44d0.1 | 4s0.23d2.84p0.54d0.1 |
| Ni1 | 3d84s2 | 4s0.63d9.14p0.3 | 4s0.63d9.14p0.2 | 4s0.53d94p0.5 |
| Ni2 | 3d84s2 | 4s0.63d9.14p0.3 | 4s0.53d94p0.5 | 4s0.53d9 4p0.5 |
| O1 | 2s22p4 | 2s1.82p5 | 2s1.82p5. | 2s1.82p5 |
| O2 | 2s22p4 | 2s1.82p5 | 2s1.82p5 | 2s1.82p5 |
| C1 | 2s22p2 | 2s1.32p2.2 | 2s1.32p2.2 | 2s1.32p2.2 |
| O1C | 2s22p4 | 2s1.72p4.8 | 2s1.72p4.7 | 2s1.72p4.7 |
| C2 | 2s22p2 | 2s1.32p2.2 | 2s1.32p2.3 | 2s1.32p2.2 |
| O2C | 2s22p4 | 2s1.72p4.8 | 2s1.72p4.8 | 2s1.72p4.7 |
| C3 | 2s22p2 | — | 2s1.22p2.4 | 2s1.22p2.4 |
| O3C | 2s22p4 | — | 2s1.72p4.7 | 2s1.72p4.7 |
| C4 | 2s22p2 | — | — | 2s1.22p2.4 |
| O4C | 2s22p4 | — | — | 2s1.72p4.7 |
4. Conclusion
By combining photoelectron imaging spectroscopy and ab initio calculations, the low-lying isomers, structural evolution, relative stabilities, and electronic structures of Ni2TiO2(CO)n− (n = 2–4) clusters are investigated. The VDEs and ADEs become larger with the increase in cluster size, meaning that the binding of electrons is stronger as the cluster size increasing. In the lowest-energy structures of Ni2TiO2(CO)n− (n = 2–4) clusters, the three transition metal atoms (one Ti atom and two Ni atoms) form a quasi-line, which is different from triangular structure for tri-metallic active center. It provides more evidence for the reaction of multicomponent alloy with CO. The adding of CO molecules makes the interaction between Ni and C weaker. The natural electron configuration shows that the strong interaction between O and Ti and the weak interaction between CO molecules and Ni2TiO2 unit lead to the absence of CO oxidation. Our results provide new light on the geometric and electronic structures of nickel–titanium dioxide carbonyl anions and will have important implications on the CO adsorbed on the surfaces/interfaces of alloy.Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
This work was supported by the National Natural Science Foundation of China (No. 21976049, 12004094, 12004095, 11904251), the Natural Science Foundation of Hebei Province (No. B2021402006, F2019402063), the Scientific and Technological Research in Higher Education Institutions of Hebei Province (No. BJK2022057, BJK2023041), and the Handan Science and Technology Bureau (No. 21422901248).References
- Y. Ishii and M. Tsutsui, Organotransition-Metal Chemistry, Science, 1975, 188, 1224–1225 CrossRef CAS PubMed.
- B. Yoon, H. Häkkinen and U. Landman, et al., Charging Effects on Bonding and Catalyzed Oxidation of CO on Au8 Clusters on MgO, Science, 2005, 307, 403–407 CrossRef CAS PubMed.
- D. K. Böhme and H. Schwarz, Gas-Phase Catalysis by Atomic and Cluster Metal Ions: The Ultimate Single-Site Catalysts, Angew. Chem., Int. Ed., 2005, 44, 2336–2354 CrossRef PubMed.
- H.-J. Freund, G. Meijer and M. Scheffler, et al., CO Oxidation as a Prototypical Reaction for Heterogeneous Processes, Angew. Chem., Int. Ed., 2011, 50, 10064–10094 CrossRef CAS PubMed.
- M. Zhou, L. Andrews and C. W. Bauschlicher, Spectroscopic and Theoretical Investigations of Vibrational Frequencies in Binary Unsaturated Transition-Metal Carbonyl Cations, Neutrals, and Anions, Chem. Rev., 2001, 101, 1931–1962 CrossRef CAS PubMed.
- A. Fielicke, P. Gruene and G. Meijer, et al., The adsorption of CO on transition metal clusters: A case study of cluster surface chemistry, Surf. Sci., 2009, 603, 1427–1433 CrossRef CAS.
- H. Schwarz and K. R. Asmis, Identification of Active Sites and Structural Characterization of Reactive Ionic Intermediates by Cryogenic Ion Trap Vibrational Spectroscopy, Chem.–Eur. J., 2019, 25, 2112–2126 CrossRef CAS PubMed.
- J.-B. Ma, Z.-C. Wang and M. Schlangen, et al., On the Origin of the Surprisingly Sluggish Redox Reaction of the N2O/CO Couple Mediated by [Y2O2]+. and [YAlO2]+. Cluster Ions in the Gas Phase, Angew. Chem., Int. Ed., 2013, 52, 1226–1230 CrossRef CAS PubMed.
- J. Zou, H. Xie and Q. Yuan, et al., Probing the bonding of CO to heteronuclear group 4 metal–nickel clusters by photoelectron spectroscopy, Phys. Chem. Chem. Phys., 2017, 19, 9790–9797 RSC.
- L. Jiang and Q. Xu, Observation of Anomalous C–O Bond Weakening on Discandium and Activation Process to CO Dissociation, J. Am. Chem. Soc., 2005, 127, 42–43 CrossRef CAS PubMed.
- Q. Xu, L. Jiang and N. Tsumori, cyclo-Ti3[η2(μ2-C,O)]3: A Side-on-Bonded Polycarbonyl Titanium Cluster with Potentially Antiaromatic Character, Angew. Chem., Int. Ed., 2005, 44, 4338–4342 CrossRef CAS PubMed.
- A. M. Ricks, Z. E. Reed and M. A. Duncan, Infrared spectroscopy of mass-selected metal carbonyl cations, J. Mol. Spectrosc., 2011, 266, 63–74 CrossRef CAS.
- A. D. Brathwaite, J. A. Maner and M. A. Duncan, Testing the Limits of the 18-Electron Rule: The Gas-Phase Carbonyls of Sc+ and Y+, Inorg. Chem., 2014, 53, 1166–1169 CrossRef CAS PubMed.
- H. Xie, J. Wang and Z. Qin, et al., Octacoordinate Metal Carbonyls of Lanthanum and Cerium: Experimental Observation and Theoretical Calculation, J. Phys. Chem. A, 2014, 118, 9380–9385 CrossRef CAS PubMed.
- J. Jin, T. Yang and K. Xin, et al., Octacarbonyl Anion Complexes of Group Three Transition Metals [TM(CO)8]− (TM = Sc, Y, La) and the 18-Electron Rule, Angew. Chem., Int. Ed., 2018, 57, 6236–6241 CrossRef CAS PubMed.
- J. Jin, S. Pan and X. Jin, et al., Octacarbonyl Anion Complexes of the Late Lanthanides Ln(CO)8− (Ln = Tm, Yb, Lu) and the 32-Electron Rule, Chem.–Eur. J., 2019, 25, 3229–3234 CrossRef CAS PubMed.
- D. T. Moore, J. Oomens and J. R. Eyler, et al., Gas-Phase IR Spectroscopy of Anionic Iron Carbonyl Clusters, J. Am. Chem. Soc., 2004, 126, 14726–14727 CrossRef CAS PubMed.
- H.-J. Zhai, B. Kiran and B. Dai, et al., Unique CO Chemisorption Properties of Gold Hexamer: Au6(CO)n− (n = 0−3), J. Am. Chem. Soc., 2005, 127, 12098–12106 CrossRef CAS PubMed.
- I. Swart, F. M. F. de Groot and B. M. Weckhuysen, et al., The Effect of Charge on CO Binding in Rhodium Carbonyls: From Bridging to Terminal CO, J. Am. Chem. Soc., 2008, 130, 2126–2127 CrossRef CAS PubMed.
- C. Chi, J. Cui and Z. H. Li, et al., Infrared photodissociation spectra of mass selected homoleptic dinuclear iron carbonyl cluster anions in the gas phase, Chem. Sci., 2012, 3, 1698–1706 RSC.
- G. Wang, J. Cui and C. Chi, et al., Bonding in homoleptic iron carbonyl cluster cations: a combined infrared photodissociation spectroscopic and theoretical study, Chem. Sci., 2012, 3, 3272–3279 RSC.
- X. Zhou, J. Cui and Z. H. Li, et al., Infrared Photodissociation Spectroscopic and Theoretical Study of Homoleptic Dinuclear Chromium Carbonyl Cluster Cations with a Linear Bridging Carbonyl Group, J. Phys. Chem. A, 2012, 116, 12349–12356 CrossRef CAS PubMed.
- J. Cui, X. Zhou and G. Wang, et al., Infrared Photodissociation Spectroscopy of Mass Selected Homoleptic Copper Carbonyl Cluster Cations in the Gas Phase, J. Phys. Chem. A, 2013, 117, 7810–7817 CrossRef CAS PubMed.
- J. Cui, X. Zhou and G. Wang, et al., Infrared Photodissociation Spectroscopy of Mass-Selected Homoleptic Cobalt Carbonyl Cluster Cations in the Gas Phase, J. Phys. Chem. A, 2014, 118, 2719–2727 CrossRef CAS PubMed.
- J. Zou, H. Xie and D. Dai, et al., Sequential bonding of CO molecules to a titanium dimer: A photoelectron velocity-map imaging spectroscopic and theoretical study of Ti2(CO)n− (n = 1–9), J. Chem. Phys., 2016, 145, 184302 CrossRef PubMed.
- N. Zhang, M. Luo and C. Chi, et al., Infrared Photodissociation Spectroscopy of Mass-Selected Heteronuclear Iron–Copper Carbonyl Cluster Anions in the Gas Phase, J. Phys. Chem. A, 2015, 119, 4142–4150 CrossRef CAS PubMed.
- Z. Liu, J. Zou and Z. Qin, et al., Photoelectron Velocity Map Imaging Spectroscopy of Lead Tetracarbonyl–Iron Anion PbFe(CO)4−, J. Phys. Chem. A, 2016, 120, 3533–3538 CrossRef CAS PubMed.
- Z. Jumei, Z. Liu and G. Li, et al., CO activation by the heterobinuclear transition metal-iron clusters: A photoelectron spectroscopic and theoretical study, J. Energy Chem., 2021, 63, 344–350 CrossRef.
- H. Qu, F. Kong and G. Wang, et al., Infrared Photodissociation Spectroscopic and Theoretical Study of Heteronuclear Transition Metal Carbonyl Cluster Cations in the Gas Phase, J. Phys. Chem. A, 2016, 120, 7287–7293 CrossRef CAS PubMed.
- Z. Liu, H. Xie and Z. Qin, et al., Structural Evolution of Homoleptic Heterodinuclear Copper–Nickel Carbonyl Anions Revealed Using Photoelectron Velocity-Map Imaging, Inorg. Chem., 2014, 53, 10909–10916 CrossRef CAS PubMed.
- H. Xie, Z. Liu and X. Xing, et al., Infrared photodissociation spectroscopy of MO(CO)5+ (M = Sc, Y, La and Ce) in the gas phase, Chem. Phys. Lett., 2015, 628, 66–70 CrossRef CAS.
- H. Xie, Z. Liu and Z. Zhao, et al., Observing the Transition from Equatorial to Axial CO Chemisorption: Infrared Photodissociation Spectroscopy of Yttrium Oxide–Carbonyls, Inorg. Chem., 2016, 55, 5502–5506 CrossRef CAS PubMed.
- Y. Chen, K. Xin and J. Jin, et al., Infrared photodissociation spectroscopic investigation of TMO(CO)n+ (TM = Sc, Y, La): testing the 18-electron rule, Phys. Chem. Chem. Phys., 2019, 21, 6743–6749 RSC.
- Z. Liu, L. Hou and Y. Li, et al., Thermodynamics and Kinetics of Gas-Phase CO Oxidation on the Scandium Monoxide Carbonyl Complexes, J. Phys. Chem. A, 2020, 124, 924–931 CrossRef CAS PubMed.
- J. Zhang, Y. Li and Z. Liu, et al., Ligand-Mediated Reactivity in CO Oxidation of Niobium–Nickel Monoxide Carbonyl Complexes: The Crucial Roles of the Multiple Adsorption of CO Molecules, J. Phys. Chem. Lett., 2019, 10, 1566–1573 CrossRef CAS PubMed.
- J. Zhang, Y. Li and Y. Bai, et al., CO oxidation on the heterodinuclear tantalum–nickel monoxide carbonyl complex anions, Chin. Chem. Lett., 2021, 32, 854–860 CrossRef CAS.
- M. Haruta, S. Tsubota and T. Kobayashi, et al., Low-Temperature Oxidation of CO over Gold Supported on TiO2, α-Fe2O3, and Co3O4, J. Catal., 1993, 144, 175–192 CrossRef CAS.
- X. Xie, Y. Li and Z.-Q. Liu, et al., Low-temperature oxidation of CO catalysed by Co3O4 nanorods, Nature, 2009, 458, 746–749 CrossRef CAS PubMed.
- B. Xu, Y.-X. Zhao and X.-L. Ding, et al., Reactions of Sc2O4− and La2O4− Clusters with CO: A comparative study, Int. J. Mass Spectrom., 2013, 334, 1–7 CrossRef CAS.
- C. Chi, H. Qu and L. Meng, et al., CO Oxidation by Group 3 Metal Monoxide Cations Supported on [Fe(CO)4]2−, Angew. Chem., Int. Ed., 2017, 56, 14096–14101 CrossRef CAS PubMed.
- L.-N. Wang, X.-N. Li and S.-G. He, Catalytic CO Oxidation by Noble-Metal-Free Ni2VO4,5− Clusters: A CO Self-Promoted Mechanism, J. Phys. Chem. Lett., 2019, 10, 1133–1138 CrossRef CAS PubMed.
- Z. Qin, X. Wu and Z. Tang, Note: A novel dual-channel time-of-flight mass spectrometer for photoelectron imaging spectroscopy, Rev. Sci. Instrum., 2013, 84, 066108 CrossRef PubMed.
- Y. Gao, W. Huang and J. Woodford, et al., Detecting Weak Interactions between Au− and Gas Molecules: A Photoelectron Spectroscopic and ab Initio Study, J. Am. Chem. Soc., 2009, 131, 9484–9485 CrossRef CAS PubMed.
- W. Huang, H.-J. Zhai and L.-S. Wang, Probing the Interactions of O2 with Small Gold Cluster Anions (Aun−, n = 1–7): Chemisorption vs. Physisorption, J. Am. Chem. Soc., 2010, 132, 4344–4351 CrossRef CAS PubMed.
- Z. Qin, R. Cong and X. Wu, et al., Photoelectron velocity-map imaging spectroscopic and theoretical study on the reactivity of the gold atom toward CH3SH, CH3OH, and H2O, J. Chem. Phys., 2013, 139, 034315 CrossRef PubMed.
- V. Jonas and W. Thiel, Theoretical study of the vibrational spectra of the transition metal carbonyls M(CO)6 [M = Cr, Mo, W], M(CO)5 [M = Fe, Ru, Os], and M(CO)4 [M = Ni, Pd, Pt], J. Chem. Phys., 1995, 102, 8474–8484 CrossRef CAS.
- A. D. Becke, Density-functional exchange-energy approximation with correct asymptotic behavior, Phys. Rev. A, 1988, 38, 3098–3100 CrossRef CAS PubMed.
- J. P. Perdew, Density-functional approximation for the correlation energy of the inhomogeneous electron gas, Phys. Rev. B: Condens. Matter Mater. Phys., 1986, 33, 8822–8824 CrossRef PubMed.
- F. Weigend and R. Ahlrichs, Balanced basis sets of split valence, triple zeta valence and quadruple zeta valence quality for H to Rn: Design and assessment of accuracy, Phys. Chem. Chem. Phys., 2005, 7, 3297–3305 RSC.
- J. Zheng, X. Xu and D. G. Truhlar, Minimally augmented Karlsruhe basis sets, Theor. Chem. Acc., 2011, 128, 295–305 Search PubMed.
- E. Papajak, J. Zheng and X. Xu, et al., Perspectives on Basis Sets Beautiful: Seasonal Plantings of Diffuse Basis Functions, J. Chem. Theory Comput., 2011, 7, 3027–3034 CrossRef CAS PubMed.
- J. Akola, M. Manninen and H. Häkkinen, et al., Photoelectron spectra of aluminum cluster anions: Temperature effects and ab initio simulations, Phys. Rev. B: Condens. Matter Mater. Phys., 1999, 60, R11297–R11300 CrossRef CAS.
- W.-J. Chen, H.-J. Zhai and X. Huang, et al., On the electronic structure of mono-rhenium oxide clusters: ReOn− and ReOn (n = 3,4), Chem. Phys. Lett., 2011, 512, 49–53 CrossRef CAS.
- H.-J. Zhai, X.-H. Zhang and W.-J. Chen, et al., Stoichiometric and Oxygen-Rich M2On− and M2On (M = Nb, Ta; n = 5–7) Clusters: Molecular Models for Oxygen Radicals, Diradicals, and Superoxides, J. Am. Chem. Soc., 2011, 133, 3085–3094 CrossRef CAS PubMed.
- H.-J. Zhai, W.-J. Chen and X. Huang, et al., On the electronic structure and conflicting d-orbital aromaticity in the Re3O3− cluster, RSC Adv., 2012, 2, 2707–2712 RSC.
- D. J. Tozer and N. C. Handy, Improving virtual Kohn–Sham orbitals and eigenvalues: Application to excitation energies and static polarizabilities, J. Chem. Phys., 1998, 109, 10180–10189 CrossRef CAS.
- J. Akola, M. Manninen and H. Häkkinen, et al., Aluminum cluster anions: Photoelectron spectroscopy and ab initio simulations, Phys. Rev. B: Condens. Matter Mater. Phys., 2000, 62, 13216–13228 CrossRef CAS.
- M. Frisch, G. Trucks, H. Schlegel, et al., Gaussian 09, Revision-D.01, Gaussian, Inc., Wallingford CT, 2013 Search PubMed.
- Q. Yuan, Z. Jumei and Z. Jinghan, et al., Photoelectron velocity map imaging spectroscopic and theoretical study of heteronuclear vanadium-nickel carbonyl anions VNi(CO)n− (n = 2–6), J. Chem. Phys., 2018, 149, 144305 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: The total energies of several anion/neutral isomers of Ni2TiO2(CO)n−1/0 (n = 2–4) clusters with different spin multiplicities. The structures, relative energies, and symmetries of the isomers with higher energies of Ni2TiO2(CO)n− (n = 3, 4) clusters. Experimental and theoretical VDEs and ADEs as well as theoretical relative energies of the higher energy structures of Ni2TiO2(CO)n− (n = 3, 4) clusters. Cartesian coordinates of all carbonyl isomers of Ni2TiO2(CO)n− (n = 2–4). See DOI: https://doi.org/10.1039/d2ra07918f |
| This journal is © The Royal Society of Chemistry 2023 |