
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceRu-based monolithic catalysts for the catalytic oxidation of chlorinated volatile organic compounds†
Yemin Zhaoabd,
Chao Xibd,
Shan Gao *bd,
Yuejun Wangbd,
Haiqiang Wangad,
Pengfei Sunc and
Zhongbiao Wuad
*bd,
Yuejun Wangbd,
Haiqiang Wangad,
Pengfei Sunc and
Zhongbiao Wuad
aDepartment of Environmental Engineering, Zhejiang University, Hangzhou 310058, P. R. China
bZhejiang Tianlan Environmental Protection Technology Co., Ltd., Hangzhou 311202, China. E-mail: gaoshan@tianlan.cn
cDepartment of Chemistry, Key Laboratory of Surface & Interface Science of Polymer Materials of Zhejiang Province, Zhejiang Sci-Tech University, Hangzhou 310018, China
dZhejiang Provincial Engineering Research Center of Industrial Boiler & Furnace Flue Gas Pollution Control, 866 Yuhangtang Road, Hangzhou 310058, P. R. China
First published on 1st March 2023
Abstract
A series of cordierite monolithic catalysts with Ru species supported on different available low-cost carriers were prepared and investigated for the elimination of CVOCs. The results suggest that the monolithic catalyst with Ru species supported on anatase TiO2 carrier with abundant acidic sites exhibited the desired catalytic activity for DCM oxidation with the T90% value of 368 °C. In addition, a pseudo-boehmite sol used as binder was introduced into the preparation of the monolithic catalysts to further improve the adhesion between the powder catalysts and cordierite honeycomb carrier. The results suggest that although the T50% and T90% of the Ru/TiO2/PB/Cor shifted to higher temperature of 376 and 428 °C, the weight loss of the coating for the Ru/TiO2/PB/Cor catalyst was improved and decreased to 6.5 wt%. Also, the as-obtained Ru/TiO2/PB/Cor catalyst exhibited ideal catalytic properties for the abatement of ethyl acetate and ethanol, indicating that the catalyst can meet the demand for the treatment of actual multi-component industrial gas.
1. Introduction
Chlorinated volatile organic compounds (CVOCs) such as dichloromethane (DCM), chlorobenzene (CB) and 1,2-dichloroethane (DCE) are widely utilized in the production processes of pharmaceuticals, synthetic resins, adhesives and coatings, and printing.1–4 Nevertheless, most CVOCs are considered as hazardous pollutants due to their severe toxicity and persistent threat to humans and the environment. Therefore, eliminating CVOC pollutants from the atmosphere becomes more urgent in order to protect the environment and promote harmonious relationships between humans and nature.5,6Among the advanced treatment technologies, catalytic oxidation, which converts chlorinated compounds into the desirable products of H2O, CO2 and HCl, has been in focus because they have the advantages of high conversion efficiency, moderate operation temperature and low energy consumption.7–9 Over the past decades, much effort has been made to improve the catalytic oxidation of CVOCs through development of efficient catalysts for the conversion process. Among the various catalytic materials, transition metal oxides, such as CeO2, MnOx, TiO2, ZrO2–TiO2, have been prepared in various strategies and investigated by modulating their reducibility, surface acidity, structure and morphology.10–17 For example, Zhan et al. prepared a series of Mn–Ti composite oxides with different surface acidity and reducibility through adjusting the Mn/Ti molar ratio, they found the catalyst surface acidity had an important effect on the oxidation of vinyl chloride.7 Zhou et al. synthesized several CeO2–ZrO2–CrOx mixed oxides and found the introduction of Zr4+ into the CeO2–CrOx can improve the catalytic oxidation of 1,2-dichloroethane remarkably since the ZrO2 doping was considered to raise the amount of the Cr6+ species and oxygen vacancies of the catalysts.13 Although the transition metal oxides exhibit superior catalytic performance, they are susceptible to inorganic chlorine species, which probably results in the quickly deactivation of the catalysts. Noble metal catalysts, such as Pt, Pd and Ru, have also attracted considerable attention in exploring their catalytic performance over the CVOCs conversion.18–28 Previous studies have indicated that Ru-based catalysts produce less polychlorinated by-products since the Cl species can be removed rapidly from the surface in form of Cl2 through the Deacon reaction in comparison with Pd and Pt-based materials. Zeolite catalysts with high specific surface area, outstanding thermal stability, intricate channels are also widely applied in CVOCs catalytic oxidation.29–33 Unfortunately, coke deposition and chlorine poisoning on zeolites surface can lead to the deactivation of catalytic performance rapidly.
In addition, the catalytic activities of noble metal supported catalysts are associated with various carriers.24 Recently, Ying and co-workers explored the catalytic oxidation of DCM over Ru-based materials with different carriers (Al2O3, TiO2, HZSM-5 SiO2/Al2O3 = 27 and 130), and they found that the Lewis acid sites on surface of carriers were more active for the C–Cl cleavage in comparison with Brønsted acid sites. Additionally, the Cl species deposited on the Al2O3 and TiO2 were more prone to generate polychlorinated by-products especially the dibenzo-p-dioxins and dibenzofurans (PCDD/Fs) through the electrophilic chlorination reactions.34
On the other hand, previous mechanism studies have indicated that the surface acidity of the catalyst affect the catalytic oxidation of CVOCs. Because the adsorption and the breakage of C–Cl bonds usually occurred on the carriers with abundant acid sites on their surface, such as TiO2.30 In addition, for Ru-based catalysts, the dissociated Cl species will be rapidly transferred to the Ru species from the active sites, and followed by the generation of Cl2 through Deacon reaction.20,34
Monolithic catalysts have been intensively developed and used in various practical industrial applications for effective abatement of the polluted waste gas such as VOCs and NOx due to their lower pressure drop, higher mechanical strength and superior heat and mass transfer property in comparison with conventional powder catalysts and granular catalysts.35–39 Cordierite honeycomb (2MgO·2Al2O3·5SiO2) is a common monolithic catalysts substrate with low cost and abundant resource, and has been explored for catalytic oxidation of non-chlorine VOCs, such as ethyl acetate, methane and toluene.40–42 For example, Ren et al. fabricated a series of MxCo3−xO4 (M = Zn, Ni, Co) nanoarray materials that supported on cordierite monolithic substrates through in situ hydrothermal synthesis for methane and carbon monoxide catalytic oxidation reaction, however, their hydrothermal approach of monolithic catalysts was time-consuming and complicated, which made it difficult for scalable fabrication.43 In general, over the past years, although catalysts with various active components and structures have been intensively synthesized and displays ideal catalytic activity and stability for the catalytic degradation of CVOCs, most of these reported catalysts are powdery, and few literature related to the performance of monolithic catalysts have been reported probably due to the complexity of preparation of catalysts and coating slurry as well as the high requirement for testing device, especially for its resistance to chlorine corrosion.15,42 Therefore, adopting feasible and facile methods to prepare catalysts and support them on monolithic substrate as well as studying their catalytic performance in fixed-bed reactor made by chlorine corrosion resistance alloy in a more applicable perspective become urgently, which may be beneficial for the guide of future pilot test or engineering applications.
Consequently, in this work, cordierite monolithic catalysts with Ru species supported on different low-cost available carriers (TiO2, P25, CeO2) are prepared through coating the powder catalysts on cordierite honeycomb monoliths and investigated their catalytic activity toward dichloromethane catalytic oxidation. On the other hand, as previous study indicates that the low-cost all-silicon beta zeolite supported Pt catalyst (Pt/Si-beta) showed the best catalytic activity for the catalytic combustion of methylcyclohexane due to its superior hydrocarbon adsorption capacity and hydrophobicity compared to the all-silicon ZSM supported Pt and SBA-15 supported Pt catalysts.44 Thus, monolithic catalyst with Ru species loaded on all-silicon beta zeolite is also explored. In addition, the properties of these as-prepared catalysts were further examined by N2 adsorption–desorption, XRD, XPS, NH3-TPD techniques, respectively. Here, the DCM was chosen as a reactant to investigate the catalytic activity of the catalysts. We believe that our work is capable of providing deep insight into designing and preparing monolithic catalysts for efficient catalytic oxidation not only for CVOCs but also for multicomponent VOCs that intensively exists in industrial exhausts from a more practical point of view.
2. Experimental
All chemicals are commercials available (Table 1).| Chemicals | Source | Purity |
|---|---|---|
| All-silicon beta zeolite (Si-beta) | Zhejiang Taide New Material Co., Ltd | ≥90% |
| Anatase titanium dioxide (TiO2) | Sichuan Huatie Vanadium and Titanium Technology Co., Ltd | ≥97% |
| Cerium dioxide (CeO2) | Yutai Qingda Fine Chemical Co., Ltd | ≥99% |
| P25 | Degussa Co., Ltd | AR |
2.1 Catalyst preparation
where M0 is the mass of pristine cordierite honeycomb monolith; M1 is the mass of the monolithic catalyst after coating and calcination.
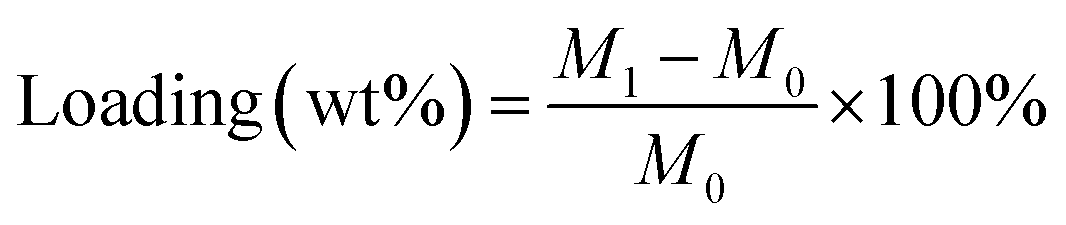
2.2 Catalyst characterization
XRD patterns were recorded on an X-ray powder diffractometer (Bruker D8 ADVANCE A25X). X-ray photoelectron spectroscopy (XPS) of the catalysts was probed on a Thermo Fisher Scientific K-Alpha instrument with a monochromatic Al Kα X-ray source (hν = 1486.8 eV). All of the binding energies were referenced to the C 1s peak (284.8 eV) corresponding to adventitious carbon. NH3-Temperature Programmed Desorption (NH3-TPD) of samples were examined in Tianjin Xianquan Industry and Trade Development Co. Ltd. Typically, 100 mg sample was pretreated in pure He at 400 °C for 1 h, then the sample was saturated with 4% NH3 in He (30 mL min−1) at 30 °C for 30 min and subsequently heated to 100 °C. Thereafter, the sample was heated at a rate of 10 °C min−1 in pure He to desorb the NH3. The weight loss of coating on monolithic catalysts is evaluated by ultrasonic vibration. Typically, the monolithic catalyst was placed in an ultrasonic bath for 30 min, and the sample was then washed by deionized water and dried at 120 °C for 3 h and weighed.where M0 is the mass of pristine cordierite honeycomb monolith; M1 is the mass of the monolithic catalyst after coating and calcination; M2 is the mass of the monolithic catalyst after ultrasonic vibration and dry.
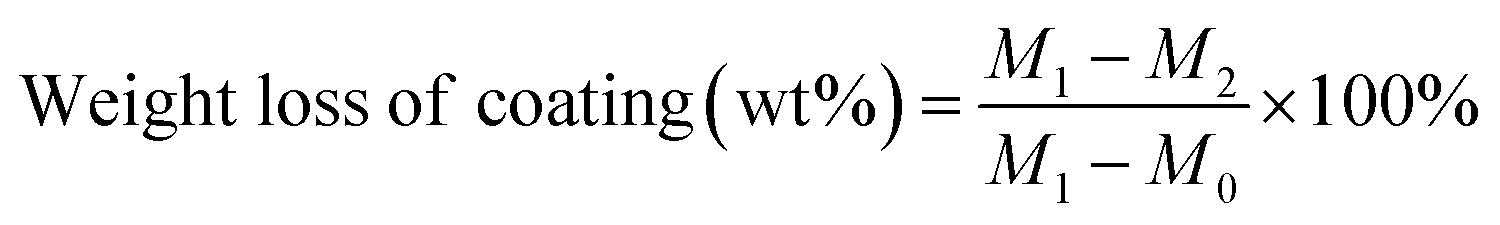
2.3 Catalytic activity evaluation
The dichloromethane, ethyl acetate (EA) and ethanol (EtOH) catalytic activities of as-prepared monolithic catalysts were tested in a conventional fixed-bed reactor, and the monolithic catalysts were placed in the constant temperature zone of the reactor. The reactor was continuously heated and the temperature of the reactor was maintained at the set temperature before analysing the VOCs concentration. The gaseous reactants (DCM, EA and EtOH) were generated by air stream through the liquid reactants in a saturator, and then diluted by another air stream. Here, the indoor air without any purification was used. The liquid reactants were maintained at 0 °C in an ice-water bath during the experiments, respectively. The mass flow controllers (Sevenstar Electronics) were applied to regulate the air intake flow. The total flow was about 6.05 L min−1 and the gas hourly space velocity (GHSV) was kept at 15![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 h−1 with an initial gaseous reactant concentration of about 1000 ppm that was tested at 250 °C in inlet. The DCM concentration in both inlet and outlet of the reactor was monitored by the gas chromatograph (Fuli, China) with flame ionization detector (FID). The schematic diagram of experimental set-up is displayed in Fig. S1.† Additionally, the ethyl acetate and ethanol concentration in both inlet and outlet of reactor was analyzed by the EXPEC 3200 portable gas chromatograph (Hangzhou PuYu Technology Development Co., Ltd. China) with flame ionization detector. The CO2 was analysed by the GC with a Ni convertor furnace. The DCM conversion (%) and CO2 yield (%) were calculated by using the following equation:
000 h−1 with an initial gaseous reactant concentration of about 1000 ppm that was tested at 250 °C in inlet. The DCM concentration in both inlet and outlet of the reactor was monitored by the gas chromatograph (Fuli, China) with flame ionization detector (FID). The schematic diagram of experimental set-up is displayed in Fig. S1.† Additionally, the ethyl acetate and ethanol concentration in both inlet and outlet of reactor was analyzed by the EXPEC 3200 portable gas chromatograph (Hangzhou PuYu Technology Development Co., Ltd. China) with flame ionization detector. The CO2 was analysed by the GC with a Ni convertor furnace. The DCM conversion (%) and CO2 yield (%) were calculated by using the following equation:where [DCM]in denotes the DCM concentration in the inlet gas. [DCM]out denotes the DCM concentration in the outlet gas.
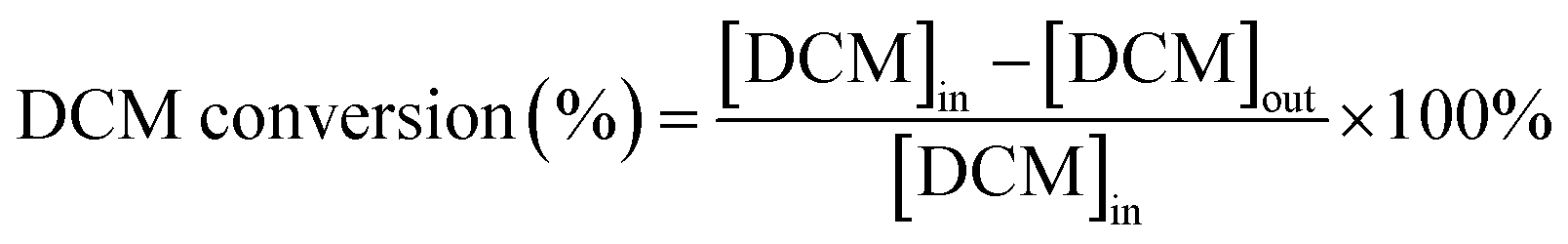
where [CO2]in is the CO2 concentration in the inlet. [CO2]out is the CO2 concentration in the outlet. [DCM]in is the DCM concentration in the inlet gas.
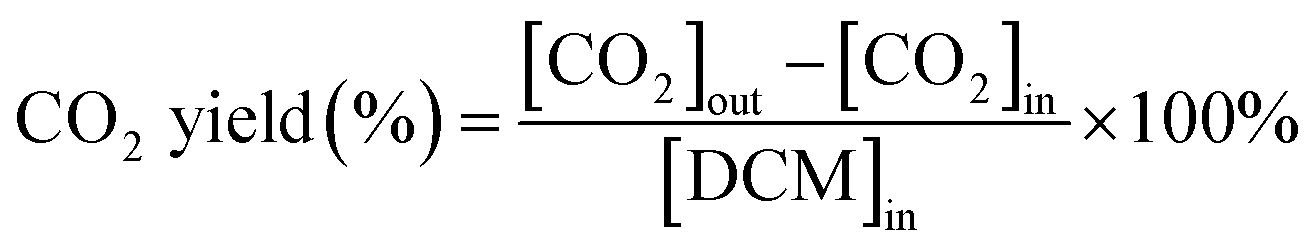
3. Results and discussion
Fig. 1 displays the preparation strategy for the formation of the monolithic catalyst. The cordierite honeycomb was immersed in the slurry containing the powder catalyst after ball milling for several minutes, then the excess slurry in pores was removed by compressed air, and the coated catalysts was dried and further subjected to calcination in air to obtain the monolithic catalyst. The powder X-ray diffraction (XRD) patterns of the various samples are shown in Fig. 2. For the Ru/TiO2 catalysts, the diffraction peaks located at 25.3°, 37.8° and 48.1° can be attributed to the (101), (004) and (200) planes of anatase TiO2 (PDF #21-1272).20,45 As for Ru/P25 catalyst, the peaks of rutile TiO2 phase at 27.5° and 36.1° are also observed and can be indexed to the (110) and (101) lattice plane of rutile TiO2 (PDF #21-1276), suggesting the existence of rutile and anatase binary titanium dioxide in Ru/P25 catalyst.34 The characteristic peak at 21.5° and 22.5° of Si-beta zeolite are observed over the Ru/Si-beta catalyst. Diffraction peaks at 28.6°, 33.1°, 47.5° and 56.3° can be observed on Ru/CeO2 catalyst ascribing to the (111), (200), (220) and (311) planes of CeO2 (PDF #34-0394), respectively.13 Additionally, the peaks at 27.9° and 35.0° corresponding to the (110) and (101) planes of RuO2 (PDF #43-1027) can be found in Ru/TiO2 and Ru/Si-beta, respectively.20,28 No obvious diffraction peaks from Ru species in Ru/CeO2 and Ru/P25, probably owing to better dispersion in these catalysts. In addition, as shown in Fig. S2,† Ru/TiO2/PB/Cor monolithic catalyst displays the characteristic peaks of anatase TiO2 and cordierite (PDF #82-1541), however, the peaks of RuO2 probably overlaps with those of cordierite, and could not be identified directly. | ||
| Fig. 1 Preparation route for the monolithic catalyst. | ||
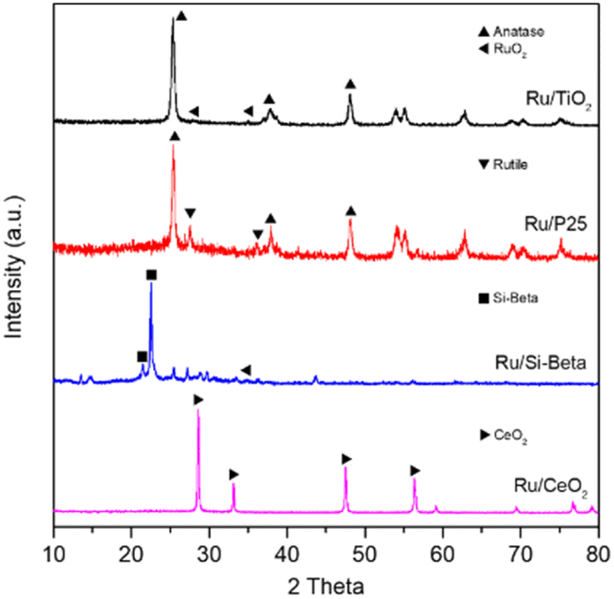 | ||
| Fig. 2 The XRD patterns of Ru/TiO2, Ru/P25, Ru/Si-beta and Ru/CeO2. | ||
X-ray photoelectron spectra (XPS) is employed to test various catalysts, and the high-resolution Ru 3d spectrum is displayed in Fig. 3. For Ru/TiO2 and Ru/P25 catalysts, the Ru 3d5/2 can be deconvoluted into two peaks with binding energy at 281.0 eV and 280.0 eV, attributed to Ru4+ and Ru0, corresponding to RuO2 and metallic Ru nanoparticle, respectively.18 Additionally, the binding energy of Ru0 shifted to higher binding energy (280.5 eV) was observed in Ru/CeO2 catalyst due to the electron transfer from Ru to Ce through Ce–O–Ru.46 The Ru/CeO2 catalyst showed one more Ru 3d5/2 peak with the binding energy at 282.5 eV, which can be assigned to Ru6+ species, probably resulting from the interaction between CeO2 and Ru, thereby promoting the transformation of Ce4+ to Ce3+, and causing the loss of electron of Ru.47 For the Ti 2p spectra in Ru/TiO2 and Ru/P25, two obvious peaks at 458.9 and 464.7 eV are in accordance with Ti 2p3/2 and Ti 2p1/2, indicating the Ti exsits in the form of Ti4+.48 For Ru/CeO2 catalyst, the complicated Ce 3d spectra can be deconvoluted into eight components. The peaks located at 884.3 and 902.6 eV can be assigned to Ce3+ species, while the peaks at 882.1, 888.8, 898.1, 900.6, 907.5 and 916.5 eV are related to Ce4+ species (Fig. S3†).49
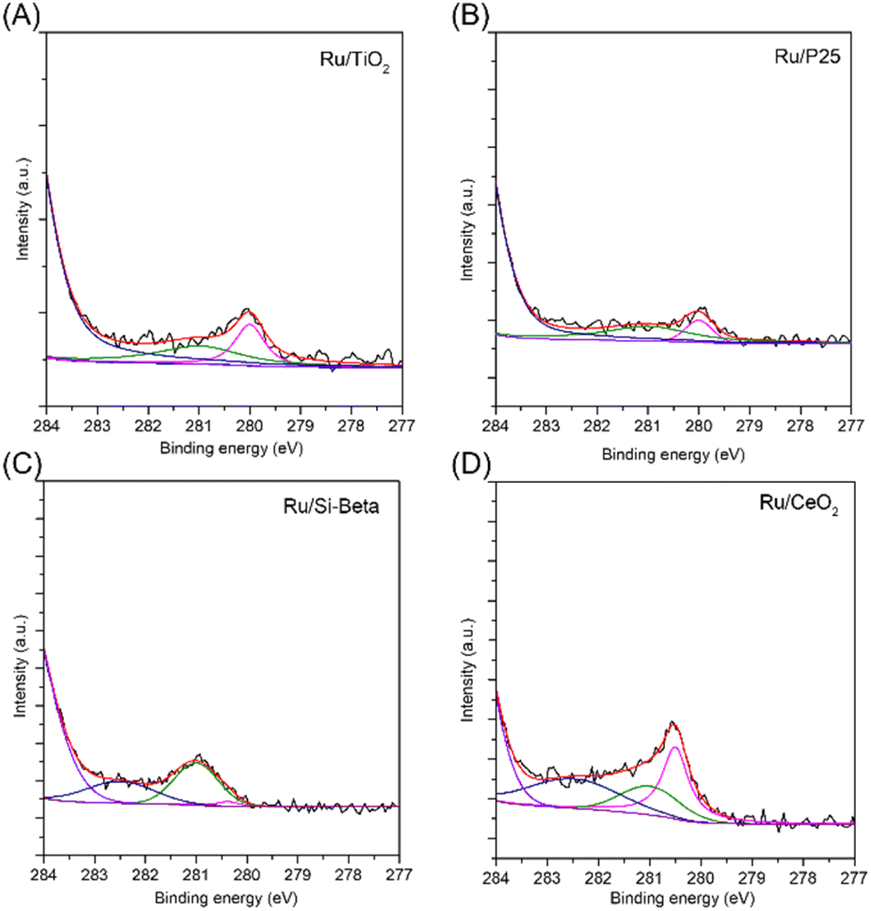 | ||
| Fig. 3 Ru 3d XPS spectra of (A) Ru/TiO2, (B) Ru/P25, (C) Ru/Si-beta and (D) Ru/CeO2. | ||
The temperature-programmed desorption of NH3 (NH3-TPD) experiments were conducted to examine the acidic properties of the as-obtained catalysts. As shown in Fig. 4, the peak that appears at a high temperature range of about 350–450 °C can be observed over Ru/TiO2 and Ru/P25 catalyst, and corresponds to the strong acid sites.50 In addition, the intensity of NH3 desorption peak of Ru/TiO2 catalyst is higher than those of Ru/P25, Ru/Si-beta and Ru/CeO2 catalysts, indicating Ru/TiO2 catalyst has abundant acid sites on its surface between 100 and 500 °C. Previous studies shows that the catalysts with abundant acid sites is beneficial for the adsorption of Cl and the cleavage of C–Cl bonds during the DCM catalytic oxidation process.49 In addition, the trend of surface acidity of various carriers is consistent with above catalysts within the same range of temperature (Fig. S4†).
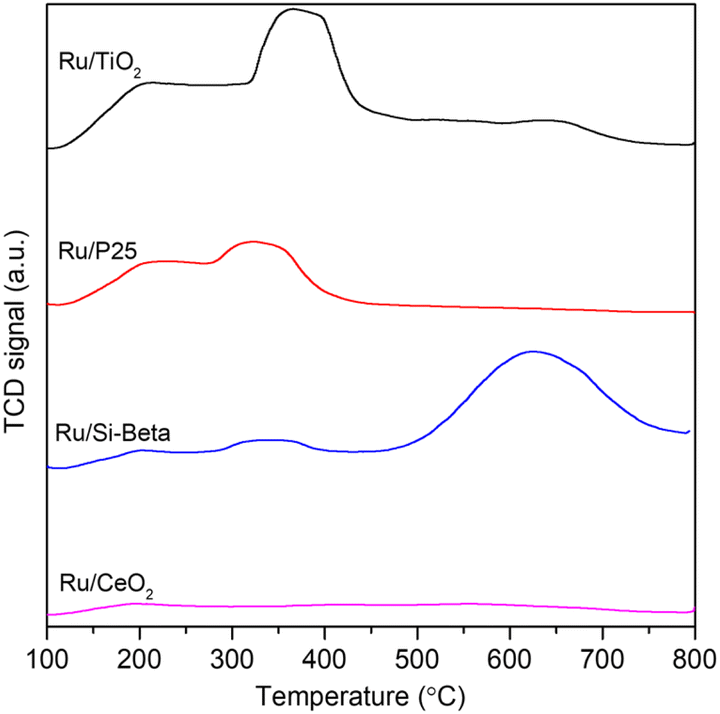 | ||
| Fig. 4 The NH3-TPD profiles of Ru/TiO2, Ru/P25, Ru/Si-beta and Ru/CeO2. | ||
The catalytic performance and CO2 yield curves of prepared monolithic catalysts with various carriers for the DCM catalytic oxidation are displayed in Fig. 5. According to T50% (reaction temperature of DCM conversion at 50%), the catalytic performance of the catalyst decreases in the order: Ru/TiO2/Cor (329 °C) > TiO2/Cor (351 °C) > Ru/P25/Cor (373 °C) > Ru/Si-beta/Cor (450 °C) > Ru/CeO2/Cor (458 °C), and the Ru/TiO2/Cor catalyst shows the superior catalytic activity in comparison with other samples, achieving almost 100% conversion of DCM at 400 °C, suggesting that the anatase TiO2 as carrier is more favourable than others in improving the catalytic activity (Fig. 5A). This can be related to the surface acidity of Ru/TiO2 which provides abundant acid sites for adsorption of Cl and the cleavage of C–Cl bonds. The DCM conversion over Ru/P25/Cor, Ru/Si-beta/Cor and Ru/CeO2/Cor catalysts reached 94%, 55% and 52% at 460 °C, respectively. The CO2 yield (Fig. 5B) of various catalysts follows similar trend as DCM conversion except for TiO2/Cor catalyst. The TiO2/Cor catalyst exhibits the worst CO2 yield among the different catalysts because pure TiO2 shows negligible activity over the cleavage of C–H bonds as well as CO oxidation (Fig. 5B). No surprisingly, the mineralization rate is elevated remarkably through the introduction of Ru onto the surface of TiO2 nanoparticles. This can be related to the active Ru species with excellent redox capability, which is capable of oxidizing the intermediates effectively. Moreover, during the oxidation of DCM, TiO2 will deactivate quickly owing to the adsorption and deposition of chlorine species onto its surface, and introducing Ru species will be more favorable in removing the chlorine species through the Deacon reaction, which eventually improving the catalytic performance. In addition, after the addition of Ru species on the surface of TiO2 carrier, both the specific surface area and pore volume decreased slightly (Table 2), which may be caused by the fact that the Ru species enter into the channel of the carrier and block some pore during preparation process of Ru/TiO2 powder catalyst.35 Such results are also found in Ru/P25 and Ru/Si-beta catalysts. However, for Ru/CeO2 catalyst, because the pore volume of CeO2 is extremely low, the Ru species mainly deposited on the surface of CeO2, as a consequence, causing a slight increase in specific surface area. The Ru/TiO2/Cor catalyst shows the highest CO2 yield of 97% at 460 °C, about 11%, 50% and 55% higher than those of Ru/P25/Cor, Ru/Si-beta/Cor and Ru/CeO2/Cor catalysts, respectively. Carbon balance was also calculated over various catalysts at 460 °C based on the inlet and outlet results (Table S1†). For TiO2/Cor catalyst, due to the weak oxidation capability of TiO2, a large number of undesired by-products (CxHyClz) could be found in the outlet gas, and is much more than those of Ru/P25/Cor, Ru/Si-beta/Cor and Ru/CeO2/Cor catalysts. For Ru/TiO2/Cor catalyst, based on the calculated result, negligible by-products (CxHyClz) could be existed in the outlet gas.
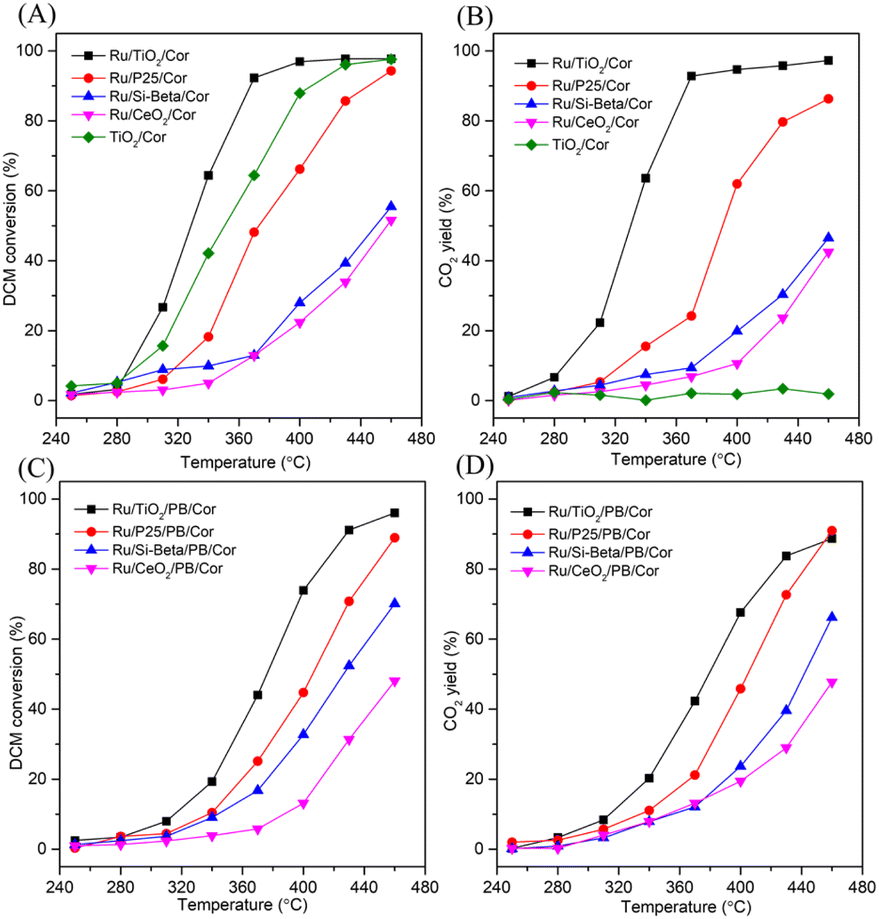 | ||
| Fig. 5 (A) DCM conversion over Ru/TiO2/Cor, Ru/P25/Cor, Ru/Si-beta/Cor, Ru/CeO2/Cor and TiO2/Cor catalysts; (B) CO2 yield during DCM oxidation over Ru/TiO2/Cor, Ru/P25/Cor, Ru/Si-beta/Cor, Ru/CeO2/Cor and TiO2/Cor catalysts; (C) DCM conversion over Ru/TiO2/PB/Cor, Ru/P25/PB/Cor, Ru/Si-beta/PB/Cor and Ru/CeO2/PB/Cor catalysts; (D) CO2 yield during DCM oxidation over Ru/TiO2/PB/Cor, Ru/P25/PB/Cor, Ru/Si-beta/PB/Cor and Ru/CeO2/PB/Cor catalysts; 1000 ppm DCM, GHSV = 15000 h−1. | ||
| Powder catalyst | SBET (m2 g−1) | Pore size (nm) | Pore volume (cm3 g−1) |
|---|---|---|---|
| TiO2 | 85.0 | 7.93 | 0.375 |
| Ru/TiO2 | 74.3 | 9.75 | 0.324 |
| P25 | 51.4 | 17.67 | 0.219 |
| Ru/P25 | 46.3 | 33.55 | 0.481 |
| Si-beta | 633.6 | 5.55 | 0.151 |
| Ru/Si-beta | 491.6 | 6.89 | 0.166 |
| CeO2 | 5.4 | 22.19 | 0.037 |
| Ru/CeO2 | 5.7 | 17.84 | 0.033 |
Moreover, the pseudo-boehmite sol used as binder was introduced into the preparation of monolithic catalysts to improve the adhesion between powder catalysts and cordierite honeycomb carrier. When the binder was introduced, the weight loss of coating on the Ru/TiO2/PB/Cor catalyst is 6.5 wt%, and lower than that of Ru/TiO2/Cor catalyst which is without binder (14.6 wt%) obviously. What's more, the light-off and CO2 yield plots over the binder-containg monolithic catalysts are also recorded. As shown in Fig. 5C, the addition of pseudo-boehmite sol can result in change of catalytic activity of monolithic catalysts in different ways. For example, a temperature of 428 °C is needed to achieve 90% conversion of DCM over the Ru/TiO2/PB/Cor catalyst, while the Ru/TiO2/Cor catalyst only needs 368 °C to achieve the same conversion. The difference may result from the effect of binder. Pseudo-boehmite sol can be gradually transformed into Al2O3 during the calcination process. It has been reported that the Ru/Al2O3 shows lower catalytic performance toward DCM oxidation than that of Ru/TiO2 because the TiO2 carrier possesses the stronger Lewis acid sites compared to Al2O3, which is more active for the DCM dissociative adsorption.34 Hence, introduction of pseudo-boehmite sol can restrict the catalytic behaviour of Ru/TiO2/PB/Cor to some extent. Interestingly, the Ru/Si-beta/PB/Cor displays the contrary result by introducing the pseudo-boehmite sol in slurry. The T50% for Ru/Si-beta/PB/Cor is 426 °C and lower than that of Ru/Si-beta/Cor (450 °C). The improved activity probably results from the Lewis acid sites that provided by Al2O3, which is considered to be favourable for DCM dissociative adsorption. Such result is different from the catalytic combustion of toluene that applying all-silicon beta zeolite supported Pd as catalysts. The authors found that the pure silica beta zeolite supported Pd catalyst with excellent hydrophobicity displayed highest catalytic activity for the catalytic oxidation of toluene than those catalysts with different SiO2/Al2O3 ratio.51 The DCM conversion over Ru/TiO2/PB/Cor, Ru/P25/PB/Cor, Ru/Si-beta/PB/Cor and Ru/CeO2/PB/Cor catalysts reached 96%, 89%, 70% and 48% at 460 °C, respectively. The CO2 yield of Ru/TiO2/PB/Cor (89%) is close to that of Ru/P25/PB/Cor (91%) at 460 °C, and about 23% and 41% higher than those of Ru/Si-beta/PB/Cor and Ru/CeO2/PB/Cor catalysts. In addition, carbon balance results shows that the Ru/TiO2/PB/Cor and Ru/Si-beta/PB/Cor produced little amounts of by-products (CxHyClz) at 460 °C. As shown in the Fig. 6A, the effect of different loading on catalytic activity of Ru/TiO2/PB/Cor catalyst was also investigated. When the coating loading increased to about 13 wt%, the T50% is 15 and 32 °C lower than those with loading of 8.5 wt% and 5.4 wt%, suggesting that the catalyst with higher loading is beneficial for the catalytic performance. Additionally, the influence of various GHSV over the Ru/TiO2/PB/Cor catalyst toward DCM degradation was examined and displayed in Fig. 6B. When the GHSV is 15000 h−1, the T50% and T90% are 376 and 428 °C, about 26 and 31 °C higher than those under the 10000 h−1. Additionally, when the GHSV increased to 20000 h−1, a temperature of 447 °C is needed to achieve 90% conversion of DCM. Such results indicate that the Ru/TiO2/PB/Cor catalyst showed ideal catalytic activity under various GHSV. Long-term stability tests of Ru/TiO2/PB/Cor catalyst at 450 °C was carried out and the result is displayed in Fig. 6C. It can be observed that the conversion of DCM over Ru/TiO2/PB/Cor catalyst still can keep at ∼90% even after 26 h, which suggests that the catalyst possesses excellent stability toward DCM catalytic oxidation. Such results can provide some valuable information for future pilot test or engineering applications. In addition, different types of VOCs, such as ethyl acetate and ethanol, are usually found to exist in industrial exhaust gas, thus, testing the adaptability of the as-obtained monolithic catalysts for the abatement of various VOCs is necessary. As shown in Fig. 6D, the T90% for ethyl acetate and ethanol oxidation over Ru/TiO2/PB/Cor catalyst is 375 °C and 344 °C, respectively, indicating that the catalyst is also applicable to the treatment of other VOCs.
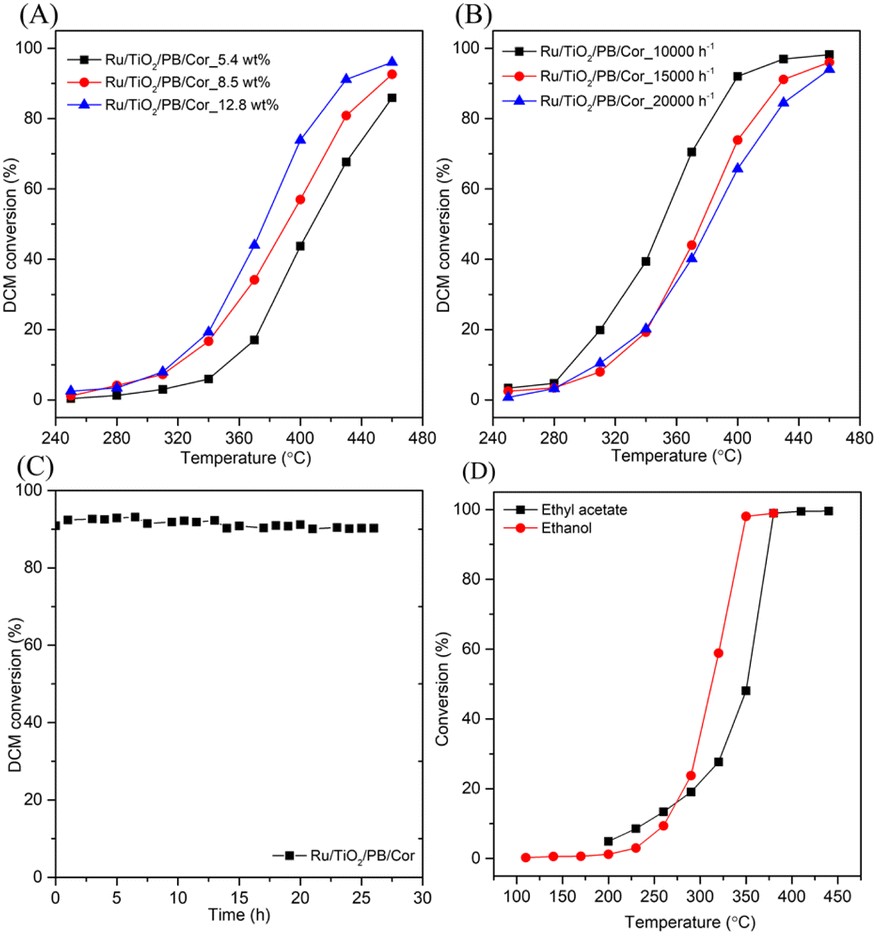 | ||
| Fig. 6 (A) Effect of the loading over the Ru/TiO2/PB/Cor catalyst; (B) effect of GHSV over the Ru/TiO2/PB/Cor catalyst; (C) stability test of the Ru/TiO2/PB/Cor catalyst for the degradation of DCM at 450 °C, 1000 ppm DCM, GHSV = 15000 h−1; (D) catalytic activity of ethyl acetate and ethanol over the Ru/TiO2/PB/Cor catalyst; 1000 ppm EA; 1000 ppm EtOH; GHSV = 15000 h−1. | ||
Based on the published studies, only few have been reported to investigate the CVOCs catalytic oxidation over the monolithic catalysts with different catalytic active components during the past years. For example, Long et al. found that the optimized Mn–Ce–Zr monolithic catalyst displayed the best activity towards chlorobenzene (CB) oxidation with T90% at 390 °C in comparison with other controlled samples.15 Zhu et al. prepared a series of multi-structured Ag/MnO2–cordierite molded catalysts that were applied in the catalytic oxidation of CB. The T90% for EA and CB are 217 °C and 385 °C respectively under the space velocity of 10000 h−1 over the optimized R–Ag/MnO2–cordierite catalyst.42 A higher temperature is usually needed to achieve the same conversion for CVOCs oxidation in comparison with other non-chlorine VOCs.
The DCM degradation mechanisms over various catalysts have been intensively studied and proposed. According to the experiments and previous researches, the mechanism over Ru/TiO2 monolithic catalyst can be deduced (Fig. 7): firstly, the DCM is adsorbed on the TiO2 surface with abundant acidic sites, then attacked by surface active oxygen species adjacent to adsorbed DCM leading to the breakage of C–Cl bonds, and formed the chloromethoxy species.49,52 In addition, the Cl of chloromethoxy species can also be adsorbed on the acidic sites, followed by the formation of bidentate methoxy species. Later the bidentate methoxy species can translate to formate species or the mixture of formate and formaldehyde species simultaneously.49,53 Finally, the formate species can further be oxidized into CO or CO2. At the same time, the dissociated adsorbed Cl species will be rapidly transferred to the Ru species, and followed by the generation of Cl2 through Deacon reaction that catalyzed by RuO2 component.20,34,54
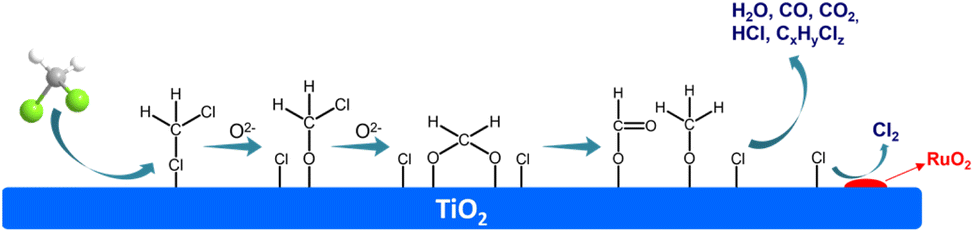 | ||
| Fig. 7 Mechanism for the catalytic oxidation of DCM over the Ru/TiO2/PB/Cor catalyst. | ||
4. Conclusion
In summary, a series of cordierite monolithic catalysts with Ru species supported on different low-cost available carriers were prepared and investigated for the degradation of CVOCs. Among them, the monolithic catalyst with Ru species supported on anatase TiO2 carrier exhibited the desired catalytic activity for DCM oxidation with T90% value of 368 °C. On the other hand, a pseudo-boehmite sol used as binder was introduced into the preparation of monolithic catalysts to further improve the adhesion between powder catalysts and cordierite honeycomb carrier. Although the T50% and T90% of the Ru/TiO2/PB/Cor shifted to higher temperature of 376 and 428 °C, the weight loss of the coating for Ru/TiO2/PB/Cor catalyst decreased to 6.5 wt%, which is significantly lower than that of Ru/TiO2/Cor catalyst (14.6 wt%). In addition, the as-obtained Ru/TiO2/PB/Cor catalyst also exhibited ideal catalytic property for the abatement of other VOCs. We believe that our work may provide new insights for design and development of high-performance catalysts through optimizing carriers and binders in a rational way to eliminate DCM as well as broaden its application to the degradation of non-chlorine VOCs.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the National Key R&D Program of China (2022YFC3701600) and the preferential funding for Postdoctoral Research Projects of Zhejiang Province, China (ZJ2021052).References
- X. Zhang, Z. Pei, H. Lu and H. Huang, RSC Adv., 2016, 6, 84209–84215 RSC
.
- H. Huang, X. Zhang, X. Jiang, K. Dou, Z. Ni and H. Lu, RSC Adv., 2016, 6, 61610–61614 RSC
- X. Zhang, Z. Pei, X. Ning, H. Lu and H. Huang, RSC Adv., 2015, 5, 79192–79199 RSC
- C. Yang, G. Miao, Y. Pi, Q. Xia, J. Wu, Z. Li and J. Xiao, Chem. Eng. J., 2019, 370, 1128–1153 CrossRef CAS
- L. Zhu, D. Shen and K. H. Luo, J. Hazard. Mater., 2020, 389, 122102 CrossRef CAS PubMed
- Q. Dai, J. Wu, W. Deng, J. Hu, Q. Wu, L. Guo, W. Sun, W. Zhan and X. Wang, Appl. Catal., B, 2019, 249, 9–18 CrossRef CAS
- H. Liu, X. Li, Q. Dai, H. Zhao, G. Chai, Y. Guo, Y. Guo, L. Wang and W. Zhan, Appl. Catal., B, 2021, 282, 119577 CrossRef CAS
- J. M. Giraudon, T. B. Nguyen, G. Leclercq, S. Siffert, J. F. Lamonier, A. Aboukaïs, A. Vantomme and B. L. Su, Catal. Today, 2008, 137, 379–384 CrossRef CAS
- R. W. van den Brink, M. Krzan, M. M. R. Feijen-Jeurissen, R. Louw and P. Mulder, Appl. Catal., B, 2000, 24, 255–264 CrossRef CAS
- T. Cai, H. Huang, W. Deng, Q. Dai, W. Liu and X. Wang, Appl. Catal., B, 2015, 166–167, 393–405 CrossRef CAS
- S. Cao, H. Wang, M. Shi, S. Chen and Z. Wu, Catal. Lett., 2016, 146, 1591–1599 CrossRef CAS
- W. Xingyi, K. Qian and L. Dao, Appl. Catal., B, 2009, 86, 166–175 CrossRef
- P. Yang, S. Yang, Z. Shi, F. Tao, X. Guo and R. Zhou, Chem. Eng. J., 2016, 285, 544–553 CrossRef CAS
- W. Deng, Q. Dai, Y. Lao, B. Shi and X. Wang, Appl. Catal., B, 2016, 181, 848–861 CrossRef CAS
- G. Long, M. Chen, Y. Li, J. Ding, R. Sun, Y. Zhou, X. Huang, G. Han and W. Zhao, Chem. Eng. J., 2019, 360, 964–973 CrossRef CAS
- J. Chen, W. Xu, M. Jiang, J. Chen and H. Jia, Appl. Catal., B, 2020, 278, 119263 CrossRef CAS
- W. Sun, B. Gong, J. Pan, Y. Wang, H. Xia, H. Zhang, Q. Dai, L. Wang and X. Wang, J. Catal., 2020, 391, 132–144 CrossRef CAS
- X. Liu, L. Chen, T. Zhu and R. Ning, J. Hazard. Mater., 2019, 363, 90–98 CrossRef CAS PubMed
- S. Cao, X. Fei, Y. Wen, Z. Sun, H. Wang and Z. Wu, Appl. Catal., A, 2018, 550, 20–27 CrossRef CAS
- Y. Yang, H. Li, S. Zhang, X. Yu, S. Liu, R. Qu, C. Zheng and X. Gao, Catal. Today, 2020, 355, 349–357 CrossRef CAS
- Q. Dai, S. Bai, J. Wang, M. Li, X. Wang and G. Lu, Appl. Catal., B, 2013, 142–143, 222–233 CrossRef CAS
- I. Maupin, L. Pinard, J. Mijoin and P. Magnoux, J. Catal., 2012, 291, 104–109 CrossRef CAS
- J. Zhao, W. Xi, C. Tu, Q. Dai and X. Wang, Appl. Catal., B, 2020, 263, 118237 CrossRef CAS
- Z. El Assal, S. Ojala, S. Pitkäaho, L. Pirault-Roy, B. Darif, J.-D. Comparot, M. Bensitel, R. L. Keiski and R. Brahmi, Chem. Eng. J., 2017, 313, 1010–1022 CrossRef CAS
- L. Ran, Z. Wang and X. Wang, Appl. Catal., A, 2014, 470, 442–450 CrossRef CAS
- L. Ran, Z. Qin, Z. Wang, X. Wang and Q. Dai, Catal. Commun., 2013, 37, 5–8 CrossRef CAS
- Q. Dai, S. Bai, X. Wang and G. Lu, Appl. Catal., B, 2013, 129, 580–588 CrossRef CAS
- Q. Dai, S. Bai, Z. Wang, X. Wang and G. Lu, Appl. Catal., B, 2012, 126, 64–75 CrossRef CAS
- R. López-Fonseca, B. de Rivas, J. I. Gutiérrez-Ortiz, A. Aranzabal and J. R. González-Velasco, Appl. Catal., B, 2003, 41, 31–42 CrossRef
- X. Fei, S. Cao, W. Ouyang, Y. Wen, H. Wang and Z. Wu, Chem. Eng. J., 2020, 387, 123411 CrossRef CAS
- Y. Shi, J. Wang, Y. Wang, F. Kong and R. Zhou, J. Environ. Chem. Eng., 2022, 10, 107629 CrossRef CAS
- Q. Dai, W. Wang, X. Wang and G. Lu, Appl. Catal., B, 2017, 203, 31–42 CrossRef CAS
- Y. Su, K. Fu, Y. Zheng, N. Ji, C. Song, D. Ma, X. Lu, R. Han and Q. Liu, Appl. Catal., B, 2021, 288, 119980 CrossRef CAS
- Q. Ying, Y. Liu, H. Li, Y. Zhang and Z. Wu, J. Colloid Interface Sci., 2022, 605, 537–546 CrossRef CAS PubMed
- X. Zhou, X. Lai, T. Lin, J. Feng, Z. Hou and Y. Chen, New J. Chem., 2018, 42, 16875–16885 RSC
- Q. Zhao, Y. Zheng, C. Song, Q. Liu, N. Ji, D. Ma and X. Lu, Appl. Catal., B, 2020, 265, 118552 CrossRef CAS
- M. Ma, R. Yang, Z. Jiang, C. Chen, Q. Liu, R. Albilali and C. He, Fuel, 2021, 303, 121244 CrossRef CAS
- V. Tomašić and F. Jović, Appl. Catal., A, 2006, 311, 112–121 CrossRef
- M. L. Rodríguez, L. E. Cadús and D. O. Borio, J. Environ. Chem. Eng., 2017, 5, 292–302 CrossRef
- C. Hao, W. Huang, J. Zhang, J. Wu, Y. Yue and G. Qian, J. Environ. Chem. Eng., 2021, 9, 106145 CrossRef CAS
- X. Lai, X. Zhou, H. Zhang, X. Jiang, T. Lin and Y. Chen, Appl. Surf. Sci., 2020, 526, 146714 CrossRef CAS
- J. Zhu, W. Zhang, Q. Qi, H. Zhang, Y. Zhang, D. Sun and P. Liang, Sci. Rep., 2019, 9, 12162 CrossRef PubMed
- Z. Ren, V. Botu, S. Wang, Y. Meng, W. Song, Y. Guo, R. Ramprasad, S. L. Suib and P.-X. Gao, Angew. Chem., Int. Ed., 2014, 53, 7223–7227 CrossRef CAS PubMed
- W. Zhao, Y. Dai, X. Cheng, S. Xu, Y. Guo and W. Fang, Combust. Sci. Technol., 2021, 193, 2009–2022 CrossRef CAS
- C. Peng, T. Zhou, P. Wei, H. Ai, B. Zhou, H. Pan, W. Xu, J. Jia, K. Zhang, H. Wang and H. Yu, Chem. Eng. J., 2022, 439, 135685 CrossRef CAS
- W. Liang, Y. Zhu, S. Ren, Q. Li, L. Song and X. Shi, Appl. Catal., A, 2021, 623, 118257 CrossRef CAS
- H. Huang, Q. Dai and X. Wang, Appl. Catal., B, 2014, 158-159, 96–105 CrossRef CAS
- D. Wang, J. Huang, F. Liu, X. Xu, X. Fang, J. Liu, Y. Xie and X. Wang, Catal. Today, 2020, 339, 220–232 CrossRef CAS
- S. Cao, H. Wang, F. Yu, M. Shi, S. Chen, X. Weng, Y. Liu and Z. Wu, J. Colloid Interface Sci., 2016, 463, 233–241 CrossRef CAS PubMed
- J.-H. Jang, S.-C. Lee, D.-J. Kim, M. Kang and S.-J. Choung, Appl. Catal., A, 2005, 286, 36–43 CrossRef CAS
- Z. Zhang, L. Xu, Z. Wang, Y. Xu and Y. Chen, J. Nat. Gas Chem., 2010, 19, 417–421 CrossRef CAS
- A. Khaleel and A. Al-Nayli, Appl. Catal., B, 2008, 80, 176–184 CrossRef CAS
- G. Busca, J. Lamotte, J. C. Lavalley and V. Lorenzelli, J. Am. Chem. Soc., 1987, 109, 5197–5202 CrossRef CAS
- K. Seki, Catal. Surv. Asia, 2010, 14, 168–175 CrossRef CAS
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d2ra07823f |
| This journal is © The Royal Society of Chemistry 2023 |