
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceInvestigation of adsorption behaviors, and electronic and magnetic properties for small gas molecules adsorbed on Pt-doped arsenene by density functional calculations
Ping Wua and
Min Huang *b
*b
aSchool of Mathematics & Physics, Shandong Advanced Optoelectronic Materials and Technologies Engineering Laboratory, Qingdao University of Science and Technology, Qingdao 266061, China
bKey Laboratory of Ferro and Piezoelectric Materials and Devices of Hubei Province, Faculty of Physics and Electronic Sciences, Hubei University, Wuhan 430062, China. E-mail: huangmin@hubu.edu.cn
First published on 26th January 2023
Abstract
Based on first-principles calculations, we compared the adsorption behaviors, electronic and magnetic properties of gas molecules (CO, NO, NO2, O2, NH3, H2O, H2 and N2) adsorbed on Pt-embedded arsenene and pristine arsenene. Our calculations show the interactions between molecules and arsenene can be enhanced by substitution of a Pt atom, suggesting the potential application of Pt-doped arsenene in gas sensing, especially for detecting NO2 gas due to the largest adsorption energy and charge transfer between NH3 and Pt-doped arsenene. Among all the molecules considered, CO, NO, NO2, O2 and NH3 molecules chemisorb on Pt-doped arsenene (these molecules physisorb on pristine arsenene) forming covalent Pt–C, Pt–N and Pt–O bonds, thus resulting in the elongation of C–O, N–O, O–O, N–H bonds in molecules. The magnetic moments of arsenene adsorbed with O2, NO, and NO2 decrease or diminish after Pt doping. Such variation on magnetism before and after Pt doping is ascribed to significant charge transfer and strong hybridization between gas molecules and the underlying Pt atoms, indicating the magnetic properties of arsenene can be tuned by molecular adsorption and Pt doping. These findings suggest Pt-doped arsenene has potential applications in spintronic devices, catalysts and gas sensors.
1 Introduction
Since the discovery of graphene in 2004,1 many other two-dimensional (2D) materials including BN,2 slicene,3 phosphorene,4,5 transition metal dichalcogenides (TMD) monolayers6,7 and MoSi2X4,8 LixBy monolayers9 and B2P6,10 as well as biphenylene,11 have attracted extensive attentions in the last two decades. Based on the intriguing physical and chemical properties, such as high carrier mobility, outstanding mechanical performance and large surface-to-volume ratio, 2D materials become important candidates for numerous promising applications in the fields of nanoelectronics,9,12 optoelectronic and spintronic devices,8,13 gas sensors,14,15 chemical catalysis11 and energy storage16 as well as photovoltaics.10,17 However, the practical application of these materials is restricted by some shortcomings, such as graphene having zero bandgap,18 MoS2 with relative low mobility19 and phosphorene susceptible to oxidation in ambient conditions.20 Therefore, in order to realize the application of 2D materials in the fields of gas sensor and molecule catalysis, it is necessary to search and synthesize new 2D materials exhibiting properties with improved performance. Motivated by expanding the applications, arsenene and its analogs have been investigated by theoretical and experimental methods.21–24 Zeng et al. predicted that arsenene monolayers show semiconducting behaviors with wide band-gap (about 2.49 eV) and high stability in ambient conditions, which is quite different from the semi-metallic characteristic of As bulk. They also observed significant transition from indirect into direct band gap semiconductor under small biaxial strain.21 Subsequently, multilayer arsenene with good stability in air has been successfully prepared by Tsai and Liang on InAs substrate using plasma assisted process.23 Zhong et al. reported the mechanically exfoliated b-As monolayer utilized to fabricate field-effect transistors (FET) shows relatively high carrier mobility, and thickness could modify efficiently the carrier transport characteristics.24 Moreover, the electronic and magnetic properties of arsenene could be modified substantially altered by selective substitution or adsorption of foreign atoms, such as transformation from a spin-nonpolarized semiconductor to metal for Ti/Mn-doped arsenene or a half-metal upon decoration of As, and the spin-polarized semiconducting state for V, Cr and Fe substituted arsenene have been observed.25–27 Additionally, our previous DFT + U calculations predicted that 3d transition metals (TM) doped arsenene (except for Cu doping) exhibits magnetism, which mainly contributed by the strong hybridization between 3d TM and surrounding As atoms.28Similar to phosphorene, buckled arsenene possess sizable band gap and naturally high surface-to-volume ratio, which is supposed to have promising applications in gas sensing, catalysis and energy storage.28–31 Liu predicted by DFT calculation that the adsorption of NO and NO2 molecules could effectively modify the electronic properties and introduce magnetism into arsenene, which is expected to be employed as a chemical nanosensor for NO and NO2 molecules.30 Chandiramouli et al. found that the resistance of armchair and zigzag arsenene varied upon adsorption of NH3 and PH3, implying arsenene nanoribbons could be utilized as nano-chemsensor to detect the poisonous gas molecules of NH3 and PH3.31 However, Ciraci et al. found that H2, O2, and H2O molecules prefer to physically adsorb at arsenene, which indicated that the atomic structures and electronic properties of both molecules and arsenene have no significant changes due to weak interaction upon gas adsorption.32 Additionally, Zheng et al. found that ferromagnetic couplings of CrI3 monolayer can be remarkably enhanced upon the adsorption of CO, H2, H2O and N2 molecules, particularly, Curie temperatures of CrI3 monolayer can be miraculously improved to about 2.6 times of that for pure CrI3 monolayer due to the adsorption of N2 molecule.33 It is known that vacancies, substitutional doping or adsorption of foreign atoms could be effective strategies to dramatically enhance the sensing ability of nanomaterials to gas molecules, such as transition metal embedded graphene34 and phospohorene substitutionally doped by alkali atom and transition metals exhibiting high-performance on catalyst and small gas sensing,35 as well as TMD monolayer decorated by Pt and Au as promising candidate for efficient gas sensors.36–38 Recently, based on First-principles calculations Ren and co-workers reported that atomic doping could significantly improve the hydrogen evolution reaction (HER) performance of biphenylene, such as the barrier for HER is only −0.03 eV for Fe doping case.39 Besides, Zheng et al. found that the introduction of Ga or N vacancies can significantly facilitated the adsorption strength of CO, NH3, NO, and NO2 on defective g-GaN with larger adsorption energy and more significant charge transfer than that on pristine g-GaN.40 Similarly, WS2 could with W defect be utilized as an ideal reversible sensor for detecting CO and NH3 owing to short recovery times, and S defect in WS2 could beneficially capture NO by enhancing the interaction between NO and substrate.41 Especially, the noble transition metals including Ag, Au, Pd, Pt and so on, display the strong catalytic behavior in gas interaction, and thus is frequently selected as a dopant to decorate the surfaces of nanomaterials in order to enhance the chemical sensitivity and selectivity of the substrate. Gao and co-workers found that NO2 favor to chemisorb on the Ag-doped WSe2 monolayer surface due to a strong interaction monolayer, which enable Ag-doped WSe2 monolayer to be as promising candidate for efficient NO2 gas sensors,36 and study reported by Wu et al. confirmed that Ta-doped WSe2 also shows high sensitivity and selectivity to NO2.38 Similarly, the MoSe2 monolayer decorated by Pd exhibits potential application as toxic gas sensor, which is due to strong adsorption ability and poor desorption performance upon four toxic gas (such as NO, NO2, SO2 and H2S).42 It also has been reported that Pd doping resulted in the transition of CO adsorption behavior from physisorption on antimonene to chemisorption after doping, as well as the CO gas sensitivity could be improved by external electronic field.43 Additionally, theoretical study confirmed that decoration of Ag, Au and Pt could drastically increase the interaction of CO and O2 molecules with bismuthene, particularly Pt embedded bismuthene shows high catalytic activity for CO oxidation with reaction barrier about 0.37 eV.44 Nevertheless, there are less theoretical study on the adsorption behaviors of common gas molecules on Pt-doped aresene, as well as the influence of Pt dopants on the electronic and magnetic properties of arsenene by comparing with these gas molecules adsorbed on pristine arsenene.
In this work, we firstly investigated the stability and electronic properties of Pt-doped arsenene, and then focused on the adsorption behaviors of various gas molecules (including CO, NO, NO2, O2, NH3, H2O, H2 and N2) on Pt doped-arsenene by using first-principles calculations. In order to clarify the influence of embedded Pt on the adsorption behaviors of exotic gas, we also investigated the adsorption of considered gas molecules on pristine arsenene for comparison. Furthermore, we examined the modification of electronic and magnetic properties of Pt-doped arsenene due to the adsorption of gas molecules by analyzing the charge density of states and charge transfer. This paper could not only help to comprehend the Pt doping effect on the structural and electronic properties of arsenene, but also provide new strategy to expand the potential applications of arsenene in the fields of spintronic device and gas sensor as well as scavenger for NO and NO2.
2 Computational methods
All our density-functional theory (DFT) calculations were performed using Vienna ab initio simulation package (VASP) code.45,46 The generalized gradient approximation (GGA) by Perdew–Burke–Ernzerhof (PBE) functional47 is chosen to express the electron exchange–correlation interaction in our calculations. And all-electron projector augmented wave (PAW) method is employed to describe the electron-ionic core interaction.48 The DFT-D2 method of Grimme49 was adopted to analyze the van der Waals interaction between considered gas molecule and substrate. An energy cutoff of 450 eV for the plane wave basis set is adopted. One arsenic atom is substituted by Pt atom in a (4 × 4) lateral supercell to simulate the doping of Pt, which is plotted in Fig. 1a. To sufficiently minimize the interactions between adjacent supercells, the thickness of vacuum layer in the z direction was set to be more than 20 Å. 5 × 5 × 1 and 9 × 9 × 1 Monkhorst–Pack grid for k-point sampling were used to sample the first Brillouin zone for structural optimizations and electronic calculations, respectively. The DFT calculations were spin-polarized to reveal the origin of magnetism upon the molecular adsorption. All the geometry structures are fully optimized until the total energy converged to 1.0 × 10−5 eV and the force on each atom was less than 0.01 eV Å−1. | ||
| Fig. 1 (a) Optimized structures for Pt-doped arsenene, and (b) and (c) the electronic band and projected density of states (PDOS) for the Pt-doped arsenene. The partial charge density distribution of impurities states for Pt-doped arsenene is shown in the insert of (c). The unit of isosurface charge density is set to 0.007e Å−3. The Fermi level (EF) is set to be 0 eV denoted by dash lines. Arsenic and platinum atoms are represented by green and silver balls, respectively. | ||
In order to verify the stability of Pt-doped arsenene, we calculated the formation energy of Pt-doped arsenene according to the following formula:
| Ef = EPt-arsenene + EAs − Earsenene − EPt, | (1) |
In order to examine the chemical reactivity of the Pt-doped arsenene, we have comparatively investigated the adsorption properties of considered gas molecules on the pristine arsenene and Pt-doped arsenene. The adsorption energy (Ead) and the amount of the charge transfer (ΔQ) are two key indicators used to describe the strength of interactions between gas molecules and adsorbent surface. We respectively defined the adsorption energy of small gas molecule on two surface as following:
| Ead = Egas@arsenene − Earsenene − Egas, | (2) |
| Ead = Egas@Pt-arsenene − EPt-arsenene − Egas, | (3) |
3 Results and discussion
3.1 Structures and electronic properties of pristine and Pt-doped arsenene monolayers
The calculated results show that the pristine arsenene adopts buckled honeycomb structures with thickness of 1.39 Å and indirect band gap of 1.61 eV, which is in good agreement with previous calculations.21,25 After full relaxation, the three bond angles of As–Pt–As are all enlarged from 92.00° for pristine arsenene to 106.05° for Pt-doped arsenene with Pt atom moved downwards by 0.67 Å related to upper As layer (shown in Fig. 1a). The bond length of Pt and surrounding As atoms significantly decreased to 2.41 Å from 2.51 Å for dAs–As of pristine arsenene. The three As–Pt–As bond angles and bond length of three Pt–As bonds are equivalent implying that the doping of Pt impurity atoms does not break the C3v symmetry, which is consistent with our previous studies about TM-doped arsenene systems.28 The decreasing of Pt–As bond length compared with As–As bond length is mainly caused by stronger ionic characteristic of Pt relative to As. And the calculated formation energy of Pt-doped arsenene is about −7.12 eV higher than −0.45 eV for Cr-doped arsenene,28 which indicated Pt-doped arsenene system show relatively higher stability.To explore the modulation effects of Pt dopants on the electronic and magnetic properties of arsenene, we calculated spin-polarized band structures (as shown in Fig. 1b) and spin-polarized projected density of states (PDOS) of Pt and surrounding As atoms in Pt-doped arsenene as depicted in Fig. 1c. As shown in Fig. 1b and c, the identical majority and minority states indicate the non-magnetic ground state of Pt-doped arsenene. Obviously, it has been observed two new gap states emerged simultaneously around the EF upon As replaced by foreign Pt atom, resulting in significant transformation from semiconducting arsenene to metallic Pt-doped arsenene. According to the analyzing the partial charge density distribution (PDOS) of impurity states shown in the insert of Fig. 1c, it is found that the impurity states is mainly contributed by Pt and surrounding As atoms. The DOS results further confirmed that the impurity states origin from the strong hybridization between 5d states of Pt dopant and 4p states of surrounding As atoms. The similar modulation mechanism of Pt doping on electronic properties of blue phosphorene also has been reported,50 the Pt-doped system shows metal behavior with impurities states crossed the Fermi level.
3.2 The adsorption behavior of gas molecules on Pt-doped arsenene
In order to explore the effect of Pt impurities on the adsorption properties and catalytic activities of arsenene, we further examine the adsorption behaviors of some small molecules (CO, NO, NO2, O2, NH3, H2O, H2 and N2) on the Pt-doped arsenene monolayer. To obtain the most favorable binding configurations, the gas molecules are initially placed at different sites or orientations above Pt-doped arsenene with different height and then relax all the structures. For considered gas molecules with free standing phase, the bond lengths are 1.14, 1.17, 1.21, 1.23, 1.02, 0.97, 0.75 and 1.15 Å for CO, NO, NO2, O2, NH3, H2O, H2 and N2 gas molecules, respectively. Optimized adsorption configurations of considered small gas molecules on Pt-doped arsenene are shown in Fig. 2. For comparison, the adsorption behaviors of gas molecules on prefect monolayer arsenene and the interactions between molecules and substrate are also explored in this work. Table 1 summaries the calculated adsorption energies Ead and the distance between the adsorbed molecule to substrate, charge transfer between gas molecules and arsenene for the most stable adsorption configurations of considered molecules on arsenene with and without Pt-doping. | ||
| Fig. 2 (a)–(h) Top (upper panel) and side views (lower panel) of the most favorable configurations of Pt-doped arsenene with gas molecules CO, NO, NO2, O2, NH3, H2O, H2 and N2 adsorbed, respectively. The pink, brown, blue and red balls represent the H, C, N and O atoms, respectively. | ||
| Molecule | Pt-doped arsenene | Prefect arsenene | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| d (Å) | Δq (e) | Doping nature | Ead (eV) | Mtot (μB) | Mgas (μB) | d (Å) | Δq (e) | Doping nature | Ead (eV) | Mtot (μB) | Mgas (μB) | |
| CO | 1.65 | −0.104 | Acceptor | −1.22 | 0 | 0 | 3.36 | −0.013 | Acceptor | −0.02 | 0 | 0 |
| NO | 1.50 | −0.252 | Acceptor | −1.48 | 0 | 0 | 2.80 | −0.077 | Acceptor | −0.18 | 1.00 | 0.99 |
| NO2 | 2.00 | −0.591 | Acceptor | −1.54 | 0 | 0 | 2.58 | −0.21 | Acceptor | −0.29 | 1.00 | 0.91 |
| O2 | 1.95 | −0.458 | Acceptor | −0.74 | 1.00 | 0.89 | 3.11 | −0.027 | Acceptor | −0.05 | 2.00 | 1.98 |
| NH3 | 1.86 | 0.165 | Donor | −0.80 | 0 | 0 | 2.69 | 0.067 | Donor | −0.22 | 0 | 0 |
| H2O | 1.99 | 0.038 | Donor | −0.37 | 0 | 0 | 2.21 | −0.037 | Acceptor | −0.18 | 0 | 0 |
| H2 | 2.01 | 0.017 | Donor | −0.11 | 0 | 0 | 2.89 | −0.011 | Acceptor | −0.04 | 0 | 0 |
| N2 | 2.65 | 0.008 | Donor | −0.15 | 0 | 0 | 3.34 | −0.013 | Acceptor | −0.02 | 0 | 0 |
It is found that CO adopts a nearly perpendicular alignment with C–Pt bond length of 1.65 Å (Fig. 2a), while NO prefers to adsorb on Pt-doped arsenene through N atom-oriented site with Pd–N bond titled to As surface (Fig. 2b). Interestingly, it can been seen form Fig. 2c that NO2 adopts an inverted V-type bend structure with both oxygen atoms connect to Pt atom with the shorter Pt–O distance of 2.29 Å, which could be explained by the interaction between the electron-rich oxygen atoms and Pt dopant. The similar V-type configuration of NO2 at pristine arsenene also has been observed that two O directly interact with As atoms, however, theoretical study carried out by Gaussian 09 reported the NO2 on arsenene adopted V-type orientation with N actively participated with arsenene surface, which is attributed to the overlapping of non-bonding lone pair orbitals of As with empty valence p-orbital of N.51 O2 molecule adopts a flat configuration almost parallel to As surface with Pt–O distance of 2.23 Å (Fig. 2d), while O–O bond length is enlarged from 1.23 Å for free O2 and O2 adsorbed at pristine arsenene to about 1.33 Å for Pt-doped arsenene. In the case of NH3 adsorption (Fig. 2e), N atom points toward to Pt dopant with hydrogen atoms on the next end, while H2O favors to lie nearly parallel to the Pt-doped arsenene with the O atom captured by Pt dopant and the O–Pt distance is measured to be 2.48 Å as shown in Fig. 2f. In Fig. 2g, H2 molecule prefers to adopt an end-on configuration with one H atom standing on the top site of Pt with a distance of 2.01 Å between H2 and Pt-doped arsenene. Similar to the case of O2, N2 molecule also tends to a flat configuration on Pt-doped arsenene with a distance of 2.65 Å towards to the substrate surface (Fig. 2h), while no significant change in N–N bond length was observed.
As shown in Table 1, the adsorption energies for all molecules considered adsorbed on Pt-doped arsenene are larger and the distance between molecules and Pt-doped arsenene are shorter than the corresponding molecule adsorbed on pristine arsenene, indicating Pt dopant can enhance the interactions between molecules and arsenene. For the adsorption of CO, NO, NO2, O2 and NH3, the adsorption energies are −1.22, −1.48, −1.54, −0.74 eV and −0.80 eV, with distance between gas molecules and substrate of 1.65, 1.50, 2.00, 1.95 and 1.86 Å, respectively, implying chemisorption between CO, NO, NO2, O2 and NH3 with Pt-doped arsenene. Differently, the calculated results shown that all considered molecules favor to physically adsorb at pristine arsenene via van der Waals interactions with the adsorption energies in the range of −0.02–−0.29 eV. Obviously, as listed in Table 1 the bond length of O–O and N–O is remarkably elongated to 1.33 Å and 1.28 Å (from 1.23 Å and 1.21 Å for free O2 and NO2 molecules, respectively) upon O2 and NO2 adsorption, and bond length of C–O or N–O increases to 1.16 Å and 1.20 Å (from 1.14 Å and 1.17 Å for free CO and NO molecules) for adsorption of CO and NO on Pt-doped arsenene, respectively. And conversely, the Ead are −0.37, −0.11 and −0.15 eV upon the adsorption of H2O, H2 and N2 on Pt-doped arsenene, which are slightly larger than these of adsorbed on pristine arsenene, indicating the physisorption for H2O, H2 and N2 molecules occurs both on pristine and Pt-doped arsenene. Meanwhile, no significant change in bond length was observed in the cases of H2O, H2 and N2 adsorption. The above calculated results predicted that the introduction of Pt dopant could enhance the adsorption of considered gas to some extent and significantly improve the reaction activity of CO, NO, NO2, O2 and NH3 instead of the weak physisorption of these upon pristine arsenene. To explore the effect of the concentrations, we consider one NO2 molecule adsorbed on the 2 × 2, 3 × 3 and 4 × 4 supercells of arsenene with one As atom replaced by Pt atom. Our calculations show that the interaction between NO2 molecule and the Pt-doped arsenene is slightly changed with coverage, resulting in similar Ead and bandgap value. So we could conclude that the concentrations of Pt doping have no significant influence on the adsorption behaviors of gas molecules on arsenene, which is similar to the previous studies reported by Safari et al.52
3.3 Electronic properties for gas adsorption systems
To further gain insight into how gas molecules adsorption affect the electronic properties of Pt-doped arsenene, we calculated the spin-polarized band structures as shown in Fig. 3 and projected density of states (PDOS) of gas molecule, Pt dopants and neighboring As atoms for Pt-doped arsenene systems shown in Fig. 4. The symmetric band structure indicated that the ground state of gas/Pt-doped arsenene systems exhibit nonmagnetic characteristic, except for the case of O2/Pt-doped arsenene (as shown in Fig. 3d) with magnetic moment of 1 μB. It is reasonable to expect that Pt-doped arsenene may be considerable polarized due to the charge transfer upon the adsorption of O2 molecule, which promoted O2 attaching to the substrate to some extent, thus induce the formation of stable configurations with higher adsorption energy. Regarding to CO adsorption on Pt-doped arsenene in Fig. 3a, there is no additional CO-induced states within the band gap, while the band that initially located at 0.0–0.5 eV above EF shifted up about 0.30 eV upon the adsorption of CO. By analyzed the DOS as shown in Fig. 4a, it can be seen that the molecule orbital of adsorbed CO is delocalized with respect to isolated CO, which is caused by mixture between 2π* antibonding of CO molecule and 5d of Pt dopant as well as 4p of surrounding As atoms. Upon the adsorption of NO and NO2 on Pt-doped arsenene (in Fig. 3b and c), obviously, new band states emerge at about 1.0 eV above the Fermi level and −0.25 eV below the Fermi level, respectively. Combined with the DOS results (shown in Fig. 4b and c), it is found that the impurities states mainly originate from the contribution of hybridization between 2p orbital of N or O attached to Pt and 5d orbital of Pt as well as 4p orbital of neighbor-nearing As atoms. However, it is observed that transformation of Pt-doped arsenene transform from metal to semiconductor upon the adsorption of NO2 and O2, such as NO2/Pt-doped arsenene exhibited an indirect band gap of 0.67 eV, while for O2 adsorption system, the majority and minority states display an indirect band gap of 0.73 and 0.64 eV, respectively, which are mainly resulted from the shift down of Fermi level caused by the loss of electron for arsenene. According to the DOS results of O2/Pt-doped arsenene system (Fig. 4d), it is significant that the impurity states (flat band) near 0.40 eV above Fermi level in spin-down channel mainly come from the contribution of strongly orbital hybridization between two O atoms and underlying Pt. By analyzing the band structure (Fig. 3e) combined with PDOS results (Fig. 4e), it is found that adsorption of NH3 affect the gap states and valence-band maximum (VBM) of Pt-doped arsenene, which is related to orbitals overlapping between N 2p and 5d of Pt dopant. This finding indicated the occupied nitrogen lone-pair orbital of N interacts with the valence bands of underlying Pt, which weaken the orbital hybridization between Pt and surrounding As atoms. Conversely, the band structure of Pt-doped arsenene systems in the range of −1.0 to 1.0 eV both did not significantly change before and after the adsorption of N2, H2 and H2O as shown in Fig. 3f–h, which is consistent with the PDOS results as plotted in Fig. 4f–h, respectively. And the charge density distribution of band gap states are plotted as shown in the insert of Fig. 4f–h for H2O, H2 and N2 adsorption, respectively. It is remarkable that the charge density distributions of gap states mainly focus on the Pt and surrounding As atoms accompanied with tiny contribution of gas molecules, which indicated that these small gas have little effects the electronic properties of Pt-doped arsenene, which is corresponding with physical adsorption characteristic of H2O, H2 and N2 molecules.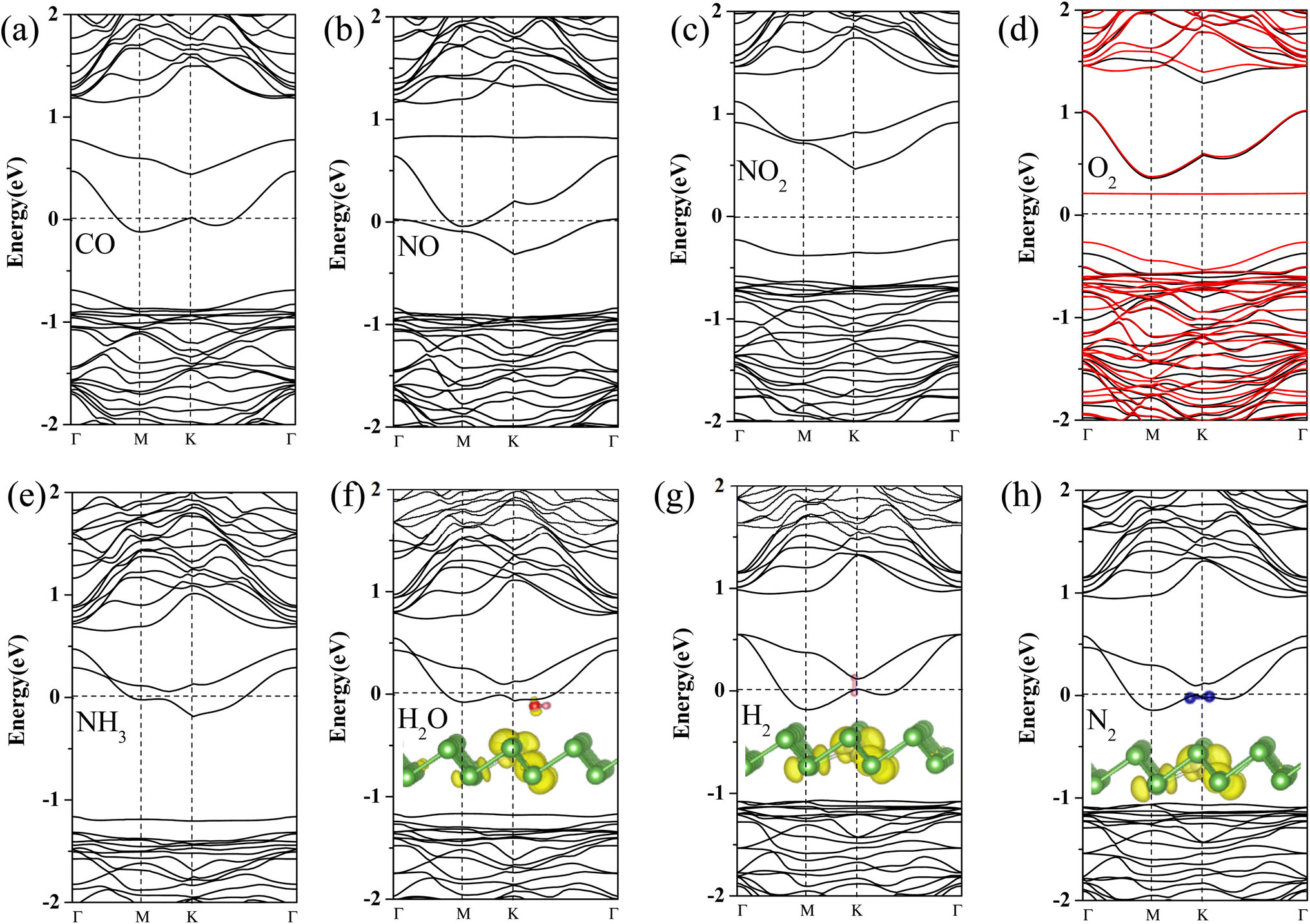 | ||
| Fig. 3 Spin-polarized band structures for (a) CO, (b) NO, (c) NO2, (d) O2, (e) NH3, (f) H2O, (g) H2 and (h) N2 adsorbed at Pt-doped arsenene, respectively. The red and black lines show spin up and spin down band structures, respectively. EF is set to be 0 eV denoted by the black dash lines. The partial charge density distribution of gap states for Pt-doped arsenene adsorbed with H2O, H2 and N2 are plotted in the insert of (f)–(h), respectively. The unit of isosurface charge density is set to 0.007e Å−3. | ||
 | ||
| Fig. 4 (a)–(h) Projected density of states (PDOS) for considered gas molecules CO, NO, NO2, O2, NH3, H2O, H2 and N2 adsorbed at Pt-doped arsenene. EF is set to be 0 eV denoted by dash lines. | ||
To quantitatively determine the amount of charge transfer from molecule to Pt-doped arsenene systems, we calculated the Bader charges53,54 and summarized the results as listed in Table 1. In here, the negative and positive values mean that the adsorbed gas obtain charge from substrate or provide charge to substrate, respectively. It is noted that CO, NO, NO2 and O2 acting as strong accepter attract 0.104, 0.252 and 0.458e from the substrate of Pt-doped arsenene, respectively, especially for NO2 obtained 0.591e, while 0.168e is transferred form NH3 molecule to the arsenene decorated by Pt. For comparison, we found most of considered gas molecules acting as weak acceptor received charge smaller than 0.08e from pristine arsenene, except for NO2 which acting as a strong acceptor gained about 0.21e from arsenene, while NH3 acting as weak donor slightly donated about 0.067e to arsenene. As known that the charge transfer mechanism and interaction between gas molecules and adsorbent surface are important characteristic as chemisorption. Compared with gas molecules adsorbed at pristine arsenene, remarkable charge transfer indicated that above five molecules interact strongly with Pt-doped arsenene surface and maybe engage in notable chemical interaction with the embedded Pt atom. That is why most considered molecules favor to chemically adsorb at Pt embedded arsenene monolayer with higher adsorption energies and shorter distance between gas molecules and adsorbent surface as listed in Table 1. For the adsorption of H2O, H2 and N2, charge transfer between gas molecules and substrate has slight change before and after the doping of Pt, which confirmed that H2O, H2 and N2 molecules still belonged to physisorption on substrate via van der Waals forces. In other words, the doping of Pt slightly enhances the interaction between H2O, H2 and N2 with substrate, which is in good agreement with the calculated adsorption energies and distance between molecules and arsenene listed in Table 1. In order to test the accuracy of DFT-D2 method for describing weak dispersion forces in current work, we took NO2, O2 and N2 molecules as examples to investigate the adsorption properties of these molecules on Pt-doped arsenene by using D3-BJ method55,56 implemented in VASP. It is found that our DFT-D2 results are generally in accordance with D3-BJ results, the charge transfers obtained by two methods are almost identical. Although the values of the adsorption energies are slightly different for the corresponding molecule by using DFT-D2 and D3-BJ, the variation trend is the same from N2 to NO2 adsorption (N2 with lowest adsorption energy and NO2 with highest adsorption energy). And we could make the same conclusion that N2 prefers to physically adsorb at Pt-doped arsenene, while O2 and NO2 favor to chemisorption with both DFT-D2 and D3-BJ. Therefore, we can expect that our DFT-D2 results and our conclusions in our work are basically reliable.
To visualize clearly the charge transfer and the bonding mechanism between gas molecules and Pt-doped arsenene, we also plotted the corresponding charge density difference images as shown in Fig. 5. According to Fig. 5a–d, we not only observed noticeable charge redistribution between molecules and Pt-doped arsenene, but also found the charge obtained by CO, NO, NO2, and O2 mainly come from underlying Pt atom and surrounding As atoms, which correspond with the noticeable change in the DOS near the Fermi level as depicted in Fig. 4a–d. This strong interaction between above four molecules and substrate is identified as the main reason for the significant elongation of bond length and enhancement of adsorption behavior of gas molecules on Pt-doped arsenene. Differently, the charge accumulation occurs around underlying Pt and surrounding As atoms, whereas charge depletion appears near NH3 molecule for NH3 adsorbed on Pt-doped arsenene as shown in Fig. 5e. The significant charge redistribution upon NH3 adsorption indicated N atom participated in the strong hybridization with underlying Pt and formed covalent bonds with ionic bond characteristics, which arising mainly from the ionization of the lone electron pair at the N atom. The total amount of transferred charge from NH3 to Pt-doped arsenene is 0.165e, while NH3 acting as a weaker donor by transferring only about 0.067e to pristine arsenene. By comparison, the increase of charge transfer mainly caused by the stronger orbital overlapping between 2p of N and 5d of underlying Pt atom, which is consistent with the results on PDOS (Fig. 4e) and band structure (Fig. 3e). Compared with the cases of CO, NO, NO2 and O2, the atomic structure of NH3 is nearly unchanged after adsorption and the adsorption energy (−0.80 eV) is moderate on Pt-doped arsenene, implying that the desorption of NH3 from Pt-doped arsenene can be readily achieved by annealing or short UV irradiation, which is an essential target for efficiency gas sensors. Therefore, Pt-doped arsenene can be expected to detect NH3 with better desorption performance than detecting CO, NO, NO2 and O2. Interestingly, the roles of gas molecules of H2O, H2 and N2 changed from weak donors when adsorbed at pristine arsenene to weak acceptors upon adsorption on Pt-doped arsenene as listed in Table 1. Upon the adsorption of H2O, H2 and N2 on Pt-doped arsenene as shown in Fig. 5f–h, the Pt dopant did not induce noticeable variation in charge density for gas molecules and substrate, implying a weak physisorption between these three gas molecules and substrate. In most of considered cases, Pt dopant acting as an electron localization center displays high carrier mobility, e.g. NO2 system, the Pt dopant accumulate electron from the arsenene and then provide to NO2, while Pt dopant obtained electron from NH3 and delivered it to arsenene, indicated that Pt dopant plays important role to bridge the charge-transfer between the arsenene and the gas molecules. Actually, embedded Pt is the intermediary of arsenene to interact with target molecules, which apparently enhances the chemical activity and sensitivity of arsenene upon CO, NO, NO2 and O2 adsorption. We can conclude that this bonding mechanism between gas molecules and arsenene suggested the embedded Pt atom is vital in stability and electronic properties of gas/Pt-arsenene systems.
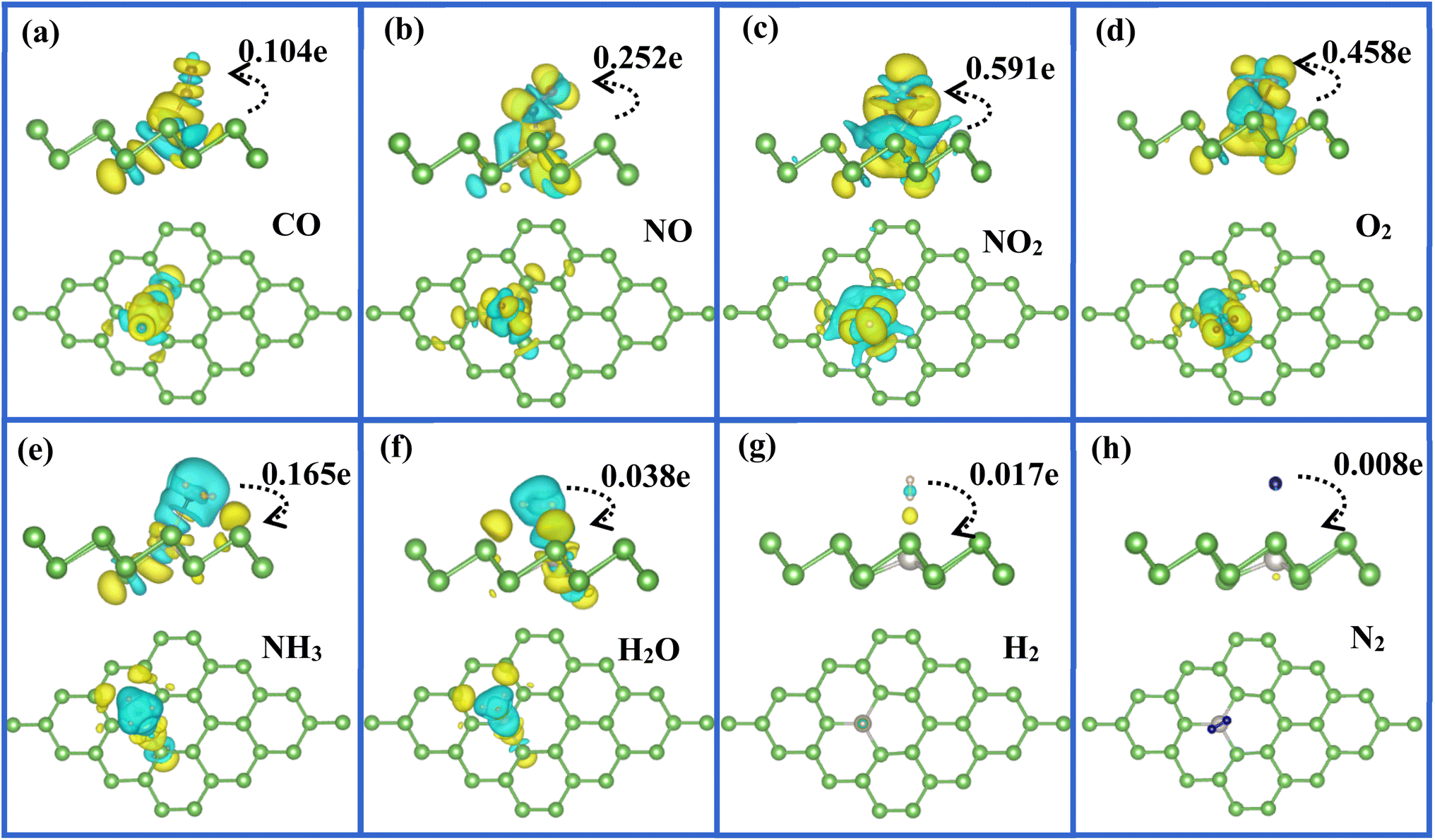 | ||
| Fig. 5 (a)–(h) Top (upper panel) and side views (lower panel) of the charge density difference plots for all considered gas molecules adsorbed at Pt-doped arsenene, respectively, in which yellow and green regions refer to the charge accumulation and depletion, respectively. The isosurface level is in the unit of 0.001e Å−3. | ||
It is demonstrated that the charge transfers occurred between gas molecules and Pt-doped arsenene are larger than those of pristine arsenene (shown in Table 1). It is well-shown that the charge carrier concentration induced by gas molecule adsorption can lead to the variation of the resistance of arsenene when it is exposed to these gases. The strong interactions between gas molecules and Pt-doped arsenene with large charge transfer compared to pristine arsenene can lead to remarkable conductivity change, which also provides basic for the potential application of making highly sensitive sensors, even with the possibility of detecting an individual molecule. On comparing the adsorption behaviors of small molecules (CO, NO, NO2, O2, NH3, H2O, H2 and N2) on pristine arsenene, it is clear that Pt impurity can enhance in varying degrees the interactions between all the molecules and arsenene with higher adsorption energy and shorter distance between molecules and arsenene. Therefore, Pt-doped arsenene can be used as gas sensors detecting such molecules with enhanced sensitivity compared with pristine arsenene, which is similar to previous reports for Ag-doped and Ta-doped WSe2 monolayer show high sensitivity and selectivity to NO2.32,34 Among all the molecules considered, Pt-doped arsenene show highest sensitivity in detecting NO2 gas than other molecules due to the largest adsorption energy and charge transfer upon NO2 adsorption.
3.4 Magnetic characteristics analysis for gas adsorption systems
We found that NO and NO2 adsorbed at pristine arsenene exhibit magnetic ground states with total magnetic moments (Mtot) of 1 μB per supercell, which is similar to NO and NO2 adsorbed at WS2,41 and arsenene with adsorption of O2 display magnetic characteristic with Mtot of 2 μB as listed in Table 1. More interestingly, Mtot of O2/Pt-doped arsenene reduced to 1 μB due to the introduction of Pt dopant, while magnetic moment nearly diminishes for NO/Pt-doped arsenene and NO2/Pt-doped arsenene systems. According to the change on magnetism of adsorption system, Pt-doped arsenene can be used to distinguish O2 from other gas molecules in the field of gas sensing. To achieve deeper insight into the origin of magnetism and influence of Pt dopant on magnetism variation of adsorption systems, the corresponding magnetization density distribution for perfect arsenene with NO, NO2 and O2 adsorbed and Pt-doped arsenene with O2 adsorbed are plotted in Fig. 6. For cases of NO adsorbed at pristine arsenene as shown in Fig. 6a, the magnetism of systems mainly comes from the contributions of gas molecules, which is caused by the unpaired electron of NO and O2 and weaker interaction between gas molecules and arsenene. Upon the adsorption of NO2 on pristine arsenene (Fig. 6b), the magnetism mainly originates from NO2 with unpaired electron, as well as underlying and second nearest-neighboring As atoms also have minor contributions to total magnetism, which is corresponding to the 0.21e charge transferred from arsenene to the NO2 molecule. It is found that magnetism of NO/Pt-doped arsenene and NO2/Pt-doped arsenene system disappeared, which is due to the orbital overlapping and dramatic charge transfer between NO and NO2 with underlying Pt atom. As shown in Fig. 6c, O2 still remains paramagnetic character with 2 μB upon adsorbed at arsenene, which is basically unaffected by the adsorbent substrate with weak interaction and slight charge transfer. As shown in Fig. 6d the magnetization density distributions of Pt-doped arsenene with O2 adsorbed mainly involved in O2 molecule and underlying Pt dopant, which is quite different with the case of O2/arsenene system (Fig. 6c). The significant decrease of magnetism from 2 μB for O2 adsorbed arsenene to 1 μB for O2 adsorbed Pt-doped arsenene could be explained by the formation of covalent bond between O and Pt atoms, which is accompanied by the elongation of O–O band and charge transfer (0.458e) between O2 and Pt dopant, further indicating that O–Pt bond is a mixture of ionic and covalent characteristics. Additionally, the redistribution of spin-charge density could further confirm the stronger hybridization between 2p orbitals of O atom and the 5d orbitals of underlying Pt atom, which is consistent with the results obtained by PDOS in Fig. 4d. Therefore, our calculations indicate that the magnetic properties of Pt-doped arsenene can be effectively tuned by the molecular adsorption thus Pt-doped arsenene can be a promising avenue to design nanoscale spintronic devices. | ||
| Fig. 6 (a)–(c) Top (upper panel) and side views (lower panel) of the magnetization charge density difference plots for NO, NO2 and O2 adsorbed on pristine arsenene, and (d) for O2 adsorbed at Pt-doped arsenene, in which blue and red regions correspond to up and down magnetization, respectively. The magnetization charge density isosurface value is 0.001e Å−3. | ||
4 Conclusions
In summary, we investigated the adsorption behaviors, electronic and magnetic properties of small gas molecules (including CO, NO, NO2, O2, NH3, H2O, H2 and N2) on pristine and Pt-doped arsenene using density-functional theory (DFT). Compared with the cases of adsorption on pristine arsenene with adsorption energy from −0.02 to −0.29 eV, most of considered gas (except H2O, H2 and N2) prefer to chemisorption at the Pt-doped arsenene with Ead higher than −0.80 eV. The calculated band structure results show that Pt-doped arsenene transforms from metal to semiconductor upon the adsorption of O2 and NO2 caused by the shifting down of Fermi level, while other adsorption systems still exhibit metallic behavior. Upon CO, NO, NO2, O2 and NH3 adsorption, the redistribution of charge density manifested the strong orbital hybridization between some gas molecules and Pt-decorated arsenene accompanied by the formation of covalent bond of gas molecules with underlying Pt dopant. Taken O2/Pt-doped arsenene as example, the 2p of O strongly hybridize with 5d orbitals of underlying Pt atom showing that O2 is highly activated, which is corresponding to the elongation of O–O bond length. Conversely, the electronic properties of Pt-doped arsenene basically unchanged before and after H2O, H2 and N2 adsorption, which is related to the physical adsorption mechanism and little charge transfer between molecules and substrate. It is noticeable that the embedded Pt in general can remarkably enhance the adsorption of gas molecules, suggesting that the Pt-doped arsenene can be used as gas (such as O2, CO, and NH3) sensor, or even as scavenger such for NO and NO2, with higher sensitivity than pristine arsenene due to the large charge transfer between molecules and substrate.Interestingly, the magnetic moment properties of gas molecules adsorbed systems also change significantly. The magnetic moment of NO, NO2 and O2 adsorbed at pristine arsenene is calculated to be about 1, 1 and 2 μB, respectively, which is caused by the unpaired electrons of gas molecule and hardly interaction between gas molecule and substrate. However, for the case of O2 adsorbed at Pt-doped Arsenene, the magnetic moment decreases drastically to 1 μB, while the magnetic moment fully diminishes for NO and NO2 adsorption. The magnetisms of O2/Pt-doped arsenene arise from the main contributions of O2 molecule and underlying Pt atom as well as minor contributions of surrounding As atoms, which is quite different with O2/arsenene. It is reasonable to explain the variation on magnetism before and after the introduction of Pt by dramatic charge transfer and strong hybridization occurred between gas molecule and arsenene by embedded Pt atoms as bridge, which indicated that magnetic properties of Pt-doped arsenene can be effectively tuned by the molecular adsorption, as well as Pt-doped arsenene may be used to realize the single O2 molecule detection by magnetic method. Therefore, our calculated results suggest that Pt-doped arsenene can be a promising avenue to design nanoscale spintronic devices, catalysis and gas sensors.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
We gratefully acknowledge the financial supported from High Education Key Program of Henan Province of China (20A140022).References
- K. S. Novoselov, A. K. Geim, S. V. Morozov, D. Jiang, Y. Zhang, S. V. Dubonos, I. V. Grigorieva and A. A. Firsov, Science, 2004, 306, 666–669 CrossRef CAS PubMed.
- W. Lei, V. N. Mochalin, D. Liu, S. Qin, Y. Gogotsi and Yi. Chen, Nat. Commun., 2015, 6, 8849 CrossRef CAS PubMed.
- S. Das, W. Zhang, M. Demarteau, A. Hoffmann, M. Dubey and A. Roelofs, Nano Lett., 2014, 14(10), 5733–5739 CrossRef CAS PubMed.
- H. Liu, A. T. Neal, Z. Zhu, Z. Luo, X. Xu, D. Tománek and P. D. Ye, ACS Nano, 2014, 8(4), 4033–4041 CrossRef CAS PubMed.
- M. Xu, T. Liang, M. Shi and H. Chen, Chem. Rev., 2013, 113, 3766–3798 CrossRef CAS PubMed.
- A. Splendiani, L. Sun, Y. Zhang, T. Li, J. Kim, C. Y. Chim, G. Galli and F. Wang, Nano Lett., 2010, 10, 1271–1275 CrossRef CAS PubMed.
- Z. Yu, Z. Y. Ong, S. Li, J. B. Xu, G. Zhang, Y. W. Zhang, Y. Shi and X. Wang, Adv. Funct. Mater., 2017, 27, 1604093 CrossRef.
- M. Sun, M. R. Fiorentin, U. Schwingenschlögl and M. Palummo, npj 2D Mater. Appl., 2022, 6, 81 CrossRef CAS.
- K. Ren, Y. Yuan, Z. Zhang, M. Sun and U. Schwingenschlögl, Appl. Surf. Sci., 2022, 604, 154317 CrossRef CAS.
- K. Ren, H. Shu, W. i. Huo, Z. Cui, J. Yu and Y. Xu, Phys. Chem. Chem. Phys., 2021, 23, 24915 RSC.
- Y. Luo, C. Ren, Y. Xu, J. Yu, S. Wang and M. Sun, Sci. Rep., 2021, 11, 19008 CrossRef CAS PubMed.
- Y. Yang, H. Hou, G. Zou, W. Shi, H. Shuai, J. Li and X. Ji, Nanoscale, 2019, 11, 16 RSC.
- W. Choi, N. Choudhary, G. H. Han, J. Park, D. Akinwande and Y. H. Lee, Mater. Today, 2017, 20(3), 116–130 CrossRef CAS.
- A. N. Abbas, B. Liu, L. Chen, Y. Ma, S. Cong, N. Aroonyadet, M. Köpf, T. Nilges and C. Zhou, ACS Nano, 2015, 9(5), 5618–5624 CrossRef CAS PubMed.
- A. Yang, D. Wang, X. Wang, D. Zhang, N. Koratkarc and M. Rong, Nano Today, 2018, 20, 13–32 CrossRef CAS.
- F. R. Fan, R. Wang, H. Zhang and W. Wu, Chem. Soc. Rev., 2021, 50, 10983–11031 RSC.
- C. Tan, X. Cao, X. Wu, Q. He, J. Yang, X. Zhang, J. Chen, W. Zhao, S. Han, G. Nam, M. Sindoro and H. Zhang, Chem. Rev., 2017, 117(9), 6225–6331 CrossRef CAS PubMed.
- S. Z. Butler, S. M. Hollen, L. Cao, Y. Cui, J. A. Gupta, H. R. Gutiérrez, T. F. Heinz, S. S. Hong, J. Huang and A. F. Ismach, ACS Nano, 2013, 7, 2898–2926 CrossRef CAS PubMed.
- Y. Cai, G. Zhang and Y.-W. Zhang, J. Am. Chem. Soc., 2014, 136, 6269–6275 CrossRef CAS PubMed.
- Y. Abate, D. Akinwande, S. Gamage, H. Wang, M. Snure, N. Poudel and S. B. Cronin, Adv. Mater., 2018, 30, 1704749 CrossRef PubMed.
- S. Zhang, Z. Yan, Y. Li, Z. Chen and H. Zeng, Angew. Chem., 2015, 127, 1–5 CrossRef.
- S. Zhang, M. Xie, F. Li, Z. Yan, Y. Li, E. Kan, W. Liu, Z. Chen and H. Zeng, Angew. Chem., Int. Ed., 2016, 55, 1666–1669 CrossRef CAS PubMed.
- H.-S. Tsai, S.-W. Wang, C.-H. Hsiao, C.-W. Chen, H. Ouyang, Y.-L. Chueh, H.-C. Kuo and J.-H. Liang, Chem. Mater., 2016, 28, 425–429 CrossRef CAS.
- M. Zhong, Q. Xia, L. Pan, Y. Liu, Y. Chen, H.-X. Deng, J. Li and Z. Wei, Adv. Funct. Mater., 2018, 28, 1802581 CrossRef.
- Z. Li, W. Xu, Y. Yu, H. Du, K. Zhen, J. Wang, L. Luo, H. Qiu and X. Yang, J. Mater. Chem. C, 2016, 4, 362–370 RSC.
- G. Li, Y. Zhao, S. Zeng and J. Ni, Appl. Surf. Sci., 2016, 390, 60–67 CrossRef CAS.
- M. Sun, S. Wang, Y. Du and J. Yu andW. Tang, Appl. Surf. Sci., 2016, 389, 594–600 CrossRef CAS.
- P. Wu and M. Huang, Appl. Surf. Sci., 2020, 506, 144660 CrossRef CAS.
- N. N. Som, V. Mankad and P. K. Jha, Int. J. Hydrogen Energy, 2018, 43, 21634–21641 CrossRef CAS.
- C. Liu, C.-S. Liu and X. Yan, Phys. Lett. A, 2017, 381, 1092–1096 CrossRef CAS.
- R. Bhuvaneswari, V. Nagarajan and R. Chandiramouli, Appl. Surf. Sci., 2019, 469, 173–180 CrossRef CAS.
- F. Ersan, E. Aktürk and S. Ciraci, J. Phys. Chem. C, 2016, 120, 14345–14355 CrossRef CAS.
- Z. Zheng, K. Ren, Z. Huang, Z. Zhu and J. Yu, Semicond. Sci. Technol., 2021, 36, 075015 CrossRef CAS.
- M. Zhou, Y.-H. Lu, Y.-Q. Cai, C. Zhang and Y.-P. Feng, Nanotechnology, 2011, 22, 385502 CrossRef PubMed.
- N. Suvansinpan, F. Hussain, G. Zhang, C. H. Chiu, Y. Cai and Y.-W. Zhang, Nanotechnology, 2016, 27, 065708 CrossRef PubMed.
- J. Ni, W. Wang, M. Quintana, F. Jia and S. Song, Appl. Surf. Sci., 2020, 514, 145911 CrossRef CAS.
- X. Lu, L. Guo, P. Wang, M. Cui, D. Kanghong and W. Peng, Appl. Surf. Sci., 2020, 513, 145860 CrossRef CAS.
- Z. Lu, Y. Zhai, Q. Liang and W. Wu, Chem. Phys. Lett., 2020, 755, 137737 CrossRef CAS.
- K. Ren, H. Shu, W. Huo, Z. Cui and Y. Xu, Nanotechnol., 2022, 33, 345701 CrossRef PubMed.
- Z. Cui, X. Wang, Y. Ding, E. Li, K. Bai, J. Zheng and T. Liu, Appl. Surf. Sci., 2020, 530, 147275 CrossRef CAS.
- Z. Cui, K. Yang, Y. Shen, Z. Yuan, Y. Dong, P. Yuan and E. Li, Appl. Surf. Sci., 2023, 613, 155978 CrossRef CAS.
- S. Ma, L. Su, L. Jin, J. Su and Y. Jin, Phys. Lett. A, 2019, 383, 125868 CrossRef CAS.
- T. T. Li, C. He and W. X. Zhang, Appl. Surf. Sci., 2018, 427, 388–395 CrossRef CAS.
- W. Pan, B. Zhao, N. Qi and Z. Chen, Mol. Catal., 2021, 501, 111379 CrossRef CAS.
- G. Kresse and J. Furthmüller, Comput. Mater. Sci., 1996, 6(1), 15–50 CrossRef CAS.
- G. Kresse and J. Furthmüller, Phys. Rev. B: Condens. Matter Mater. Phys., 1996, 54(16), 11169–11186 CrossRef CAS PubMed.
- J. P. Perdew, K. Burke and M. Ernzerhof, Phys. Rev. Lett., 1996, 77(18), 3865–3868 CrossRef CAS PubMed.
- G. Kresse and D. Joubert, Phys. Rev. B: Condens. Matter Mater. Phys., 1999, 59(3), 1758–1775 CrossRef CAS.
- S. Grimme, J. Antony, S. Ehrlich and H. Krieg, J. Chem. Phys., 2010, 132(15), 154104 CrossRef PubMed.
- B. Su and N. Li, J. Magn. Magn. Mater., 2019, 469, 236–244 CrossRef CAS.
- M. S. Khan, A. Srivastava and R. Pandey, RSC Adv, 2016, 6, 72634 RSC.
- F. Safari, M. Moradinasab, M. Fathipour and H. Kosina, Appl. Surf. Sci., 2019, 464, 153–161 CrossRef CAS.
- E. Sanville, S. D. Kenny, R. Smith and G. Henkelman, J. Comput. Chem., 2007, 28, 899–908 CrossRef CAS PubMed.
- G. Henkelman, A. Arnaldsson and H. Jonsson, Comput. Mater. Sci., 2006, 36, 354–360 CrossRef.
- W. Reckien, F. Janetzko, M. F. Peintinger and T. Bredow, J. Comput. Chem., 2012, 33, 2023–2031 CrossRef CAS PubMed.
- N. V. Ilawe, J. A. Zimmerman and B. M. Wong, J. Chem. Theory Comput., 2015, 11, 5426–5435 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2023 |