
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceAlteration of the central core of a DF-PCIC chromophore to boost the photovoltaic applications of non-fullerene acceptor based organic solar cells†
Amna Zahoora,
N. M. A. Hadia *b,
Sahar Javaid Akrama,
Rana Farhat Mehmoodc,
Sonia Sadiqa,
Ahmed M. Shawky*d,
Naifa S. Alatawie,
Asma Ahmedf,
Javed Iqbal*a and
Rasheed Ahmad Khera*a
*b,
Sahar Javaid Akrama,
Rana Farhat Mehmoodc,
Sonia Sadiqa,
Ahmed M. Shawky*d,
Naifa S. Alatawie,
Asma Ahmedf,
Javed Iqbal*a and
Rasheed Ahmad Khera*a
aDepartment of Chemistry, University of Agriculture, Faisalabad 38000, Pakistan. E-mail: Javed.iqbal@uaf.edu.pk; javedkhattak79@gmail.com; rasheed.ahmad.khera@uaf.edu.pk; rasheedahmadkhera@yahoo.com
bPhysics Department, College of Science, Jouf University, P.O. Box 2014, Sakaka, Al-Jouf, Saudi Arabia. E-mail: nmhadia@ju.edu.sa
cDepartment of Chemistry, Division of Science and Technology, University of Education, Township, Lahore 54770, Pakistan
dScience and Technology Unit (STU), Umm Al-Qura University, Makkah, 21955, Saudi Arabia
ePhysics Department, Faculty of Science, University of Tabuk, Tabuk, 71421, Saudi Arabia
fDepartment of Computer Science, Faculty of Computer and Information Technology, University of Tabuk, Tabuk, Saudi Arabia
First published on 24th February 2023
Abstract
Modifying the central core is a very efficient strategy to boost the performance of non-fullerene acceptors. Herein five non-fullerene acceptors (M1–M5) of A–D–D′–D–A type were designed by substituting the central acceptor core of the reference (A–D–A′–D–A type) with different strongly conjugated and electron donating cores (D') to enhance the photovoltaic attributes of OSCs. All the newly designed molecules were analyzed through quantum mechanical simulations to compute their optoelectronic, geometrical, and photovoltaic parameters and compare them to the reference. Theoretical simulations of all the structures were carried out through different functionals with a carefully selected 6-31G(d,p) basis set. Absorption spectra, charge mobility, dynamics of excitons, distribution pattern of electron density, reorganization energies, transition density matrices, natural transition orbitals and frontier molecular orbitals, respectively of the studied molecules were evaluated at this functional. Among the designed structures in various functionals, M5 showed the most improved optoelectronic properties, such as the lowest band gap (2.18 e V), highest maximum absorption (720 nm), and lowest binding energy (0.46 eV) in chloroform solvent. Although the highest photovoltaic aptitude as acceptors at the interface was perceived to be of M1, its highest band gap and lowest absorption maxima lowered its candidature as the best molecule. Thus, M5 with its lowest electron reorganization energy, highest light harvesting efficiency, and promising open-circuit voltage (better than the reference), amongst other favorable features, outperformed the others. Conclusively, each evaluated property commends the aptness of designed structures to augment the power conversion efficiency (PCE) in the field of optoelectronics in one way or another, which reveals that a central un-fused core having an electron-donating capability with terminal groups being significantly electron withdrawing, is an effective configuration for the attainment of promising optoelectronic parameters, and thus the proposed molecules could be utilized in future NFAs.
1 Introduction
Giving consideration to the current dilemma of energy shortage and climate change, it is understood that there should be an alternative to the existing unsustainable energy sources. Such contemplations can come true with fruitful results by installing a photovoltaic setup.1–3 Photovoltaic units absorb sunlight and convert its energy into electrical current. Because these units are independent of the utility grid, this technique is exclusively cost-effective for remote sites.4,5 Furthermore, the fuel source of solar energy, the sun, requires no drilling, refining, or delivery to the site, like that of petroleum-based fuels.6The first photovoltaic panel was invented by Charles Fritts in 1883 using a sheet of selenium, coated with a thin layer of gold, which achieved an efficiency of only 1%. This efficiency was increased up to 4% in the 1900s by replacing selenium with other materials (like silicon), however, this came with the disadvantage of a high cost.7–9 Now more than a century after the invention of the primeval solar cells, third generation-based organic solar cells (OSCs) are replacing the previous ones, due to their large storage capacity, flexibility, and high optical absorption coefficient.10,11 Moreover, they are quite affordable and easy to install, due to their films being 1000 times more slender than inorganic SCs.12 These solar cells are comprised of both an electron-donating (polymer, etc.) and an electron-accepting layer. For the later layer, non-fullerene acceptors (NFAs) have received more attention in recent years, as compared to their fullerene counterparts due to facile synthesis methods, lower fabrication costs, as well as easy tunability of band gap, which shifts the absorption to the maximum value (in UV, visible, and IR ranges).13–16
Till now, 18% PCE has been achieved with A–D–A′–D–A type Y-series NFAs, and it is seen that the push–pull effect between the acceptor and donor fragments is generally the reason behind their better efficiencies.17,18 The fused ring backbone of these NFAs allows for prominent planarity, which facilitates efficient charge transfer, better π–π stacking19,20, and increased absorption in the UV-vis region.21 However, the complex fabrication of these structures with a minimum of five to fifteen steps leads to a reduced yield and an increased cost in their production.22–24 In addition, due to the phenomenon of photo-oxidation, they are seen to have poor photostability when exposed to sunlight and air. Thus, non-fused ring based NFAs were introduced, which are simply designed (in two to four steps) by separating the donor–acceptor components of the molecule with a single bond, which allows for higher yield, as well as lower cost. Though the A–D–A′–D–A configuration of these NFAs seems to be quite proficient, other sequences are also being explored. In this regard, a recent study conducted on hundreds of NFAs showed that 57.1% of the studied A–D–D′–D–A sequenced NFAs performed better upon comparison with other analogues, especially A–D–A′–D–A ones. This sequence in conjunction with A–A–D–A–A one also exhibited the highest theoretical PCEs, which negates the need for the alternating sequence of A–D building blocks in the molecule. This sequence of the A–D components also agrees with our understanding of the push–pull effect mentioned above.25
So, it seemed to us that NFAs with an A–D–D′–D–A configuration might perform better than their corresponding A–D–A′–D–A counterparts in OSCs. To test this assumption, the central electron-accepting core (A') of the 2,2'-((2Z,2′Z)-(((2,5-difluoro-1,4-phenylene)bis(4,4-bis(2-ethylhexyl)-4H-cyclopenta[2,1-b:3,4-b′]dithiophene-6,2-diyl))bis(methanylylidene))bis(3-oxo-2,3-dihydro-1H-indene-2,1-diylidene))dimalononitrile (DF-PCIC) molecule was replaced with different strongly conjugated and electron rich donor groups. As a result, various A–D–D′–D–A based molecules were formulated for their possible higher photovoltaic performance (open-circuit voltage, fill factor, band gap, absorption, etc.) than the reference (DF-PCIC). The reason behind the selection of DF-PCIC as the reference molecule was the weak and significantly low conjugated electron-withdrawing accepting core (which is to be substituted)26 attached to the strongly electron-donating donors.27,28
In A–D–A′–D–A type geometry of this remarkable DF-PCIC molecule, the central donor (A') core (2,5-difluorobenzene (DFB)) is linked to two electron-accepting end groups (2-(3-oxo-2,3-dihydro-1H-inden-1-ylidene)malonitrile) via two electron-donating cyclopentadithiophene CPDT (D) units. DF-PCIC molecule reveals a low optical band gap of 1.59 eV in film, a reduced value of band gap of 1.72 eV,29 as well as a sharp peak in its absorption spectrum at 671 nm in the chloroform solvent. Its calculated values for open-circuit voltage; Voc, fill factor, short circuit current, and power conversion efficiency were 0.91 V, 72.62%, 15.66 mA cm−2, and 10.14%, respectively, when computed with the PBDB-T polymer donor.28 The relatively low Voc of DF-PCIC molecule was attributed to the electron-withdrawing fluoro groups bonded to the core ring of the molecule, which significantly lowered the LUMO value and thus reduced the relative Voc.28 With this in mind, the root cause of the low Voc, i.e., the electron accepting core (A') of the reference was replaced with various strongly conjugated and electron rich donor cores, so that possible higher Voc can be achieved while retaining the exceptional properties of the rest of the structure.
2 Computational methodolgy
To execute quantum mechanical calculations, the Gaussian 09,30 program was used, while to visualize the attained outputs, a well-known GaussView 6.0 software31 was employed. For the determination of the method to be utilized in this theoretical work, density functional theory (DFT)32 was first used to geometrically optimize the reference (R) at the ground state using four hybrid functionals; CAM-B3LYP,33 B3LYP,34 ωB97XD,35 and MPW1PW91,36 with carefully selected basis set; 6-31G(d,p). After the geometrical optimization of R, its time-dependent-DFT37 calculations were made in the excited state, specifically the cited chloroform solvent. Chloroform was used as the solvent of choice due to its implementation on the reference molecule in the cited literature, so that a relatable replication of environment of reference can be made possible.28 The consequence of solvent (chloroform) on the optical and electronic parameters of the molecule was determined with the help of the Polarizable Continuum Model (PCM) using the integral equation formalism variant, i.e., IEFPCM.38,39 The thus obtained wavelength of maximum absorption (λmax) of R molecule obtained by the afore-stated functional, in solvent phase was 527, 700, 651, and 500 nm, accordingly (Fig. 1; attained through Origin 6.0,40 software for ten excited states). Amongst which, MPW1PW91 was perceived to be the best functional for further computations, due to its proximity to the literature value of 671 nm for R molecule.41 However, upon further validation according to the frontier molecular orbitals and corresponding band gap values, it was seen that the B3LYP functional gave the closest results to the experimental values (Table S1†). Thus, it was also utilized for calculation of the some of the opto-electronic properties of the studied molecules. Additionally, in order to avoid any functional related error, the long range functional-ωB97XD was also used to evaluate the excited state properties of the molecules in the phases of study.42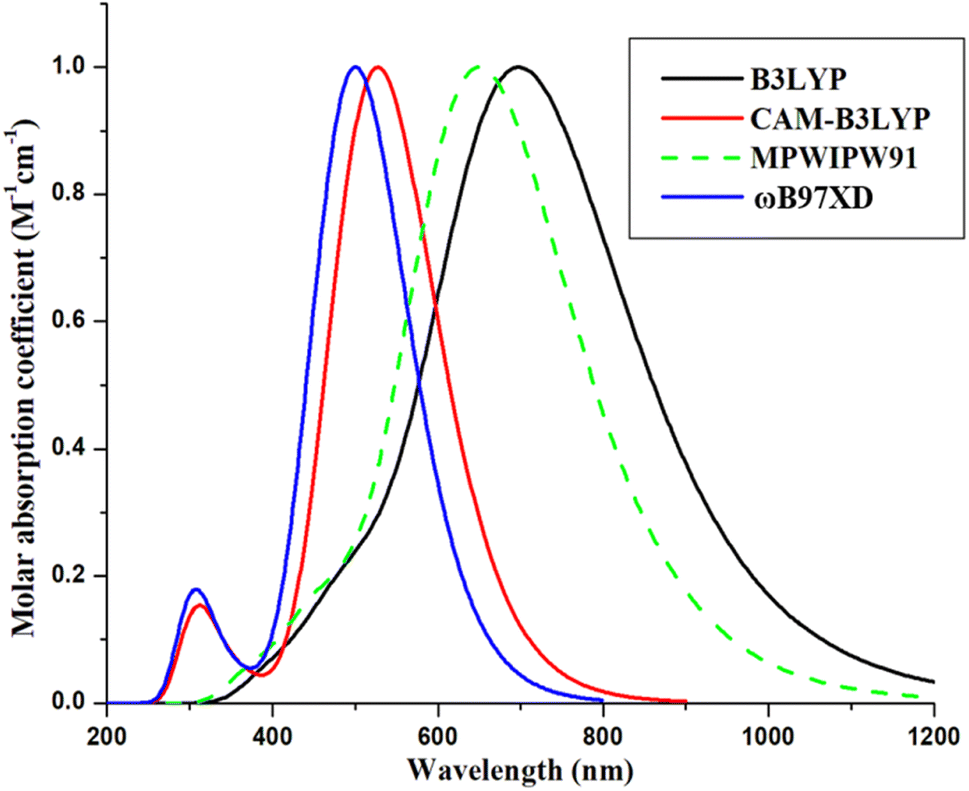 | ||
| Fig. 1 Absorption spectra of reference with four distinct functionals using chloroform. | ||
After the selection of the method for computation, all the molecules were optimized at their ground state, in order to evaluate their planarity and reactivity parameters, such as frontier molecular orbitals (highest occupied-HOMO and lowest unoccupied-LUMO molecular orbitals), molecular electrostatic potentials, natural transition orbitals, and some relative planarity measurements. After this, their evaluation in the excited states of the gas and chloroform mediums was performed using three different functionals to attain their absorption parameters, dipole moments, light harvesting efficiencies, etc. Additionally, for the generation of the graphs of absorption, transition density matrices, and density of states, the software utilized were Origin 6.0,40 Multiwfn 3.7,43 and PyMOlyze 1.1,44 respectively.
Moreover, reorganization energy values for hole (λhole) and electron (λelectron) were computed using Marcus rate equation.45
| λelectron = [E0− − E−] + [E−0 − E0] | (1) |
| λhole = [E0+ − E+] + [E+0 − E0] | (2) |
In these equations, E+0 and E−0 signifies the energies at neutral charge conditions of optimized cation and anion. E0− and E0+ are the energies at −1 and +1 charge of optimized molecules, E0 is the energy of neutral optimized structures. E+ is the energy of optimized cation and E− is the energy of optimized anion.46
Finally, the open-circuit voltage values, in addition to the fill factor, were also computed for the molecules, concerning the prediction of their possible aptitude to generate higher power conversion efficiencies in the active layers.
3 Results and discussion
For the design of the five A–D–D′–D–A molecules, the DFB central acceptor core of the reference (A–D–A′–D–A type) was replaced with five different strongly electron-donating cores.47 The newly formulated molecules were then analyzed and paralleled to the reference molecule, in order to scrutinize the effect of substituted donor cores. These cores are known to provide significant results in various reported experimental literature and include 5,10-di-thiophene-2-yl-3,8-dithia-dicyclopentanaphthalene (M1), 4,8-dithiophene-2-yl-1,5-dithiaindacine (M2), 4,8-bis-(3-fluoro-4-methylesulfanyl-phenyl)-1,5-dithiaindacine (M3), 1,4-dimethoxybenzene (M4), and 4,10-dimethyl-1-10,11-dihydro-4H-1,7-dithia-4,10-diaza-dicyclopentaneanthracene-5-one (M5). It should be noted that except for reference molecule, which was taken from the existing literature, all the proposed molecules are novel and have not been reported before. However, the various fragments in different molecules have been taken from existing literature. Thus, despite being designed theoretically in this research, their already reported donor cores have shown significant photovoltaic attributes when utilized in experimental synthesis of various chromophoric molecules and thus these molecules are also assumed to be capable of being synthesized accordingly.48–50 The sketched scheme of all compounds (using ChemDraw 7.0) of this study are presented in Fig. 2, while their optimized ground state structures are shown in Fig. S1.† | ||
| Fig. 2 Scheme of design of M1–M5 from R. | ||
Generally, for computational work, side alkyl chains are replaced with simple methyl ones, due to their insignificant effect on the absorption spectra and frontier molecular orbitals, but some studies show that they have a major impact on the crystallinity, photovoltaic attributes, and charge mobilities of the molecule, which is the reason behind us not following the conventional time-saving approach.51,52
Using computational investigations at selected DFT level of theory (i.e., MPW1PW91), we have looked into the geometries, along with the various optoelectronic properties, and photovoltaic features of R and M1–M5. This theoretical work might prove itself to be extremely beneficial in comprehending the performance of these molecules prior to their use in practical applications.
3.1 Method selection and optimized geometries
The ground state geometry is ideal for studying the optoelectronic attributes of designed chromophores before their characterization in the excited state. So all created chromophores were optimized using the 6-31G(d,p) basis set and MPW1PW91 functional at the ground state. The molecules were optimized globally and thus it is assumed that they can be synthesized experimentally if needed and are the most stable conformation of the molecules. The better stability of the molecules with respect to the reference can be seen according to their potential energies in the optimized ground states provided in Table S2.† We used different parameters to estimate the charge transfer and planarity of molecules, i.e., bond length, bond angle, span of deviation from plane (SDP), and molecular planarity parameter (MPP).The calculated value of bond length between carbon atoms of the central cores and CPDT units of all our molecules lies in between 1.41 Å and 1.45 Å, which is among the bond length of single (1.54 Å) and double bond (1.34 Å) between the atoms of carbon. This emphasizes the presence of notable conjugation between the newly introduced cores and CPDT units that can help the molecule in exhibiting increased values of absorption and reduced excitation energies.53 Further study showed that the dihedral angles between the cores and CPDT units of all studied molecules lie in the range of 5.8° to 25.7°, which hints towards their somewhat planar configuration, and might be helpful in effective charge transfer between donating and accepting moieties. The slight deviation within the dihedral angle of M1–M4 molecules from that of R might be helpful to the molecules in retaining the charge density within their respective fragments after effective intramolecular charge transfer.54 Individually, the higher dihedral angles of M1–M3 could be because of the vertical expanse of the attached large donor (D') cores, while the lowered one of M5 despite its bulky core, could be as a result of its horizontal expansion, which leads to a greater distance between the terminals of the molecule. The larger dihedral angle of M4 in spite of its smaller donor core (somewhat similar to R) could be due to the lowered space between the bulky alkyl chains of CPDT donors, which might have caused the rotation of the molecule about the single bond. The reason behind the absence of this anomaly in R, despite its somewhat similar structure to M4, could be due to the non-covalent interactions between the fluoro on the DFB (D') core and hydrogen on the CPDT (D) donors, explained in the literature.28
The dihedral angle only helps to determine the orientation of the D' core with respect to the CPDT donors. Thus, to investigate the impact of the donor cores on the overall planarity of the molecules, span of deviation from plane (SDP) and molecular planarity parameter (MPP) were computed using Multiwfn 3.8,55 and their structures were displayed by applying VMD 1.9.3.56 Table 1 demonstrates the outcome of all these parameters, where MPP provides an estimate of the general deviation of the whole structure from the plane. Actually, a lower MPP value corresponds to the planar structure of molecules and vice versa. The parts that are above the plane are shown in blue, and those that are below the plane are shown in red in this respect (Fig. 3). According to Table 1, M5 has the lowest MPP value, which reveals its superior planar configuration, and M4 has the least planarity as shown by its higher MPP value. The lowest overall planarity of M4 might be attributed to the most reduced distance between the bulky alkyl chains of the molecule due to its smallest central core, which might have caused steric hindrance between the core and peripheral donors of the molecule. Finally, SDP is helpful in understanding the range of deviations from the fitted plane of various components of a compound. In the case of SDP, M5's lowest value indicates its lowest deviation from the fitted plane, whereas the highest one for M3 indicates a greater deviation from the plane. However, according to Fig. 3, this deviation shown by M3 is due to the perpendicular fluoro-phenyl rings of its core, while the rest of the core is in plane with the CPDT donors.
| Molecules | Bond length (Lc–c) (Å) | Dihedral angle (θ°) | Molecular planarity parameter (MPP) Å | Span of deviation from plane (SDP) Å |
|---|---|---|---|---|
| R | 1.45 | 14.8 | 0.924 | 3.487 |
| M1 | 1.44 | 25.7 | 0.802 | 4.143 |
| M2 | 1.44 | 19.1 | 0.655 | 4.343 |
| M3 | 1.44 | 21.5 | 0.949 | 5.680 |
| M4 | 1.45 | 22.1 | 1.017 | 3.950 |
| M5 | 1.43 | 5.8 | 0.311 | 1.833 |
 | ||
| Fig. 3 Representation of span of deviation from planarity (SDP) and molecular planarity parameter (MPP) of reference and investigated molecules (R, M1–M5). | ||
3.2 Absorption profile
The absorption spectra of studied structures revealed that they all have absorption bands in UV-visible area, with their sharp peaks of maximum absorption being in the visible region. To observe the optical characteristics of the molecules, in both gaseous and solution states, their wavelength with maximum absorption (λmax) has been determined by observing their absorption bands in both these mediums (Fig. S2 and S5).† As mentioned above, MPW1PW91, B3LYP, and ωB97XD were used to carry all the computations for ten excited states using 6-31G(d,p) basis set. However, the absorption properties of the molecules were also computed with an added diffused state, i.e., MPW1PW91/6-31G+(d,p), so that appropriate and sufficient values in this phase can be determined, the values for which are provided in Table S3† in chloroform solvent.The λmax of R and M1–M5 in the solvent at MPW1PW91 is 651 nm, 638 nm, 680 nm, 682 nm, 694 nm, and 719 nm, at B3LYP functional the values are 700 nm, 692 nm, 735 nm, 739 nm, 742 nm, 781 nm, and finally in ωB97XD the values are 500 nm, 493 nm, 507 nm, 505 nm, 512 nm, and 519 nm (Table 2). In gas (Table S4†), the λmax for R is 613 nm for MPW1PW91, 656 nm for B3LYP, and finally 479 nm for ωB97XD. While for M1–M5, the values ranges as; 603–681 nm (MPW1PW91), 653–736 nm (B3LYP), and 472–500 nm (ωB97XD). It is clear from the above values that, except for M1, all the proposed molecules show a bathochromic shift in their absorption maxima in either of the two phases with regards to their corresponding functional or diffused state. The highest one observed by M5 might be attributed to the planar configuration of its donor cores with respect to the terminals of the molecule, which with the help of the greater planarity might have increased the overall conjugation in the molecule. While the lowest observed value for this parameter seen for M1 could be ascribed to its highest dihedral angle discussed above. On a side note, the relatively higher values of the molecules in the solvent, as opposed to the gas medium, illustrate their better compatibility with chloroform and their suitability for futuristic solution processing fabrications of non-fullerene OSCs.
| Compounds | Experimental λmax (nm) | Functional | Computed λmax (nm) | Excitation energies Ex (eV) | Oscillator strength (f) | Major transition character |
|---|---|---|---|---|---|---|
| R | 671 | MPW1PW91 | 651 | 1.90 | 3.32 | HOMO → LUMO |
| — | B3LYP | 700 | 1.77 | 3.19 | HOMO → LUMO | |
| ωB97XD | 500 | 2.47 | 3.43 | HOMO → LUMO | ||
| M1 | — | MPW1PW91 | 638 | 1.94 | 3.30 | HOMO → LUMO |
| — | B3LYP | 692 | 1.78 | 2.79 | HOMO → LUMO | |
| ωB97XD | 493 | 2.51 | 3.68 | HOMO → LUMO | ||
| M2 | — | MPW1PW91 | 680 | 1.82 | 3.53 | HOMO → LUMO |
| — | B3LYP | 735 | 1.68 | 3.26 | HOMO → LUMO | |
| ωB97XD | 507 | 2.44 | 4.14 | HOMO → LUMO | ||
| M3 | — | MPW1PW91 | 682 | 1.82 | 3.54 | HOMO → LUMO |
| — | B3LYP | 739 | 1.67 | 3.31 | HOMO → LUMO | |
| ωB97XD | 505 | 2.45 | 4.08 | HOMO → LUMO | ||
| M4 | — | MPW1PW91 | 694 | 1.78 | 3.12 | HOMO → LUMO |
| — | B3LYP | 742 | 1.66 | 2.95 | HOMO → LUMO | |
| ωB97XD | 512 | 2.40 | 4.18 | |||
| M5 | — | MPW1PW91 | 720 | 1.72 | 3.79 | HOMO → LUMO |
| — | B3LYP | 781 | 1.58 | 3.45 | HOMO → LUMO | |
| ωB97XD | 519 | 2.38 | 4.54 | HOMO → LUMO |
3.3 Excitation energy
Another convincing factor, important for assessing the photovoltaic features of an OSC is the first excitation energy, attained by TD-DFT simulations, in both the gas and solvent excited mediums. It is the energy needed for transition between two orbitals of different energy levels. First excitation energy is strongly associated with the band gap, which is the difference between the HOMO and LUMO energy levels. This assertion is rationally supported by the fact that the lowest band gap makes it easier for the charge carrier to transition from the ground-HOMO to the excited-LUMO energy state, resulting in effective charge transfer.57 First excitation energy is also inversely linked to the maximum absorption wavelength.According to Tables 2 and S4,† first excitation energies of reference and designed molecule for all three functionals follow the sequence; M5 < M4 < M2 < M3 < R < M1 in both the mediums of study, with M2 being equal to M3 only in case of MPW1PW91 in the solvent phase. Opposite to the maximum absorption values, the highest excitation in both the phases is M1, which solidifies our assertion of the inverse relation between both these parameters. Thus according to Table S5†, the lowest excitation energy of M5, along with its highest absorption maxima, might make this molecule to be the best one so far, with M2–M4 being close behind. Based on these values M2–M5 molecules can be better utilized for future OSCs to boost their photovoltaic performance.
Additionally, in order to validate the results of vertical excitation energy, its values for CIS/6-31G(d,p) were also evaluated in our chosen solvent, and the outputs are compiled in Table S5†, along with that of all other methodologies used in this research work for a proper comparison. The CIS methodology was selected due to its reported accuracy with respect to the experimental values.58 According to Table S5†, despite the varied values from all the four methodologies with that of CIS being at one extreme and B3LYP at other, the order of them all remained the same. Thus, M1 gave the highest value of Ex and M5 demonstrated the lowest excitation energy values. On a side note, though the values from CIS methodology were quite higher than others, but it is rather important to know that this methodology is known to have a slight positive standard deviation with respect to the experimental values.59
3.4 Frontier molecular orbitals
Quantum chemical descriptors, mainly FMOs, are one of the prime determinants of the optoelectronic and chemical parameters of a molecule. These are the highest occupied molecular orbital (HOMO) and lowest unoccupied molecular orbital (LUMO). In OSCs, the aim of employing this approach is to explain the position of charge dispersion in the orbitals with major transitions, i.e., the FMOs, which is helpful to assess the probable charge transfer in a molecule.60 In accordance with energy band theory, electron transfer can be described by taking HOMO as the valence band for the donor of electrons and LUMO as the conduction band for the acceptance of those electrons.61 Thus for the superior transfer of charges, the HOMO shall be over the donor with LUMO over the acceptors. The distribution of charge density in the frontier molecular orbitals of M1–M5 and R molecules, along with their values and corresponding difference (band gap) is represented in Fig. 4. Upon examining the HOMO densities, it's perceived that all the molecules have high charge density upon their donors (D) and cores, either the acceptor one (A') of reference or the donor (D') of M1–M4. Here, the acceptor core (A') of the reference doesn't actually follow the afore-stated distribution strategy, while all the derived molecules do so in case of their HOMO densities. Similarly, except for M4, all the derived molecules have a shift of charge density from their donor core (D') towards their terminal electron-accepting moieties (A), with the CPDT (D) donors working as bridges between them. While the reason behind the somewhat similar distribution of charge density of R and M4 is their similar cores with only the difference of fluoro and methoxy group between them. Here, it can be said that effective charge density dispersal is seen in all molecules, except M4, despite their relatively planar geometry, and the reason could be attributed to their prominent donor cores. Additionally, the absence of charge density in the alkyl chains of the CPDT core could be due to their perpendicular orientation relative to the plane of the molecule. | ||
| Fig. 4 FMOs of reference molecule R and all designed molecules (M1–M5). | ||
The values of FMOs and their band gap for all the molecules of our study were evaluated with respect to three diverse basis sets at MPW1PW91 functional. The reason being the proper validation of results, and the basis sets used were 6-31G(d,p), 6-311G(d,p), as well as LanL2DZ. The band gap values from 6-311G(d,p) were quite higher, while that from LanL2DZ were relatively the lowest ones. However, this basis set was not chosen as the one for further evaluation due to its effect on the LUMO values. From Table S6†, it can be seen that the LUMO values from LanL2DZ are quite low, which means that this basis set would also give quite low values of the open-circuit voltage if utilized as an NFA in organic solar cells. Additionally, this basis set is generally used for heavy metal atoms and not for organic molecules.62 Thus, 6-31G(d,p) was used as the one due to its relatively closer HOMO values to the experimental value (5.49 eV) of reference molecule in Table S1†.
Thus according to 6-31G(d,p) basis set, the HOMO values of designed molecules vary with that of reference in the arrangement M5 > M4 > M2 > M2 > M1 > R and their LUMO values vary in the arrangement M1 > M4 > M2 > M3 > M5 > R. Here, the relatively upshifted LUMO value of the derived molecules might help achieve high open-circuit voltage value by them, if assumed to be acceptors in the photo-active interface. Moving on to the band gap, when compared with reference, its value decreased to a reasonable level in all designed molecules except M1. With that of M1 (2.42 eV) being relatively comparable to the 2.41 eV of reference. M5 proved to be the best candidate so far, in terms of the lowest band gap.
Individually, M5 proved to be the best candidate so far, in terms of the lowest band gap and might be helpful in boosting the photovoltaic performance of respective OSCs. It could be due to its most planar confirmation, increased conjugation, as well as the high electron donating ability of the nitrogen atoms of its donor (D') core, which through collaboration with CPDT (D) donors might effectively transfer electrons towards the terminals. The highest band gap of M1 could be due to the non-planar confirmation of its donor core (D'), which also decreased its absorption maxima and increased its excitation energy.
3.5 Density of states (DOS)
DOS helps in verifying the results of FMOs and demonstrating the involvement of the core, donor, and acceptor in the formation of different energy levels (especially HOMO and LUMO), and helps in the evaluation of possible intramolecular charge transfer. It tells us about the availability of the positions that can be occupied by an electron at a certain energy level, i.e., charge distribution.63 The density of state plots for R and designed compounds is provided in Fig. 5. For the sake of convenience, molecules were fragmented as acceptors, donors, and cores. The reason for such fragmentation was to check the participation of each portion in the production of molecular orbitals. These fragments, representing the partial DOS of donor (green), core (black), and acceptor (red) can be seen in Fig. 5. Finally, the blue line in each plot represents the total density of states. Furthermore, the first peak on the left side of the central planar area represents the HOMO region and the first one on the right side signifies the LUMO region. The central area between them demonstrates the band gap of LUMO and HOMO, which is similar to the Eg calculated by FMO analysis. Finally, the scales were set as energy in eV on the x-axis and relative intensity set at 21 on the y-axis. Here, the TDOS of all the derived molecules on the right side is greater than R, which signifies prominent charge transportation within the molecule towards the LUMO region. Likewise, the black peaks of the core in all the proposed molecules are more prominent than in R. Though the red ones of the acceptors are all significantly similar, due to the identical acceptor group in all the studied molecules, which is also the case with the green peaks of donors. | ||
| Fig. 5 FMOs of reference molecule R and all designed molecules (M1–M5). | ||
According to the data of Table 3 about the percentage contribution of various fragments in FMOs' formation, in the production of R's HOMO, the contribution of the core unit is very little due to its accepting nature. Conversely, regarding the electron-donating capabilities of the cores of M1–M5, they all have significant contributions in HOMO generation. This trend is especially seen in M1, M2, M3, and M5. The lower contribution of M4, which is still much higher than R, might be due to its comparatively similar structure to the reference molecule, but with an electron-donating methoxy group instead of an electron-withdrawing fluoro one. Moving on towards the contribution of terminal acceptors in HOMO formation, it is seen that all the proposed molecules have lower contributions in this respect, which illustrates the effective HOMO generation in them as opposed to R. On the other hand, with the assumption that the core of the reference molecule is an accepting one, it should have had a strong contribution to the LUMO generation, but that is not the case. On a side note, all these revelations match well with the charge density distribution in the FMOs investigation. Thus, it could be said that the FMOs in the derived molecules are much more effective for charge transfer than the reference, according to both the FMO and DOS analysis.
| Molecules | Excitation energy state | Percentage contribution of core | Percentage contribution of acceptor | Percentage contribution of donor |
|---|---|---|---|---|
| R | HOMO | 12.7 | 24.1 | 63.2 |
| LUMO | 8.4 | 52.3 | 39.4 | |
| M1 | HOMO | 44.6 | 15.1 | 40.3 |
| LUMO | 8.5 | 56.3 | 35.2 | |
| M2 | HOMO | 39.2 | 16.2 | 44.6 |
| LUMO | 11.0 | 52.0 | 37.0 | |
| M3 | HOMO | 38.9 | 16.3 | 44.8 |
| LUMO | 11.1 | 51.6 | 37.3 | |
| M4 | HOMO | 21.4 | 21.1 | 57.5 |
| LUMO | 6.9 | 54.2 | 38.8 | |
| M5 | HOMO | 52.7 | 12.1 | 35.2 |
| LUMO | 14.2 | 49.4 | 36.4 |
3.6 Natural transition orbitals
Many distinct molecular orbitals (MOs) transitions have non-negligible contributions to the transition of electronic state, which may be assessed as the square of the associated configuration coefficient. This characteristic makes it difficult to analyse transition character by viewing only one pair of MOs. The NTO approach seeks to alleviate this issue by performing unitary transformation for occupied MOs and virtual MOs, such that only one or a small number of orbital pairings have dominant contributions, making assessment of orbital a lot easier.64,65Natural transition orbitals (NTOs) have been studied to better explain the percentage ETC (electron transport contribution) from ground to excited state. The electrical and optical features of devised molecules are governed by the charge density distribution throughout the overall NTOs. The capacity to absorb and donate electrons is reflected by the distribution of LUMO and HOMO. As shown in Fig. 6, the designed molecules% ETC is more than R, ranging as 91–97%.
 | ||
| Fig. 6 Natural transition orbitals of all the studied molecules. | ||
3.7 Molecular-electrostatic potential (MEP)
In order to access the reactivity of a compound, MEP study was made by analyzing the electron-poor and electron-rich regions at different positions on the molecule. It aids in determining the degree to which charge transports occur from donor to acceptor. MEP surface refers to the colored counters that surround the molecules, showing electron distribution at different regions of the molecule. The electron-rich region on the molecule is susceptible to attack by an electrophile and is characterized by the red color on the MEP plot, i.e., it has a negative potential. Whereas the blue region represents the electron deficiency at that region and its susceptibility is to be attacked by a nucleophile, hence with positive potential. The green color on the map shows the neutral region, which can neither be attacked by an electrophile nor a nucleophile.66 Thus, the MEP analysis can help in determining the possible reactivity of the future NFAs with respect to electrophile and nucleophile.Fig. S3† is presenting the MEP maps of all concerned molecules. In all the molecules the distribution of charge density is somewhat similar at the terminals, with deep red colors present at strongly electron-withdrawing unsaturated oxygen and nitrogen atoms. While, the difference is in the central regions of the molecules, i.e., the core. All the molecules, except for the unsaturated oxygen atoms of M5, have deep blue color in the central region, which manifests the positive potential in this region. Here, it should be noted that generally, the donor regions garner a blue hue, with the accepting ones having a red one, and M1–M4, all follow this trend. However, the accepting core of R, instead of having red colors around it, is surrounded by blue hues, which depicts its inconsistent behavior. On the other hand, despite the red hues in the core of M5, its deep blue zone around the saturated electron-donating nitrogen atoms makes it a prominent donor core. On a side note, the presence of greenish hues around the CPDT donors of the molecules hints towards their charge transferring attributes.
3.8 Exciton binding energy (Eb)
A crucial parameter for studying the coulombic interactions among charge transporters/carriers produced in conjugated molecules upon photo-excitation is binding energy. It is the least extent of energy required to separate an electron and a hole produced by light absorption as a bound exciton. Organic chromophores generate excitons in the active layer upon light absorption, which must be separated into electrons and holes, so that they can move to relevant electrodes where current is generated.67–69 The ease with which electrons and holes separate depends upon the electron-accepting abilities of the end groups. Powerful electron acceptors at the terminals lessen the coulombic forces of attraction in excitons, resulting in easy separation and lowering of the binding energy values, and this effect is enhanced ten folds by the presence of strong donors in the center for an effective push–pull effect.70 Lower binding energy leads to quicker dissociation, which is the ground for a higher current generation. The following equation was availed to calculate the values of exciton binding energy:69| Eb = Eg − Ex | (3) |
| Molecules | EH–L (eV) | Eb (eV) | Eb (eV) | Interaction coefficient |
|---|---|---|---|---|
| Gaseous | Solvent | |||
| R | 2.41 | 0.39 | 0.51 | 0.68520 |
| M1 | 2.42 | 0.37 | 0.48 | 0.65342 |
| M2 | 2.30 | 0.36 | 0.48 | 0.68162 |
| M3 | 2.29 | 0.35 | 0.47 | 0.68185 |
| M4 | 2.30 | 0.38 | 0.52 | 0.68936 |
| M5 | 2.18 | 0.36 | 0.46 | 0.67647 |
Table 4 also tabulates the interaction coefficient of all the molecules in the studied solvent. It is actually a measure of the mobility of the charge carriers and is perceived to be the lower the better. Because a low interaction coefficient lowers the interaction between the excitons at the interface and as a result increases their transfer toward respective electrodes.71 Overall, except for the slightly higher one of M4, all the proposed molecules have a lower value for this parameter if compared to the reference.
3.9 Reorganization energy
Another notable parameter that explains the charge transfer attributes of scrutinized chromophores by evaluating their charge mobilities. Reorganization energy inversely influences the charge mobility i.e., the lower its value more will be the mobility of charge.72 Following the exciton generation, after light absorption, exciton dissociation results in the generation of electrons and holes as distinct entities. These electrons and holes rapidly set themselves in motion towards their corresponding electrodes before they could recombine.73–75 As a result of this movement, distortion takes place inside the molecular structure, thus the molecule requires some energy to reorganize its structure after distortion. This energy is called internal reorganization energy (λint). Another type of reorganization energy is the external one (λext), which is related to external variables, i.e., the polarization of neighboring environment, sometimes solvent. As the environment utilized here is a constant one of either the chloroform solvent or the gas one, we have excluded this energy.76 On the other hand, the λint, including the one for the hole (λhole) and the one for the electron (λelectron), was computed using eqn (1) and (2), taken from the Marcus theory.77 These values of the reorganization energy of designed molecules and R are given in Table 5.| Molecules | λe (electron) | λh (hole) |
|---|---|---|
| R | 0.1913679 | 0.2129563 |
| M1 | 0.1191389 | 0.1930686 |
| M2 | 0.1596601 | 0.2361311 |
| M3 | 0.1673931 | 0.2428248 |
| M4 | 0.1723481 | 0.2312414 |
| M5 | 0.1189349 | 0.1735589 |
According to Table 5, the λelectron of all the derived molecules is contrastingly reduced compared to the reference. The order of this reduction by all the molecules is; R > M4 > M3 > M2 > M1 > M5. Here, the lowest value of M5 molecule helps retain its candidature as the best-formulated chromophore. While the second lowest value for M1 with a difference of only 0.0002 eV depicts its enhanced electron mobility as well. Moving on towards the λhole, which except for M1 and M5, is higher for all the proposed molecules when compared to R, making the reducing order of M3 > M2 > M4 > R > M1 > M5. This depicts the prominent hole mobilities in both M1 and M5, in addition to their significant electron mobilities, which proves their better capabilities to enhance the photovoltaic performance of future NFAs. Upon comparison between the values of both these energies, the lower λelectron of all the molecules than their λhole, leads to the conclusion that these molecules might act as better electron transporters than hole, and thus are assumed to be acceptors in the interface of active layers.
3.10 Transition density matrix (TDM)
It is a convenient parameter for interpreting and assessing the electronic excitations within molecular systems and also the electron–hole coherence.78–80 Besides this, calculations of TDM divulge the participation of different moieties of molecules in charge movement, which provide us with information about the acceptor–donor interactions in the first excited state.81 The hydrogen is ignored in the transition density matrix calculations because of its negligible contribution to transition.82 Here, Multiwfn software was used to plot the graphs of R and M1–M5. All molecules are classified into three sections; acceptor, core, and donor (A, C, and D, respectively). In TDM plots, the left y- and horizontal x-axis show the number of atoms (with the exclusion of hydrogen), and the y-axis on the right, has a band of colors, showing the electron density, which ranges from blue (being the least) to red (the possible highest one). The arrows showing electron on left y- and hole on lower x-axes helps in determination of their possible off-diagonal charge coherence or transfer. TDM maps of purposed compounds are presented in Fig. 7. The TDM map of R clearly shows that charge distribution is present in the core of the molecule, however, the acceptors and CPDT donors have comparatively lower charge density spaces. This could be due to the electron-accepting nature of the core of the reference, which shifted the charge density significantly towards itself and reduced its flow toward the terminal acceptors. Additionally, the off-diagonal coherences in the opposite direction is quite low in case of R, while its diagonal bands seems to somewhat present in the acceptor region. | ||
| Fig. 7 TDM plots of all investigated molecules (R, M1–M5). | ||
Conversely, the cores of the derived molecules being densely electron rich have brightly colored diagonal and off-diagonal charge transfer as well as electronic coherences towards the acceptors at the peripheries while traveling from the CPDT donors. The central core (C) of all the derived molecules is brightly shaded with both diagonal and off diagonal bands, showing significant charge coherence and transfer with the molecule. Here, the relatively low charge off diagonal charge density in the donors (D) of the molecules justifies their rule as bridges in the derived molecule.83 The dim regions of charge density in all CPDT donors of the molecules exhibit the peripheral alkyl chains, showing their inferior contribution to charge transfer. Specifically, the brightest cores are M1 and M5, which shows their prominence in terms of being effective chromophores for future OSCs.
3.11 Dipole moment
Another vital parameter that justifies the photovoltaic aptitude of an OSC is the dipole moment (μ), which provides us with some knowledge about the electron allocations, solubility, and polarity of a molecule. Due to its relation to the polar atoms in a molecule, it is directly related to how soluble a compound is in a polar organic solvent. This is why generally the dipole moment of a polar molecule is greater in the polar solvent than in the gaseous medium.84 Moreover, a greater value of dipole moment improves the self-assembly, which in turn minimizes the chances of disorders. This is due to the fact that in polar molecules, the charges arrange themselves in an opposite manner. Actually, when molecules get close to each other, they organize themselves so that their opposite poles attract each other via intermolecular forces of attraction.85 The dipole moment also elucidates the adept movement of holes and electrons among acceptor and donor portions.The computed values of dipole moment of M1–M5 and R, are given in Table S7† for MPW1PW91. Here, the dipole moment for R and M1–M5 follows the sequence: M3 > M2 > M1 > M5 > R > M4 in both the studied phases. However, due to the polar nature of the studied molecules and that of the chloroform solvent, greater values of studied dipole moment are seen in this phase than in the gas one. The difference between the values of both these mediums is also given in Table S7†. Here, the lowest difference is of R, which elaborates its insignificant interaction with the solvent in terms of dipole moment, just like the M4 molecule. The lowest dipole moment amongst all is seen to be of M4 molecule, which could be due to the somewhat non-polar symmetry of its core. Nevertheless, the highest value of M3 might be due to the off-centered polar fluoro groups present in its substituted core. Despite the presence of the fluoro groups in the core of the reference molecule, their symmetry with respect to the plane of the core seemed to cancel each other's polar natures. So, it could be concluded that in addition to the polar nature of the atoms, their symmetry concerning the deliberated fragment also affects the dipole moment in a chromophore.
3.12 Open-circuit voltage
The open circuit voltage (Voc) is one of the most important factors to determine the effectiveness of organic photovoltaic (OPV) systems, which is the maximum voltage that an organic solar cell can supply to an external circuit after separating electrons and holes from each other. By increasing the HOMO of the donor moiety and decreasing the LUMO of the acceptor moiety, maximum Voc can be achieved.86 For efficient charge movement, the designed acceptors are blended with relevant polymer donor material; PTB7-Th, which has a HUMO and LUMO of 5.20 eV and 3.59 eV.71 This donor is excessively used in theoretical research to evaluate the computational Vocs. In this research, the values of Voc are examined by utilizing the LUMO level of designed molecules (assumed to be acceptors) and HOMO of donor material PTB7-Th, by utilizing the following equation:87
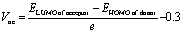 | (4) |
 | ||
| Fig. 8 Computed Vocs for reference R and designed M1–M5 acceptors with donor; PTB7-Th. | ||
3.13 Oscillator strength and LHE
Oscillator strength is another crucial tool that justifies the power conversion ability of an OSC. It represents the intensity of electromagnetic energy emitted upon electronic excitation between two energy states.88 It is associated with the absorption of radiation in the UV-vis area, as the compounds that exhibit strong absorption in the UV-vis region (200–800 nm) tend to have greater oscillator strength.89 All of our designed molecules absorb strongly in the visible region and thus have good oscillator strength (fos), as can be seen from the GaussView graphs of absorption (at MPW1PW91/6-31G(d,p)) depicting the oscillator strength at right y-axis in Fig. S4 and S5.† The computed oscillator strength of reference and M1–M5 in the gas phase follows the order as M4 < M1 < R < M2 < M3 < M5 at MPW1PW91, R = M1 < M4 < M2 < M3 < M5 at B3LYP, and R < M1 < M4 < M3 < M2 < M5 at ωB97XD (Table S4†). While, in the solvent phase a rather different trend is seen according to the functionals. However, a general trend could be seen that regardless of the significant absorption maxima and excitation energy values attained from B3LYP, it gave poor values of oscillator strength in comparison to other functionals. On the other hand, ωB97XD gave the highest oscillator strength amongst all the studied functionals. It should be noted that M5, irrespective of the functional, exhibited greater oscillator strength among all, due to the presence of unsaturated heteroatoms at the central core that results in extensive conjugation. With the help of oscillator strength, the light-harvesting efficiencies (LHE) of M1–M5 and R were also calculated using following equation53 and the values for all the three functionals are given in Table 6:| LHE = 1 − 10−f | (5) |
| Molecules | LHE (MPW1PW91) | LHE (B3LYP) | LHE (ωB97XD) |
|---|---|---|---|
| R | 0.99952 | 0.99935 | 0.99962 |
| M1 | 0.99950 | 0.99838 | 0.99979 |
| M2 | 0.99945 | 0.99992 | 0.99992 |
| M3 | 0.99971 | 0.99951 | 0.99991 |
| M4 | 0.99924 | 0.99888 | 0.99993 |
| M5 | 0.99984 | 0.99965 | 0.99997 |
This LHE has a direct relation with the creation of charge carriers, it is also in direct relation with short-circuit current density, which is a very important parameter in determining the productivity of OSCs, or in other words their PCE. The trend of increasing LHE could be written as similar to that for the oscillator strength, ascribed to the direct relation between these two parameters. M2, M3, and M5 showed higher LHE than R irrespective of the functional, emphasizing their superior ability to produce charge carriers by absorbing light in the gas phase.
3.14 Fill factor
It is an important tool to evaluate the performance of photovoltaic cells which we calculated through the following equation:90
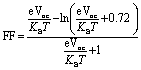 | (6) |
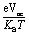 is normalized Voc which is an important factor to calculate the fill factor, where KB and T represent Boltzmann's constant (8.61 × 10−5 eV) and constant temperature (300 K), respectively. Without going into details, it is clear from Table 7 that FF follows the same trend as that of Voc. This depicts the dependence of FF on open-circuit voltage, which illustrates the significance of Voc in terms of the calculation of PCE.
is normalized Voc which is an important factor to calculate the fill factor, where KB and T represent Boltzmann's constant (8.61 × 10−5 eV) and constant temperature (300 K), respectively. Without going into details, it is clear from Table 7 that FF follows the same trend as that of Voc. This depicts the dependence of FF on open-circuit voltage, which illustrates the significance of Voc in terms of the calculation of PCE.
| Molecules | Voc (V) | Normalized (Voc) | FF |
|---|---|---|---|
| R | 1.61 | 62.22 | 0.9187 |
| M1 | 1.75 | 67.63 | 0.9239 |
| M2 | 1.70 | 65.70 | 0.9221 |
| M3 | 1.67 | 64.54 | 0.9201 |
| M4 | 1.72 | 66.47 | 0.9228 |
| M5 | 1.64 | 63.38 | 0.9198 |
The power conversion efficiency (PCE) of OSCs is associated mainly with these crucial factors; fill factor (FF), short circuit current density (Jsc), power of incident light Pin, and open circuit voltage Voc. By knowing the values of all mentioned parameters we can calculate the PCE by following equation;91
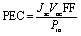 | (7) |
The Voc and FF values were computationally determined for this work. While the Jsc was not computed (due to the limitation of resources and this being a pure theoretical research work), several parameters that are directly related to it have been calculated, including band gaps, maximum absorption wavelengths, binding energies, excitation energies, and most importantly the light-harvesting efficiencies. So, despite not directly calculating the value of Jsc, a reliable estimate of Jsc can be given based on these results. These results indicate that M5 could show better results than R in all calculated parameters, with others not being far behind, due to the efficiency of the newly introduced core in them and thus could be the best molecule for future NFAs in OSCs.
4 Conclusion
In the present study, we designed five new chromophores (with A–D–D′–D–A confirmation) to explore their photovoltaic potential as in light-capturing device by performing modifications at the central electron accepting core of reference, synthesized experimentally. A DFT study was used to thoroughly investigate the optoelectronic parameters and structure–performance relationship with carefully selected three different functionals. Among all modified structures M5 seemed to be the best due to its various photovoltaic and optoelectronic properties, such as the least energy gap, a broader absorption band in the solvent (chloroform), highest oscillator strengths, and lowest binding energies in both studied phases, least studied interaction coefficient, lowest electron reorganization energy (0.1189349 eV), and lowest excitation energy in all the studied functionals. Its open-circuit voltage, dipole moment, and fill factor, though not the highest but are still higher than the reference molecule. Moreover, M5 is denoted for improved charge transfer from HOMO towards LUMO due to the planar configuration of its donor core with the rest of the molecule.Nonetheless, reasonable Voc values with respect to HOMOPTB7-Th − LUMOacceptor showed that all our modified molecules are best-suited non-fullerene acceptors for future utilization in OSCs. Additionally, all the considered molecules have planar configuration, which shows the extended conjugation in them. It is concluded from above discussion that our investigated molecules are much better than the reported molecule (R) in terms of device performance, spectral absorption, and charge transfer rate with special reference to M5. Thus, the modeled molecules should be employed as non-fullerene acceptor molecules in active layer of organic solar cells after careful analysis of photovoltaic parameters.
Data availability
Data will be made available on request.Author contributions
Amna zahoor: methodology, investigation, writing original draft. N. M. A. Hadia: methodology, investigation, validation. Sonia Sadiq: formal analysis, conceptualization, Javed Iqbal: project administration, formal analysis, conceptualization, writing & editing. Ahmed M. Shawky: investigation, validation. Naifa S. Alatawi: resources, visualization, writing, funding acquisition. Muhammad Umar Saeed: programming, software development; designing computer programs, Rasheed Ahmad Khera: project administration, funding acquisition, supervision.Conflicts of interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.Acknowledgements
The authors acknowledge the technical support from the chemistry department, University of Agriculture, Faisalabad, Pakistan. The authors would like to thank the Deanship of Scientific Research at Umm Al-Qura University for supporting this work by Grant Code: 22UQU4331174DSR53.References
- T. Abbasi and S. Abbasi, Crit. Rev. Environ. Sci. Technol., 2012, 42, 99–154 CrossRef.
- C. Lu, M. Paramasivam, K. Park, C. H. Kim and H. K. Kim, ACS Appl. Mater. Interfaces, 2019, 11(15), 14011–14022 CrossRef CAS PubMed.
- S. A. H. Zaidi, F. Hou and F. M. Mirza, Environ. Sci. Pollut. Res., 2018, 25, 31616–31629 CrossRef CAS PubMed.
- D. Kajela and M. S. Manshahia, Int. J. Sci. Res. Sci. Technol., 2017, 3, 769–795 Search PubMed.
- N. Hagumimana, J. Zheng, G. N. O. Asemota, J. D. D. Niyonteze, W. Nsengiyumva, A. Nduwamungu and S. Bimenyimana, Int. J. Photoenergy, 2021, 2021 Search PubMed.
- M. Z. Jacobson and M. A. Delucchi, Energy Policy, 2011, 39, 1154–1169 CrossRef CAS.
- K. Kumar, H. Finnegan, G. Farabow and L. Dunner, Intellectual Property Magazine, 2020 Search PubMed.
- I. Hadar, T. B. Song, W. Ke and M. G. Kanatzidis, Adv. Energy Mater., 2019, 9, 1802766 CrossRef.
- P. Mahalingavelar, Energy Fuels, 2022, 36(4), 2095–2107 CrossRef CAS.
- A. Khatibi, F. Razi Astaraei and M. H. Ahmadi, Energy Sci. Eng., 2019, 7, 305–322 CrossRef.
- E. U. Rashid, N. Hadia, J. Iqbal, R. F. Mehmood, H. Somaily, S. J. Akram, A. M. Shawky, M. I. Khan, S. Noor and R. A. Khera, RSC Adv., 2022, 12, 21801–21820 RSC.
- S. Kim, M. Jahandar, J. H. Jeong and D. C. Lim, Curr. Altern. Energy, 2019, 3, 3–17 CrossRef.
- D. Luo, W. Jang, D. D. Babu, M. S. Kim, D. H. Wang and A. K. K. Kyaw, J. Mater. Chem. A, 2022, 10(7), 3255–3295 RSC.
- H. Yao, Y. Cui, R. Yu, B. Gao, H. Zhang and J. Hou, Angew. Chem., 2017, 129, 3091–3095 CrossRef.
- A. Rasool, S. Zahid, R. A. Shehzad, M. S. Akhter and J. Iqbal, Comput. Theor. Chem., 2021, 1203, 113359 CrossRef CAS.
- E. U. Rashid, J. Iqbal, M. I. Khan, Y. A. El-Badry, K. Ayub and R. A. Khera, RSC Adv., 2022, 12, 12321–12334 RSC.
- C. Yan, S. Barlow, Z. Wang, H. Yan, A. K.-Y. Jen, S. R. Marder and X. Zhan, Nat. Rev. Mater., 2018, 3, 1–19 CrossRef.
- Y. Lin and X. Zhan, Adv. Energy Mater., 2015, 5, 1501063 CrossRef.
- G. Sivakumar, M. Paramasivam and V. J. Rao, New J. Chem., 2019, 43(13), 5173–5186 RSC.
- M. Paramasivam, R.K. Chitumalla, J. Jang and J.H. Youk, Phys. Chem. Chem. Phys., 2018, 20(35), 22660–22673 RSC.
- Q. Bai, Q. Liang, H. Li, H. Sun, X. Guo and L. Niu, Aggregate, 2022, e281 CAS.
- M. Yang, W. Wei, X. Zhou, Z. Wang and C. Duan, Energy Mater., 2021, 1, 100008 CrossRef.
- S. Pang, X. Zhou, S. Zhang, H. Tang, S. Dhakal, X. Gu, C. Duan, F. Huang and Y. Cao, ACS Appl. Mater. Interfaces, 2020, 12, 16531–16540 CrossRef CAS PubMed.
- X. Li, Z. Xu, X. Guo, Q. Fan, M. Zhang and Y. Li, Org. Electron., 2018, 58, 133–138 CrossRef CAS.
- B. L. Greenstein, D. C. Hiener and G. R. Hutchison, J. Chem. Phys., 2022, 156, 174107 CrossRef CAS PubMed.
- N. Wang, L. Zhan, S. Li, M. Shi, T.-K. Lau, X. Lu, R. Shikler, C.-Z. Li and H. Chen, Mater. Chem. Front., 2018, 2, 2006–2012 RSC.
- D. He, F. Zhao, L. Jiang and C. Wang, J. Mater. Chem. A, 2018, 6, 8839–8854 RSC.
- S. Li, L. Zhan, F. Liu, J. Ren, M. Shi, C. Z. Li, T. P. Russell and H. Chen, Adv. Mater., 2018, 30, 1705208 CrossRef PubMed.
- Y. Li, J. Yu, Y. Zhou and Z. a. Li, Chem.–Eur. J., 2022, 28, e202201675 CAS.
- M. Frisch, et al., Gaussian 09, revision D.01, Gaussian, Inc., Wallingford, CT, 2009 Search PubMed.
- R. Dennington, T. A. Keith and J. M. Millam, GaussView, version 6.0.16, Semichem Inc., Shawnee Mission, KS, 2016 Search PubMed.
- R. G. Parr, in Horizons of quantum chemistry, Springer, 1980, pp. 5–15 Search PubMed.
- T. Yanai, D. P. Tew and N. C. Handy, Chem. Phys. Lett., 2004, 393, 51–57 CrossRef CAS.
- B. Civalleri, C. M. Zicovich-Wilson, L. Valenzano and P. Ugliengo, CrystEngComm, 2008, 10, 405–410 RSC.
- J.-D. Chai and M. Head-Gordon, Phys. Chem. Chem. Phys., 2008, 10, 6615–6620 RSC.
- C. Adamo and V. Barone, J. Chem. Phys., 1998, 108, 664–675 CrossRef CAS.
- M. D. Hack and D. G. Truhlar, J. Phys. Chem. A, 2000, 104, 7917–7926 CrossRef CAS.
- J. Tomasi, B. Mennucci and R. Cammi, Chem. Rev., 2005, 105, 2999–3094 CrossRef CAS PubMed.
- E. Cances, B. Mennucci and J. Tomasi, J. Chem. Phys., 1997, 107, 3032–3041 CrossRef CAS.
- L. Deschenes and A. David, Origin 6.0: Scientific Data Analysis and Graphing Software Origin Lab Corporation (Formerly Microcal Software, Inc.), Vanden Bout University of Texas, 2000, p. 595, http://www.originlab.com Search PubMed.
- J. Huang, S. Li, J. Qin, L. Xu, X. Zhu and L.-M. Yang, ACS Appl. Mater. Interfaces, 2021, 13, 45806–45814 CrossRef CAS PubMed.
- C. Halsey-Moore, P. Jena and J. T. McLeskey Jr, Comput. Theor. Chem., 2019, 1162, 112506 CrossRef.
- T. Lu and F. Chen, J. Comput. Chem., 2012, 33, 580–592 CrossRef CAS PubMed.
- A. L. Tenderholt, PyMOlyze: A Program to Analyze Quantum Chemistry Calculations, 2019 Search PubMed.
- D.-W. Yan, X.-D. Li, P.-C. Li, W.-L. Tang, H.-H. Ren and Y.-G. Yan, Polymer, 2021, 237, 124355 CrossRef CAS.
- E. U. Rashid, N. Hadia, O. Alaysuy, J. Iqbal, M. Hessien, G. A. Mersal, R. F. Mehmood, A. M. Shawky, M. I. Khan and R. A. Khera, RSC Adv., 2022, 12, 28608–28622 RSC.
- M. Ans, J. Iqbal, B. Eliasson, M. J. Saif, H. M. A. Javed and K. Ayub, J. Mol. Model., 2019, 25, 1–12 CrossRef CAS PubMed.
- J. Melidonie, E. Dmitrieva, K. Zhang, Y. Fu, A. A. Popov, W. Pisula, R. Berger, J. Liu and X. Feng, J. Org. Chem., 2019, 85, 215–223 CrossRef PubMed.
- J. Guo, B. Qiu, D. Yang, C. Zhu, L. Zhou, C. Su, U. S. Jeng, X. Xia, X. Lu and L. Meng, Adv. Funct. Mater., 2022, 32, 2110159 CrossRef CAS.
- M. Rani, J. Iqbal, R. F. Mehmood, S. J. Akram, K. Ghaffar, Z. M. El-Bahy and R. A. Khera, Chem. Phys. Lett., 2022, 139750 CrossRef CAS.
- X. Jiang, W. Xue, W. Lai, D. Xia, Q. Chen, W. Ma and W. Li, J. Mater. Chem. C, 2021, 9, 16240–16246 RSC.
- H. Hu, K. Jiang, G. Yang, J. Liu, Z. Li, H. Lin, Y. Liu, J. Zhao, J. Zhang and F. Huang, J. Am. Chem. Soc., 2015, 137, 14149–14157 CrossRef CAS PubMed.
- S. J. Akram, J. Iqbal, M. Ans, Y. A. El-Badry, R. F. Mehmood and R. A. Khera, Sol. Energy, 2022, 237, 108–121 CrossRef CAS.
- S. J. Akram, J. Iqbal, R. F. Mehmood, S. Iqbal, Y. A. El-Badry, M. I. Khan, M. Ans and R. A. Khera, Sol. Energy, 2022, 240, 38–56 CrossRef CAS.
- T. Lu, J. Mol. Model., 2021, 27, 1–6 CrossRef PubMed.
- W. Humphrey, A. Dalke and K. Schulten, J. Mol. Graphics, 1996, 14, 33–38 CrossRef CAS PubMed.
- S. Liu, W. Su, X. Zou, X. Du, J. Cao, N. Wang, X. Shen, X. Geng, Z. Tang and A. Yartsev, J. Mater. Chem. A, 2020, 8, 5995–6003 RSC.
- C. Fang, B. Oruganti and B. Durbeej, J. Phys. Chem. A, 2014, 118, 4157–4171 CrossRef CAS PubMed.
- S. Grimme and E. I. Izgorodina, Chem. Phys., 2004, 305, 223–230 CrossRef CAS.
- R. Tariq, R. A. Khera, H. Rafique, U. Azeem, A. Naveed, A. R. Ayub and J. Iqbal, Comput. Theor. Chem., 2021, 1203, 113356 CrossRef CAS.
- S. Bibi, R. A. Khera, A. Farhat and J. Iqbal, Comput. Theor. Chem., 2021, 1198, 113176 CrossRef CAS.
- S. N. Upadhyay and S. Pakhira, Phys. Chem. Chem. Phys., 2022, 24, 22823–22844 RSC.
- N. Naeem, T. Tahir, M. Ans, A. Rasool, R. A. Shehzad and J. Iqbal, Comput. Theor. Chem., 2021, 1204, 113416 CrossRef CAS.
- F. Abbas, U. Ali, A. Tallat, H. M. R. Ahmad, S. A. Siddique, Z. Zeb and M. B. A. Siddique, Chem. Pap., 2022, 76, 4977–4987 CrossRef CAS.
- F. Plasser and H. Lischka, J. Chem. Theory Comput., 2012, 8, 2777–2789 CrossRef CAS PubMed.
- H. Yao, Y. Cui, D. Qian, C. S. Ponseca Jr, A. Honarfar, Y. Xu, J. Xin, Z. Chen, L. Hong and B. Gao, J. Am. Chem. Soc., 2019, 141, 7743–7750 CrossRef CAS PubMed.
- T. M. Clarke and J. R. Durrant, Chem. Rev., 2010, 110, 6736–6767 CrossRef CAS PubMed.
- M. U. Khan, M. Khalid, R. A. Khera, M. N. Akhtar, A. Abbas, M. F. ur Rehman, A. A. C. Braga, M. M. Alam, M. Imran and Y. Wang, Arabian J. Chem., 2022, 15, 103673 CrossRef CAS.
- M. I. Khan, J. Iqbal, S. J. Akram, Y. A. El-Badry, M. Yaseen and R. A. Khera, J. Mol. Graphics Modell., 2022, 113, 108162 CrossRef CAS PubMed.
- A. Farhat, R. A. Khera, S. Iqbal and J. Iqbal, Opt. Mater., 2020, 107, 110154 CrossRef CAS.
- S. Sabir, N. Hadia, J. Iqbal, R. F. Mehmood, S. J. Akram, M. I. Khan, A. M. Shawky, M. Raheel, H. Somaily and R. A. Khera, Chem. Phys. Lett., 2022, 806, 140026 CrossRef CAS.
- V. Vehmanen, N. V. Tkachenko, H. Imahori, S. Fukuzumi and H. Lemmetyinen, Spectrochim. Acta, Part A, 2001, 57, 2229–2244 CrossRef CAS PubMed.
- M. Waqas, J. Iqbal, R. F. Mehmood, S. J. Akram, A. M. Shawky, M. Raheel, E. U. Rashid and R. A. Khera, J. Mol. Graphics Modell., 2022, 116, 108255 CrossRef CAS PubMed.
- M. Waqas, N. Hadia, M. Hessien, S. J. Akram, A. M. Shawky, J. Iqbal, M. A. Ibrahim and R. A. Khera, Comput. Theor. Chem., 2022, 1217, 113904 CrossRef CAS.
- M. Waqas, N. Hadia, M. Hessien, J. Iqbal, G. A. Mersal, S. Hameed, A. M. Shawky, Z. Aloui, M. A. Ibrahim and R. A. Khera, J. Mol. Liq., 2022, 120770 CrossRef CAS.
- S. J. Akram, N. Hadia, J. Iqbal, R. F. Mehmood, S. Iqbal, A. M. Shawky, A. Asif, H. Somaily, M. Raheel and R. A. Khera, RSC Adv., 2022, 12, 20792–20806 RSC.
- M. Adeel, M. Khalid, M. A. Ullah, S. Muhammad, M. U. Khan, M. N. Tahir, I. Khan, M. Asghar and K. S. Mughal, RSC Adv., 2021, 11, 7766–7778 RSC.
- L. van Dijk, PhD thesis, Eindhoven University of Technology, 2010 Search PubMed.
- M. U. Saeed, J. Iqbal, R. F. Mehmood, M. Riaz, S. J. Akram, H. Somaily, A. M. Shawky, M. Raheel, M. I. Khan and E. U. Rashid, J. Phys. Chem. Solids, 2022, 170, 110906 CrossRef CAS.
- M. U. Saeed, J. Iqbal, R. F. Mehmood, S. J. Akram, Y. A. El-Badry, S. Noor and R. A. Khera, Surf. Interfaces, 2022, 30, 101875 CrossRef CAS.
- F. B. Kooistra, J. Knol, F. Kastenberg, L. M. Popescu, W. J. Verhees, J. M. Kroon and J. C. Hummelen, Org. Lett., 2007, 9, 551–554 CrossRef CAS PubMed.
- I. Yousaf, R. A. Khera, J. Iqbal, S. Gul, S. Jabeen, A. Ihsan and K. Ayub, Mater. Sci. Semicond. Process., 2021, 121, 105345 CrossRef CAS.
- S. Tretiak and S. Mukamel, Chem. Rev., 2002, 102, 3171–3212 CrossRef CAS PubMed.
- H. D. de Gier, R. Broer and R. W. Havenith, Phys. Chem. Chem. Phys., 2014, 16, 12454–12461 RSC.
- M. Khalid, R. A. Khera, S. Jabeen, P. Langer and J. Iqbal, Comput. Theor. Chem., 2020, 1183, 112848 CrossRef CAS.
- J. Yoon, Y. Hou, A. M. Knoepfel, D. Yang, T. Ye, L. Zheng, N. Yennawar, M. Sanghadasa, S. Priya and K. Wang, Chem. Soc. Rev., 2021, 50(23), 12915–12984 RSC.
- U. Azeem, R. A. Khera, A. Naveed, M. Imran, M. A. Assiri, M. Khalid and J. Iqbal, ACS Omega, 2021, 6, 28923–28935 CrossRef CAS PubMed.
- M. Rafiq, R. A. Khera, M. Salim, M. Khalid, K. Ayub and J. Iqbal, Chem. Phys. Lett., 2021, 782, 139018 CrossRef CAS.
- L. Zheng, N. F. Polizzi, A. R. Dave, A. Migliore and D. N. Beratan, J. Phys. Chem. A, 2016, 120, 1933–1943 CrossRef CAS PubMed.
- B. Qi and J. Wang, Phys. Chem. Chem. Phys., 2013, 15, 8972–8982 RSC.
- M.-H. Jao, H.-C. Liao and W.-F. Su, J. Mater. Chem. A, 2016, 4, 5784–5801 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d2ra08091e |
| This journal is © The Royal Society of Chemistry 2023 |