
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Screening of electrode materials for ammonium ion batteries by high throughput calculation†
Sheqiang Han a,
Xiaodong Zhanga,
Qi Songa,
Bo Zhou*a and
Shangwu Fanb
a,
Xiaodong Zhanga,
Qi Songa,
Bo Zhou*a and
Shangwu Fanb
aInstitute of Modern Physics, Shaanxi Key Laboratory for Theoretical Physics Frontiers, Northwest University, Xi'an, Shaanxi 710069, People's Republic of China. E-mail: zhoubo@nwu.edu.cn
bScience and Technology on Thermostructural Composite Materials Laboratory, Northwestern Polytechnical University, Xi'an, Shaanxi 710072, People's Republic of China
First published on 24th February 2023
Abstract
Ammonium-ion batteries (AIBs) have attracted intense interest lately as promising energy storage systems due to their light weight, safe, inexpensive, and widely available advantages. It is of great significance to find a fast ammonium ion conductor for the electrode of AIBs that directly affects the electrochemical performance of the battery. Using high-throughput bond-valence calculation, we screened the electrode materials of AIBs with a low diffusion barrier from more than 8000 compounds in the ICSD database. Twenty-seven candidate materials were finally identified by the bond-valence sum method and density functional theory. Their electrochemical properties were further analyzed. Our results, which give the relationship between the structure and electrochemical properties of various important electrode materials which are suitable for AIBs development, may pave the way for next-generation energy storage systems.
1. Introduction
With the worldwide emphasis on renewable energy sources such as solar and wind, battery energy storage has become an essential solution for grid stability and reliability. Traditional metal ion batteries (Li+,1 Na+,2 K+,3 etc.) are relatively expensive in terms of abundance in Earth's crust and mining cost. Emerging non-metallic batteries (proton,4 NH4+,5 Cl−,6 etc.) have developed rapidly in recent years. Their cost, capacity, operating voltage, rate performance, and cycle stability are better than metal batteries. Among them, ammonium-ion batteries (AIBs) are promising candidates for large-scale energy storage systems. Compared with metal batteries, ammonium ions have the merit of low molar mass (18 g mol−1), rich reserves and being renewable (only containing N and H essential elements), and having smaller hydrated ion sizes.7,8 Compared with acidic electrolytes, electrolytes containing NH4+ show weak acidity and are less corrosive, will not lead to the dissolution of electrode materials and have higher HER (hydrogen evolution reaction) overpotential than electrolytes containing H+.9,10The progress of ammonium-ion batteries (AIBs)5,9,11–15 is still in its infancy. Ji's team systematically studied the insertion of NH4+ into layered V2O5. It was found that ammonium-ion forms strong hydrogen bonds with layered V2O5, and the diffusion mode of NH4+ is similar to monkey swing.11 Wang also developed NH4+ storage based on supercapacitor. The research indicates that the ammoniation/deamination mechanism is dominated by non-diffusion controlled pseudo capacitance behavior.14 So far, the reported ammonium storage materials are basically MXene,16 Prussian blue analogues,17 conductive polymers (PANI)18 and metal oxides (MoO3, V2O5, MnOx, Fe5V15O39(OH)9·9H2O).12,19,20 However, the performances achieved to date remain inferior to what lithium-ion batteries can do, showing limited capacity and cycling stability. Therefore, to achieve rechargeable AIBs with excellent performance, it is necessary to explore new electrodes for NH4+ storage.
At the current stage, the discovery of these energy materials relies predominantly on experimental serendipity and the try-and-error experimental process that are inefficient and time-consuming. Nevertheless, much evidence has shown that the discovery process of new energy materials could be significantly accelerated by the data mining process and high throughput calculation.21,22 Now, various experimental and computational repositories such as Inorganic Crystal Structure Database (ICSD),23 Crystallography Open Database (COD),24 the Materials Project,25 and others store thousands of experimental and calculated structures, which already serve as potential batteries electrodes or still wait for such an opportunity.
Meanwhile, A. O. Boev et al. extracted 2800 initial structures from the material database and identified 33 potential compounds for cathode materials of lithium-oxygen batteries with low normalized surface energy.26 Max Avdeev used high-throughput screening to conduct a detailed analysis of the 13![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 materials in ICSD, and identified the crystal structures featuring infinite networks of pathways suitable for Li+, Na+, K+, Ag+, and Cu+ ionic transport.27 However, there is no report on the high-throughput screening method to determine the candidate electrode materials for ammonium-ion batteries.
000 materials in ICSD, and identified the crystal structures featuring infinite networks of pathways suitable for Li+, Na+, K+, Ag+, and Cu+ ionic transport.27 However, there is no report on the high-throughput screening method to determine the candidate electrode materials for ammonium-ion batteries.
As inspired by the ion exchange experiments, the compounds from ICSD (inorganic crystal structures database)23 which contain K+ ion were chosen. We replaced the K+ ions in these compounds with NH4+ to work as the input of our following screening process, since the K+ and NH4+ ions have the same valence and almost the same ionic radii (149 pm for K+ and 148 pm for NH4+).28 Further, we used the bond valence energy landscape method (BVEL),29–33 density functional theory (DFT) method and high-throughput screening to find new electrode materials for AIBs. 27 candidate materials were identified. Our theoretical results may open new opportunities for the development of high-performance AIBs.
2. Methods
2.1 Screening algorithm
The approach can be considered as multi-level screening, where at each higher level the complexity of computational methods increases, while the number of candidates reduces. A total of 120481 database entries were screened. We selected the world's largest database ICSD for completely identified inorganic crystal structures as the input to the screening framework. The main steps of the filtering algorithm are as follows: (I) Screening compounds containing K+ in the database and replacing K+ with NH4+, about 8221 compounds were obtained. (II) From bond valence energy landscape (BVEL) calculation, 441 compounds with diffusion barriers less than 0.3 eV were screened. (III) Considering that structures which are suitable for chemical ion exchange route should have robust framework. 166 compounds with general formula of (NH4)aMb(XYc)d (M – metal. in which inactive metals such as Zn, Zr, Ga, Ge, Mg, Al, etc., for electrolyte and active transition metal such as Fe, Mn, Ti, Cr, V, Co, Ni, Nb, Mo, etc. for electrode material; XYc – polyanion groups, X – metalloids such as B, Si, Ge, As and Mo, Nb, etc., Y – O, F, S, etc.) were screened.34 (IV) 108 compounds were obtained by a filter of specific capacity greater than 100 mA h g−1. (V) After the first principle calculation, the 72 compounds were screened using the condition that the open circuit voltage (OCV) was higher than 3 V. (VI) The final screening condition is that the volume expansion rate of the optimized structure is less than 10% compared with the K-contained analog, and 27 compounds are selected (Fig. 1).
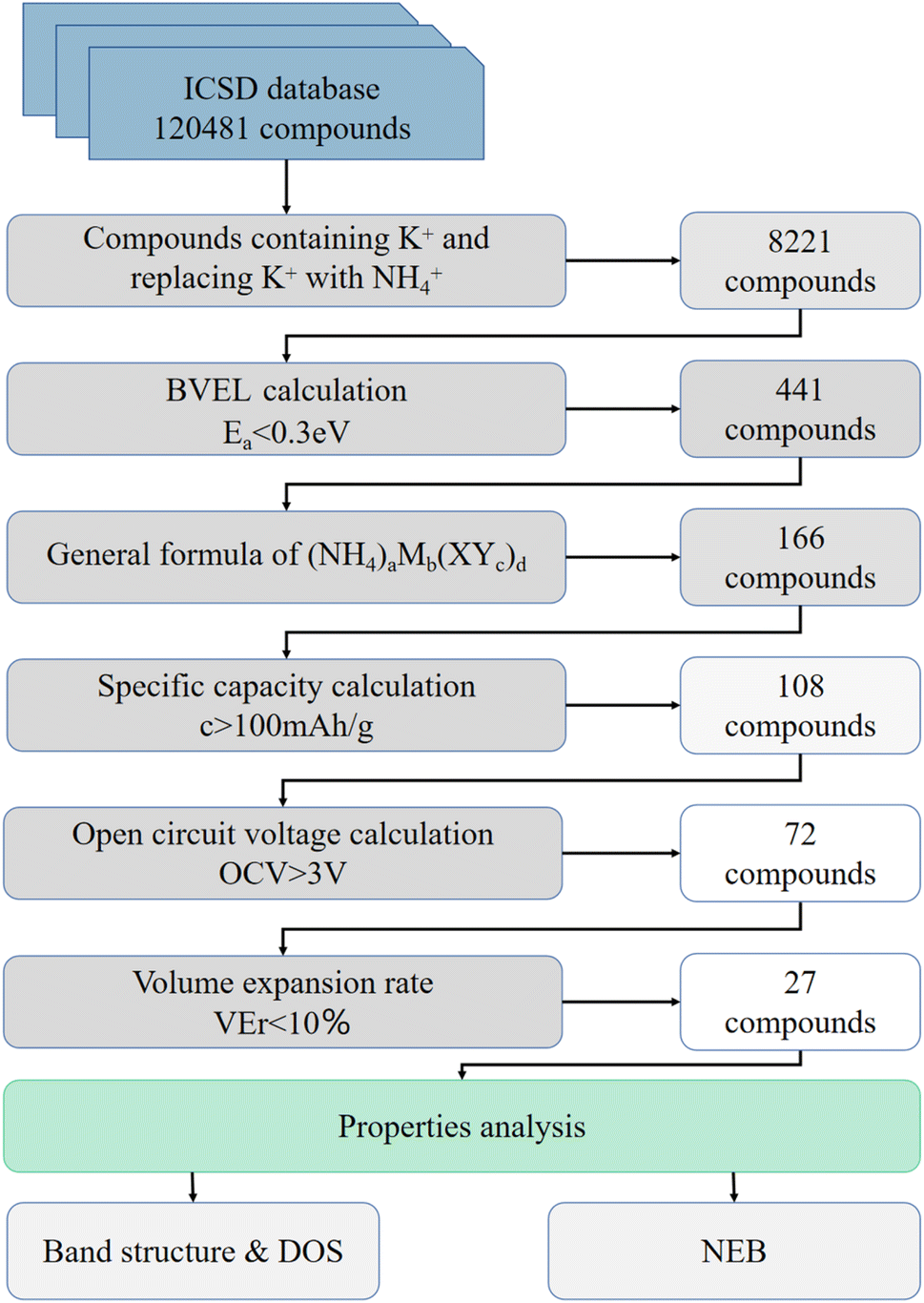 | ||
| Fig. 1 The schema diagram of the screening process. All data relating to screening process are included in the ESI. | ||
2.2 Bond valence energy landscape (BVEL) method
The bond-valence method35 is the development of the principle of local charge neutrality (first proposed by Pauling). The valence of an atom is the sum of the individual bond valences surrounding the atom. The results show that the dependence of Sij on the distance between the ith and jth ions Rij can be well described by exponential function.where R0 and b are empirical parameters.36
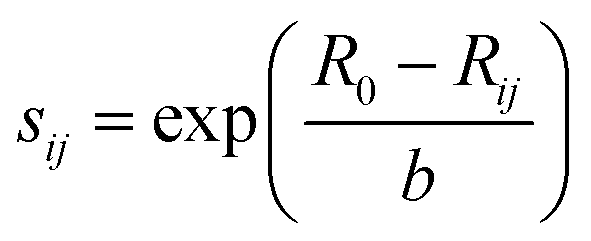
As shown in S. Adams's work,31 the bond valence site energy of i can then be interpreted as a result of a Morse-type interaction with the adjacent anions j, it can be combined with the Coulomb repulsion Erepulsion of i with other immobile cations.
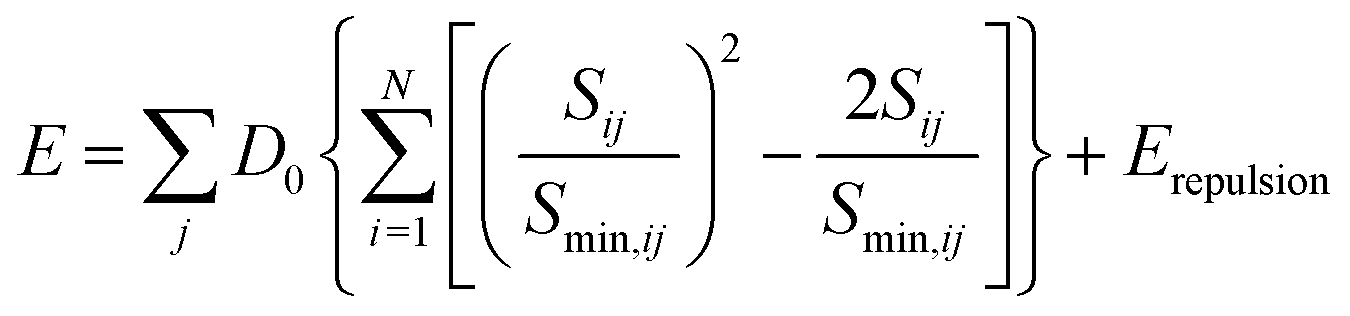
where ri and rj are the covalent radii of the respective cation (or anion) pair i, j and f is a screening factor (typically of the order f ≈ 0.75).37 It may be noted that for the purpose of analyzing E landscapes, Coulomb repulsions are considered only between mobile and immobile cations to reveal continuous pathways, while coulomb attraction terms are generally integrated into the Morse attraction term. The charge screening is based on an error function complement term Erepulsion.
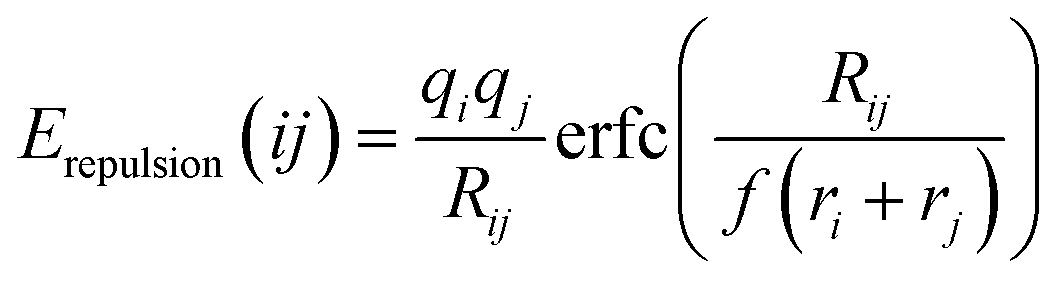
The console utilities CrysFML (Crystallographic Fortran 95 Modules Library) which are developed by N. A. Katcho et al.38 are used for bond-valence energy landscapes calculations. Small Python scripts have been written to bond the input and output between programs to reduce human intervention and facilitate data management. Within the theoretical framework of bond-valence sum, the diffusion barriers (Ea) in three directions of a, b and c of the cell are obtained. The fundamental process that enables the functioning of most of today's rechargeable batteries is the reversible intercalation of ions into electrode. Ionic conductivity emerges as a paramount criterion. We abandon the structures with diffusion barrier higher than 0.3 eV.
Specific capacity is an indicator to the amount of electric charge stored by the electroactive materials in a unit mass. The formula of the specific capacity is as follows:
| C = nF/M |
801 mA h mol−1). We selected compounds with theoretical specific capacity more than 100 mA h g−1 for the next round of screening. 108 kinds of materials were screened from 8221 types of materials.
Open circle voltage (OCV)47,48 is determined by the chemical potential difference between cathode and anode, and can be calculated as
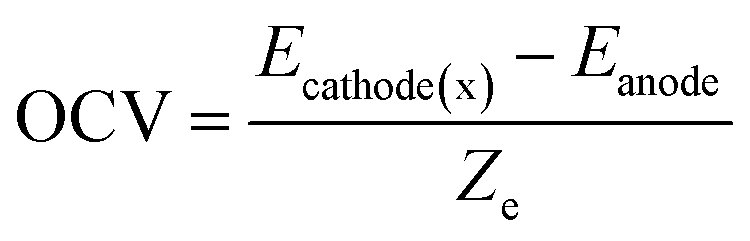
To further evaluate the ionic conductivities, the nudged elastic band (NEB)49,50 method based on DFT is adopted to identify ion migration paths and compute their corresponding energy barriers. Despite the success of the NEB method in characterizing the diffusion process in electrode, the significant computational expense of NEB relative to standard DFT calculations, e.g., geometry relaxations and static energy calculations, has hampered its application to cover all the filtered compounds. Only the final screening compounds are calculated to verify the diffusion barrier calculated by the BVEL method.
3. Results and discussion
3.1 Screening overview
In the screening algorithm, only the first screening method BVEL diffusion barrier calculation is empirical method. The rest of the filtering is based on first principle calculations. The diffusion barrier, open circuit voltage, and specific capacity are designed for better battery performance. While the general formula of (NH4)aMb(XYc)d and smaller volume expansion rate compounds are selected to obtain more stable structures. Moreover, the smaller volume expansion rate means that the compound can be more likely synthesized by the ion exchange experiment.From 8221 compounds containing K+ in the ICSD database, we obtained 27 potential electrode materials for AIBs (Table 1). The calculated OCV is in the range of 3.39 V to 6.85 V. The maximum OCV is obtained from NH4Zn(BeF3)3 which contains fluorine atoms. The OCV of (NH4)aMb(PO4)d derivatives are about 5.0 V depending on the different metal ion involved. The specific capacity of selected compounds is in the range of 100.33 to 336.97 mA h g−1. The maximum specific capacity is calculated from NH4Li2(BO3) which contains only light elements. We also explored their electronic structures within the theoretical framework of density functional theory. Band structures of the screened compounds have been obtained, and the band gaps are shown in Table S6 in ESI.† Further, we have also performed DFT-based NEB calculations for 27 candidate compounds to analyze the feasibility of the screening algorithm and the diffusion properties of candidate materials (Table 2).
| ID | Formula | E | D | c | OCV | B_g | V | V′ | Ver |
|---|---|---|---|---|---|---|---|---|---|
| 16557 |
NH4TiNbO5 | 0.09 | 2 | 121.22 | 5.24 | 2.88 | 452.43 | 483.36 | 6.84 |
| 18022 |
NH4Zn(BeF3)3 | 0.30 | 3 | 101.86 | 6.85 | 6.14 | 361.15 | 390.72 | 8.19 |
| 30910 |
NaNH4GeO3 | 0.17 | 2 | 186.03 | 4.37 | 3.31 | 353.52 | 378.93 | 7.19 |
| 39440 |
NH4CrPO4F | 0.29 | 3 | 161.38 | 4.80 | 2.57 | 855.76 | 910.87 | 6.44 |
| 39697 |
(NH4)2(Zr0.86Ti0.14)(Si3O9)(H2O) | 0.23 | 3 | 171.19 | 3.76 | 0.04 | 969.57 | 1060.22 | 9.35 |
| 39735 |
NH4GeOPO4 | 0.26 | 3 | 145.59 | 5.04 | 3.15 | 794.77 | 853.99 | 7.45 |
| 48177 |
NH4Li2(BO3) | 0.26 | 3 | 336.97 | 3.39 | 4.34 | 331.05 | 356.73 | 7.76 |
| 59281 |
(NH4)2MgWO2(PO4)2 | 0.29 | 3 | 124.60 | 5.06 | 3.55 | 898.89 | 968.91 | 7.79 |
| 59285 |
NH4(TiO)(PO4) | 0.24 | 3 | 168.48 | 5.16 | 3.07 | 866.17 | 919.61 | 6.17 |
| 65260 |
NH4Li4(AlO4) | 0.15 | 1 | 273.36 | 3.95 | 4.16 | 889.14 | 968.35 | 8.91 |
| 69429 |
NH4(SbO)(SiO4) | 0.17 | 3 | 116.47 | 5.33 | 2.67 | 893.75 | 954.77 | 6.83 |
| 74591 |
NH4(TiO)(AsO4) | 0.19 | 3 | 131.97 | 5.13 | 3.13 | 928.52 | 989.23 | 6.54 |
| 79650 |
NH4SnO(PO4) | 0.22 | 3 | 116.47 | 5.30 | 3.02 | 925.30 | 983.87 | 6.33 |
| 79651 |
NH4(VO)(PO4) | 0.24 | 3 | 165.36 | 3.98 | 1.55 | 864.21 | 909.35 | 5.22 |
| 79702 |
(NH4)2Ni(WO2(PO4)2) | 0.19 | 3 | 123.45 | 4.93 | 3.38 | 896.14 | 964.34 | 7.61 |
| 80023 |
NH4(TiO)((P0.56As0.44)O4) | 0.22 | 3 | 150.20 | 5.15 | 3.12 | 894.19 | 960.44 | 7.41 |
| 80893 |
NH4GaF(PO4) | 0.30 | 3 | 145.59 | 5.18 | 3.98 | 835.98 | 895.06 | 7.07 |
| 82398 |
(NH4)2(Be2Si3O9) | 0.28 | 3 | 217.80 | 4.92 | 5.03 | 829.99 | 871.74 | 5.03 |
| 82457 |
NH4Li(Si2O5) | 0.28 | 2 | 187.34 | 4.99 | 4.98 | 233.78 | 255.75 | 9.40 |
| 91534 |
NH4(Ti0.936Sn0.064)O(PO4) | 0.24 | 3 | 163.80 | 4.90 | — | 870.50 | 913.43 | 4.93 |
| 200310 |
(NH4)2Ni(MoO4)2 | 0.27 | 2 | 141.37 | 5.16 | 3.85 | 1747.49 | 955.38 | 9.34 |
| 200613 |
NH4Fe(PO4)F | 0.28 | 3 | 157.58 | 5.17 | 2.33 | 874.62 | 925.15 | 5.78 |
| 203214 |
(NH4)2(Mn3(OH)2(VO4)2) | 0.30 | 2 | 124.89 | 3.48 | 1.96 | 487.51 | 261.09 | 7.11 |
| 203218 |
NH4(MoO2)(AsO4) | 0.28 | 1 | 100.33 | 5.24 | 2.86 | 493.16 | 542.13 | 9.93 |
| 280327 |
(NH4)2(TiSi3O9)(H2O) | 0.16 | 3 | 194.12 | 5.19 | 3.46 | 920.71 | 985.68 | 7.06 |
| 400850 |
(NH4)2(TiO)2(As0.43P0.57O4)2 | 0.21 | 3 | 150.57 | 5.15 | 3.16 | 895.69 | 959.36 | 7.11 |
| 411501 |
(NH4)3(BSb4O13) | 0.21 | 3 | 113.67 | 5.22 | 1.78 | 598.67 | 642.27 | 7.28 |
| ID | Formula | E_a | E_b | E_c | E_neb | OCV |
|---|---|---|---|---|---|---|
| 16557 |
NH4TiNbO5 | 0.09 | 0.65 | — | 0.11 | 5.24 |
| 18022 |
NH4Zn(BeF3)3 | 3.06 | 3.06 | 0.30 | 1.18 | 6.85 |
| 30910 |
NaNH4GeO3 | — | 0.17 | 2.00 | 0.67 | 4.37 |
| 39440 |
NH4CrPO4F | 1.00 | 0.29 | 1.00 | 0.69 | 4.80 |
| 39697 |
(NH4)2(Zr0.86Ti0.14)(Si3O9)(H2O) | 0.85 | 0.85 | 0.23 | 0.26 | 3.76 |
| 39735 |
NH4GeOPO4 | 0.91 | 0.91 | 0.26 | 1.24 | 5.04 |
| 48177 |
NH4Li2(BO3) | 1.72 | 0.26 | 1.72 | 0.12 | 3.39 |
| 59281 |
(NH4)2MgWO2(PO4)2 | 0.53 | 0.49 | 0.29 | 0.46 | 5.06 |
| 59285 |
NH4(TiO)(PO4) | 0.67 | 0.67 | 0.24 | 0.79 | 5.16 |
| 65260 |
NH4Li4(AlO4) | 0.15 | — | — | 0.60 | 3.95 |
| 69429 |
NH4(SbO)(SiO4) | 0.72 | 0.72 | 0.17 | 0.83 | 5.33 |
| 74591 |
NH4(TiO)(AsO4) | 0.51 | 0.51 | 0.19 | 0.14 | 5.13 |
| 79650 |
NH4SnO(PO4) | 0.67 | 0.67 | 0.22 | 1.67 | 5.30 |
| 79651 |
NH4(VO)(PO4) | 0.65 | 0.65 | 0.24 | 0.14 | 3.98 |
| 79702 |
(NH4)2Ni(WO2(PO4)2) | 0.44 | 0.44 | 0.19 | 0.48 | 4.93 |
| 80023 |
NH4(TiO)((P0.56As0.44)O4) | 0.58 | 0.58 | 0.22 | 0.62 | 5.15 |
| 80893 |
NH4GaF(PO4) | 1.17 | 1.17 | 0.30 | 0.21 | 5.18 |
| 82398 |
(NH4)2(Be2Si3O9) | 0.77 | 0.28 | 0.77 | 0.49 | 4.92 |
| 82457 |
NH4Li(Si2O5) | 3.17 | 0.28 | — | 0.50 | 4.99 |
| 91534 |
NH4(Ti0.936Sn0.064)O(PO4) | 0.67 | 0.67 | 0.24 | 0.14 | 4.90 |
| 200310 |
(NH4)2Ni(MoO4)2 | 0.27 | — | 0.47 | 0.54 | 5.16 |
| 200613 |
NH4Fe(PO4)F | 0.28 | 1.03 | 1.03 | 0.12 | 5.17 |
| 203214 |
(NH4)2(Mn3(OH)2(VO4)2) | — | 0.30 | 2.31 | 0.12 | 3.48 |
| 203218 |
NH4(MoO2)(AsO4) | — | 0.28 | — | 0.69 | 5.24 |
| 280327 |
(NH4)2(TiSi3O9)(H2O) | 0.16 | 0.50 | 0.49 | 0.63 | 5.19 |
| 400850 |
(NH4)2(TiO)2(As0.43P0.57O4)2 | 0.57 | 0.57 | 0.21 | 0.63 | 5.15 |
| 411501 |
(NH4)3(BSb4O13) | 0.21 | 0.23 | 0.67 | 0.23 | 5.22 |
We take (NH4)2Ni(MoO4)2 as an example. (NH4)2Ni(MoO4)2 (Fig. 2(b)) is a compound obtained by replacing K+ from K2Ni(MoO4)2 (Fig. 2(a)). The space group is P1, and NH4+ can migrate along the a-axis and c-axis. Therefore, after BVEL calculation, as shown in Fig. 2(c), the low-valued isosurface identifies areas with a low energy barrier and therefore represents the most likely pathway for ion transport. In addition, Fig. 2(d) shows the charge density after the DFT calculation. When we replace K+ with NH4+, and after structural relaxation, it is found that the unit cell has a small expansion (Fig. 2(a) and (b)), the unit cell volume of K2Ni(MoO4)2 expanded from 1747.49 Å3 to 1910.76 Å3 (Tables S4 and S5 in ESI†), At the same time, the symmetry of the structure also changed because of the difference between the tetrahedral structure of NH4+ and the spherical structure of K+. This expansion distortion will not cause the structure of the compound to collapse. From NEB calculation, it is revealed that the strong hydrogen bond makes the diffusion pattern of NH4+ ions similar to the monkey swing (Fig. 2(f)), which is consistent with the study of Ji's team.11 And this diffusion mode is accompanied by the continuous formation and breaking of hydrogen bonds, which can show rapid electrode dynamics and further improve the electrochemical performance of AIBs.
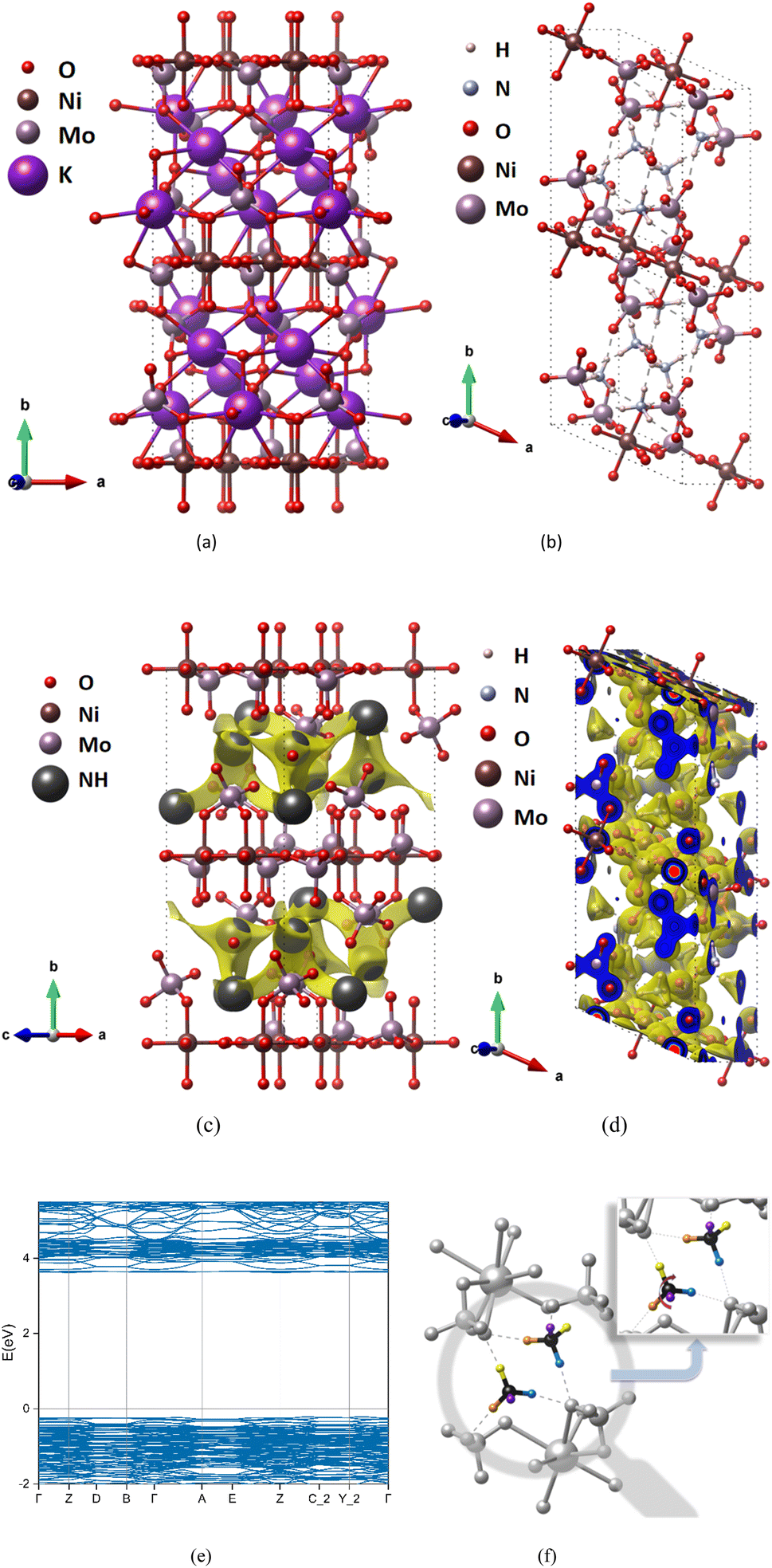 | ||
| Fig. 2 Structure and charge distribution of (K/NH4)2Ni(MoO4)2, the atomic type is marked in the upper left corner of the figures, where “NH” represents ammonium-ion. Unoptimized K precursor K2Ni(MoO4)2 (a) and optimized structure after replacing K+ with NH4+ (NH4)2Ni(MoO4)2 (b). BVEL data (c), charge density diagram (d) and band structure calculated by DFT (e). The diffusion mode of ammonium ion is revealed by NEB calculation (f), in which ammonium is a colored atom, and the same color represents the same atom before and after diffusion. | ||
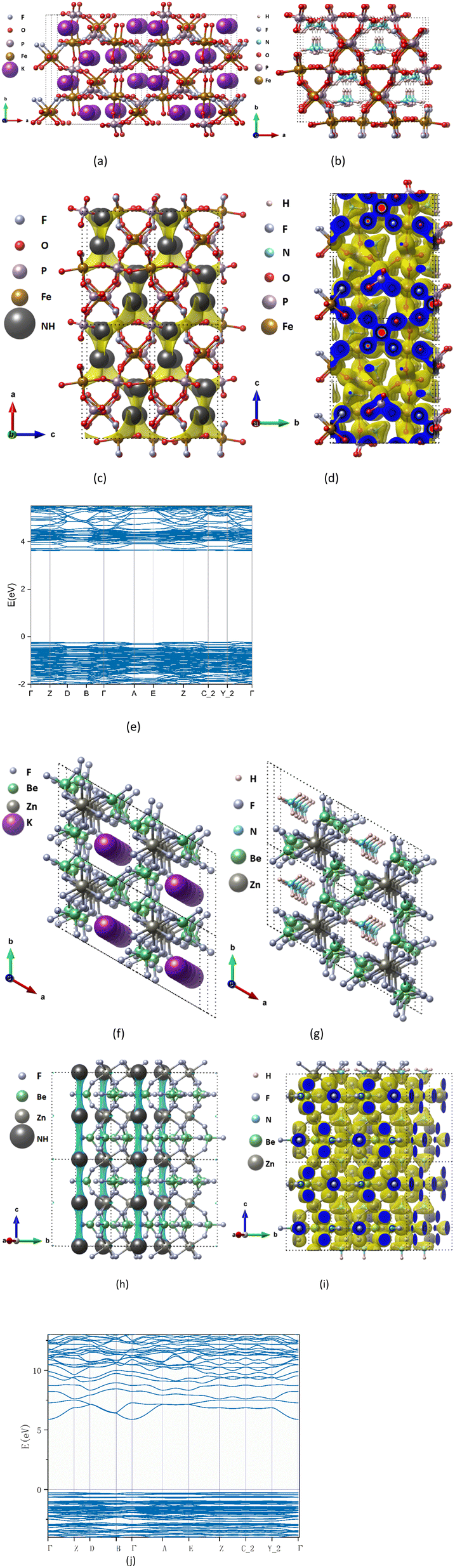 | ||
| Fig. 3 Structure and charge distribution of (K/NH4)Fe(PO4)F and (K/NH4)Zn(BeF3)3 (2 × 2 × 2 supercell), the atomic type is marked in the upper left corner of the figures, where “NH” represents ammonium-ion. Unoptimized K precursor KFe(PO4)F (a), KZn(BeF3)3 (f) and optimized structure after replacing K+ with NH4+ NH4Fe(PO4)F (b) NH4Zn(BeF3)3 (g). BVEL data (c and h) and charge density diagram (d and i) and band structure calculated by DFT (e and j). | ||
3.2 Electronic structure
The preliminary idea to calculate the electronic band structure of a potential electrode material is to determine its metallic, semiconducting or insulating character. The structures and density of states diagrams are listed in Fig. S1–S27 and Tables S7–S33 of the ESI.† Almost all the compounds have band gaps, suggesting poor electronic conductivity. Therefore, further work is needed to improve electronic conductivity.Among the 27 selected compound, there are eleven (NH4)aMb(PO4)d derivatives. The band gap of NH4CrPO4F, NH4GeOPO4, (NH4)2MgWO2(PO4)2, NH4(TiO)(PO4), NH4SnO(PO4), NH4(VO)(PO4), (NH4)2Ni(WO2(PO4)2), NH4(TiO)((P0.56As0.44)O4), NH4GaF(PO4), NH4Fe(PO4)F and (NH4)2(TiO)2(As0.43P0.57O4)2 is 2.57, 3.15, 3.55, 3.07, 3.02, 1.55, 3.38, 3.12, 3.98, 2.33 and 3.16 eV respectively. The band gap of LiFePO4 determined with the ab initio GGA+U method is 3.8 eV.55 Incorporating of metal ion can efficiently modulate the band gap of (NH4)aMb(PO4)d derivatives. The only exception is NH4(VO)(PO4) which has much smaller band gap (1.55 eV). Also, there are five (NH4)aMb(SicOd)e derivatives. The band gap of these compound is in the range 0.04 eV to 5.03 eV. The maximum band gap is 6.14 eV calculated from fluorid NH4Zn(BeF3)3. The theoretical band gap of (NH4)2(Zr0.86Ti0.14)(Si3O9)(H2O) calculated by virtual crystal approximation is very small (0.04 eV) and NH4(Ti0.936Sn0.064)O(PO4) does not show band gap. These two values are abnormal requiring further calculation under more complex methodological architectures.
3.3 Diffusion barrier calculation
Further, we have also carried out more accurate NEB calculations of the screened 27 compounds. The layered structure of NH4TiNbO5 and (NH4)2Ni(MoO4)2 has small diffusion barrier (0.11 and 0.54 eV) which indicates fast NH4+ kinetics. From a structural point of view, the compounds with small diffusion barrier have obvious diffusing channel. The selected bulk materials shown in ESI,† have diverse diffusion pattern.The results (Table 2) show that there is small error between the NEB method based on DFT and the BVEL calculation results for most cases. The main cause of the deviation is that a “static” structure is used during the BVEL calculation. And the cell parameters of the compound remain unchanged after NH4+ substitution, which further leads to the difference between BVEL and the experimental (or DFT) barrier. For instance, the diffusion barrier of NH4Zn(BeF3)3 and NH4(SnO)(PO4) using NEB based on DFT optimization shows ∼1 eV higher than the BVEL barrier, which is due to the significant changes in structural parameters compared with its K precursor (specific data are shown in Tables S4 and S5 of ESI†). Despite deviations in the computed values, the high-throughput screening is capable to predict trends in structure–property relationships. The magnitude of deviations can differ substantially for different materials, environments, and thermodynamic factors. The results from the NEB method are more trustable and should have the quantitative agreement of dynamical properties with experimental results. Nevertheless, the BVEL method is still reliable as a rough selection of candidate battery electrode materials. Considering the complexity of the diffusion process of these compounds and the NEB paths considered may be unexhaustive, all the 27 compounds are reserved for further comprehensive study.
4. Summary
In this work, we have scanned 8221 K+-contained compounds from ICSD to find the proper electrode material for ammonium-ion batteries. The compounds with the exchange of K+ ions with NH4+ ions are used as the input of the following screening process. After the BVEL diffusion barrier filter, general formula filter, OCV screening, specific capacity screening, and volume expansion rate screening, we have successfully predicted 27 potential fast ammonium ion conductors for AIBs, all of which can stimulate further experimental studies and thorough theoretical investigations.Our research is not exhaustive due to the rich structural chemistry of these filtered compounds, it gives an idea of the research interest in electrode materials for ammonium-ion batteries applications. Due to the simplicity of the BVEL method, its straightforward application to high-throughput schemes has some shortcomings. The empirical parameters of the BVEL method for NH4+ ions are incomplete. This is the main factor that causes the miss. Although the ionic radii and valence of K+ and NH4+ ions are similar, the stability analysis after the replacement would probably be insufficient, and additional phonon dispersion calculation would be required. Structures with partial occupations need to be properly treated, which requires more complex methodological architectures. Continued effort is undertaken to expand the databases used for screening and solving associated algorithmic and computational problems.
Data availability
All data generated or analyzed during this study are included in this article and its ESI.†Conflicts of interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.Acknowledgements
This research is supported by the Double First-class University Construction Project of Northwest University, National Natural Science Foundation of China under grants (no. 11947301, 12047502).References
- R. Xiao, H. Li and L. Chen, High-throughput design and optimization of fast lithium ion conductors by the combination of bond-valence method and density functional theory, Sci. Rep., 2015, 5, 14227 CrossRef CAS PubMed.
- V. Palomares, P. Serras, I. Villaluenga, K. B. Hueso, J. Carretero-González and T. Rojo, Na-ion batteries, recent advances and present challenges to become low cost energy storage systems, Energy Environ. Sci., 2012, 5, 5884–5901 RSC.
- X. Wu, D. P. Leonard and X. Ji, Emerging Non-Aqueous Potassium-Ion Batteries: Challenges and Opportunities, Chem. Mater., 2017, 29, 5031–5042 CrossRef CAS.
- R. Emanuelsson, M. Sterby, M. Strømme and M. Sjödin, An All-Organic Proton Battery, J. Am. Chem. Soc., 2017, 139, 4828–4834 CrossRef CAS PubMed.
- Y. Wang and S. F. Kuchena, Recent Progress in Aqueous Ammonium-Ion Batteries, ACS Omega, 2022, 7, 33732–33748 CrossRef CAS PubMed.
- X. Zhao, S. Ren, M. Bruns and M. Fichtner, Chloride ion battery: a new member in the rechargeable battery family, J. Power Sources, 2014, 245, 706–711 CrossRef CAS.
- V. Sidey, On the effective ionic radii for ammonium, Acta Crystallogr., Sect. B: Struct. Sci., Cryst. Eng. Mater., 2016, 72, 626–633 CrossRef CAS PubMed.
- C. D. Wessells, S. V. Peddada, M. T. McDowell, R. A. Huggins and Y. Cui, The Effect of Insertion Species on Nanostructured Open Framework Hexacyanoferrate Battery Electrodes, J. Electrochem. Soc., 2011, 159, A98–A103 CrossRef.
- X. Wu, Y. Qi, J. J. Hong, Z. Li, A. S. Hernandez and X. Ji, Rocking-Chair Ammonium-Ion Battery: A Highly Reversible Aqueous Energy Storage System, Angew. Chem., Int. Ed., 2017, 56, 13026–13030 CrossRef CAS PubMed.
- G. Liang, F. Mo, X. Ji and C. Zhi, Non-metallic charge carriers for aqueous batteries, Nat. Rev. Mater., 2020, 6, 109–123 CrossRef.
- S. Dong, W. Shin, H. Jiang, X. Wu, Z. Li, J. Holoubek, W. F. Stickle, B. Key, C. Liu, J. Lu, P. A. Greaney, X. Zhang and X. Ji, Ultra-fast NH4+ Storage: Strong H Bonding between NH4+ and Bi-layered V2O5, Chem, 2019, 5, 1537–1551 CAS.
- J. Han, A. Varzi and S. Passerini, The Emergence of Aqueous Ammonium-Ion Batteries, Angew. Chem., Int. Ed., 2022, 61, e202115046 CAS.
- H. Li, J. Yang, J. Cheng, T. He and B. Wang, Flexible aqueous ammonium-ion full cell with high rate capability and long cycle life, Nano Energy, 2020, 68, 104369 CrossRef CAS.
- P. Wang, Y. Zhang, Z. Feng, Y. Liu and C. Meng, A dual-polymer strategy boosts hydrated vanadium oxide for ammonium-ion storage, J. Colloid Interface Sci., 2022, 606, 1322–1332 CrossRef CAS PubMed.
- X. Wu, Y. Xu, H. Jiang, Z. Wei, J. J. Hong, A. S. Hernandez, F. Du and X. Ji, NH4+ Topotactic Insertion in Berlin Green: An Exceptionally Long-Cycling Cathode in Aqueous Ammonium-Ion Batteries, ACS Appl. Energy Mater., 2018, 1, 3077–3083 CrossRef CAS.
- M. R. Lukatskaya, O. Mashtalir, C. E. Ren, Y. Dall'Agnese, P. Rozier, P. L. Taberna, M. Naguib, P. Simon, M. W. Barsoum and Y. Gogotsi, Cation Intercalation and High Volumetric Capacitance of Two-Dimensional Titanium Carbide, Science, 2013, 341, 1502–1505 CrossRef CAS PubMed.
- K. Itaya, T. Ataka and S. Toshima, Spectroelectrochemistry and electrochemical preparation method of Prussian blue modified electrodes, J. Am. Chem. Soc., 1982, 104, 4767–4772 CrossRef CAS.
- S. F. Kuchena and Y. Wang, Superior Polyaniline Cathode Material with Enhanced Capacity for Ammonium Ion Storage, ACS Appl. Energy Mater., 2020, 3, 11690–11698 CrossRef CAS.
- Y. Song, Q. Pan, H. Lv, D. Yang, Z. Qin, M. Y. Zhang, X. Sun and X. X. Liu, Ammonium-Ion Storage Using Electrodeposited Manganese Oxides, Angew. Chem., Int. Ed., 2021, 60, 5718–5722 CrossRef CAS PubMed.
- W. Xu, L. Zhang, K. Zhao, X. Sun and Q. Wu, Layered ferric vanadate nanosheets as a high-rate NH4+ storage electrode, Electrochim. Acta, 2020, 360, 137008 CrossRef CAS.
- A. Gandomi and M. Haider, Beyond the hype: big data concepts, methods, and analytics, J. Int. Inf. Manag., 2015, 35, 137–144 CrossRef.
- M. J. Shaw, C. Subramaniam, G. W. Tan and M. E. Welge, Knowledge management and data mining for marketing, Decis Support Syst, 2001, 31, 127–137 CrossRef.
- A. Belsky, M. Hellenbrandt, V. L. Karen and P. Luksch, New developments in the Inorganic Crystal Structure Database (ICSD): accessibility in support of materials research and design, Acta Crystallogr., Sect. B: Struct. Sci., Cryst. Eng. Mater., 2002, 58, 364–369 CrossRef PubMed.
- S. Grazulis, D. Chateigner, R. T. Downs, A. F. T. Yokochi, M. Quiros, L. Lutterotti, E. Manakova, J. Butkus, P. Moeck and A. Le Bail, Crystallography Open Database – an open-access collection of crystal structures, J. Appl. Crystallogr., 2009, 42, 726–729 CrossRef CAS PubMed.
- B. Hourahine, B. Aradi, V. Blum, F. Bonafe, A. Buccheri, C. Camacho, C. Cevallos, M. Y. Deshaye, T. Dumitrica, A. Dominguez, S. Ehlert, M. Elstner, T. van der Heide, J. Hermann, S. Irle, J. J. Kranz, C. Kohler, T. Kowalczyk, T. Kubar, I. S. Lee, V. Lutsker, R. J. Maurer, S. K. Min, I. Mitchell, C. Negre, T. A. Niehaus, A. M. N. Niklasson, A. J. Page, A. Pecchia, G. Penazzi, M. P. Persson, J. Rezac, C. G. Sanchez, M. Sternberg, M. Stohr, F. Stuckenberg, A. Tkatchenko, V. W. Yu and T. Frauenheim, DFTB+, a software package for efficient approximate density functional theory based atomistic simulations, J. Chem. Phys., 2020, 152, 124101 CrossRef CAS PubMed.
- A. O. Boev, S. S. Fedotov, K. J. Stevenson and D. A. Aksyonov, High-throughput computational screening of cathode materials for Li–O2 battery, Comput. Mater. Sci., 2021, 197, 110592 CrossRef CAS.
- M. Avdeev, M. Sale, S. Adams and R. P. Rao, Screening of the alkali-metal ion containing materials from the Inorganic Crystal Structure Database (ICSD) for high ionic conductivity pathways using the bond valence method, Solid State Ionics, 2012, 225, 43–46 CrossRef CAS.
- A. G. Volkov, S. Paula and D. W. Deamer, Two mechanisms of permeation of small neutral molecules and hydrated ions across phospholipid bilayers, Bioelectrochem. Bioenerg., 1997, 42, 153–160 CrossRef CAS.
- I. D. Brown, Recent developments in the methods and applications of the bond valence model, Chem. Rev., 2009, 109, 6858–6919 CrossRef CAS PubMed.
- L. L. Wong, K. C. Phuah, R. Dai, H. Chen, W. S. Chew and S. Adams, Bond Valence Pathway Analyzer—An Automatic Rapid Screening Tool for Fast Ion Conductors within softBV, Chem. Mater., 2021, 33, 625–641 CrossRef CAS.
- S. Adams and R. P. Rao, Understanding Ionic Conduction and Energy Storage Materials with Bond-Valence-Based Methods, Bond Valences, 2014, pp. 129–159 Search PubMed.
- S. Adams and R. P. Rao, High power lithium ion battery materials by computational design, Phys. Status Solidi A, 2011, 208, 1746–1753 CrossRef CAS.
- S. Adams, From bond valence maps to energy landscapes for mobile ions in ion-conducting solids, Solid State Ionics, 2006, 177, 1625–1630 CrossRef CAS.
- S. S. Fedotov, N. A. Kabanova, A. A. Kabanov, V. A. Blatov, N. R. Khasanova and E. V. Antipov, Crystallochemical tools in the search for cathode materials of rechargeable Na-ion batteries and analysis of their transport properties, Solid State Ionics, 2018, 314, 129–140 CrossRef CAS.
- I. D. Brown, Recent developments in the methods and applications of the bond valence model, Chem. Rev., 2009, 109(12), 6858–6919 CrossRef CAS PubMed.
- S. Adams, softBV ver. 0.96, 2004 Search PubMed.
- S. Adams, Practical Considerations in Determining Bond Valence Parameters, 2013, pp. 91–128 Search PubMed.
- N. A. Katcho, J. Carrete, M. Reynaud, G. Rousse, M. Casas-Cabanas, N. Mingo, J. Rodríguez-Carvajal and J. Carrasco, An investigation of the structural properties of Li and Na fast ion conductors using high-throughput bond-valence calculations and machine learning, J. Appl. Crystallogr., 2019, 52, 148–157 CrossRef CAS.
- W.-J. Zhang, Structure and performance of LiFePO4 cathode materials: a review, J. Power Sources, 2011, 196, 2962–2970 CrossRef CAS.
- C. Li, H. P. Zhang, L. J. Fu, H. Liu, Y. P. Wu, E. Rahm, R. Holze and H. Q. Wu, Cathode materials modified by surface coating for lithium ion batteries, Electrochim. Acta, 2006, 51, 3872–3883 CrossRef CAS.
- G. Kresse and J. Furthmüller, Efficient iterative schemes for ab initio total-energy calculations using a plane-wave basis set, Phys. Rev. B: Condens. Matter Mater. Phys., 1996, 54, 11169–11186 CrossRef CAS PubMed.
- P. E. Blöchl, Projector augmented-wave method, Phys. Rev. B: Condens. Matter Mater. Phys., 1994, 50, 17953–17979 CrossRef PubMed.
- S. P. Ong, W. D. Richards, A. Jain, G. Hautier, M. Kocher, S. Cholia, D. Gunter, V. L. Chevrier, K. A. Persson and G. Ceder, Python Materials Genomics (pymatgen): A robust, open-source python library for materials analysis, Comput. Mater. Sci., 2013, 68, 314–319 CrossRef CAS.
- A. Jain, S. Ong, G. Hautier, W. Chen, W. Richards, S. Dacek, S. Cholia, D. Gunter, D. Skinner, G. Ceder and K. Persson, Commentary: The Materials Project: A materials genome approach to accelerating materials innovation, APL Mater., 2013, 1, 011002 CrossRef.
- J. P. Perdew, K. Burke and M. Ernzerhof, Generalized Gradient Approximation Made Simple, Phys. Rev. Lett., 1996, 77, 3865 (Phys. Rev. Lett., 1997, 78, 1396) CrossRef CAS PubMed.
- D. A. Blom and T. Vogt, Probing Compositional Order in Atomic Columns: STEM Simulations Beyond the Virtual Crystal Approximation, Microsc. Microanal., 2020, 26, 46–52 CrossRef CAS PubMed.
- M. K. Aydinol, A. F. Kohan, G. Ceder, K. Cho and J. Joannopoulos, Ab initio study of lithium intercalation in metal oxides and metal dichalcogenides, Phys. Rev. B: Condens. Matter Mater. Phys., 1997, 56, 1354–1365 CrossRef CAS.
- X. Rocquefelte, F. Boucher, P. Gressier and G. Ouvrard, First-Principle Study of the Intercalation Process in the LixV2O5 System, Chem. Mater., 2003, 15, 1812–1819 CrossRef CAS.
- Z. Rong, R. Malik, P. Canepa, G. Sai Gautam, M. Liu, A. Jain, K. Persson and G. Ceder, Materials Design Rules for Multivalent Ion Mobility in Intercalation Structures, Chem. Mater., 2015, 27, 6016–6021 CrossRef CAS.
- Y. Mo, S. P. Ong and G. Ceder, Insights into Diffusion Mechanisms in P2 Layered Oxide Materials by First-Principles Calculations, Chem. Mater., 2014, 26, 5208–5214 CrossRef CAS.
- O. A. Drozhzhin, V. V. Grigoryev, A. M. Alekseeva, R. R. Samigullin, D. A. Aksyonov, O. V. Boytsova, D. Chernyshov, V. V. Shapovalov, A. A. Guda, A. V. Soldatov, K. J. Stevenson, A. M. Abakumov and E. V. Antipov, Revisited Ti2Nb2O9 as an Anode Material for Advanced Li-Ion Batteries, ACS Appl. Mater. Interfaces, 2021, 13, 56366–56374 CrossRef CAS PubMed.
- A. D. Dembitskiy, D. A. Aksyonov, A. M. Abakumov and S. S. Fedotov, NH+-based frameworks as a platform for designing electrodes and solid electrolytes for Na-ion batteries: a screening approach, Solid State Ionics, 2022, 374, 115810 CrossRef CAS.
- L. Zheng, Y. Xu, D. Jin and Y. Xie, Novel Metastable Hexagonal MoO3 Nanobelts: Synthesis, Photochromic, and Electrochromic Properties, Chem. Mater., 2009, 21, 5681–5690 CrossRef CAS.
- G. Liang, Y. Wang, Z. Huang, F. Mo, X. Li, Q. Yang, D. Wang, H. Li, S. Chen and C. Zhi, Initiating Hexagonal MoO3 for Superb-Stable and Fast NH4 (+) Storage Based on Hydrogen Bond Chemistry, Adv. Mater., 2020, 32, e1907802 CrossRef PubMed.
- F. Zhou, K. Kang, T. Maxisch, G. Ceder and D. Morgan, The electronic structure and band gap of LiFePO4 and LiMnPO4, Solid State Commun., 2004, 132, 181–186 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3ra00284e |
| This journal is © The Royal Society of Chemistry 2023 |