
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSynthesis of tetrahydrochromenes and dihydronaphthofurans via a cascade process of [3 + 3] and [3 + 2] annulation reactions: mechanistic insight for 6-endo-trig and 5-exo-trig cyclisation†
Yeruva Pavankumar Reddya,
V. Srinivasadesikana,
Rengarajan Balamurugan b,
M. C. Linc and
Shaik Anwar*a
b,
M. C. Linc and
Shaik Anwar*a
aDepartment of Chemistry, School of Applied Sciences and Humanities, Vignan's Foundation for Science Technology and Research-VFSTR (Deemed to be University), Vadlamudi-522213, Guntur, Andhra Pradesh, India. E-mail: shaikanwarcu@gmail.com; drsa_sh@vignan.ac.in; Web: https://www.vignan.ac.in/bshanwar.php Tel: +91-8632344700
bSchool of Chemistry, University of Hyderabad, Gachibowli, Hyderabad, India 500046
cDepartment of Applied Chemistry, National Yang Ming Chiao Tung University, Hsinchu 30010, Taiwan
First published on 16th February 2023
Abstract
Substituted tetrahydrochromenes and dihydronaphthofurans are easily accessible by the treatment of β-tetralone with trans-β-nitro styrene derived Morita–Baylis–Hillman (MBH) acetates through a formal [3 + 3]/[3 + 2] annulation. The reaction proceeds through a cascade Michael/oxa-Michael pathway with moderate to good yields. A DFT study was carried out to account for the formation of the corresponding six and five-membered heterocycles via 6-endo-trig and 5-exo-trig cyclization.
The ability to synthesize diverse molecules utilizing nitro allylic MBH acetates in various cascade reactions has received considerable interest.1 A few molecules synthesized using nitro allylic acetates have shown promising cytotoxic, trypanocidal and AchE inhibition2 activity in pharmaceutical and medicinal chemistry. Nitro allylic MBH acetates have been used as main precursors in organocatalysis3 and heterocyclic chemistry,4 and as bicyclic skeletons5 for the construction of elegant building blocks like tetrahydro-pyranoquinolinones,6 sulfonyl furans,7 pyranonaphthoquinones,8 arenopyrans/arenylsulfanes,9 triazoles,10a tetrasubstituted furans,10b fused furans,10c,d tetrasubstituted pyrroles,11a benzofuranones,11b and tetrahydropyrano scaffolds/pyranocoumarins.12 The nitro allylic MBH-acetates can also undergo asymmetric benzylic13a and allylic alkylation13b reactions as well as kinetic resolution [KR]13c,d under normal conditions. These acetates undergo a range of cascade [2 + 3],14 [3 + 2]15 and [3 + 3]16 ring annulation reactions using different substrates. They have been widely utilized in [3 + 2]17a and [3 + 3]17b annulation reactions due to their unique nature of 1,2-/1,3-biselectrophilic reactivity to form either five or six membered rings depending on the nature of nucleophiles employed in the reaction17c,d These adducts are also stable under NHC catalytic conditions to yield cyclopentanes.18
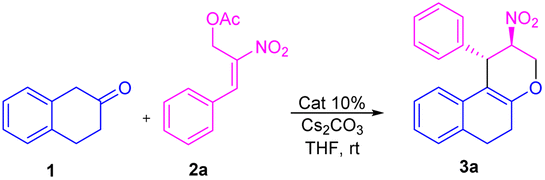
Peng-Fei Xu et al. (Scheme 1, eqn (a)) synthesised tetrahydropyranoindoles through organocatalytic asymmetric C–H functionalization of indoles via [3 + 3] annulation through 6-endo trig cyclization.19 The Namboothiri group recently developed a metal free regioselective synthesis of α-carbolines via [3 + 3] annulation involving secondary MBH acetate (Scheme 1, eqn (b)).20 Previously, our group carried out a [3 + 3] cyclization reaction of β-naphthol with primary MBH acetate to study the scope of SN2′ vs. SN2 reaction.21 With our ongoing interest in using nitro styrene derived MBH adducts22 explored the reactivity of primary and secondary MBH acetate with β-tetralone 1 as our model reaction. Initially, the reaction carried out using β-tetralone 1 with primary MBH-acetate 2, predominantly gave a tetrahydrochromene 3 via [3 + 3] annulation involving 6-endo trig cyclization through Michael/oxa-Michael cascade process. The possible dihydronaphthofuran product was not observed under the present conditions as primary MBH acetate 2 acts as 1,3-biselectrophile instead of 1,2-biselectrophile (Scheme 1, eqn (c)).
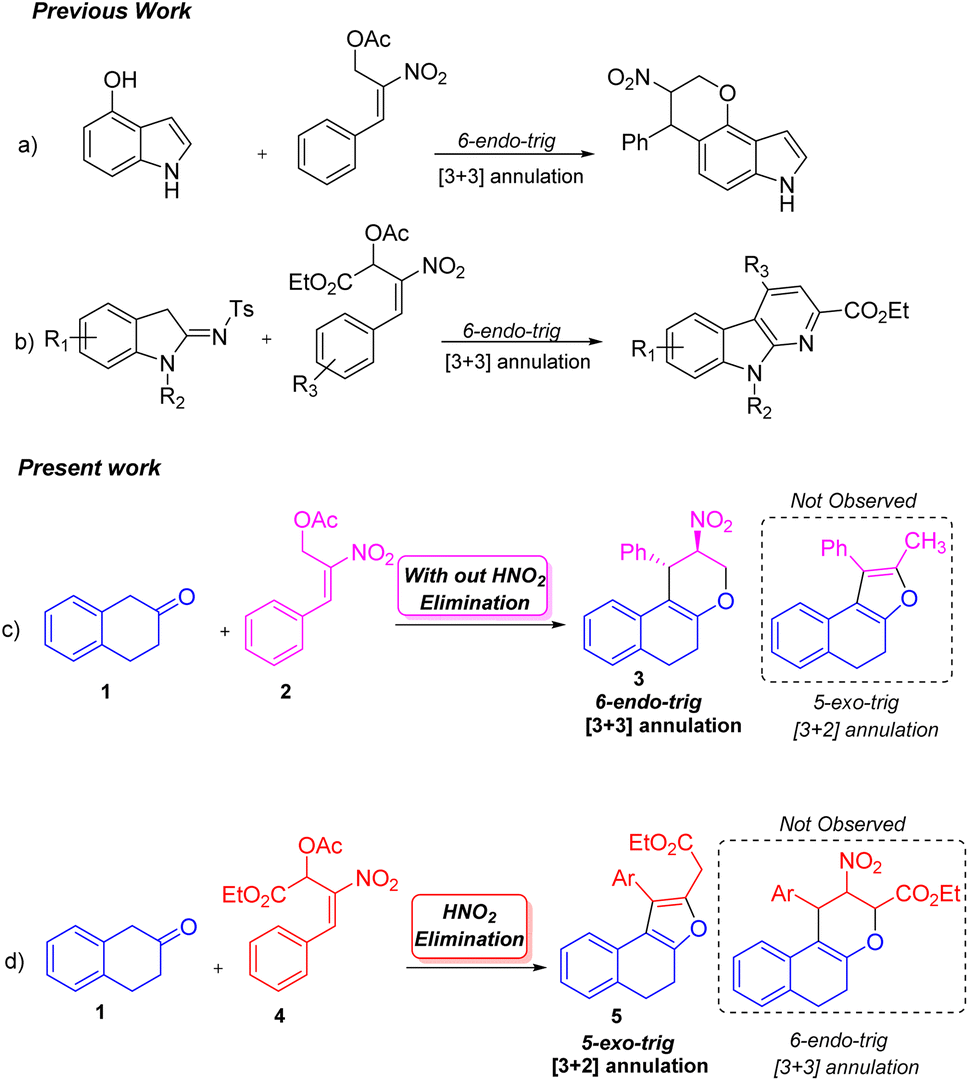 | ||
| Scheme 1 [3 + 3] and [3 + 2] annulation reactions using 1°- and 2°-nitro allylic MBH acetate. | ||
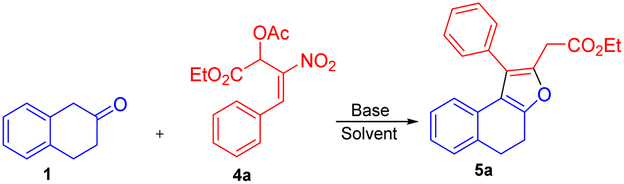
On the other hand, the reaction of β-tetralone 1 with secondary MBH acetate 4 gave dihydronaphthofuran instead of the possible tetrahydrochromene product due to the 1,2-biselectrophile nature of secondary MBH acetate (Scheme 1, eqn (d)). The formation of dihydronaphthofuran 5 occurs in an SN2′ fashion via [3 + 2] annulation involving 5-exo-trig cyclization through Michael followed by intramolecular oxa-Michael reaction with the elimination of HNO2. Subsequently, we have carried out a DFT calculation to prove the formation of tetrahydrochromene 3 using primary MBH acetate 2 and dihydronaphthofuran 5 in the case of secondary MBH acetate 4.
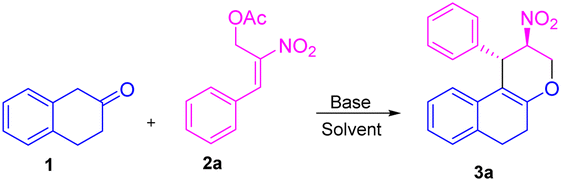
Initially, we carried out the optimization conditions for constructing tetrahydrochromenes 3a using β-tetralone 1 with MBH nitro allylic primary acetate 2a with different bases and solvents. Reaction with organic base, i.e. DABCO using a polar aprotic solvent such as acetonitrile at room temperature gave the desired product in 27% (Table 1, entry 1). Using chlorinated solvents like CHCl3 and CH2Cl2 resulted in meagre yield (i.e., 22 and 19%, respectively) with the recovery of starting material (Table 1, entries 2 and 3). The yield of the product was increased to 40% in the presence of DMAP and THF solvent (Table 1, entry 4). We obtained similar yields i.e. 45 and 44% in the presence of triethylamine, triphenylphosphine respectively (Table 1, entries 5 and 6). The reaction carried out using an inorganic base such as K2CO3 in THF solvent led to an increase in yield i.e., 60% (Table 1, entry 7). A maximum yield of 77% was observed when the reaction was carried out in the presence of Cs2CO3 as base and THF as a solvent at room temperature for 4 h (Table 1, entry 8). The reaction with reduced base equivalents led to reduced yields concluded that 2.0 equiv. of Cs2CO3 is desirable to yield tetrahydrochromene 3a under the present reaction conditions (Table 1, entries 9–11). Further, increase of Cs2CO3 with 3.0 equiv. led to unclean reaction mixture with decreased yield of the product 3a (Table 1, entry 12). Notably, excellent diastereoselectivity of 99![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 dr was retained in all the cases of solvent/base optimisation studies. The stereocenter of the compound 3a was further confirmed by 1H, 13C NMR, HRMS, and single crystal XRD23 (CCDC-2149875) (Table 2).
:1 dr was retained in all the cases of solvent/base optimisation studies. The stereocenter of the compound 3a was further confirmed by 1H, 13C NMR, HRMS, and single crystal XRD23 (CCDC-2149875) (Table 2).
|
|||||
|---|---|---|---|---|---|
| Entry | Base | Solvent | Time (h) | Yield (%) | drb |
| a Unless otherwise noted, reactions were carried out by and (0.11 mmol) of 1 with (0.11 mmol) of 2a using 0.22 mmol of a base in 1 ml of THF solvent.b Determined by 1H-NMR analysis of crude reaction mixture.c Reaction was carried out using 0.5 equiv. of Cs2CO3.d Reaction was carried out using 1.0 equiv. of Cs2CO3.e Reaction was carried out using 1.5 equiv. of Cs2CO3.f Reaction was carried out using 3.0 equiv. of Cs2CO3. | |||||
| 1 | DABCO | CH3CN | 5 | 27 | 99:1 |
| 2 | DABCO | CH2Cl2 | 5 | 22 | 99:1 |
| 3 | DABCO | CHCl3 | 5 | 19 | 99:1 |
| 4 | DMAP | THF | 5 | 40 | 99:1 |
| 5 | TEA | THF | 5 | 45 | 99:1 |
| 6 | PPh3 | THF | 5 | 44 | 99:1 |
| 7 | K2CO3 | THF | 4 | 60 | 99:1 |
| 8 | Cs2CO3 | THF | 4 | 77 | 99:1 |
| 9c | Cs2CO3 | THF | 8 | 50 | 99:1 |
| 10d | Cs2CO3 | THF | 5 | 61 | 99:1 |
| 11e | Cs2CO3 | THF | 4 | 65 | 99:1 |
| 12f | Cs2CO3 | THF | 4 | 60 | n.d |
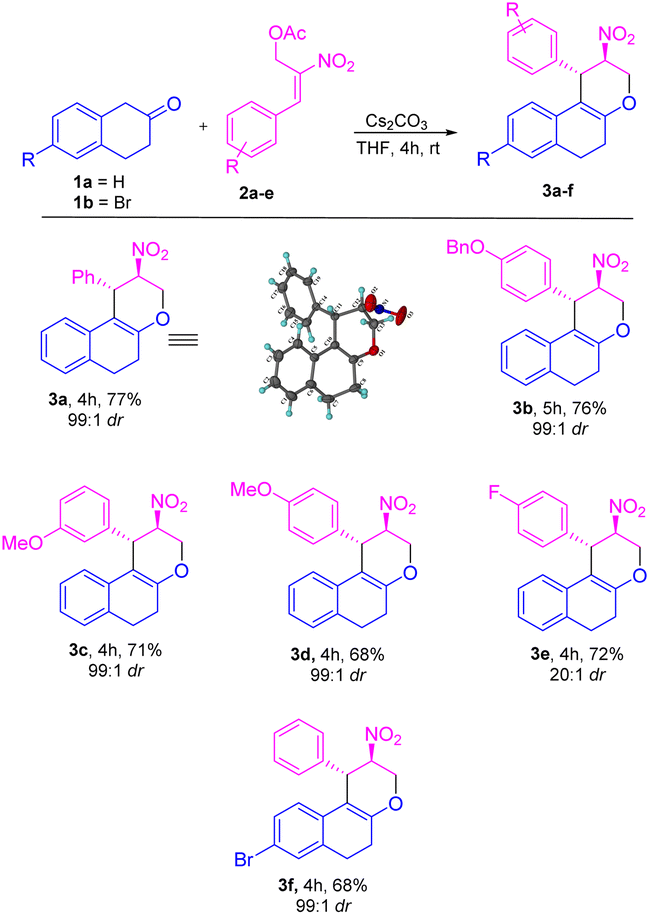 |
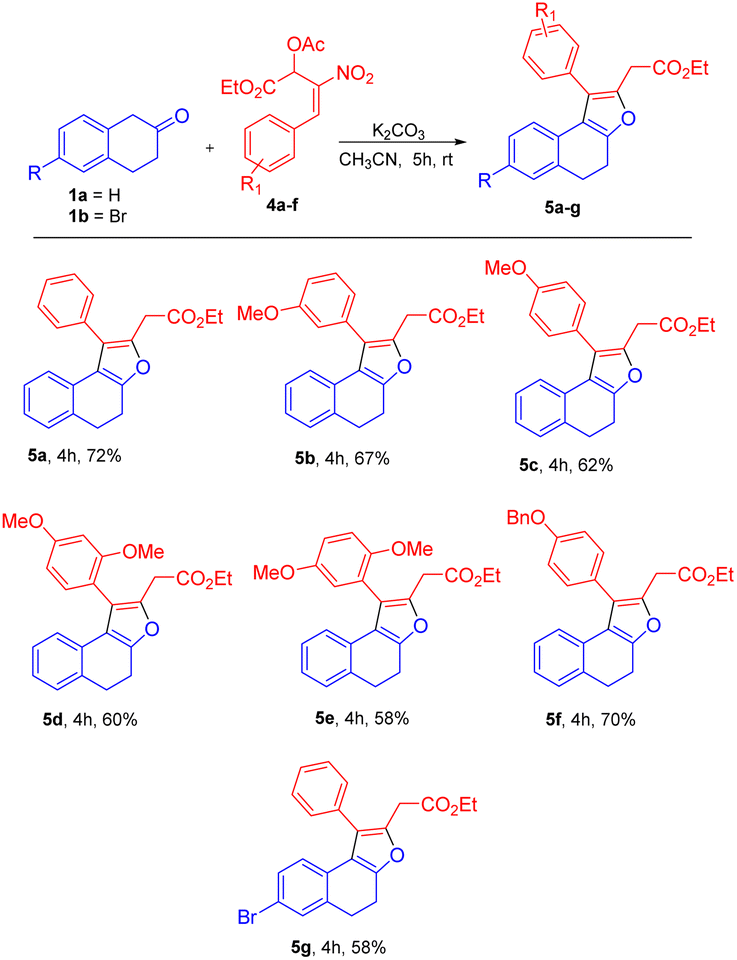
Based on the best optimized conditions, we studied the scope of different nitro allylic MBH primary acetates (2a–e) with β-tetralone 1. The reaction accommodates various electron rich substituents on the primary MBH acetates (2a–e). The electron rich substituent contaning 2b gave 76% yield for the benzyloxy product 3b. The substrate having meta-OMe and para–OMe gave the desired product 3c and 3d with 71 & 68% of yield, respectively. Furthermore, using fluoro substituent at para position of the MBH adduct gave the product 3e with 72% yield. Reaction carried out using 6-bromo tetralone 1b gave the corresponding product 3f in 68% of yield. Notably, remarkable diastereoselectivity of 99:1 dr was observed in all the cases of base screening and substrate scope of MBH primary acetate.
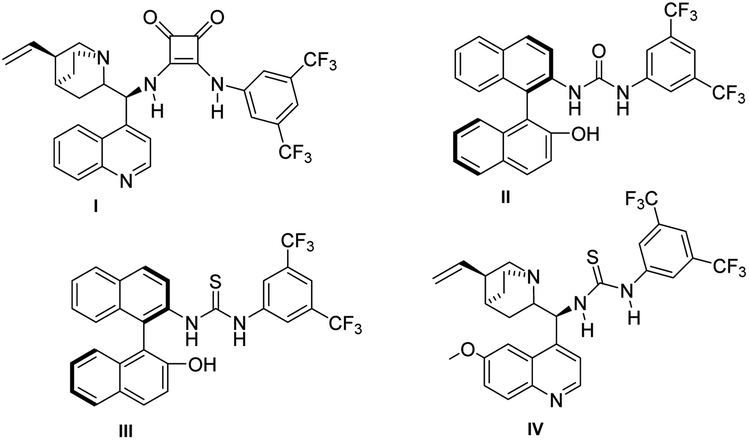
Encouraged, by the high diastereoselectivity for various tetrahydrochromenes derivatives 3a–e, we pursued our studies towards asymmetric synthesis of 3a using different chiral catalysts (I–IV). We observed the poor ee for the product formation in the presence of cinchona based squaramide catalyst I & BINAM based urea catalyst II (Table 3, entries 1 and 2). BINAM derived thiourea catalyst III resulted in a 10% enantiomeric excess (Table 3, entry 3). We obtained a moderate enantiomeric excess (49% ee) with good diastereoselectivity (99:1) in the presence of thiourea based hydrogen bonding catalyst IV (Table 3, entry 4). The use of various Lewis base catalysts also could not enhance the enantioselectivity.
|
||||
|---|---|---|---|---|
| Entry | Catalyst | Time (h) | Yield (%) | ee (%) |
| a All the reactions were carried out with (0.11 mmol) of 1, (0.11 mmol) of 2a, (0.22 mmol) of base and 10 mol% in 1 ml of THF solvent. | ||||
| 1 | I | 4 | 59 | <2 |
| 2 | II | 4 | 60 | <2 |
| 3 | III | 4 | 60 | 10 |
| 4 | IV | 4 | 63 | 49 |
We next focused our studies on understand the reactivity of secondary MBH acetate 4a using β-tetralone 1. Interestingly, the reaction followed an SN2′ Michael/intramolecular oxa-Michael pathway to form dihydronaphthofuran 5a via [3 + 2] annulation instead of an alternate path resulting in the formation of chromene product via 3 + 3 annulation (i.e., Scheme 1; eqn (d)). To recognize the optimal reaction condition, we carried out the reaction in the presence of Cs2CO3 in THF solvent to get the desired dihydronaphthofuran 5a in 35% yield (Table 4, entry 1). Next, we investigated the influence of chlorinated solvents on the product formation 5a. Solvents such as CH2Cl2, CHCl3, and CCl4 failed to enhance the yield for the product formation (Table 4, entries 2–4). Furthermore, an organic base such as TEA in THF gave the required product 5a with 46% of yield (Table 4, entry 5). A drop in the yield was observed using DABCO in presence of a polar aprotic solvent such as acetonitrile (Table 4, entry 6). Unfortunately, the yield drastically dropped to 27% using triphenylphosphine as a base (Table 4, entry 7). Finally, a good yield was observed with an inorganic base, i.e. K2CO3 using acetonitrile as a solvent to furnish the desired product 5a in 72% of yield (Table 4, entry 8).
|
||||
|---|---|---|---|---|
| Entry | Base | Solvent | Time (h) | Yield (%) |
| a Unless otherwise noted, reactions were carried out with (0.11 mmol) of 1 with (0.11 mmol) of 4a using 0.22 mmol% of base in 1 ml of acetonitrile solvent.b Reaction was carried out using 0.5 equiv. of K2CO3.c Reaction was carried out using 1.0 equiv. of K2CO3.d Reaction was carried out using 1.5 equiv. of K2CO3.e Reaction was carried out using 3.0 equiv. of K2CO3. | ||||
| 1 | Cs2CO3 | THF | 6 | 35 |
| 2 | Cs2CO3 | CH2Cl2 | 4.5 | 31 |
| 3 | Cs2CO3 | CHCl3 | 4.5 | 27 |
| 4 | Cs2CO3 | CCl4 | 4.5 | 16 |
| 5 | TEA | THF | 5 | 46 |
| 6 | DABCO | CH3CN | 6 | 41 |
| 7 | PPh3 | CH3CN | 5 | 27 |
| 8 | K2CO3 | CH3CN | 4 | 72 |
| 9b | K2CO3 | CH3CN | 7 | 59 |
| 10c | K2CO3 | CH3CN | 6 | 63 |
| 11d | K2CO3 | CH3CN | 5 | 68 |
| 12e | K2CO3 | CH3CN | 4 | 65 |
The reaction with reduced base equivalents, led to reduced yields confirming that 2.0 equiv. of K2CO3 is desirable to yield dihydronaphthofuran 5a at room temperature within 4 h (Table 4, entries 9–11). It was observed that a low yield was obtained when the reaction was carried out with 3.0 equiv. of base (Table 4, entries 12).
Utilizing the established optimized reaction conditions, i.e. (Table 4, entry 8), the substrate scope of this formal [3 + 2] annulation was explored towards the construction of substituted dihydronaphthofurans 5b–g (Table 5). The meta-OMe and para-OMe on the phenyl ring gave the good yield of 67 and 62%, respectively, for products 5b and 5c. The di substituted 2,4-dimethoxy, and 2,5-dimethoxy containing substrates gave a moderate yield of 60 and 58% for the desired products 5d and 5e. The substrate with benzyloxy substituent at the para position of the phenyl ring also gave the desired product 5f in 70% of yield. A reaction carried out using substrate 1b resulted in product 5g in 58% yield (Table 5).
 |
To further demonstrate our protocol's practical and scalable utility, we have carried out the gram scale preparation of tetrahydrochromene 3a and dihydronaphthofuran 5a in 66 and 70% of yield. We observed the retention of diastereoselectivity i.e., 99:1 of tetrahydrochromene 3a, even at the gram scale condition (Scheme 2).
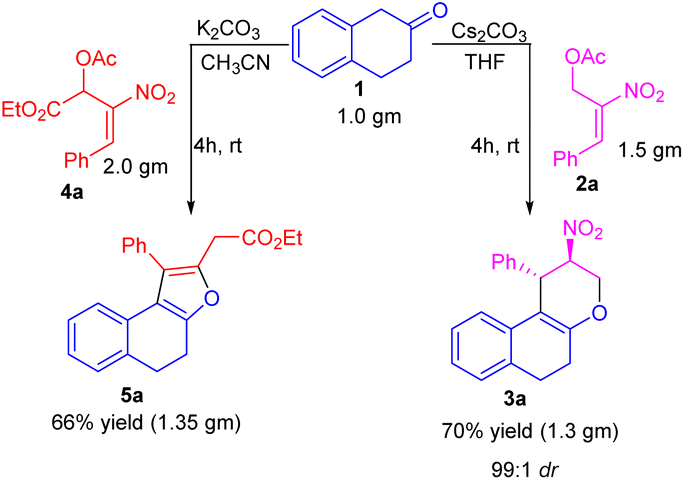 | ||
| Scheme 2 Gram scale synthesis of tetrahydrochromene 3a and dihydronaphthofuran 5a. | ||
We have successfully applied the synthetic utility for dihydronaphthofuran 5a. Reduction of the ester group in 5a was feasible using LAH in THF to afford the desired alcohol product 6 with 60% of yield. Using KOH, the ester group in dihydronaphthofuran 5a was hydrolysed to the corresponding acid derivative 7 in 70% yield. The amidation reaction of 7 with aniline accomplished oxidation of the tetralone ring providing the N-phenyl-2-(1-phenylnaphtho[2,1-b]furan-2-yl)acetamide product 8 in 67% of yield (Scheme 3).
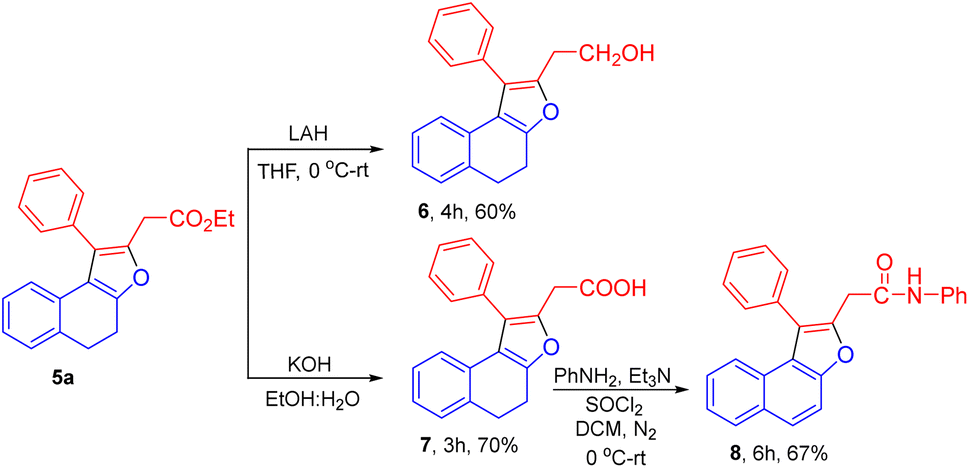 | ||
| Scheme 3 Synthetic utility for the dihydronaphthofuran 5a. | ||
DFT studies
Computational methods
The mechanistic pathway has been studied using the state-of-the-art density functional theory (DFT). All the computations were carried out at the B3LYP24 level of theory using the Gaussian 0925 suite of program. The reactants, intermediates, and products were optimized at the B3LYP level of theory and confirmed no imaginary frequency. Furthermore, a minimum of one imaginary frequency were confirmed for all transition states.
Results and discussions
The reaction of β-tetralone 1(A) with MBH nitro allylic primary acetate 2a has produced tetrahydrochromene 3a in this experiment. The reactant 1 was considered as an anion, where one of the protons of alpha carbon was deprotonated in the presence of base, and was allowed to interact with 2a, resulting in the complex formation with the binding energy of −19.56 kcal mol−1 as shown in the potential energy surface diagram Fig. 1 (i.e. P-cmplx). The respective anion attacked the alpha carbon of primary acetate 2a via SN2′ fashion to form the intermediate (P-int1) with the C–C bond formation via P-TS1 with the activation of energy of 10.56 kcal mol−1. Due to the double bond migration, the OAc was dissociated from P-int1 via P-TS2 with the activation energy of 2.87 kcal mol−1 and produced P-int2 with the lowest energy of −10.35 kcal mol−1 compared with P-int1. The unstable P-int2 immediately undergone for six-member ring cyclisation with the activation energy of 30.92 kcal mol−1. The activation energy for the six-member ring cyclisation using the DFT method was in line with the previous literature report.26 The product formation P-int3 with enthalpy of activation 13.37 kcal mol−1. The proton transfer has occurred on P-int3 via P-TS4 with the activation energy of 30.85 kcal mol−1 has produced the final product of naphthopyran (3a). The higher amount of activation energy was observed for proton transfer due to the proton migration from rich nucleophilic center to a weak electrophilic center. The Mulliken charge for the electron rich and poor centers have been shown in ESI Fig. 1.† The geometrical parameters of hydrogen bond length have been observed to be 2.161 and 1.505 Å shown in Fig. 1 (see ESI†). The exothermicity of naphthopyran (3a) was obtained with the −10.49 kcal mol−1, exothermically. The lowest energy shows the stability of the final product 3a. Here, we studied the five-member ring formation also to understand the stability and possibility of formation. The β-tetralone oxygen atom attacks the tertiary carbon and produced five membered cyclized product. However, the energy barrier has observed to be 41.28 kcal mol−1 and the energy of five-member ring product has observed to be endothermic and shown the instability (Fig. 1 and 2).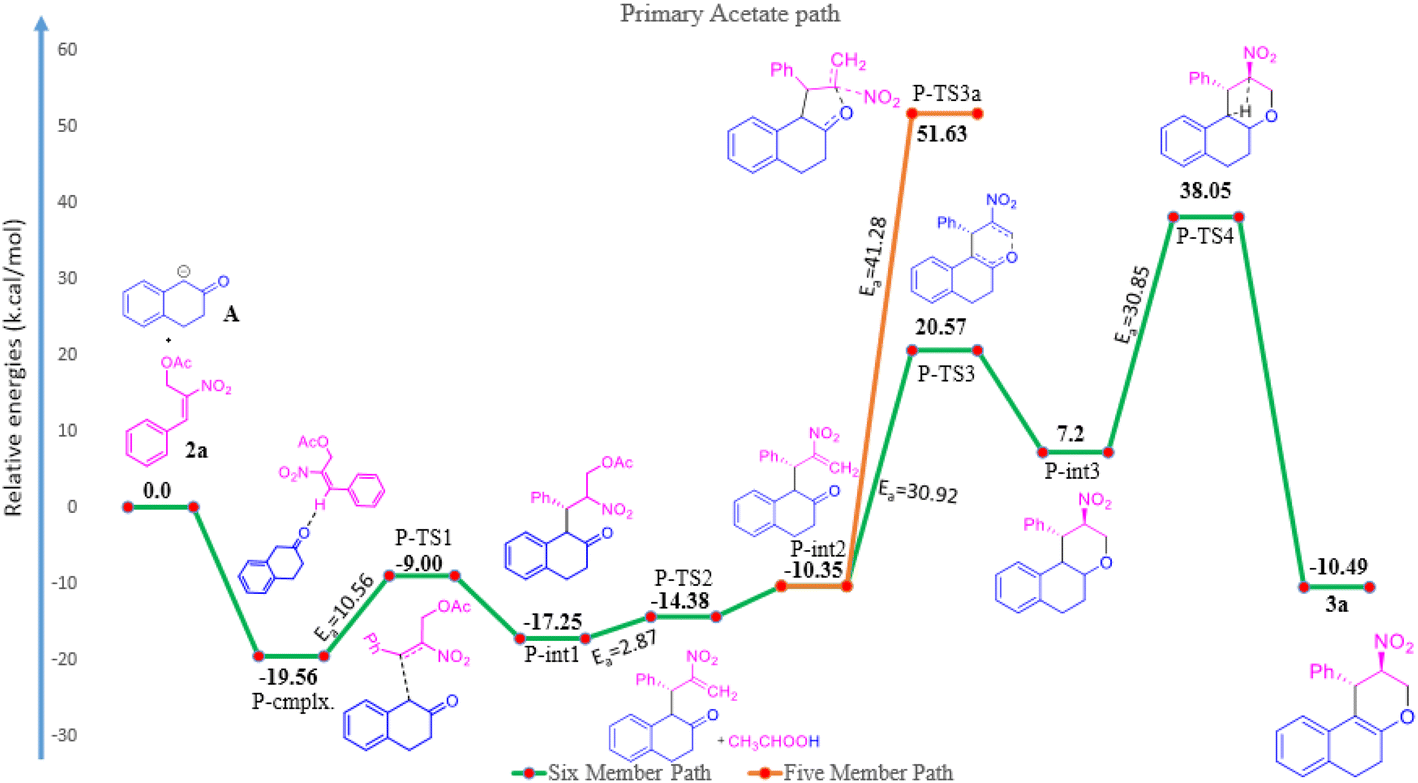 | ||
| Fig. 1 Potential energy surface mechanism of primary acetate 2a with β-tetralone 1. | ||
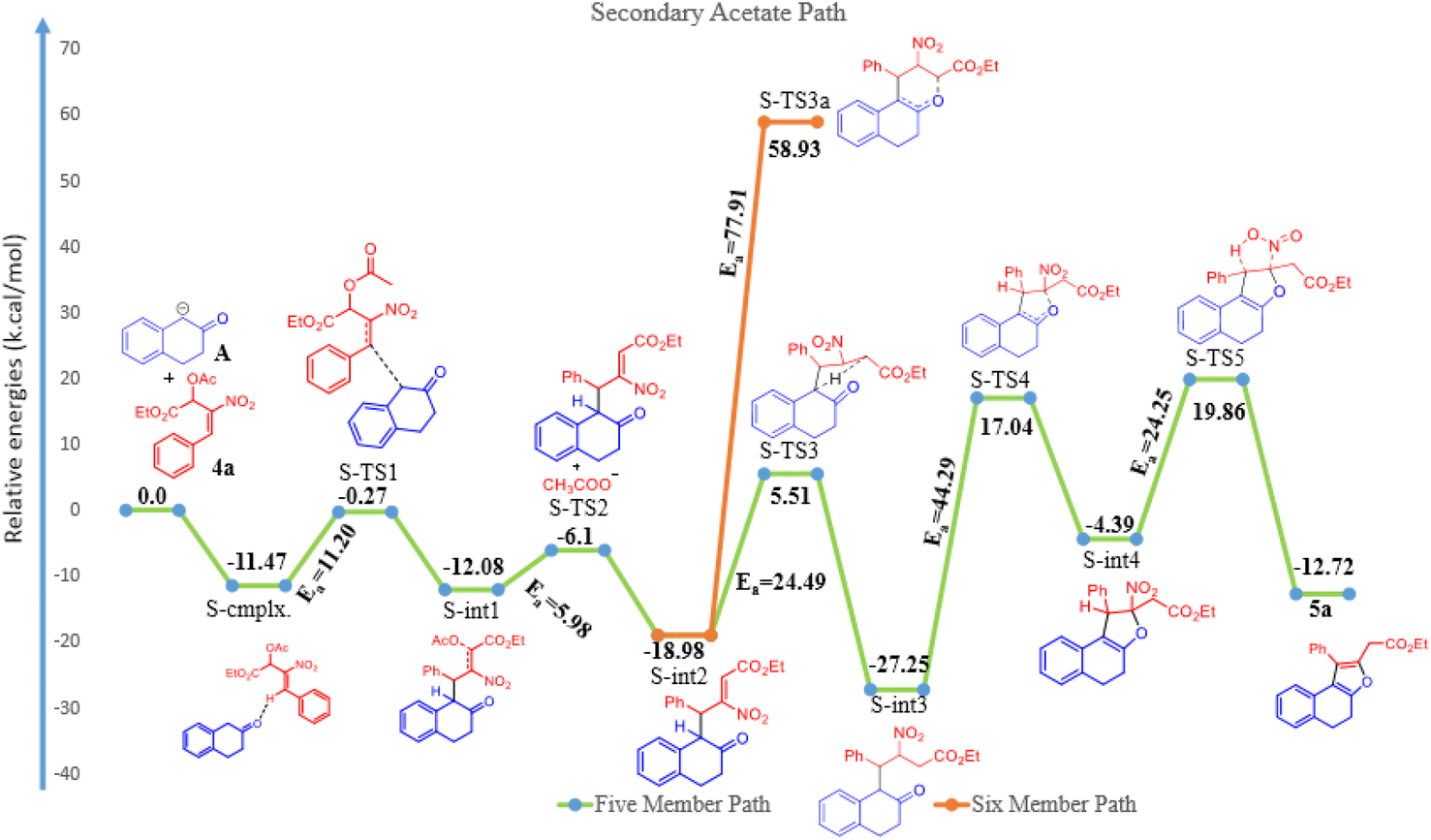 | ||
| Fig. 2 Potential energy surface mechanism of secondary acetate 4a with β-tetralone 1. | ||
The β-tetralone 1 reacting with nitro allylic secondary MBH acetate 4a produced the dihydronaphthofuran derivatives, 5a–g, which is observed experimentally as shown in Table 5. The anion of β-tetralone 1 was considered for the DFT calculation. Here, an anion of β-tetralone interacts with the MBH nitro allylic secondary acetate 4a has produced a complex with a binding energy of −11.47 kcal mol−1. The anion path was considered for the theoretical calculation due to the abstraction of a proton by the base, initiating the reaction observed experimentally. The anion is displacing the acetate via SN2′ fashion has produced the intermediate (S-int1) as C–C bond formation with the barrier energy of 11.20 kcal mol−1. The formation of S-int1 is stable exothermically than the complex (S-cmplx). The S-TS2 was shown to be an acetate leaving group with a barrier energy of 5.98 kcal mol−1 and produced the product with new C–C bond formation and with dissociation the acetate group. The S-int2 initiate the proton transfer to produce the S-int3 with the stable intermediate. The barrier energy for the proton transfer has been observed as 24.49 kcal mol−1 showed in Fig. 2. The formation of S-int3, readily undergo for five-member cyclisation. As shown in Fig. 2, the S-int3 has produced S-int4 via S-TS4 with a barrier energy of 44.29 kcal mol−1. The higher barrier energy observed for cyclisation is accountable for the concern carbon has the electron-withdrawing nitro group. The highest energy barrier for cyclisation has considered as the rate determining step (RDS) for the secondary acetate path. Thus, the nitro group leave immediately after five-member ring cyclisation as HONO with the barrier energy of 24.25 kcal mol−1 and produced the observed product 5a. The relative energy of 5a was observed to be −12.72 kcal mol−1 showing the stability of the product. In the secondary acetate path, we have studied the possibility of a six-member ring formation. However, the barrier energy was observed to be 77 kcal mol−1 (S-Ts3a) and it observed as unfavourable path.
We have explained a plausible mechanism for the formation of tetrahydrochromene 3a and dihydronaphthofuran 5a in correlation to density functional theory (Fig. 1 and 2). The β-tetralone 1 generates the nucleophile A in the presence of a base. The nucleophile A attacks at α-position on the benzylic carbon atom of primary MBH-acetate 2a to generate intermediate B. This initial Michael attack involves the migration of double bond with the elimination of acetic acid via SN2′ reaction. Further deprotonation in the form of keto–enol tautomerism generates a nucleophilic oxygen atom to facilitate the oxa-Michael attack at γ-carbon atom of intermediate B. This intramolecular oxa-Michael addition favors 6-endo trig cyclization with [3 + 3] annulation to furnish the desired product 3a.
The possible alternate pathway involving oxa-Michael attack at the β-carbon atom of B′ is disfavoured for forming fused furan ring. Due to the 1,3-biselectrophilic nature of primary MBH acetate there exists a ring strain associated with undergoing the 5-exo-trig cyclization via [3 + 2] annulation (Scheme 4). The formation of 5a initiates with the attack of nucleophile A at the α-position of the benzylic carbon atom of secondary MBH-acetate 4a. This initial Michael addition involves migration of double bond and elimination of acetic acid through effective SN2′ reaction to generate intermediate C. Further deprotonation followed by tautomerism results in nucleophilic oxygen which undergoes intramolecular oxa-Michael reaction at the β-position of 4a to give intermediate D with [3 + 2] annulation via 5-exo-trig cyclization. Further elimination of HONO under basic conditions results in 5a as the favoured product. Alternate oxa-Michael attack on γ-position of intermediate C′ was disfavoured for a possible [3 + 3] annulation via 6-endo-trig cyclization. This alternate pathway towards the 4H-pyran precedes through D′ with 1,2-elimination of HONO is disfavoured.
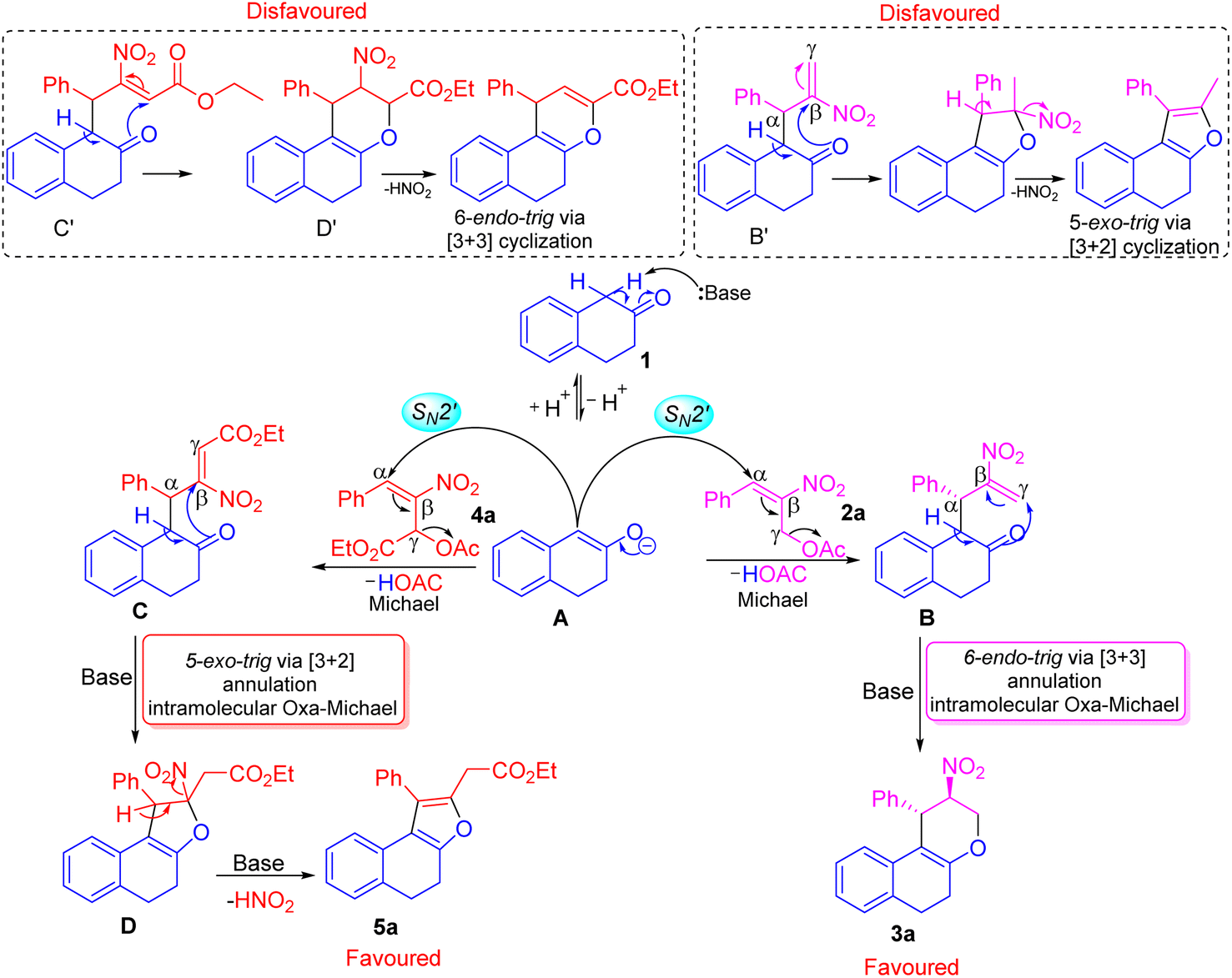 | ||
| Scheme 4 Plausible mechanism for the construction of tetrahydrochromene 3a and dihydronaphthofuran 5a. | ||
Conclusions
In conclusion, β-tetralone in reaction with primary MBH acetate results in six membered tetrahydrochromenes via [3 + 3] annulation. On the other hand, the reaction with secondary MBH acetate results in the formation of five membered dihydronaphthofuran derivatives via [3 + 2] annulation. The DFT result shows that the β-tetralone attacks MBH nitro allylic primary acetate through SN2′ and intramolecular oxa-Michael reaction via 6-endo-trig with a low barrier energy of 1.47 kcal mol−1 which favors the six membered cyclization compared with five membered ring cyclization with the barrier energy of 52.63 kcal mol−1. Similarly, secondary MBH acetate follows the SN2′ reaction and intramolecular oxa-Michael reaction via 5-exo-trig occurs with a low barrier energy of 55.29 kcal mol−1 to obtain the 5-member ring cyclization product disfavouring 6-endo-trig cyclization with a barrier energy of 56 kcal mol−1.Author contribution
All authors contributed to the conception and design of the study. Material preparation, data collection, and analysis were performed by Y. P. Reddy. S. A. and R. B. contributed on additional analysis required to address the comments and issues from the reviewers. MCL and VSD contributed DFT calculations. All authors read and approved the final manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
SA and RB thanks DST-SERB for providing the financial support under TARE (file number: TAR/2022/000207). MCL and VSD thanks the National Centre for High-performance Computing, Hsinchu for providing computer time. We are indebted to Vignan's Foundation for Science, Technology and Research for providing central instrumentation facilities at CoExAMMPC.Notes and references
- For reactions using primary MBH acetate references (a) W. Xiao, X. Yin, Z. Zhou, W. Du and Y.-C. Chen, Org. Lett., 2016, 18, 116 CrossRef CAS PubMed; (b) Y.-L. Ji, X.-H. He, G. Li, Y.-Y. Ai, H.-P. Li, C. Peng and B. Han, Org. Chem. Front., 2020, 7, 563 RSC; (c) J. Xie, F. Sha and X.-Y. Wu, Tetrahedron, 2016, 72, 4047 CrossRef CAS; (d) K. Divya, T. Sudheesh, T. S. Sivanandan, K. Pravin and I. N. N. Namboothiri, Tetrahedron, 2019, 75, 130761 CrossRef; (e) S. Anwar, L.-T. Lin, V. Srinivasadesikan, V. B. Gudise and K. Chen, RSC Adv., 2021, 11, 38648 RSC; (f) V. B. Gudise, P. C. Settipalli, Y. P. Reddy and S. Anwar, ChemistrySelect, 2021, 6, 13589 CrossRef CAS ; for reactions using secondary MBH acetate references; (g) S. Anwar, W.-Y. Huang, C.-H. Chen, Y. S. Cheng and K. Chen, Chem. Eur. J., 2013, 19, 4344 CrossRef CAS PubMed; (h) J.-Q. Zhang, J.-J. Liu, C.-L. Gu, D. Wang and L. Liu, Eur. J. Org. Chem., 2014, 2014, 5885 CrossRef CAS; (i) S. Roy, Eur. J. Org. Chem., 2019, 2019, 765 CrossRef CAS; (j) E. Gopi and I. N. N. Namboothiri, J. Org. Chem., 2014, 79, 7468 CrossRef CAS PubMed; (k) D. Majee, S. Biswas, S. Mobin and S. Samanta, J. Org. Chem., 2016, 81, 4378 CrossRef CAS PubMed; (l) J. Liu, Q. Li, Z.-M. Cao, Y. Jin, J. Lin and S.-J. Yan, J. Org. Chem., 2019, 84, 1797 CrossRef CAS PubMed; (m) K. Divya, M. Mobin and I. N. N. Namboothiri, Org. Lett., 2012, 14, 4580 CrossRef PubMed; (n) T. Kumar, M. Mobin and I. N. N. Namboothiri, Tetrahedron, 2013, 69, 4964 CrossRef CAS; (o) T. Chen, N. Shao, H. Zhu, B. Zhang and H. Zou, Tetrahedron, 2013, 69, 10558 CrossRef CAS; (p) N. Shao, T. Chen, T. Zhang, H. Zhu, Q. Zheng and H. Zou, Tetrahedron, 2014, 70, 795 CrossRef CAS; (q) P. C. Settipalli, Y. P. Reddy, V. B. Gudise and S. Anwar, ChemistrySelect, 2021, 6, 47 CrossRef CAS; (r) S. Guin, D. Majee, S. Biswas and S. Samanta, Asian J. Org. Chem., 2018, 7, 1810 CrossRef CAS , for MBH acetates review; (s) W.-Y. Huang, S. Anwar and K. Chen, Chem. Rec., 2017, 17, 363 CrossRef CAS PubMed.
- (a) T. Baiju, R. Almeida, S. Sivanandan, C. Simone, L. Brito, B. Cavalcanti, C. Pessoa, I. N. N. Namboothiri and E. Silvajunior, Eur. J. Med. Chem., 2018, 151, 686 CrossRef CAS PubMed; (b) T. Kumar, D. Verma, R. Menna-Barreto, W. Valença, E. Silva Júnior and I. N. N. Namboothiri, Org. Biomol. Chem., 2015, 13, 1996 RSC; (c) E. Gopi, T. Kumar, F. S. Rubem, O. Wagner, N. Eufrânio and I. N. N. Namboothiri, Org. Biomol. Chem., 2015, 13, 9862 RSC; (d) E. K Reddy, C. Remya, A. Sajith, K. Dileep, C. Sadasivan and S. Anwar, RSC Adv., 2016, 6, 77431 RSC.
- J. Raju and K. Chen, Org. Lett., 2011, 13, 1458 CrossRef PubMed.
- (a) H. Zhu, N. Shao, T. Chen and H. Zou, Chem. Commun., 2013, 49, 7738 RSC; (b) M. Yaqub, C.-Y. Yu, Y.-M. Jia and Z.-T. Huang, Synlett, 2008, 9, 1357 Search PubMed; (c) T. Zhang, N. Shao, H. Zhu, T. Chen, Q. Zheng and H. Zou, Tetrahedron, 2014, 70, 7454 CrossRef CAS; (d) Q.-W. Gui, B.-B. Wang, S. Zhu, F.-L. Li, M.-X. Zhu, M. Yi, Z.-L. Yu, Z.-L. Wu and W.-M. He, Green Chem., 2021, 23, 4430 RSC.
- V. Mane, S. T. Sivanandan, R. Santana, A. Beatriz, E. Silvajúnior and I. N. N. Namboothiri, J. Org. Chem., 2020, 85, 8825 CrossRef CAS PubMed.
- J. Li, Q.-L. Hu, X.-P. Chen, K.-Q. Hou, A. S. C. Chan and X.-F. Xiong, Chin. Chem. Lett., 2020, 31, 697 CrossRef CAS.
- C. Cao, Y. Zhou, J. Zhou, X. Sun, Y. Tang, Y. Li, G. Li and J. Sun, Chem. Eur. J., 2009, 15, 11384 CrossRef CAS PubMed.
- (a) O.-P. Laura, R. -E. Carles and A. Miquel, Catal. Sci. Technol., 2016, 6, 4686 RSC; (b) K. Divya, F. S. Rubem, E. Silva Júnior, M. Mobin and I. N. N. Namboothiri, Chem. Commun., 2014, 50, 6973 RSC.
- P. Basu, R. Sikdar, T. Kumar and I. N. N. Namboothiri, Eur. J. Org. Chem., 2018, 2018, 5735 CrossRef CAS.
- (a) J. Raju, M. Waheed, T. Karthik and A. Shankar, New J. Chem., 2018, 42, 980 RSC; (b) W.-Y. Huang, Y.-C. Chen and K. Chen, Chem.–Asian J., 2012, 7, 688 CrossRef CAS PubMed; (c) V. Mane, T. Kumar, S. Pradhan, K. Savita and I. N. N. Namboothiri, RSC Adv., 2015, 5, 69990 RSC; (d) K. Divya, S. Mobin and I. N. N. Namboothiri, Tetrahedron Lett., 2012, 53, 3349 CrossRef.
- (a) R. Dhananjay, Y.-J. Ke and K. Chen, Asian J. Org. Chem., 2013, 2, 330 CrossRef; (b) L. Zhao, G. Raabe and D. Enders, Synthesis, 2019, 51, 1391 CrossRef CAS.
- J. Zhang, G. Yin, Y. Du, Z. Yang, Y. Li and L. Chen, J. Org. Chem., 2017, 82, 13594 CrossRef CAS PubMed.
- (a) X.-L. He, H.-R. Zhao, C.-Q. Duan, X. Han, W. Du and Y.-C. Chen, Chem.–Eur. J., 2018, 24, 6277 CrossRef CAS PubMed; (b) J. Wang, P. Wang, L. Wang, D. Li, K. Wang, Y. Wang, H. Zhu, D. Yang and R. Wang, Org. Lett., 2017, 19, 4826 CrossRef CAS PubMed; (c) J. Raju, P.-H. Lee, R. Dhananjay, J.-H. Chen and K. Chen, Eur. J. Org. Chem., 2012, 2012, 353 CrossRef; (d) L. Yeh, S. Anwar and K. Chen, Tetrahedron, 2012, 68, 7317 CrossRef CAS.
- Y.-Y. Ai, D.-A. Li, G. Li, H.-P. Li, X.-H. He, X.-J. Fu, Y.-T. Wang, G. Zhan and B. Han, Adv. Synth. Catal., 2021, 363, 3283 CrossRef CAS.
- R. Chen, X. Fan, J. Gong and Z. He, Asian J. Org. Chem., 2014, 3, 877 CrossRef CAS.
- (a) S. Lakshminarayana and I. N. N. Namboothiri, J. Org. Chem., 2018, 83, 9471 CrossRef PubMed; (b) J.-Y. Liu, J. Zhao, J.-L. Zhang and P.-F. Xu, Org. Lett., 2017, 19, 1846 CrossRef CAS PubMed.
- (a) A. Pareek, S. T. Sivanandan, S. Bhagat and I. N. N. Namboothiri, Tetrahedron, 2022, 108, 132650 CrossRef CAS; (b) P. Basu, C. Hazra, T. Baiju and I. N. N. Namboothiri, New J. Chem., 2020, 44, 1389 RSC; (c) Y. Zheng, L. Cui, Y. Wang and Z. Zhou, J. Org. Chem., 2016, 81, 4340 CrossRef CAS PubMed; (d) A. A. Kostenko, A. Kseniya, S. Alexander, N. Andrey, B. V. Lichitsky and S. G. Zlotin, Org. Biomol. Chem., 2021, 19, 1780 RSC.
- T. Shu, Q. Ni, X. Song, K. Zhao, T. Wu, P. Rakesh, K. Rissanen and D. Enders, Chem. Commun., 2016, 52, 2609 RSC.
- J.-Y. Liu, X.-C. Yang, H. Lu, Y.-C. Gu and P.-F. Xu, Org. Lett., 2018, 20, 2190 CrossRef CAS PubMed.
- S. T. Sivanandan and I. N. N. Namboothiri, J. Org. Chem., 2021, 86, 8465 CrossRef CAS PubMed.
- V. S. Kumar, V. B. Gudise, P. C. Settipalli, E. K. Reddy, S. F. Basha, Y. P. Reddy, V. Srinivasadesikan, S.-L. Lee and S. Anwar, ChemistrySelect, 2020, 5, 3080 CrossRef CAS.
- (a) Y. P. Reddy, V. B. Gudise, P. C. Settipalli and S. Anwar, ChemistrySelect, 2021, 6, 4456 CrossRef CAS; (b) V. B. Gudise, P. C. Settipalli, E. K. Reddy and S. Anwar, Eur. J. Org. Chem., 2019, 2019, 2234 CrossRef CAS; (c) Y. P. Reddy and S. Anwar, RSC Adv., 2022, 12, 34634 RSC.
- CCDC 2149875 for 3a, possess the crystallographic data for this manuscript.
- (a) A. D. Becke, Phys. Rev. A, 1993, 98, 5648 CAS; (b) C. Lee, W. Yang and R. G. Parr, Phys. Rev. B: Condens. Matter Mater. Phys., 1988, 37, 785 CrossRef CAS PubMed.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, B. Mennucci, G. A. Petersson, H. Nakatsuji, M. Caricato, X. Li, H. P. Hratchian, A. F. Izmaylov, J. Bloino, G. Zheng, J. L. Sonnenberg, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, J. A. Montgomery, J. E. Peralta Jr, F. Ogliaro, M. Bearpark, J. J. Heyd, E. Brothers, K. N. Kudin, V. N. Staroverov, R. Kobayashi, J. Normand, K. Raghavachari, A. Rendell, J. C. Burant, S. S. Iyengar, M. Tomasi, N. Cossi, J. M. Rega, M. Millam, J. E. Klene, J. B. Knox, V. Cross, C. Bakken, J. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, R. L. Martin, K. Morokuma, V. G. Zakrzewski, G. A. Voth, P. Salvador, J. J. Dannenberg, S. Dapprich, A. D. Daniels, O. Farkas, J. B. Foresman, J. V. Ortiz, J. Cioslowski and D. J. Fox, Revision A.02, Gaussian Inc., Walling ford CT, 2009 Search PubMed.
- A. Pareek, S. T. Sivanandan, S. Bhagat and I. N. N. Namboothiri, Tetrahedron, 2022, 108, 132650 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 2149875. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d2ra08163f |
| This journal is © The Royal Society of Chemistry 2023 |