
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceMachine learning based implicit solvent model for aqueous-solution alanine dipeptide molecular dynamics simulations†
Songyuan Yaoa,
Richard Vana,
Xiaoliang Pan a,
Ji Hwan Parkb,
Yuezhi Mao*c,
Jingzhi Pu*d,
Ye Mei*efg and
Yihan Shao*a
a,
Ji Hwan Parkb,
Yuezhi Mao*c,
Jingzhi Pu*d,
Ye Mei*efg and
Yihan Shao*a
aDepartment of Chemistry and Biochemistry, University of Oklahoma, Norman, OK 73019, USA. E-mail: yihan.shao@ou.edu
bSchool of Computer Science, University of Oklahoma, Norman, OK 73019, USA
cDepartment of Chemistry and Biochemistry, San Diego State University, San Diego, CA 92182, USA. E-mail: ymao2@sdsu.edu
dDepartment of Chemistry and Chemical Biology, Indiana University-Purdue University Indianapolis, Indianapolis, IN 46202, USA. E-mail: jpu@iupui.edu
eState Key Laboratory of Precision Spectroscopy, School of Physics and Electronic Science, East China Normal University, Shanghai 200062, China. E-mail: ymei@phy.ecnu.edu.cn
fNYU-ECNU Center for Computational Chemistry at NYU Shanghai, Shanghai 200062, China
gCollaborative Innovation Center of Extreme Optics, Shanxi University, Taiyuan, Shanxi 030006, China
First published on 3rd February 2023
Abstract
Inspired by the recent work from Noé and coworkers on the development of machine learning based implicit solvent model for the simulation of solvated peptides [Chen et al., J. Chem. Phys., 2021, 155, 084101], here we report another investigation of the possibility of using machine learning (ML) techniques to “derive” an implicit solvent model directly from explicit solvent molecular dynamics (MD) simulations. For alanine dipeptide, a machine learning potential (MLP) based on the DeepPot-SE representation of the molecule was trained to capture its interactions with its average solvent environment configuration (ASEC). The predicted forces on the solute deviated only by an RMSD of 0.4 kcal mol−1 Å−1 from the reference values, and the MLP-based free energy surface differed from that obtained from explicit solvent MD simulations by an RMSD of less than 0.9 kcal mol−1. Our MLP training protocol could also accurately reproduce combined quantum mechanical molecular mechanical (QM/MM) forces on the quantum mechanical (QM) solute in ASEC environment, thus enabling the development of accurate ML-based implicit solvent models for ab initio-QM MD simulations. Such ML-based implicit solvent models for QM calculations are cost-effective in both the training stage, where the use of ASEC reduces the number of data points to be labelled, and the inference stage, where the MLP can be evaluated at a relatively small additional cost on top of the QM calculation of the solute.
1. Introduction
One of the central tasks in the field of Computational Chemistry is to simulate various chemical processes and conformational changes of molecules and macromolecules in aqueous solutions.1–5 In general, molecular mechanics (MM), quantum mechanics (QM),6 or combined quantum mechanics molecular mechanics (QM/MM)7 molecular dynamics (MD) simulations incorporate the aqueous environment through explicit or implicit solvent models.8–10 In the explicit solvent models, the solvent molecules are accounted for explicitly, where solvent–solute interactions can be calculated with atomistic resolution. Popular models for explicit solvents include TIPnP,11–13 SPC,14 and AMOEBA.15,16 On the other hand, the implicit solvent models (also known as the continuum solvent models)17–20 treat the solvent implicitly as a dielectric continuum, thus reducing the number of explicit particle–particle interactions. Popular methods include the Poisson–Boltzmann (PB),21 COSMO/polarized continuum model (PCM),22–33 or Generalized Born (GB) models.34–37With fewer atoms involved in the calculation, the implicit solvent models offer an advantage in terms of its lower computational cost.38 However, the accuracy of implicit solvent models can be limited by its inadequacy to capture specific solute–solvent interactions and solvent structure fluctuations.39 In MM-based modeling, implicit solvent model based simulations do not always accurately reproduce folded40–42 and unfolded structural distributions.8,38,43,44 For QM-based modeling, pKa predictions of the solute can sometimes be off by a couple of pH units, if the hydrogen bonding between solute and the nearby solvent molecules is completely ignored in implicit solvent calculations.45 In principle, such deficiencies in implicit solvent based MM and QM modelings can be mitigated by including one or multiple solvent molecules or the entire first solvation shell into the solute region of implicit solvent calculations.46–48 However, such a treatment might inadvertently reduce the mobility of those solvent molecules and thus over-stabilize the short-range solute–solvent interactions.
To guide the development of more accurate implicit solvent models, it would be highly desirable if one can “derive” an implicit solvent model directly from explicit solvent MD simulations of the system. A breakthrough along this line was made recently by Noé and coworkers.49,50 Specifically, after collecting configurations from an explicit-solvent MD simulation of alanine dipeptide and chignolin, they successfully trained the first machine learning (ML) based implicit solvent model for peptides. The ML model reproduced the forces on solute atoms from the explicit-solvent calculation and could thus be employed to drive solution-phase MD simulations. Their pioneering work follows the recent, rapidly growing use of ML techniques51 in the development of computational chemistry models.52–54 As far as the solvent effect is concerned, ML models have been developed to capture the effect on chemical reactions,55,56 spectral properties,57–60 identify solvation characteristics in general molecular environment,50,61,62 and explore the solvent effect on mixture solvent system.63–65
Inspired by the work of Noé and coworkers,42,49,50,66 in this paper we will also use the solvated alanine dipeptide67–69 as an example and further explore the possibility of “deriving” an implicit solvent model directly from explicit solvent MD simulations. As Noé and coworkers pointed out, for a system with solute atoms at positions r, solvent atoms at positions w = {w1, w2, …}, and a total potential expressed as V(r, w), an implicit solvent model should correspond to the solute potential-of-mean-force (PMF),70,71
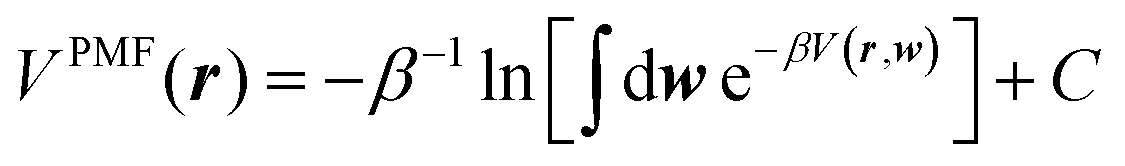 | (1) |
Clearly, this opens the possibility of acquiring the corresponding forces on the solute atoms (at geometry r) by extensively sampling solvent configurations, w(m) (m = 1, 2, …, M), around the solute,
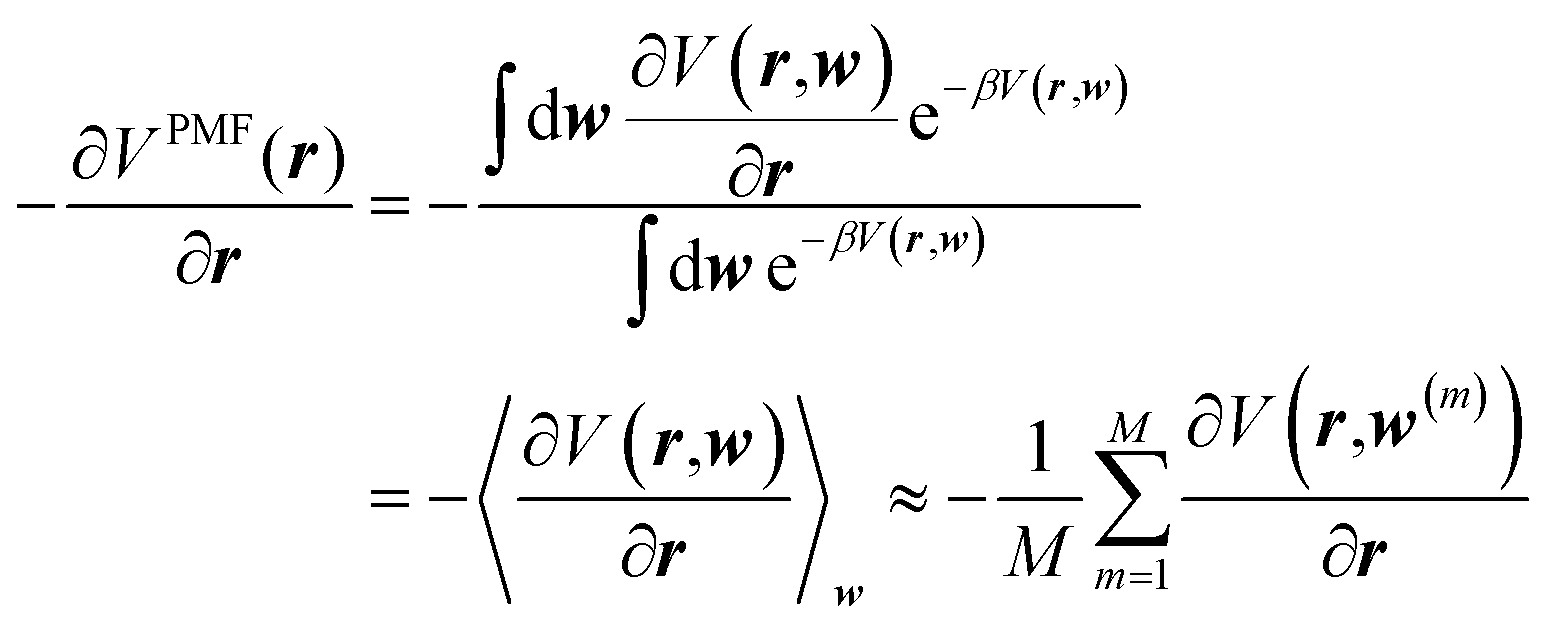 | (2) |
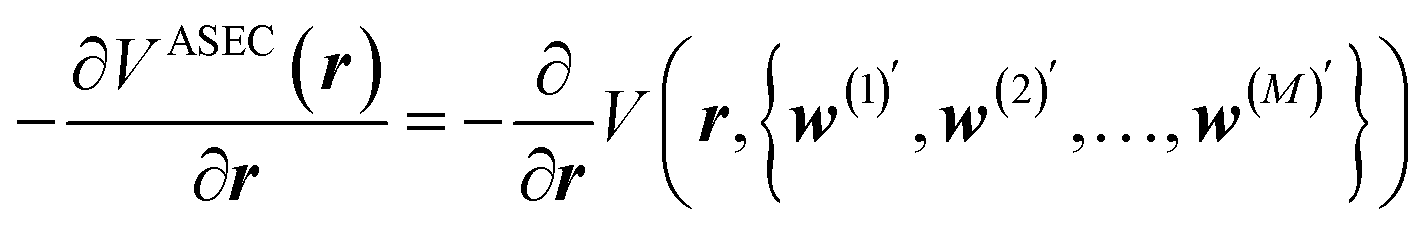 | (3) |
 and van der Waals epsilon values by
and van der Waals epsilon values by 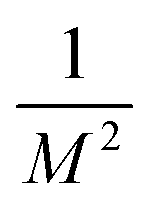 (due to the geometric combination rule). For a solute that is described by a QM method or a polarizable force field, the ASEC description (with scaled charge and epsilon values) still holds approximately despite the fact the polarization effect from solvent molecules has nonlinear components. Formally, for a single solvent configuration, w(m) with K solvent molecules, the many-body expansion81–83 of the force on the solute is
(due to the geometric combination rule). For a solute that is described by a QM method or a polarizable force field, the ASEC description (with scaled charge and epsilon values) still holds approximately despite the fact the polarization effect from solvent molecules has nonlinear components. Formally, for a single solvent configuration, w(m) with K solvent molecules, the many-body expansion81–83 of the force on the solute is
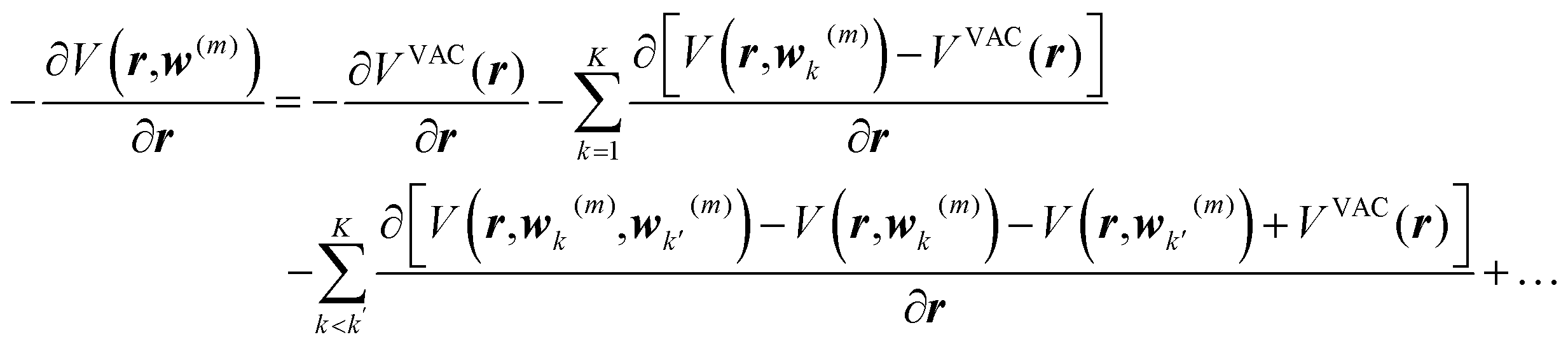 | (4) |
In this work, we will thus seek to build a Δ machine learning potential (MLP) to capture the solute–solvent interactions within the ASEC environment, which is acquired from explicit solvent MD simulations. In the spirit of widely-used force-matching techniques,85,86 the MLP will be trained to minimize a loss function,
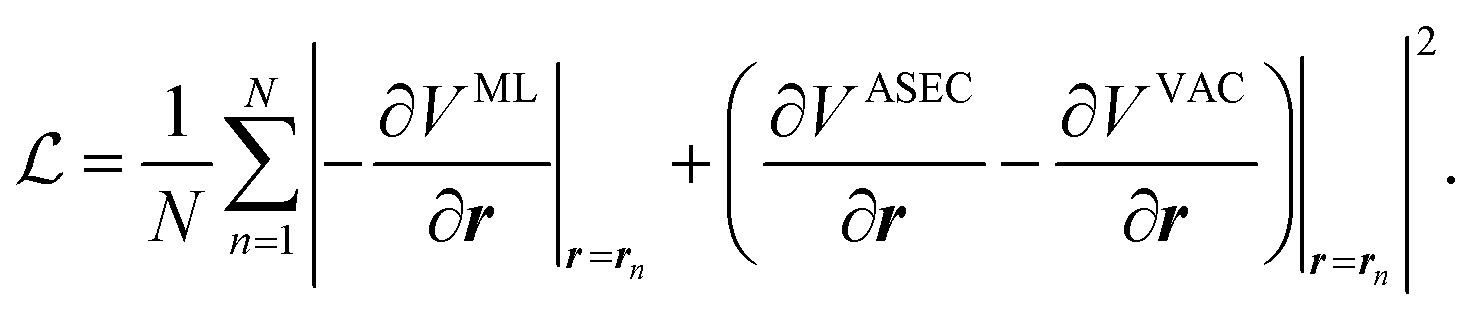 | (5) |
It should be noted that, as far as the solvent effect is concerned, there exists a separate line of efforts towards an accurate ML-based prediction of solvation free energies.48,91–93 While progress along that line has been rather encouraging (with the average error often falling below 0.5 kcal mol−1), those ML models are usually based on SMILES and other descriptors that do not explicitly depend on the coordinates of solute atoms. So, even with neural network automatic differentiation, most of those ML models do not readily provide the forces on solute atoms to drive their dynamical motions, which is the main objective of our work.
This paper is structured as follows. Section 2 summarizes the procedure for training our MLP model, with the corresponding simulation details provided in Section 3. Results and discussion on the MLP implicit solvent models for MM and QM simulations of alanine dipeptide in an aqueous solution will be presented in Section 4. Concluding remarks are made in Section 5.
2. Machine learning based implicit solvent model for MM and QM modeling of solute molecules
For the construction of our machine-learning based implicit solvent model, we will adopt the Deep Potential-Smooth Edition (DeepPot-SE).87,89,90 The overall workflow for our training of the MLP implicit solvent model is shown in Fig. 1, which closely resembles our previous use of DeepPot-SE for the simulation of enzyme reactions.94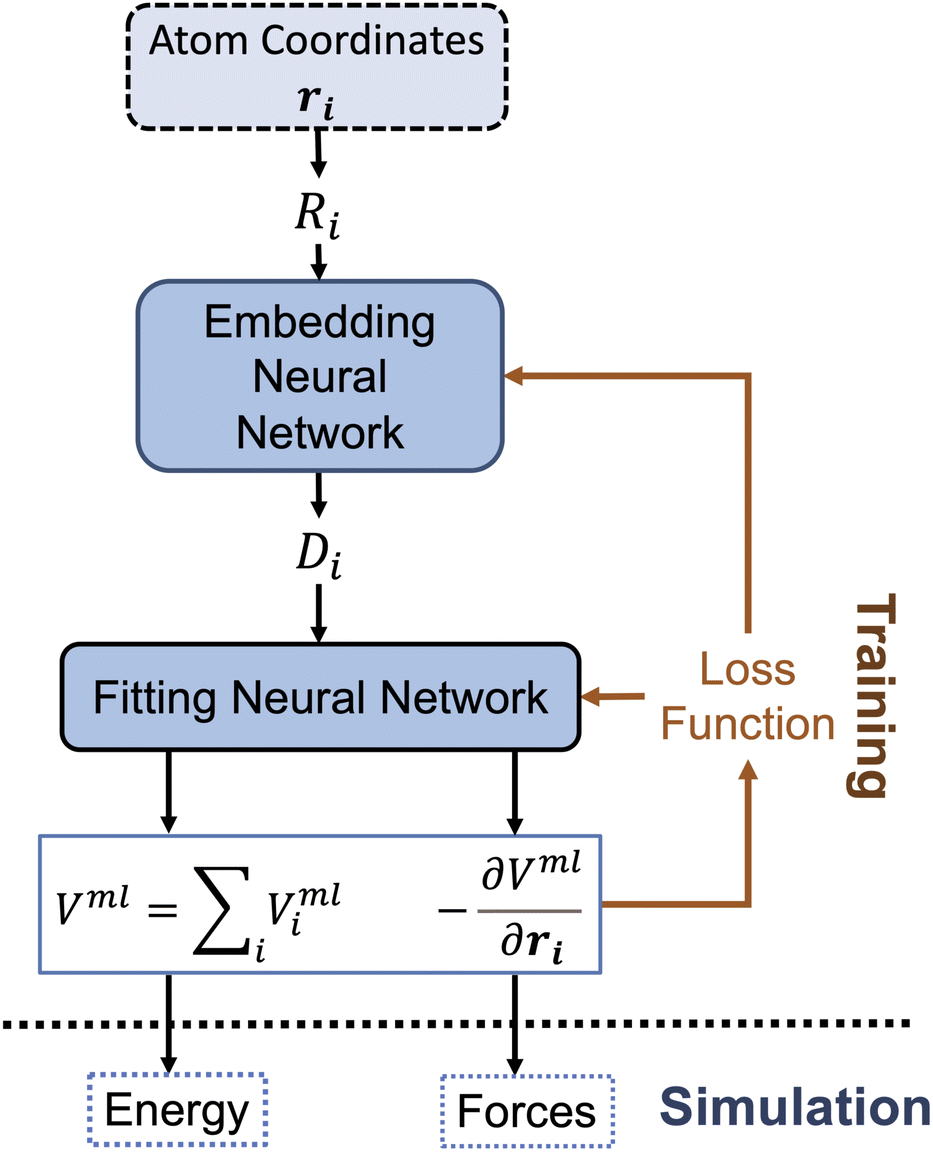 | ||
| Fig. 1 Workflow for the training of the MLP for implicit solvation of alanine dipeptide. The neural networks are optimized to predict the solute–solvent interaction energy V and the corresponding forces on each solute atom. | ||
2.1 Descriptor for the solute molecule
The first step of feature abstraction within this presentation is to build an environment matrix, Ri, for each atom (labelled i),
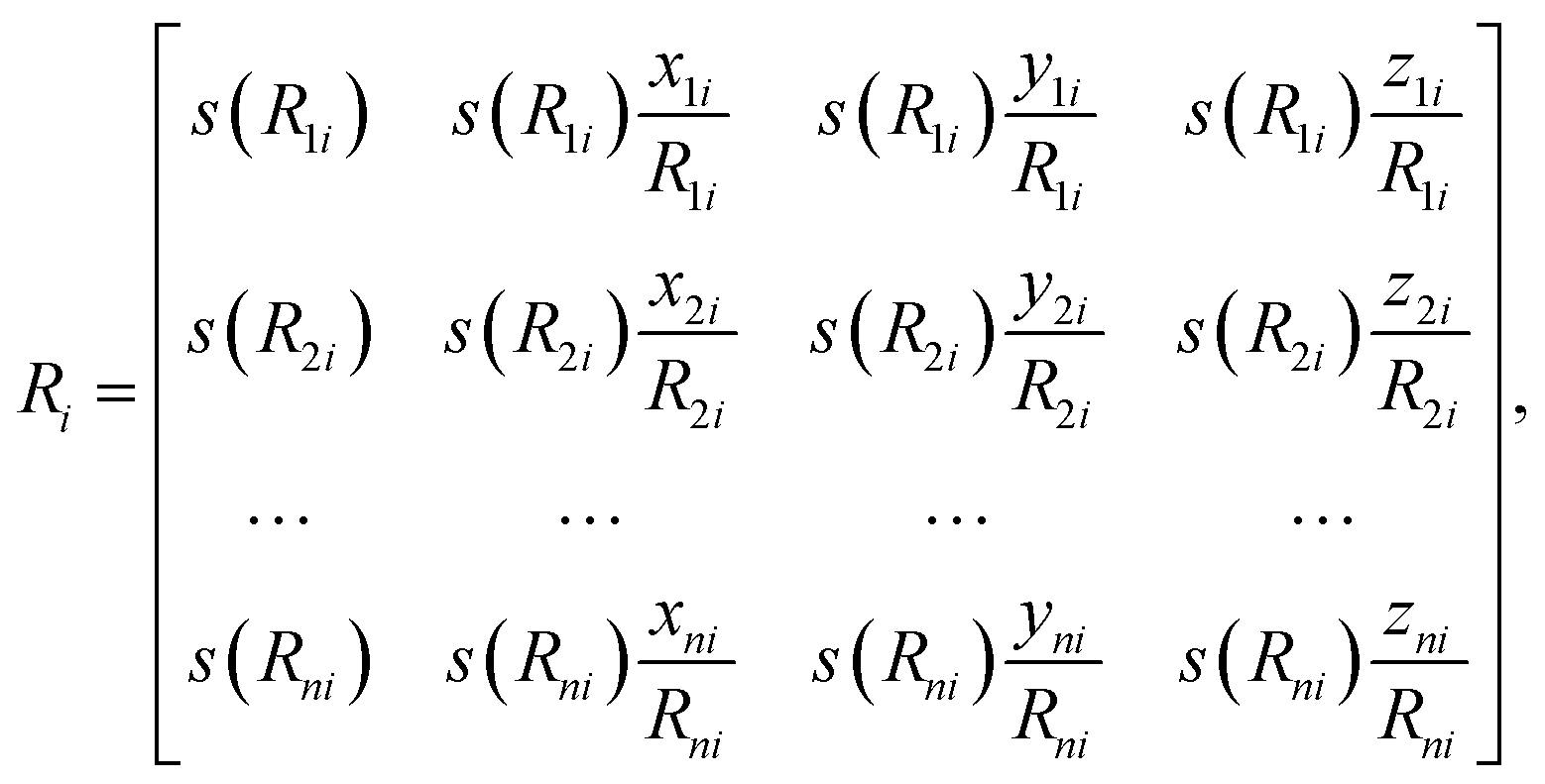 | (6) |
In the next step of feature abstraction, an embedding neural network (ENN), G, is used to map each s(Rji) value through multiple hidden layers into m1 outputs, which form the j-th row of the embedding matrix gi.
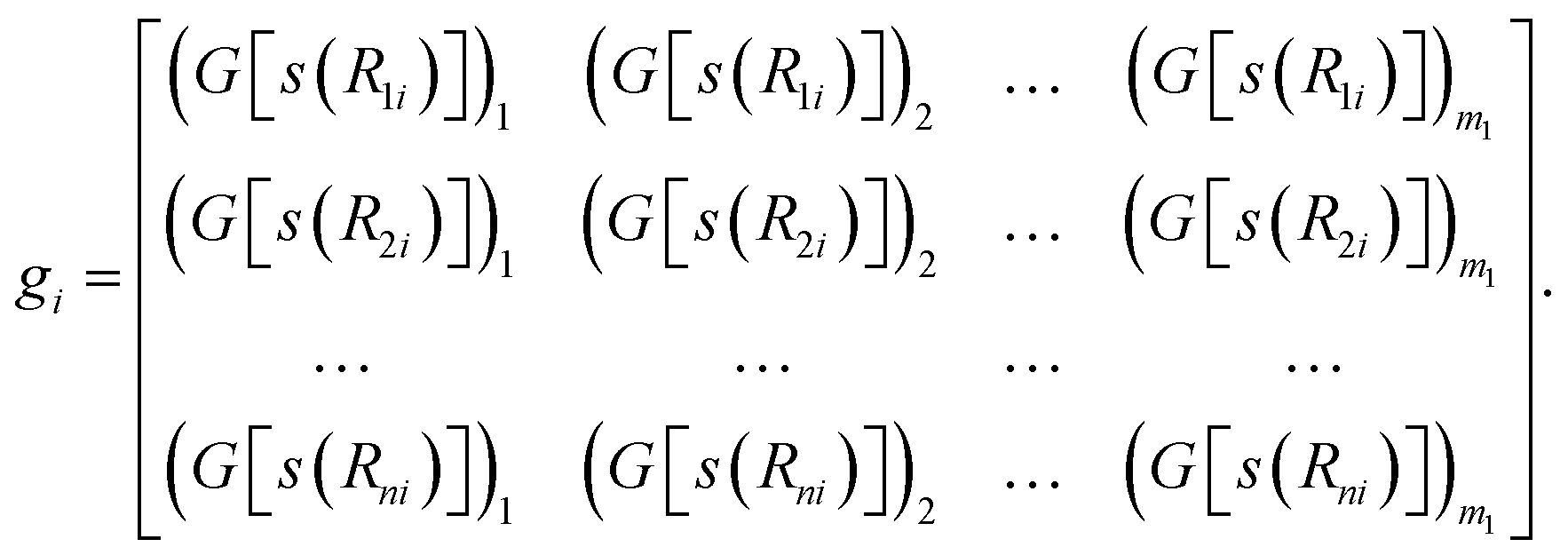 | (7) |
To ensure the permutational symmetry of the MLP, all s(RHC) values for a carbon atom connecting to a hydrogen atom neighbor are fed into the same embedding neural network, GHC. The same goes with RCC, RNC, ROC, RHH, RCH, RNH, ROH and other distances. For a system with Nelements different elements, there would be Nelements2 distinct embedding neural networks, each fully-connected with one input, three hidden layers of neurons, and an output layer with m1 neurons.
In the next step, an encoded feature matrix Di of size m1 by m2 is computed
| Di = (g1i)TRiRTig2i, | (8) |
2.2 The fitting neural networks (FNN) and loss function
As shown in Fig. 1, the next key component of our machine learning architecture are fitting neural networks (FNN). Specifically, a fitting neural network will be used for each element (C, H, O, N for the alanine dipeptide test system in our work). For each FNN, its input is the feature matrix defined in eqn (8). As the output, FNN predicts the solute–solvent interaction energy for each solute atom,
VMLi = FNN(![[scr D, script letter D]](https://www.rsc.org/images/entities/char_e523.gif) i) i)
| (9) |
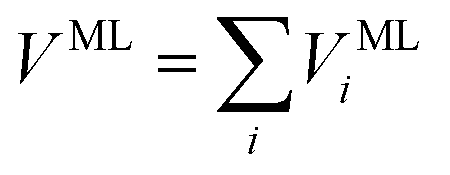 | (10) |
Through automatic differentiation, it also produces the corresponding force on each atom, 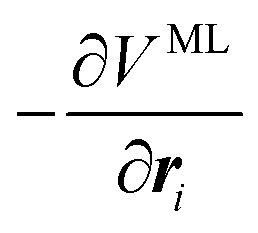 . The loss function in eqn (5) measures the mean square difference (MSD) between the predicted forces and reference values,
. The loss function in eqn (5) measures the mean square difference (MSD) between the predicted forces and reference values,
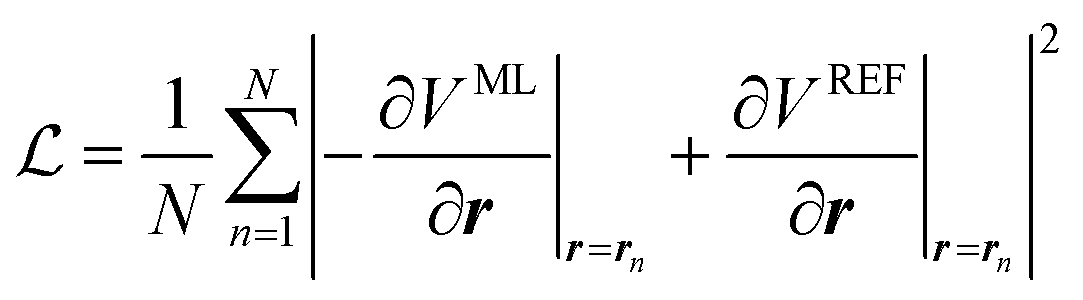 | (11) |
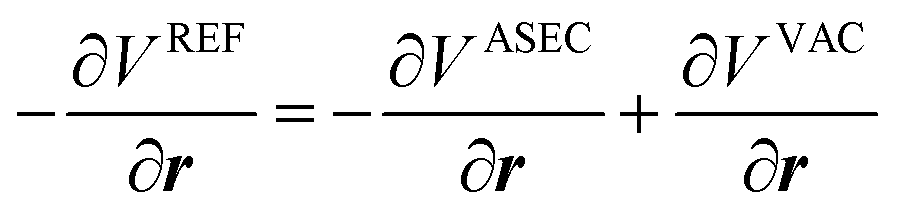 | (12) |
Our detailed procedure for the collection of the training/validation configurations and the computation of the corresponding reference forces will be provided in the next Section.
3. Computational details
Alanine dipeptide was used in this work as a test molecule for exploring the possibility of building machine learning based implicit solvent models for MD simulations. This molecule, which is shown in Fig. 2, consists of 22 atoms and has two key torsion angles ϕ(C–N–Cα–C) and ψ(N–Cα–C–N). This system has been widely used in the development of simulation methodologies, such as free energy analyses,95,96 conformational analysis,49,67,97,98 reaction coordinates,99–101 and free energy surface computation.68 | ||
| Fig. 2 Structure of alanine dipeptide. | ||
3.1 Training/validation configurations
Instead of employing a single MD trajectory for explicitly solvated alanine dipeptide like Noé and coworkers,49,102 we collected the training/validation configurations at various combinations of (ϕ, ψ) dihedral angles of the alanine dipeptide solute molecule. Furthermore, we utilized two slightly different ways to generate these configurations, depending on whether the solute molecule adopted a minimized energy structure (with the two dihedral angles restrained to their respective values). This would lead to two sets of machine learning potentials, which were termed “MLP” and “MLP-O”.For the training of MLP, short MD simulations using the  program in AmberTools21 (ref. 103) were performed at 362 = 1296 combinations of dihedral angles, with a 10-degree increment for each angle. Specifically, for each of these combinations, the solute molecule (as described with the ff14SB force field104) was solvated in TIP3P water molecules11 in a simulation box of ∼33 Å × 33 Å × 33 Å. Then each system was equilibrated with 50 ps NPT simulations, where the two dihedral angles were restrained to their target values using the restraint potential (shown later in eqn (13)) with a force constant of 1000 kcal mol−1 rad−2. The NPT simulations were performed at 1.0 atm (with Berendsen barostat) and 300 K (with Langevin thermostat) using a leapfrog integration (at 1.0 fs time step) under periodic boundary conditions. Once equilibrated, the solute molecule was frozen at its final geometry, and the entire system underwent another NVT simulation for 500 ps at 300 K. During this simulation, the configurations were saved every 0.1 ps, which resulted in 5000 solvent configurations for each solute structure for subsequent ASEC calculations.
program in AmberTools21 (ref. 103) were performed at 362 = 1296 combinations of dihedral angles, with a 10-degree increment for each angle. Specifically, for each of these combinations, the solute molecule (as described with the ff14SB force field104) was solvated in TIP3P water molecules11 in a simulation box of ∼33 Å × 33 Å × 33 Å. Then each system was equilibrated with 50 ps NPT simulations, where the two dihedral angles were restrained to their target values using the restraint potential (shown later in eqn (13)) with a force constant of 1000 kcal mol−1 rad−2. The NPT simulations were performed at 1.0 atm (with Berendsen barostat) and 300 K (with Langevin thermostat) using a leapfrog integration (at 1.0 fs time step) under periodic boundary conditions. Once equilibrated, the solute molecule was frozen at its final geometry, and the entire system underwent another NVT simulation for 500 ps at 300 K. During this simulation, the configurations were saved every 0.1 ps, which resulted in 5000 solvent configurations for each solute structure for subsequent ASEC calculations.
For the training of MLP-O, restrained geometry optimizations of the gas-phase solute molecule was carried out using the  program in AmberTools21, again with the force constant for each dihedral angle set to be 1000 kcal mol−1 rad−2. The optimization continued for 500 steepest descent and 1500 conjugated gradient steps. The relative energy of these configurations was shown in Fig. S1,† which closely resembled the vacuum Ramachandran plot in ref. 49. Subsequently, with the solute retaining a fixed geometry, each system was first solvated in the water box, equilibrated with NPT simulations at 300 K and 1.0 atm and then subjected to a solvent configuration simulation, which also involved a 500 ps NVT simulation at 300 K. With the configurations saved every 0.1 ps, 5000 configurations were also obtained for each solute structure.
program in AmberTools21, again with the force constant for each dihedral angle set to be 1000 kcal mol−1 rad−2. The optimization continued for 500 steepest descent and 1500 conjugated gradient steps. The relative energy of these configurations was shown in Fig. S1,† which closely resembled the vacuum Ramachandran plot in ref. 49. Subsequently, with the solute retaining a fixed geometry, each system was first solvated in the water box, equilibrated with NPT simulations at 300 K and 1.0 atm and then subjected to a solvent configuration simulation, which also involved a 500 ps NVT simulation at 300 K. With the configurations saved every 0.1 ps, 5000 configurations were also obtained for each solute structure.
3.2 Labelling data
 program was used to compute the force on solute atoms for each of the 5000 solvent configurations. Then the total force was averaged over the 5000 solvent configurations, and the reference force was obtained by subtracting the corresponding vacuum force.
program was used to compute the force on solute atoms for each of the 5000 solvent configurations. Then the total force was averaged over the 5000 solvent configurations, and the reference force was obtained by subtracting the corresponding vacuum force.3.3 Training of the machine-learning models
In the training of our ENN and FNN, the labelling dataset was composed of 1296 solute configurations, each containing the coordinate data as input for the ENN and the reference force from ASEC calculations as label. This dataset was randomly split into a training set of 1245 configuration and a validation set of 51 configurations. An additional 500 configurations were collected from an explicit-solvent simulation trajectory of alanine dipeptide and used as the testing set to assess the accuracy our new models. The distribution of the testing set were shown in Fig. S2.† For these configurations, the ASEC forces were obtained with the same procedure as in Section 3.1.The ENN/FNN were optimized using the training set and then tested on the validation set to determine the accuracy of the prediction. The neural networks for learning the reference ASEC forces from MM and QM/MM simulations were trained for 200 epochs. Other hyperparameters used for the training are listed in Table 1.
| Hyperparameter | Value |
|---|---|
| ENN layer size | [5, 10, 20] |
| FNN layer size | [240, 240] |
| Activation function | tanh |
| Optimizer | Adam |
| Initial learning rate | 5 × 10−4 |
| Decay rate (per epoch) | 0.95 |
| Batch size | 32 |
| (m1, m2) | (20, 4) |
3.4 Umbrella sampling
The umbrella sampling technique was used to produce the 2-dimensional free energy surface of alanine dipeptide.111 Specifically, a sequence of sampling windows with a harmonic biasing potential,
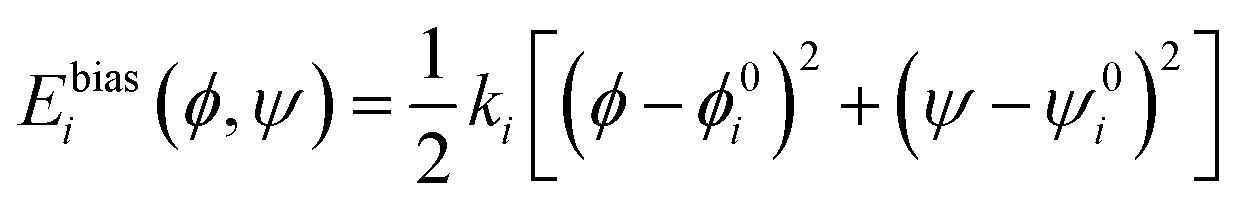 | (13) |
Three sets of umbrella sampling simulations (vacuum, implicit solvation with the trained ML potential, and explicit solvations) were performed with same setting. For vacuum and explicit solvations, the  and
and  programs were used, respectively, while the implicit solvent simulation with the ML potential were performed using the
programs were used, respectively, while the implicit solvent simulation with the ML potential were performed using the  plug-in for the OpenMM package.112 To construct the 2-dimensional free energy surface profile, the 2-d Weighted Histogram Analysis method (WHAM) was employed.113 The free energy surface was calculated with a convergence tolerance of 1.0 × 10−6 kcal mol−1, where the results in the next section (shown later in Fig. 4) are nearly indistinguishable from those obtained using the 100–300 ps segments of the trajectories (Fig. S3,† left column) and the 300–500 ps segments (Fig. S3,† right column).
plug-in for the OpenMM package.112 To construct the 2-dimensional free energy surface profile, the 2-d Weighted Histogram Analysis method (WHAM) was employed.113 The free energy surface was calculated with a convergence tolerance of 1.0 × 10−6 kcal mol−1, where the results in the next section (shown later in Fig. 4) are nearly indistinguishable from those obtained using the 100–300 ps segments of the trajectories (Fig. S3,† left column) and the 300–500 ps segments (Fig. S3,† right column).
4. Results and discussion
4.1 MLP implicit solvent model for MM modeling
For the training of our MLP models, the optimization learning curves were shown in Fig. S5a and b.† To examine the accuracy of the optimized MLP model for handling the implicit solvation of alanine dipeptide, the predicted solvation forces were plotted against the reference ASEC values for the training and validation configurations (whose ϕ and ψ values were constrained to the grid values) in the top row of Fig. 3. Clearly, our MLP reproduced the ASEC solvation forces on the molecule within the chemical accuracy: the RMSE value of the force difference was 0.342 kcal mol−1 Å−1 for the training set and 0.327 kcal mol−1 Å−1 for the validation set. This can be alternatively seen from the narrow distributions in the force differences in the bottom row of Fig. 3. As shown in Fig. S4,† the MLP-O model, which was trained with an energy-optimized geometry for the solute at each given (ϕ, ψ) combination (the same set of torsional angle values were used as for the training of the MLP model), the RMSE value for the predicted force was 0.268 kcal mol−1 Å−1 and 0.259 kcal mol−1 Å−1 for the training and validation sets, respectively.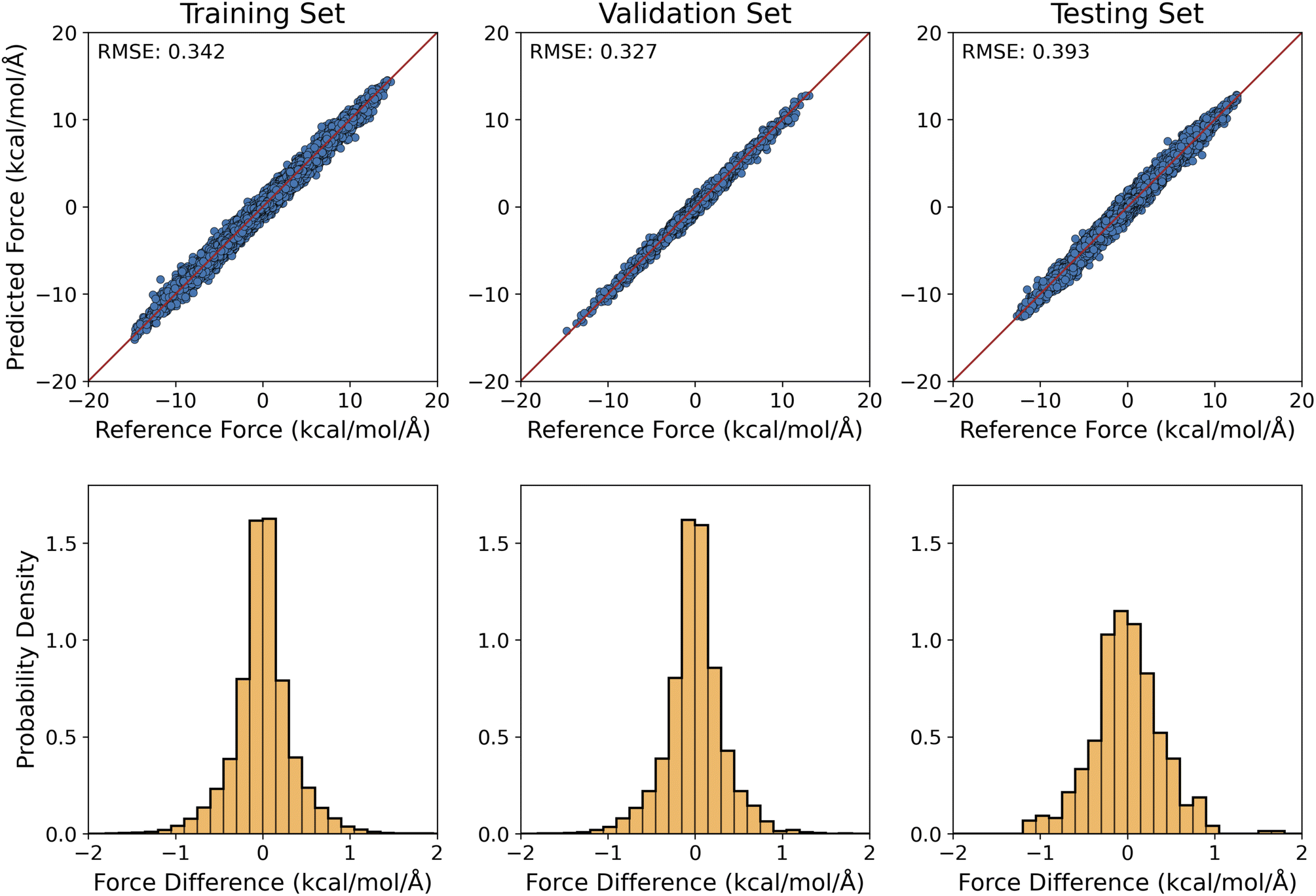 | ||
| Fig. 3 ML predicted solvation forces acquired from ASEC simulation using trajectory configurations. Top: ML-predicted solvation forces versus reference values (in kcal mol−1 Å−1) for the training, validation, and testing sets, which consist of 1245, 51, and 500 configurations, respectively. Bottom: the distribution of errors in the ML-predicted forces. | ||
For the 500 testing set configurations (Section 3.3.), our MLP and MLP-O models predicted the solvation forces with an RMSE of 0.393 kcal mol−1 Å−1 and 0.415 kcal mol−1 Å−1, respectively. With these force differences comparable to those of the training and validation sets, it indicated that our MLP and MLP-O models could accurately predict the interaction of alanine dipeptide with the aqueous environment and can be readily employed to model the solvation dynamics of alanine peptides.
Henceforth, umbrella sampling trajectories were generated using the MLP and MLP-O models and then subject to WHAM analysis to produce the free energy surface (i.e., Ramachandran plots) for solvated alanine dipeptide. A comparison of the free energy landscapes between the MLP and explicit solvent models in Fig. 4 indicated that the MLP model replicated the critical free energy minima (such as C5, PII, αR, αD, and αL). Our MLP Ramachandran plot (or the MLP-O one in Fig. S6b†) was also similar to the explicit-solvent free energy landscape using the same ff14SB force field reported in Fig. S1B of ref. 104. Specifically, our MLP model as shown in Fig. 4a accurately captured the PII, αR, αL and αD regions, with slightly larger errors for the C5 and α′ regions. The surfaces revealing the free energy differences between predictions of the (a) MLP, (b) MLP-O, and (c) vacuum models and the explicit solvent result were shown in Fig. S7.† Panels a and b indicated that the MLP and MLP-O models can yield a free energy surface with the maximum deviation from the explicit solvent result smaller than 0.9 kcal mol−1. This was substantially lower than the difference between gas-phase and explicit solvent simulations (panel c, Fig. S7†).
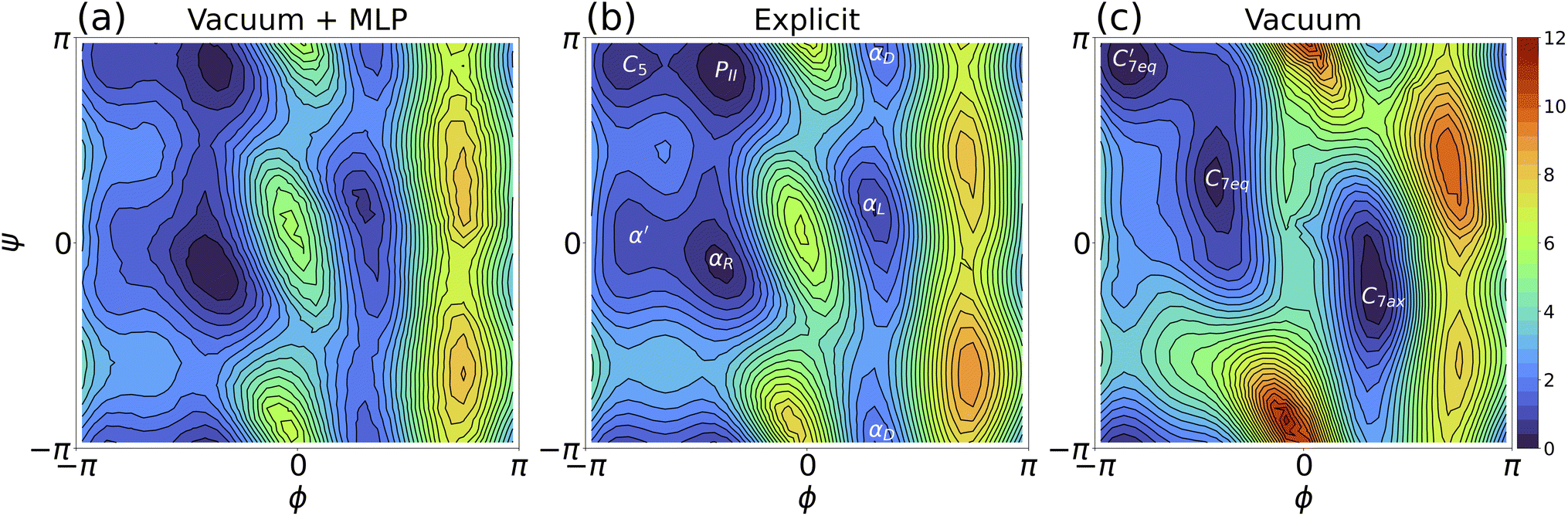 | ||
| Fig. 4 Two-dimensional free energy surfaces of alanine dipeptide: (a) implicit solvation with MLP trained by non-minimized solute configurations; (b) explicit solvation; and (c) solute in vacuum. The color bar is in kcal mol−1. | ||
There results clearly demonstrated that our MLP models can reliably predict the solute–solvent interaction forces for alanine dipeptide. Most importantly, by including solute configurations with all possible combinations of torsional angles in the training set, we could reproduce the high-energy regions, which are essential for studying the transition between different peptide conformations.
4.2 MLP implicit solvent model for QM modeling
So far, our MLP models have been used to predict the solute–solvent interaction forces with the solute described at the MM level. However, with the large number of neuron in our current PyTorch implementation (see Table 1 for details), the anticipated cost to compute MLP (i.e. the force for driving the dynamics) at each time step is higher than to that of an explicit solvent calculation (with a MM description for both the solute and solvent). This could be clearly seen from Table 2, which collected the timings for 10 ns MD trajectories for alanine dipeptide with vacuum, explicit solvent, and MLP models. This suggests that a substantial optimization of the neural network model would be needed before such MLP-based implicit solvent models can be routinely adopted in MM MD simulations.| Solvent model (package) | Time (ns per day) |
|---|---|
| MM vacuum (SANDER) | 450 |
| Explicit solvent (PMEMD) | 90 |
| MM + MLP (OpenMM-Torch) | 11 |
| QM vacuum (Q-Chem) | ∼0.03 |
However, as is well known, QM calculations of the solute molecule have a much higher cost, which can be seen from the timing in Table 2 for gas-phase ab initio QM MD simulation using the Q-Chem software110 at the B3LYP/6-31G* level of theory. Therefore, it would be more attractive to employ our MLP training protocol to learn the QM/MM solute–solvent forces and thus to build ML-based implicit solvent models for the QM modeling.
Our preliminary results for training MLPs to produce QM/MM-quality interaction forces between alanine dipeptide (QM region) and water molecules (MM region) were summarized in Fig. 5. The RMSE values of the predicted QM/MM forces were 0.559, 0.567, and 0.795 kcal mol−1 Å−1, respectively, for the training, validation, and testing sets. To this extent, it also reached the target accuracy level of 1 kcal mol−1 Å−1 for ML force predictions.94,114
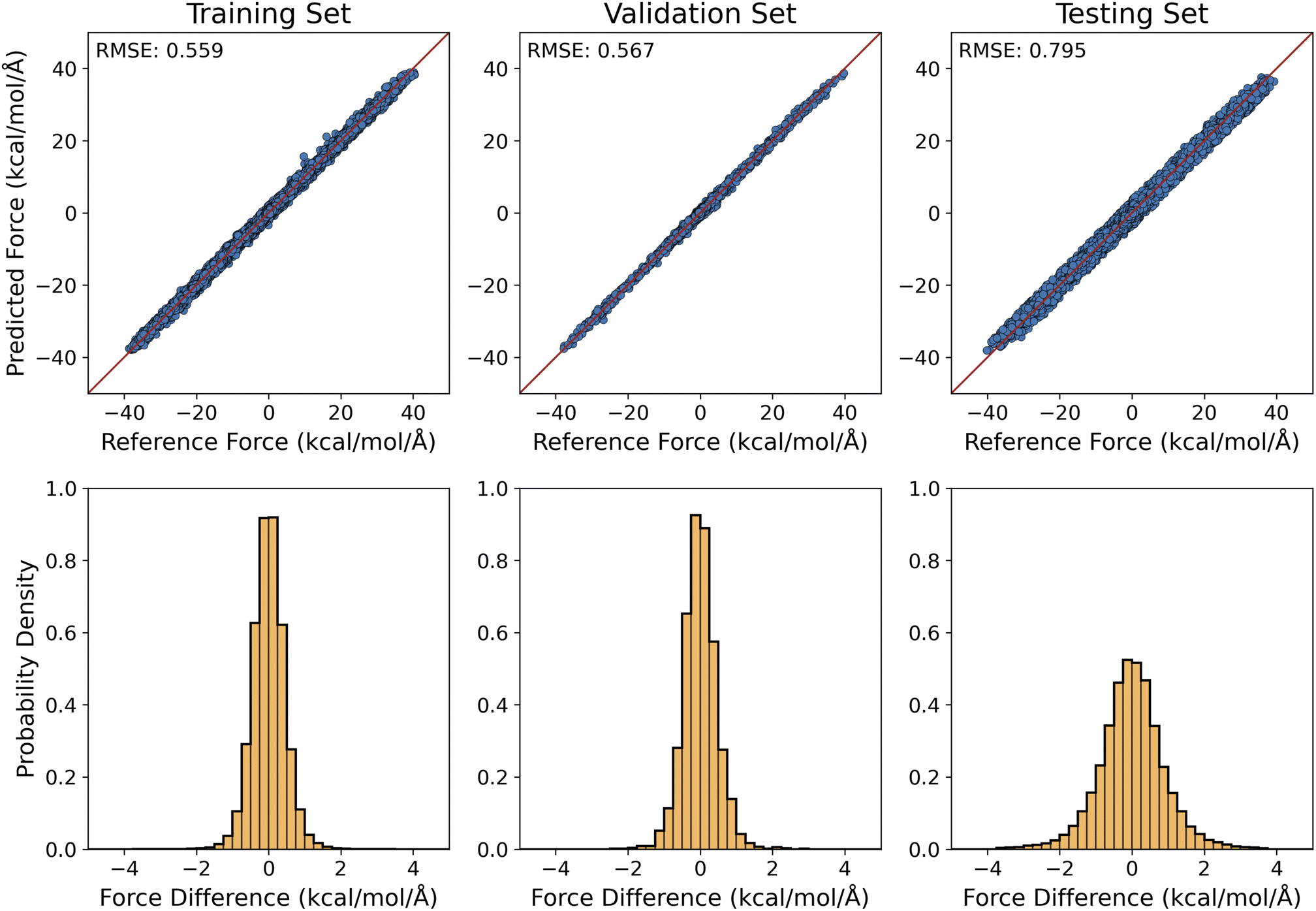 | ||
| Fig. 5 Scatter plots (top row) and histograms (bottom row) displaying the differences between predicted and reference forces for the QM/MM MLP model. The training set, validation set, and testing set all remain the same as for the MM MLP model. The RMSE values in kcal mol−1 Å−1 are also shown. | ||
Recently, there has been a great success in the construction of QM-quality MLPs for small to medium size molecules.115–124 Our work constitutes a natural extensions of these MLP models to offer an implicit description for the interaction of a QM molecule with their solvent environments. Cost-wise, the results above showed that the QM/MM-quality MLP interaction forces could be predicted using the same ML architecture for the prediction of the MM-quality solute–solvent interaction MLP forces, which was far less expensive than the QM calculation of the solute molecule (see Table 2). Therefore, it pointed to the feasibility of utilizing ML to construct QM/MM-quality implicit solvent models.
5. Conclusions
In this work, we employed the DeepPotential descriptors to learn the solute–solvent interaction forces on the alanine dipeptide molecule within an average solvent environment configuration (ASEC). Here are the highlights:• Compared to the inspiring work by Chen et al.,49 our training/validation sets consisted of 1296 ASEC configurations, in each of which a fixed-geometry solute of varying (ϕ, ψ) angles was embedded in thermally-fluctuating water molecules (as described by the TIP3P model). This contrasted with the employment of long MM trajectories with a flexible-geometry solute by Chen et al. From our point of view, our ML training against the ASEC forces more closely aligned with the goal of the implicit solvent models, which was to integrate over the solvent degrees of freedom.
• Our MLP predicted the ASEC solute–solvent interaction forces on alanine dipeptide with RMSEs below 0.5 kcal mol−1 Å−1 for MM-level simulations and below 1.0 kcal mol−1 Å−1 for QM/MM-level simulations. It opened the possibility of developing accurate ML based “implicit” solvation models for simulating biomolecules in solvent environments. This is especially encouraging for implicit-solvent QM simulations, where the MLP adds little extra computational cost to the simulation.
On the other hand, there are a couple of issues that need to be further addressed:
• Our MLP training protocol, which is based on restrained MD simulations at grid points of the Ramanchandran plot, was not necessarily transferable to other biomolecules. We imagine that, for other molecules, a 2-D or higher-dimensional scan of the most relevant dihedral angles (like ours) can produce an initial training set. However, the dataset will need to be augmented on-the-fly using active learning techniques to detect (and account for) solute configurations out of the trust region.
• Unlike commonly-used implicit solvent models in QM calculations, our MLP implicit solvent model for QM simulations in Section 4.2 did not affect the electronic structure of the QM solute. This can be overcome by developing a different MLP, which is trained to reproduce the external electrostatic potential and field at QM solute atom positions arising from the ASEC environment. The machine-learning potential/field can then be used to polarized the QM solute wavefunction.
These issues will be explored in our future work.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
YS and JP were supported by the National Institutes of Health through Grant R01GM135392. YS is also supported by Grant P30GM145423. YM acknowledges the support from San Diego State University Startup Fund. The authors thank the OU Supercomputing Center for Education & Research (OSCER) for the computational resources.References
- W. L. Jorgensen and J. Tirado-Rives, Potential Energy Functions for Atomic-level Simulations of Water and Organic and Biomolecular Systems, Proc. Natl. Acad. Sci. U. S. A., 2005, 102, 6665–6670 CrossRef CAS PubMed.
- X. Liu, G. Zhou, F. Huo, J. Wang and S. Zhang, Unilamellar Vesicle Formation and Microscopic Structure of Ionic Liquids in Aqueous Solutions, J. Phys. Chem. C, 2016, 120, 659–667 CrossRef CAS.
- D. E. Smith and L. X. Dang, Computer Simulations of NaCl Association in Polarizable Water, J. Chem. Phys., 1994, 100, 3757–3766 CrossRef CAS.
- X. Liu, G. Zhou, H. He, X. Zhang, J. Wang and S. Zhang, Rodlike Micelle Structure and Formation of Ionic liquid in Aqueous Solution by Molecular Simulation, Ind. Eng. Chem. Res., 2015, 54, 1681–1688 CrossRef CAS.
- T. W. Walker, A. K. Chew, H. Li, B. Demir, Z. C. Zhang, G. W. Huber, R. C. V. Lehn and J. A. Dumesic, Universal Kinetic Solvent Effects in Acid-catalyzed Reactions of Biomass-derived Oxygenates, Energy Environ. Sci., 2018, 11, 617–628 RSC.
- P. Cieplak, P. Bash, U. C. Singh and P. A. Kollman, A Theoretical Study of Tautomerism in the Gas Phase and Aqueous Solution: A Combined Use of State-of-the-art ab initio Quantum Mechanics and Free Energy-perturbation Methods, J. Am. Chem. Soc., 1987, 109, 6283–6289 CrossRef CAS.
- J. Gao and X. Xia, A Priori Evaluation of Aqueous Polarization Effects through Monte Carlo QM-MM Simulations, Science, 1992, 258, 631–635 CrossRef CAS PubMed.
- H. Nymeyer and A. E. Garcia, Simulation of the Folding Equilibrium of α-Helical Peptides: A Comparison of the Generalized Born Approximation with Explicit Solvent, Proc. Natl. Acad. Sci. U.S.A., 2003, 100, 13934–13939 CrossRef CAS PubMed.
- R. Anandakrishnan, A. Drozdetski, R. C. Walker and A. V. Onufriev, Speed of Conformational Change: Comparing Explicit and Implicit Solvent Molecular Dynamics Simulations, Biophys. J., 2015, 108, 1153–1164 CrossRef CAS PubMed.
- A. Cumberworth, J. M. Bui and J. Gsponer, Free Energies of Solvation in the Context of Protein Folding: Implications for Implicit and Explicit Solvent Models, J. Comput. Chem., 2016, 37, 629–640 CrossRef CAS PubMed.
- W. L. Jorgensen, J. Chandrasekhar, J. D. Madura, R. W. Impey and M. L. Klein, Comparison of Simple Potential Functions for Simulating Liquid Water, J. Chem. Phys., 1983, 79, 926–935 CrossRef CAS.
- C. Vega, J. L. F. Abascal and I. Nezbeda, Vapor-liquid Equilibria from the Triple Point up to the Critical Point for the New Generation of TIP4P-like Models: TIP4P/Ew, TIP4P/2005, and TIP4P/Ice, J. Chem. Phys., 2006, 125, 034503 CrossRef CAS PubMed.
- M. W. Mahoney and W. L. Jorgensen, Diffusion Constant of the TIP5P Model of Liquid Water, J. Chem. Phys., 2001, 114, 363–366 CrossRef CAS.
- H. J. C. Berendsen, J. R. Grigera and T. P. Straatsma, The Missing Term in Effective Pair Potentials, J. Phys. Chem., 1987, 91, 6269–6271 CrossRef CAS.
- P. Ren and J. W. Ponder, Temperature and Pressure Dependence of the AMOEBA Water Model, J. Phys. Chem. B, 2004, 108, 13427–13437 CrossRef CAS.
- J. W. Ponder, C. Wu, P. Ren, V. S. Pande, J. D. Chodera, M. J. Schnieders, I. Haque, D. L. Mobley, D. S. Lambrecht, R. A. DiStasio, M. Head-Gordon, G. N. I. Clark, M. E. Johnson and T. Head-Gordon, Current Status of the AMOEBA Polarizable Force Field, J. Phys. Chem. B, 2010, 114, 2549–2564 CrossRef CAS PubMed.
- B. Roux and T. Simonson, Implicit Solvent Models, Biophys. Chem., 1999, 78, 1–20 CrossRef CAS PubMed.
- C. J. Cramer and D. G. Truhlar, Implicit Solvation Models: Equilibria, Structure, Spectra, and Dynamics, Chem. Rev., 1999, 99, 2161–2200 CrossRef CAS PubMed.
- J. Tomasi, B. Mennucci and R. Cammi, Quantum Mechanical Continuum Solvation Models, Chem. Rev., 2005, 105, 2999–3094 CrossRef CAS PubMed.
- J. Ho, A. Klamt and M. L. Coote, Comment on the Correct Use of Continuum Solvent Models, J. Phys. Chem. A, 2010, 114, 13442–13444 CrossRef CAS PubMed.
- X. Yang, H. Lei, P. Gao, D. G. Thomas, D. L. Mobley and N. A. Baker, Atomic Radius and Charge Parameter Uncertainty in Biomolecular Solvation Energy Calculations, J. Chem. Theory Comput., 2018, 14, 759–767 CrossRef CAS PubMed.
- S. Miertuš, E. Scrocco and J. Tomasi, Electrostatic Interaction of a Solute with a Continuum. A Direct Utilization of AB initio Molecular Potentials for the Prevision of Solvent Effects, Chem. Phys., 1981, 55, 117–129 CrossRef.
- A. Klamt and G. Schüürmann, COSMO: A New Approach to Dielectric Dcreening in Solvents with Explicit Expressions for the Screening Energy and Its Gradient, J. Chem. Soc., Perkin Trans. 2, 1993, 2, 799–805 RSC.
- T. N. Truong and E. V. Stefanovich, A New Method for Incorporating Solvent Effect into the Classical, ab initio Molecular Orbital and Density Functional Theory Frameworks for Arbitrary Shape Cavity, Chem. Phys. Lett., 1995, 240, 253–260 CrossRef CAS.
- E. Cancès, B. Mennucci and J. Tomasi, A New Integral Equation Formalism for the Polarizable Continuum Model: Theoretical Background and Applications to Isotropic and Anisotropic Dielectrics, J. Chem. Phys., 1997, 107, 3032–3041 CrossRef.
- B. Mennucci, E. Cancès and J. Tomasi, Evaluation of Solvent Effects in Isotropic and Anisotropic Dielectrics and in Ionic Solutions with a Unified Integral Equation Method: Theoretical Bases, Computational Implementation, and Numerical Applications, J. Phys. Chem. B, 1997, 101, 10506–10517 CrossRef CAS.
- D. M. York and M. Karplus, A Smooth Solvation Potential Based on the Conductor-Like Screening Model, J. Phys. Chem. A, 1999, 103, 11060–11079 CrossRef CAS.
- D. M. Chipman, Reaction Field Treatment of Charge Penetration, J. Chem. Phys., 2000, 112, 5558–5565 CrossRef CAS.
- A. V. Marenich, C. J. Cramer and D. G. Truhlar, Universal Solvation Model Based on Solute Electron Density and on a Continuum Model of the Solvent Defined by the Bulk Dielectric Constant and Atomic Surface Tensions, J. Phys. Chem. B, 2009, 113, 6378–6396 CrossRef CAS PubMed.
- A. W. Lange and J. M. Herbert, A Smooth, Nonsingular, and Faithful Discretization Scheme for Polarizable Continuum Models: The Switching/Gaussian Approach, J. Chem. Phys., 2010, 133, 244111 CrossRef PubMed.
- B. Mennucci, Polarizable Continuum Model, Wiley Interdiscip. Rev.: Comput. Mol. Sci., 2012, 2, 386–404 CAS.
- A. Klamt, C. Moya and J. Palomar, A Comprehensive Comparison of the IEFPCM and SS(V)PE Continuum Solvation Methods with the COSMO Approach, J. Chem. Theory Comput., 2015, 11, 4220–4225 CrossRef CAS PubMed.
- J. Ho and M. Z. Ertem, Calculating Free Energy Changes in Continuum Solvation Models, J. Phys. Chem. B, 2016, 120, 1319–1329 CrossRef CAS PubMed.
- A. V. Marenich, R. M. Olson, C. P. Kelly, C. J. Cramer and D. G. Truhlar, Self-Consistent Reaction Field Model for Aqueous and Nonaqueous Solutions Based on Accurate Polarized Partial Charges, J. Chem. Theory Comput., 2007, 3, 2011–2033 CrossRef CAS PubMed.
- A. W. Lange and J. M. Herbert, Improving Generalized Born Models by Exploiting Connections to Polarizable Continuum Models. I. An Improved Effective Coulomb Operator, J. Chem. Theory Comput., 2012, 8, 1999–2011 CrossRef CAS PubMed.
- A. V. Marenich, C. J. Cramer and D. G. Truhlar, Generalized Born Solvation Model SM12, J. Chem. Theory Comput., 2013, 9, 609–620 CrossRef CAS PubMed.
- M. S. Lee and M. A. Olson, Comparison of Volume and Surface Area Nonpolar Solvation Free Energy Terms for Implicit Solvent Simulations, J. Chem. Phys., 2013, 139, 044119 CrossRef PubMed.
- A. Onufriev, D. Bashford and D. Case, Exploring Protein Native States and Large-scale Conformational Changes with a Modified Generalized Born Model, Proteins, 2004, 55, 383–394 CrossRef CAS PubMed.
- F. Lipparini and V. Barone, Polarizable Force Fields and Polarizable Continuum Model: A Fluctuating Charges/PCM Approach. 1. Theory and Implementation, J. Chem. Theory Comput., 2011, 7, 3711–3724 CrossRef CAS PubMed.
- R. Zhou and B. J. Berne, Can a Continuum Solvent Model Reproduce the Free Energy Landscape of a β-Hairpin Folding in Water?, Proc. Natl. Acad. Sci. U.S.A., 2002, 99, 12777–12782 CrossRef CAS PubMed.
- F. Noé, C. Schütte, E. Vanden-Eijnden, L. Reich and T. R. Weikl, Constructing the Equilibrium Ensemble of Folding Pathways from Short Off-equilibrium Simulations, Proc. Natl. Acad. Sci. U. S. A., 2009, 106, 19011–19016 CrossRef PubMed.
- F. Noé, G. De Fabritiis and C. Clementi, Machine Learning for Protein Folding and Dynamics, Curr. Opin. Struct. Biol., 2020, 60, 77–84 CrossRef PubMed.
- E. J. M. Lang, E. G. Baker, D. N. Woolfson and A. J. Mulholland, Generalized Born Implicit Solvent Models Do Not Reproduce Secondary Structures of De Novo Designed Glu/Lys Peptides, J. Chem. Theory Comput., 2022, 18, 4070–4076 CrossRef CAS PubMed.
- Q. Shao and W. Zhu, Assessing AMBER Force Fields for Protein Folding in an Implicit Solvent, Phys. Chem. Chem. Phys., 2018, 20, 7206–7216 RSC.
- J. Chen, Y. Shao and J. Ho, Are Explicit Solvent Models More Accurate than Implicit Solvent Models? A Case Study on the Menschutkin Reaction, J. Phys. Chem. A, 2019, 123, 5580–5589 CrossRef CAS PubMed.
- B. Thapa and H. B. Schlegel, Improved pKa Prediction of Substituted Alcohols, Phenols, and Hydroperoxides in Aqueous Medium Using Density Functional Theory and a Cluster-Continuum Solvation Model, J. Phys. Chem. A, 2017, 121, 4698–4706 CrossRef CAS PubMed.
- J. Ho, Are Thermodynamic Cycles Necessary for Continuum Solvent Calculation of pKas and Reduction Potentials?, Phys. Chem. Chem. Phys., 2015, 17, 2859–2868 RSC.
- S. T. Hutchinson and R. Kobayashi, Solvent-Specific Featurization for Predicting Free Energies of Solvation through Machine Learning, J. Chem. Inf. Model., 2019, 59, 1338–1346 CrossRef CAS PubMed.
- Y. Chen, A. Krämer, N. E. Charron, B. E. Husic, C. Clementi and F. Noé, Machine Learning Implicit Solvation for Molecular Dynamics, J. Chem. Phys., 2021, 155, 084101 CrossRef CAS PubMed.
- F. Noé, A. Tkatchenko, K.-R. Müller and C. Clementi, Machine Learning for Molecular Simulation, Annu. Rev. Phys. Chem., 2020, 71, 361–390 CrossRef PubMed.
- I. H. Sarker, Machine Learning: Algorithms, Real-World Applications and Research Directions, SN Comput. Sci., 2021, 2, 160 CrossRef PubMed.
- O. T. Unke and M. Meuwly, PhysNet: A Neural Network for Predicting Energies, Forces, Dipole Moments, and Partial Charges, J. Chem. Theory Comput., 2019, 15, 3678–3693 CrossRef CAS PubMed.
- M. Gastegger, L. Schwiedrzik, M. Bittermann, F. Berzsenyi and P. Marquetand, wACSF-Weighted Atom-centered Symmetry Functions as Descriptors in Machine Learning Potentials, J. Chem. Phys., 2018, 148, 241709 CrossRef CAS PubMed.
- S. Grimme and P. R. Schreiner, Computational Chemistry: The Fate of Current Methods and Future Challenges, Angew. Chem., Int. Ed. Engl., 2018, 57, 4170–4176 CrossRef CAS PubMed.
- M. Meuwly, Machine Learning for Chemical Reactions, Chem. Rev., 2021, 121, 10218–10239 CrossRef CAS PubMed.
- A. Khorshidi and A. Peterson, Amp: A Modular Approach to Machine Learning in Atomistic Simulations, Comput. Phys. Commun., 2016, 207, 310–324 CrossRef CAS.
- M. Gastegger, K. T. Schütt and K.-R. Müller, Machine Learning of Solvent Effects on Molecular Spectra and Reactions, Chem. Sci., 2021, 12, 11473–11483 RSC.
- Q. He, W. Yang, W. Luo, S. Wilhelm and B. Weng, Label-Free Differentiation of Cancer and Non-Cancer Cells Based on Machine-Learning-Algorithm-Assisted Fast Raman Imaging, Biosensors, 2022, 12, 250 CrossRef CAS PubMed.
- M. Gastegger, J. Behler and P. Marquetand, Machine Learning Molecular Dynamics for the Simulation of Infrared Spectra, Chem. Sci., 2017, 8, 6924–6935 RSC.
- A. Chandra and T. Ichiye, Dynamical Properties of the Soft Sticky Dipole Model of Water: Molecular Dynamics Simulations, J. Chem. Phys., 1999, 111, 2701–2709 CrossRef CAS.
- I.-B. Magdău and T. F. Miller, Machine Learning Solvation Environments in Conductive Polymers: Application to ProDOT-2Hex with Solvent Swelling, Macromolecules, 2021, 54, 3377–3387 CrossRef.
- Y. Basdogan, M. C. Groenenboom, E. Henderson, S. De, S. B. Rempe and J. A. Keith, Machine Learning-Guided Approach for Studying Solvation Environments, J. Chem. Theory Comput., 2020, 16, 633–642 CrossRef PubMed.
- T. W. Walker, A. K. Chew, R. C. Van Lehn, J. A. Dumesic and G. W. Huber, Rational Design of Mixed Solvent Systems for Acid-Catalyzed Biomass Conversion Processes Using a Combined Experimental, Molecular Dynamics and Machine Learning Approach, Top. Catal., 2020, 63, 649–663 CrossRef CAS.
- A. K. Chew, T. W. Walker, Z. Shen, B. Demir, L. Witteman, J. Euclide, G. W. Huber, J. A. Dumesic and R. C. Van Lehn, Effect of Mixed-Solvent Environments on the Selectivity of Acid-Catalyzed Dehydration Reactions, ACS Catal., 2020, 10, 1679–1691 CrossRef CAS.
- A. M. Maldonado, Y. Basdogan, J. Berryman, S. Rempe and J. Keith, First-principles Modeling of Chemistry in Mixed Solvents: Where to Go from Here?, J. Chem. Phys., 2020, 152, 130902 CrossRef CAS PubMed.
- J. Wang, N. Charron, B. Husic, S. Olsson, F. Noé and C. Clementi, Multi-body Effects in a Coarse-grained Protein Force Field, J. Chem. Phys., 2021, 154, 164113 CrossRef CAS PubMed.
- D. J. Tobias and C. L. Brooks, Conformational Equilibrium in the Alanine Dipeptide in the Gas Phase and Aqueous Solution: a Comparison of Theoretical Results, J. Phys. Chem., 1992, 96, 3864–3870 CrossRef CAS.
- J. Apostolakis, P. Ferrara and A. Caflisch, Calculation of Conformational Transitions and Barriers in Solvated Systems: Application to the Alanine Dipeptide in Water, J. Chem. Phys., 1999, 110, 2099–2108 CrossRef CAS.
- V. Mironov, Y. Alexeev, V. K. Mulligan and D. G. Fedorov, A Systematic Study of Minima in Alanine Dipeptide, J. Comput. Chem., 2019, 40, 297–309 CrossRef CAS PubMed.
- E. Guàrdia, R. Rey and J. Padró, Potential of Mean Force by Constrained Molecular Dynamics: A Sodium Chloride Ion-pair in Water, Chem. Phys., 1991, 155, 187–195 CrossRef.
- E. Guàrdia, R. Rey and J. Padró, Na+–Na+ and Cl−–Cl− Ion Pairs in Water: Mean Force Potentials by Constrained Molecular Dynamics, J. Chem. Phys., 1991, 95, 2823 CrossRef.
- Y. Orozco-Gonzalez, M. Manathunga, M. d. C. Marín, D. Agathangelou, K.-H. Jung, F. Melaccio, N. Ferré, S. Haacke, K. Coutinho, S. Canuto and M. Olivucci, An Average Solvent Electrostatic Configuration Protocol for QM/MM Free Energy Optimization: Implementation and Application to Rhodopsin Systems, J. Chem. Theory Comput., 2017, 13, 6391–6404 CrossRef CAS PubMed.
- D. M. Nikolaev, M. Manathunga, Y. Orozco-Gonzalez, A. A. Shtyrov, Y. O. Guerrero Martínez, S. Gozem, M. N. Ryazantsev, K. Coutinho, S. Canuto and M. Olivucci, Free Energy Computation for an Isomerizing Chromophore in a Molecular Cavity via the Average Solvent Electrostatic Configuration Model: Applications in Rhodopsin and Rhodopsin-Mimicking Systems, J. Chem. Theory Comput., 2021, 17, 5885–5895 CrossRef CAS PubMed.
- M. L. Sanchez, M. A. Aguilar and F. J. O. del Valle, Study of Solvent Effects by Means of Averaged Solvent Electrostatic Potentials Obtained from Molecular Dynamics Data, J. Comput. Chem., 1997, 18, 313–322 CrossRef CAS.
- M. Mendoza, M. Aguilar and F. del Valle, A Mean Field Approach that Combines Quantum Mechanics and Molecular Dynamics Simulation: The Water Molecule in Liquid Water, J. Mol. Struct., 1998, 426, 181–190 CrossRef.
- K. Coutinho, H. Georg, T. Fonseca, V. Ludwig and S. Canuto, An Efficient Statistically Converged Average Configuration for Solvent Effects, Chem. Phys. Lett., 2007, 437, 148–152 CrossRef CAS.
- X. Zhou, J. W. Kaminski and T. A. Wesolowski, Multi-scale Modelling of Solvatochromic Shifts from Frozen-density Embedding Theory with Non-uniform Continuum Model of the Solvent: The Coumarin 153 Case, Phys. Chem. Chem. Phys., 2011, 13, 10565 RSC.
- A. Laktionov, E. Chemineau-Chalaye and T. A. Wesolowski, Frozen-density Embedding Theory with Average Solvent Charge Densities from Explicit Atomistic Simulations, Phys. Chem. Chem. Phys., 2016, 18, 21069–21078 RSC.
- I. Brandão, T. L. Fonseca, H. C. Georg, M. A. Castro and R. B. Pontes, Assessing the Structure and First Hyperpolarizability of Li@B10H14 in Solution: A Sequential QM/MM Study Using the ASEC–FEG Method, Phys. Chem., 2020, 22, 17314–17324 RSC.
- C. E. González-Espinoza, C. A. Rumble, D. Borgis and T. A. Wesolowski, Quantifying Fluctuations of Average Solvent Environments for Embedding Calculations, J. Chem. Theory Comput., 2022, 18, 1072–1088 CrossRef PubMed.
- M. S. Gordon, D. G. Fedorov, S. R. Pruitt and L. V. Slipchenko, Fragmentation Methods: A Route to Accurate Calculations on Large Systems, Chem. Rev., 2012, 112, 632–672 CrossRef CAS PubMed.
- R. M. Richard, K. U. Lao and J. M. Herbert, Understanding the Many-body Expansion for Large Systems. I. Precision Considerations, J. Chem. Phys., 2014, 141, 014108 CrossRef PubMed.
- M. A. Collins and R. P. A. Bettens, Energy-Based Molecular Fragmentation Methods, Chem. Rev., 2015, 115, 5607–5642 CrossRef CAS PubMed.
- G. Koenig, Y. Mei, F. C. Pickard, A. C. Simmonett, B. T. Miller, J. M. Herbert, H. L. Woodcock, B. R. Brooks and Y. Shao, Computation of Hydration Free Energies Using the Multiple Environment Single System Quantum Mechanical/Molecular Mechanical Method, J. Chem. Theory Comput., 2016, 12, 332–344 CrossRef PubMed.
- Y. Zhou and J. Pu, Reaction Path Force Matching: A New Strategy of Fitting Specific Reaction Parameters for Semiempirical Methods in Combined QM/MM Simulations, J. Chem. Theory Comput., 2014, 10, 3038–3054 CrossRef CAS PubMed.
- B. Kim, R. Snyder, M. Nagaraju, Y. Zhou, P. Ojeda-May, S. Keeton, M. Hege, Y. Shao and J. Pu, Reaction Path-Force Matching in Collective Variables: Determining Ab Initio QM/MM Free Energy Profiles by Fitting Mean Force, J. Chem. Theory Comput., 2021, 17, 4961–4980 CrossRef CAS PubMed.
- L. Zhang, J. Han, H. Wang, W. Saidi, R. Car, W. E, End-to-end Symmetry Preserving Inter-atomic Potential Energy Model for Finite and Extended Systems, Advances in Neural Information Processing Systems, 2018, vol. 31, pp. 4436–4446 Search PubMed.
- L. Zhang, J. Han, H. Wang, R. Car and W. E, Deep Potential Molecular Dynamics: A Scalable Model with the Accuracy of Quantum Mechanics, Phys. Rev. Lett., 2018, 120, 143001 CrossRef CAS PubMed.
- H. Wang, L. Zhang, J. H. Han and W. E, DeePMD-kit: A Deep Learning Package for Many-body Potential Energy Representation and Molecular Dynamics, Comput. Phys. Commun., 2018, 228, 178–184 CrossRef CAS.
- J. Han, L. Zhang, R. Car and W. E, Deep Potential: A General Representation of a Many-body Potential Energy Surface, Commun. Comput. Phys., 2017, 23, 629–639 Search PubMed.
- F. H. Vermeire and W. H. Green, Transfer Learning for Solvation Free Energies: From Quantum Chemistry to Experiments, Chem. Eng. J., 2021, 418, 129307 CrossRef CAS.
- H. Lim and Y. Jung, Delfos: Deep Learning Model for Prediction of Solvation Free Energies in Generic Organic Solvents, Chem. Sci., 2019, 10, 8306–8315 RSC.
- D. Zhang, S. Xia and Y. Zhang, Accurate Prediction of Aqueous Free Solvation Energies Using 3D Atomic Feature-Based Graph Neural Network with Transfer Learning, J. Chem. Inf. Model., 2022, 62, 1840–1848 CrossRef CAS PubMed.
- X. Pan, J. Yang, R. Van, E. Epifanovsky, J. Ho, J. Huang, J. Pu, Y. Mei, K. Nam and Y. Shao, Machine-Learning-Assisted Free Energy Simulation of Solution-Phase and Enzyme Reactions, J. Chem. Theory Comput., 2021, 17, 5745–5758 CrossRef CAS PubMed.
- M. Feig, Kinetics from Implicit Solvent Simulations of Biomolecules as a Function of Viscosity, J. Chem. Theory Comput., 2007, 3, 1734–1748 CrossRef CAS.
- J. Behler, Representing Potential Energy Surfaces by High-Dimensional Neural Network Potentials, J. Condens. Matter Phys., 2014, 26, 183001 CrossRef CAS PubMed.
- F. Nüske, L. Boninsegna and C. Clementi, Coarse-graining Molecular Systems by Spectral Matching, J. Chem. Phys., 2019, 151, 044116 CrossRef PubMed.
- W. Wang and R. Gómez-Bombarelli, Coarse-graining Auto-encoders for Molecular Dynamics, Npj Comput. Mater., 2019, 5, 125 CrossRef.
- P. G. Bolhuis, C. Dellago and D. Chandler, Reaction Coordinates of Biomolecular Isomerization, Proc. Natl. Acad. Sci. U.S.A., 2000, 97, 5877–5882 CrossRef CAS PubMed.
- S. Wu and A. Ma, Mechanism for the Rare Fluctuation that Powers Protein Conformational Change, J. Chem. Phys., 2022, 156, 054119 CrossRef CAS.
- S. Wu, H. Li and A. Ma, A Rigorous Method for Identifying a One-Dimensional Reaction Coordinate in Complex Molecules, J. Chem. Theory Comput., 2022, 18, 2836–2844 CrossRef CAS PubMed.
- J. Wang, S. Olsson, C. Wehmeyer, A. Pérez, N. E. Charron, G. de Fabritiis, F. Noé and C. Clementi, Machine Learning of Coarse-Grained Molecular Dynamics Force Fields, ACS Cent. Sci., 2019, 5, 755–767 CrossRef CAS PubMed.
- D. A. Case, H. M. Aktulga, K. Belfon, I. Ben-Shalom, S. R. Brozell, D. S. Cerutti, T. E. Cheatham III, V. W. D. Cruzeiro, T. A. Darden, R. E. Duke, G. Giambasu, M. K. Gilson, H. Gohlke, A. W. Goetz, R. Harris, S. Izadi, S. A. Izmailov, C. Jin, K. Kasavajhala, M. C. Kaymak, E. King, A. Kovalenko, T. Kurtzman, T. Lee, S. LeGrand, P. Li, C. Lin, J. Liu, T. Luchko, R. Luo, M. Machado, V. Man, M. Manathunga, K. M. Merz, Y. Miao, O. Mikhailovskii, G. Monard, H. Nguyen, K. A. O'Hearn, A. Onufriev, F. Pan, S. Pantano, R. Qi, A. Rahnamoun, D. R. Roe, A. Roitberg, C. Sagui, S. Schott-Verdugo, J. Shen, C. L. Simmerling, N. R. Skrynnikov, J. Smith, J. Swails, R. C. Walker, J. Wang, H. Wei, R. M. Wolf, X. Wu, Y. Xue, D. M. York, S. Zhao, and P. A. Kollman, Amber 2021, University of California, San Francisco, p. 2021 Search PubMed.
- J. A. Maier, C. Martinez, K. Kasavajhala, L. Wickstrom, K. E. Hauser and C. Simmerling, ff14SB: Improving the Accuracy of Protein Side Chain and Backbone Parameters from ff99SB, J. Chem. Theory Comput., 2015, 11, 3696–3713 CrossRef CAS.
- A. D. Becke, Density-functional Exchange-Energy Approximation with Correct Asymptotic Behavior, Phys. Rev. A, 1988, 38, 3098–3100 CrossRef CAS.
- A. D. Becke, A New Mixing of Hartree-Fock and Local Density-Functional Theories, J. Chem. Phys., 1993, 98, 1372–1377 CrossRef CAS.
- C. Lee, W. Yang and R. G. Parr, Development of the Colle-Salvetti Correlation-Energy Formula into a Functional of the Electron Density, Phys. Rev. B: Condens. Matter Mater. Phys., 1988, 37, 785–789 CrossRef CAS PubMed.
- P. C. Hariharan and J. A. Pople, The Influence of Polarization Functions on Molecular Orbital Hydrogenation Energies, Theor. Chim. Acta, 1973, 28, 213–222 CrossRef CAS.
- X. Pan, K. Nam, E. Epifanovsky, A. C. Simmonett, E. Rosta and Y. Shao, A Simplified Charge Projection Scheme for Long-range Electrostatics in ab initio QM/MM Calculations, J. Chem. Phys., 2021, 154, 024115 CrossRef CAS PubMed.
- E. Epifanovsky, A. T. B. Gilbert, X. Feng, J. Lee, Y. Mao, N. Mardirossian, P. Pokhilko, A. F. White, M. P. Coons, A. L. Dempwolff, Z. Gan, D. Hait, P. R. Horn, L. D. Jacobson, I. Kaliman, J. Kussmann, A. W. Lange, K. U. Lao, D. S. Levine, J. Liu, S. C. McKenzie, A. F. Morrison, K. D. Nanda, F. Plasser, D. R. Rehn, M. L. Vidal, Z.-Q. You, Y. Zhu, B. Alam, B. J. Albrecht, A. Aldossary, E. Alguire, J. H. Andersen, V. Athavale, D. Barton, K. Begam, A. Behn, N. Bellonzi, Y. A. Bernard, E. J. Berquist, H. G. A. Burton, A. Carreras, K. Carter-Fenk, R. Chakraborty, A. D. Chien, K. D. Closser, V. Cofer-Shabica, S. Dasgupta, M. de Wergifosse, J. Deng, M. Diedenhofen, H. Do, S. Ehlert, P.-T. Fang, S. Fatehi, Q. Feng, T. Friedhoff, J. Gayvert, Q. Ge, G. Gidofalvi, M. Goldey, J. Gomes, C. E. González-Espinoza, S. Gulania, A. O. Gunina, M. W. D. Hanson-Heine, P. H. P. Harbach, A. Hauser, M. F. Herbst, M. Hernández Vera, M. Hodecker, Z. C. Holden, S. Houck, X. Huang, K. Hui, B. C. Huynh, M. Ivanov, A. Jász, H. Ji, H. Jiang, B. Kaduk, S. Kähler, K. Khistyaev, J. Kim, G. Kis, P. Klunzinger, Z. Koczor-Benda, J. H. Koh, D. Kosenkov, L. Koulias, T. Kowalczyk, C. M. Krauter, K. Kue, A. Kunitsa, T. Kus, I. Ladjánszki, A. Landau, K. V. Lawler, D. Lefrancois, S. Lehtola, R. R. Li, Y.-P. Li, J. Liang, M. Liebenthal, H.-H. Lin, Y.-S. Lin, F. Liu, K.-Y. Liu, M. Loipersberger, A. Luenser, A. Manjanath, P. Manohar, E. Mansoor, S. F. Manzer, S.-P. Mao, A. V. Marenich, T. Markovich, S. Mason, S. A. Maurer, P. F. McLaughlin, M. F. S. J. Menger, J.-M. Mewes, S. A. Mewes, P. Morgante, J. W. Mullinax, K. J. Oosterbaan, G. Paran, A. C. Paul, S. K. Paul, F. Pavošević, Z. Pei, S. Prager, E. I. Proynov, A. Rák, E. Ramos-Cordoba, B. Rana, A. E. Rask, A. Rettig, R. M. Richard, F. Rob, E. Rossomme, T. Scheele, M. Scheurer, M. Schneider, N. Sergueev, S. M. Sharada, W. Skomorowski, D. W. Small, C. J. Stein, Y.-C. Su, E. J. Sundstrom, Z. Tao, J. Thirman, G. J. Tornai, T. Tsuchimochi, N. M. Tubman, S. P. Veccham, O. Vydrov, J. Wenzel, J. Witte, A. Yamada, K. Yao, S. Yeganeh, S. R. Yost, A. Zech, I. Y. Zhang, X. Zhang, Y. Zhang, D. Zuev, A. Aspuru-Guzik, A. T. Bell, N. A. Besley, K. B. Bravaya, B. R. Brooks, D. Casanova, J.-D. Chai, S. Coriani, C. J. Cramer, G. Cserey, A. E. DePrince, R. A. DiStasio, A. Dreuw, B. D. Dunietz, T. R. Furlani, W. A. Goddard, S. Hammes-Schiffer, T. Head-Gordon, W. J. Hehre, C.-P. Hsu, T.-C. Jagau, Y. Jung, A. Klamt, J. Kong, D. S. Lambrecht, W. Liang, N. J. Mayhall, C. W. McCurdy, J. B. Neaton, C. Ochsenfeld, J. A. Parkhill, R. Peverati, V. A. Rassolov, Y. Shao, L. V. Slipchenko, T. Stauch, R. P. Steele, J. E. Subotnik, A. J. W. Thom, A. Tkatchenko, D. G. Truhlar, T. Van Voorhis, T. A. Wesolowski, K. B. Whaley, H. L. Woodcock, P. M. Zimmerman, S. Faraji, P. M. W. Gill, M. Head-Gordon, J. M. Herbert and A. I. Krylov, Software for the Frontiers of Quantum Chemistry: An Overview of Developments in the Q-Chem 5 Package, J. Chem. Phys., 2021, 155, 084801 CrossRef CAS PubMed.
- P. Virnau and M. Müller, Calculation of Free Energy through Successive Umbrella Sampling, J. Chem. Phys., 2004, 120, 10925–10930 CrossRef CAS PubMed.
- P. Eastman, J. Swails, J. D. Chodera, R. T. McGibbon, Y. Zhao, K. A. Beauchamp, L.-P. Wang, A. C. Simmonett, M. P. Harrigan, C. D. Stern, R. P. Wiewiora, B. R. Brooks and V. S. Pande, OpenMM 7: Rapid Development of High Performance Algorithms for Molecular Dynamics, PLoS Comput. Biol., 2017, 13, e1005659 CrossRef PubMed.
- S. Kumar, J. M. Rosenberg, D. Bouzida, R. H. Swendsen and P. A. Kollman, The Weighted Histogram Analysis Method for Free-energy Calculations on Biomolecules. I. The Method, J. Comput. Chem., 1992, 13, 1011–1021 CrossRef CAS.
- R. Ramakrishnan, P. O. Dral, M. Rupp and O. A. von Lilienfeld, Big Data Meets Quantum Chemistry Approximations: The Δ-Machine Learning Approach, J. Chem. Theory Comput., 2015, 11, 2087–2096 CrossRef CAS PubMed.
- J. Behler, Perspective: Machine Learning Potentials for Atomistic Simulations, J. Chem. Phys., 2016, 145, 170901 CrossRef PubMed.
- K. T. Butler, D. W. Davies, H. Cartwright, O. Isayev and A. Walsh, Machine Learning for Molecular and Materials Science, Nature, 2018, 559, 547–555 CrossRef CAS PubMed.
- P. O. Dral, Quantum Chemistry in the Age of Machine Learning, J. Phys. Chem. Lett., 2020, 11, 2336–2347 CrossRef CAS PubMed.
- J. Zhang, Y.-K. Lei, Z. Zhang, J. Chang, M. Li, X. Han, L. Yang, Y. I. Yang and Y. Q. Gao, A Perspective on Deep Learning for Molecular Modeling and Simulations, J. Phys. Chem. A, 2020, 124, 6745–6763 CrossRef CAS PubMed.
- V. L. Deringer, A. P. Bartók, N. Bernstein, D. M. Wilkins, M. Ceriotti and G. Csányi, Gaussian Process Regression for Materials and Molecules, Chem. Rev., 2021, 121, 10073–10141 CrossRef CAS PubMed.
- J. Westermayr, M. Gastegger, K. T. Schütt and R. J. Maurer, Perspective on Integrating Machine Learning into Computational Chemistry and Materials Science, J. Chem. Phys., 2021, 154, 230903 CrossRef CAS PubMed.
- T. Zubatiuk and O. Isayev, Development of Multimodal Machine Learning Potentials: Toward a Physics-Aware Artificial Intelligence, Acc. Chem. Res., 2021, 54, 1575–1585 CrossRef CAS PubMed.
- N. Fedik, R. Zubatyuk, M. Kulichenko, N. Lubbers, J. S. Smith, B. Nebgen, R. Messerly, Y. W. Li, A. I. Boldyrev, K. Barros, O. Isayev and S. Tretiak, Extending Machine Learning beyond Interatomic Potentials for Predicting Molecular Properties, Nat. Rev. Chem, 2022, 6, 653–672 CrossRef CAS.
- H. Gokcan and O. Isayev, Learning Molecular Potentials with Neural Networks, Wiley Interdiscip. Rev.: Comput. Mol. Sci., 2022, 12, e1564 Search PubMed.
- H. J. Kulik, T. Hammerschmidt, J. Schmidt, S. Botti, M. A. L. Marques, M. Boley, M. Scheffler, M. Todorović, P. Rinke, C. Oses, A. Smolyanyuk, S. Curtarolo, A. Tkatchenko, A. P. Bartók, S. Manzhos, M. Ihara, T. Carrington, J. Behler, O. Isayev, M. Veit, A. Grisafi, J. Nigam, M. Ceriotti, K. T. Schütt, J. Westermayr, M. Gastegger, R. J. Maurer, B. Kalita, K. Burke, R. Nagai, R. Akashi, O. Sugino, J. Hermann, F. Noé, S. Pilati, C. Draxl, M. Kuban, S. Rigamonti, M. Scheidgen, M. Esters, D. Hicks, C. Toher, P. V. Balachandran, I. Tamblyn, S. Whitelam, C. Bellinger and L. M. Ghiringhelli, Roadmap on Machine Learning in Electronic Structure, Electron. Struct., 2022, 4, 023004 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d2ra08180f |
| This journal is © The Royal Society of Chemistry 2023 |