
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceThe electronic and mechanical properties of (U, Th)O2 compounds: a first-principles study
Jianguo Zhu a and
Diwei Shi*b
a and
Diwei Shi*b
aInstitute of Pharmaceutical and Biomaterials, Lianyungang Normal College, Sheng Hu Lu 28, Lianyungang 222006, China
bSchool of Naval Architecture and Maritime, Zhejiang Ocean University, Haida South Road 1, Zhoushan 316022, China. E-mail: shidiwei@zjou.edu.cn
First published on 3rd March 2023
Abstract
Alloying is widely acknowledged as an effective strategy for enhancing the performance of UO2 nuclear fuel. Herein, the thermodynamic stability and kinetic stability of U–Th–O ternary compounds are used to clarify the hidden stable structures. The calculation results of the total and the partial density of states indicated that there is significant orbital hybridization between the added Th and O atoms at −5 eV. Furthermore, the mechanical anisotropy was evaluated by means of the three-dimensional Young's modulus, revealing that the U–Th–O ternary compound exhibits a high degree of isotropy, with the Young's modulus reaching approximately 200 GPa in all three directions. In our upcoming work, our focus will be on studying the changes in properties, such as thermal conductivity of the U–Th–O ternary compound, which may provide a data basis for the application of ternary U–Th–O fuel in reactors.
1. Introduction
The most efficient, economical and environmentally friendly commercial light-water reactor fuel currently used is UO2, which is the world's main source of electricity.1–3 Traditional UO2 fuel has very high melting point and good water corrosion resistance, however, its thermal conductivity is not high enough to meet industrial demand.4,5 After the Fukushima nuclear accident in 2011, one of the focuses on the fault-tolerant fuel system is to modify UO2 fuel or to find new fuels in place of UO2 to ensure that the improved fuel has higher thermal conductivity and mechanical stability at high temperatures.5 Among them, it is an effective solution to improve the overall performance by adding other elements to form new configurations.6Various properties of U–O–X compounds have been studied extensively in recent years. Rittman D. R. et al.7 studied the phase behavior of ternary compounds formed by doping UO2 with different concentrations of lanthanides (La, Nd) under high-pressure conditions of 0–50 GPa. The results show that the pressure of phase transformations increases with the doping concentration, while the volume modulus changes in inversely proportional to the radius and concentration of doped atoms. S. C. Finkeldei et al.8 explored the preparation of monolithic UO2 and UO2–Mo composite materials using molybdenum metal powder and UO3 spheres as raw materials. The results prove that by adding 10% Mo as the second phase to UO2 will increase the thermal conductivity by 30%, and different sizes of molybdenum raw materials will lead to different microstructures, which will affect the thermal conductivity of the composite material. Be element was introduced into the system to form the UO2–Mo–Be system, the thermal conductivity of the system increased by 128.65 percent and 157.71 percent at the temperature of 298 K and 1273 K, respectively, made it become one of the accident fault tolerance candidate materials.9 Rahman M.10 performed the classical molecular dynamics (MD) simulation method to prove that the number of residual defects in (U0.5Th0.5)O2 are significantly higher than the number of residual defects in UO2 and ThO2. The research results of Evitts L. et al. showed the existence of Th can increase the ratio of 242 Pu/239 Pu by 3.5% when UO2 is low fuel consumption.11
Various studies have shown that adding other elements to UO2 fuel can significantly improve its overall performance. Thorium fuel has the advantages of abundant reserves, less waste, and anti-nuclear proliferation.12 Thorium-based materials has excellent properties as viable nuclear fuel candidates in fourth-generation nuclear reactors, and are considered to be one of the most easily commercialized reactor types.13 So far, researchers have done a series of work on UO2 and Th-based nuclear fuels, while the research on (Th, U)O ternary nuclear fuels has not yet been fully developed. More intriguingly, a mixture of ThO2 and U in recent article demonstrated that (Th, U)O ternary nuclear fuels can be comparable in energy generation with existing UO2 fuels while own long-term cost effectiveness than UO2 fuels.14 In addition, K. Rickert et al. found that by tune the content of Th in the (U, Th)O2 solid solution, the oxidation of UO2 to U3O8 can be completely inhibited which originated from the fact that Th has a stable oxidation state of 4+.15 Therefore, this work is to systematically study the electronic and mechanical properties of Th element as a dopant and UO2 basis to form a new thorium–uranium–oxygen ternary compound based on the first-principles method. The research results may be helpful for the design and application of Th-doped UO2-based modified nuclear fuel to new A generation of commercial reactors provides reference.
2. Method
The theoretical calculations in this part of the work are based on density functional theory,16,17 using the Vienna ab initio calculation package for first-principles calculations.18,19 In the calculation process, the projected enhanced wave electric potential (PAW) method is used.20 The exchange-correlation functional uses the generalized gradient approximation (GGA) function combined with the PBE functional21 and the local density approximation (LDA). The PBE + U approximation method is proven to effectively improve the accuracy of the performance calculation of actinide materials.22,23 Therefore, U = 3.0 is used as a calculation parameter considering the strong correlation effect of 5f orbital, which is consistent with the calculation parameters of Torres and Pegg et al.24–26 The calculation uses the Monkhorst–Pack method to divide the simple Briyuan area by 5 × 3 × 3 K points.27 The truncation can be 500 eV, and when the atomic energy volume converges to 1.0 × 10−5 eV per atom, and the atomic force converges below 0.01 eV s−1, the structural relaxation optimization is completed. All calculations are done under periodic boundary conditions. Considering the computational cost, a supercell of size 2 × 2 × 1 is used to calculate properties such as energy and electrons.3. Results and discussion
3.1. Structure optimization and design
UO2 belongs to the cubic crystal system, with a space group of Fm![[3 with combining macron]](https://www.rsc.org/images/entities/char_0033_0304.gif) m, contains three atoms (including two O atoms and one U atom) in a unit cell. The specific structure is shown in Fig. 1(a) (some models used in calculations are based on this structure, as shown in Fig. 1(b), but due to the requirements of this work, the lattice structure shown in Fig. 1(a) is adopted). To eliminate the boundary effect, all calculations are performed in the 2 × 2 × 1 supercell. In the optimization process of the initial structure, we considered the strong correlation effect of the 5f orbital of U atoms, and the DFT + U method was used through the calculations. For the U atom, U = 3.0 and J = 0 were used as the calculation parameters, which is tested by the simulation research of Torres and Pegg et al.24–26 and has been proved to be reliable. For Th atoms, Wang28 et al. verified through research that the 5f orbitals in thorium atoms are empty, so it is reasonable to set the U value of thorium atoms to 0 in our calculations. At the same time, considering that the U atom itself has a certain degree of magnetism, we will consider the spin in the following calculations. It should be pointed out that the lattice parameter of UO2 calculated in this work is 5.462 Å (Table 1), which is consistent with previous experimental value 5.474 (ref. 29) and 5.468 Å.30
m, contains three atoms (including two O atoms and one U atom) in a unit cell. The specific structure is shown in Fig. 1(a) (some models used in calculations are based on this structure, as shown in Fig. 1(b), but due to the requirements of this work, the lattice structure shown in Fig. 1(a) is adopted). To eliminate the boundary effect, all calculations are performed in the 2 × 2 × 1 supercell. In the optimization process of the initial structure, we considered the strong correlation effect of the 5f orbital of U atoms, and the DFT + U method was used through the calculations. For the U atom, U = 3.0 and J = 0 were used as the calculation parameters, which is tested by the simulation research of Torres and Pegg et al.24–26 and has been proved to be reliable. For Th atoms, Wang28 et al. verified through research that the 5f orbitals in thorium atoms are empty, so it is reasonable to set the U value of thorium atoms to 0 in our calculations. At the same time, considering that the U atom itself has a certain degree of magnetism, we will consider the spin in the following calculations. It should be pointed out that the lattice parameter of UO2 calculated in this work is 5.462 Å (Table 1), which is consistent with previous experimental value 5.474 (ref. 29) and 5.468 Å.30
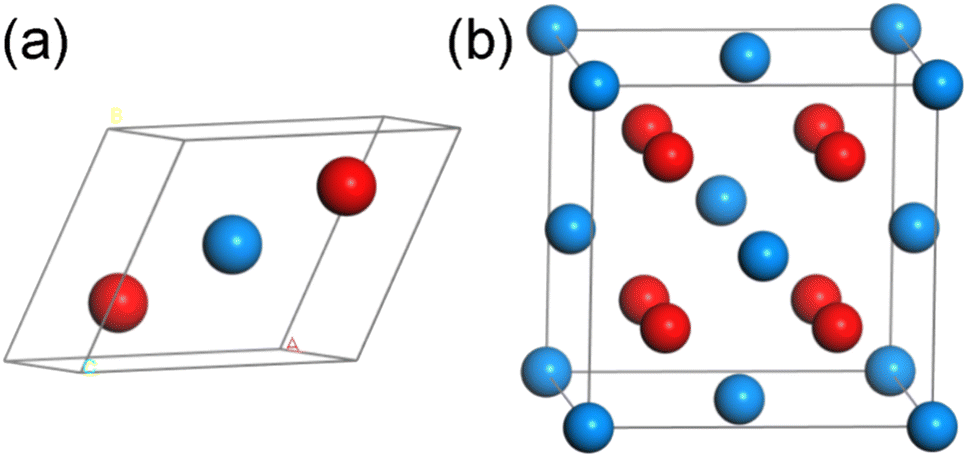 | ||
| Fig. 1 The schematic diagrams for the (a) unit cell and (b) supercell of UO2 (blue atom: U, red atom: O). | ||
| Phase | Lattice parameter (Å) |
|---|---|
| UO2 | 5.462 |
| U3O8Th | 5.473 |
| U2O8Th2 | 5.539 |
| UO8Th3 | 5.550 |
Based on the above optimized primitive cell, we designed four configurations of Th–U–O ternary compounds in the 2 × 2 × 1 supercell by substituting the U and O with Th element. As shown in Fig. 2, they are named U4O7Th, U3O8Th, U2O8Th2 and UO8Th3, and their structures are optimized respectively. Furthermore, as can be seen from Table 1, the lattice constants change very little after alloying and therefore structural distortions are not considered. In the next step, we will calculate and analyze their thermodynamic stability and kinetic stability.
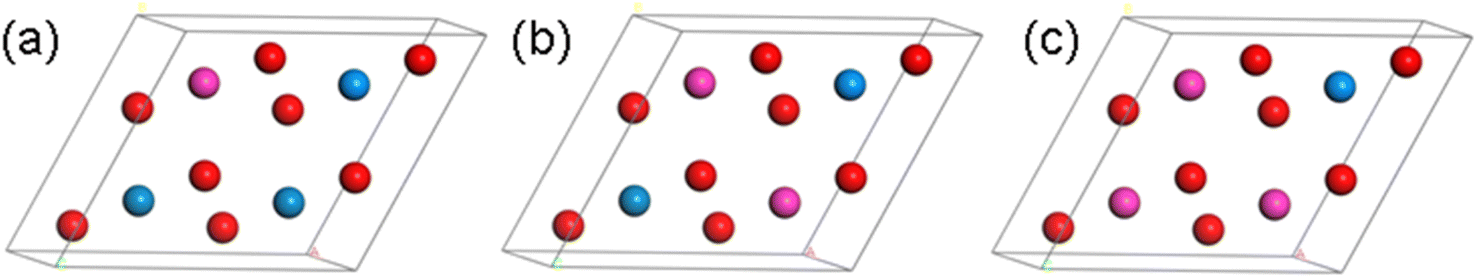 | ||
| Fig. 2 Crystal structure of (a) U3O8Th, (b) U2O8Th2 and (c) UO8Th3 (blue atom: U; red atom: O; pink atom: Th). | ||
3.2. Thermodynamic stability
To judge whether the designed four ternary compounds are thermodynamically stable, the cohesive energy are used to evaluate. The calculation formula of cohesive energy is as follows:12| Ef = Etot − EU4O8 + nETh − nEU/O |
Generally speaking, the cohesive energy of a compound turns to be negative should indicate that the structure is thermodynamically stable under 0 K. While the formation can be positive, indicating this compound is thermodynamically unstable. At the same time, the smaller the negative value is, the more stability the structure possess, and the total energy and cohesive energy of the Th–U–O ternary compounds are listed in Table 1. From the calculation results shown in Table 2, it can be concluded that the cohesive energy of the compound formed by the replacement of the Th atom with one O atom is 4.36 eV, indicates that U4O7Th possesses a thermodynamically unstable structure. Other constructions are thermodynamically stable at 0 K, and as the number of substituted uranium atoms increases, the structural stability is further improved, and the formation energies are −7.50, −9.2 and −11.14 eV, respectively. To further understand their dynamic stability, we calculated the phonon dispersion curves of three thermodynamically stable structures and UO2.
| Phase | Etotal (eV) | ΔH |
|---|---|---|
| U4O7Th | −115.58 | 4.36 |
| U3O8Th | −121.51 | −7.50 |
| U2O8Th2 | −119.52 | −9.21 |
| UO8Th3 | −117.75 | −11.14 |
3.3. Dynamic stability
The phonon dispersion curves of three thermodynamically stable structures are calculated, among which U3O8Th is a dynamically stable structure, U2O8Th2 and UO8Th3 are dynamically unstable structures, they reach negative frequencies of −0.5 and −2.6 in the K-path and Y-path, respectively. The calculation results of U3O8Th are shown in Fig. 3(b). If the phonon dispersion curve appears in the Brillouin region with a frequency lower than 0 THz, that is, “imaginary frequency”, it means that the structure is dynamically unstable.31 By calculating the phonon spectra of UO2 and U–Th–O ternary compounds, it is found that the structures of U2O8Th2 and UO8Th3 have obvious imaginary frequencies. Therefore, we believe that these structures are relatively dynamically unstable. Under the slight influence, the atoms in these structures may shift, that is, the structures may undergo phase changes. Therefore, these dynamically unstable structures will not be considered in the following calculations. The phonon spectrum of U3O8Th do not show any imaginary frequencies, indicating that this structure occupies the lowest point of the energy curve of the compound, so it is the focus of the next calculation.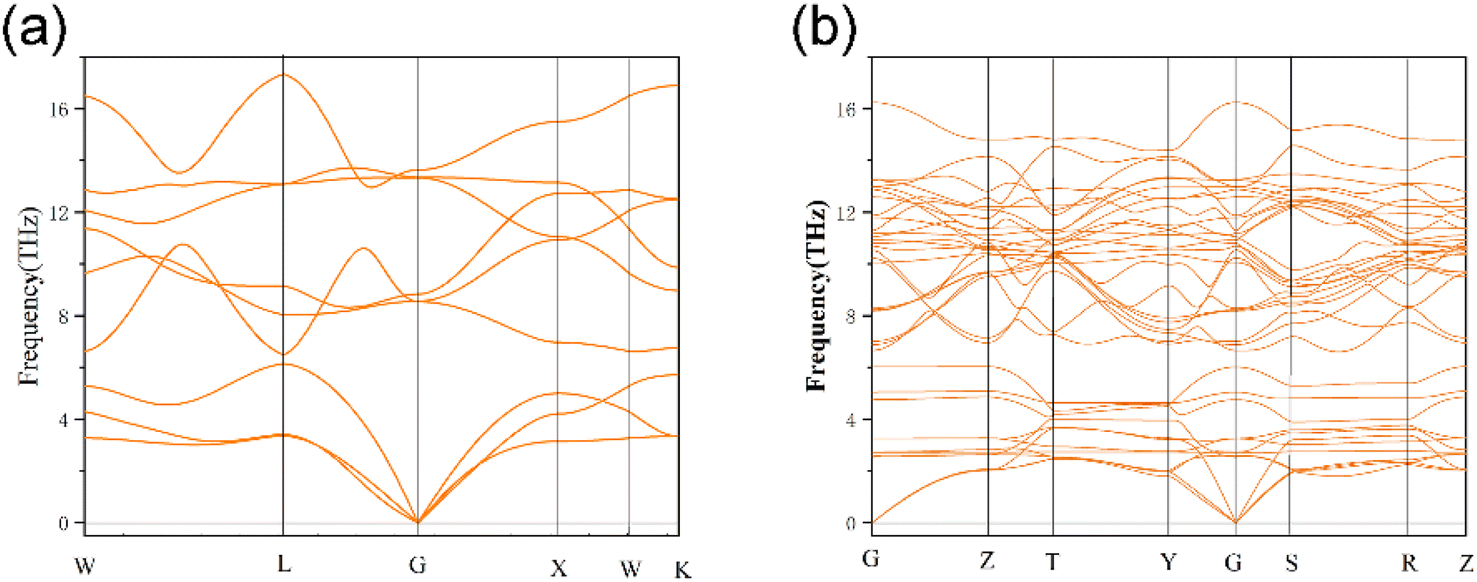 | ||
| Fig. 3 Phonon dispersion curve of (a) UO2 and (b) U3O8Th. | ||
3.4. The density of states including the total (TDOS) and partial (PDOS) parts
In order to further explore the charge distribution and bonding characteristics of U3O8Th, we calculated the total density of states (TDOS) and partial density of states (PDOS) of UO2 and U3O8Th, and the calculation results are displayed in Fig. 4. From the TDOS of UO2 and U3O8Th in Fig. 4, it can be seen that all curves of UO2 and U3O8Th cross the Fermi level, which may be inferred that these two uranium fuels have a certain degree of metallicity. At the same time, the TDOS diagrams of UO2 and U3O8Th show a slight upward and downward asymmetry, indicating that these two structures have certain magnetic properties. The magnetic moment of the uranium atom itself is asymmetrical, hence both structures exhibit a certain degree of magnetism.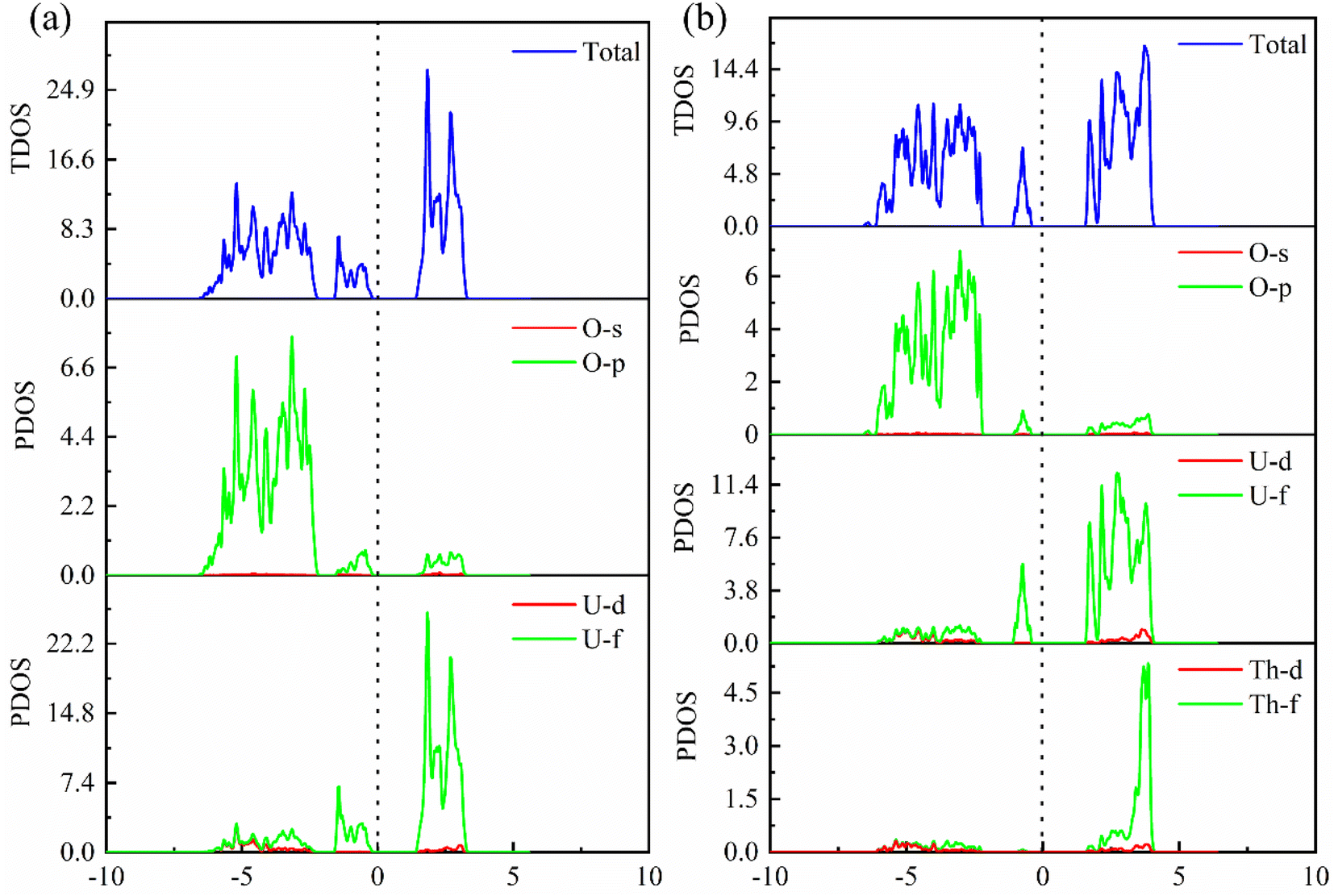 | ||
| Fig. 4 The TDOS and PDOS curves of (a) UO2 and (b) U3O8Th. | ||
The main peak of UO2 is mainly determined by the 5f orbital of the U atom above the Fermi level, while the contribution of the O element can be ignored; in addition, the main peak of the TDOS of U3O8Th is contributed by the 3p orbital of the O element and the 5f orbital of the uranium element while the contribution from Th element is very small. The density of states around the Fermi level of U3O8Th is mainly contributed by the 5f orbital of U atom, the 3p orbital of O atom while the 6d orbital of Th atom contribute a small amount, the density of states around the Fermi level of UO2 is mainly contributed by the 5f orbital of U atom, while the 3p orbital of O atom contributes a small amount. In addition, the TDOS of UO2 shows that the s-, p-orbitals of O atoms and the 5f orbitals of U atoms have peaks at −3 eV at the same time, indicating that O atoms have obvious hybrid bonding effects, while in U3O8Th at −5 eV, all three kinds of atoms have peaks, which proves that Th atoms form new bonds with O atoms.
3.5. Mechanical properties
The condition stability of nuclear fuel is very important, and the material may appear fracture failure with the action of the mechanical and thermal stresses. Therefore, there are certain requirements for mechanical anisotropy. To further understand the changes in the mechanical anisotropy of the compound after the addition of Th atoms, we use the three-dimensional Young's modulus diagram to more intuitively show the anisotropic changes in elasticity. For cubic crystal form and orthorhombic crystal form, the calculation formula of the three-dimensional Young's modulus diagram is as follows:32| Eo = 1/(l14S11+2l12l22S12+2l12l32S13 + l24S22+2l22l32S23 + l34S33 + l22l32S44 + l12l32S55 + l12l22S66) |
The calculation result is shown in Fig. 5, and the two-dimensional projection of the three-dimensional Young's modulus diagram in the three directions of (100), (010) and (001) is also added. For the length of each axial direction of the Young's modulus for the nuclear fuel, the more its shape tends to be circular, the more mechanical isotropy turns up, and vice versa. From the overall point of view, the two compounds are cylindrical, indicating that the addition of Th almost has no effect on the overall Young's modulus of UO2 and maintains its mechanical properties to a certain extent.
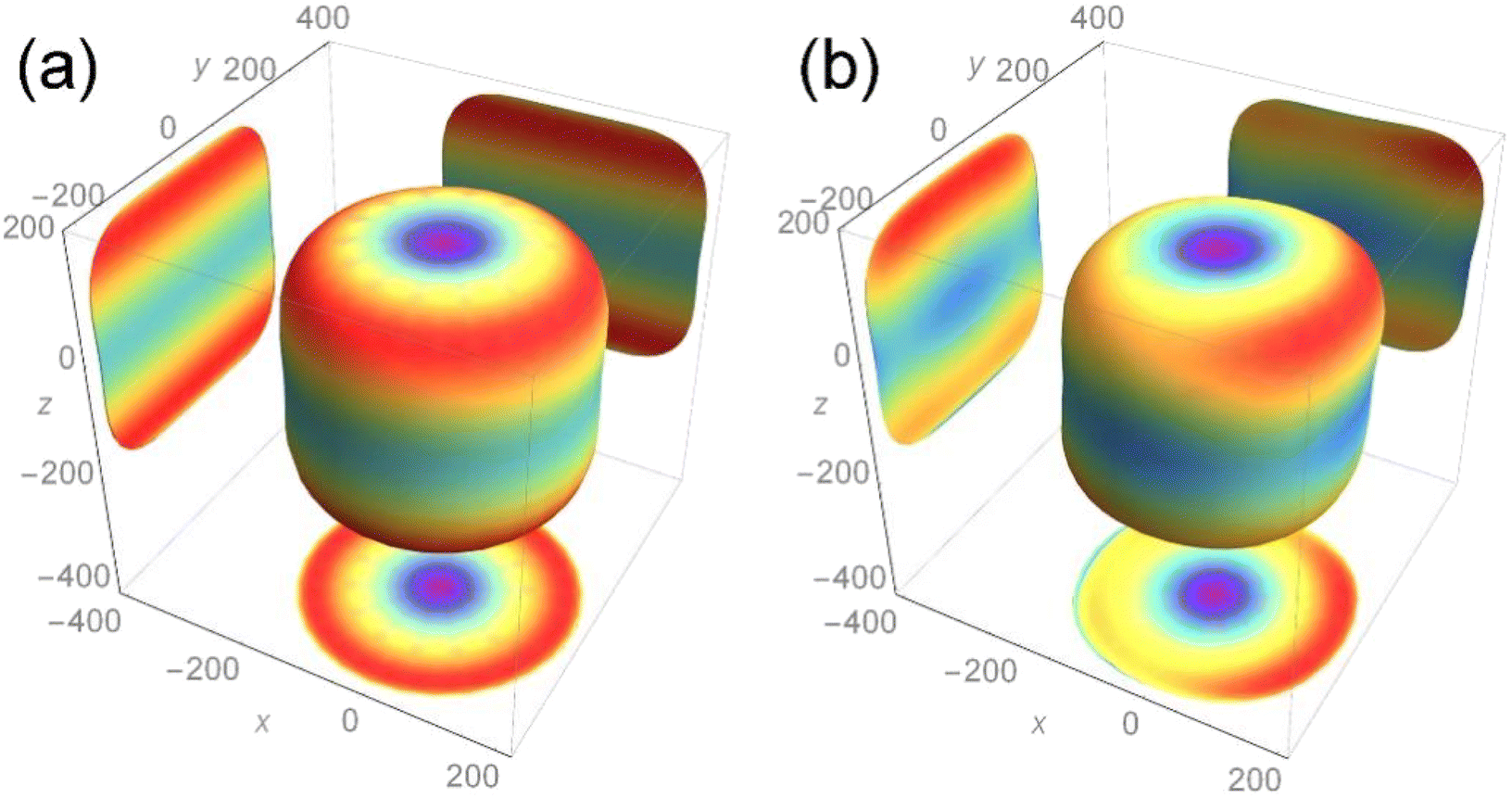 | ||
| Fig. 5 The three-dimensional Young's modulus for (a) UO2 and (b) U3O8Th. | ||
It can be seen from the projection diagram that the projections of the two compounds on the x–y plane are circular, but U3O8Th is slightly elliptical compared to UO2, indicating that Young's modulus of U3O8Th in the 001 direction is A slight change. At the same time, the projections of the two compounds upon the x–z and y–z planes are square, in addition, the maximum Young's modulus of the two configurations appears in the (011) and (101) directions. The maximum Young's modulus of U3O8Th reaches 200 GPa, which is almost equal to that of UO2, and the calculated results show that the formation of U–Th–O compound formed with the Th element doped in the U–O compound will not reduce Young's modulus for the fuel. Hence, we can conclude that the U–Th–O compound designed in this work behaves good mechanical behavior and isotropy of the mechanical property.
4. Conclusion
In this work, the thermodynamic stability and kinetic stability of U–Th–O ternary compounds were carried out to screen the promising new stable configurations. The calculation results of the density of states including total and partial parts indicated that obvious orbital hybridization emerges between the addition of Th and O atoms at −5 eV. The three-dimensional Young's modulus of the U–Th–O ternary compound demonstrates that the mechanical properties of the Th-doped compound remain highly isotropic, reaching around 200 GPa in all three directions. In the future work, we will focus on the changes in properties such as the thermal conductivity of the U–Th–O ternary compound, hoping to provide a data basis for the application of ternary U–Th–O fuel to the light water reactor.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors acknowledge the financial support from the National Key Research and Development Program of China (no. 2019YFB1901003), National Natural Science Foundation of China (grant no. 21875271 and U20B2021), the Entrepreneurship Program of Foshan National Hi-tech Industrial Development Zone and Zhejiang Province Key Research and Development Program (no. 2019C01060), “Pioneer” and “Leading Goose” R&D Program of Zhejiang (grant no. 2022C01236), Leading Innovative and Entrepreneur Team Introduction Program of Zhejiang (grant no. 2019R01003), and Research Foundation for Talented Scholars of Zhejiang Ocean University (no. 11185091722).References
- J. Roleček, Š. Foral and K. Katovský, et al., A feasibility study of using CeO2 as a surrogate material during the investigation of UO2 thermal conductivity enhancement, Adv. Appl. Ceram., 2017, 116(3), 123–131 CrossRef.
- T. Wiss, J.-P. Hiernaut and D. Roudil, et al., Evolution of spent nuclear fuel in dry storage conditions for millennia and beyond, J. Nucl. Mater., 2014, 451(1–3), 198–206 CrossRef CAS.
- S. J. Zinkle and G. Was, Materials challenges in nuclear energy, Acta Mater., 2013, 61(3), 735–758 CrossRef CAS.
- X. Chen, Y. Qin and D. Shi, et al., First-principles investigations on the anisotropic elasticity and thermodynamic properties of U3Si2–Al, RSC Adv., 2020, 10(58), 35049–35056 RSC.
- X. Chen, Y. Qin and D. Shi, et al., Investigations of the stability and electronic structures of U3Si2–Al: A first-principles study, Chem. Phys., 2021, 543, 111088 CrossRef CAS.
- E. Wu, N. Qiu and K. Luo, et al., The studies of electronic structure, mechanical properties and ideal fracture behavior of U3Si1.75Al0.25: first-principle investigations, J. Mater. Res. Technol., 2021, 15, 1356–1369 CrossRef CAS.
- D. R. Rittman, S. Park and C. L. Tracy, et al., Structure and bulk modulus of Ln-doped UO2 (Ln = La, Nd) at high pressure, J. Nucl. Mater., 2017, 490, 28–33 CrossRef CAS.
- S. C. Finkeldei, J. Kiggans and R. D. Hunt, et al., Fabrication of UO2-Mo composite fuel with enhanced thermal conductivity from sol-gel feedstock, J. Nucl. Mater., 2019, 520, 56–64 CrossRef CAS.
- L. Cheng, R. Gao and Q. Xu, et al., UO2–Mo–Be composites for Accident Tolerant Fuel: SPS fabrication, microcracks-free in as-fabricated state and superior thermal conductivity, Ceram. Int., 2020, 46(18), 28939–28948 CrossRef CAS.
- M. Rahman, M. W. D. Cooper and B. Szpunar, et al., Primary radiation damage on displacement cascades in UO2, ThO2 and (U0.5Th0.5)O2, Comput. Mater. Sci., 2018, 154, 508–516 CrossRef CAS.
- L. Evitts, M. Gilbert and S. Middleburgh, et al., Utilizing neutronics modelling to predict changing Pu ratios in UO2 in the presence of Th, Prog. Nucl. Energy, 2021, 137, 103762 CrossRef CAS.
- D. Shi, Y. Guo and Y. Qin, et al., The mechanical and thermal properties of (Th, U)Si compounds: A systematic density functional theory research, Comput. Mater. Sci., 2021, 188, 110148 CrossRef CAS.
- A. Alemberti, K. Tuček and T. Obara, et al., Status of Generation-IV Lead Fast Reactor Activities, 2017 Search PubMed.
- J. S. Herring, P. E. Macdonald and K. D. Weaver, et al., Low cost, proliferation resistant, uranium–thorium dioxide fuels for light water reactors, Nucl. Eng. Des., 2001, 203(1), 65–85 CrossRef CAS.
- K. Rickert, T. A. Prusnick and E. Hunt, et al., Inhibiting laser oxidation of UO2 via Th substitution, J. Nucl. Mater., 2019, 517, 254–262 CrossRef CAS.
- A. D. Becke, Density-functional thermochemistry I. The effect of the exchange-only gradient correction, J. Chem. Phys., 1992, 96(3), 2155–2160 CrossRef CAS.
- D. R. Kripalani, A. A. Kistanov and Y. Cai, et al., Strain engineering of antimonene by a first-principles study: mechanical and electronic properties, Phys. Rev. B, 2018, 98(8), 085410 CrossRef CAS.
- G. Kresse and D. Joubert, From ultrasoft pseudopotentials to the projector augmented-wave method, Phys. Rev. B: Condens. Matter Mater. Phys., 1999, 59(3), 1758 CrossRef CAS.
- D. R. Kripalani, P.-P. Sun and P. Lin, et al., Strain-driven superplasticity of ultrathin tin (II) oxide films and the modulation of their electronic properties: a first-principles study, Phys. Rev. B, 2019, 100(21), 214112 CrossRef CAS.
- P. E. Blöchl, Projector augmented-wave method, Phys. Rev. B: Condens. Matter Mater. Phys., 1994, 50(24), 17953 CrossRef PubMed.
- J. P. Perdew, K. Burke and M. Ernzerhof, Generalized gradient approximation made simple, Phys. Rev. Lett., 1996, 77(18), 3865 CrossRef CAS PubMed.
- J. T. Pegg, X. Aparicio-Angles and M. Storr, et al., DFT + U study of the structures and properties of the actinide dioxides, J. Nucl. Mater., 2017, 492, 269–278 CrossRef CAS.
- B. Ao, R. Qiu and S.-X. Hu, Plutonium oxidation states in complex molecular solids, J. Phys. Chem. C, 2019, 123(19), 12096–12103 CrossRef CAS.
- E. Torres, I. Cheiknjifon and T. Kaloni, et al., A comparative analysis of the phonon properties in UO2 using the Boltzmann transport equation coupled with DFT + U and empirical potentials, Comput. Mater. Sci., 2020, 177, 109594 CrossRef CAS.
- J. T. Pegg, A. E. Shields and M. T. Storr, et al., Magnetic structure of UO2 and NpO2 by first-principle methods, Phys. Chem. Chem. Phys., 2019, 21(2), 760–771 RSC.
- E. Torres and T. Kaloni, Thermal conductivity and diffusion mechanisms of noble gases in uranium dioxide: a DFT + U study, J. Nucl. Mater., 2019, 521, 137–145 CrossRef CAS.
- H. J. Monkhorst and J. D. Pack, Special points for Brillouin-zone integrations, Phys. Rev. B: Solid State, 1976, 13(12), 5188 CrossRef.
- B.-T. Wang, H. Shi and W.-D. Li, et al., First-principles study of ground-state properties and high pressure behavior of ThO2, J. Nucl. Mater., 2010, 399(2–3), 181–188 CrossRef CAS.
- B. Frazer, G. Shirane and D. Cox, et al., Neutron-Diffraction Study of Antiferromagnetism in UO2, Phys. Rev., 1965, 140(4A), A1448 CrossRef.
- A. Padel and C. De Novion, Constantes elastiques des carbures, nitrures et oxydes d'uranium et de plutonium, J. Nucl. Mater., 1969, 33(1), 40–51 CrossRef CAS.
- M. Born, K. Huang and M. Lax, Dynamical theory of crystal lattices, Am. J. Phys., 1955, 23(7), 474 CrossRef.
- D. Shi, J. Song and Z. Liu, et al., Defective structures in FeCrAl alloys from first principles calculations, Jpn. J. Appl. Phys., 2020, 59(4), 046003 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2023 |