DOI:
10.1039/D3RA00167A
(Paper)
RSC Adv., 2023,
13, 8577-8585
Three new coordination geometries of homoleptic Zn complexes of curcuminoids and their high antiproliferative potential†
Received
9th January 2023
, Accepted 1st March 2023
First published on 15th March 2023
Abstract
To our previously reported first crystal structure of a homoleptic zinc curcuminoid complex with square pyramidal geometry, we add herein three new geometries of homoleptic type complexes i.e. octahedral, trigonal-pyramidal, and trigonal-bipyramidal. Octahedral geometry was observed in the new pseudo-polymorph of the DAC–Zn complex resulting from crystallization in DMF, while square-pyramidal geometry was obtained in DMSO. Improving crystallinity involved suppressing the phenolic interactions by etherification and esterification. The complete characterization of these complexes was carried out using SCXRD, IR, MS, EA, liquid, and solid-state NMR. Moreover, the cytotoxic activity of all complexes was evaluated. The IC50 values for the DiMeOC-Zn (7) complex were 8 or 22 times higher than for cisplatin in the U251 and HCT-15 cell lines, indicating a high antiproliferative and therapeutic potential.
Introduction
Turmeric is the generic name given to the rhizome of the perennial herb Curcuma longa, a spice widely cultivated in India.1 The main biologically active components of turmeric are curcumin, demethoxycurcumin, and bis-demethoxycurcumin, although approximately 13 curcuminoids have been identified from the rhizome.2 Milobedzka and Lampe first proposed the chemical structure of curcumin in 1910, which was isolated for the first time by Vogel and Pelletier.3 In recent years, numerous studies have been performed to better understand the medicinal properties of curcumin, curcuminoids, and their metal complexes, with purported antitumor, antimicrobial, anti-inflammatory, antioxidant, antiviral, anti-Alzheimer, and anti-cancer potential.1,4–7 Curcumin and curcuminoids are chelating agents due to the β-diketone functionality, which allows them to form stable complexes with a series of metal ions.1 In the literature, the biological properties of these metal complexes are described,8–20 although their structural characterization often lacks a detailed description.8,9,13 The relative scarcity of reports on crystal structures of curcuminoid metal complexes has been attributed to a generalized low crystallinity of these compounds, making their characterization impossible by the single crystal X-ray diffraction technique.1,4,8,9,13 This feature is attributable to the formation of polymer matrices and the inherent low solubility in most common organic solvents. In recent works, we reported the crystal structures of various complexes of different curcuminoids and metal ions (Meza-Morales et al., 2019), and described eight crystal structures and two polymorphs of Zn complexes.21,22 The relevance of the presence of Zn as the complexing metal was first suggested from our previous studies by the high in vitro and low in vivo cytotoxicity exerted by the diacetylcurcumin–zinc complex,21 possibly related to the role of Zn(II) in regulating mitochondrial apoptosis as occurs with many mammalian cells.19
The geometries resulting from the complexation of 2,4-pentanedione with Zn are basically i.e., octahedral, square pyramid, and trigonal-bipyramid.23–29 Therefore, part of our interest focuses in the geometries that are obtained when using curcuminoid as ligands.
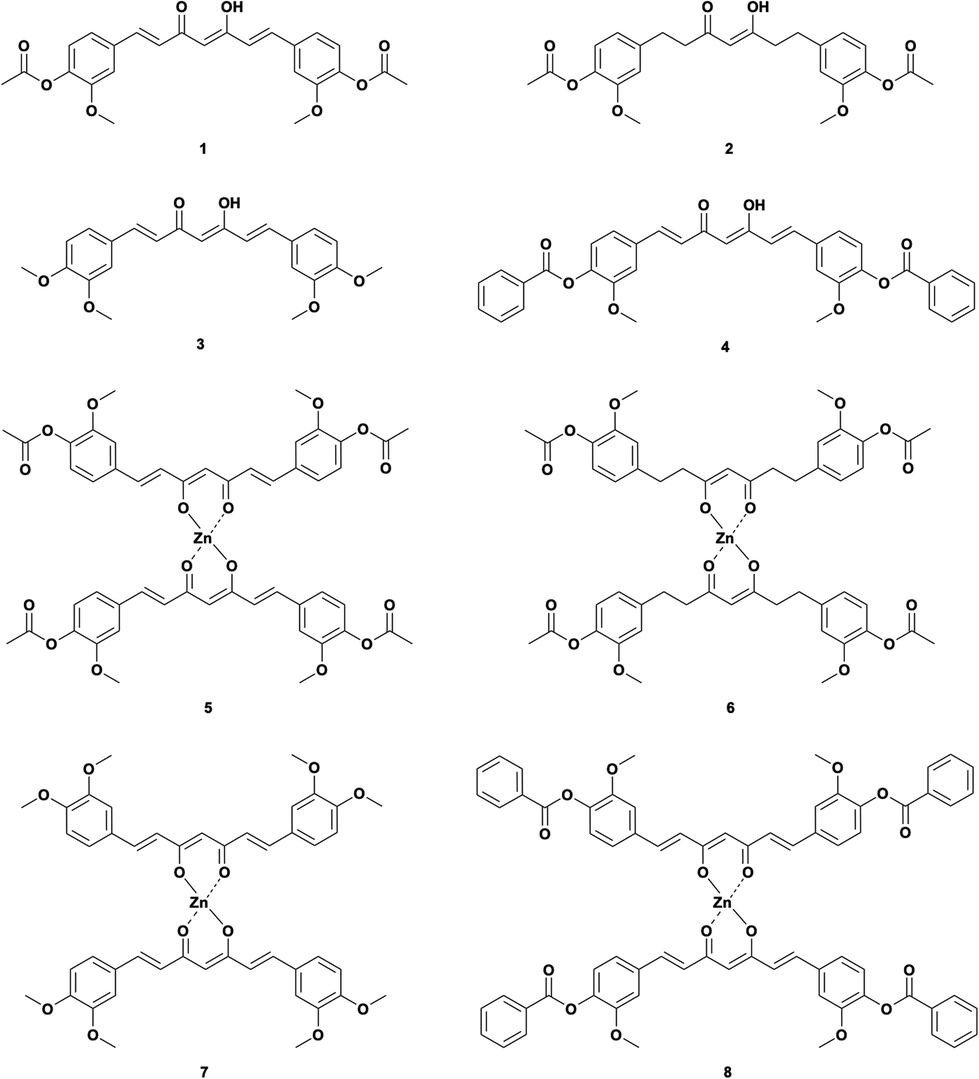
Thus, we used several curcuminoids as ligands for complexation with zinc(II), i.e., acetylated curcumin (1,7-bis(3-methoxyl-4-acetoxyphenyl)-1,6-heptadiene-3,5-diketone, DAC) 1, hydrogenated acetylated curcumin (1,7-bis(3-methoxyl-4-acetoxy)-phenol-1,6-heptane-3,5-diketene, DACH) 2, methoxylated curcumin (1,7-bis(3,4-dimethoxyphenyl)-1,6-heptadiene-3,5-diketone, DiMeOC) 3 and benzoylated curcumin (1,7-bis(3-methoxy 4-benzoatephenyl)-1,6-heptadiene-3,5-diketone, DiBzOC) 4 (Fig. 1). The characterization of all synthesized homoleptic complexes was carried out using IR, NMR in liquid and solid state, MS, EA as well as single crystal X-ray diffraction techniques. Also, a preliminary profile of the biological activity of these complexes was obtained through a cytotoxicity assay in vitro using several cancerous cell lines.
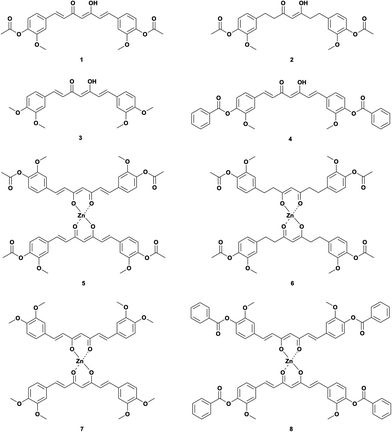 |
| | Fig. 1 Curcuminoids and their zinc complexes. | |
Results and discussion
NMR liquid state
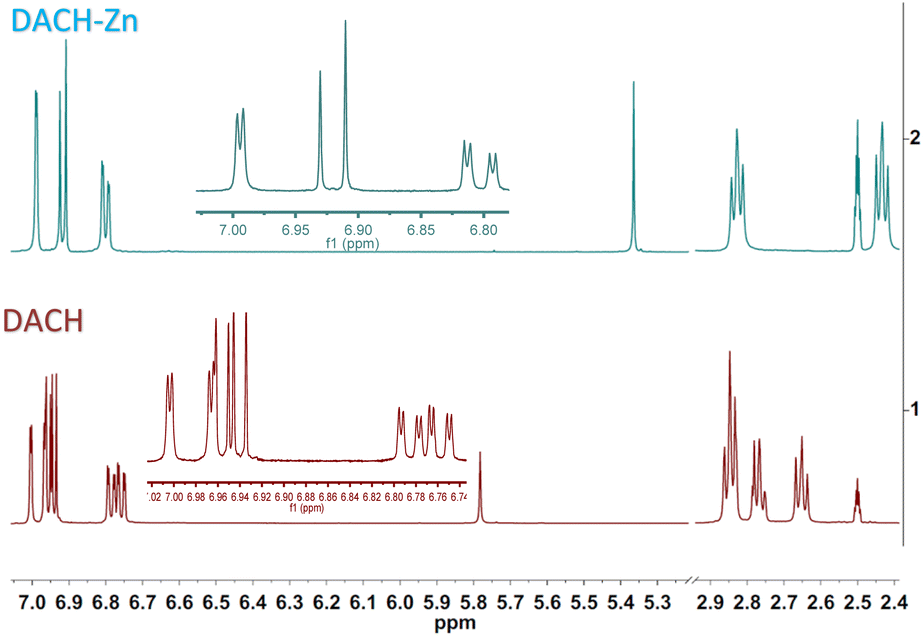
The 1H NMR spectrum of ligand DAC (1) shows one singlet for the OH proton at 16.12 ppm (strong intramolecular hydrogen bond) and one singlet for the methine proton at 6.20 ppm (vinylic proton). Protons α to the diketone appear at 6.99 ppm, and protons β to the diketone at 7.66 ppm, with a trans coupling constant of 15.9 Hz. Methoxyl and acetyl protons appear as singlets at 3.85 ppm and 2.28 ppm, respectively. The 1H NMR spectrum of DACH (2) shows keto–enol equilibrium with a ca. 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 ratio. The enol tautomer shows one broad singlet for the OH proton at 15.53 ppm and a narrow singlet for the methine proton at 5.78 ppm; both protons are involved in a strong intramolecular hydrogen bridge. The keto tautomer shows one singlet for the methylene proton at 3.74 ppm. Protons α to the diketone group appear at 2.65 and 2.78 ppm, while protons β appear at 2.85 ppm. Methoxyl and acetyl protons appear as singlets at 3.74 ppm and 2.23 ppm, respectively (Fig. 2). The 1H NMR spectrum of DiMeOC (3) shows one singlet for the OH proton at 16.25 ppm and one singlet for the methine proton at 6.10 ppm; both protons are involved in a strong intramolecular hydrogen bond (enol tautomer). Protons α to the diketone appear at 6.82 ppm, and protons β to the diketone at 7.59 ppm, with a trans coupling constant of ca. 15.9 Hz. Methoxyl protons are singlets at 3.81 ppm and 3.83 ppm. The 1H NMR spectrum of DiBzOC (4) shows one singlet for the methine proton at 6.24 ppm. Unsaturated protons α to the diketone group appear at 7.04 ppm and the corresponding β protons at 7.70 ppm, with a trans coupling constant of ca. 15.9 Hz. Methoxyl protons are singlets at 3.85 ppm. Aromatics ring protons are from 7.39 to 8.13 ppm.
:1 ratio. The enol tautomer shows one broad singlet for the OH proton at 15.53 ppm and a narrow singlet for the methine proton at 5.78 ppm; both protons are involved in a strong intramolecular hydrogen bridge. The keto tautomer shows one singlet for the methylene proton at 3.74 ppm. Protons α to the diketone group appear at 2.65 and 2.78 ppm, while protons β appear at 2.85 ppm. Methoxyl and acetyl protons appear as singlets at 3.74 ppm and 2.23 ppm, respectively (Fig. 2). The 1H NMR spectrum of DiMeOC (3) shows one singlet for the OH proton at 16.25 ppm and one singlet for the methine proton at 6.10 ppm; both protons are involved in a strong intramolecular hydrogen bond (enol tautomer). Protons α to the diketone appear at 6.82 ppm, and protons β to the diketone at 7.59 ppm, with a trans coupling constant of ca. 15.9 Hz. Methoxyl protons are singlets at 3.81 ppm and 3.83 ppm. The 1H NMR spectrum of DiBzOC (4) shows one singlet for the methine proton at 6.24 ppm. Unsaturated protons α to the diketone group appear at 7.04 ppm and the corresponding β protons at 7.70 ppm, with a trans coupling constant of ca. 15.9 Hz. Methoxyl protons are singlets at 3.85 ppm. Aromatics ring protons are from 7.39 to 8.13 ppm.
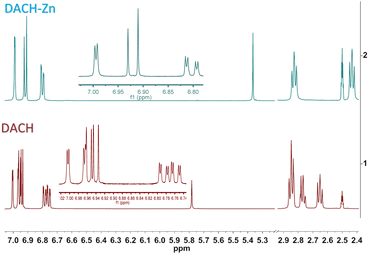 |
| | Fig. 2 NMR liquid state of DACH and DACH-Zn. | |
The 1H NMR spectrum of ligand DAC-Zn (5) shows one singlet for the methine proton at 5.84 ppm (vinylic proton). Protons α to the diketone appear at 6.91 ppm, and protons β to the diketone at 7.53 ppm, with a trans coupling constant of 15.7 Hz. Methoxyl and acetyl protons appear as singlets at 3.83 ppm and 2.26 ppm, respectively. The 1H NMR spectrum of DACH-Zn (6) shows the enol tautomer. The one singlet signal for the methine proton is at 5.37 ppm. Protons α to the diketone group appear at 2.44 ppm, while protons β appear at 2.83 ppm. Methoxyl and acetyl protons appear as singlets at 3.73 ppm and 2.21 ppm, respectively (Fig. 2). It can be appreciated (Fig. 2) that in the 1H NMR spectra of DACH, the former approaches a near 1:1 keto–enol equilibrium, while in the DACH-Zn such equilibrium is completely shifted to the enolic complexed form. The 1H NMR spectrum of DiMeOC-Zn (7) shows one singlet for the methine proton at 5.76 ppm. Protons α to the diketone appear at 6.77 ppm, and protons β to the diketone at 7.46 ppm, with a trans coupling constant of ca. 15.6 Hz. Methoxyl protons are singlets at 3.82 and 3.79 ppm. The 1H NMR spectrum of DiBzOC-Zn (8) shows one singlet for the methine proton at 5.90 ppm. Unsaturated protons α to the diketone group appear at 6.98 ppm with a trans coupling constant of ca. 15.7 Hz. Methoxyl protons are singlets at 3.84 ppm. The methine protons of complexes consistently appear at lower frequencies, vinyl, and aromatic groups of metal complexes is presumably because DMSO is a highly coordinating molecule (being part of the complex)1 and thus provides electronic density, which produces a shielding effect at the coordination region (Table 1).
Table 1 1H NMR chemical shifts of the methine proton of ligand and their zinc complexes
| Compounds |
δ (ppm) of methine in DMSO-d6 |
| DAC |
6.20 |
| DACH |
5.78 |
| DiMeOC |
6.10 |
| DiBzOC |
6.24 |
| DAC-Zn |
5.84 |
| DACH-Zn |
5.37 |
| DiMeOC-Zn |
5.76 |
| DiBzOC-Zn |
5.90 |
NMR solid state
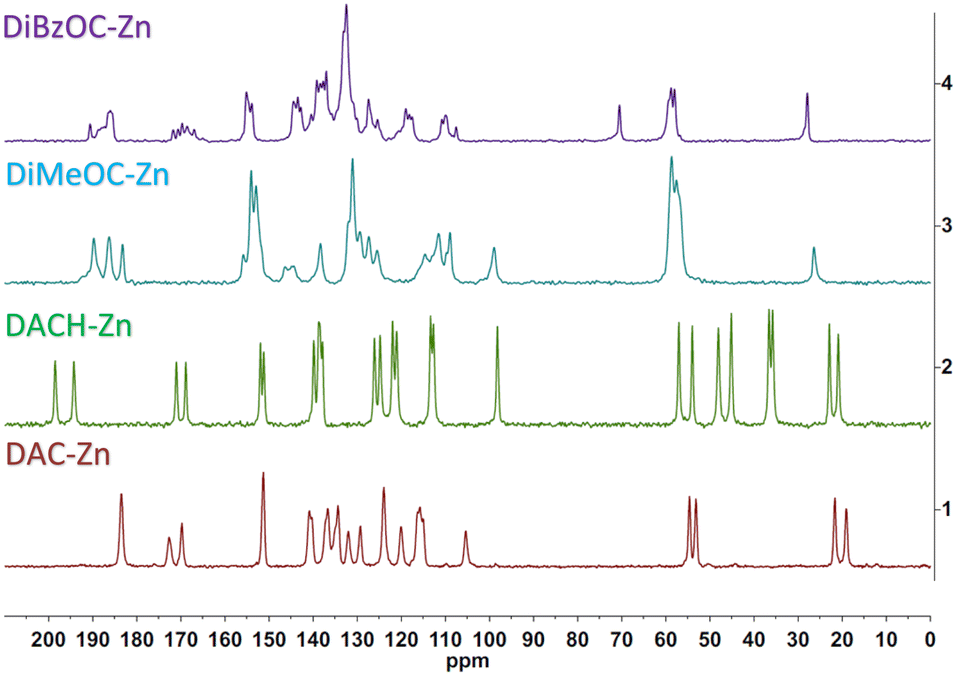
The Cross Polarized Magic Angle Spinning (CP-MAS) 13C NMR spectrum of ligand DAC and its complex is already described in our previous work.22 The carbonyls overlap into a single signal, which can be attributed to an increased symmetry at the plane of coordination about the metal atom; a different behavior was found for the DiMeOC-Zn (7) and DiBzOC-Zn (8) complexes, observing three singlets for the carbonyls of the dicarbonyl system of DiMeOC-Zn (7) and a group of signals for DiBzOC-Zn (8), indicating that the geometry found in the SCXRD structures and the signals found for the carbonyls in ssNMR can be related to some extent (Fig. 3). In the case of DACH-Zn, two signals are observed for the carbonyl groups of β-diketone, indicating a loss of symmetry in the coordination plane concerning DAC-Zn, which shows a singlet for these four carbons.
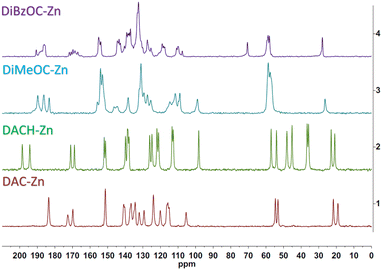 |
| | Fig. 3 ssNMR of zinc complexes. | |
SCXRD
The curcuminoid ligands used afforded four homoleptic bis-chelated complexes upon complexation with zinc(II). Although all crystal structures contain DMF molecules, crystallization yields uncoordinated (7), mono-coordinated (8), and two-coordinated DMF (5a) crystal structures giving rise to three different coordination geometries. Their crystallographic data are summarized in Table S1,† and selected distances and angles are in Table 2.
Table 2 Selected distances and angles of selected bonds
| Comp. |
5a |
5b |
7 |
8 |
| 1 − X, −Y, 1 − Z. 1 − X, +Y, 3/2 − Z. Oxygen atom in part B, *degrees. |
| Zn–Oequatorial (Å) |
1.986(4) |
2.021(2) |
1.938(3) |
2.0355(16) |
| 1.986(4)a |
1.987(2) |
1.938(3)b |
1.9909(16) |
| 2.037(4) |
2.031(2) |
1.930(3) |
1.9830(16) |
| 2.037(4)a |
1.991(2) |
1.930(3)b |
2.0216(15) |
| Zn–Oaxial (Å) |
2.252(6) |
2.014(2) |
— |
2.056(5) |
| 2.252(6)a |
2.054(7)c |
| O–Zn–Ochelate (°) |
90.60(16) |
89.50(10) |
97.68(18) |
91.35(7) |
| 90.60(16) |
88.97(10) |
97.73(18) |
91.48(6) |
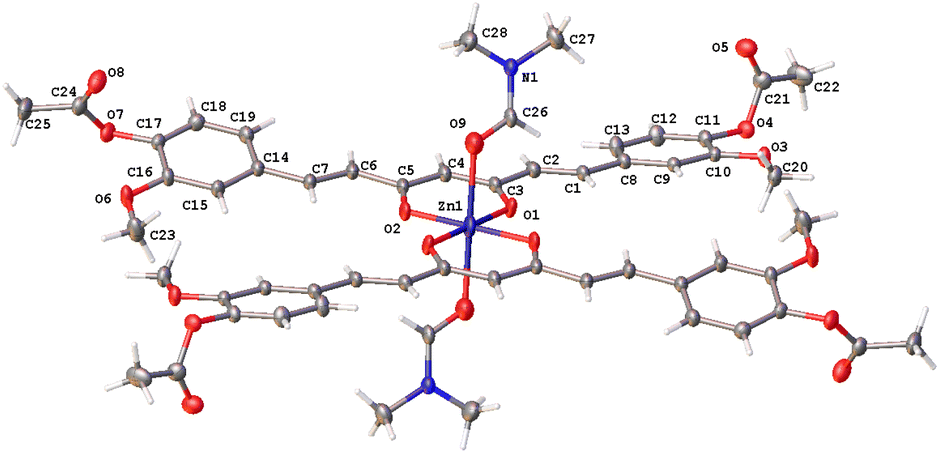
The curcuminoids framework is fully extended and essentially planar in the three complexes with specific orientations of the phenyl rings. The methoxyl substituents of complexes 5a and 7 point to the same side, while in complex 8, both groups point to opposite sides with respect to the conjugated planar (heptanoid) chain fragment.
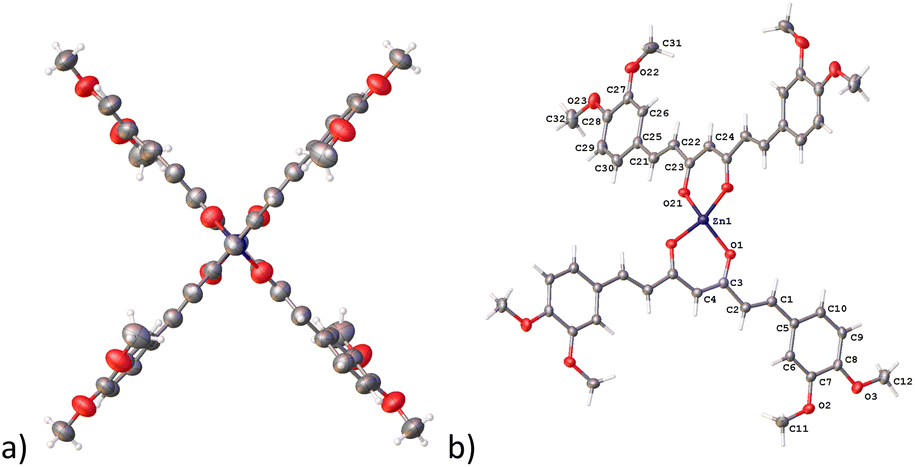
In complex 7, the Zn atom resides in a position that defines a twofold axe with a nearly perfect D2d point group (Fig. 4a). Such arrangement defines a geometry around the zinc atom best described as a distorted trigonal pyramid with a τ4 value of 0.84 supporting the assignment (Fig. 4b).30
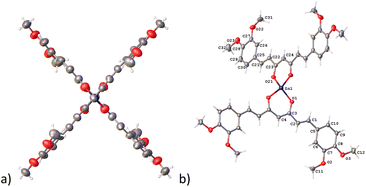 |
| | Fig. 4 Compound 7, thermal ellipsoids at 50% probability level. (a) D2d point group and (b) trigonal pyramid geometry. | |
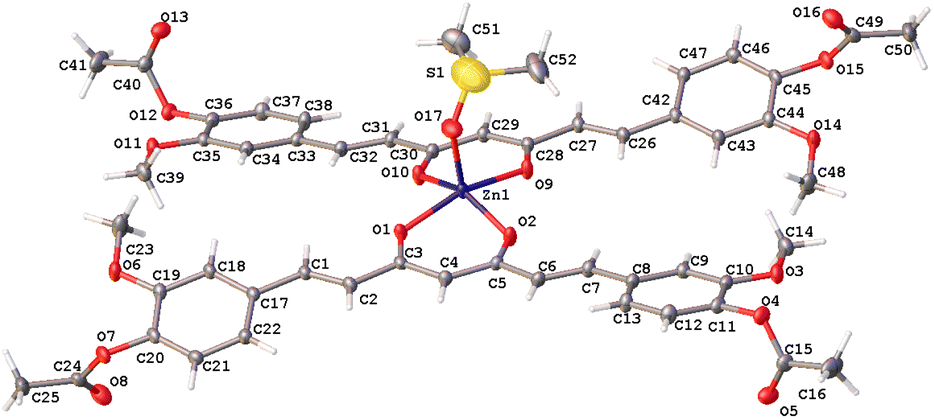
Complex 8 has a pentacoordinate Zn(II) ion that adopts a distorted trigonal-bipyramidal geometry where O2, O9, and O17 oxygen atoms define the base of the pyramid and O1, O10 oxygen atoms, the apical positions of this polyhedron (τ5 = 0.85) (Fig. 5).31
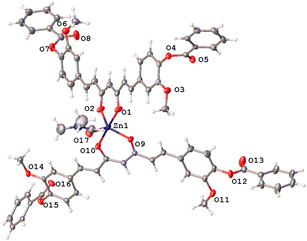 |
| | Fig. 5 Thermal ellipsoids of compound 8, at 50% probability level. | |
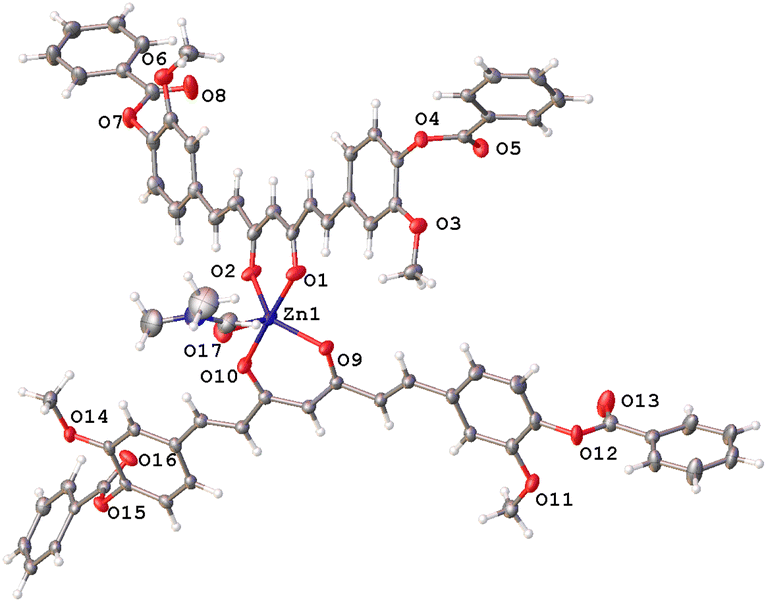
The hexacoordinated zinc complex 5a is shown in Fig. 6 and has different axial and equatorial bonds. Thus, the length of the equatorial Zn–Ocurcuminoid bond varies within 1.986(3)–2.037(5) Å, while the axial Zn–ODMF bond is 2.252(6). Angular deviations from the ideal octahedron (∼6°) correspond to an inclination of the axial bonds referred to as the basal plane. Interestingly, complex 5a exhibits the same ligands as in the DAC–zinc(II) complex reported by us and numbered herein 5b for comparison purposes.22 The coordination geometry of complex 5b corresponds to a square pyramidal conformation (τ5 = 0.16, Fig. 7), while 5a is octahedral (Fig. 6). The O–Zn–O bite angles of oxygens atom from the diketone chelate vary significantly in compound 7 (97.68°), while in the other compounds, such angles go from 89.47 to 91.48°.
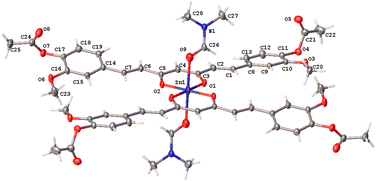 |
| | Fig. 6 Thermal ellipsoids of compound 5a at 50% probability level. | |
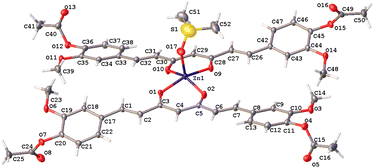 |
| | Fig. 7 Thermal ellipsoids of compound 5b at 50% probability level. | |
Non-covalent interactions
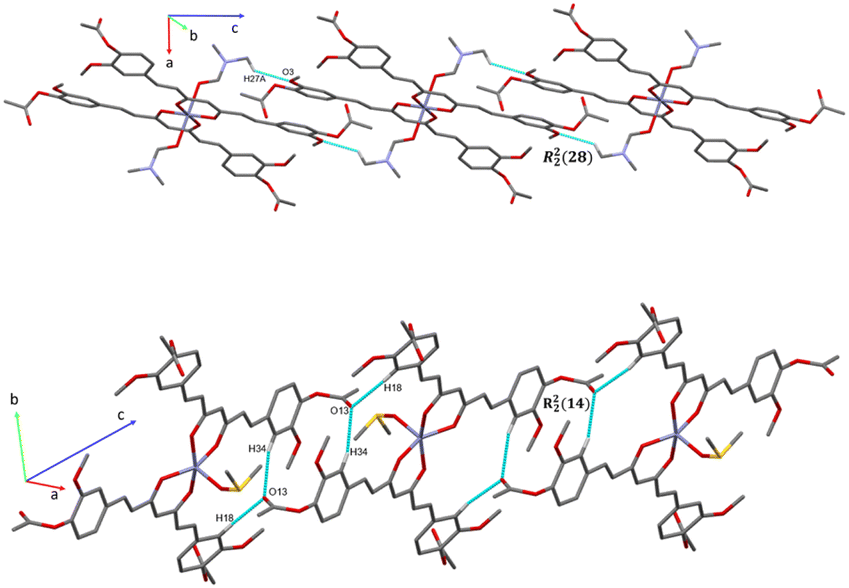
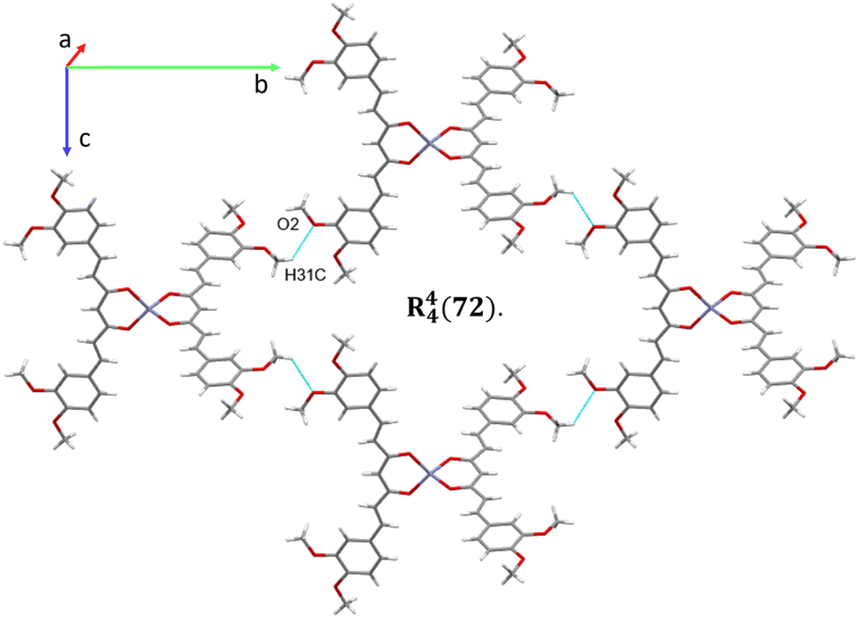
The analysis of the crystallographic arrangement is based on CH⋯O and CH⋯π types of interactions and the distances of non-covalent bonds are summarized in Table 3. The interactions involving the curcuminoid moieties fall within previously reported values.32–34 Compound 5a shows a polymeric interaction mediated through C15H15⋯O5 (2.631 Å) and C27H27A⋯O3 (2.537 Å) between a methoxy group and coordinated dimethylformamide R22(28) (Fig. 8, upper trace). Furthermore, 5b forms a polymeric species by two bifurcated hydrogen bonding between two phenyl rings and an oxygen atom of the ester group, forming two supramolecular rings of 14 and 16 members [R22(14) and R12(16)] (Fig. 8, lower trace). Compound 7 forms a conspicuous 2D mesh and defines supramolecular tetrameric cycles of 72 members [R44(72)] within the crystal lattice through C13H13C⋯O2 (2.620 Å) interactions with the two methoxy groups (Fig. 9). A similar effect was found for compound 8 with C–H⋯O and C–H⋯π interactions forming rings of 20 and 52 members [R22(20)] and [R22(52)], respectively (Fig. S1†).
Table 3 No covalent interaction in compoundsa
| Comp. |
Interaction |
D–H⋯A (Å) |
D⋯A (Å) |
D–H–A (°) |
Symmetry |
| Cg = C70, C71, C72, C73, C74, C75. |
| 5a |
C15H15⋯O5 |
2.631 |
3.495 |
154.74 |
x, y, 1 + z |
| C27H27A⋯O3 |
2.537 |
3.274 |
133.68 |
1 − x, −y, 1 − z |
| 5b |
C9H9⋯O5 |
2.658 |
3.506 |
151.91 |
1 − x, 2 − y, 1 − z |
| C43H43⋯O5 |
2.662 |
3.540 |
157.73 |
1 − x, 2 − y, 1 − z |
| C18H18⋯O13 |
2.631 |
3.531 |
163.05 |
1 − x, 1 − y, 2 − z |
| C34H34⋯O13 |
2.690 |
3.538 |
151.85 |
1 − x, 1 − y, 2 − z |
| 7 |
C13H13C⋯O2 |
2.620 |
3.315 |
129.56 |
1 − x, 1 + y, 1/2 − z |
| 8 |
C33H33⋯Cg |
2.829 |
3.711 |
154.82 |
2 − x, −y, 2 − z |
| C25H25⋯O3 |
2.545 |
3.197 |
125.93 |
2 + x, −1 + y, z |
| C73H73⋯O14 |
2.553 |
3.203 |
125.79 |
−2 + x, −1 + y, 1 + z |
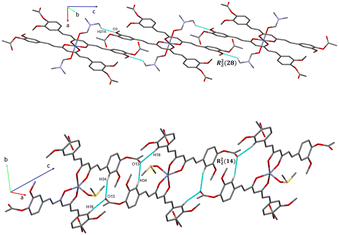 |
| | Fig. 8 Intermolecular C–H⋯O interaction in compound 5a (upper trace) showing a polymeric arrangement and a bifurcate C–H⋯O interaction for compound 5b (lower trace) forming two rings extended for 1D chain (axe C). Note: Hydrogen atoms not involved in hydrogen bonds were omitted for clarity. | |
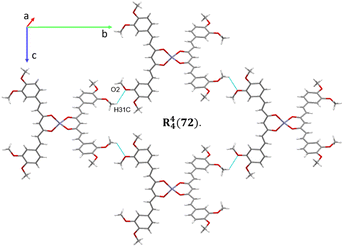 |
| | Fig. 9 2D supramolecular interactions of compound 7 supported via CH⋯O forming a large macrocycle R44(72). | |
Hirshfeld surface
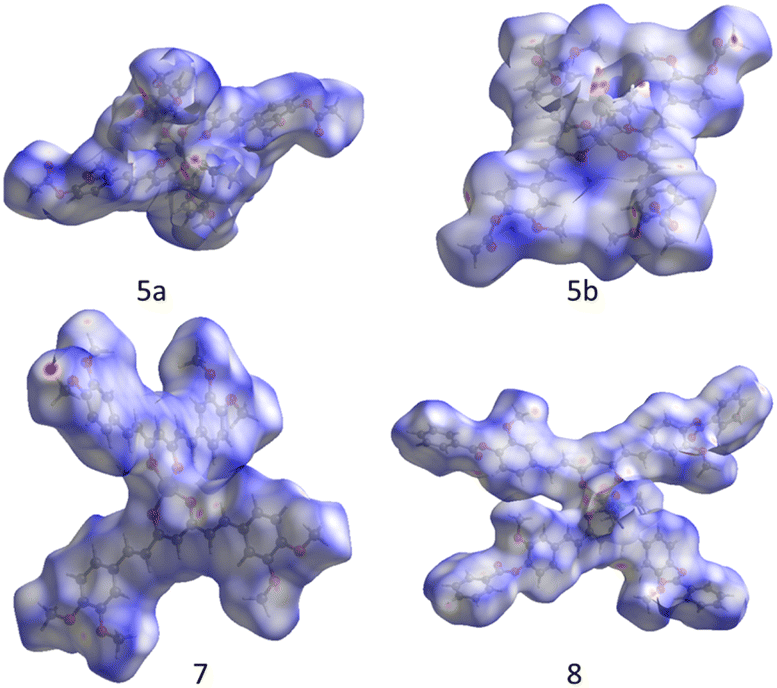
A deeper description of intermolecular interactions was achieved by visually analyzing the Hirshfeld surface to distinguish strong from weak interactions.35 This renders a picture of the molecular shape within the crystalline environment, which enhances similarities and differences between neighboring molecules in all curcuminoid compounds studied. We employed CrystalExplorer software based on a single crystal from CIF files to perform both analyses.36
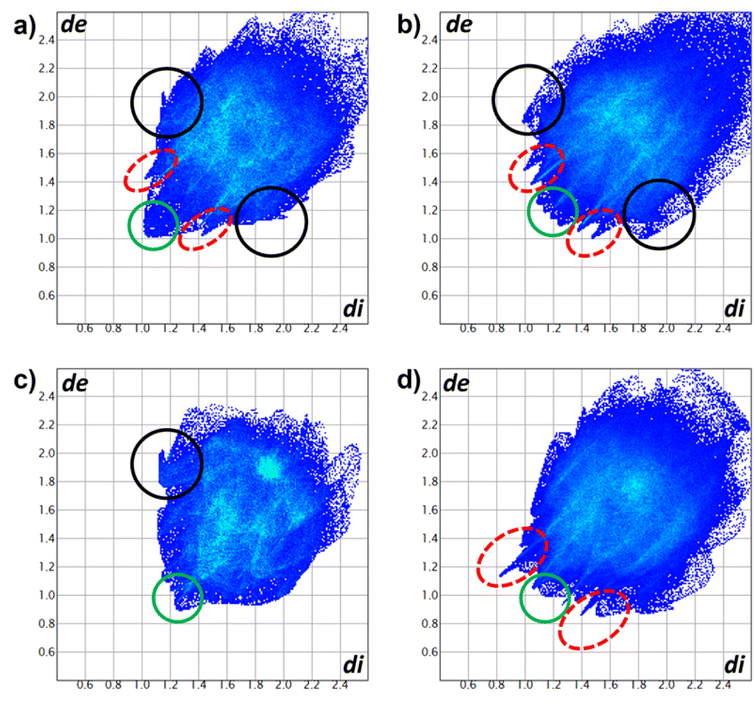
The Hirshfeld surfaces of compounds (Fig. 10) were mapped using the dnorm parameter. Solvent molecules were excluded in all cases. In the special case of compound 8, the original disorder from the crystal structure was considered to avoid nonrelevant contacts with neighboring molecules. The red regions (closest contacts) found over oxygen atoms and blue regions (van der Waals contacts) were predominant. 2D fingerprints plots show, in most cases, the characteristic of C⋯H/H⋯C contact (dark circle) or two symmetric peaks corresponding to O⋯H/H⋯O (red dashed ellipsoid) (Fig. 11). The greater percentages of contacts originate from H⋯H, followed by C⋯H/H⋯C and O⋯H/H⋯O contacts. In contrast, other contacts represent nearly 7 percent (Tables 4 and S2†).
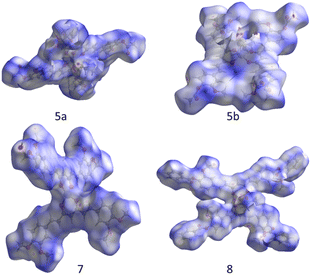 |
| | Fig. 10 Hirshfeld surface over dnorm for compounds. | |
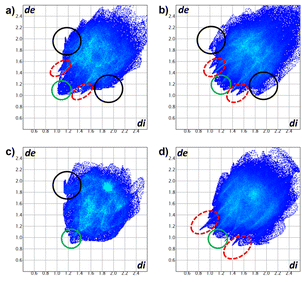 |
| | Fig. 11 2D fingerprints of compounds (a) 5a, (b) 5b, (c) 7, and (d) 8. O⋯H/H⋯O (red dashed ellipsoids), C⋯H/H⋯C (black circle) and H⋯H (green circle) contacts. | |
Table 4 Percentage of contacts in compounds
| Compound |
O⋯H/H⋯O |
C⋯H/H⋯C |
H⋯H |
Other |
| 5a |
28.4% |
20.8% |
46.2% |
4.6% |
| 5b |
30.1% |
21.5% |
42.1% |
6.3% |
| 7 |
22.3% |
21.3% |
50.0% |
6.4% |
| 8 |
24.6% |
26.3% |
42.3% |
6.8% |
Cytotoxic activity
DiMeOC (3) is an important ligand possessing higher cytotoxicity than curcumin, a noteworthy fact since the phenolic groups are blocked.21 A significant decrease in cytotoxic activity occurs from DAC (1) to DACH (2), indicating the importance of conjugated double bonds in the heptanoid fragment of curcumin or curcuminoids. In general, a significant increase in the cytotoxic activity of the zinc complexes 5–7 with respect to the free ligands is observed (Table 5). DACH-Zn (6) and DiBzOC-Zn (8) are the only complexes with lower cytotoxicity than curcumin.
Table 5 Cytotoxic screening of ligands and their zinc complexes 1–8a
| Compounds |
% of growth inhibition by the cell line |
| U251 |
PC-3 |
K562 |
HCT-15 |
MCF-7 |
SKLU-1 |
|
| NC: no cytotoxic. |
| [ ] 25 μM |
| CurCu |
67.1 |
94.4 |
80.1 |
80.0 |
82.4 |
94.5 |
|
| DAC |
46.2 |
77.5 |
67.8 |
46.9 |
41.7 |
41.7 |
|
| DACH |
22.8 |
26.1 |
39.4 |
31.1 |
7.2 |
7.2 |
|
| DiMeOC |
92.6 |
100 |
89.4 |
96.9 |
100 |
100 |
|
| DiBzOC |
1.2 |
4.1 |
11.8 |
NC |
NC |
NC |
|
| DAC-Zn |
91.5 |
100 |
82.8 |
84.44 |
100 |
100 |
|
| DACH-Zn |
NC |
60.5 |
55.7 |
57.3 |
49.0 |
48.5 |
|
| DiMeOC-Zn |
91.0 |
100 |
83.3 |
94.0 |
86.2 |
86.3 |
|
| DiBzOC-Zn |
NC |
13.3 |
12.5 |
NC |
NC |
3.8 |
|
These results show that complexes (5) and (7) have significant cytotoxic effects against cell lines U251, PC-3, K562, HCT-15, MCF-7, and SKLU-1 (Table 6). Furthermore, the IC50 values of the two homoleptic complexes DAC-Zn and DiMeOC-Zn are similar to or lower than cisplatin and much lower than their corresponding free ligands. Thus, the DiMeOC-Zn (7) complex shows 8 and 22 times higher potency than cisplatin in the U251 and HCT-15 cell lines, respectively. These results agree with several studies of antitumoral activity with homoleptic metal complexes of curcuminoids, where authors suggest that the metal presence in the complex preserves the therapeutic potential of curcuminoids.21,22
Table 6 IC50 (μM) for cancer cell lines with compounds 1–8 and cisplatin
| Compounds |
U251 |
PC-3 |
K562 |
HCT-15 |
MCF-7 |
SKLU-1 |
| Curc |
20.5 ± 1.7 |
22.5 ± 1.9 |
16.4 ± 0.04 |
21.3 ± 0.9 |
18.4 ± 0.9 |
12.9 ± 0.7 |
| DAC |
24.8 ± 2.4 |
33.3 ± 2.3 |
15.2 ± 0.4 |
21.0 ± 1.3 |
27.2 ± 2.2 |
16.6 ± 2.0 |
| DACH |
— |
60.0 ± 4.5 |
44.9 ± 2.1 |
48.0 ± 1.0 |
68.0 ± 6.0 |
— |
| DiMeOC |
2.02 ± 0.07 |
5.9 ± 0.6 |
6.5 ± 1.4 |
6.6 ± 0.3 |
6.9 ± 1.0 |
7.2 ± 0.4 |
| DAC-Zn |
11.0 ± 0.8 |
14.9 ± 1.3 |
7.4 ± 0.9 |
7.3 ± 0.9 |
5.4 ± 0.5 |
5.1 ± 0.5 |
| DACH-Zn |
— |
— |
33.2 ± 1.7 |
36.3 ± 1.5 |
38.4 ± 0.6 |
— |
| DiMeOC-Zn |
0.6 ± 0.04 |
1.7 ± 0.2 |
4.4 ± 0.7 |
0.46 ± 0.09 |
3.4 ± 0.4 |
10.4 ± 1.1 |
| Cisplatin |
4.7 ± 0.4 |
8.94 ± 0.9 |
8.6 ± 0.9 |
10.0 ± 0.9 |
9.4 ± 1.0 |
4.3 ± 0.5 |
Experimental
All chemicals used were available commercially and the solvents purified by conventional methods before use.
Materials and methods
Melting points were determined in electrothermal engineering IA9100X1 melting point apparatus and are uncorrected. IR absorption spectra were recorded in the range of 4000–230 cm−1 as KBr pellets on a BRUKER Tensor 27 spectrophotometer. 1H and 13C NMR spectra were recorded in dimethyl sulfoxide (DMSO-d6) on a Bruker Fourier 300 MHz and Varian Unity Inova 500 MHz spectrometers using TMS as the internal reference. 13C ssNMR spectra were recorded on a Bruker 400 MHz and a Jeol 600 MHz spectrometer (14.0 and 15.0 kHz of MAS, respectively) using adamantane as the reference (298 K). Mass spectra were recorded in a JEOL, SX 102 spectrometer Bruker Microflex instrument equipped with MALDI-Flight time and MStation JMS-700 equipment in the fast atom bombardment (FAB+) mode. EA were recorded in a Thermo Scientific/Flash 2000. Single-crystal X-ray diffractions (XRD) were obtained in a Bruker diffractometer, model Smart Apex, equipped with Mo radiation (λ = 0.71073 Å), CCD two-dimensional detector, and low-temperature device. Data collection and data reduction were performed by APEX and SAINT-Plus software. The structures were solved by direct methods using SHELXS-2013 software and refined by Full-matrix least-squares procedure on F2 using SHELXL-2019/2 program.37
Cytotoxic activity in human tumour cells
Cytotoxicity of all compounds was tested against three cancer cell lines: U251 (human glioblastoma cell line), PC-3 (human Caucasian prostate adenocarcinoma), K562 (human Caucasian chronic myelogenous leukemia), HCT-15 (human colon adenocarcinoma), MCF-7 (human mammary adenocarcinoma), and SKLU-1 (human lung adenocarcinoma). Cell lines were supplied by the U.S. National Cancer Institute (NCI). The cell lines were cultured in RPMI-1640 medium, supplemented with 10% fetal bovine serum, 2 mM L-glutamine, 10000 units per mL penicillin G sodium, 10000 μg mL−1 streptomycin sulfate, 25 μg mL−1 amphotericin B (Invitrogen/Gibco™, Thermo Fisher Scientific, Waltham, MA, USA) and 1% non-essential amino acids (Gibco). They were maintained at 37 °C in a humidified atmosphere with 5% CO2. The cell viability in the experiments exceeded 95%, as determined with trypan blue. The human tumor cytotoxicity was determined using the protein-binding dye sulforhodamine B (SRB) in a microculture assay to measure cell growth, as described in the protocols established by the NaCl.38–40
Results were expressed as inhibitory concentration 50 (IC50) values. They were calculated according to the protocol of Monks,38 where a dose–response curve was plotted for each compound and the concentration (IC50), resulting in an inhibition of 50% estimated through non-linear regression analysis.
Synthetic procedures
Compound 1. 14 g of curcumin in 70 mL of dichloromethane (CH2Cl2) reacted with 2.6 mL of pyridine (Py) and 1.6 mL of acetic anhydride at room temperature for approximately 3 hours. The reaction media was stirred at room temperature following the disappearance of the starting material by TLC (4:6 ethyl acetate:hexane elute mixture). The solvent was removed under reduced pressure. The product was repeatedly extracted with ethyl acetate (EtOAc)–water (3:7) to remove pyridine entirely from the organic phase. The product was crystallized in EtOAc (1), 70.1% yield. 1H NMR (600 MHz DMSO-d6): δ 2.28 (s, 6Haliph), 3.85 (s, 6Haliph), 6.20 (s, 1H), 6.99 (d, 2Hvinyl, J 15.9 Hz), 7.16 (d, 2Haryl, J 8.1 Hz), 7.33 (dd, 2Haryl, J 8.2; 1.9 Hz), 7.52 (d, 2Haryl, J 2 Hz), 7.66 (d, 2Hvinyl, J 15.9 Hz), 16.1 (br s, 1H) ppm. 13C NMR (13C {1H} 150 MHz, DMSO-d6): δ 20.17 (Caliph), 55.72 (Caliph), 101.62 (C–H), 111.93 (Caryl), 121.27 (Caryl), 123.23 (Caryl), 124.58 (Cvinyl), 133.59 (Caryl), 139.77 (Cvinyl), 140.93 (Caryl), 151.11 (Caryl), 168.30 (C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O), 183.10 (CO) ppm. IR 1755 (CO, ester) cm−1, 1596 cm−1 (CO, β-diketone), 1506 (CO and CC, arom) cm−1, 1295 (C–C, ester) cm−1, 966 (C–O, β-diketone) cm−1. LRMS: M+ 453.746. mp of yellow crystal: 170.5 °C. T. C: 66.36 H: 5.35; E. C: 66.27 H: 5.33.
O), 183.10 (CO) ppm. IR 1755 (CO, ester) cm−1, 1596 cm−1 (CO, β-diketone), 1506 (CO and CC, arom) cm−1, 1295 (C–C, ester) cm−1, 966 (C–O, β-diketone) cm−1. LRMS: M+ 453.746. mp of yellow crystal: 170.5 °C. T. C: 66.36 H: 5.35; E. C: 66.27 H: 5.33.
Compound 2. 3.8 g of DAC was dissolved in 60 mL of EtOAc and reacted in a hydrogen atmosphere with 380 mg of Pd/C-10%. The reaction mixture was stirred at room temperature until the disappearance of the starting material was monitored by TLC (3:7 ethyl acetate:hexane elute mixture). The reaction was completed after 4 hours, filtered off through Celite, and solvent removed in vacuo. The product was purified by SiO2 column chromatography eluting with a 7:3 hexane–EtOAc solvent mixture, and the product was dried under a high vacuum (2), 80.4% yield. 1H NMR (500 MHz DMSO-d6): δ 2.23 (s, 12H), 2.65 (t, 4Haliph, J 7.86 Hz), 2.78 (m, 4Haliph), 2.85 (t, 8Haliph, J 7.02 Hz), 3.74 (d, 14H), 5.78 (s, 1H), 6.76 (dd, 2Haryl, J 8.09; 1.83 Hz), 6.79 (dd, 2Haryl, J 8.09; 1.83 Hz), 6.93 (s, 1Haryl), 6.95 (d, 2Haryl, J 2.44 Hz), 6.96 (m, 3Hvinyl, Hz), 7.00 (d, 2Haryl, J 1.83 Hz), 15.51 (br s, 1H) ppm. 13C NMR (13C {1H} 125 MHz, DMSO-d6): δ 20.17 (C–H), 29.03 (Caliph), 31.02 (Caliph), 39.44 (Caliph), 39.50 (Caliph), 44.72 (Caliph), 56.19 (C–H), 56.65 (Caliph), 100.11 (C–H), 113.25 (Caryl), 113.32 (Caryl), 120.49 (Caryl), 120.54 (Caryl) 122.90 (Caryl), 122.94 (Caryl), 137.91 (Caryl), 138.03 (Caryl), 140.05 (Caryl), 140.35 (Caryl), 151.02 (Caryl), 169.07 (CO), 193.57 (CO), 204.87 (CO) ppm. IR 2974 (C–H, aliph) cm−1, 2939 (C–H, aliph) cm−1, 2841 (C–H, aliph) cm−1, 1795 (CO, ester) cm−1, 1757 (CO, ester) cm−1, 1597 (CO, β-diketone) cm−1, 1510 cm−1 (CO and CC, arom), 1271 cm−1 (C–C, ester), 930 (C–O, β-diketone) cm−1. LRMS: M+ 455.857; white solid, mp 68.3 °C. T. C: 65.78 H: 6.18; E. C: 65.71 H: 6.11.
Compound 3. 4 g of curcumin dissolved in 120 mL of anhydrous acetone were reacted with 0.75 g of potassium carbonate (K2CO3), 1.2 mL of dimethyl sulphate (SO2(OCH3)2) and refluxed with stirring for 48 hours until the disappearance of the starting material. The solvent was removed under reduced pressure, and the product was extracted with a 3:7 mixture of EtOAc–H2O and NaOH 10% until SO2(OCH3)2 was removed from the organic phase. The product was purified by SiO2 column chromatography eluting with a mixture of hexane–CH2Cl2–methanol (MeOH) 50:45:5, and the product was dried under high vacuum (3), 63.2% yield. 1H RMN (500 MHz DMSO-d6): δ 3.81 (s, 6Haliph) 3.83 (s, 6Haliph), 6.10 (s, 1H), 6.82 (d, 2Hvinyl, J 15.84 Hz), 7.01 (d, 2Haryl, J 8.42 Hz), 7.26 (dd, 2Haryl, J 8.42, 1.92 Hz), 7.35 (d, 2Haryl, J 1.92 Hz), 7.59 (d, 2Hvinyl, J 15.93 Hz), 16.25 (br s, 1H) ppm. 13C NMR (13C {1H} 125 MHz, DMSO-d6): δ 55.57 (Caliph), 100.99 (C–H), 110.47 (Caryl), 111.67 (Caryl), 122.03 (Caryl), 122.90 (Cvinyl), 127.56 (Caryl), 140.40 (Cvinyl), 149.02 (Caryl), 150.96 (Caryl), 183.19 (CO) ppm. IR 3005 (C–H, aliph) cm−1, 2926 (C–H, aliph) cm−1, 2831 (C–H, aliph) cm−1, 1624 (CO, β-diketone) cm−1, 1504 (CO and CC, Arom) cm−1, 1134 (C–C, ester) cm−1, 965 (C–O, β-diketone) cm−1. LRMS: M+ 396.770. mp of orange crystal: 133.5 °C. T. C: 69.68 H: 6.10; E. C: 69.98 H: 6.31.
Compound 4. 4 g of curcumin in 70 mL of CH2Cl2 was reacted with 2.6 mL of Py and 2.6 mL of benzoyl chloride (BzCl) at room temperature for approximately 2 hours. The reaction mixture was stirred at room temperature following the disappearance of the starting material by TLC (1:1 ethyl acetate:hexane elute mixture). The reaction solvent was removed under reduced pressure, and the product was repeatedly extracted with a 3:7 proportion of EtOAc–H2O until pyridine was removed from the organic phase. The product was crystallized in EtOAc (4) with a 90.7% yield. 1H NMR (500 MHz DMSO-d6): δ 3.85 (s, 6Haliph), 6.24 (s, 1H), 7.05 (d, 2H, J 15.97 Hvinyl), 7.33 (d, 2H, J 8.16 Haryl), 7.41 (dd, 2H, J 8.30, 1.83 Haryl), 7.59 (d, 2H, J 1.85 Haryl), 7.62 (t, 4H, J 7.80 Haryl), 7.70 (d, 2H, J 15.96 Hvinyl), 7.76 (m, 2H, Haryl), 8.13 (dd, 4H, J 7.35, 1.36 Haryl) ppm. IR 1730 (CO, ester) cm−1, 1591 (CO, β-diketone) cm−1, 1510 (CO and CC, arom) cm−1, 1300 (C–C, ester) cm−1, 1118 (C–C, ester) cm−1, 969 (C–O, β-diketone) cm−1. LRMS: M+ 576.897. mp of yellow crystal: 234.6 °C. T. C: 72.91 H: 4.89; E. C: 72.36 H: 4.81.
Compound 5. 1 mmol of DAC was dissolved in a mixture of 25 mL ethyl acetate, and later, a solution of zinc acetate in methanol (0.5 mmol) was added dropwise. After 2 h of stirring at room temperature, a yellow powder was formed in the flask, which was filtered off and crystallized in DMSO and DMF (5), 95.6% yield. 1H NMR (600 MHz DMSO-d6): δ 2.26 (s, 6Haliph), 3.83 (s, 6Haliph), 5.84 (s, 1H), 6.91 (d, 2H, J 15.7 Hvinyl), 7.11 (d, 2H, J 8.1 Haryl), 7.26 (dd, 2H, J 8.3; 1.8 Haryl), 7.47 (d, 2H, J 1.9 Haryl), 7.53 (d, 2H, J 15.6 Hvinyl) ppm. 13C NMR (13C {1H} 150 MHz, DMSO-d6): δ 20.40 (Haliph), 55.87 (Haliph), 103.02 (C–H), 111.59 (Caryl), 120.69 (Caryl), 123.18 (Caryl), 129.89 (Cvinyl), 134.44 (Caryl), 137.07 (Cvinyl), 140.16 (Caryl), 151.11 (Caryl), 168.47 (CO), 183.50 (CO) ppm. IR 3326 (C–H, aliph) cm−1, 1758 (CO, ester) cm−1, 1587 (CO, β-diketone) cm−1, 1509 (CO and CC, arom) cm−1, 1204 (C–C, ester) cm−1, 632 (C–O, β-diketone) cm−1. LRMS: M+ 967.381. mp of yellow powder: 258.9 °C. H2O T. C: 60.23 H: 5.09; E. C: 60.89 H: 4.91.
Compound 6. 1 mmol of DACH was dissolved in a mixture of 25 mL ethyl acetate, and further, a solution of zinc acetate in methanol (0.5 mmol) was added dropwise. After 5 h of stirring at room temperature, a yellow powder was formed in the flask (6), 63.6% yield. 1H NMR (600 MHz DMSO-d6): δ 2.21 (s, 6Haliph), 2.44 (dd, 4H, J 8.92; 6.68 Haliph), 2.83 (dd, 4H, J 8.91; 6.63 Haliph), 3.73 (s, 6Haliph), 5.37 (s, 1H), 6.79 (dd, 2H, J 8.10, 1.87 Haryl), 6.92 (d, 2H, J 8.06 Haryl), 6.99 (d, 2H, J 1.92 Haryl) ppm. 13C NMR (13C {1H} 150 MHz, DMSO-d6): δ 20.40 (Haliph), 31.69 (Haliph), 42.32 (Haliph), 55.60 (Haliph), 98.25 (C–H), 112.90 (Caryl), 120.17 (Caryl), 122.35 (Caryl), 137.34 (Caryl), 140.63 (Caryl), 150.47 (Caryl), 168.47 (CO), 183.50 (CO) ppm. IR 3300 (C–H, aliph) cm−1, 1758 (CO, ester) cm−1, 1589 (CO, β-diketone) cm−1, 1509 (CO and CC, arom) cm−1, 1205 (C–C, ester) cm−1, 633 (C–O, β-diketone) cm−1. HRMS FAB + O: 975.2803 E: 975.2782. mp of white powder: 98.3 °C. T. C: 61.51 H: 5.58; E. C: 61.29 H: 5.62.
Compound 7. 1 mmol of DiMeOC was dissolved in a mixture of 25 mL ethyl acetate, and later, a solution of zinc acetate in methanol (0.5 mmol) was added slowly. After 2 h of stirring at room temperature, an orange powder was formed, filtered off, and crystallized in DMF (7), 46.4% yield. 1H NMR (500 MHz DMSO-d6): δ 3.79 (s, 6Haliph) 3.82 (s, 6Haliph), 5.76 (s, 1H), 6.77 (d, 2H, J 15.67 Hvinyl), 6.97 (d, 2H, J 8.41 Haryl), 7.19 (dd, 2H, J 8.42, 1.97 Haryl), 7.30 (s, 2Haryl), 7.46 (d, 2H, J 15.57 Hvinyl) ppm. 13C NMR (13C {1H} 125 MHz, DMSO-d6): δ 55.51 (Caliph), 102.82 (C–H), 110.16 (Caryl), 111.69 (Caryl), 121.99 (Caryl), 127.28 (Cvinyl), 128.24 (Caryl), 137.89 (Cvinyl), 148.97 (Caryl), 150.21 (Caryl), 183.49 (CO) ppm. IR 1632 (CO, β-diketone) cm−1, 1503 (CO and CC, Arom) cm−1, 1262 (C–C, ester) cm−1, 979 (C–O, β-diketone) cm−1. HRMS FAB+ O: 855.2322 E: 855.2359. mp of orange powder: 220.1 °C.
Compound 8. 1 mmol of DiBzOC was dissolved in a mixture of 50 mL tetrahydrofuran (THF), and a zinc acetate solution in methanol (0.5 mmol) was added dropwise. After 2 h of stirring at room temperature, a yellow powder was formed in the flask, which was filtered off, and crystallized in DMF (8), 41.3% yield. 1H NMR (500 MHz DMSO-d6): δ 3.84 (s, 6Haliph), 5.90 (s, 1H), 6.98 (d, 2H, J 15.68 Hvinyl), 7.31 (m, 4H), 7.59 (m, 8H), 7.75 (t, 2H, J 7.43 Haryl), 8.13 (d, 4H, J 7.34 Haryl) ppm. 13C NMR (13C {1H} 125 MHz, DMSO-d6): δ 55.96 (Caliph), 103.17 (C–H), 111.71 (Caryl), 120.79 (Caryl), 123.34 (Caryl), 128.55 (Cvinyl), 129.03 (Caryl), 129.85 (Cvinyl), 130.05 (Caryl), 134.11 (Caryl), 134.72 (Caryl), 140.21 (Caryl), 151.19 (Caryl), 163.95 (CO), 183.49 (CO) ppm. IR 1737 (CO, ester) cm−1, 1597 (CO, β-diketone) cm−1, 1508 (CO and CC, arom) cm−1, 1253 (C–C, ester) cm−1, 983 (C–O, β-diketone) cm−1. LRMS: M2+ 1216.657. mp of yellow powder: 291.3 °C. T. C: 69.11 H: 4.47; E. C: 67.96 H: 4.44.
Conclusions
The synthesis of 3 new homoleptic complexes with zinc was achieved with 3 different curcuminoid ligands. Their crystal structures reveal octahedral geometry, square pyramidal, trigonal pyramid, and trigonal-bipyramidal geometries for the zinc complexes. We contribute to this work with the first three different crystal geometries reported for zinc complexes of curcuminoids. Therefore, using DMSO and DMF as crystallization solvents was of capital importance in the obtention of single crystals. Of particular significance is the remarkable cytotoxic effect found for DAC-Zn (5) and DiMeOC-Zn (7) against U251, PC-3, K562, HCT-15, MC-7, and SKLU-1 human cancer cell lines, which surprisingly equated or significantly surpassed that of cisplatin. Among them, the DiMeOC-Zn (7) complex stands out because it is almost 22 times more cytotoxic than cisplatin in the human colon cancer cell line (HCT-15), revealing its great potential as a therapeutic agent.
Author contributions
Conceptualization, RGE and WMM; methodology, RGE and WMM; validation, RAT, MTRA and JCPJ; formal analysis, RGE and WMM; investigation, WMM, YAR and MAOM; resources, RGE; data curation, RGE, WMM, RAT, AAC, JCPJ and MTRA; writing—original draft preparation, WMM; writing—review and editing RGE and WMM; supervision, RGE; project administration, RGE; funding acquisition, RGE.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
We gratefully acknowledge financial support from CONACyT (CB 252524 and CONACYT-FOINS-PRONACES 307152) and PAPIIT (DGAPA, UNAM, IN208516 and DGAPA, UNAM, IT200720). The payment of fees from CONACYT-FOINS-PRONACES 307152 to WMM, YAR, and MAOM. is gratefully acknowledged. We are indebted to María de la Paz Orta Perez (AE), María del Rocío Patiño (R.I.P.) (IR), Isabel Chávez (NMR), María del Carmen Garcia González (FAB+) and Lucero Rios Ruiz (MALDI-TOF) from Instituto de Química, UNAM. We acknowledge the funding support provided to Dr Meza-Morales by the CAWT under NSF grant OIA-1849243.
References
- S. Wanninger, V. Lorenz, A. Subhan and F. T. Edelmann, Chem. Soc. Rev., 2015, 44, 4986–5002 RSC.
- W. Li, S. Wang, J. Feng, Y. Xiao, X. Xue, H. Zhang, Y. Wang and X. Liang, Magn. Reson. Chem., 2009, 47, 902–908 CrossRef CAS PubMed.
- S. Ghosh, S. Banerjee and P. C. Sil, Food Chem. Toxicol., 2015, 83, 111–124 CrossRef CAS PubMed.
- P. Sanphui and G. Bolla, Cryst. Growth Des., 2018, 18, 5690–5711 CrossRef CAS.
- A. Goel, A. B. Kunnumakkara and B. B. Aggarwal, Biochem. Pharmacol., 2008, 75, 787–809 CrossRef CAS PubMed.
- O. Naksuriya, S. Okonogi, R. M. Schiffelers and W. E. Hennink, Biomaterials, 2014, 35, 3365–3383 CrossRef CAS PubMed.
- G. Liang, S. Yang, H. Zhou, L. Shao, K. Huang, J. Xiao, Z. Huang and X. Li, Eur. J. Med. Chem., 2009, 44, 915–919 CrossRef CAS PubMed.
- J. Wang, D. Wei, B. Jiang, T. Liu, J. Ni and S. Zhou, Transition Met. Chem., 2014, 39, 553–558 CrossRef CAS.
- N. Aliaga-Alcalde, P. Marqués-Gallego, M. Kraaijkamp, C. Herranz-Lancho, H. Den Dulk, H. Görner, O. Roubeau, S. J. Teat, T. Weyhermüller and J. Reedijk, Inorg. Chem., 2010, 49, 9655–9663 CrossRef CAS PubMed.
- Y. Sumanont, Y. Murakami, M. Tohda, O. Vajragupta, H. Watanabe and K. Matsumoto, Biol. Pharm. Bull., 2007, 30, 1732–1739 CrossRef CAS PubMed.
- X. Z. Zhao, T. Jiang, L. Wang, H. Yang, S. Zhang and P. Zhou, J. Mol. Struct., 2010, 984, 316–325 CrossRef CAS.
- M. Asti, G. Atti, S. Rubagotti, M. Iori, P. C. Capponi, A. Versari, E. Ferrari, M. Saladini, S. Croci and A. Zerbini, Inorg. Chem., 2014, 53, 4922–4933 CrossRef CAS PubMed.
- N. Aliaga-Alcalde, L. Rodríguez, M. Ferbinteanu, P. Höfer and T. Weyhermüller, Inorg. Chem., 2012, 51, 864–873 CrossRef CAS PubMed.
- S. Zhou, X. Xue, B. Jiang and Y. Tian, Sci. China: Chem., 2012, 55, 334–340 CrossRef CAS.
- T. Sarkar, R. J. Butcher, S. Banerjee, S. Mukherjee and A. Hussain, Inorg. Chim. Acta, 2016, 439, 8–17 CrossRef CAS.
- J. Rajesh, A. Gubendran, G. Rajagopal and P. Athappan, J. Mol. Struct., 2012, 1010, 169–178 CrossRef CAS.
- Z. Pi, J. Wang, B. Jiang, G. Cheng and S. Zhou, Mater. Sci. Eng., C, 2015, 46, 565–571 CrossRef CAS PubMed.
- S. Musmade Deepak, R. Pattan Shashikant and S. Y. Manjunath, Int. J. Pharm. Chem., 2015, 5, 11–20 Search PubMed.
- D. Pucci, T. Bellini, A. Crispini, I. D'Agnano, P. F. Liguori, P. Garcia-Orduña, S. Pirillo, A. Valentini and G. Zanchetta, MedChemComm, 2012, 3, 462–468 RSC.
- W. Zhang, C. Chen, H. Shi, M. Yang, Y. Liu, P. Ji, H. Chen, R. X. Tan and E. Li, Phytomedicine, 2016, 23, 1–8 CrossRef PubMed.
- W. Meza-Morales, J. C. Machado-Rodriguez, Y. Alvarez-Ricardo, M. A. Obregón-Mendoza, A. Nieto-Camacho, R. A. Toscano, M. Soriano-García, J. Cassani and R. G. Enríquez, Molecules, 2019, 24, 910 CrossRef PubMed.
- W. Meza-Morales, M. Mirian Estévez-Carmona, Y. Alvarez-Ricardo, M. A. Obregón-Mendoza, J. Cassani, M. T. Ramírez-Apan, C. Escobedo-Martínez, M. Soriano-García, W. F. Reynolds and R. G. Enríquez, Molecules, 2019, 24, 1598 CrossRef CAS PubMed.
- S. Brahma, H. P. Sachin, S. A. Shivashankar, T. Narasimhamurthy and R. S. Rathore, Acta Crystallogr., Sect. C: Cryst. Struct. Commun., 2008, 64, 140–143 CrossRef PubMed.
- S. Brahma, M. Srinidhi, S. A. Shivashankar, T. Narasimhamurthy and R. S. Rathore, Acta Crystallogr., Sect. E: Struct. Rep. Online, 2011, 984, 819 CrossRef PubMed.
- R. Bennet, M. J. Cotton and F. A. Eiss, Acta Crystallogr., Sect. B: Struct. Crystallogr. Cryst. Chem., 1968, 24, 904–913 CrossRef.
- M. Hamid, A. Ali and A. D. Hunter, Acta Crystallogr., Sect. E: Struct. Rep. Online, 2005, 2, 1539–1541 CrossRef.
- D. I. Rio and R.A Gossage, Acta Crystallogr., Sect. E: Struct. Rep. Online, 2009, 200, 103–104 CrossRef PubMed.
- A. W. Fronczek, F. R. Ivie and M. L. Mawrickk, Acta Crystallogr., Sect. C: Cryst. Struct. Commun., 1990, 2, 3–8 Search PubMed.
- X. Yang, Acta Crystallogr., Sect. E: Struct. Rep. Online, 2007, 2, 284–285 CrossRef.
- L. Yang, D. R. Powell and R. P. Houser, Dalton Trans., 2007, 955–964 RSC.
- D. Trans, A. W. Addison and T. N. Rao, J. Chem. Soc., Dalton Trans., 1984, 1349–1356 Search PubMed.
- M. Nishio, Y. Umezawa, K. Honda and H. Suezawa, CrystEngComm, 2009, 11, 1757–1788 RSC.
- M. Anioła, Z. Dega-szafran, A. Katrusiak and M. Szafran, New J. Chem., 2014, 38, 3556–3568 RSC.
- G. Desiraju, Acc. Chem. Res., 1996, 29, 441–449 CrossRef CAS PubMed.
- F. L. Hirshfeld, Theor. Chim. Acta, 1977, 138, 129–138 CrossRef.
- P. R. Spackman, M. J. Turner, J. J. Mckinnon, S. K. Wolff, D. J. Grimwood and M. A. Spackman, J. Appl. Crystallogr., 2021, 54, 1006–1011 CrossRef CAS PubMed.
- G. M. Sheldrick, Acta Crystallogr., Sect. A: Found. Adv., 2008, 64, 112–122 CrossRef CAS PubMed.
- A. Monks, D. Scudiero, P. Skehan, R. Shoemaker, K. Paull, D. Vistica, C. Hose, J. Langley, P. Cronise, A. Vaigro-wolff, M. Gray-goodrich, H. Campbell, J. Mayo and M. Boyd, J. Natl. Cancer Inst., 1991, 83, 757–766 CrossRef CAS PubMed.
- J. Van Meerloo, G. J. L. Kaspers and J. Cloos, Methods Mol. Biol., 2011, 731, 237–245 CrossRef CAS PubMed.
- M. Concepción Lozada, O. Soria-Arteche, M. Teresa Ramírez Apan, A. Nieto-Camacho, R. G. Enríquez, T. Izquierdo and A. Jiménez-Corona, Bioorg. Med. Chem., 2012, 20, 5077–5084 CrossRef PubMed.
|
| This journal is © The Royal Society of Chemistry 2023 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence c and
Raúl G. Enríquez
c and
Raúl G. Enríquez