
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceComputational studies on functionalized Janus MXenes MM′CT2, (M, M′ = Zr, Ti, Hf, M ≠ M′; T = –O, –F, –OH): photoelectronic properties and potential photocatalytic activities†
Kuangwei Xiong *a,
Ziqiang Chenga,
Jianpeng Liua,
Peng-Fei Liub and
Zhenfa Zic
*a,
Ziqiang Chenga,
Jianpeng Liua,
Peng-Fei Liub and
Zhenfa Zic
aDepartment of Physics, East China Jiaotong University, Nanchang 330013, P.R. China. E-mail: xiongkw@ecjtu.edu.cn
bInstitute of High Energy Physics, Chinese Academy of Sciences, Beijing 100049, P.R. China
cSchool of Physics and Materials Engineering, Hefei Normal University, Hefei 230601, P.R. China
First published on 9th March 2023
Abstract
Motivated by the successful synthesis of Janus monolayers of transition metal dichalcogenides (i.e., MoSSe), we computationally investigated the structural, electronic, optical, and transport properties of functionalized Janus MXenes, namely MM′CT2 (M, M′ = Zr, Ti, Hf, M ≠ M′, T = –O, –F, –OH). The results of the calculations demonstrate that five stable O-terminated Janus MXenes (ZrTiCO2-I, ZrHfCO2-I, ZrHfCO2-III, HfTiCO2-I, and HfTiCO2-III), exhibit modest bandgaps of 1.37–1.94 eV, visible-light absorption (except for ZrHfCO2-I), high carrier mobility, and promising oxidization capability of photoinduced holes. Additionally, their indirect-gap, spatially separated electron–hole pairs, and the dramatic difference between the mobilities of electrons and holes could significantly limit the recombination of photoinduced electron–hole pairs. Our results indicate that the functionalized Janus MXene monolayers are ideal and promising materials for application in visible light-driven photocatalysis.
1. Introduction
Since the discovery of graphene by Novoselov et al.1 two-dimensional (2D) materials have attracted tremendous interest in experimental and theoretical studies, owing to the unique properties of these 2D free-standing crystals. Remarkably, a new class of 2D transition-metal carbides and nitrides, called MXenes,2 has received increasing attention due to their potential applications in photocatalysts,3–5 supercapacitors,6 and storage systems.7,8 Generally, MXenes are synthesized by selectively etching A layers from a fascinating family of MAX phases presenting chemically as Mn+1AXn using hydrofluoric acid (HF) solutions.9,10 MAX phases11 are a family of more than 80 ternary carbides, nitrides and borides in hexagonal crystal symmetry. Here, M is an early transition metal, A is generally a group IIIA or IVA element (e.g., Al, Si), X represents C, N, and/or B, n = 1, 2, or 3. After the etching process, the surfaces of MXene are always covered with functional groups (–O, –OH, or –F) to passivate the outer-layer metal atoms. The surface groups may serve as anchor sites for the reactants, resulting in more efficient adsorption and oxidation of organic molecules. Many theoretical studies show that bare MXenes are metallic, but their band-gap can be opened upon appropriate surface functionalization.12,13 For example, 2D single-layer Ti2CO2, Zr2CO2, and Hf2CO2 MXenes transform from metals to semiconductors with band gaps of 0.92, 1.54, 1.75 eV, respectively, upon surface functionalization with –O, –OH, or –F.4 The suitable band-gaps of surface-functionalized MXenes strongly support their visible light absorption and possible photocatalytic applications.Structural symmetry is an important factor affecting the properties of 2D materials. Structural symmetry-breaking may introduce many novel properties in these materials. 2D Janus materials containing different atoms on the upper and lower facets, breaking the mirror symmetry, have been extensively studied in recent years.14–19 In 2017, Janus monolayers of transition metal dichalcogenides (TMDs, i.e., MoSSe) have been synthesized through the modified chemical vapor deposition (CVD) method.20,21 A room-temperature synthetic strategy for 2D Janus TMDs and their vertical and lateral heterostructures has also been reported.22 Previous studies on MXenes with symmetrical configurations have demonstrated their excellent photo- and electrocatalytic properties.23–25 The successful synthesis of Janus MoSSe monolayers inspires further studies of MXenes with Janus structures. In some recent studies,26–28 Janus MXenes were obtained by introducing asymmetric surface functional groups, and their electronic, magnetic, mechanical, and photocatalytic properties were also investigated. Moreover, considering that M2C MXenes are composed of two layers of the same transition metal atoms, one of them could be replaced with a layer of another transition metal, forming the so-called MM′C Janus structure. For instance, Akgenc investigated the structural, electronic, magnetic and vibrational properties of the Janus MXenes MoWC, MoCrC, WCrC, and VNbC (pristine and terminated with T = –O, –F, or –OH). The results suggested that the surface-functionalized Janus MXenes are promising candidates for spintronic applications.29 Su et al. studied the effect of different transition metal atoms on the structural stability as well as magnetic and electronic properties of MM′CO2 (M and M′ = V, Cr, and Mn). Janus MXenes can regulate the size of band gap, magnetic ground state, and net magnetic moment.30 To date, a few studies have discussed the effects of surface groups on the electronic and optical properties of MXenes with Janus structure. Therefore, it is essential to systematically investigate their physicochemical properties and further discuss the feasibility of their photocatalytic applications before trial-and-error experiments.
In this paper, first-principles calculations were performed to explore the structural, electronic, optical, and transport properties of functionalized Janus MXenes, namely MM′CT2 (M, M′ = Zr, Ti, Hf, M ≠ M′, T = –O, –F, –OH). We summarized the crucial characteristics of these materials, including thermodynamic stabilities, band structures, optical absorptions, redox potentials, and carrier mobilities. The results of our calculations show that the five oxygen-functionalized Janus MXenes exhibit appropriate band-gaps of 1.37–1.94 eV, visible light absorption, oxidization capability of photo induced holes, high hole carrier mobility, as well as efficient separation of electron–hole pairs. The good thermodynamic stabilities and promising characteristics of the functionalized Janus MXenes modeled in this study highlight their experimental feasibility and great potential for photocatalytic applications.
2. Computational methods
All calculations were performed using in the vienna ab initio simulation package (VASP)31,32 based on density functional theory (DFT). The Perdew–Burke–Ernzerhof (PBE)33 functional within generalized gradient approximation (GGA) was applied to deal with electron exchange–correlation effects. The electron–nuclei interaction was treated with the projector augmented wave (PAW) method.34 The energy cutoff was set to 600 eV and the energy precision of the calculations was 10−6 eV. The atomic positions were fully relaxed until the maximum force on each atom was less than 0.01 eV Å−1. In the geometry optimization and self-consistent calculations, the first Brillouin zone was sampled with 11 × 11 × 1 and 17 × 17 × 1 Γ-centered Monkhorst–Pack k-point grids due to their 2D structures. To compensate for the underestimation of band gaps by the PBE functional, the band structures and dielectric constants of functionalized Janus MXenes were calculated using the Heyd–Scuseria–Ernzerhof (HSE06) hybrid functional,35 which is a reliable method for the calculation of electronic and optical properties. The DFT-D2 correction method proposed by Grimme36 was adopted to describe the weak van der Waals (vdW) interactions between adjacent layers. Phonon dispersion spectra were calculated with a 4 × 4 × 1 supercell and 5 × 5 × 1 k-point mesh using the Phonopy code,37 interfaced with the density functional perturbation theory (DFPT)38 module implemented in VASP. Atomic structures were visualized using the VESTA code.39 Moreover, a vacuum space of 20 Å was confirmed to be large enough to eliminate interactions between adjacent layers.The optical absorption properties were investigated by calculating the complex dielectric constants (ε) at a given frequency (ω) using the HSE06 hybrid functional with a 21 × 21 × 1 k-point mesh. The dielectric constant can be defined as ε(ω) = ε1(ω) + iε2(ω). The absorption coefficient I(ω) can be calculated through the following equation:40
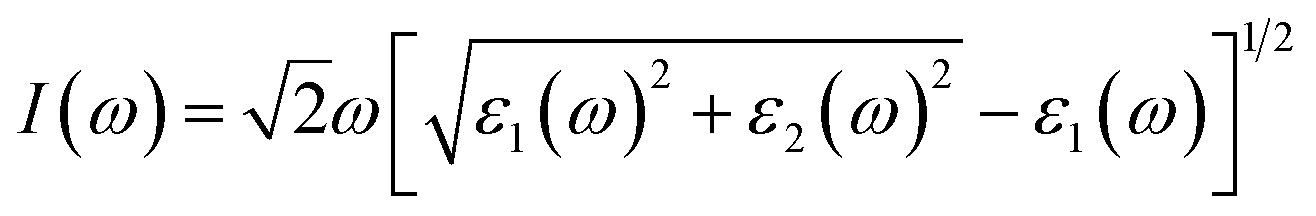 | (1) |
As shown in eqn (1), I(ω) will be positive only if the imaginary part ε2(ω) > 0; therefore, ε2(ω) reflects the light absorption at a given frequency. The imaginary component can be determined as:41
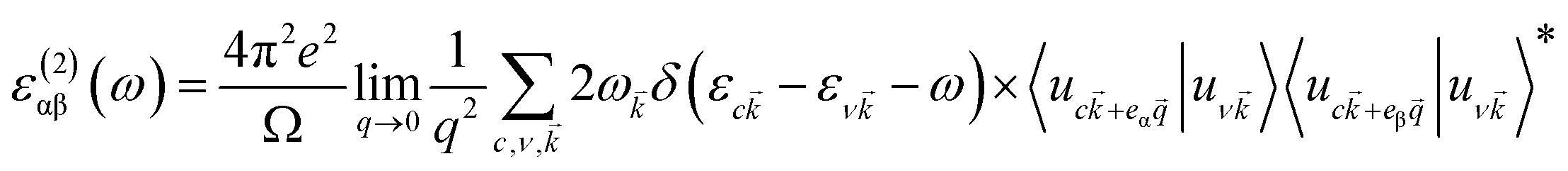 | (2) |
![[k with combining right harpoon above (vector)]](https://www.rsc.org/images/entities/i_char_006b_20d1.gif) is the cell periodic part of the wavefunctions at the k-point. The summation of eqn (2) includes empty conduction band states with twice the number of valence band states.
is the cell periodic part of the wavefunctions at the k-point. The summation of eqn (2) includes empty conduction band states with twice the number of valence band states.
The deformation potential theory42 was utilized to evaluate the carrier mobility of functionalized Janus MXenes, which were derived from the following analytical expression:43,44
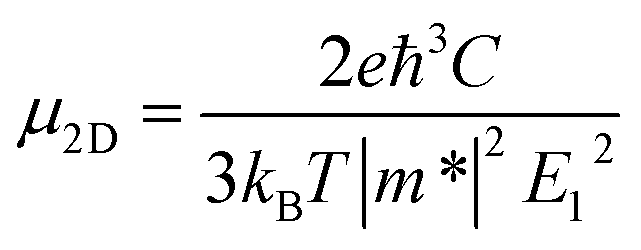 | (3) |
3. Results and discussion
3.1 Geometries and stability
A conventional M2C MXene (M = Zr, Ti, Hf) consists of trilayer sheets with a hexagonal unit cell, where one carbon layer is sandwiched between two layers of the same transition-metal atoms.4 We replaced one layer with another transition metal layer, forming the MM′C Janus structure. The functionalized Janus MXene MM′CT2 (T = –O, –F, –OH) has three possible geometries. Taking the O-terminated Janus MXene MM′CO2 as an example for illustration, the top and side views of its structure are shown in Fig. 1. The structures of MM′C(OH)2 and MM′CF2 are shown in Fig. S1 (ESI†). As shown in Fig. 1a, the bare Janus MM′C MXene contains one carbon layer sandwiched by two transition metal layers. The functionalized MM′CO2 MXenes have three possible configurations: geometry I with surface O atoms located above the opposite-side metal atoms (Fig. 1b, MM′CO2-I), geometry II with two terminal O atoms located on the middle C atoms (Fig. 1c, MM′CO2-II), and geometry III with one terminal O atom lying on the top of the opposite metal atom and another one lying on the top of the middle C atom (Fig. 1d, MM′CO2-III).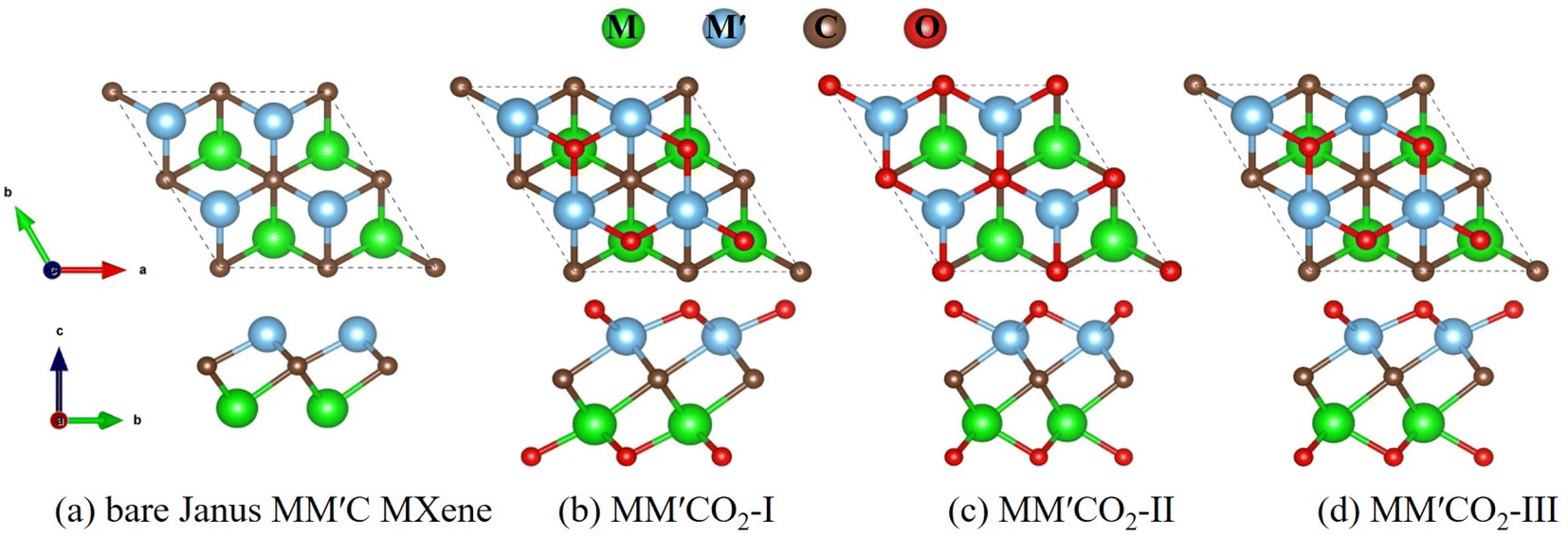 | ||
| Fig. 1 Top view (top) and side view (bottom) of geometries of (a) bare Janus MM′C MXene; (b) MM′CO2-I; (c) MM′CO2-II; (d) MM′CO2-III. M, M′ = Zr, Ti, Hf; M ≠ M′. | ||
We first calculated the cohesive energies (Ecoh) to evaluate the thermodynamic stabilities of the functionalized Janus MXenes with different geometries. Ecoh was calculated as:
| Ecoh = (EM + EM′ + EC+ 2ET − EMM′CT2)/5 | (4) |
To assess the lattice dynamic stabilities of the surface-functionalized Janus MXenes, phonon dispersions were also calculated along high-symmetry lines in the first Brillouin zone, as illustrated in Fig. S2–S4.† Among all O-terminated Janus MXene, no distinct imaginary frequency was observed in the phonon spectrum of ZrTiCO2 with geometry I (Fig. S2a†), as well as ZrHfCO2 (Fig. S3a and c†) and HfTiCO2 with geometry I and III (Fig. S4a and c†), demonstrating the kinetic stability of the five oxygen-functionalized Janus MXenes. The highest frequencies in their phonon spectra could reach up to 22 THz (734 cm−1), indicating the strong connection among the M, M′, C, and O atoms. Except for five O-functionalized MXenes mentioned above, no imaginary frequency was present in the phonon spectra of any of the –OH passivated MXenes (Fig. S2g–i, S3g–i, and S4g–i†), which demonstrates that the –OH termination was also dynamically stable. However, the negative frequencies in the phonon spectra of the F-terminated Janus MXenes reflect their kinetic instability.
3.2 Electronic structures and optical properties
Based on the cohesive energies and phonon spectra discussed above, ZrTiCO2-I, ZrHfCO2-I, ZrHfCO2-III, HfTiCO2-I, HfTiCO2-III among the O-terminated Janus MXenes, and all OH-functionalized MXenes had good thermodynamic and dynamical stabilities, confirming the feasibility of their experimental preparation. Therefore, we calculated the band structures of these functionalized Janus MXenes with higher thermodynamic stabilities, in order to assess whether they have appropriate characteristics for photocatalytic applications.Although there are transition metals in the Janus MXenes monolayers, the effect of spin orbital coupling (SOC)48,49 on their band structures can be negligible due to the relatively forward positions of these metal atoms in the periodic table. Taking ZrTiCO2-I as an example, we calculate its band structures by PBE functional with and without SOC effects in Fig. S5.† The results show slight differences. Due to the computational cost, the SOC effect is not considered in the subsequent calculations.
The band structures of the five stable O-terminated Janus MXene monolayers (Fig. 2) and all OH-functionalized MXene sheets (Fig. S6†) were calculated with the HSE06 hybrid functional (Table S2†). As shown in Fig. 2, ZrTiCO2-I, ZrHfCO2-I, ZrHfCO2-III, HfTiCO2-I, and HfTiCO2-III are all indirect-gap semiconductors with the VBM at the Γ point and the CBM at the M point, along with bandgaps of 1.37, 1.71, 1.77, 1.43, and 1.94 eV, respectively. In general, the visible light wavelength window for photocatalysis ranges from 390 to 760 nm.50 Therefore, the typical band gap of the semiconductor should be in the range of 1.6–3.1 eV to effectively utilize visible light. Most of the bandgaps (1.37–1.94 eV) of the five O-terminated Janus MXenes fall within the visible spectrum, which indicates that these materials can potentially serve as visible light driven photocatalysts. In terms of photocatalytic activity, indirect bandgaps are more advantageous than direct ones for limiting the recombination of photogenerated electrons with holes, due to the different positions of electrons and holes in momentum space.51,52 In additions, all the OH-functionalized Janus MXenes exhibited metallic characteristics, due to their band curves crossing the Fermi level (as shown in Fig. S6†). Therefore, the following analysis does not include further calculations of the optical absorptions, band edge positions, and carrier mobility of the OH-functionalized Janus MXene monolayers.
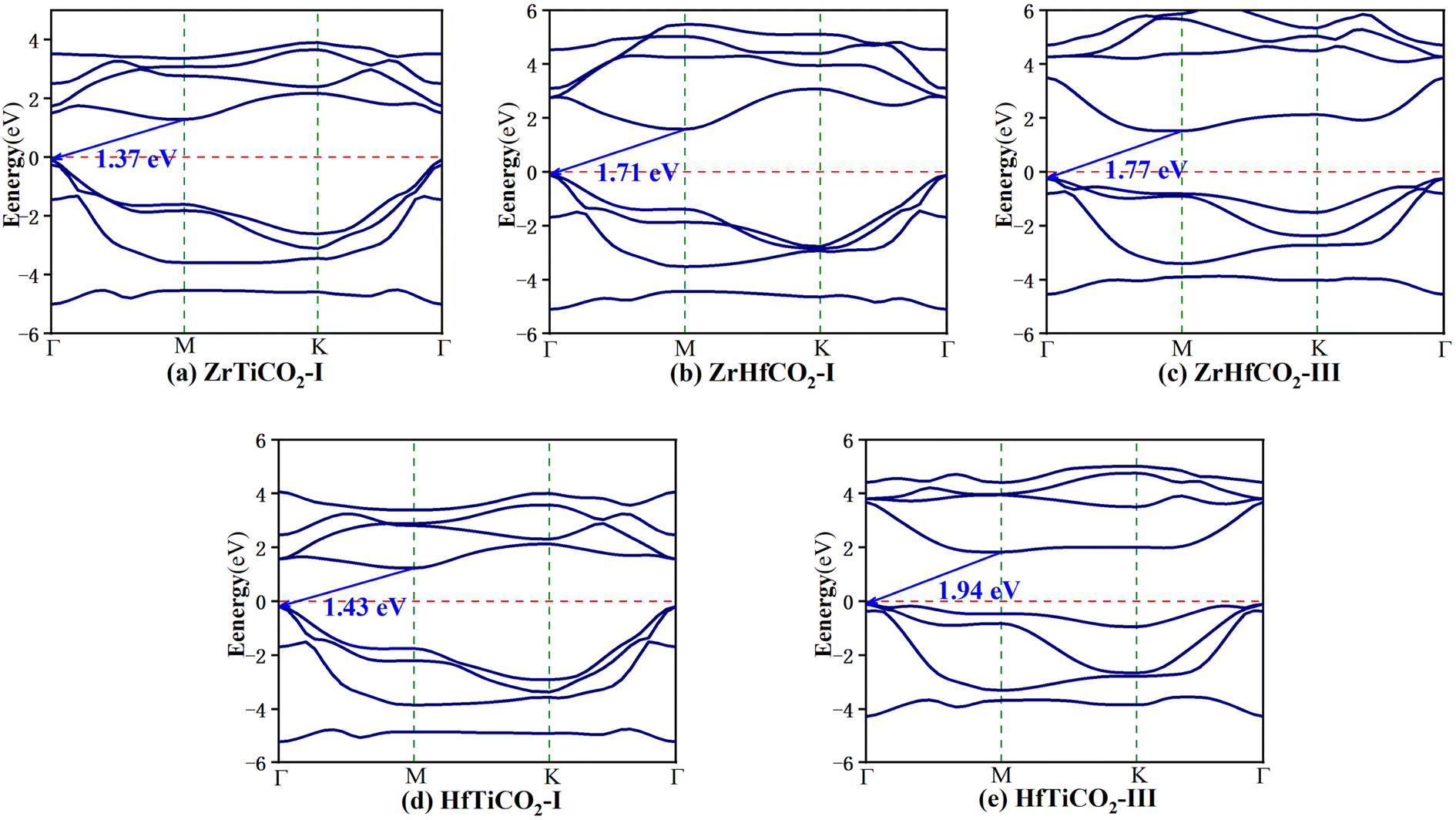 | ||
| Fig. 2 Band structures of (a) ZrTiCO2-I, (b) ZrHfCO2-I, (c) ZrHfCO2-III, (d) HfTiCO2-I, and (e) HfTiCO2-III. The red dashed lines mark the Fermi level at 0 eV. | ||
The band structures of the five stable O-terminated Janus MXene monolayers suggest that they can absorb in the visible region, highlighting their potential in photocatalytic applications. Accordingly, we calculated the dielectric constants ε(ω) = ε1(ω) + iε2(ω) to investigate their optical absorption properties. The imaginary part ε2(ω) of the dielectric constants consists of εxy(ω) and εzz(ω) contributions, which are the components perpendicular and parallel to the z direction, respectively. As shown in Fig. 3, obvious absorption peaks in the visible-light region (<3.2 eV) were observed for ZrTiCO2-I (2.06, 2.28, 2.57, 3.14 eV), ZrHfCO2-III (2.65, 3.03 eV), HfTiCO2-I (1.98, 2.24, 2.55, 3.07 eV), and HfTiCO2-III (2.85 eV), but not for ZrHfCO2-I (3.26 eV). These high absorption peaks resulted in a large area under the ε2 curves, implying that these materials can harvest a significant portion of visible light spectrum and have potential application in visible light-driven photocatalysis. In contrast, the absorption peak of ZrHfCO2-I was located at 3.26 eV, which is slightly larger than 3.2 eV, indicating a relatively low-efficiency response to visible light.
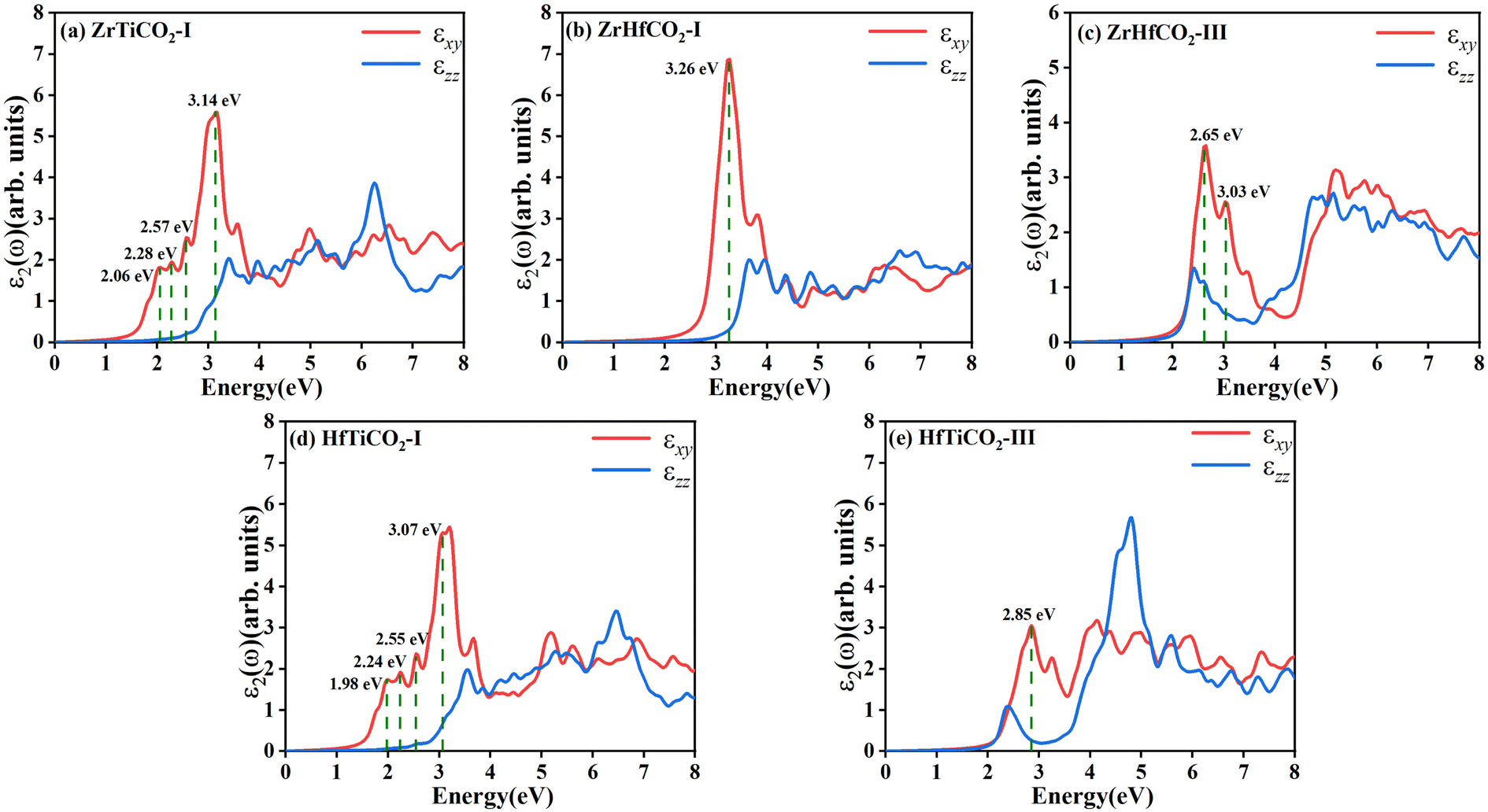 | ||
| Fig. 3 Imaginary parts of dielectric constants of five O-terminated Janus MXenes: (a) ZrTiCO2-I, (b) ZrHfCO2-I, (c) ZrHfCO2-III, (d) HfTiCO2-I, and (e) ZrHfCO2-III. | ||
3.3 Redox capabilities
Besides an appropriate bandgap and visible light response, a decent band edge position is another crucial factor with significant impact on the activity of a photocatalyst. The band edge positions were determined by calculating the CBM/VBM energy levels relative to the vacuum levels at 0 eV. Additionally, in order to evaluate the oxidation/reduction ability of the photocatalysts, the normal hydrogen electrode (NHE, ENHE = −4.5 eV vs. the vacuum level at 0 eV) is usually selected as a reference for comparing the redox potentials of the band edges. Generally, a photocatalytic material will have a stronger reduction capability of photoinduced electrons when its CBM level (vs. NHE) is higher (more negative), along with a greater oxidation capability of photoactivated holes when its VBM level (vs. NHE) is lower (more positive). Valence band holes with chemical potentials of +1.0 to +3.5 eV (vs. NHE) are considered excellent oxidants, while conduction band electrons with chemical potentials between +0.5 to −1.5 eV (vs. NHE) are good reductants.53The band alignment is shown in Fig. 4 and Table S3.† The VBM levels (vs. NHE) of ZrTiCO2-I, ZrHfCO2-I, ZrHfCO2-III, HfTiCO2-I, and HfTiCO2-III (located at 1.59, 1.22, 2.43, 1.58, and 2.87 eV, respectively) were in the range of +1.0 to +3.5 eV (vs. NHE). These results indicate that the photoinduced valence band holes in these catalysts possess promising oxidation capabilities. Moreover, the VBM levels of the above five materials are also comparable to or even more positive than those of other high-performance photocatalysts, such as Hf2CO2, Zr2CO2, Ti2CO2 MXenes with symmetrical configurations (1.49, 1.69, 2.02 eV, respectively),4 g-C3N4 (1.97 eV),54 and holey C2N (2.05 eV).55 Therefore, these catalysts can efficiently degrade organic pollutants, such as rhodamine B and methyl orange, on their surface.
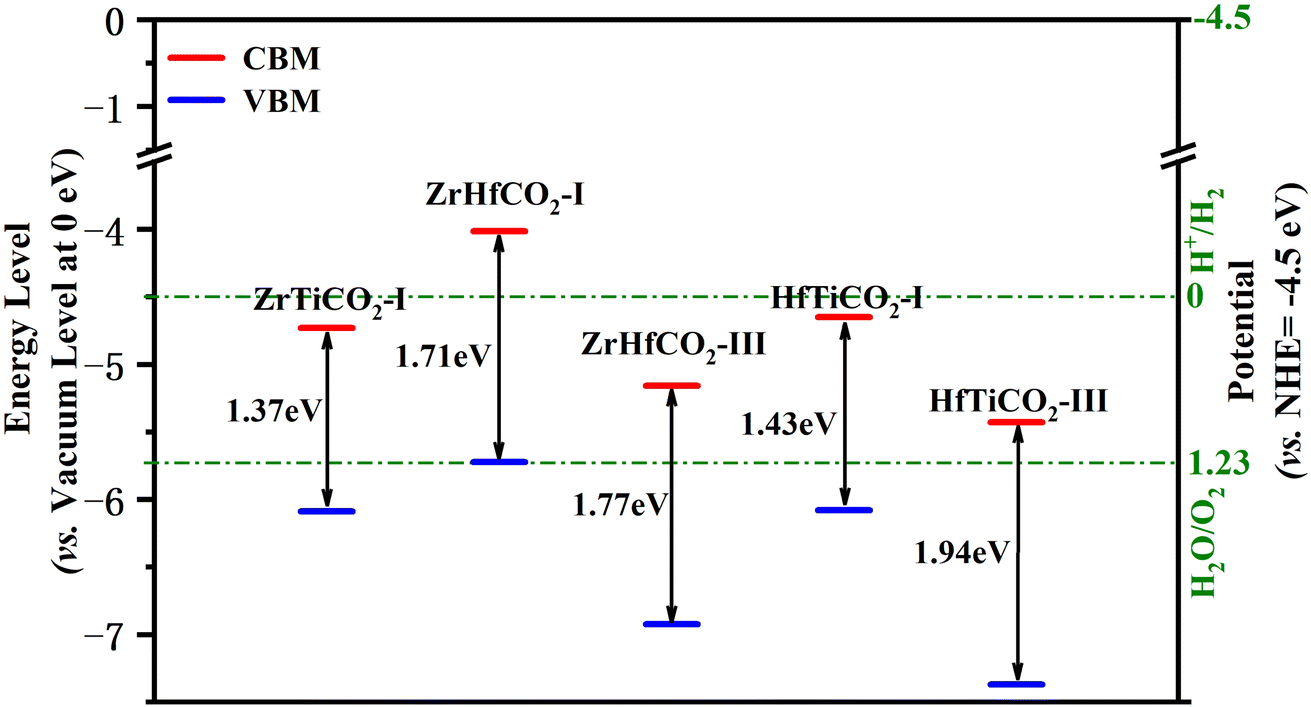 | ||
| Fig. 4 Band edge positions of ZrTiCO2-I, ZrHfCO2-I, ZrHfCO2-III, HfTiCO2-I, and HfTiCO2-III. The H+/H2, H2O/O2, and CO2/CH4 redox potentials at pH = 0 are also provided as a reference (green dashed lines). | ||
Compared with the excellent oxidation capability of holes, the reduction capabilities of the conduction band electrons were not as impressive. The CBM levels (vs. NHE) of ZrTiCO2-I, ZrHfCO2-I, ZrHfCO2-III, HfTiCO2-I, and HfTiCO2-III were 0.23, −0.49, 0.66, 0.15, and 0.93 eV, respectively, suggesting that ZrTiCO2-I (0.23 eV), ZrHfCO2-I (−0.49 eV), and HfTiCO2-I (0.15 eV) can reduce heavy metal ions or other reactants, while ZrHfCO2-III (0.66 eV) and HfTiCO2-III (0.93 eV) are not suitable for photocatalytic reduction reactions, due to their inappropriate CBM level positions. Nevertheless, the band alignment could be effectively modulated through doping,56 strain and electric field engineering,57 and van der Waals heterostructures formation58 to meet the requirements of the photocatalytic process.
3.4 Carrier migration mechanism
After the photocatalyst is activated, the photogenerated electrons and holes must be separated efficiently and transferred to the photocatalyst surface in a short time, to participate in the photocatalytic reactions. In this process, due to the attractive force between positive and negative charges, the electrons and holes tend to aggregate around positively and negatively charged areas, respectively. The electron localization function (ELF)59 is a useful tool to describe electron localization in molecules or solids. Therefore, we calculated the ELFs of five O-terminated Janus MXene monolayers to identify possible capturing centers of activated electrons and holes. As shown in Fig. 5, the electrons were mainly distributed around the C and O atoms, while less electrons were located around M/M′ atoms in all five O-terminated Janus MXenes, as also evidenced by the different colored regions in the ELF map. Based on the ELF map in Fig. 5, the red-colored region around the C or O atoms indicates a marked electron transfer from metal to C/O atoms. As a consequence, C/O atoms acted as negatively charged centers, while M/M′ atoms served as positively charged centers, which would facilitate the separation and effectively limit the recombination of photogenerated electron–holes pairs.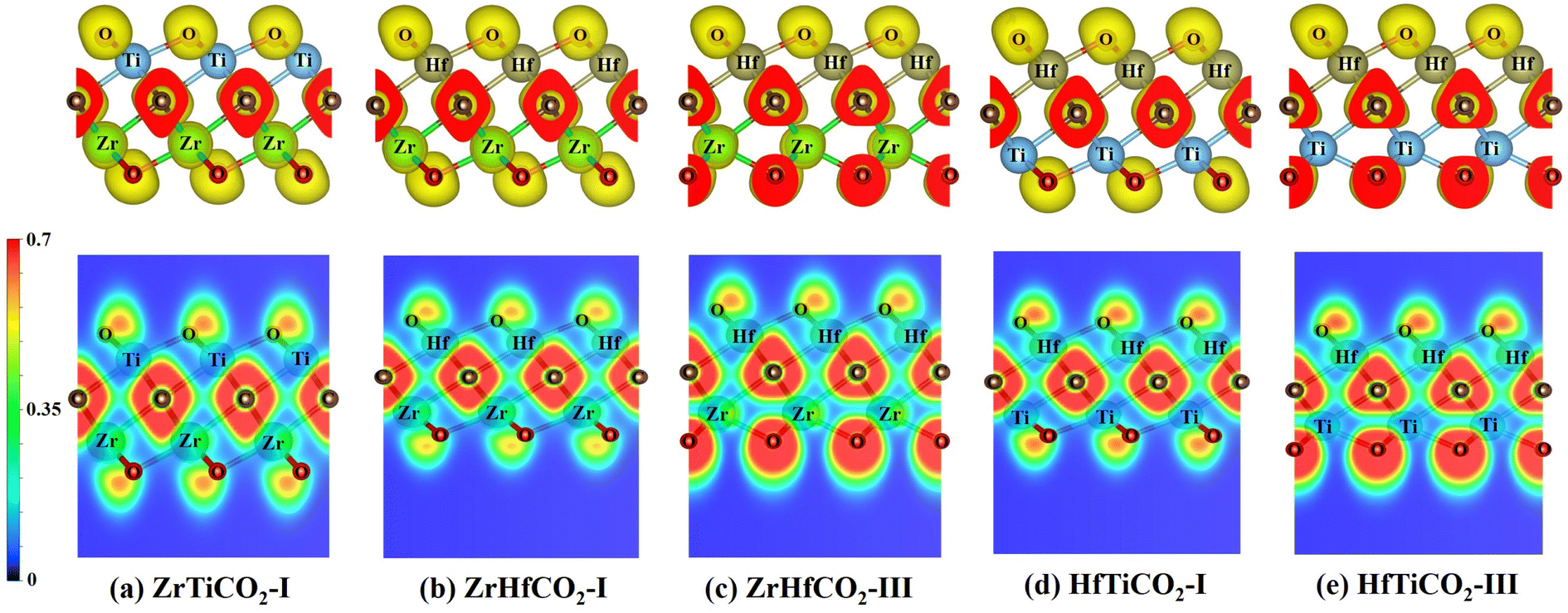 | ||
| Fig. 5 ELF isosurfaces (top) plotted with the value of 0.7 au and ELF maps (bottom) sliced perpendicular to the (100) direction for (a) ZrTiCO2-I, (b) ZrHfCO2-I, (c) ZrHfCO2-III, (d) HfTiCO2-I, and (e) ZrHfCO2-III. In the ELF map, the red and blue colors represent the highest (0.7) and lowest (0) values of the ELF, indicating accumulation and depletion of electrons, respectively. | ||
The carrier mobility is another critical factor for evaluating the transport characteristic of photocatalytic materials. The carrier mobilities of the investigated Janus MXenes along the armchair (x) and zigzag (y) directions of the hexagonal cell are listed in Table S4,† and the related calculation details are presented in Fig. S7.† As shown in Table S4,† the electron mobilities were in the range of 3–19649 cm2 V−1 s−1, and the hole mobilities ranged from 24 to 21![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 621 cm2 V−1 s−1. In particular, except for HfTiCO2-I (64 and 24 cm2 V−1 s−1 for the x- and y-direction, respectively), all hole mobilities along both the x- and y-directions were comparable to or even higher than those of some well-known 2D materials, such as MoS2 single-layer transistors (200 cm2 V−1 s−1),60 Janus MoSSe monolayers (530 cm2 V−1 s−1),61 and transition metal trichalcogenide MnPSe3 monolayers (625.9 cm2 V−1 s−1).62 The high hole mobility would allow the photoinduced holes to rapidly migrate to the reactive sites of the catalyst and then participate in the oxidation reaction. Furthermore, it is noteworthy that there is the dramatic difference between the μ values of electrons and holes in most of the O-terminated Janus MXenes. For instance, the hole mobility in ZrTiCO2-I along the x-direction was almost 10 times larger than that of electrons, which could significantly suppress the recombination of photoinduced electron–hole pairs and thus ensure a high photocatalytic performance of the investigated photocatalyst. This property is expected to favor the separation of electrons and holes during the photocatalytic process.
621 cm2 V−1 s−1. In particular, except for HfTiCO2-I (64 and 24 cm2 V−1 s−1 for the x- and y-direction, respectively), all hole mobilities along both the x- and y-directions were comparable to or even higher than those of some well-known 2D materials, such as MoS2 single-layer transistors (200 cm2 V−1 s−1),60 Janus MoSSe monolayers (530 cm2 V−1 s−1),61 and transition metal trichalcogenide MnPSe3 monolayers (625.9 cm2 V−1 s−1).62 The high hole mobility would allow the photoinduced holes to rapidly migrate to the reactive sites of the catalyst and then participate in the oxidation reaction. Furthermore, it is noteworthy that there is the dramatic difference between the μ values of electrons and holes in most of the O-terminated Janus MXenes. For instance, the hole mobility in ZrTiCO2-I along the x-direction was almost 10 times larger than that of electrons, which could significantly suppress the recombination of photoinduced electron–hole pairs and thus ensure a high photocatalytic performance of the investigated photocatalyst. This property is expected to favor the separation of electrons and holes during the photocatalytic process.
4. Conclusions
Using first-principles calculations, we have systematically investigated the functional group-dependent structural, electronic, optical, and transport characteristics of Janus MXenes, namely MM′CT2 (M, M′ = Zr, Ti, Hf, M ≠ M′, T = –O, –F, –OH). We first evaluated the thermal and dynamical stabilities of these Janus MXene monolayers through cohesive energy and phonon dispersion calculations. Based on these results, we found that the five O-terminated Janus MXenes (ZrTiCO2-I, ZrHfCO2-I, ZrHfCO2-III, HfTiCO2-I, HfTiCO2-III), had appropriate indirect bandgaps of 1.37–1.94 eV, visible light absorption ability (except for ZrHfCO2-I), photoinduced hole oxidization capability, spatially separated electron–hole pairs, as well as high hole mobilities, indicating their possible application in high-efficiency photocatalytic processes.The experimental preparation of Janus MXenes remains challenging. However, similar to other successfully synthesized Janus 2D materials, such as Janus graphene63 and Janus TMDs,20,21 the synthesis of Janus MXenes is expected to feasible. Our calculations can provide some guidance for future experimental studies as well as practical applications of Janus MXenes.
Author contributions
Kuangwei Xiong: conceptualization, data curation, formal analysis, investigation, funding acquisition, writing – original draft, writing – review & editing. Ziqiang Cheng: conceptualization, methodology, formal analysis, funding acquisition, validation. Jianpeng Liu: investigation, validation, visualization. Peng-Fei Liu: resources, software, writing – review & editing. Zhenfa Zi: supervision, funding acquisition, writing – review & editing.Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
This work was supported by the Natural Science Foundation of China (Nos. 22262013, 12004108, 12104458), Talent Project of Anhui Provincial Department of Education (No. gxbjZD2022050). The calculations were performed at Supercomputer Centre in China Spallation Neutron Source.References
- K. S. Novoselov, A. K. Geim, S. V. Morozov, D. Jiang, Y. Zhang, S. V. Dubonos, I. V. Grigorieva and A. A. Firsov, Science, 2004, 306, 666–669 CrossRef CAS PubMed
.
- M. Naguib, M. Kurtoglu, V. Presser, J. Lu, J. Niu, M. Heon, L. Hultman, Y. Gogotsi and M. W. Barsoum, Adv. Mater., 2011, 23, 4248–4253 CrossRef CAS PubMed
- Z. Guo, J. Zhou, L. Zhu and Z. Sun, J. Mater. Chem. A, 2016, 4, 11446–11452 RSC
- H. Zhang, G. Yang, X. Zuo, H. Tang, Q. Yang and G. Li, J. Mater. Chem. A, 2016, 4, 12913–12920 RSC
- N. Van-Huy, N. Ba-Son, C. Hu, N. Chinh Chien, N. Dang Le Tri, D. Minh Tuan Nguyen, D.-V. N. Vo, T. Quang Thang, M. Shokouhimehr, A. Hasani, S. Y. Kim and L. Quyet Van, Nanomaterials, 2020, 10, 602 CrossRef PubMed
- Y. Xia, T. S. Mathis, M. Q. Zhao, B. Anasori, A. Dang, Z. H. Zhou, H. Cho, Y. Gogotsi and S. Yang, Nature, 2018, 557, 409 CrossRef CAS PubMed
- X. Wu, Z. Wang, M. Yu, L. Xiu and J. Qiu, Adv. Mater., 2017, 29, 1607017 CrossRef PubMed
- P. A. Shinde, A. M. Patil, S. Lee, E. Jung and S. Chan Jun, J. Mater. Chem. A, 2022, 10, 1105–1149 RSC
- M. Naguib, O. Mashtalir, J. Carle, V. Presser, J. Lu, L. Hultman, Y. Gogotsi and M. W. Barsoum, ACS Nano, 2012, 6, 1322–1331 CrossRef CAS PubMed
- M. R. Lukatskaya, O. Mashtalir, C. E. Ren, Y. Dall'Agnese, P. Rozier, P. L. Taberna, M. Naguib, P. Simon, M. W. Barsoum and Y. Gogotsi, Science, 2013, 341, 1502–1505 CrossRef CAS PubMed
- M. A. Hadi, J. Phys. Chem. Solids, 2020, 138, 109275 CrossRef CAS
- M. Khazaei, M. Arai, T. Sasaki, C. Y. Chung, N. S. Venkataramanan, M. Estili, Y. Sakka and Y. Kawazoe, Adv. Funct. Mater., 2013, 23, 2185–2192 CrossRef CAS
- M. Khazaei, M. Arai, T. Sasaki, M. Estili and Y. Sakka, Phys. Chem. Chem. Phys., 2014, 16, 7841–7849 RSC
- A. C. Riis-Jensen, T. Deilmann, T. Olsen and K. S. Thygesen, ACS Nano, 2019, 13, 13354–13364 CrossRef CAS PubMed
- M. Yagmurcukardes, C. Sevik and F. M. Peeters, Phys. Rev. B, 2019, 100, 045415 CrossRef CAS
- Z. Wang and G. Zhou, J. Phys. Chem. C, 2020, 124, 167–174 CrossRef CAS
- T. V. Vu, V. T. T. Vi, C. V. Nguyen, H. V. Phuc and N. N. Hieu, J. Phys. D: Appl. Phys., 2020, 53, 455302 CrossRef CAS
- P. Wang, Y. X. Zong, H. Liu, H. Y. Wen, H. X. Deng, Z. M. Wei, H. B. Wu and J. B. Xia, J. Phys. Chem. C, 2020, 124, 23832–23838 CrossRef CAS
- T. V. Vu, C. V. Nguyen, H. V. Phuc, A. A. Lavrentyev, O. Y. Khyzhun, N. V. Hieu, M. M. Obeid, D. P. Rai, H. D. Tong and N. N. Hieu, Phys. Rev. B, 2021, 103, 085422 CrossRef CAS
- A. Y. Lu, H. Y. Zhu, J. Xiao, C. P. Chuu, Y. M. Han, M. H. Chiu, C. C. Cheng, C. W. Yang, K. H. Wei, Y. M. Yang, Y. Wang, D. Sokaras, D. Nordlund, P. D. Yang, D. A. Muller, M. Y. Chou, X. Zhang and L. J. Li, Nat. Nanotechnol., 2017, 12, 744–749 CrossRef CAS PubMed
- J. Zhang, S. Jia, I. Kholmanov, L. Dong, D. Q. Er, W. B. Chen, H. Guo, Z. H. Jin, V. B. Shenoy, L. Shi and J. Lou, ACS Nano, 2017, 11, 8192–8198 CrossRef CAS PubMed
- D. B. Trivedi, G. Turgut, Y. Qin, M. Y. Sayyad, D. Hajra, M. Howell, L. Liu, S. Yang, N. H. Patoary, H. Li, M. M. Petric, M. Meyer, M. Kremser, M. Barbone, G. Soavi, A. V. Stier, K. Muller, S. Yang, I. S. Esqueda, H. Zhuang, J. J. Finley and S. Tongay, Adv. Mater., 2020, 32, 2006320 CrossRef CAS PubMed
- K. Hantanasirisakul and Y. Gogotsi, Adv. Mater., 2018, 30, 1804779 CrossRef PubMed
- A. D. Handoko, K. D. Fredrickson, B. Anasori, K. W. Convey, L. R. Johnson, Y. Gogotsi, A. Vojvodic and Z. W. Seh, ACS Appl. Energy Mater., 2018, 1, 173–180 CrossRef CAS
- Z. W. Seh, K. D. Fredrickson, B. Anasori, J. Kibsgaard, A. L. Strickler, M. R. Lukatskaya, Y. Gogotsi, T. F. Jaramillo and A. Vojvodic, ACS Energy Lett., 2016, 1, 589–594 CrossRef CAS
- J. He, P. Lyu, L. Z. Sun, A. M. Garcia and P. Nachtigall, J. Mater. Chem. C, 2016, 4, 6500–6509 RSC
- W. Jin, S. Wu and Z. Wang, Phys. E, 2018, 103, 307–313 CrossRef CAS
- Y. Zhang, B. Sa, N. Miao, J. Zhou and Z. Sun, J. Mater. Chem. A, 2021, 9, 10882–10892 RSC
- B. Akgenc, Comput. Mater. Sci., 2020, 171, 109231 CrossRef CAS
- P. Gao, M. Song, X. Wang, Q. Liu, S. He, Y. Su and P. Qian, Nanomaterials, 2022, 12, 556 CrossRef CAS PubMed
- G. Kresse and J. Hafner, Phys. Rev. B: Condens. Matter Mater. Phys., 1993, 47, 558–561 CrossRef CAS PubMed
- G. Kresse and J. Furthmuller, Phys. Rev. B: Condens. Matter Mater. Phys., 1996, 54, 11169–11186 CrossRef CAS PubMed
- J. P. Perdew, K. Burke and M. Ernzerhof, Phys. Rev. Lett., 1996, 77, 3865–3868 CrossRef CAS PubMed
- G. Kresse and D. Joubert, Phys. Rev. B: Condens. Matter Mater. Phys., 1999, 59, 1758–1775 CrossRef CAS
- J. Heyd, G. E. Scuseria and M. Ernzerhof, J. Chem. Phys., 2006, 124, 219906 CrossRef
- S. Grimme, J. Comput. Chem., 2006, 27, 1787–1799 CrossRef CAS PubMed
- A. Togo, F. Oba and I. Tanaka, Phys. Rev. B: Condens. Matter Mater. Phys., 2008, 78, 134106 CrossRef
- X. Gonze and C. Lee, Phys. Rev. B: Condens. Matter Mater. Phys., 1997, 55, 10355–10368 CrossRef CAS
- K. Momma and F. Izumi, J. Appl. Crystallogr., 2011, 44, 1272–1276 CrossRef CAS
- S. Saha and T. P. Sinha, Phys. Rev. B: Condens. Matter Mater. Phys., 2000, 62, 8828–8834 CrossRef CAS
- M. Gajdos, K. Hummer, G. Kresse, J. Furthmuller and F. Bechstedt, Phys. Rev. B: Condens. Matter Mater. Phys., 2006, 73, 045112 CrossRef
- J. Bardeen and W. Shockley, Phys. Rev., 1950, 80, 72–80 CrossRef CAS
- R. X. Fei and L. Yang, Nano Lett., 2014, 14, 2884–2889 CrossRef CAS PubMed
- J. S. Qiao, X. H. Kong, Z. X. Hu, F. Yang and W. Ji, Nat. Commun., 2014, 5, 4457 CrossRef PubMed
- H. J. Zhang, Y. F. Li, J. H. Hou, K. X. Tu and Z. F. Chen, J. Am. Chem. Soc., 2016, 138, 5644–5651 CrossRef CAS PubMed
- Y. F. Li, Y. L. Liao and Z. F. Chen, Angew. Chem., Int. Ed., 2014, 53, 7248–7252 CrossRef CAS PubMed
- X. H. Zha, K. Luo, Q. W. Li, Q. Huang, J. He, X. D. Wen and S. Y. Du, Europhys. Lett., 2015, 111, 26007 CrossRef
- C. Si, K.-H. Jin, J. Zhou, Z. Sun and F. Liu, Nano Lett., 2016, 16, 6584–6591 CrossRef CAS PubMed
- H. Weng, A. Ranjbar, Y. Liang, Z. Song, M. Khazaei, S. Yunoki, M. Arai, Y. Kawazoe, Z. Fang and X. Dai, Phys. Rev. B, 2015, 92, 075436 CrossRef
- R. Peng, Y. Ma, B. Huang and Y. Dai, J. Mater. Chem. A, 2019, 7, 603–610 RSC
- H. Zhang, L. Liu and Z. Zhou, Phys. Chem. Chem. Phys., 2012, 14, 1286–1292 RSC
- H. J. Zhang, L. Liu and Z. Zhou, RSC Adv., 2012, 2, 9224–9229 RSC
- M. R. Hoffmann, S. T. Martin, W. Choi and E. W. Bahnemann, Chem. Rev., 1995, 95, 69–96 CrossRef CAS
- H. J. Zhang, X. Q. Zuo, H. B. Tang, G. Li and Z. Zhou, Phys. Chem. Chem. Phys., 2015, 17, 6280–6288 RSC
- H. J. Zhang, X. Zhang, G. Yang and X. M. Zhou, J. Phys. Chem. C, 2018, 122, 5291–5302 CrossRef CAS
- L. N. Wang, L. H. Xiao, Q. Jin and Q. Chang, J. Environ. Inform., 2022, 40, 30–40 Search PubMed
- L. H. Zhang, X. Y. Guo and S. P. Huang, Int. J. Hydrogen Energy, 2021, 46, 26950–26960 CrossRef CAS
- M. Munawar, M. Idrees, I. Ahmad, H. U. Din and B. Amin, RSC Adv., 2021, 12, 42–52 RSC
- A. Savin, R. Nesper, S. Wengert and T. F. Fässler, Angew. Chem., Int. Ed. Engl., 1997, 36, 1808–1832 CrossRef CAS
- B. Radisavljevic, A. Radenovic, J. Brivio, V. Giacometti and A. Kis, Nat. Nanotechnol., 2011, 6, 147–150 CrossRef CAS PubMed
- X. C. Ma, X. Wu, H. D. Wang and Y. C. Wang, J. Mater. Chem. A, 2018, 6, 2295–2301 RSC
- X. Zhang, X. D. Zhao, D. H. Wu, Y. Jing and Z. Zhou, Adv. Sci., 2016, 3, 1600062 CrossRef PubMed
- L. M. Zhang, J. W. Yu, M. M. Yang, Q. Xie, H. L. Peng and Z. F. Liu, Nat. Commun., 2013, 4, 1–7 Search PubMed
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3ra00303e |
| This journal is © The Royal Society of Chemistry 2023 |