
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceTowards electrochemical iridium recycling in acidic media: effect of the presence of organic molecules and chloride ions†
L. Moriau *ab,
K. Stojanovskic,
P. Jovanoviča,
D. Escalera-Lópezc,
S. Cherevkoc and
N. Hodnikad
*ab,
K. Stojanovskic,
P. Jovanoviča,
D. Escalera-Lópezc,
S. Cherevkoc and
N. Hodnikad
aDepartment of Materials Chemistry, National Institute of Chemistry, 1000 Ljubljana, Slovenia. E-mail: leonard.moriau@ki.si
bCenter of Excellence Low-Carbon Technologies, 1000 Ljubljana, Slovenia
cHelmotz-Institute Erlangen Nümberg for Renewable Energy (IEK-11), Forschunszentrum Jülich GmbH, Erlangen, Germany
dUniversity of Nova Gorica, 5000 Nova Gorica, Slovenia
First published on 9th March 2023
Abstract
The utilization of iridium is expected to surge in the next few years, notably due to the rising implementation of water electrolyzer devices in the energy transition. However, the natural resources of this noble metal are extremely limited and thus its recycling will become of high importance. Unfortunately, iridium is also the most corrosion resistant platinum group metal, making its recovery from waste a difficult and energy-demanding process. Hereby, we study the impact of organics and chloride ions on the electrochemical dissolution of iridium in order to pave the way towards green recycling of this precious metal. We present a 40 times increased dissolution when cycling iridium in presence of HCl and 1 M ethanol compared to HClO4. Our results point towards the direction of destabilizing Ir at relatively mild conditions in acidic media.
Introduction
Platinum group metals (PGMs) have been employed in many applications as biomedical and electronic devices, jewellery, alloying agents for hardening platinum and osmium, in the car industry and glass industry, for catalysis and electrocatalysis (e.g., proton exchange membrane fuel cells (PEMFCs) and water electrolyzers (WE)), etc.1–5 The latter ones will be particularly important in the transition towards clean energy sources; with platinum (Pt), iridium (Ir) and ruthenium (Ru) being the state-of-the-art electrocatalysts in these technologies.6,7 Although PGM-free electrocatalysts are currently being developed for PEMFC and WE applications, they are still at a development stage, requiring short/middle-term solutions to be developed based on PGM catalysts.8 However, the annual worldwide consumption of PGMs has steadily increased since the 1980s and is expected to continue accordingly in the next decades.9–11 Besides this, the mining production of PGMs is difficult, requires strong chemicals and is energy-intensive.12,13 Their natural sources are geologically unevenly distributed (mostly located in the South African Republic and Russia) and thus present an additional supply risk for the production of PGM-containing devices. For this reason, the European Union declared PGMs to be critical raw materials (CRMs).14 It is expected that the supply from natural sources will not meet the demand, and, in the future, the recycling of these precious metals will take even bigger importance in the world supply.3,15,16Nowadays, the recycling processes of PGMs are still costly and energy-intensive (e.g., pyrometallurgy), using toxic chemicals (e.g., aqua regia) which are not environmentally friendly and usually need to be adjusted to the specific waste origin.9,17,18 Therefore, new and greener recycling processes for PGMs need to be developed.
Considering the (future) importance of iridium in the hydrogen economy, as the state-of-the-art oxygen evolution catalyst and its extremely low mining yield (less than 10T per year worldwide),19,20 it is crucial to investigate an economically viable recycling process. Indeed, the current WE technology requires 1–2 g of iridium per kW of electrical energy, which means that with a target of 200 GW (energy needed for Germany alone),21 100 to 200 tons of iridium would be needed.20,22 Even if some breakthrough solutions are found to reduce (if not fully replace) the amount of Ir needed in the future, its recycling from waste and end-of-life catalysts is of utmost importance. Unfortunately, iridium is one of the most corrosion resistant of all the PGMs, which makes it difficult to recycle, even more under mild hydrometallurgical conditions.23 The state-of-the-art recycling technology for PGMs/iridium is either using a combination of aqua regia and heating,17,24 a Smelter-Leaching-Electro Winning-Refining process developed by Umicore25,26 or the Rose process used by Tanaka (utilization of metal oxide as PbO to extract PGMs at high temperature).27 All these processes employ toxic chemicals and exposure to high temperatures that increase the energy/environmental cost.
One promising way to achieve energy-efficient recycling of iridium is by using electrochemical processes. Compared to pyrometallurgy, electrochemical dissolution or leaching can be considered a type of hydrometallurgy, which is known to have many benefits such as being environmentally friendly. Indeed, Cherevko et al. showed that all the PGMs transiently dissolved when cycled in a potential window sufficiently broad to undergo oxidation (onset of significant dissolution is 0.9 VRHE) and subsequent oxide reduction.28,29 During the anodic sweep, metallic iridium Ir (0) is firstly oxidized to Ir (3+), forming in acidic media intermediate surface oxides/hydroxides and dissolved species [Ir(H2O)6]3+. Afterwards, the oxidation continues until stable IrO2 is formed. Similarly, when the iridium oxide is reduced back to metallic iridium, Ir (3+) is formed and dissolved during the reaction.30 Therefore, by imposing a sufficient number of cycles between reductive and oxidative potentials, any precious metal could theoretically be completely dissolved as successfully demonstrated by Hodnik et al. for Pt and Pd.31 In addition, iridium is prone to dissolution when catalysing the oxygen evolution reaction (OER). When OER occurs on iridium catalysts, different reaction pathways are possible. One of the plausible OER mechanisms proceeds via oxygen consumption from the Ir-oxide lattice, inducing dissolution of iridium.32 Therefore, the two possible approaches to electrochemically dissolve iridium are by either alternating surface Ir redox chemistry or by pushing the lattice oxygen evolution reaction, which could be used for recycling under mild conditions.
In industrial recycling reactors, disparate waste containing PGMs are mixed with the lixiviant solution (electrolyte) without any physical contact which allows recycling of large quantities. But to gather insights on impact of any electrochemical parameters on the iridium dissolution,33,34 controlled experiments are required, and thus, laboratory conditions. In this case, a film of the powder catalyst/material of interest is deposited on a conductive working electrode on which a potential is applied. In addition, it can be coupled to various analytical method as inductively coupled plasma mass spectrometry (ICP-MS), identical location transmission electron microscopy (IL-TEM), Raman spectroscopy, etc35–38. However, it can be difficult to transfer the electrochemical parameters from the laboratory experiment to an industrial reactor. Nonetheless, Hodnik et al. proposed an elegant solution to mimic the oxidation and reduction cycles of electrochemical cell by using oxidative and reductive reactive gases.31 In a study of Pt dissolution, the authors varied the saturated gas by purging O3 to impose oxidative potential and create precious metal oxides, followed by a purge with a reductive gas as CO or H2 to reduce the oxides previously formed. Accordingly, the authors showed that Pt was dissolving with the same transient dissolution mechanism as in electrochemical cycling.39–41 The use of gases such as O3 and H2 in diluted acidic electrolyte correspond to a “mild and green” alternative to the existing recycling processes used such as aqua regia. One of the limitation of this technology is that the upper potential is lowered by the relatively low saturation concentration of ozone in the electrolyte. The theoretical oxidizing potential of O3 is 2.07 VNHE, which would be more than enough to oxidize all PGM, and in particular also Ir.42,43 However in practice, the open circuit potential (OCP) under ozone in aqueous electrolyte is between 1.385 VRHE and 1.6 VRHE for a Pt working electrode31,44 as it can vary depending on the electrolyte composition and/or the working electrode material as well as the ozone concentration in the case of only one redox reactions (following Nernst equation). Another possibility for the lower observed OCP is the presence of other electrochemical reactions like the splitting of water in the cell, leading to a mixed potential for the OCP. In order to reduce the oxidized metals hydrogen or CO can be used. In addition to gases, other compounds can be used as reducing agents, i.e. organics were shown to act like this during the synthesis of nanoparticles,45–47 and they could be used alone or in complementarity with H2 to reduce nanoparticles.
One way to increase the dissolution of metals is by using a complexing agent. It is well known that the presence of organic molecules, ions, gases, etc, can increase the dissolution of precious metals either by preventing redeposition or by “chelating” the metal ions. For example, the dissolution of Pt was shown to be higher in presence of organics48,49 or chlorides;50–52 similarly, gold dissolves more in presence of chloride, bromide, and cyanides.53–56 Therefore, organics molecules could have the double benefit of acting as reducing agents and complexing compounds.
However, in the case of iridium, only a few studies about its stability in presence of organics have been performed.57,58 Interestingly, it is often used as a catalyst for the oxidation of organics (organics oxidation reaction, OOR) and several studies, firstly by Comninellis et al., were conducted on the mechanism of oxidation.59–63 The authors reported that iridium has a typical active catalyst behaviour in these reactions. Hence, the oxidation of organics that occurs on such oxide anodes (MOx) is selective as opposed to the non-selective oxidation occurring on “non-active” electrodes and leading to CO2.60,61 Therefore, the OOR driven by active MOx involves a reaction pathway leading to the formation of a higher oxide MOx+1. At the potential of OOR and/or OER, metallic iridium is already oxidized (generally to IrO2), and thus the formation of higher oxide during the oxidation of organics involves the formation of IrO3. This compound is also an intermediate participating in the oxygen evolution reaction pathway dominating at high potentials as shown by Kasian et al.30 The authors also suggested that it can further be oxidized to IrO42− in presence of H2O.30 Thus, organics oxidation, OER and iridium dissolution could be conceptually linked. Several authors stated that organics oxidation seems to facilitate the OER and vice versa.64–66 Therefore, the iridium dissolution during OER could be in principle facilitated and be achieved at lower potentials, opening a path towards “green” electrochemical recycling of iridium.
In our study, we investigate different electrolyte additives and conditions for the dissolution of iridium. First, the impact of different organic molecules on iridium dissolution was studied in 0.1 M HClO4. Namely, the dissolution in presence of 0.05, 0.1, 0.5 and 1 M of isopropanol (IPA), formic acid (FA) and ethanol was tested. Isopropanol and ethanol were chosen over methanol based on a previous study by Simond and Comninellis where they showed the higher reactivity for the chosen organics, which is favorable to trigger iridium dissolution during the organic oxidation.59 However, the authors specified that longer carbon chain of primary alcohols would be detrimental for reactivity on iridium and thus slower organic oxidation reaction is expected. Afterwards, the dissolution in presence of the most impactful organics was performed in HCl electrolyte. The impact of chlorides on iridium dissolution was proven. Finally, hypotheses for the observed enhanced dissolution are provided.
Experimental
H-cell electrochemical experiments
Fluorine doped tin oxide (FTO)-coated glass slides were used as working electrode support. First, the FTO substrates were sequentially cleaned by 10 minutes sonication in Hellamnex, MilliQ water and Isopropanol, respectively. The FTO was abundantly rinsed with MilliQ water between each sonication step. To ensure good electrical contact, a silver paste layer was added on the top of the FTO. Next, a small layer of silver epoxy was put around the silver paster layer to avoid any electrolyte to touch the electrical contact by capillary rise during the experiment. Once the FTO support was fully conditioned, a 2.2 mg mL−1 commercial Ir-black (Alfa Aesar, extensively characterized in the literature67–69) ink was sonicated with an ultrasonication horn (Branson, SFX 150) in an ice bath before 10 μL of the suspension was deposited on the FTO. The final Ir loadings were estimated to be of ca. 75–90 μg cm−2, based on drop-casted spot diameter analysis by laser microscopy (Keyence VK-X250). This concentration was chosen to be close to values commonly reported for Ir rotating disk electrode (RDE) testing. Next, the Ir-black on FTO was transferred in a H-cell: a glassy carbon counter electrode (CE) was placed in the opposite cell compartment, separated by a glass frit, to avoid any redeposition of iridium on it during the experiment. To monitor the dissolution rates of Ir, an auto-sampler (Liquid handler, Gilson GX-271 Prep LH) was extracting electrolyte aliquots during electrochemical treatment. One cycle consists in 2 minutes at 0 VRHE (simulation of hydrogen saturated) followed by a linear sweep to 1.385 VRHE at 20 mV s−1, 2 minutes at 1.385 VRHE (simulation of ozone saturation) and a final linear sweep to 0 VRHE at 20 mV s−1. Electrolyte aliquots were extracted at the end of the second linear sweep after the cycles number 0, 2, 4, 6, 8, 10, 15, 20, 25, 30, 50, 75, and 100. In order to monitor potential diffusion of dissolved Ir across the H-cell compartments, aliquot samples were also extracted from the CE compartment. The potential was controlled by a Gamry potentiostat (Interface 1000B ZRA). The electrolyte solutions were made with ultrapure deionized water (Merck, MilliQ IQ 7000) and commercially available chemicals (Table S1†).Liquid sample analysis: offline ICP-MS
Dissolved iridium content in aliquots was analysed with an Inductively Coupled Plasma – Mass Spectrometer (ICP-MS, PerkinElmer NexION 300×). The samples were diluted in order to reach organics concentration lower than 5 wt% (0.05 M maximum concentration), the tolerable limit of the instrument. The instrument was calibrated with a set of standard solutions of a known amount of metal using the same electrolyte matrix as studied (HClO4 or HCl + organics). An internal standard of 187Re (for Ir) and 103Rh (for Sn) was run through the ICP-MS during the experiment to ensure the stability of the system during the measurement time.Online-ICP-MS
Online-ICP-MS study was performed by connecting a scanning flow cell to the ICP-MS (PerkinElmer NexION 300×) as described in previous articles.70,71 The working electrode was a polycrystalline bulk iridium crystal while the counter and reference electrodes were a carbon rod and a double junction Ag/AgCl (Metrohm), respectively. Prior to the start of the electrochemical protocol, the potential was held at 0.8 VRHE for 2 min to allow electrochemical contact with the scanning flow cell (cell opening diameter = 2 mm). Afterwards, a slow cycle (5 mV s−1) was performed up to 1.385 VRHE, where the potential was then held for 30 s to mimic an ozone-induced potential. Finally, the electrolyte was saturated with H2 and the potential was allowed to go to OCP during 3 min. Subsequently, two additional cycles (slow linear ramp, 30 s hold and 3 min at OCP) were performed.Results and discussion
First, the impact of organics on dissolution was studied in a chloride-free electrolyte (0.1 M HClO4) in an H-cell configuration. Ir-black was deposited on FTO and cycled in 0.1 M HClO4 electrolyte containing different concentrations of the organic compounds: 0.05 M, 0.1 M, 0.5 M and 1 M of isopropanol (IPA), formic acid (FA), and ethanol (for further details see experimental).In all the cases, the dissolution of iridium increases with the concentration of organics (Fig. 1). After 100 cycles, the iridium concentration in solution is 0.56, 0.79, 1.27 and 1.88 μgIr L−1 in presence of 0.05, 0.1, 0.5 and 1 M of formic acid, respectively (Fig. 1a). Similar trends are observed in presence of IPA (Fig. 1b) and ethanol (Fig. 1c), albeit a slower improvement for low concentrations of IPA. Therefore, this leads us to believe that the presence of organics promotes iridium dissolution. However, the amount of dissolved iridium varies highly depending on the organic compound. Namely, in presence of 1 M ethanol an iridium dissolution of 9.48 μg L−1 was recorded after 100 cycles, which corresponds to 10 and 5 times the dissolution in presence of 1 M IPA and 1 M FA with 0.87 μg L−1 and 1.88 μg L−1, respectively (Fig. S1† and Table 1).
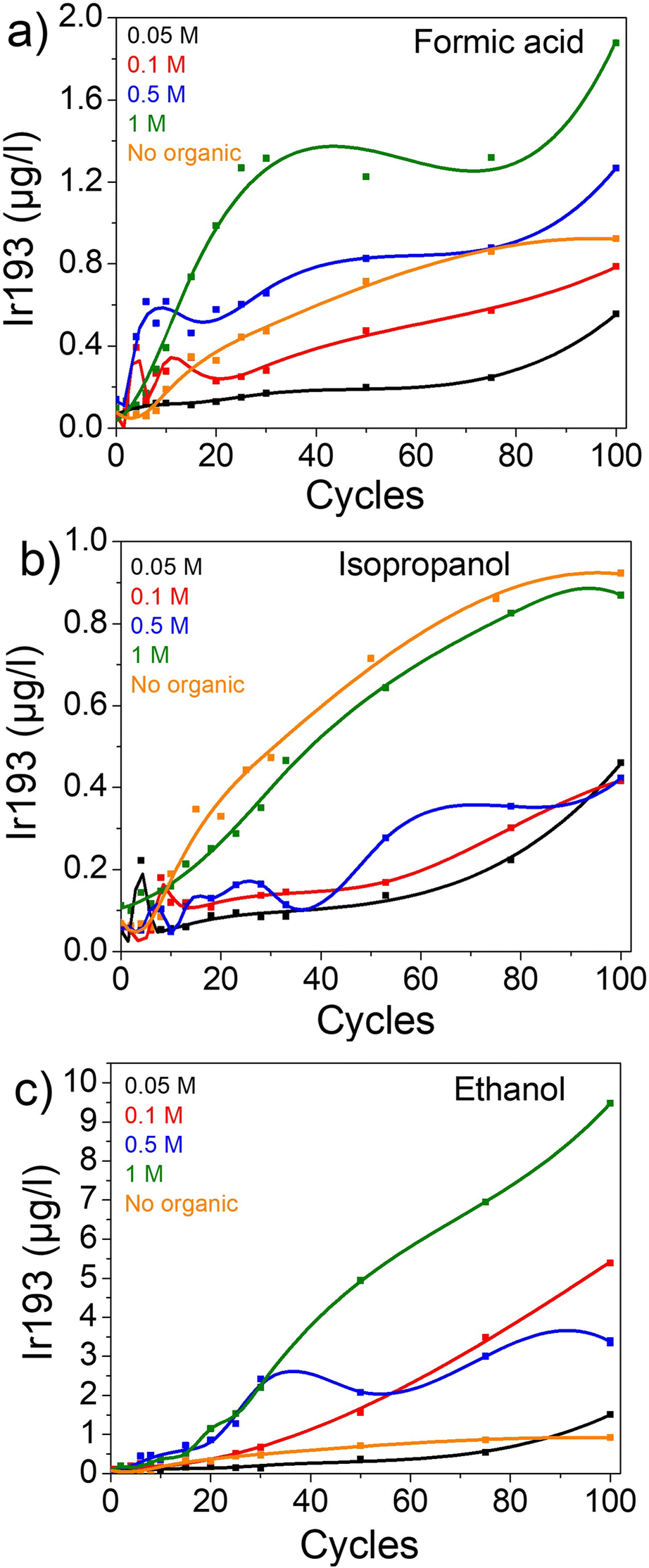 | ||
| Fig. 1 Dissolution of iridium in different concentrations of organics in 0.1 M HClO4. (a) In formic acid. (b) In isopropanol. (c) In ethanol. | ||
| Electrolyte concentration (M) | Formic acid | Isopropanol | Ethanol |
|---|---|---|---|
| 0 (HClO4) | 0.92 ± 0.13 | 0.92 ± 0.13 | 0.92 ± 0.13 |
| 0.05 | 0.56 ± 0.08 | 0.46 ± 0. 06 | 1.51 ± 0.22 |
| 0.1 | 0.79 ± 0.11 | 0.42 ± 0.06 | 5.39 ± 1.12 |
| 0.5 | 1.27 ± 0.09 | 0.42 ± 0.06 | 3.34 ± 0.49 |
| 1 | 1.88 ± 0.08 | 0.87 ± 0.04 | 9.48 ± 0.42 |
Nonetheless, it is surprising to see a rise in dissolution with increasing isopropanol concentration but still an overall lower dissolution than in 0.1 M HClO4 alone (0.92 μg L−1) (Fig. 1b).
One explanation is the formation of strongly adsorbing species and consequent blockage of the surface. Indeed, the oxidation of isopropanol can lead to acetone, which is known to block the surface of other PGMs.57,72,73 As an increase in high isopropanol concentration is correlated with an increase in iridium dissolution, isopropanol oxidation induces this concomitant iridium degradation. Thus, on one hand there is less dissolution than in pure HClO4 due to the process of blockage of the transient oxidation/reduction reaction of Ir surface by acetone or possibly isopropanol (IPA). On the other hand, when a significantly higher amount of isopropanol is present, the process of oxidation of an organic compound is promoted which seems to induce iridium dissolution and counterbalance the blocking ability of IPA. Therefore, two separate processes are occurring at the same time, where the dissolution of Ir is accelerated only when the IPA concentration is sufficiently increased.
Similarly, low amounts of formic acid seem to hinder the oxidation/reduction of iridium and thus decrease the following iridium dissolution. However, a higher amount of the organic compound results in boosted dissolution compared to HClO4. Hence, during formic acid oxidation no more iridium oxide is left to be further reduced and thus transiently dissolved. Indeed, the reaction with formic acid happens following an two steps electrochemical-chemical mechanism (see ESI and Fig. S3† for more details) which consumes all the iridium oxides. It also explaines why higher amount of FA, and thus higher FA oxidation, leads to more iridium dissolution. Therefore, at sufficient FA concentration the degradation induced by the FA oxidation overcomes the decrease of dissolution from the transient processes. A similar phenomenon has been previously observed for the dissolution of Pt in presence of different concentrations of Cl−.74 In addition, the presence of adsorbed organics (reactant and products) on the iridium surface could limit the iridium redeposition, and thus increase the iridium concentration in the solution.70
In the case of ethanol, its complete oxidation yields CO2 and H2O while the partial oxidation, expected on Ir-based catalysts, yields acetic acid (CH3COOH).61 Thus, and evidently from the absence of blocking ability, no poisoning species are formed.59 Therefore, neither the oxidation/reduction processes are hindered nor its associated dissolution of iridium. This mechanism, in addition to some dissolution induced during the oxidation of ethanol, could explain the higher degradation observed for ethanol-containing electrolytes.
As ethanol has the most impact on the dissolution, the same experiment was conducted with 0.1 M HCl as a supporting electrolyte instead of HClO4 (Fig. 2). In addition, a hold experiment was conducted to prove the importance of cycling. Thus, the potential was held at 1.385 VRHE for the same amount of time as 100 cycles. The electrolyte aliquots were taken after a time equivalent to the number of cycles on x axis in Fig. 2a and b. It is clearly visible that during the hold experiment iridium is much more stable, emphasizing the importance of potential cycling between oxidizing and reducing potentials to trigger transient dissolution (Fig. 2a). In other words, during the hold potential an iridium oxide layer is formed with a first dissolution. Afterwards, the iridium layer is stable and the dissolution is heavily hindered. In the case of cycling, the switch between oxidative and reductive potential induces a transient dissolution as previously observed by Cherevko et al.28,29 Moreover, the impact of Cl− on iridium dissolution is noticeable with a dissolution around 10 times higher for 0.1 M HCl compared to 0.1 M HClO4 with 9.72 μgIr L−1 vs. 0.92 μgIr L−1 after 100 cycles, respectively. After the addition of ethanol, the same trend is observed as previously with an increase in dissolution with increasing organic concentration (Fig. 2a).
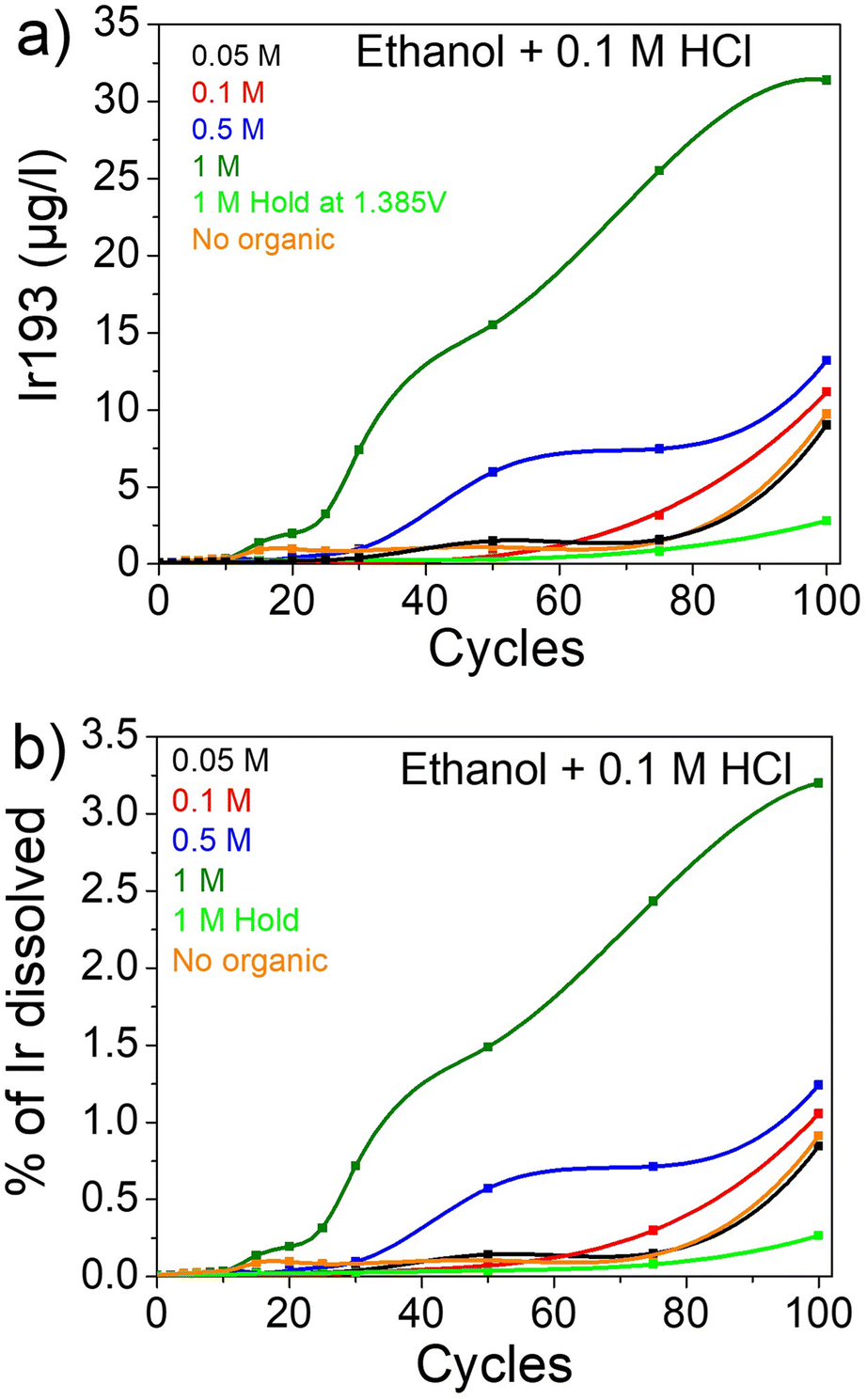 | ||
| Fig. 2 Dissolution of iridium in presence of ethanol in HCl. (a) μgIr L−1 and (b) % of Ir dissolved. | ||
In this experiment, both the importance of Cl− and of ethanol for enhancing dissolution are clearly highlighted. It was shown that Cl−does not chemically attack Ir films but it facilitates the formation of IrxCly soluble complexes.71 Unfortunately, even if the leaching of iridium is greatly enhanced, the overall dissolution is still low with around 0.91% of the iridium dissolved in 0.1 M HCl (corresponding to 5.19% of an iridium monolayer, see Discussion S1† for details) and only 0.08% in 0.1 M HClO4 (0.5% of a monolayer) after 100 cycles (Fig. 2b and S2†). In the same way, when 1 M ethanol was added to both electrolytes, the amount of Ir dissolved after 100 cycles increases from 0.91% and 0.08% to 3.2% (16.87% of a monolayer) and 0.9% (5.1% of a monolayer) for HCl and HClO4, respectively. Thus, the dissolution of iridium is enhanced 40 times when 1 M ethanol and chlorides are presents in solution compared to a HClO4 electrolyte, even when cycled to “low” potentials where iridium is mostly stable.
The mechanism of iridium dissolution in presence of organics was then investigated with an online ICP-MS set-up.70,71 First, a slow oxidative ramp was applied to mimic the introduction of ozone in the system (I in Fig. 3a). Afterwards, the potential is held at 1.385 VRHE (ozone simulated potential, II) before the introduction of H2 in the system which produces a rapid drop of the potential to around 0.05 VRHE (III). Fig. 3a shows the dissolution of iridium in HCl and 0.05 M of ethanol. The low organic concentration is necessary to avoid damage to the ICP-MS. The low concentration could explain the similar dissolution observed for 0.1 M HCl and 0.1 M HCl + 0.05 M ethanol, as was the case in the H-cell experiments (Fig. 2a black and orange curves).
 | ||
| Fig. 3 (a) Online dissolution of iridium during cycling in presence of 0.1 M HCl and 0.1 M HCl + 0.05 M ethanol. (b) Online dissolution of iridium during three cycles in presence of organics (0.05 M) in 0.1 M HCl. (c) Zoom in one cycle. | ||
Additionally, the experiment was also conducted in presence of FA and IPA (Fig. 3b and c). The higher dissolution observed compared to HCl alone while there is a flow of electrolyte hints that the blockage of the surface could indeed be the limiting factor in the H-cells experiments.
Qualitatively, the dissolution process is similar in all the cases (Fig. 3b). A small anodic peak is observed during the slow ramp to 1.385 VRHE (onset of 1.04 ± 0.02 VRHE) corresponding to the oxidation process of iridium (Ir0/Ir3+ redox couple)28,29. Afterwards, a passivating oxide layer is formed, and the dissolution decreases during the “ozone-potential” hold (II). Then, a higher cathodic peak is visible in agreement with the introduction of H2 to the system and the reductive atmosphere. It corresponds to the reduction of the previously formed iridium oxide layer through the formation of soluble Ir3+ before reaching metallic state.30 While looking at only one cycle (Fig. 3c), it can be observed that the anodic peak is higher in presence of organics, possibly due to the OOR. However, the cathodic peak can have a lower maximum intensity. This kind of mechanism in presence of iridium and IPA has already been observed.57
Moreover, a longer time is needed to reach the background after the reduction step (III) in presence of organics which could indicate a destabilization of the iridium surface or some reaction with organics. Therefore, the overall dissolution during the three cycles is higher with organics (Table 2), even with the lower amounts used in this experiment (0.05 M) compared to the H-Cell (up to 1 M).
| Electrolyte | Dissolved iridium (ngIr) | Dissolution compared to HCl (%) |
|---|---|---|
| 0.1 M HCl | 2.77 ± 0.13 | 100 |
| +0.05 M ethanol | 3.07 ± 0.08 | 111 ± 2.6% |
| +0.05 M FA | 3.65 ± 0.07 | 132 ± 1.9% |
| +0.05 M IPA | 3.39 ± 0.16 | 125 ± 4.7% |
Fig. 4 shows a scheme of the plausible ongoing reaction mechanisms, based on the work of Comninellis et al.59–62 and Kasian et al.30 In absence of organics and at low potentials, iridium will have the tendency to catalyst OER trough the formation of HIrO2 (steps 1; 4 and 6 in green) with the consequent dissolution mechanism due to the formation of Ir3+ (step 5). At higher potential without organics (above 1.6 VRHE),30 the mechanism involving IrO3 starts to be preferred (steps 1; 2 and 3 in red). The latter is linked with higher stability due to the higher kinetics of the formation of O2 and regeneration of IrO2 over the formation of IrO42− as shown with online electrochemical mass spectrometer (OLEMS) by Kasian et al.30 In our particular case, the OOR has a common intermediate with the “high potentials” pathway, IrO3. Therefore, this pathway (steps 1; 2 and 3) could be promoted in presence of organics, even at low potentials. This could lead to higher formation of the soluble IrO42−. In addition, more IrO3 is formed in presence of organics and thus more oxides are present. Consequently, more oxide can be reduced when going to the reductive environment, linked to a higher transient dissolution process. Based on CVs in presence of ethanol (Fig. S3†), the iridium oxide is additionally reduced during the organics oxidation as hypothesized in the step 3 and accordingly to the work of Comninellis,60 even at low potentials.
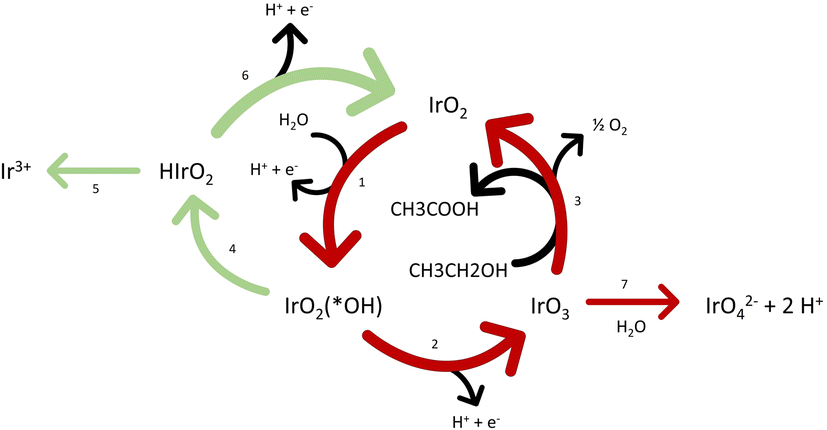 | ||
| Fig. 4 Scheme of the combined OOR and OER pathways for ethanol. | ||
These phenomena, in addition to some ligand effect in solution (Cl− is promoting the formation of soluble complexes) which limits the redeposition and possible destabilization of the surface, could explain the enhanced iridium dissolution.
Conclusions and outlook
Our study showcased that an enhanced dissolution of iridium under mild conditions was presented as a first step toward the green electrochemical recycling process. The use of organic compounds was proven to be efficient to increase iridium leaching as a higher concentration of organics in solution was correlated with an increased iridium dissolution. The presence of chlorides was also shown to be beneficial for the leaching of iridium with a 10-fold increase for HCl electrolyte over HClO4. In addition, a 10-fold and 3.5-fold increase of iridium leaching was observed in 0.1 M HClO4 and 0.1 M HCl, respectively, after the addition of 1 M ethanol. In the case of 1 M ethanol in 0.1 M HCl, the dissolution was enhanced 40 times compared to just 0.1 M HClO4.In a closed system (H-Cell), other organics like formic acid and isopropanol are not efficient, due to the blockage effect of the iridium surface by products of the OOR. This is in line with the fact that when the iridium dissolution was recorded with a flow of electrolyte (online ICP-MS), all three organics showed improved dissolution over pure 0.1 M HCl, even with low concentration (0.05 M). Moreover, the importance of cycling between oxidative and reductive environments was proven with almost no dissolution in the hold experiments, even when both HCl and ethanol were used.
Theoretical studies should be pursued in the future to confirm our proposed Ir leaching mechanisms. Namely, three different processes could participate to the enhanced dissolution. First, the formation of soluble complexes with iridium by Cl− and organics compounds can prevent the redeposition and drive the dissolution. Secondly, the formation of more iridium oxide or formation of less stable oxide could explain the higher dissolution during transient processes. Finally, a possible dissolution during the OOR itself or driving the formation of soluble IrO42− could participate in the increased dissolution.
Altogether, an increased dissolution of iridium was achieved under relatively mild conditions. The synergistic impact of Cl− and of organic compounds on the iridium dissolution was shown for the first time. Nonetheless, theoretical studies as well as investigations under realistic recycling reactor conditions to reach complete Ir dissolution still need to be performed to fully understand and enhance the proof-of-concept presented here. Finally, other parameters such as acid concentration, chloride ions concentration, type of acid or other halides (Br− or I−) could also influence the iridium dissolution and will be investigated in future work.
Author contributions
L. Moriau: investigation, methodology, formal analysis, writing – original draft, visualization. K. Stojanovski: investigation, resources. P. Jovanovič: writing – review and editing. D. Escalera-López: resources, methodology, writing – review and editing, validation. S. Cherevko: conceptualization, funding acquisition, writing – review and editing, supervision. N. Hodnik: conceptualization, funding acquisition, writing – review and editing, supervision.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was financially supported by the Slovenian Research Agency for the research programs P2-0393 and I0-0003, projects Z2-8161 and N2-0155, ERA-MIN2 project Reduction/Oxidation Recycling (RedOxRec, EU H2020) and a bilateral exchange project between Germany and Slovenia BI-DE/18-19-001.Notes and references
- J. Emsley, Nature's Building Blocks: An A-Z Guide to the Elements, 2nd edn, Oxford University Press, New York, 2011 Search PubMed.
- T. Biggs, S. S. Taylor and E. van der Lingen, Platinum Met. Rev., 2005, 49, 2 CrossRef CAS.
- C. Hageluken, Platinum Met. Rev., 2012, 56, 29 CrossRef.
- E. Antolini, ACS Catal., 2014, 4, 1426 CrossRef CAS.
- J. G. Vos, Z. Liu, F. D. Speck, N. Perini, W. Fu, S. Cherevko and M. T. M. Koper, ACS Catal., 2019, 9, 8561 CrossRef CAS.
- I. Katsounaros and M. T. M. Koper, “Electrocatalysis for the hydrogen economy” in Electrochemical Science for a Sustainable Society, Springer, Cham., 2017, vol. 23 Search PubMed.
- I. Katsounaros, S. Cherevko, A. R. Zeradjanin and K. J. J. Mayrhofer, Angew. Chem., Int. Ed., 2014, 53, 102 CrossRef CAS PubMed.
- C. S. Gittleman, A. Kongkanand, D. Masten and W. Gu, Curr. Opin. Electrochem., 2019, 18, 81 CrossRef CAS.
- J. He and A. Kappler, Microb. Biotechnol., 2017, 10, 1194 CrossRef PubMed.
- C. Mack, B. Wilhelmi, J. R. Duncan and J. E. Burgess, Biotechnol. Adv., 2007, 25, 264 CrossRef CAS PubMed.
- L. S. Morf, R. Gloor, O. Haag, M. Haupt, S. Skutan, F. Di Lorenzo and D. Boni, J. Waste Manage., 2013, 33, 634 CrossRef CAS PubMed.
- J. O. Marsden and C. I. House, The Chemistry of Gold Extraction, 2nd edn, The Society for Mining Metallurgy and Exploration Inc., USA, 2006 Search PubMed.
- R. J. Seymour and J. I. Farrelly, Kirk–Othmer encyclopedia of chemical technology, 5th edn Wiley-Interscience, John Wiley & Sons, Hoboken, New Jersey, vol. 19, 2006 Search PubMed.
- E. Commision, Critical raw materials, European Commision, 2020, available, https://ec.europa.eu/growth/sectors/raw-materials/specific-interest/critical_en, accessed 30 October 2022 Search PubMed.
- T. E. Graedel, J. Allewood, J.-P. Birat, B. K. Reck, S. F. Sibley, G. Sonnemann, M. Buchert and C. Hageluken, Recycling rates of Metals– a Status Report, a Report of the Working Group on the Global Metal Flows to the International Resource Panel, UNEP, 2011 Search PubMed.
- H. Dong, J. Zhao, J. Chen, Y. Wu and B. Li, Int. J. Miner. Process., 2015, 145, 108 CrossRef CAS.
- E. van Westing, V. Savran and J. Hofman, Recycling of metals from coatings – a desk study, NL Agency, 2013 Search PubMed.
- A. J. Chandler, T. Eighmy, O. Hjelmar, D. Kosson, S. Sawell, J. Vehlow, H. A. van der Sloot and J. Hartlen, Municipal Solid Waste Incinerator Residues, Elsevier, Amsterdam, 1997 Search PubMed.
- A. E. Hughes, N. Haque, S. A. Northey and S. Giddey, Resources, 2021, 10, 93 CrossRef.
- Milestone for green hydrogen: Heraeus launches new electrocatalysts, September 2020, Hanau, https://www.heraeus.com/en/hpm/hpm_news/2020_hpm_news/09_milestone_for_green_hydrogen.html Accessed 5th September 2022 Search PubMed.
- B. Burger, Installierte Leistung zur Stromerzeugung fossil/nuklear (linke Balken) und erneuerbar (rechte Balken), Öffentliche Nettostromerzeugung in Deutschland im Jahr 2021, 2022 Search PubMed.
- M. Bernt, A. Hartig-Weiss, M. F. Tovini, H. A. El-Sayed, C. Schramm, J. Schroter, C. Gebauer and H. A. Gasteiger, Chem. Ing. Tech., 2019, 92, 31 CrossRef.
- M. Pourbaix, in Aqueous Solution, ed. National Association of Corrosion Engineers, 1974, ch. IV, section 13, vol. 5, pp. 373–377 Search PubMed.
- Y. J. Park and D. J. Fray, J. Hazard. Mater., 2009, 164, 1152 CrossRef CAS PubMed.
- C. Hageluken, in Tackling e-waste towards efficient management techniques, ed. L. V. Rajeshwari, S. Basu and R. Johri, TERI Press, New Delhi, 2007, pp.97–104 Search PubMed.
- C. Hagelüken and C. W. Corti, Gold Bull., 2010, 43, 209 CrossRef.
- T. Nagai, H. Kumakura, M. Abe, K. Seki and D. Noguchi, in The Minerals, Metals & Materials Series, ed. Springer, 2017, Cham Search PubMed.
- S. Cherevko, J. Electroanal. Chem., 2017, 787, 11 CrossRef CAS.
- S. Cherevko, in Encyclopedia of Interfacial Chemistry, ed. K. Wandelt, Elsevier, 2018 Search PubMed.
- O. Kasian, J.-P. Grote, S. Geiger, S. Cherevko and K. J. J. Mayrhofer, Angew. Chem., Int. Ed., 2018, 57, 2488 CrossRef CAS PubMed.
- N. Hodnik, C. Baldizzone, G. Polymeros, S. Geiger, J.-P. Grote, S. Cherevko, A. Mingers, A. Zeradjanin and K. J. J. Mayrhofer, Nat. Commun., 2016, 7, 13164 CrossRef CAS PubMed.
- O. Kasian, S. Geiger, T. Li, J.-P. Grote, K. Schweinar, S. Zhang, C. Scheu, D. Raabe, S. Cherevko, B. Gaut and K. J. J. Mayrhofer, Energy Environ. Sci., 2019, 12, 3548 RSC.
- S. Cherevko, S. Geiger, O. Kasian, A. Mingers and K. J. J. Mayrhofer, J. Electroanal. Chem., 2016, 773, 69 CrossRef CAS.
- S. Cherevko, S. Geiger, O. Kasian, A. Mingers and K. J. J. Mayrhofer, J. Electroanal. Chem., 2016, 774, 102 CrossRef CAS.
- P. Jovanovic, N. Hodnik, F. Ruiz-Zepeda, I. Arcon, B. Jovanovic, M. Zorko, M. Bele, M. Sala, V. S. Selih, S. Hocevar and M. Gaberscek, J. Am. Chem. Soc., 2017, 139, 12837 CrossRef CAS PubMed.
- L. Moriau, M. Bele, Z. Marinko, F. Ruiz-Zepeda, G. Koderman Podborsek, M. Sala, A. K. Surca, J. Kovac, I. Arcon, P. Jovanovic, N. Hodnik and L. Suhadolnik, ACS Catal., 2021, 11, 670 CrossRef CAS PubMed.
- S. Cherevko and K. J. J. Mayrhofer, On-line inductively coupled plasma spectrometry in electrochemistry: Basic principles and applications, 2018, pp. 326–335 Search PubMed.
- K. Schlogl, K. J. J. Mayrhofer, M. Hanzlik and M. Arenz, J. Electroanal. Chem., 2011, 662, 355 CrossRef.
- A. A. Topalov, S. Cherevko, A. R. Zeradjanin, J. C. Meier, I. Katsounaros and K. J. J. Mayrhofer, Chem. Sci., 2014, 5, 631 RSC.
- S. Cherevko, G. P. Keeley, S. Geiger, A. R. Zeradjanin, N. Hodnik, N. Kulyk and K. J. J. Mayrhofer, ChemElectroChem, 2015, 2, 1471 CrossRef CAS PubMed.
- P. Jovanovic, A. Pavlisic, V. S. Selih, M. Sala, N. Hondik, M. Bele, S. Hocevar and M. Gaberscek, ChemCatChem, 2014, 6, 449 CrossRef CAS.
- M. R. Viera, M. Fernandez Lorenzo de Mele and H. A. Videla, J. Appl. Electrochem., 2001, 31, 591 CrossRef CAS.
- R. C. Weast, Handbook of Chemistry and Physics, The Chemical Rubber Co., Cleveland OH, 1970 Search PubMed.
- C. Baldizzone, S. Mezzavilla, N. Hodnik, A. R. Zeradjanin, A. Kostka, F. Schuth and K. J. J. Mayrhofer, Chem. Commun., 2015, 51, 1226 RSC.
- R. S. Varma, ACS Sustainable Chem. Eng., 2016, 4, 5866 CrossRef CAS PubMed.
- Y. Wang, Z. Zhu, F. Xu and X. Wei, J. Nanopart. Res., 2012, 14, 755 CrossRef.
- J. Kou and R. J. Varma, RSC Adv., 2012, 2, 10283 RSC.
- M. Fayette, J. Nutariya, N. Vasiljevic and N. Dimitrov, ACS Catal., 2013, 3, 1709 CrossRef CAS.
- P. Jovanovic, F. Ruiz-Zepeda, M. Sala and N. Hodnik, J. Phys. Chem. C, 2018, 122, 10050 CrossRef CAS.
- A. Pavlisic, P. Jovanovic, V. S. Selih, M. Sala, N. Hodnik, S. Hocevar and M. Gaberscek, Chem. Commun., 2014, 50, 3732 RSC.
- S. Geiger, S. Cherevko and K. J. J. Mayrhofer, Electrochim. Acta, 2015, 179, 24 CrossRef CAS.
- R. Torres and G. T. Lapidus, Hydrometallurgy, 2016, 166, 185 CrossRef CAS.
- O. Kasian, N. Kulyk, A. Mingers, A. R. Zeradjanin, K. J. J. Mayrhofer and S. Cherevko, Electrochim. Acta, 2016, 222, 1056 CrossRef CAS.
- J. N. Gaur and G. M. Schmid, J. Electroanal. Chem. Interfacial Electrochem., 1970, 24, 279 CAS.
- R. B. E. Trindade, P. C. P. Rocha and J. P. Barbosa, in Hydrometallurgy ’94, Springer, Dordrecht, 1994 Search PubMed.
- D. W. Kirk and F. R. Foulkes, J. Electrochem. Soc., 1980, 127, 1993 CrossRef CAS.
- A. Kormanyos, A. Savan, A. Ludwig, F. D. Speck, K. J. J. Mayrhofer and S. Cherevko, J. Catal., 2021, 396, 387 CrossRef CAS.
- W. Chen and S. Chen, J. Mater. Chem., 2011, 21, 9169 RSC.
- O. Simmond and C. Comninellis, Electrochim. Acta, 1997, 42, 2013 CrossRef.
- O. Simmond, V. Schaller and C. Comninellis, Electrochim. Acta, 1997, 42, 2009 CrossRef.
- O. Simmond, C. Comninellis and G. Foti, in Novel Trends in Electro organic Synthesis, ed. S. Torii. Springer, Japan, 1998 Search PubMed.
- G. Foty, D. Gandini, C. Comninellis, A. Perret and W. Haenni, Electrochem. Solid-State Lett., 1999, 2, 228 CrossRef.
- A. Capon and R. Parson, J. Electroanal. Chem. Interfacial Electrochem., 1973, 44, 1 CrossRef CAS.
- Y.-Y. Hou, J.-M. Hu, L. Liu, J.-Q. Zhang and C.-N. Cao, Electrochim. Acta, 2006, 51, 6258 CrossRef CAS.
- A. Rossi and J. F. C. Boodts, J. Appl. Electrochem., 2002, 32, 735 CrossRef CAS.
- J. C. Forti, P. Olivi and A. R. de Andrade, J. Electrochem. Soc., 2003, 150, E222 CrossRef CAS.
- M. Bele, K. Stojanovski, P. Jovanovic, L. Moriau, G. Koderman Podborsek, J. Mozkon, P. Umek, M. Sluban, G. Drazic, N. Hodnik and M. Gaberscek, ChemCatChem, 2019, 11, 5038 CrossRef CAS.
- E. Antolini, ACS Catal., 2014, 4, 1426 CrossRef CAS.
- M. G. Chourashiya and A. Urakawa, J. Mater. Chem. A, 2017, 5, 4774 RSC.
- A. K. Schuppert, A. A. Topalov, I. Katsounaros, S. O. Klemm and K. J. J. Mayrhofer, J. Electrochem. Soc., 2012, 159, F670 CrossRef CAS.
- D. Escalera-Lopez, S. Czioska, J. Geppert, A. Boubnov, P. Rose, E. Saraci, U. Krewer, J.-D. Grunwaldt and S. Cherevko, ACS Catal., 2021, 11, 9300 CrossRef CAS.
- L. D. Burke and W. A. O'Leary, Chem. Sci., 1989, 89B, 389 Search PubMed.
- Y. Liu, Y. Zeng, R. Liu, H. Wu, G. Wang and D. Cao, Electrochim. Acta, 2012, 76, 174 CrossRef CAS.
- S. Geiger, S. Cherevko and K. J. J. Mayrhofer, Electrochim. Acta, 2015, 179, 24–31 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d2ra07142h |
| This journal is © The Royal Society of Chemistry 2023 |