DOI:
10.1039/D3RA00310H
(Paper)
RSC Adv., 2023,
13, 7929-7938
Development and validation of an UPLC-ESI-MS/MS method for quantification of duvelisib in plasma: application to pharmacokinetic study in rats
Received
15th January 2023
, Accepted 1st March 2023
First published on 10th March 2023
Abstract
Duvelisib (DUV) is a new oral phosphoinositide-3-kinase (PI3K)-δ and PI3K-γ inhibitor. It is used for the treatment of relapsed or refractory chronic lymphocytic leukemia (CLL) and small lymphocytic lymphoma (SLL). This study describes the development and validation of a new highly sensitive and efficient UPLC-ESI-MS/MS method for quantitation of DUV in plasma samples and its application to the pharmacokinetic study of DUV in rats. The method employed a very simple step for plasma sample pretreatment via precipitation of protein using methanol. DUV and ceritinib (CRB) as an internal standard (IS) were separated on a porous Hypersil BDS-C18 column (125 mm × 2 mm, 3 μm) using a mobile phase consisting of ammonium formate (10 mM, pH 4.2):acetonitrile (42![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :58, v/v), pumped isocratically at a flow rate of 0.3 mL min−1. DUV and CRB were eluted at 0.58 and 1.10 min, respectively. The mass spectrometric analysis was performed using an ESI in positive mode with multiple reaction monitoring (MRM). The technique was validated in accordance with the standards for validating bioanalytical methods established by the International Conference on Harmonization (ICH). The method's linear range was 5–500 ng mL−1, and its correlation coefficient was satisfactory as it is almost unity (0.9999). The limit of quantitation (LOQ) was 5 ng mL−1, while the limit of detection (LOD) was 1.7 ng mL−1. The recovery of the spiking DUV was between 94.95 and 102.21%, and the relative standard deviation (RSD) was less than 2.70%, confirming the method's accuracy and precision. The specificity/carryover of the method was proved. The robustness and ruggedness of the method was proved as the recovery values were 97.6–101.96% (±01.17–2.20%) and 98.74–102.00 (±1.18–4.02%) for robustness and ruggedness, respectively. The stability of DUV under the different analytical conditions were documented as the recovery values were in the range of 95.89–103.28% and the RSD values did not exceed 7.36%. The method was efficiently used to analyze DUV in human plasma samples that had been spiked with DUV and to conduct pharmacokinetic investigations of DUV in rats after giving them a single oral dosage of 25 mg kg−1 of the drug. The methodology is distinguished by excellent sensitivity, accuracy, and ease of sample pretreatment. Furthermore, it is efficient and has a short run time, which makes it high throughput and accordingly enables faster processing of many samples in clinical laboratories.
:58, v/v), pumped isocratically at a flow rate of 0.3 mL min−1. DUV and CRB were eluted at 0.58 and 1.10 min, respectively. The mass spectrometric analysis was performed using an ESI in positive mode with multiple reaction monitoring (MRM). The technique was validated in accordance with the standards for validating bioanalytical methods established by the International Conference on Harmonization (ICH). The method's linear range was 5–500 ng mL−1, and its correlation coefficient was satisfactory as it is almost unity (0.9999). The limit of quantitation (LOQ) was 5 ng mL−1, while the limit of detection (LOD) was 1.7 ng mL−1. The recovery of the spiking DUV was between 94.95 and 102.21%, and the relative standard deviation (RSD) was less than 2.70%, confirming the method's accuracy and precision. The specificity/carryover of the method was proved. The robustness and ruggedness of the method was proved as the recovery values were 97.6–101.96% (±01.17–2.20%) and 98.74–102.00 (±1.18–4.02%) for robustness and ruggedness, respectively. The stability of DUV under the different analytical conditions were documented as the recovery values were in the range of 95.89–103.28% and the RSD values did not exceed 7.36%. The method was efficiently used to analyze DUV in human plasma samples that had been spiked with DUV and to conduct pharmacokinetic investigations of DUV in rats after giving them a single oral dosage of 25 mg kg−1 of the drug. The methodology is distinguished by excellent sensitivity, accuracy, and ease of sample pretreatment. Furthermore, it is efficient and has a short run time, which makes it high throughput and accordingly enables faster processing of many samples in clinical laboratories.
Introduction
Chronic lymphocytic leukemia (CLL) and small lymphocytic lymphoma (SLL) are cancers of blood and bone marrow, in which there is an excess of immature lymphoid stem cells.1,2 Chemotherapy with various drugs is the standard approach for the treatment of CLL and SLL. These drugs belong to different pharmacological classes including nucleoside analogues, alkylating agents, immunomodulators, and monoclonal antibodies. For some of these, the therapeutic regimes with these drugs have achieved an overall response rate of ≥90%, with median survival >10 years. However, eventually relapse with low response rates and short survival duration occurs.3,4 Therefore, the researchers undertook the discovery of new drugs with higher efficacy with better safety and innovative mechanisms of therapeutic action. Through their work, compounds that block the phosphoinositide 3-kinase (PI3K) enzyme, a component of intracellular signal transmission, have been discovered. There are four isoforms of the catalytic subunit of PI3K; those are: α, β, δ and γ.5,6 Malignant-cell proliferation and migration are reduced when both PI3K-δ and PI3K-γ isoforms are inhibited.7–9 Additionally, dual isoform contributions to tumor development and survival are complementary.10,11 As a result, simultaneous inhibition of PI3K-δ and γ enhances the therapeutic effects of these compounds in CLL/SLL patients.12 The first drug in its class, idelalisib, is used to treat CLL, relapsed SLL, and relapsed follicular lymphoma.12 Additionally, it has been approved as the first-line treatment for CLL patients with a poor prognosis and in patients who cannot receive chemoimmunotherapy.12
Duvelisib (DUV) is a small drug molecule chemically named as: 8-chloro-2-phenyl-3-[(1S)-1-(7H-purin-6-ylamino)ethyl]isoquinolin-1-one. It has dual inhibitory action on both PI3K-δ and PI3K-γ. It reveals potent selective antiproliferative activity against leukemia cells.13 On September 24, 2018, the FDA approved the use of DUV for the treatment of adult patients with relapsed or refractory CLL or SLL. DUV was developed by Verastem, Inc. (Maryland, USA).14 Additionally, expedited approval for the treatment of refractory or relapsed follicular lymphoma has been given.15 Under the brand name Copiktra® capsules, DUV is sold (Verastem, Inc., Massachusetts, USA). DUV is to be administered twice daily at a dosage of 25 mg for a total of 28 days of therapy.14 DUV treatment must be monitored therapeutically by estimating its plasma concentrations in order to be effective and secure by proper analytical method. Few methods exist in literature for quantitation of DUV in biological fluids or tissues.16–18 These techniques include HPLC with UV detector for assessing DUV in rat plasma16 and UPLC-MS/MS for predicting tissue-to-plasma distribution ratios of basic substances, such as DUV, in mice17 and in beagle dogs.18 Obviously, for therapeutic monitoring of DUV in human plasma during patient therapy, none of these approaches have been verified. Also, the analysis run time was long as to obtain a thorough chromatographic separation of DUV from the other co-administered drugs.16,17 In addition, the precision and accuracy of some of these reported methods were poor as the relative standard deviation values and determination errors were high (∼12.6 and 14.1%, respectively).18 Therefore, there is a serious and urgent need for new method with high sensitivity, simple extraction procedure and high accuracy for quantitation of DUV in human plasma samples. In a previous study,19 our laboratory reported a highly sensitive nonextraction-assisted HPLC method with fluorescence detection for quantification of DUV in human plasma samples and described its application to pharmacokinetic study. UPLC-MS/MS is an increasingly important technique in therapeutic monitoring of drugs as it offers increased sensitivity and specificity.20,21 In this investigation, a highly sensitive UPLC-ESI-MS/MS methodology for quantifying DUV in plasma samples with a LOQ of 5 ng mL−1 was developed and validated. The methodology entailed employing a rapid, easy, non-extractive protein precipitation procedure to prepare plasma samples. Pharmacokinetic investigations of DUV in rats were successfully conducted using the technique. The method described herein is the first report describing the details of the procedures that can be easily applicable in clinical laboratories. The method was validated using human plasma samples as to simulate the matrix effect upon its real applications for therapeutic monitoring and pharmacokinetic studies of DUV. The method is superior to the reported method in terms of its high accuracy and precision.
Experimental
Materials
Duvelisib (DUV), docmitinib, nadolol, procainamide and glibenclamide were purchased from LC Laboratories (Woburn, USA). Ceritinib (CRB) was purchased from MedChemExpress (Woburn, MA, USA). Acquity UPLC Hypersil BDS-C18 column (125 mm × 2 mm, 3 μm) was a product of Agilent Technologies (Saugus, MA, USA). King Khaled University Hospital's Blood Bank in Riyadh, Saudi Arabia, provided the human plasma, which was then kept in a freezer at −20 °C till the analysis. The other chemicals were of analytical quality (Fisher Scientific, USA), and all of the solvents were of chromatographic grade (Merck KGaA, Darmstadt, Germany).
Experimental animals
A healthy male Wistar rat weighing 250 ± 30 g was procured from the King Saud University College of Pharmacy's animal facility (Riyadh, Saudi Arabia). Under typical laboratory circumstances (well-ventilation, a regular twelve-hour day/night cycle, a range of temperature of 24–27 °C, and a humidity level of 40–60%), the animals were housed in cages. All rats had unlimited access to water at all times, and the experiment was carried out after a 12 hour diet halt. Before beginning the research, the rats spent 7 days becoming familiar with the lab environment.
Preparation of standard solutions
Separately, properly weighed portions (25 mg) of each DUV and CRB (IS) were added to a 25 mL calibrated flask, dissolved in 1 mL of dimethyl sulfoxide, and then topped up with acetonitrile to volume to create stock solutions of 1 mg mL−1. When maintained in a refrigerator (8 °C), these stock solutions were discovered to remain stable for at least one month. To create working solutions with concentrations between 5 and 500 ng mL−1 for DUV and 200 ng mL−1 for CRB, the solutions were diluted with acetonitrile.
Preparation of calibration standards and quality control samples
Drug-free human plasma (blank) was spiked with DUV and CRB (IS) to produce final concentrations of DUV in the range of 5–500 ng mL−1 and a set concentration of CRB (200 ng mL−1) in all the solutions. This was done to prepare the calibration standards. The spiked samples were combined with equal amounts of the mobile phase, vortexed for 30 seconds, and then centrifuged using a Biofuge Pico centrifuge for 10 minutes at 13000 rpm (Heraeus Instruments, Germany). The supernatants were aspirated using syringes and filtered through 0.2 μm Millipore filters. Filtered supernatants (5 μL) were injected into the UPLC-ESI-MS/MS system.
Quality control (QC) samples at four different levels; limit of quantitation (LOQ: 7 ng mL−1), low quality control sample (LQC: 15 ng mL−1), medium quality control sample (MQC: 150 ng mL−1) and high-quality control sample (HQC: 400 ng mL−1). DUV QC samples received the same processing as the calibration standards. These samples were evaluated on several days, and on each day of the experiments, the system suitability parameters were assessed.
Samples preparation procedure
Aliquots (1 mL) of plasma samples (obtained from the Blood Bank at King Khalid Hospital of King Saud University, Riyadh, Saudi Arabia) were spiked with therapeutic level of DUV. The spiked samples were mixed with 1 mL of the mobile phase, vortexed for 30 seconds, and then centrifuged for 10 minutes at 13000 rpm. The supernatants were aspirated using syringes and filtered through 0.2 μm Millipore filters. Filtered supernatants (5 μL) were injected into the UPLC-ESI-MS/MS system.
UPLC system and analysis conditions
The chromatography was performed on an ACQUITY™ UPLC system (Waters Corp., Milford, MA, USA). The UPLC system includes a column heater–cooler, a degasser, an autosampler with a 10 μL injection loop, a quaternary solvent regulator, and a binary pump. The UPLC separation conditions and MS detection parameters are summarized in Table 1. The UPLC-MS/MS system was controlled by Mass Lynx software (SCN 805; Version 4.1).
Table 1 UPLC system and analysis conditions
| Parameter/condition |
Optimum |
| Column |
UPLC hypersil BDS™ C18 column (125 × 2 mm, i.d., 3 μm) manufactured by Waters Corp. (Milford, MA, USA) maintained at 25 ± 2 °C |
| Mobile phase |
Ammonium formate buffer (pH 4.2):acetonitrile (42:58, v/v) |
| Flow rate |
0.3 mL min−1 |
| Injection volume |
5 μL in partial loop mode |
| Reaction mode |
Multiple reaction monitoring (MRM) |
| Mass interface |
Electrospray interface (ESI) |
| Desolvation |
Nitrogen gas at a flow rate of 650 L h−1 at a desolvation temperature of 350 °C with a temperature source of 150 °C |
| Capillary voltage |
4 kV |
| Collision gas |
Argon at a flow rate of 0.1 mL min−1 |
| Parameters of MS analyzer |
HM1 and LM1 resolution 14.4 and 11.0; HM2 and LM2 resolution 14.8 and 12.0 respectively |
| Dwell time |
0.025 s; ion energy 1, 0.4 V; ion energy 2, 1.4 V |
Method validation procedure
To confirm that the proposed UPLC-ESI-MS/MS technique is acceptable in terms of its linearity, sensitivity, precision, accuracy, selectivity, robustness, and stability of DUV in its samples, the method was validated in accordance with the ICH recommendations for validation of bioanalytical procedure.22
Assessment of linearity and sensitivity
By constructing three separate calibration curves, which served as the basis for the regression equations and their correlation coefficients, linearity was examined. The degree of method linearity was expressed using the correlation coefficient value of the calibration line. The sensitivity was expressed as LOD and LOQ. The formula used was: LOD or LOQ = XSDa/b, where X = 3.3 for LOD and 10 for LOQ, SDa is the standard deviation of the intercept, and b is the slope of the calibration line.
Determination of precision and accuracy
Each QC sample was subjected to repeated (n = 6) analysis at each of the four QC levels (LOQ, LQC, MQC, and HQC) as a batch in a single run to determine the intra-day precision and accuracy. By conducting repeated (n = 3) analyses of each QC sample at each level over the course of three days, the inter-day precision and accuracy were evaluated. The accuracy was represented as a percentage of the recovery values, while the precisions were represented as a percentage of the relative standard deviation (RSD, %).
Determination of specificity and carryover
To investigate the specificity of the proposed UPLC-ESI-MS/MS method, drug-free plasma, plasma spiked with DUV at concentrations of 7, 15, 150 and 400 ng mL−1; each was sample spiked with CRB (200 ng mL−1) were treated for protein precipitation and then injected into the UPLC system to identify any potential peaks at elution times of DUV and CRB.
Assessment of robustness and ruggedness
Minor changes have been made to the analytical conditions (mobile phase composition and flow rate) and the effects on recovery and accuracy were observed in order to evaluate the method's robustness. The operating settings of the methodology were used for the analysis of DUV samples on two separate UPLC instruments at two different laboratories and at various time intervals in order to evaluate the robustness of the method. RSD (%) was employed to depict the results.
Stability studies of DUV in samples
The stability of DUV was investigated under various circumstances and locations (autosampler, bench-top, freeze–thaw cycles, and long-term storage). QC samples were kept under autosampler settings for around 48 hours before being injected into the UPLC system in order to examine the stability in autosampler. For studying the stability onto the bench-top, plasma samples were retained at room temperature for ∼6 h and subsequently subjected to the analysis. Three cycles of freeze (at −80 °C)–thaw (at room temperature) were carried out in order to explore the stability during freeze–thaw cycle. For assessing the stability at long-term storage conditions, samples were kept at −80 °C for 60 days and then subjected to analysis.
Pharmacokinetic study in the rats
Each rat received oral gavage dosing with DUV (25 mg kg−1, dissolved in 1% dimethyl sulfoxide/saline). Approximately 300 μL of blood were drawn and placed in heparinized tubes. Prior to the administration of DUV, and after DUV administration at predetermined time points at 0.25, 0.5, 1, 1.5, 2, 2.5, 3, 4, 6, 8 and 12 h. Blood samples were centrifuged at 4500 rpm and 4 °C for 30 minutes, and the supernatants (plasma) were stored at −20 °C until the analysis. The experimental investigation was carried out in accordance with the requirements of the King Saud University's Research Ethics Committee (RCE) for performing studies on living creatures, Riyadh, Saudi Arabia, with reference number KSU-SE-20-51.
Statistical analysis
The statistical analysis was conducted using Microsoft Excel software, version 2018 (Microsoft Corporation, Washington, USA). All values were given as mean ± SD or RSD (%). The Data Analysis Package integrated in the Excel Software performed regression analysis on the calibration data for the UPLC-ESI-MS/MS methodology at a probability value (p value) <0.05. The intercept of the line, slope, correlation coefficient, and variance were all calculated during the regression analysis. The non-compartmental model was exploited to calculate the pharmacokinetic parameters. The maximum plasma concentration (Cmax), time to reach the maximum concentration (Tmax), half-life time (t½), and mean residence time (MRT) were computed, as well as the area under the curve from zero to last and infinity (AUC0–24; AUC0–∞). Microsoft-Excel 2018 was used to determine the means, SD, and RSD (%) values (Microsoft Corporation, Washington, USA).
Results and discussion
Strategy for method development
DUV has piqued our interest because of its invention and effective usage in the treatment of CLL and SLL and expansion of its use in the treatment of T-cell lymphoma,23 solid tumors,24 and non-Hodgkin's lymphoma.25 Our selection of DUV as a target analyte in the present study was also supported by the frequent appearance of some adverse effects leading to discontinuation of treatment with DUV was reported. The therapeutic monitoring of DUV levels in patient's plasma could be useful in adjusting the most appropriate dose for achieving the highest therapeutic benefits with minimal side effects. Therefore, refining the pharmacokinetic profile of DUV is important for achieving such goal. Accordingly, a proper analytical method is necessary. The current work focused on developing a sensitive UPLC-ESI-MS/MS methodology for analyzing DUV in human plasma samples.
Optimization of the analysis conditions
The optimal settings of the proposed UPLC-MS/MS for the chromatographic separation of DUV and CRB (IS), were thoroughly explored utilizing a mixture of ammonium formate (10 mM) and acetonitrile as the mobile phase and different columns, additionally, the multiple reaction monitoring (MRM) mode was elected during the entire experiment to eliminate probable intrusive signals and at the same time augment the specificity of the method during the development. At the beginning of the experimental trials, a sample containing only DUV was injected on a HILIC column (150 mm × 2 mm, 3 μm) using ammonium formate (10 mM):acetonitrile (65:35, v/v, pH 3.5) at flow rate of 0.2 mL min−1. The resulted chromatogram showed no peaks; therefore, the column was replaced with Hypersil BDS-C18 column (125 mm × 2 mm, 3 μm) and another sample was injected on the column with the aforementioned conditions. Under these conditions, DUV was eluted at ∼5 min. In order to enhance the elution time of DUV (shorten the run time), the mobile phase components ratio, pH and flow rate were readjusted to ammonium formate (10 mM):acetonitrile (30:70, v/v, pH 4.2) at flow rate of 0.3 mL min−1. The resulted chromatogram using these conditions displayed a distorted peak with poor symmetry at 0.5 min. Accordingly, the percentage of acetonitrile was lowered to be 58% (v/v) to improve the resolution and symmetry of the peak. The optimum conditions at which sharp and symmetric peaks were achieved were found to be Hypersil BDS-C18 column (125 mm × 2 mm, 3 μm) with ammonium formate (10 mM):acetonitrile (42:58, v/v, pH 4.2) as the mobile phase pumped at 0.3 mL min−1 and the retention time of DUV was 0.58 min.
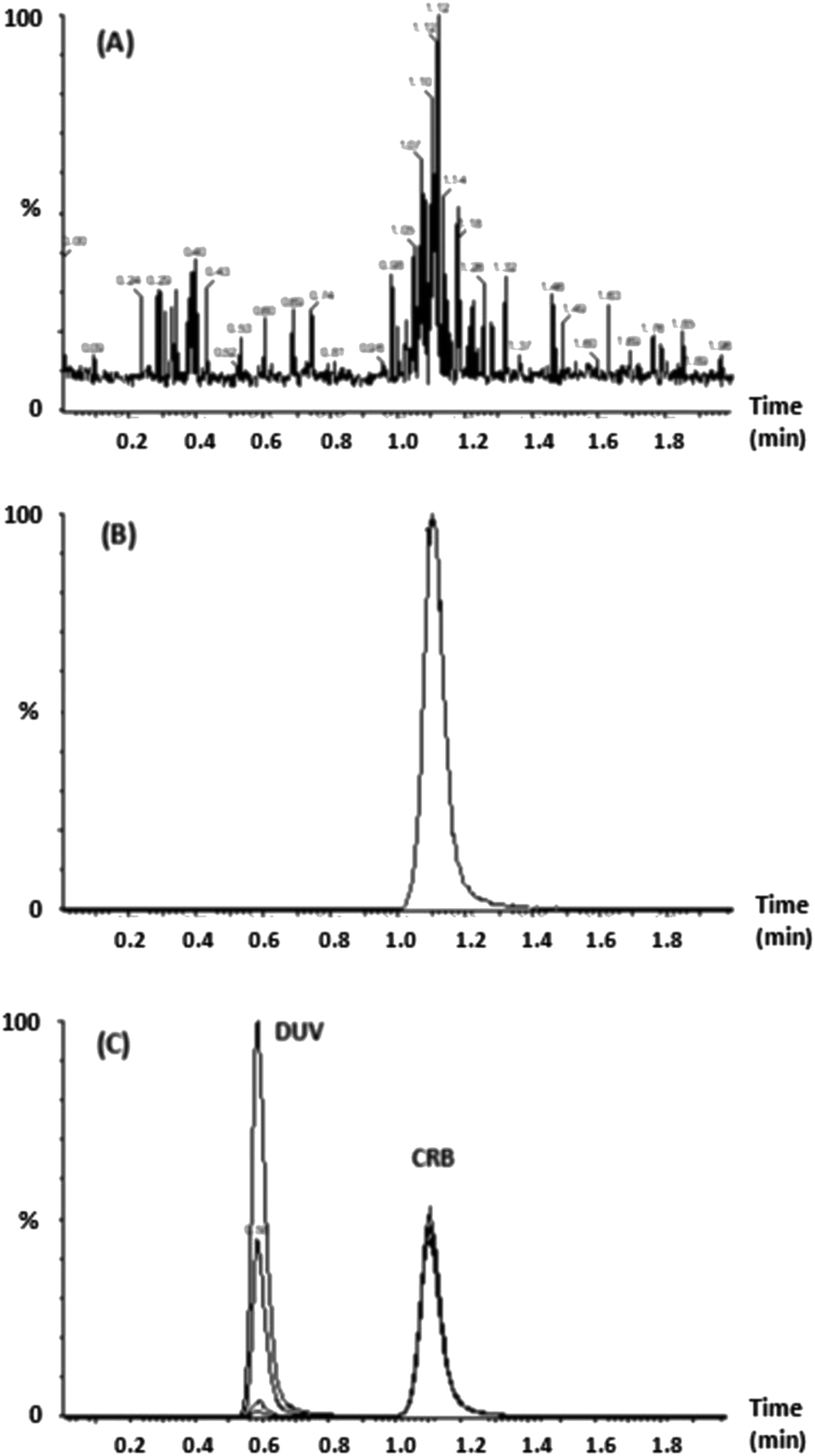
Different compounds (CRB, docmitinib, nadolol, procainamide, and glibenclamide) were tested for their use as internal standards with DUV; the obtained chromatogram is shown in (Fig. 1). The retention times for these compounds were 0.7, 0.95, 1.1, 1.5, and 1.9 min for procainamide, nadolol, CRB, glibenclamide, and docmitinib, respectively. CRB was chosen as IS as it provides suitable resolution from DUV peak, appropriate peak shape, and short run time without the need to change the chromatographic conditions. The positive product ion scan of DUV (m/z 417) yielded two major ions at [M + H]+ m/z 136 and 282. Similarly, the ion scan of CRB as IS (m/z 558) showed two major ions at [M + H]+ m/z 84 and 433 (Fig. 2).
 |
| | Fig. 1 (A) Chromatogram of a mixture containing DUV, docmitinib, CRB, nadolol, procainamide, and glibenclamide; concentration of each is 200 ng mL−1. (B) Chromatogram of DUV (150 ng mL−1) with CRB (200 ng mL−1). | |
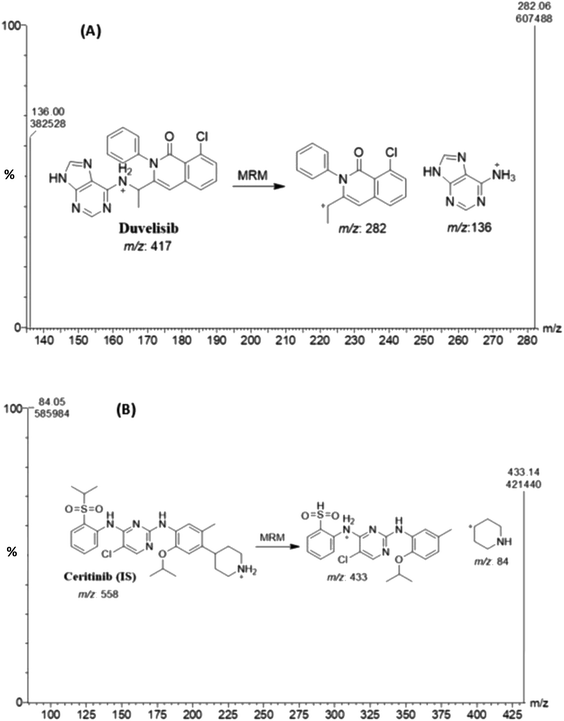 |
| | Fig. 2 Mass spectra of duvelisib (A) and ceritinib (B). | |
Validation of the method
The proposed methodology was entirely validated in accordance with the requirements of the ICH for validation of bioanalytical procedure22 to ensure acceptability of the method in terms of its specificity, linearity, limits of detection and quantitation, precision, accuracy, robustness, and ruggedness. By constructing three separate calibration curves, deriving the regression equations, and calculating the correlation coefficients of the calibration lines, linearity was examined.
Specificity and carryover
To investigate the specificity of the proposed UPLC-MS/MS method, drug-free plasma, plasma samples spiked with DUV at concentrations of 5, 15, 150 and 400 ng mL−1; each was sample spiked with IS (CRB, 200 ng mL−1) were treated for protein precipitation and then injected into the UPLC-MS/MS system to identify any peaks at elution times of DUV and CRB (Fig. 3). The total ion chromatogram of the MRM technique revealed that no comparable peaks were observed close to the DUV and CRB retention times in the human plasma samples. As a result, extracting DUV from human plasma using the mobile phase ammonium formate (10 mM):acetonitrile as the protein precipitant worked well. Furthermore, neither DUV nor IS carryover was seen in plasma samples.
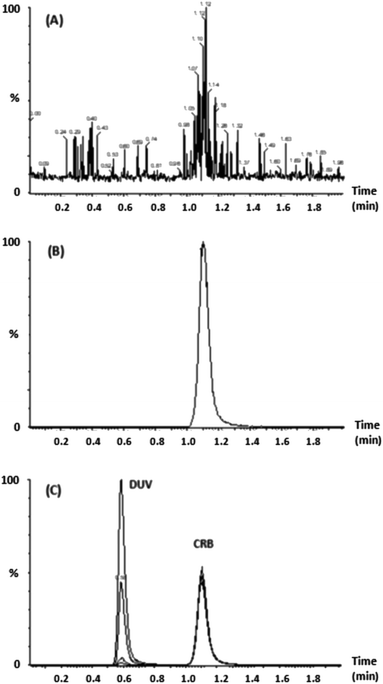 |
| | Fig. 3 Chromatogram of (A) blank plasma sample. (B) 5, 15, 150 and 400 ng mL−1 DUV spiked plasma (C) CRB 200 ng mL−1 spiked plasma. | |
Linearity and sensitivity
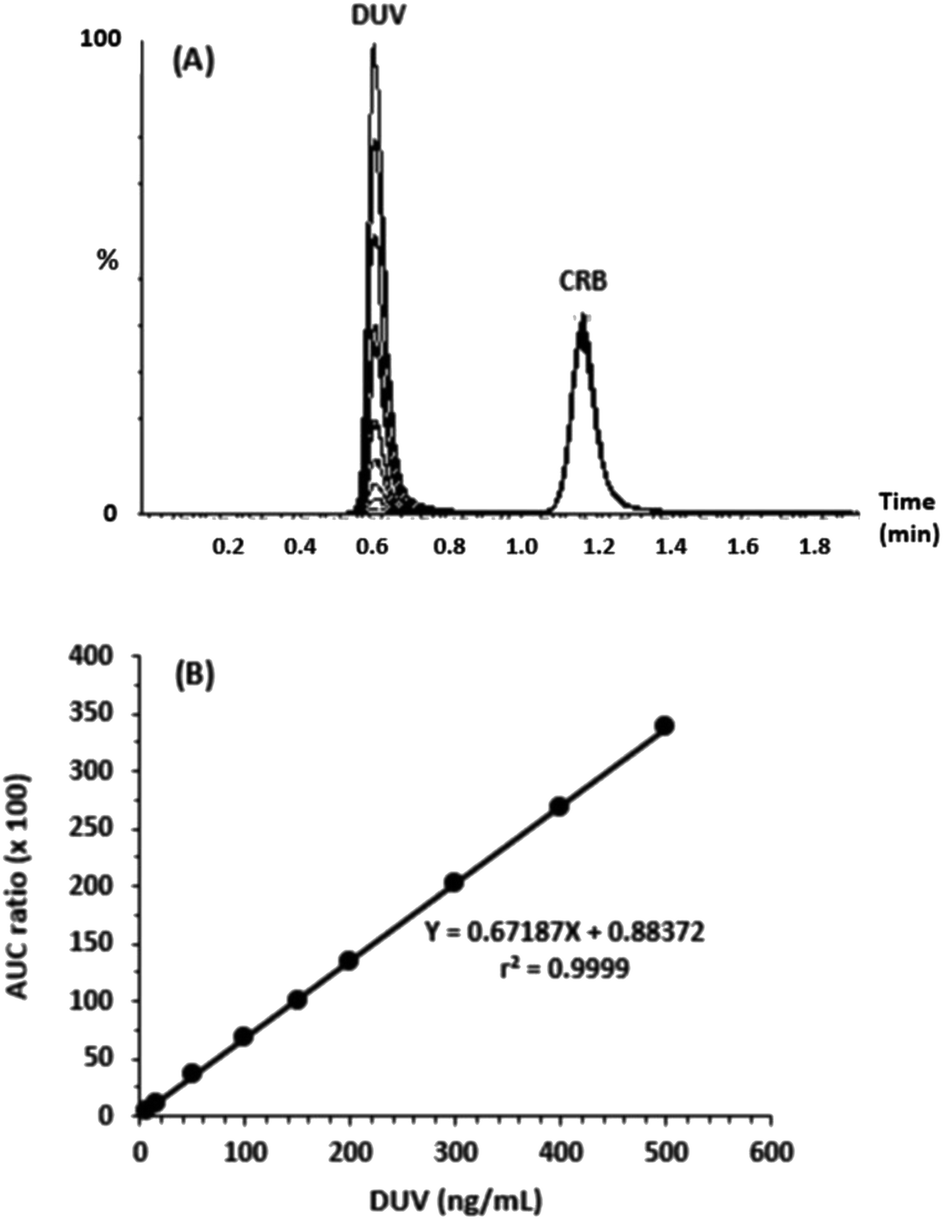
For linearity evaluation of the suggested methodology, a calibration graph was created (n = 9) by putting the peak-area ratio of DUV to IS (Y-axis) as a function of the concentration of DUV (X-axis) in the range of 5–500 ng mL−1 in human plasma (Fig. 4). A linear relationship with excellent determination coefficient (r2 = 0.99991) was found. The regression equation of the calibration curve was Y = 0.88372 + 0.67187X. The RSD values of all points did not exceed 2.69%; whereas, the SD ranged from 0.06 to 3.86. The calculated LOD and LOQ were 1.7 and 5 ng mL−1, respectively.
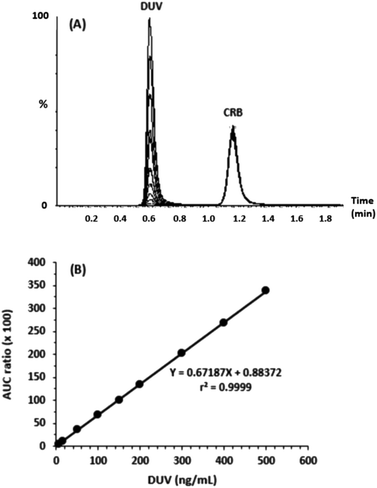 |
| | Fig. 4 Panel (A): chromatogram of 5, 15, 50, 100, 150, 200, 300, 400, 500 ng mL−1 DUV and 200 ng mL−1 CRB. Panel (B): calibration curve of DUV. | |
Precision and accuracy
Using four levels of quality control (QC) samples, the precision and accuracy of the examined technique were tested at the calibration range (Table 2). These levels were: LOD (5 ng mL−1), LQC (15 ng mL−1), MQC (150 ng mL−1), and HQC (400 ng mL−1). Intra-day recovery values were in the range of 95.12–102.21% (with a mean value of 98.60 ± 3.02%), whereas those of the inter-day were in the range of 94.95–100.88% (with a mean value of 98.26 ± 2.58%). These high recovery values validated the method's accuracy. RSD values for intra- and inter-day precisions were lied the ranges of 0.24–2.15 and 0.75–2.69%, respectively. These results indicated that the method has acceptable precision. It is important to state that the precision of accuracy of the present method were superior to those of the reported method,18 which demonstrated precisions and accuracy at approximately 12.63% and 86.9%, respectively.
Table 2 Intra-assay and inter-assay precision and accuracy for determination of DUV in spiked human plasma
| Nominal DUV (ng mL−1) |
Mean DUV (ng mL−1) |
Recovery (%) |
RSD (%) |
| Intra-day |
| 5 |
4.76 |
95.12 |
1.15 |
| 15 |
15.33 |
102.21 |
2.15 |
| 150 |
146.19 |
97.46 |
0.70 |
| 400 |
398.37 |
99.59 |
0.24 |
|
| Inter-day |
| 5 |
4.75 |
94.95 |
2.69 |
| 15 |
15.13 |
100.88 |
1.21 |
| 150 |
146.47 |
97.64 |
1.38 |
| 400 |
398.23 |
99.56 |
0.75 |
Robustness and ruggedness
The method's robustness was assessed by examining the effect of slight changes in the experimental chromatographic conditions (mobile phase composition and pH) on the analytical performance of the method in terms of recovery and precision. As shown in (Table 3), these minor changes did not distress the method's accuracy and precision as recovery values were in the range of 97.60–101.96% and the range of RSD values were 1.17–2.20%.
Table 3 Robustness and ruggedness study of the proposed UPLC-MS/MS method for determination of DUV in human plasma samples
| Parameters |
Recoverya (% ± RSD) |
| Values are mean of 3 determinations. |
| Robustness |
Ratio of acetonitrile![[thin space (1/6-em)]](https://www.rsc.org/images/entities/b_char_2009.gif) :ammonium formate (10 mM) :ammonium formate (10 mM) |
| 40:60 |
98.44 ± 1.17 |
| 45:55 |
97.60 ± 1.38 |
| pH |
| 4 |
100.49 ± 2.20 |
| 4.1 |
101.96 ± 1.26 |
|
| Ruggedness |
| Instrument-to-instrument |
| Instrument-1 |
99.56 ± 1.18 |
| Instrument-2 |
98.94 ± 3.05 |
| Analyst-to-analyst |
| Analyst-1 |
99.83 ± 3.47 |
| Analyst-2 |
102.00 ± 4.02 |
| Day-to-day |
| Day-1 |
99.76 ± 2.38 |
| Day-2 |
98.74 ± 3.85 |
The suggested approach was used to analyze DUV under identical operational settings, but two separate instruments from two different laboratories were used, and varied elapsed periods were used to demonstrate the robustness of the method. Results were expressed as RSD (%). The recovery values were in the range of 98.74–102.00% and the RSD values were in the range of 1.18–4.02%, indicating the ruggedness and reproducibility of the method (Table 3).
Stability studies of DUV in samples
DUV was shown to be stable in plasma samples for at least 6 hours at room temperature on the bench and 48 hours when stored under autosampler storage settings, according to the findings of stability tests (Table 4). DUV was also discovered to be stable at −80 °C for 60 days and during the three freeze–thaw cycles. It was discovered that the stock and working standard solutions of DUV were stable for 30 days at refrigerated conditions (at 8 °C). These outcomes are clear from the recovery values that were attained, which ranged from 95.89 to 103.28%.and the RSD values which did not exceed 7.36%.
Table 4 Data of stability studies of DUV in human plasma samples
| Stability |
Spiked conc. (ng mL−1) |
Recoverya (%) |
Precisiona (RSD, %) |
| Values are mean of 3 determinations. |
| Bench top (6 h) |
5 |
99.52 |
7.05 |
| 15 |
100.21 |
3.12 |
| 150 |
101.43 |
5.82 |
| 400 |
99.26 |
2.63 |
| Autosampler (48 h) |
5 |
100.12 |
6.45 |
| 15 |
95.89 |
4.08 |
| 150 |
98.42 |
5.14 |
| 400 |
103.28 |
2.28 |
| Freeze–thaw (3 cycle) |
5 |
96.17 |
7.36 |
| 15 |
99.89 |
6.51 |
| 150 |
101.13 |
5.81 |
| 400 |
97.28 |
1.19 |
| 60 days at −80 °C |
5 |
101.51 |
6.27 |
| 15 |
99.84 |
4.18 |
| 150 |
101.19 |
3.05 |
| 400 |
99.87 |
2.18 |
| 30 days at 8 °C |
5 |
96.94 |
6.07 |
| 15 |
103.24 |
3.29 |
| 150 |
101.02 |
2.16 |
| 400 |
96.27 |
1.42 |
Pharmacokinetic study in rats
As aforementioned, the main goal for the development and validation of the present method was its application for routine use in clinical laboratories for the for therapeutic monitoring of DUV levels in human plasma during patient therapy and conducting pharmacokinetic studies, it was validated using human plasma samples to simulate the real circumstances of its application and achieving its purpose. However, for the law's restrictions on the academic/research universities to use human subjects in conducting research, therefore it was decided to assess the applicability of the present method using plasma samples of experimental animals. The applicability of the UPLC-ESI-MS/MS method described herein for the quantitation of DUV in plasma samples, a pharmacokinetic profile of DUV after its oral administration in rats at a dose of 25 mg kg−1 was investigated using rats. Rats were selected for the study because they were verified to be the most suitable animal model to investigate human biology because of to their high similarities in their genomic and physiologic to humans.26 Pharmacokinetic study was conducted using non-compartmental analysis as described previously.27 The mean plasma DUV concentrations (in ng mL−1) versus time (h) is given in Fig. 5, and the main pharmacokinetic parameters explored are summarized in Table 5.
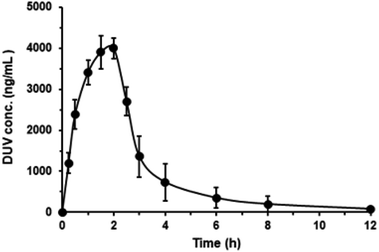 |
| | Fig. 5 Concentration-time profile of DUV in rats after single oral administration at a dose of 25 mg kg−1. Concentrations are presented in ng mL−1 (means of 5 rats ± SD). | |
Table 5 The pharmacokinetic parameters of DUV in rat plasma after oral administration of 25 mg kg−1
| Parameter |
Unit |
Valuea |
| Values are mean of 5 determinations. |
| Dose (D) |
mg kg−1 |
25 |
| Maximum plasma concentration (Cmax) |
ng mL−1 |
3804.24 |
| Time required for maximum plasma conc. (Tmax) |
h |
1.83 |
| Volume of distribution (Vd) |
L kg−1 |
0.88 |
| Elimination rate constant (Kel) |
h−1 |
0.97 |
| Elimination half-life time (t1/2) |
h−1 |
0.72 |
| Clearance (CL) |
L h−1 kg−1 |
0.85 |
| Area under curve from time 0 to last conc. (AUC0–t) |
ng h mL−1 |
28271.33 |
| Area under curve at infinite time (AUC0–∞) |
ng h mL−1 |
29544.19 |
| Area under curve ration (AUC0–t/AUC0–∞) |
% |
95.69 |
| Mean residence time (MRT) |
h |
1.04 |
DUV was quickly absorbed following oral treatment in rats, reaching its peak plasma concentration (Cmax) of 3804.24 ± 500 ng mL−1 after 1.83 h (tmax), and having an elimination half-life (t½) of 0.72 h. The current UPLC-ESI-MS/MS technique was shown to be sensitive enough to cover the elimination phase of DUV. This was confirmed by computing the ratios of AUC (AUC0–t/AUC0–∞), which were established to be 95.69%. Adult patients with advanced hematologic malignancies who received oral DUV as a single dosage of 25 mg twice daily showed similar drug plasma profiles. After administration to humans, it was noted that DUV exhibits a quick absorption with a Tmax of 1 to 2 hours.14
Conclusions
This article demonstrates the detailed description for the development and validation of a new UPLC-ESI/MS/MS method for the quantitation of DUV in both human and animal plasma samples. The proposed method combined many advantages which include the simple straightforward one-step protein precipitation for the plasma sample preparation, employing a simple isocratic mode for the chromatographic separation and a short run time (2 min). The validation results confirmed that with concentrations as low as 5 ng mL−1, the suggested UPLC-ESI-MS/MS method is appropriate for the accurate quantitation of DUV in plasma samples and has a wide linear range of 5–500 ng mL−1. The simple extraction procedure and the short run time of the method makes it high throughput which facilitates the processing of many samples in clinical laboratories. The method is valuable for the combined pharmacokinetic studies and therapeutic monitoring of DUV in human subjects after oral administration of therapeutic its dose.
Abbreviations
| DUV | Duvelisib |
| PI3K | Phosphoinositide 3-kinase |
| CLL | Chronic lymphocytic leukemia |
| FDA | U.S. Food and Drug Administration |
| HPLC | High performance liquid chromatography |
| FD | Fluorescence detection |
| UPLC | Ultraperformance liquid chromatography |
| ESI | Electrospray ionization |
| MS/MS | Tandem mass spectrometry |
| MRM | Multiple reaction monitoring |
| CRB | Ceritinib |
| IS | Internal standard |
| ICH | The International Conference on Harmonization |
| LOD | Limit of detection |
| LOQ | Limit of quantification |
| QC | Quality control |
| RSD | Relative standard deviation |
| AUC | Area under curve |
Ethics statement
Human plasma was obtained from the Blood Bank of King Khaled Hospital of King Saud University (Riyadh, Saudi Arabia). Samples were collected from a healthy volunteer after receiving the consent, and the guidelines outlined in the Helsinki were followed. All procedures performed in studies involving experimental animals were in accordance with the ethical standards for conducting studies on Living Creatures at King Saud University (Riyadh, Saudi Arabia). The study, presented in this manuscript, was approved by the Research Ethics Committee (RCE) of King Saud University with Ethics Reference No. KSU-SE-20-51.
Conflicts of interest
The authors report no conflicts of interest for this work.
Acknowledgements
The authors extend their appreciation to the Deputyship for Research & Innovation, Ministry of Education in Saudi Arabia for funding this research work through the project no. (IFKSURG-2-1066).
References
- Canadian Cancer Society, Chronic lymphocytic leukemia, Ontario, 2019, Available from: https://www.cancer.ca/en/cancer-information/cancer-type/leukemia-chronic-lymphocytic-cll/chronic-lymphocytic-leukemia/?region=on, accessed on November 20, 2022 Search PubMed.
- The American Chemical Society, Key statistics for chronic lymphocytic leukemia, Washington, 2019, available from: https://www.cancer.org/cancer/chronic-lymphocytic-leukemia/about/key-statistics.html, accessed on November 20, 2022 Search PubMed.
- S. D. Kotiah, Chronic lymphocytic leukemia treatment protocols, New York, 2019, available from: https://emedicine.medscape.com/article/2005390-overview, accessed on November 20, 2022 Search PubMed.
- C. Bello, L. Zhang and M. Naghashpour, Follicular lymphoma: current management and future directions, Cancer Control, 2012, 19(3), 187–195 CrossRef PubMed.
- E. Clayton, G. Bardi, S. E. Bell, D. Chantry, C. P. Downes, A. Gray, L. A. Humphries, D. Rawlings, H. Reynolds, E. Vigorito and M. Turner, A crucial role for the p110 delta subunit of phosphatidylinositol 3-kinase in B cell development and activation, J. Exp. Med., 2002, 196(6), 753–763 CrossRef CAS PubMed.
- W. P. Fung-Leung, Phosphoinositide 3-kinase delta (PI3Kδ) in leukocyte signaling and function, Cell. Signalling, 2011, 23(4), 603–608 CrossRef CAS PubMed.
- M. Peluso, K. Faia, D. Winkler, N. Patel, E. Brophy, K. White, M. Douglas, H. M. Stern, V. Palombella, K. McGovern and J. L. Kutok, Duvelisib (IPI-145) inhibits malignant B-cell proliferation and disrupts signaling from the tumor microenvironment through mechanisms that are dependent on PI3K-δ and PI3K-γ, Blood, 2014, 124(21), 328 CrossRef.
- J. Hoellenriegel, S. A. Meadows, M. Sivina, W. G. Wierda, H. Kantarjian, M. J. Keating, N. Giese, S. O'Brien, A. Yu, L. L. Miller, B. J. Lannutti and J. A. Burger, The phosphoinositide 30-kinase delta inhibitor, CAL-101, inhibits B-cell receptor signaling and chemokine networks in chronic lymphocytic leukemia, Blood, 2011, 118(13), 3603–3612 CrossRef CAS PubMed.
- K. Okkenhaug, A. Bilancio, G. Farjot, H. Priddle, S. Sancho, E. Peskett, W. Pearce, S. E. Meek, A. Salpekar, M. D. Waterfield, A. J. H. Smith and B. Vanhaesebroeck, Impaired B and T cell antigen receptor signaling in p110 delta PI 3-kinase mutant mice, Science, 2002, 297(5583), 1031–1034 CrossRef CAS PubMed.
- B. Vanhaesebroeck, J. Guillermet-Guibert, M. Graupera and B. Bilanges, The emerging mechanisms of isoform-specific PI3K signalling, Nat. Rev. Mol. Cell Biol., 2010, 11(5), 329–341 CrossRef CAS PubMed.
- K. Faia, K. White, J. Proctor, E. Murphy, J. Proctor, M. Pink, N. Kosmider, K. McGovern and J. Kutok, The phosphoinositide-3 kinase (PI3K)-δ,γ inhibitor, duvelisib shows preclinical synergy with multiple targeted therapies in hematologic malignancies, PLoS One, 2018, 13(8), 1–14 CrossRef PubMed.
- Y. Qingshan, M. Prexy, N. Terry, Q. Christophe and G. Varsha, Idelalisib: first-in-class PI3K delta inhibitor for the treatment of chronic lymphocytic leukemia, small lymphocytic leukemia, and follicular lymphoma, Clin. Cancer Res., 2015, 21(7), 1537–1542 CrossRef PubMed.
- K. Balakrishnan, M. Peluso, M. Fu, N. Y. Rosin, J. A. Burger, W. G. Wierda, M. J. Keating, K. Faia, S. O'Brien, J. L. Kutok and V. Gandhi, The phosphoinositide-3-kinase (PI3K)-delta and gamma inhibitor, IPI-145 (Duvelisib), overcomes signals from the PI3K/AKT/S6 pathway and promotes apoptosis in CLL, Leukemia, 2015, 29(9), 1811–1822 CrossRef CAS PubMed.
- U.S. Food & Drug Administration (FDA), Duvelisib (Copiktra, Verastem, Inc.) for adult patients with relapsed or refractory chronic lymphocytic leukemia (CLL) or small lymphocytic lymphoma
(SLL), Maryland, 2018, available from: https://www.fda.gov/Drugs/InformationOnDrugs/ApprovedDrugs/ucm621503.htm, accessed on November 20, 2022 Search PubMed.
- L. Leah, Duvelisib granted priority review for R/R CLL/SLL and FL, Cancer Network, 2018, available from: https://www.cancernetwork.com/view/duvelisib-granted-priority-review-rr-cllsll-and-fl, accessed on November 20, 2022 Search PubMed.
- A. Siddesh, D. Sriram, A. Zakkula, R. Kumar, S. Dittakavi, M. Zainuddin, R. K. Trivedi and R. Mullangi, Validated HPLC-UV method for simultaneous quantification of phosphatidylinositol 3-kinase inhibitors, copanlisib, duvelisib and idelalisib, in rat plasma: application to a pharmacokinetic study in rats, Biomed. Chromatogr., 2020, 35(4), e5015 Search PubMed.
- P. B. Nigade, J. Gundu, K. S. Pai and K. V. S. Nemmani, Prediction of tissue-to-plasma ratios of basic compounds in mice, Eur. J. Drug Metab. Pharmacokinet., 2017, 42(5), 835–847 CrossRef CAS PubMed.
- Y. Shao, S. Xie, H. Zhu, X. Du and R. Xu, Development of a novel and quick LCMS/MS method for the pharmacokinetic analysis of duvelisib in beagle dogs, J. Pharm. Biomed. Anal., 2020, 187, 113355 CrossRef CAS PubMed.
- A. Y. Sayed, N. Y. Khalil, A. Almomen, N. Z. Alzoman, A. A. Almehizia and I. A. Darwish, A highly sensitive nonextraction-assisted HPLC method with fluorescence detection for quantification of duvelisib in plasma samples and its application to pharmacokinetic study in rats, Drug Des., Dev. Ther., 2021, 15, 2667–2677 CrossRef PubMed.
- J. E. Adaway and B. G. Keevil, Therapeutic drug monitoring and LC–MS/MS, J. Chromatogr. B: Anal. Technol. Biomed. Life Sci., 2012, 883–884, 33–49 CrossRef CAS PubMed.
- V. Avataneo, A. D'Avolio, J. Cusato, M. Cantù and A. De Nicolò, LC-MS application for therapeutic drug monitoring in alternative matrices, J. Pharm. Biomed. Anal., 2019, 166, 40–51 CrossRef CAS PubMed.
- The International Conference on Harmonization (ICH), Q2(R1): validation of analytical procedure: text and methodology, ICH, Geneva, 2005 Search PubMed.
- S. M. Horwitz, R. Koch, P. Porcu, Y. Oki, A. Moskowitz, M. Perez, P. Myskowski, A. Officer, J. D. Jaffe, S. N. Morrow, K. Allen, M. Douglas, H. Stern, J. Sweeney, P. Kelly, V. Kelly, J. C. Aster, D. Weaver, F. M. Foss and D. M. Weinstock, Activity of the PI3K-δ,γ inhibitor duvelisib in a phase 1 trial and preclinical models of T-cell lymphoma, Blood, 2018, 131(8), 888–898 CrossRef CAS PubMed.
- H. A. Blair, Duvelisib: first global approval, Drugs, 2018, 78(17), 1847–1853 CrossRef PubMed.
- I. W. Flinn, M. Patel, Y. Oki, S. Horwitz, F. F. Foss, K. Allen, M. Douglas, H. Stern, J. Sweeney, J. Kharidia, P. Kelly, V. M. Kelly and B. Kahl, Duvelisib, an oral dual PI3K-δ, γ inhibitor, shows clinical activity in indolent non-Hodgkin lymphoma in a phase 1 study, Am. J. Hematol., 2018, 93(11), 1311–1317 CrossRef CAS.
- E. M. Blais, K. D. Rawls, B. V. Dougherty, Z. I. Li, G. L. Kolling, P. Ye, A. Wallqvist and J. A. Papin, Reconciled rat and human metabolic networks for comparative toxicogenomics and biomarker predictions, Nat. Commun., 2017, 8, 14250 CrossRef CAS PubMed.
- A. Almomen, H. M. Maher, N. Z. Alzoman, S. M. Shehata and A. Alsubaie, Flavoured water consumption alters pharmacokinetic parameters and increases exposure of erlotinib and gefitinib in a preclinical study using Wistar rats, PeerJ, 2020, 8, e9881 CrossRef PubMed.
|
| This journal is © The Royal Society of Chemistry 2023 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence *,
Nourah Z. Alzoman,
Aliyah Almomen,
Abdulrahman A. Almehizia
*,
Nourah Z. Alzoman,
Aliyah Almomen,
Abdulrahman A. Almehizia