
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicencePropranolol induces large-scale remodeling of lipid bilayers: tubules, patches, and holes†
Ni He and
Tao Zhao *
*
College of Chemistry and Chemical Engineering, Shanghai University of Engineering Science, Shanghai 201620, China. E-mail: zhaotao1982@126.com; Fax: +86-021-67791214
First published on 8th March 2023
Abstract
Herein, we report fluorescence microscopy analysis of the interaction between propranolol (PPN), a beta-adrenergic blocking agent, and planar supported lipid bilayers (SLBs), as model membranes. The results indicate that PPN can remarkably promote largescale remodeling in SLBs with various lipid compositions. It was found that PPN insertion induces the formation of long microtubules that can retract into hemispherical caps on the surface of the bilayer. These transformations are dynamic, partially reversible, and dependent upon the drug concentration. Quantitative analysis revealed a three-step model for PPN–lipid bilayer interaction, with the first step involving interfacial electrostatic adsorption, the second step centered on hydrophobic insertion, and the third step associated with membrane disruption and hole formation. By introducing cholesterol, phosphoethanolamine, phosphatidylglycerol, and phosphatidylserine lipids into the phosphocholine SLBs, it was illustrated that both the chemistry of the lipid headgroups and the packing of lipid acyl chains can substantially affect the particular steps in the interactions between PPN and lipid bilayers. Our findings may help to elucidate the possible mechanisms of PPN interaction with lipid membranes, the toxic behavior and overdosage scenarios of beta-blockers, and provide valuable information for drug development and modification.
Introduction
Currently, more than 60% of clinically important drugs target either integral or peripheral membrane-bound proteins, such as receptors and ion channels.1 Meanwhile, drugs need to pass through one or more cell membranes to reach their targets of action. A great many studies have shown that membrane interactions can either result in better efficacy, selectivity and longer duration of action or cause undesirable toxicities through off-target interactions, reducing or even inhibiting the biological function of membrane proteins, and giving rise to possible serious cell damage.2,3 Therefore, it is of critical importance to understand the intrinsic influence of a drug on the lipid membrane in pharmacological science.4,5Propranolol (PPN) (Fig. 1a) is a beta-adrenergic blocking drug used in the treatment of several conditions, such as hypertension, angina pectoris, cardiac arrhythmias, myocardial infarction, anxiety disorders and migraine.6 It prevents binding of epinephrine and norepinephrine to cell surface receptors and inhibits intracellular signaling cascades.7 Extensive studies of PPN-membrane interactions have been made over the last several decades, including the antihypertensive mechanism for this drug,8,9 its location within the lipid bilayer,10,11 and its influence on membrane physicochemical characteristics and the activity of membrane-bound enzymes.12–14 It has been reported that PPN is readily to interact with cellular membranes and induce functional alterations in a wide variety of membrane systems.14–18 For example, it was found that the addition of PPN can change membrane fluidity,19 membrane curvature,20 as well as membrane elasticity and viscosity.21 Moreover, when the PPN concentration reached the mM range, it may lead to formation of PPN–lipid micelles and subsequently solubilization of the membrane.17 Although the effective concentration of PPN in the plasma at the proper dose of the drug is usually at μM range (about 0.03–600 μM),22–24 the peak concentrations of PPN in the cytoplasm are found up to 10–25 mM.7,25 Although it seems to be crucial for better understanding the mechanisms of drug action, systematic studies on PPN binding at lipid membrane interface over a wide range of concentrations is still lacking. Besides, in spite of qualitative insights into the interactions between PPN and lipid membranes, there is little information about the kinetics of membrane solubilization induced by PPN binding.19,20,26–29
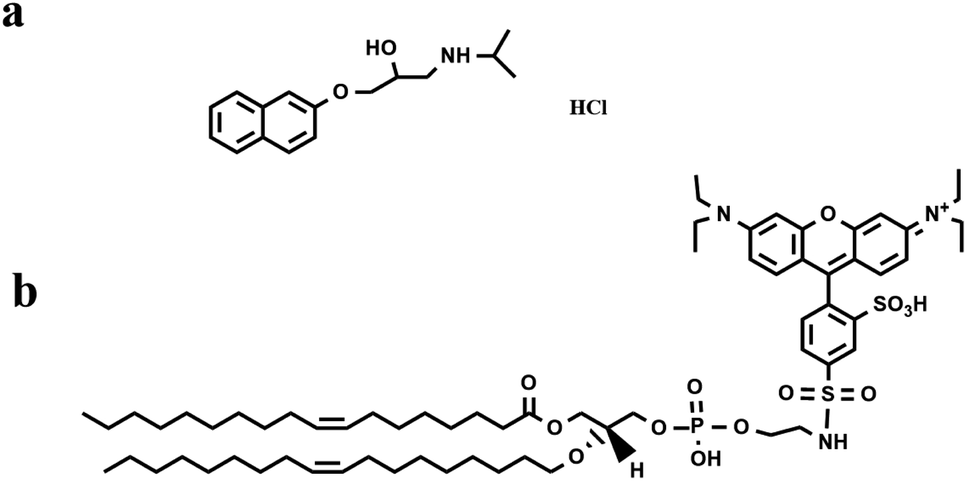 | ||
| Fig. 1 Molecular structures of (a) PPN and (b) Liss-Rhod-DOPE. | ||
In the current study, we carried out fluorescence analysis to investigate the PPN (concentration range from μM to mM) interactions with supported lipid bilayers (SLBs), as model membranes. The SLBs were coated inside a polydimethylsiloxane (PDMS) well or a microfluidic device, allowing for various concentrations of PPN to be introduced over the bilayer surface in a buffer solution. As one of the most popular model systems used for studying surface biochemistry, SLBs have relatively simple geometry, maintain appropriate lateral mobility (two-dimensional fluidity) for the lipid molecule, and offer high bilayer stability.30 Moreover, for studying drug–membrane interactions, it is advantageous to employ SLBs instead of liposomes, as SLBs require far less lipid material and sample volume. The fluorescent probe used in the study is 1,2- dioleoyl-sn-glycero-3-phosphoethanolamine-N-(lissamine rhodamine B sulfonyl) (Liss-Rhod-DOPE) (Fig. 1b) which is known to prefer the high-curvature structures because of its bulky head group.31 The strategy for our detection is based upon the notion that binding of PPN to lipid bilayers will induce an increase in the local curvature of the membrane20,32 and thus leads to an uneven partitioning of the probe in the membrane. Besides, as the drug–membrane interaction may eventually lead to membrane disruption at high drug concentration,17,28,33 the release of the fluorescent probe can be detected by monitoring the change in fluorescence intensity.
The results show that PPN binds with phosphatidylcholine membranes and induces significant membrane reorganization, including the formation of highly curved surface features as well as membrane solubilization. Moreover, by varying the drug concentration, it was possible to obtain a three-step binding profile of PPN with lipid membranes. In addition, it was found that PPN/membrane interaction could be modulated by varying the lipid composition in the membrane.
Experimental
Materials
The phospholipids 1-palmitoyl-2-oleoyl-sn-glycero-3-phosphocholine (POPC), 1-hexadecanoyl-2-(9-Z-octadecenoyl)-sn-glycero-3-phosphoethanolamine (POPE), 1-palmitoyl-2-oleoyl-sn-glycero-3-phospho-(1′-rac-glycerol) (POPG), 1-palmitoyl-2-oleoyl-sn-glycero-3-phosphoserine (POPS), and Liss-Rhod-DOPE were purchased from Avanti Polar Lipids (Alabaster, AL, USA). Cholesterol was obtained from Sigma Aldrich (Shanghai, China). Sodium chloride, chloroform, 4-hydroxyethyl piperazine sulfonic acid (HEPES), hydrochloric acid, sodium hydroxide and propranolol hydrochloride were of analytical grade and were purchased from Titan (Shanghai, China). Microscope coverslips (22 × 40 mm, no. 1.5) was supplied by Fisher Scientific (Pittsburgh, Pennsylvania, USA). PDMS (Dow Corning Sylgard Silicone Elastomer-184) was provided from Krayden, Inc. (El Paso, TX). Purified water (18.25 mΩ cm) was produced from a Direct-pure UP Water System (RephiLe Bioscience, Ltd, China).Small unilamellar vesicles (SUVs) preparation
Lipids of desired molar composition were mixed with 0.5 mol% of the fluorescent probe in chloroform in a glass vial and then dried under a flow of high purity nitrogen gas. The resulted lipid film was further dried under vacuum for 3 h to completely remove any residual organic solvent. The desiccated lipid film was hydrated with 10 mM HEPES buffer solution containing 150 mM NaCl (pH = 7.4) to obtain a final lipid concentration of 0.5 mg mL−1. The suspension was sonicated using an ultrasonic bath for about 10 s at 25 °C. The suspension was then subjected to at least 10 freezing–thawing cycles with liquid nitrogen and water (25 °C) and extruded for 10 times through two stacked 100 nm polycarbonate membrane (Whatman) using a Lipex thermobarrel extruder (Northern Lipids, Inc., Vancouver, Canada). The size of the lipid vesicles was about 110 ± 10 nm as determined by dynamic light scattering (Zetasizer Nano S90, Malvern Panalytical, United Kingdom). The resulted SUV solutions were stored at 4 °C until use.Microfluidic device and PDMS well setup
Microfluidic device was fabricated according to procedures reported elsewhere.34,35 The glass slides were cleaned in a boiling 7× cleaning solution (MP Biomedicals, Solon, OH) for 1.5 h, rinsed copiously with purified water, and then dried with high purity nitrogen gas. The photolithographically patterned photoresist (Microposit S1813, Shipley Corp., Marlborough, MA) was deposited on the glass surface. A patterned glass slide was HF-etched to form a model glass slide and washed with acetone. PDMS was degassed under vacuum for at least 1 h and was then poured onto a clean model glass slide and cured in incubator at 70 °C for 3 h. Small holes were poked with a needle in PDMS mold, serving as inlet and outlet ports for flowing liquid. The PDMS mold and glass slide were placed in a plasma cleaner (PDC-32 G, Harrick, Pleasantville, NY) for 1 min of plasma treatment. Immediately after plasma treatment, PDMS mold and glass slide were pressed together, and annealed at 108 °C for 1 min. PDMS well setup was fabricated by placing a PDMS film (∼0.5 mm) with a 6 mm diameter hole in the middle.Supported lipid bilayer (SLB) formation
To form a SLB, the desired SUV solution was added into the circular hole of a PDMS well or the microchannels of a microfluidic device and allowed to incubate for 20 min. SLBs formed spontaneously on the glass coverslip via vesicle fusion.36,37 Excess lipids were removed from the surface of SLB by rinsing with HEPES buffer. All SLBs were formed at room temperature (25 ± 1 °C).Fluorescence microscopy
Fluorescent images were captured by Nikon Eclipse Ti-U inverted microscope (Tokyo, Japan) equipped with an Andor iKon-M 934 Back Illuminated CCD camera (Andor, Belfast, United Kingdom) and SHI-130 N1 (Nikon Yokohama, Japan) light source. A Texas Red filter set (Nikon, Tokyo, Japan) was used for imaging along with 4× (N.A. = 0.13), 10× (N.A. = 0.45×) and 40× air objectives (N.A. = 0.95). The NIS-Elements BR software (Nikon, Tokyo, Japan) was employed to process the fluorescent images.Fluorescence recovery after photobleaching (FRAP)
The lateral diffusion of SLBs was determined by FRAP experiments.38–41 Herein, a 560 nm light beam (28 mm radius) was used to bleach the fluorescent probes within SLBs. Immediately after photobleaching, a series of images were captured and the fluorescence recovery (F(t)) was calculated using the following equation (eqn (1)):
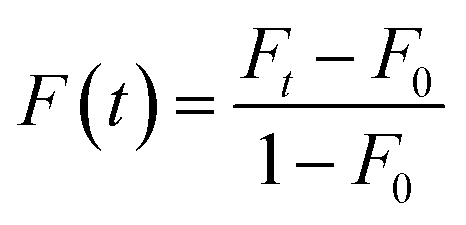 | (1) |
| y = A(1 − e−kt) | (2) |
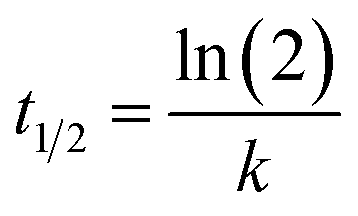 | (3) |
Use t1/2 to calculate the lateral diffusion coefficient (D) (eqn (4)):
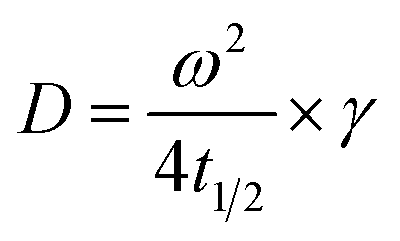 | (4) |
Membrane hole identification
To identify membrane holes, the threshold value was set by using the signal/noise ratio option in NIS-Elements BR software. The setting used was the result of subtracting 6 of standard deviations from average fluorescence intensity of the bilayer areas between holes. This threshold was automatically applied for analysis of the areas and the sizes of membrane holes.Results and discussion
PPN induces reorganization of POPC SLBs
To explore the structural effects of PPN on SLBs, incubation experiments were carried out in PDMS wells. The work was performed with 10 mM HEPES buffer solution containing 150 mM NaCl at pH 7.4 unless otherwise specified. The lateral mobility of SLBs at room temperature was confirmed by FRAP measurements (data not shown).Phosphatidylcholine (PC) is one of the essential components of all mammalian cell types and subcellular organelles. It accounts for more than 50% of total cellular phospholipids.42 In an initial series of experiments, SLBs containing 99.5 mol% POPC and 0.5 mol% Liss-Rhod-DOPE were used. Time-lapse fluorescence images obtained after incubation of PPN with SLBs are shown in Fig. 2. As can be seen, bright fluorescent spots started to decorate on the surface of homogeneous bilayers immediately (within few seconds) after PPN addition (Fig. 2b). Fluorescence intensity of the bright spots is roughly double that of the surrounding area, suggesting an uneven distribution of the dye in the bilayer. According to the curvature preference of Liss-Rhod-DOPE,43 it can be inferred that the bright spots are areas of high membrane curvature induced by PPN. Strand like protrusions that we term lipid tubules then grew from the bright spots in several minutes (Fig. 2c). These lipid tubules retain a point of association to the bilayer surface and may extend up to hundreds of micrometers at higher concentrations of PPN (15 mM–40 mM). The floating ends of the lipid tubules possess high mobility and many of them were consequently out of focus (see movies S1 and S2†).
 | ||
| Fig. 2 Time-lapse fluorescence micrographs of structural reformation in POPC SLBs treated with 20 mM PPN. Each image represents the SLB after PPN incubation for: (a) 0 min, (b) 0.12 min, (c) 5 min, (d) 60 min, (e) 80 min, (f) 110 min, (g) 120 min, and (h) 140 min. The micrographs were observed at 40× magnification. Scale bar: 20 μm. | ||
Curiously, over the course of minutes, numerous lipid tubules were observed to shrink in length followed by the formation of large circular patches (Fig. 2d–f). These patches may gradually increase in size and eventually disappear from bilayer surface in a short time (Fig. 2h). Fluorescence characteristics of the patch were then investigated. Line scan of the image shows that fluorescence profile of the patch approaches a Gaussian distribution, indicating higher curvature in the center of the patch (Fig. 3). Moreover, the fluorescence intensity of the patch levels off at first and then decreased sharply at the same time with the disappearance of the patch (Fig. 4). Besides, FRAP experiments revealed that lipids maintained lateral mobility after disappearance of the patch. The diffusion coefficient was consistent before (0.786 ± 0.022 μm2 s−1) and after (0.979 ± 0.031 μm2 s−1) PPN treatment. The data are consistent with the literature value for POPC SLB doped with 1 mol% of fluorescent Texas Red DHPE (0.8–1.2 μm2 s−1).44 Moreover, the mobile fraction for the SLB is 93 ± 1%, confirming the two-dimensional fluidity of the bilayer that enables the recovery of fluorescence intensity after photobleaching.
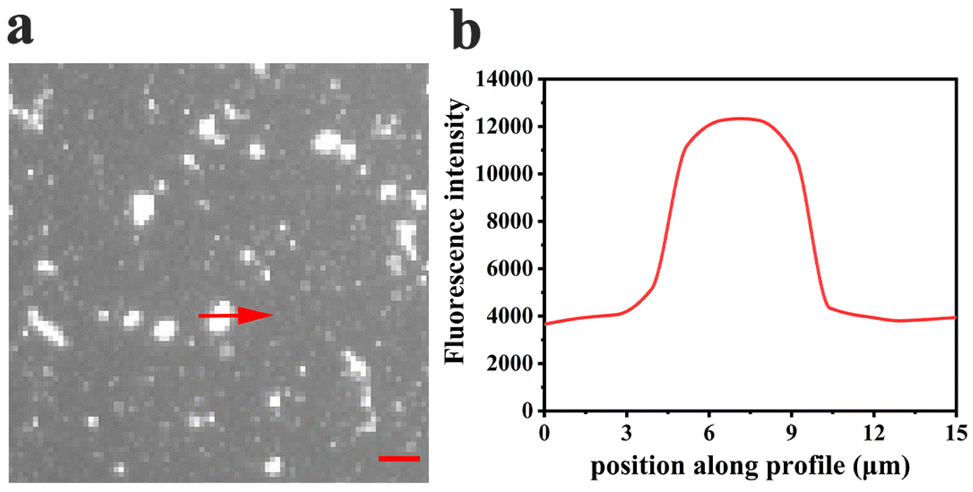 | ||
| Fig. 3 (a) A patch observed in POPC SLBs treated with 20 mM PPN. The red arrow traces the fluorescence intensity profile across the centre of the patch, shown in (b). The micrographs were observed at 40× magnification. Scale bar: 10 μm. | ||
 | ||
| Fig. 4 Fluorescence intensity of the patch changes with time after introduction of 20 mM PPN. The red arrows in the sequential images point to the patch. The fluorescence images were observed at 40× magnification. Scale bar: 10 μm. | ||
These observations, taken together, indicate a transition from high-curvature structures (i.e., tubules) to low-curvature structures (i.e., patches) and ultimately, to a flat bilayer. We therefore propose that the patches are hemispherical caps which caused by tubule collapse,45,46 rather than spherical buds composed of entangled tubules.47 In addition to the tubules and patches described above, the floating fluorescent materials across the field of view were observed (Fig. 2g), suggesting that some local removal of bilayer material could occur during PPN incubation.
Additional experiments revealed that formation of these surface features was, at least partially reversible upon the removal of the substances loosely attached to the surface. Fig. 5 shows images of SLBs after PPN treatment followed by thoroughly rinsing with HEPES buffer solution. As can be seen, SLBs treated with lower concentrations of PPN (<5 mM) could recover to be uniform after surface washing (Fig. 5a and b). However, darker areas of decreased fluorescence were observed on the SLBs treated with higher concentrations of PPN (5 mM–40 mM) (Fig. 5c–f). The size of the darker areas varied from <1 μm2 up to 14.5 μm2 and the size distribution gradually shifted to larger area with increasing concentrations of the drug (Fig. 6). FRAP experiments showed that the lipids had lateral mobility within the remaining membrane, with dark areas unperturbed (Fig. 7). This supports the notion of static holes within an otherwise continuous fluid lipid bilayers.33,48 Therefore, the dark areas are holes formed in the bilayer due to partial solubilization of the bilayer induced by PPN. The observation of these darker areas is consistent with our observations, as discussed above, of fluorescent material floating across the field of view when PPN was introduced to the bilayers.
 | ||
| Fig. 5 Hole formation in POPC SLBs after PPN (>5 mM) treatment and washing. (a–f) Fluorescence images showing SLBs after treatment with different concentrations of PPN followed by washing the surface. The fluorescence images were observed at 40× magnification. Scale bar: 20 μm. | ||
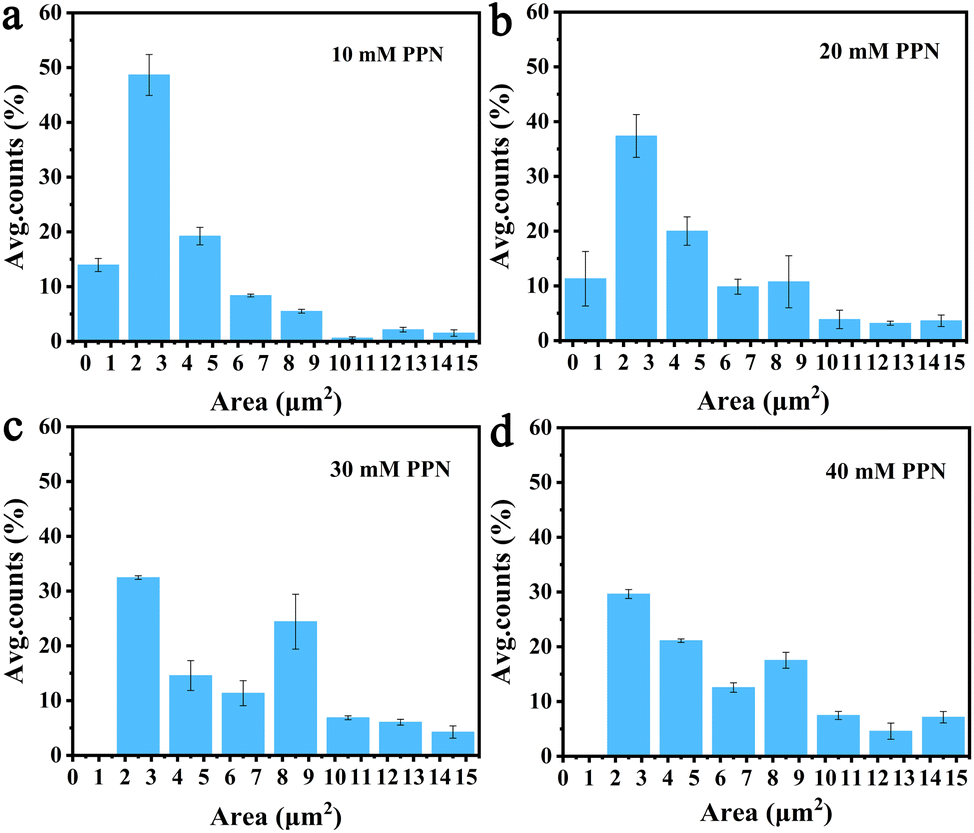 | ||
| Fig. 6 Histogram of the hole size on the surface of POPC SLBs after 1 h incubation with different concentrations of PPN: (a) 10 mM, (b) 20 mM, (c) 30 mM, and (d) 40 Mm. The y-axis shows the percentage of average count numbers from different bilayers. The error bars represent the standard deviations obtained by averaging at least 4 independent measurements. | ||
 | ||
| Fig. 7 FRAP experiments of POPC SLBs treated with 20 mM PPN followed by washing the surface. The red line is single exponential fits to the data. The error bars represent the standard deviations obtained by averaging at least 4 independent measurements. | ||
Analogous phenomena have been observed both in modeling experiments and biological processes such as endocytosis, tabulation, and vesiculation.49–52 Generally, the transition of a relative planar structure to a more curved structure may arise from the adsorption of various substances, such as macromolecules, ions, as well as small molecules, which differ in their sizes and properties.43,53–59 It has been reported that amphiphile-induced bilayer remodeling are strongly correlated to the thermodynamic barrier of the interacting amphiphile to translocation across the bilayer.45,46,60 Specifically, when amphiphiles are incubated with a bilayer, they may impart an asymmetric, lateral stress on the outer leaflet that is not immediately matched by the inner leaflet. If the bilayer area is fixed, as in the current SLB experiment, the PPN-induced asymmetrical stress would overwhelm the adhesive strength of the underlying solid support, leading to the formation of regions with high curvature (such as lipid tubules).45 However, if the translocation, i.e., “flip-flop”, of amphiphiles across the bilayer occurs, the asymmetrical stress would be expected to be relieved, leading to the decreasing of curvature, i.e. the formation of lower-curvature structures (such as hemispherical caps) or restoration of planar bilayers. Therefore, the shrinking and disappearance of tubules and caps observed in our experiments can be interpreted in terms of accumulation and relief of asymmetric stresses in the inner and outer leaflets of a bilayer upon the insertion and translocation of PPN in ways that are similar to previous experiments using other amphiphiles. It should be noted that desorption of PPN from the bilayer upon washing with buffer or bilayer dissolution may also equilibrate stresses in the bilayer and promotes a transition from high-curvature tubules to low-curvature hemispherical caps and ultimately, in the limit of complete PPN desorption, to a flat bilayer.
Moreover, as a short molecule whose non-polar moieties are not as long as the hydrocarbon chains of phospholipids, PPN may act as a wedge in the membrane, causing energetically unfavorable voids between hydrocarbon chains in the bilayer interior.61 Thus, bending of membrane is energetically favored to eliminate the voids. Indeed, due to the asymmetric distribution of PPN, the bilayer will prefer to curve toward the inserted PPN, expanding the upper leaflet with the larger coverage of drug molecules and compressing the lower leaflet with less drug molecules.58
Besides, the observation of substantial disruption and dissolution of bilayers may be related to the detergent-like characteristic of PPN.4,8 Although the PPN concentration used in this study is lower than its cmc (∼108 mM),62 local concentration of the drug in the bilayer may be much higher than that in the bulk aqueous phase, which possibly promotes the formation of lipid-drug mixed micelles that may consequently shed from the bilayer into the bulk solution. Indeed, tubulation expands lipid headgroup–headgroup spacing in the upper leaflet of bilayer which in turn promotes accumulation of PPN in the tubules compared with the surrounding planar bilayers. Actually, the onset of micellization occurs at the concentration lower than the cmc of pure detergents has been reported elsewhere.63
Kinetics of interaction between PPN and POPC SLBs
To further explore the intrinsic mechanism of interaction between PPN and lipid bilayers, microfluidic experiments were conducted. HEPES buffer solution was flowed into the microfluidic channels in which lipid bilayers were formed. After fluorescence intensity reaches equilibrium, PPN (0–40 mM) was introduced into the channels. Time-lapse images were acquired every 5 min and fluorescence stabilization took about 1 h at the lowest drug concentration. Buffer solution was then introduced to rinse away PPN and any other loosely attached lipid materials. The individual fluorescence line scans were then exerted. As can be seen in Fig. 8, the fluorescence intensity of lipid bilayers decreased with increasing concentration of PPN. At the highest concentration of PPN employed, 40 mM, the fluorescence intensity decreased to 70% of its initial level. This attenuation in the fluorescence intensity confirms the PPN-induced disruption of SLBs, i.e., the release of bilayer lipids into the medium.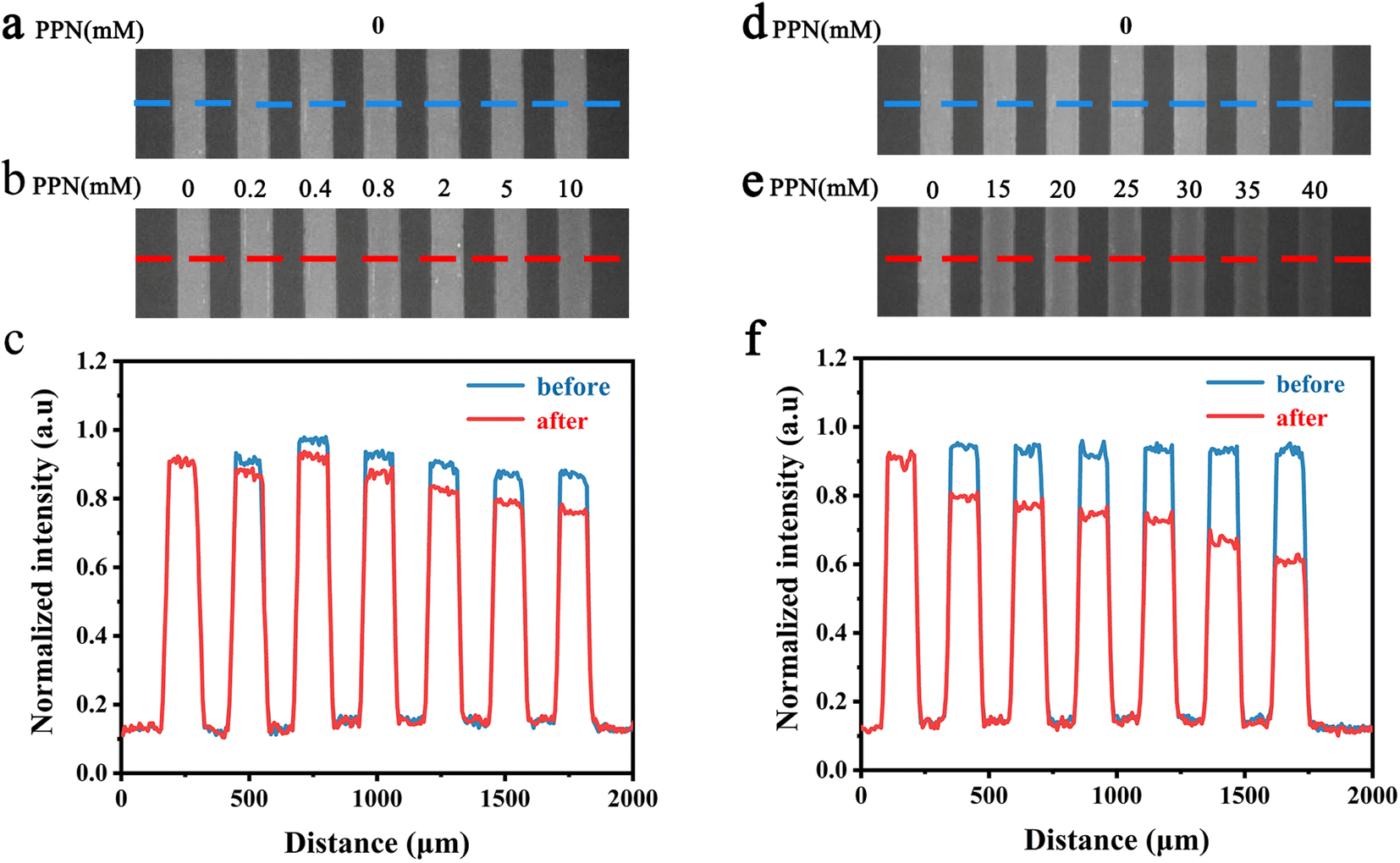 | ||
| Fig. 8 Fluorescence images from microfluidic devices with POPC SLBs before (a and d) and after (b and e) introduction of PPN. The blue and red lines represent the regions used to obtain the line profiles shown in (c and f). | ||
By plotting the fluorescence intensity change as a function of drug concentration, the profile of PPN-bilayer interaction can be obtained (Fig. 9). The y-axis represents change of fluorescence intensity at equilibrium, ΔF, which is calculated as (F/F0 − 1). Here, F corresponds to the fluorescence intensity of the bilayer at a specific concentration of PPN in the bulk solution, whereas F0 is the fluorescence intensity of the bilayer in pure buffer. Significantly, the interaction profile showed a complex shape, consisting of three distinct steps, corresponding to three separate concentration ranges (0–10 mM, 10–30 mM and 30–40 mM). The interaction profiles for step 1 and step 2 fit well to a Langmuir isotherm (eqn (5)):
 | (5) |
 | ||
| Fig. 9 Binding profile of POPC SLBs with PPN concentration of 0–40 mM. The data (step 1 and 2) can be fit well to a Langmuir isotherm. At high concentrations (>30 mM), the data (step 3) can be fit to a straight line (r2 = 0.995). The error bars represent the standard deviations obtained by averaging at least 4 independent measurements. | ||
Our current data showing a three-step interaction profile of PPN-bilayer is consistent with the notion that binding of amphiphiles with lipid membranes undergoes a consecutive stepwise process.53,65,66 Generally, the first binding step was dominated by electrostatics, which involves the interaction between the amine group of PPN and the carbonyl group of phospholipids, as well as the interaction between the hydroxyl group of PPN and the carbonyl group of phospholipids.61 This can be confirmed by an additional experiment using higher buffer concentrations. Presumably, the increase in the ionic strength of the solution leads to the decrease in Debye length which, in turn, enlarge the electrostatic screening and therefore the drug–membrane interactions should weaken.67,68 Herein, 50 mM HEPES buffer solution was utilized and the assays were otherwise carried out under the same conditions as those in Fig. 9. The resulted binding curve was provided in Fig. 10. As can be seen, increasing buffer concentration decreased the affinity between PPN and the bilayers by approximately 3-fold (Kd = 3.68 ± 0.36 mM), verifying that the first step of PPN/membrane binding is mainly electrostatic. In fact, there is a competition between strong electrostatic interactions and van der Waals interactions between the naphthalene ring of PPN and non-polar lipid tails.61 Although electrostatic interactions prevail in the first step, the adsorption of PPN to the bilayer may gradually increase the fluidity of the lipids, lower its area stretch modulus and decrease the interfacial potential, which helps to facilitate the second step, in which hydrophobic insertion and deeper penetration of the drug into the lipid bilayer occurs.33,69 Moreover, PPN not only intercalated into lipid bilayer or expanded the membrane area, but also disrupt the packing of PC lipids.61 In that case, the third step associated with membrane disruption and hole formation can be considered a direct consequence of PPN binding.62,70 These results are in agreement with the observation obtained from the PDMS well experiments. It should be noted that the attenuation in the fluorescence intensity in the first and the second step, suggesting PPN-containing bilayer is likely to be unstable even at lower concentrations, although major damage or hole formation on the membrane is not detectable in the incubation experiments.
 | ||
| Fig. 10 Binding curve of PPN (0–10 mM) and POPC lipid bilayer in 50 mM HEPES buffer at pH 7.4. The error bars represent the standard deviations obtained by averaging at least 4 independent measurements. | ||
The dissociation constants of PPN–membrane interaction were compared with those for the interaction of other small drugs, including ibuprofen (IBU) and tetracaine (TTC), with lipid membranes.33,71 As expected, the membrane affinities of the drug are substantially different, although they have similar amphiphilic properties and similar multiple consecutive binding steps with lipid membranes. The dissociation constant values of Kd follow the order: PPN > TTC > IBU. This is consistent with the notion that the interaction of a bioactive compound across lipid membranes is correlated with its lipophilicity.21,72 Supposedly, the drugs with higher lipophilicity have relative stronger ability to interact with lipid membranes. In the current study, the weaker membrane affinity of PPN could be due to the fact that PPN has relatively lower lipophilicity as compared to TTC and IBU.
Interaction between PPN and binary mixed SLBs
In the next set of experiments, cholesterol, uncharged POPE, negatively charged POPG and POPS was respectively introduced into POPC SLBs and tested for PPN interactions. The buffer for POPS contained 0.8 mM EDTA due to the potential quenching ability of trace concentrations of divalent metal ions such as Cu2+.64 The experimental conditions were otherwise identical to those in POPC experiments.A representative image from flow cell experiment after incubation with 15 mM PPN is shown in Fig. 11. As can be seen, the formation of surface features considerably differs upon adding other membrane lipids into POPC SLBs, suggesting PPN/SLB interaction is significantly dependent on membrane composition. Binding profiles of PPN to bilayers are plotted in Fig. 12 and the extracted apparent Kd values are provided in Table 1.
 | ||
| Fig. 11 Fluorescence images of various SLBs treated with 20 mM PPN followed by washing the surface. (a–e) Each column represents the SLB containing a specific lipid composition. The fluorescence images were observed at 40× magnification. Scale bar: 20 μm. | ||
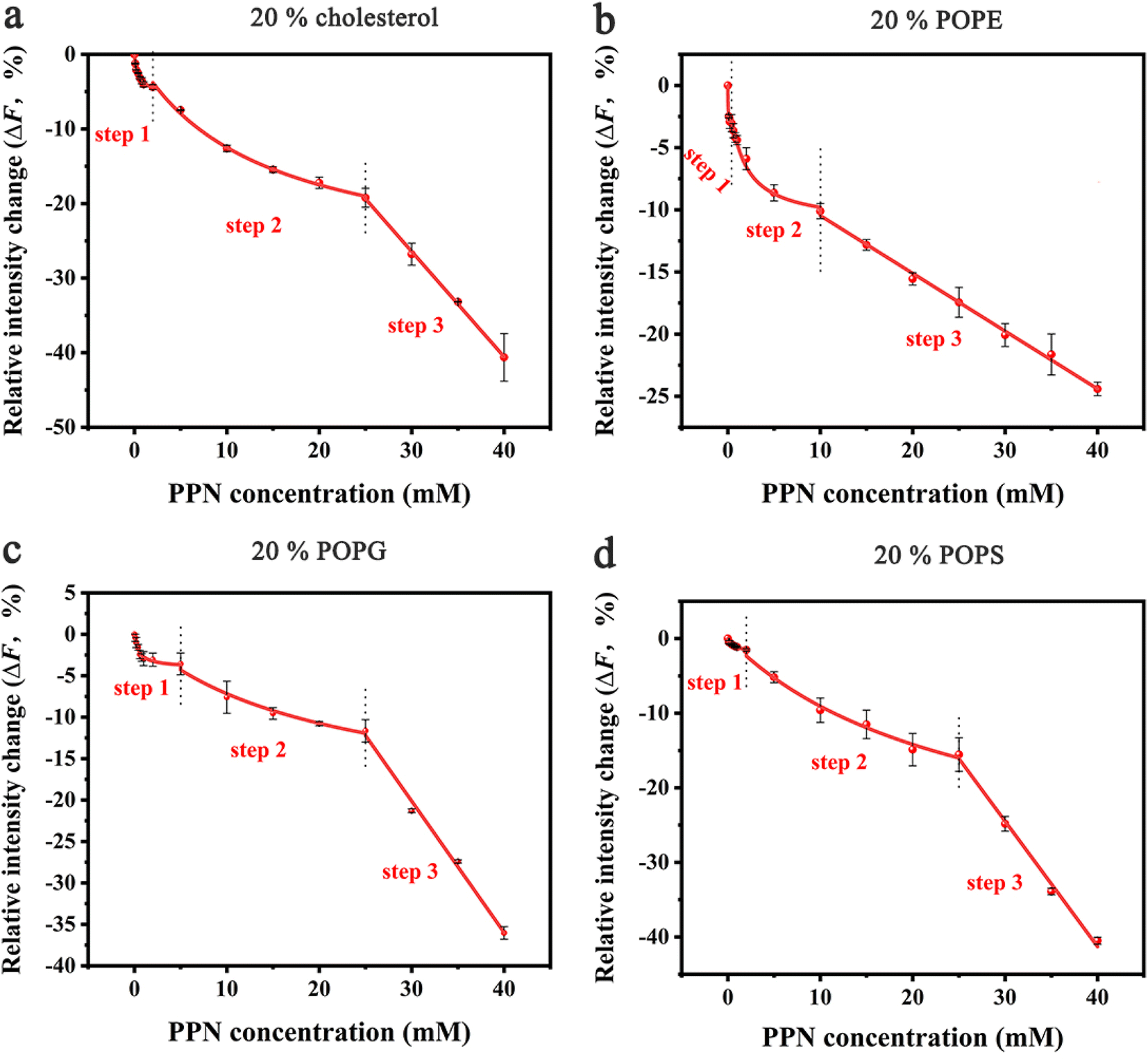 | ||
| Fig. 12 Binding profiles of PPN to SLBs with different lipid compositions. All the membranes contained 0.5 mol% Liss-Rhod-DOPE and 79.5 mol% POPC: (a) 20 mol% cholesterol; (b) 20 mol% POPE; (c) 20 mol% POPG; (d) 20 mol% POPS. The red curve represents the best fit for step 1, 2 and 3. The error bars represent the standard deviations obtained by averaging at least 4 independent measurements. | ||
| POPC | POPC + 20% cholesterol | POPC + 20% POPE | POPC + 20% POPG | POPC + 20% POPS | |
|---|---|---|---|---|---|
| Kd1 (mM) | 1.40 ± 0.12 | 0.35 ± 0.05 | 0.03 ± 0.004 | 0.49 ± 0.10 | 0.52 ± 0.13 |
| Kd2 (mM) | 28.60 ± 7.71 | 13.45 ± 1.50 | 1.42 ± 0.20 | 20.00 ± 5.86 | 26.02 ± 7.38 |
Cholesterol is an essential structural component of mammalian cells, varying significantly in concentration in the membranes of different organelles. It affects the structure and dynamics of membranes by interacting with lipids and membrane-related proteins.73–75 To test the effect of cholesterol on PPN/bilayer interaction, 20 mol% cholesterol was introduced into POPC SLBs. As is shown in Fig. 12a and Table 1, the Kd1 value for the first binding step decreased approximately 4-fold (form 1.40 ± 0.12 mM to 0.35 ± 0.05 mM) compared to pure POPC bilayer membrane. This is likely the consequence of hydrogen bonding between cholesterol and PPN. Several studies have demonstrated the ability of cholesterol to form hydrogen bond with various small molecules.76–79 Liang Chen et al. demonstrated hydrogen bond formation between hydroxyl hydrogen on the A-ring of epicatechin and hydroxyl oxygen of cholesterol.76 Yuan et al. proposed hydrogen bond formation between POPC and cholesterol between either the hydroxyl and phosphate or hydroxyl and carbonyl.77 By analogy, a hydrogen bond is likely to form between the amine groups on PPN and the hydroxyl from cholesterol. In the second binding step, Kd2 decreased approximately 2-fold (from 28.60 ± 7.71 mM to 13.45 ± 1.50 mM) compared with pure POPC bilayer. This strengthening of the PPN/SLB interaction is in agreement with the notion that cholesterol can induce expansion of lipid–lipid distance that facilitates insertion of various amphiphilic molecules into the bilayer.77,80–83 This observation therefore confirms that the second binding step is dominated by hydrophobic interaction. Moreover, previous studies have shown that cholesterol generates intrinsic negative curvature in lipid bilayers and lower the formation energy of highly curved membrane structures such as lipid stalks that are proposed as lipid intermediates in membrane fusion. Cholesterol thereby has the potential of promoting membrane fusion.84–89 As such, it is reasonable that cholesterol helps formation of lipid tubules, hence facilitating membrane disruption and hole formation.
Phosphatidylethanolamine (PE) is the second most abundant phospholipid in eukaryotic cells membranes, accounting for 20–50 mol% of total phospholipid, which can comprise up to 45 mol% phospholipids in the brain.90,91 Therefore, it is of great importance to understand the influence of PE on drug/membrane interaction. For the current experiment, POPE was used since it has the same tail group as POPC. The binding constants Kd1 and Kd2 were both significantly decreased for SLBs containing 20% POPE compared to pure POPC bilayers (Fig. 12b and Table 1). This increase of PPN affinity with bilayer is probably due to the formation of hydrogen bonding between POPE lipids and PPN. There has been evidence that the amine group of PPN interacts with carbonyl group of the lipid acyl chains; on average, amino group have two hydrogen bonds that are provided to carbonyl oxygen atoms.61 Also, a hydrogen bond is likely to form between the hydroxyl group of PPN and the phosphate group of the lipids and generally one hydrogen bond is donated by the hydroxyl group to phosphates.61,92 Moreover, compared to most other cylindrical-shaped phospholipids such as PC, PE lipids adopt a conical shape due to the relative volume difference between its small ethanolamine head group and long, unsaturated acyl chains.93 PE thereby induces negative spontaneous curvature in membranes, which is reported to make the planar bilayers unstable.31 In fact, when PPN inserts into the POPE-containing bilayers, it acts as a wedge, relieving the lipid packing constraints caused by the presence of POPE.94 Thus, it is energetically favorable for PPN to fill up the POPE-induced voids between phospholipid headgroups.
Negatively charged lipids are integral components of eukaryotic cell membranes and organelles. Although they are not as abundant as PE or cholesterol in biological membranes, negatively charged lipids play essential roles in biological processes, such as apoptosis and cell signaling.95–99 Herein, two negatively charged lipids, POPS and POPG with concentration of 20 mol%, are introduced into POPC SLBs. As can be seen in Fig. 12c, d and Table 1, the influence of POPS and POPG on the Kd1 and Kd2 values is essentially identical, suggesting the effect should be electrostatic rather than chemically specific. Compared with pure POPC SLBs, the first binding step was significantly strengthened by the addition of POPS and POPG. This is expected since the incorporation of any of these two lipids into the bilayer will make the surface potential more negative and in turn promotes the electrostatic interactions between the positively charged PPN and the bilayer.100 Moreover, it was observed that the insertion ability of PPN into the bilayer was identical between the negatively charged bilayers and pure POPC SLBs. Indeed, as cylindrical lipids, with hydrophobic tails and hydrophilic heads of similar cross section PS and PG are likely to form lamellar bilayers lacking curvature as the size of polar head groups rather than the acyl chain determines the intrinsic shape of the lipid.84 Therefore, it is logical that PS and PG do not affect the insertion of PPN into the bilayer as they do not influence membrane curvature.
Conclusions
The work reported here demonstrates that PPN, because of its structure and of the presence of charged heads, may interact with lipid membranes to induce large-scale membrane remodeling, i.e., the formation of three-dimensional surface structures with high curvature followed by membrane disruption and solubilization (Fig. 13). Moreover, the kinetic analysis revealed that PPN has concentration-dependent binding behaviour with lipid membranes. The interaction between PPN and lipid membranes shows three consecutive steps, involving the initial electrostatically adsorption onto the bilayer surface, the consequent hydrophobic insertion of the drug, and the final membrane solubilization. The profiles of the first two steps fit well to a Langmuir isotherm, while the final step is associated with the detergent characteristic of PPN, i.e., unsaturable interaction that induces membrane solubilization. Besides, the affinity between PPN and lipid bilayer could be significantly modulated by incorporating other lipids into POPC SLBs, indicating the dependence of membrane physical properties on drug–membrane interactions. | ||
| Fig. 13 Schematic illustration of the structural remodeling of POPC bilayers induced by the addition of propranolol. | ||
Conflicts of interest
There are no conflicts to declare.Acknowledgements
We are grateful to Danling Cheng at the Shanghai University of Engineering Science for technical assistance with small unilamellar vesicles preparation and supported lipid bilayer formation.Notes and references
- H. Yin and A. D. Flynn, Annu. Rev. Biomed. Eng., 2016, 18, 51 CrossRef CAS PubMed.
- M. Lucio, J. Lima and S. Reis, Curr. Med. Chem., 2010, 17, 1795–1809 CrossRef CAS PubMed.
- M. J. Saxton, Biophys. J., 1987, 52, 989–997 CrossRef CAS PubMed.
- J. K. Seydel, E. A. Coats, H. P. Cordes and M. Wiese, Arch. Pharm., 1994, 327, 601–610 CrossRef CAS PubMed.
- G. P. van Balen, C. a. M. Martinet, G. Caron, G. Bouchard, M. Reist, P. A. Carrupt, R. Fruttero, A. Gasco and B. Testa, Med. Res. Rev., 2004, 24, 299–324 CrossRef CAS PubMed.
- M. R. Bristow, Circulation, 2000, 101, 558–569 CrossRef CAS PubMed.
- R. Bisby, S. Botchway, A. Crisostomo, J. Karolin, A. Parker and L. Schröder, Spectroscopy, 2010, 24, 137–142 CrossRef CAS.
- E. K. Jackson and W. B. Campbell, Hypertension, 1981, 3, 23–33 CrossRef CAS PubMed.
- R. Grewal and C. Kaul, Br. J. Pharmacol., 1970, 38, 771 CrossRef CAS PubMed.
- K. Azizi and M. G. Koli, J. Mol. Graphics Modell., 2016, 64, 153–164 CrossRef CAS PubMed.
- G. Först, L. Cwiklik, P. Jurkiewicz, R. Schubert and M. Hof, Eur. J. Pharm. Biopharm., 2014, 87, 559–569 CrossRef PubMed.
- K. W. Surewicz, I. Fijałkowska and W. Leyko, Biochem. Pharmacol., 1981, 30, 839–842 CrossRef CAS PubMed.
- D. V. Godin, T. W. Ng and J. M. Tuchek, Biochim. Biophys. Acta, Biomembr., 1976, 436, 757–773 CrossRef CAS PubMed.
- A. Ekman, V. Manninen and S. Salminen, Acta Physiol. Scand., 1969, 75, 333–344 CrossRef CAS PubMed.
- W. K. Surewicz and W. Leyko, Biochim. Biophys. Acta, 1981, 643, 387–397 CrossRef CAS PubMed.
- D. V. Godin, T. W. Ng and J. M. Tuchek, Biochim. Biophys. Acta, 1976, 436, 757–773 CrossRef CAS PubMed.
- H. Porzig, J. Physiol., 1975, 249, 27–49 CrossRef CAS PubMed.
- H. Komai and H. A. Berkoff, Biochem. Pharmacol., 1979, 28, 1501–1504 CrossRef CAS PubMed.
- C. Pereira-Leite, C. Carneiro, J. X. Soares, C. Afonso, C. Nunes, M. Lúcio and S. Reis, Eur. J. Pharm. Biopharm., 2013, 84, 183–191 CrossRef CAS PubMed.
- G. Mangiapia, M. Gvaramia, L. Kuhrts, J. Teixeira, A. Koutsioubas, O. Soltwedel and H. Frielinghaus, Phys. Chem. Chem. Phys., 2017, 19, 32057–32071 RSC.
- K. Kannisto, L. Murtomäki and T. Viitala, Colloids Surf., B, 2011, 86, 298–304 CrossRef CAS PubMed.
- L. Li, L. Yu, H. He, L. Wei, Z. Xu, L. Zhao, Y. Sun, B. Zhang, Y. Liu and R. He, Frontiers in Pediatrics, 2022, 10, 849496 CrossRef PubMed.
- J. Wójcicki, V. Sulżyc-Bielicka, J. Kutrzeba, B. Gawrońska-Szklarz, M. Droździk and Z. Sterna, J. Clin. Pharmacol., 1999, 39, 826–833 CrossRef PubMed.
- M. N. Kalam, M. F. Rasool, A. U. Rehman and N. Ahmed, Curr. Drug Metab., 2020, 21, 89–105 CrossRef CAS PubMed.
- R. Bisby, A. Crisostomo, S. Botchway, A. Parker and S. Watts, Central Laser Facility Annual Report, 2009, pp. 147–148 Search PubMed.
- M. Mizogami, K. Takakura and H. Tsuchiya, Eur. J. Anaesthesiol., 2010, 27, 829–834 CrossRef CAS PubMed.
- G. Castronuovo and M. Niccoli, Bioorg. Med. Chem., 2006, 14, 3883–3887 CrossRef CAS PubMed.
- S. De Carlo, H. Fiaux and C. A. Marca-Martinet, J. Liposome Res., 2004, 14, 61–76 CrossRef CAS PubMed.
- M. Ikonen, L. Murtomäki and K. Kontturi, Colloids Surf., B, 2010, 78, 275–282 CrossRef CAS PubMed.
- H. Li, T. Zhao and Z. Sun, Rev. Anal. Chem., 2017, 37, 20170012 Search PubMed.
- A. M. Sendecki, M. F. Poyton, A. J. Baxter, T. Yang and P. S. Cremer, Langmuir, 2017, 33, 13423–13429 CrossRef CAS PubMed.
- W. K. Surewicz and W. Leyko, Biochim. Biophys. Acta, Biomembr., 1981, 643, 387–397 CrossRef CAS PubMed.
- S. Sun, A. M. Sendecki, S. Pullanchery, D. Huang, T. Yang and P. S. Cremer, Langmuir, 2018, 34, 10782–10792 CrossRef CAS PubMed.
- H. Jung, A. D. Robison and P. S. Cremer, J. Am. Chem. Soc., 2009, 131, 1006–1014 CrossRef CAS PubMed.
- M. F. Poyton, A. M. Sendecki, X. Cong and P. S. Cremer, J. Am. Chem. Soc., 2016, 138, 1584–1590 CrossRef CAS PubMed.
- A. A. Brian and H. M. Mcconnell, Proc. Natl. Acad. Sci. U. S. A., 1984, 81, 6159–6163 CrossRef CAS PubMed.
- P. S. Cremer and S. G. Boxer, J. Phys. Chem. B, 1999, 103, 2554–2559 CrossRef CAS.
- D. Axelrod, D. E. Koppel, J. Schlessinger, E. Elson and W. W. Webb, Biophys. J., 1976, 16, 1055–1069 CrossRef CAS PubMed.
- M. D. Soumpasis, Biophys. J., 1983, 41, 95–97 CrossRef PubMed.
- R. Richter, A. Mukhopadhyay and A. Brisson, Biophys. J., 2003, 85, 3035–3047 CrossRef CAS PubMed.
- D. Shengjuler, S. Sun, P. S. Cremer and C. E. Cameron, J. Visualized Exp., 2017, e55869 Search PubMed.
- J. N. van der Veen, J. P. Kennelly, S. Wan, J. E. Vance, D. E. Vance and R. L. Jacobs, Biochim. Biophys. Acta, Biomembr., 2017, 1859, 1558–1572 CrossRef CAS PubMed.
- P. G. Adams, L. Lamoureux, K. L. Swingle, H. Mukundan and G. A. Montaño, Biophys. J., 2014, 106, 2395–2407 CrossRef CAS PubMed.
- A. M. Brozell, M. A. Muha, B. Sanii and A. N. Parikh, J. Am. Chem. Soc., 2006, 128, 62–63 CrossRef CAS PubMed.
- M. Staykova, M. Arroyo, M. Rahimi and H. A. Stone, Phys. Rev. Lett., 2013, 110, 028101 CrossRef PubMed.
- C. G. Gahan, S. J. Patel, L. M. Chen, D. E. Manson, Z. J. Ehmer, H. E. Blackwell, R. C. Van Lehn and D. M. Lynn, Langmuir, 2021, 37, 9120–9136 CrossRef CAS PubMed.
- B. K. Yoon, J. A. Jackman, M. C. Kim and N.-J. Cho, Langmuir, 2015, 31, 10223–10232 CrossRef CAS PubMed.
- P. Rangamani, K. K. Mandadap and G. Oster, Biophys. J., 2014, 107, 751–762 CrossRef CAS PubMed.
- J. C. Stachowiak, C. C. Hayden and S. Whitesides, Proc. Natl. Acad. Sci. U. S. A., 2010, 107, 7781–7786 CrossRef CAS PubMed.
- N. Wang, L. D. Clark, Y. Gao, M. M. Kozlov, T. Shemesh and T. A. Rapoport, Nat. Commun., 2021, 12, 1–15 CrossRef PubMed.
- S. Jones, A. Huynh, Y. Gao and Y. Yu, Mater. Chem. Front., 2018, 2, 603–608 RSC.
- S. Zuraw-Weston, D. A. Wood, I. K. Torres, Y. Lee, L.-S. Wang, Z. Jiang, G. R. Lázaro, S. Wang, A. A. Rodal and M. F. Hagan, Nanoscale, 2019, 11, 18464–18474 RSC.
- S. Hu, T. Zhao, H. Li, D. Cheng and Z. Sun, Biochim. Biophys. Acta, Biomembr., 2020, 1862, 183351 CrossRef CAS PubMed.
- R. Lipowsky, Faraday Discuss., 2012, 161, 305–331 RSC.
- V. Nikolov, R. Lipowsky and R. Dimova, Biophys. J., 2007, 92, 4356–4368 CrossRef CAS PubMed.
- H. T. McMahon and J. L. Gallop, Nature, 2005, 438, 590–596 CrossRef CAS PubMed.
- J. Zimmerberg and M. M. Kozlov, Nat. Rev. Mol. Cell Biol., 2006, 7, 9–19 CrossRef CAS PubMed.
- R. Lipowsky and H.-G. Döbereiner, Europhys. Lett., 1998, 43, 219 CrossRef CAS.
- R. Lipowsky, Europhys. Lett., 1995, 30, 197 CrossRef CAS.
- H. Heerklotz, Q. Rev. Biophys., 2008, 41, 205–264 CrossRef CAS PubMed.
- G. Först, L. Cwiklik, P. Jurkiewicz, R. Schubert and M. Hof, Eur. J. Pharm. Biopharm., 2014, 87, 559–569 CrossRef PubMed.
- D. Attwood and S. Agarwal, J. Pharm. Pharmacol., 1979, 31, 392–395 CrossRef CAS PubMed.
- D. Lichtenberg, H. Ahyayauch and F. M. Goñi, Biophys. J., 2013, 105, 289–299 CrossRef CAS PubMed.
- D. Huang, T. Zhao, W. Xu, T. Yang and P. S. Cremer, Anal. Chem., 2013, 85, 10240–10248 CrossRef CAS PubMed.
- N. B. Last and A. D. Miranker, Proc. Natl. Acad. Sci. U. S. A., 2013, 110, 6382–6387 CrossRef CAS PubMed.
- H. W. Huang, Biochemistry, 2000, 39, 8347–8352 CrossRef CAS PubMed.
- N. Ac, Annu. Rev. Biophys. Biomol. Struct., 1993, 22, 1–25 CrossRef PubMed.
- S. Li and N. Malmstadt, Soft Matter, 2013, 9, 4969–4976 RSC.
- L. Picas, F. Rico and S. Scheuring, Biophys. J., 2012, 102, L01–L03 CrossRef CAS PubMed.
- H. Porzig, J. Physiol., 1975, 249, 27–49 CrossRef CAS PubMed.
- S. Hu, T. Zhao, H. Li, D. Cheng and Z. Sun, Biochim. Biophys. Acta, Biomembr., 2020, 1862, 183351 CrossRef CAS PubMed.
- T. T. Nguyen, K. Rembert and J. C. Conboy, J. Am. Chem. Soc., 2009, 131, 1401–1403 CrossRef CAS PubMed.
- J. L. Pike, J. Lipid Res., 2008, 50, S323–S328 CrossRef PubMed.
- R. M. Epand, Prog. Lipid Res., 2006, 45, 279–294 CrossRef CAS PubMed.
- J. Fantini, Curr. Med. Chem., 2007, 14, 2911–2917 CrossRef CAS PubMed.
- K. Zheng, K. Guo, J. Xu, W. Liu and L. Chen, Open Chem., 2020, 18, 357–368 CAS.
- J. Yuan and F. Meng, J. Mol. Model., 2021, 27, 1–10 CrossRef PubMed.
- M. Markiewicz and M. Pasenkiewicz-Gierula, Langmuir, 2011, 27, 6950–6961 CrossRef CAS PubMed.
- T. E. Oliver, S. Piantavigna, P. C. Andrews, S. A. Holt and C. T. Dillon, Langmuir, 2021, 37, 1337–1352 CrossRef CAS PubMed.
- M. Egashira, G. Gorbenko, M. Tanaka, H. Saito, J. Molotkovsky, M. Nakano and T. Handa, Biochemistry, 2002, 41, 4165–4172 CrossRef CAS PubMed.
- A. Jp, B. Ad and B. Ls, Biochim. Biophys. Acta, Biomembr., 2019, 1861, 201–209 CrossRef PubMed.
- A. Martyna, B. Bahsoun, J. J. Madsen, F. S. J. Jackson, M. D. Badham, G. A. Voth and J. S. Rossman, J. Phys. Chem. B, 2020, 124, 6738–6747 CrossRef CAS PubMed.
- A. Khajeh and H. Modarress, Biochim. Biophys. Acta, Biomembr., 2014, 1838, 2431–2438 CrossRef CAS PubMed.
- W. Wang, L. Yang and H. W. Huang, Biophys. J., 2007, 92, 2819–2830 CrossRef CAS PubMed.
- A. Ivankin, I. Kuzmenko and D. Gidalevitz, Phys. Rev. Lett., 2012, 102, 293a Search PubMed.
- Y. Kozlovsky and M. M. Kozlov, Biophys. J., 2002, 82, 882–895 CrossRef CAS PubMed.
- V. S. Markin and J. P. Albanesi, Biophys. J., 2002, 82, 693–712 CrossRef CAS PubMed.
- L. V. Chernomordik and M. M. Kozlov, Cell, 2005, 123, 375–382 CrossRef CAS PubMed.
- L. V. Chernomordik and M. M. Kozlov, Annu. Rev. Biochem., 2003, 72, 175–207 CrossRef CAS PubMed.
- J. E. Vance, J. Lipid Res., 2008, 49, 1377–1387 CrossRef CAS PubMed.
- N. R. Deleault, J. R. Piro, D. J. Walsh, F. Wang, J. Ma, J. C. Geoghegan and S. Supattapone, Proc. Natl. Acad. Sci. U. S. A., 2012, 109, 8546–8551 CrossRef CAS PubMed.
- C. D. Graaf, C. Oostenbrink, P. H. J. Keizers, B. M. A. V. Vugt-Lussenburg and N. P. E. Vermeulen, Eur. Biophys. J., 2007, 36, 589–599 CrossRef PubMed.
- N. D. Ridgway, Biochemistry of Lipids Lipoproteins & Membranes, 2016, 209–236 Search PubMed.
- A. P. Hornby and P. R. Cullis, Biochim. Biophys. Acta, 1981, 647, 285–292 CrossRef CAS PubMed.
- D. G. Grenache and A. M. Gronowski, Clin. Biochem., 2006, 39, 1–10 CrossRef PubMed.
- A. M. Seddon, M. Lorch, O. Ces, R. H. Templer, F. Macrae and P. J. Booth, J. Mol. Biol., 2008, 380, 548–556 CrossRef CAS PubMed.
- Y. Ohkawa, Y. Ohmi, O. Tajima, Y. Yamauchi, K. Furukawa and K. Furukawa, Biochem. Biophys. Res. Commun., 2011, 411, 483–489 CrossRef CAS PubMed.
- R. K. Yu, Y. T. Tsai and T. Ariga, Neurochem. Res., 2012, 37, 1230–1244 CrossRef CAS PubMed.
- T. Yeung, G. E. Gilbert, J. Shi, J. Silvius, A. Kapus and S. Grinstein, Science, 2008, 319, 210–213 CrossRef CAS PubMed.
- M. Nakagaki, I. Katoh and T. Handa, Biochemistry, 1981, 20, 2208–2212 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Additional figures and movies. Movie S1, remodeling of POPC SLBs after 2.5 h incubation with 20 mM PPN. Movie S2, remodeling of POPC SLBs after 1.5 h incubation with 35 mM PPN. See DOI: https://doi.org/10.1039/d3ra00319a |
| This journal is © The Royal Society of Chemistry 2023 |