
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceUltra-small Co3O4 particles embedded into N-doped carbon derived from ZIF-9 via half-pyrolysis for activating peroxymonosulfate to degrade sulfamethoxazole†
Bin Suab,
Lu Zhangab,
Yifan Wangab,
Yuxin Liab,
Tianyu Zhouab,
Bo Liu b,
Wei Jiang*ab,
Linlin Liu*b and
Chunhong Ma*ab
b,
Wei Jiang*ab,
Linlin Liu*b and
Chunhong Ma*ab
aCollege of Engineering, Jilin Normal University, Siping, 136000, P. R. China. E-mail: jiangwjlnu@163.com; jlspmch@163.com; Fax: +86-434-3290623
bKey Laboratory of Preparation and Application of Environmental Friendly Materials, Ministry of Education, Jilin Normal University, Changchun 130103, P. R. China. E-mail: jlsdlll@163.com
First published on 7th March 2023
Abstract
The fabrication of novel and efficient transition metal-based catalysts for peroxymonosulfate (PMS) activation is of great significance for environmental remediation. Concerning energy consumption, the Co3O4@N-doped carbon (Co3O4@NC-350) was constructed via a half-pyrolysis strategy. The relatively low calcination temperature (350 °C) caused Co3O4@NC-350 to exhibit ultra-small Co3O4 nanoparticles, rich functional groups, uniform morphology, and a large surface area. For PMS activation, Co3O4@NC-350 could degrade 97% of sulfamethoxazole (SMX) in 5 min with a high k value of 0.73364 min−1, which was superior to the ZIF-9 precursor and other derived materials. Besides, Co3O4@NC-350 could be re-used over 5 times without obvious performance and structure change. The investigation of the influencing factors containing co-existing ions and organic matter demonstrated the Co3O4@NC-350/PMS system has satisfactory resistance. The quenching experiments and electron paramagnetic resonance (EPR) tests showed ˙OH, SO4˙−, ˙O2− and 1O2 participated in the degradation process. Moreover, the structure and toxicity of intermediates during the SMX decomposing process have been evaluated. Overall, this research provides new prospects for exploring efficient and recycled MOF-based catalysts for PMS activation.
1. Introduction
The continuous emergence of various stubborn pollutants is endangering human health and hindering the development of a low-carbon society.1–3 Thus, exploring highly-efficient water treatment technology is a concern. Compared with traditional physical, biological, and chemical treatment methods, advanced oxidation processes (AOPs) based on free radicals exhibit a strong oxidation ability, which can achieve deep mineralization of refractory pollutants.4–7 Among them, sulfate radicals generated with activating peroxymonosulfate (PMS) technology have a higher oxidation potential (2.6–3.1 V), wider pH application range (2–9), and longer half-life (30–40 μs) than the hydroxyl radical in the Fenton reaction.8–10Up to now, many metals such as Co, Fe, Cu, and Mn have been reported to rapidly activate PMS to generate abundant free radicals, among which cobalt ions have been regarded as the most effective.11–13 Although the homogeneous catalyst is more efficient, it is difficult to recover and has the possibility of secondary pollution after the activation process. Thus, the development of efficient, stable, and recyclable transition metal-based heterogeneous catalysts is still the focus of current research. Recently, some Co-based heterogeneous catalysts including cobalt oxide, cobalt hydroxides, and cobalt phosphide have been extensively investigated.14,15 Among them, Co3O4 has been demonstrated to be more favorable for PMS activation, resulting from the co-existence of Co2+ and Co3+ in the crystal structure.16,17 The mixed valence characteristic makes the Co site efficiently take part in the redox process without structural change. However, Co3O4 always demonstrates sphere or cluster morphology, which has defects of low dispersibility and large particle size, leading to a dissatisfied performance.18 Therefore, it is urgent to explore new methods to construct efficient and stable Co3O4-based catalysts.
Metal–organic framework (MOF), as a kind of coordination functional material, is uniformly assembled with metal centers and ligands, which have the advantages of porosity, homogeneous morphology, rich active sites, and modifiability.19–22 Until now, MOFs have been widely employed in the field of environmental remediation and energy conversion.23–26 However, metal leakage and weight loss of MOFs in continuous redox processes perplex researchers in the field of environmental chemistry. Utilizing MOFs as precursors to construct transition metal oxide/carbon composites can inherit partial advantages and further improve the metal dispersity, surface area, stability, and PMS activation efficiency.27–29 Zeolitic imidazole frameworks (ZIFs), as classical MOFs, are recognized as good self-sacrifice templates for building carbon-based materials, due to their facile preparation, unique topology, large specific surface area, and regular morphology.30–33 Among them, Co3O4/nitrogen-doped carbon composites obtained by pyrolysis of ZIF-67 constructed from 2-methylimidazole can effectively activate PMS to purify wastewater. Based on the above, it is of great significance to widely develop ZIF-based derived Co3O4-based materials to summarize the structure–activity relationship and explore the activation mechanism.
In this work, ZIF-9 constructed with carbon-rich benzimidazole and Co ions was employed as a self-sacrificial template. Concerning energy consumption, ZIF-9 was calcined under a relatively low temperature (350 °C) to obtain Co3O4@N-doped carbon (Co3O4@NC-350). Various characterization techniques showed Co3O4@NC-350 had ultra-small Co3O4 nanoparticles, uniform morphology, rich organic groups, and large surface area. Co3O4@NC-350 exhibited high and recycled efficiency for activating PMS to degrade sulfamethoxazole (SMX). The influence of catalyst dosage, PMS usage, pH, temperature, co-existing ions, and organic matter have been studied. In addition, the reactive oxygen species (ROS), SMX degradation intermediates, toxicity prediction, and structural stability were evaluated.
2. Experimental
2.1 Preparation of ZIF-9
ZIF-9 was prepared based on the previous report.34 Typically, benzimidazole (1.2 g, 10 mmol) was dissolved in ethanol (69 g), followed by dropping ammonium hydroxide (0.6 g, 10 mmol) with continuous stirring. Then, Co(CH3COO)2·4H2O (1.25 g, 5 mmol) was added into the solution and stirred for 3 h at room temperature. After that, the ZIF-9 powder could be collected by filtration and washed with ethanol, and dried at room temperature.2.2 Preparation of Co3O4@NC-x
The 500 mg of ZIF-9 powder was heated to x °C (x = 350, 400, 500, 600) in a muffle with a rate of 5 °C min−1 and kept at x °C for 2 h. After cooling down to room temperature, the Co3O4@NC-x was obtained.2.3 Catalytic degradation of SMX
All the SMX degradation experiments were carried out in a water bath. Typically, 5 mg of Co3O4@NC-x and 6.4 mg of PMS were added into 10 mg L−1 SMX solution at 25 °C with a stirring rate of 300 rpm. At regular time intervals, 1 mL samples were taken out, filtered with 0.22 μm membrane, and then quenched with saturated Na2S2O3 solution. The degradation efficiency of SMX was determined by HPLC equipped with a C18 column. Finally, the used catalysts were recovered with filtration.3. Results and discussion
3.1 Chemical and physical characterizations
For the fabricating highly efficient catalysts, Co3O4@NC-350 was obtained by half-pyrolysis of ZIF-9 under 350 °C (Fig. 1a). For comparison, the ZIF-9 was also calcinated under 400, 500, and 600 °C to construct Co3O4@NC-400, Co3O4@NC-500, and Co3O4@NC-600. As shown in the X-ray diffraction (XRD) patterns, the diffraction peaks at 19, 31.2, 36.9, 38.6, 44.7, 59.4, and 65.3° could be indexed into the (1 1 1), (2 2 0), (3 1 1), (2 2 2), (4 0 0), (5 1 1), and (4 4 0) crystal planes of Co3O4 (JCPDS No. 43-1003), respectively, indicating the metal species for all derived material were the same (Fig. 1b).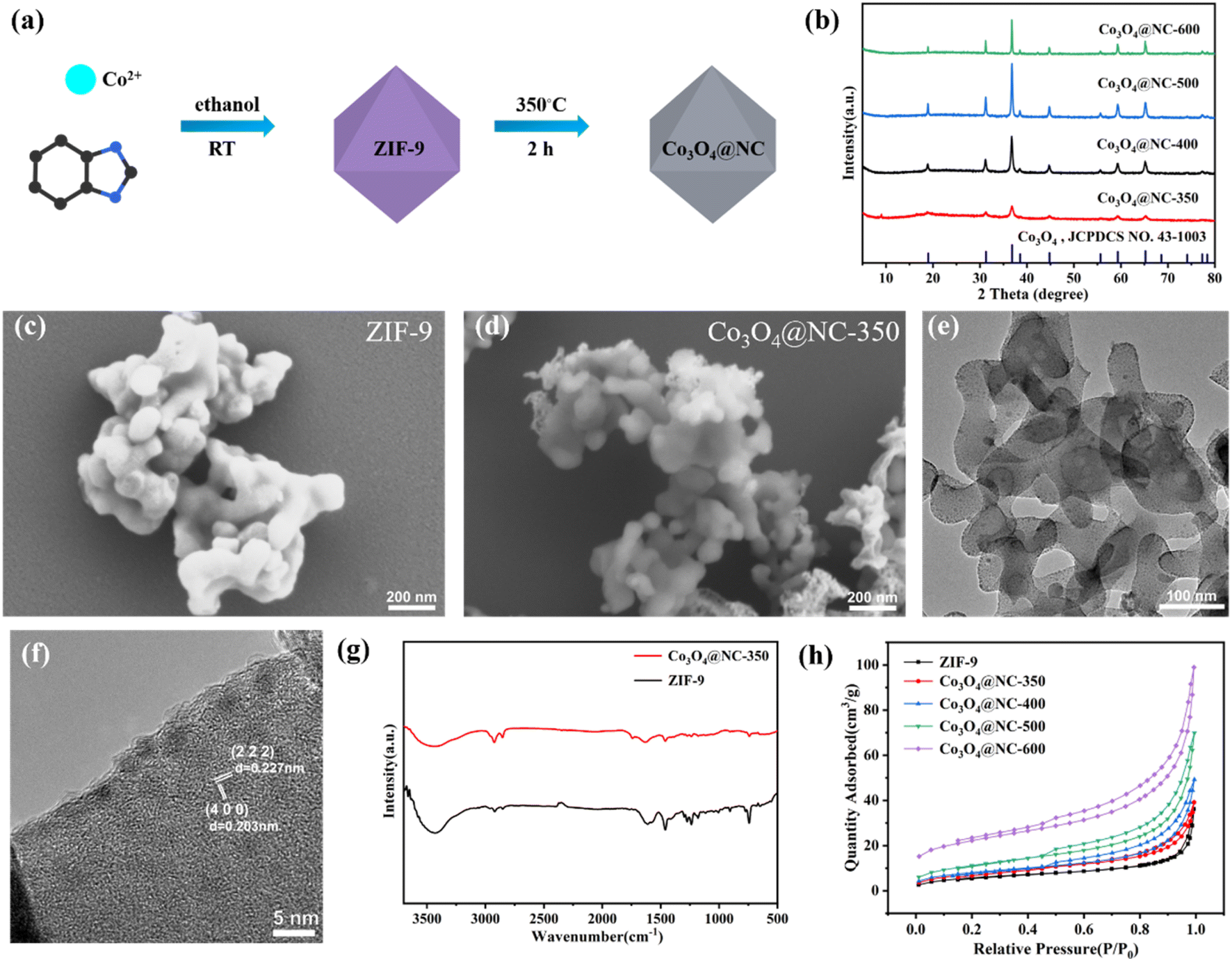 | ||
| Fig. 1 (a) Schematic illustration for the preparation of Co3O4@NC-350. (b) XRD patterns of Co3O4@NC-x. (c) SEM image of ZIF-9. (d) SEM, (e) TEM and (f) HRTEM images of Co3O4@NC-350. (g) FTIR spectra of ZIF-9 and Co3O4@NC-350. (h) N2 adsorption/desorption isotherms of ZIF-9 and Co3O4@NC-x. | ||
The morphology of ZIF-9 and the derived materials were examined by scanning electronic microscopy (SEM). As shown in Fig. 1c, the pristine ZIF-9 was polyhedral particles with an average size of 100 nm. After calcination at 350 °C, the morphology and size of Co3O4@NC-350 were maintained well as the precursor, but the particles stuck together due to the carbonization process (Fig. 1d). In the EDS-mapping image, the Co, C, N, O elements were distributed uniformly on the surface of Co3O4@NC-350 (Fig. S1†). With the increase of calcination temperature, the morphology of Co3O4@NC-400, Co3O4@NC-500, and Co3O4@NC-600 have changed obviously due to the rapid oxidation reactions (Fig. S2†).
Fig. 1e illustrated in the transmission electron microscopy (TEM) image of Co3O4@NC-350. It could be observed that Co3O4@NC-350 displayed chiffon-like morphology at high magnification after half-pyrolysis, and the ultra-small Co3O4 particles were embedded in the carbon base with an average size of 5 nm. In the HRTEM image, the lattice fringes with 0.227 and 0.203 nm spacing were identified, corresponding to the (2 2 2) and (4 0 0) crystal planes of Co3O4, respectively (Fig. 1f). The rich and ultra-small metal particles and unique morphology could significantly enhance the catalytic activity.
To validate the organic composition, the Fourier transform infrared (FTIR) spectra of ZIF-9 and Co3O4@NC-x were measured and shown in Fig. 1g. For ZIF-9, the bands in the range of 600–1500 cm−2 could be ascribed to the stretching and bending modes of the imidazole ring. And the peaks around 1634 cm−2 could be attributed to the stretching mode of C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N and CC bonding in benzimidazole. Besides, the peaks at 2854 and 2924 cm−2 were derived from characteristic C–H bonds in the aromatic ring.35 The obvious band around 3340 cm−2 should be responsible for the adsorbed water molecules. Notably, the characteristic bands of Co3O4@NC-350 were consistent with those of the ZIF-9 precursor, indicating a lot of functional groups in benzimidazole could be maintained via the half-pyrolysis process. The partially degraded organic ligands could provide more structural defects and active sites to promote the catalytic process.
N and CC bonding in benzimidazole. Besides, the peaks at 2854 and 2924 cm−2 were derived from characteristic C–H bonds in the aromatic ring.35 The obvious band around 3340 cm−2 should be responsible for the adsorbed water molecules. Notably, the characteristic bands of Co3O4@NC-350 were consistent with those of the ZIF-9 precursor, indicating a lot of functional groups in benzimidazole could be maintained via the half-pyrolysis process. The partially degraded organic ligands could provide more structural defects and active sites to promote the catalytic process.
The porosity of ZIF-9 and derived materials was examined by N2 adsorption/desorption isotherms. As shown in Fig. 1h, all the isotherms were close to Type IV with an H3 hysteresis loop. The Brunauer–Emmett–Teller (BET) surface area of ZIF-9, Co3O4@NC-350, Co3O4@NC-400, Co3O4@NC-500, Co3O4@NC-600 were 21.1042, 27.9117, 29.2132, 41.0585, 75.9440 m2 g−1, respectively. With the increase of calcination temperature, the N2 adsorption ability and BET surface area increased, indicating high temperature improved the porosity. The enhanced porosity could facilitate the interaction between active sites and pollutants.
The chemical composition and valence state of Co3O4@NC-350 was analyzed by X-ray photoelectron spectroscopy (XPS). As shown in the survey spectrum, Co, C, O, and N could be identified (Fig. S3†). The high-resolution Co 2p spectrum could be deconvoluted into six peaks (Fig. 2a). The peaks at 780.3 and 781.8 eV should be ascribed to the 2p3/2 of Co(III) and Co(II), respectively, while those at 795.4 and 797.1 eV were attributed to Co(III) and Co(II) at 2p1/2, respectively, confirming the existence of multivalent Co in Co3O4.36 Besides, the satellite peaks of Co3O4 were located at 787.1 and 803.7 eV. Moreover, the oxidation state of Co species was not significantly affected by calcination temperature (Fig. S4†). In the C 1s spectrum, the binding energies at 284.7, 285.8, and 288.7 eV belong to sp2–C, C–N, and C–O bonds, respectively (Fig. 2b). The N 1s spectrum could be fitted into three peaks at 399.3, 400.9, and 402.1 eV, which were assigned to pyridinic N, pyrrolic N, and graphitic N, respectively (Fig. 2c). The active N species were favorable for the catalytic process. In the O 1s spectrum, the three peaks at 530.4, 532, and 533.6 eV were derived from Co–O, C–O, and adsorbing water, respectively (Fig. 2d).
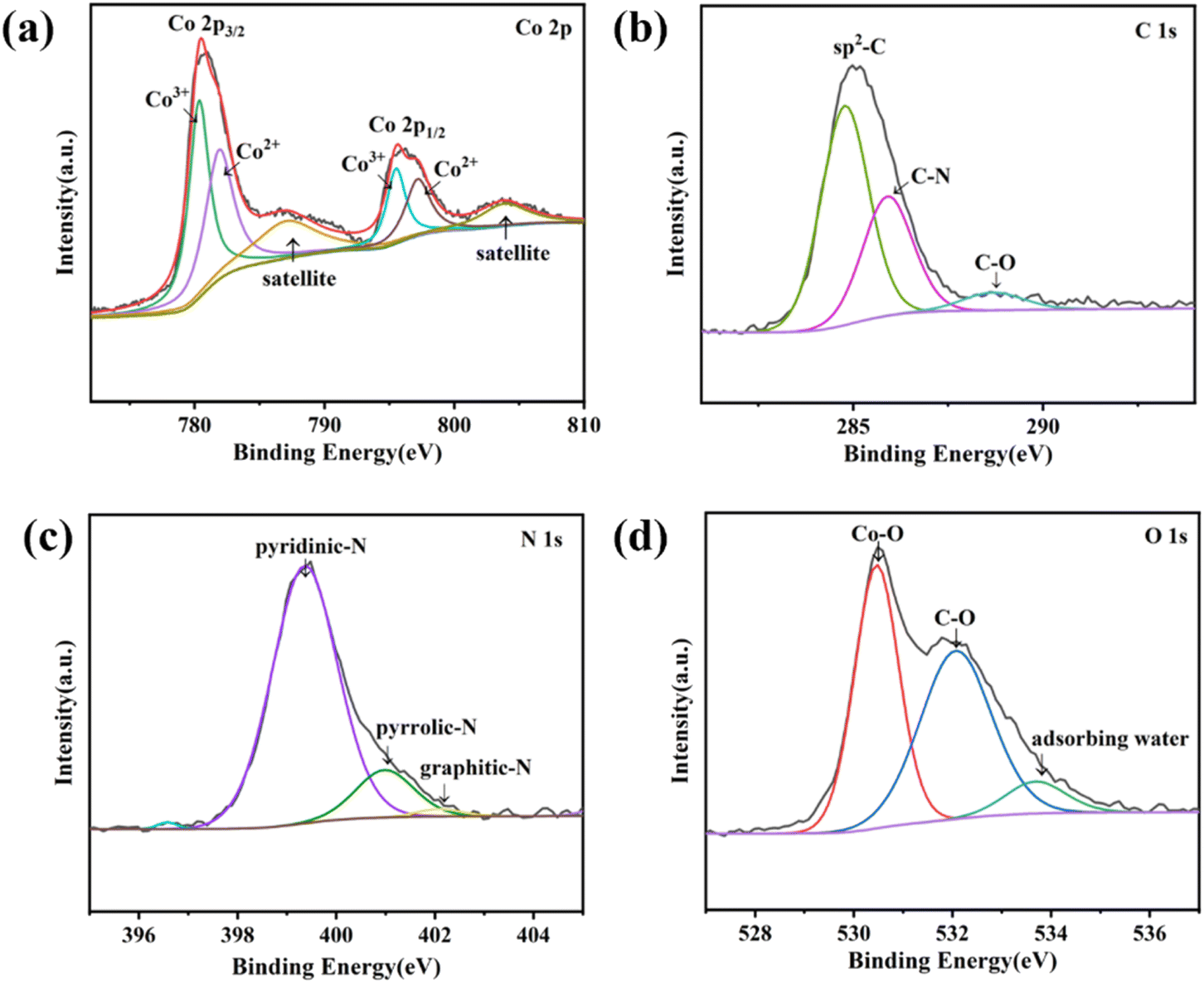 | ||
| Fig. 2 The high-resolution XPS spectra of Co3O4@NC-350: (a) Co 2p, (b) C 1s, (c) N 1s, (d) O 1s. | ||
3.2 Catalytic performance
The catalytic behavior of Co3O4@NC-350 for degradation of SMX by activating PMS was evaluated in detail. As illustrated in Fig. 3a, the degradation rate of SMX by employing PMS alone was only 13.2% after 30 min, demonstrating PMS had a weak catalytic ability without activation. When Co3O4@NC-350 was used to activate PMS, the SMX removal efficiency can be achieved over 97% within reacting 5 min. To rule out the influence of adsorption, the removal efficiency of Co3O4@NC-350 for SMX without PMS was verified. The result showed only 16.1% of SMX could be removed after 30 min. Moreover, the SMX was hard to self-decompose in 30 min. The degradation kinetics of SMX with various catalysts were further calculated. As shown in Fig. 3b, the reaction rate constant (k) value of Co3O4@NC-350/PMS system was 0.73364 min−1, which was 221 and 168.7 times of Co3O4@NC-350 (0.00332 min−1) and PMS (0.00435 min−1) alone, respectively, demonstrating the strong synergistic effect during the PMS activation process.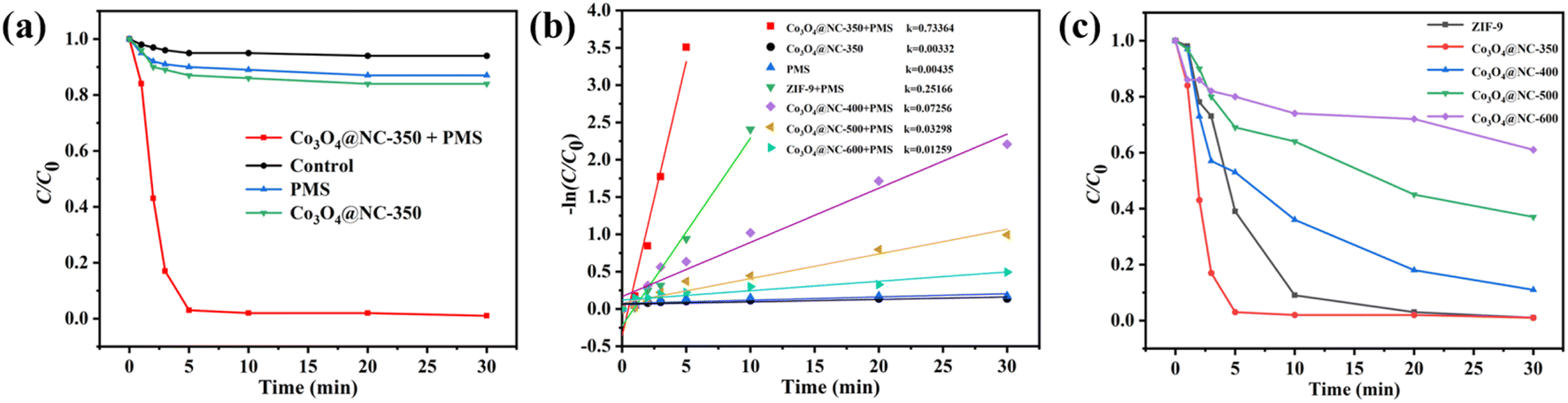 | ||
| Fig. 3 (a) and (c) The degradation efficiency and (b) reaction rate constant in various systems. | ||
To verify the advantage of the half-pyrolysis strategy for constructing an efficient catalyst. The ability of PMS activation for pristine ZIF-9 and Co3O4@NC-x calcinated under 400–600 °C was investigated. As shown in Fig. 3c, Co3O4@NC-400, Co3O4@NC-500, and Co3O4@NC-600 have shown SMX degradation efficiencies of 89, 64, and 39% in 30 min, respectively, which were much lower than that of Co3O4@NC-350. Meanwhile, even though the pristine ZIF-9 showed a good catalytic performance for degradation SMX with PMS, the coordination bonds of Co–O in MOFs always displayed poor stability in the violent oxidation reaction, leading to a weight loss. In addition, the k value of ZIF-9, Co3O4@NC-400, Co3O4@NC-500, and Co3O4@NC-600 was 0.25166, 0.07256, 0.03298, and 0.01259, respectively. It could be concluded that Co3O4@NC-350 displayed the optimal k value among ZIF-9 (0.25166 min−1) and all the derived materials, indicating the half-pyrolysis (350 °C) could achieve high catalytic performance with lower energy consumption.
To further understand the influence of various catalytic conditions on PMS activation, the effects of catalyst usage, PMS usage, pH value, and temperature were investigated. As shown in Fig. 4a, the degradation efficiency was increased from 41% to 98% in 10 min with the increase of Co3O4@NC-350 usage from 20 mg L−1 to 100 mg L−1. However, the catalytic activity was relatively stable when the Co3O4@NC-350 was over 100 mg L−1. The results demonstrated that the excess catalyst could not promote the reaction in the fast process of activating PMS. As we know, more PMS usage would produce more ROS. When the PMS usage increased from 0.4 mM to 0.8 mM, the catalytic degradation reactions were accelerated (Fig. 4b). In the degradation reactions with 1.0 mM and 1.2 mM of PMS, the catalytic efficiencies showed no obvious change, which could be ascribed to the high concentration of PMS would consume and quench SO4˙− by itself. The temperature was another important factor affecting catalytic efficiency. As displayed in Fig. 4c, within 5 min, the degradation rates were 71.1% at 15 °C and 97.9% at 35 °C. The hastening phenomenon could be attributed to the accelerated interactions among catalyst, PMS, and SMX. It should be noted that Co3O4@NC-350/PMS system could efficiently degrade SMX under room temperature (25 °C). In the practical wastewater, the pH value was various, thus, the SMX aqueous solutions with initial pH of 3.98, 6.86, and 8.97 were employed. As shown in Fig. 4d, the degradation efficiency could reach 97% and 99% in the weak acidic and neutral conditions, respectively. However, the catalytic efficiency was suppressed obviously at a pH of 8.97, which might be resulted from PMS being unstable in an alkaline environment.
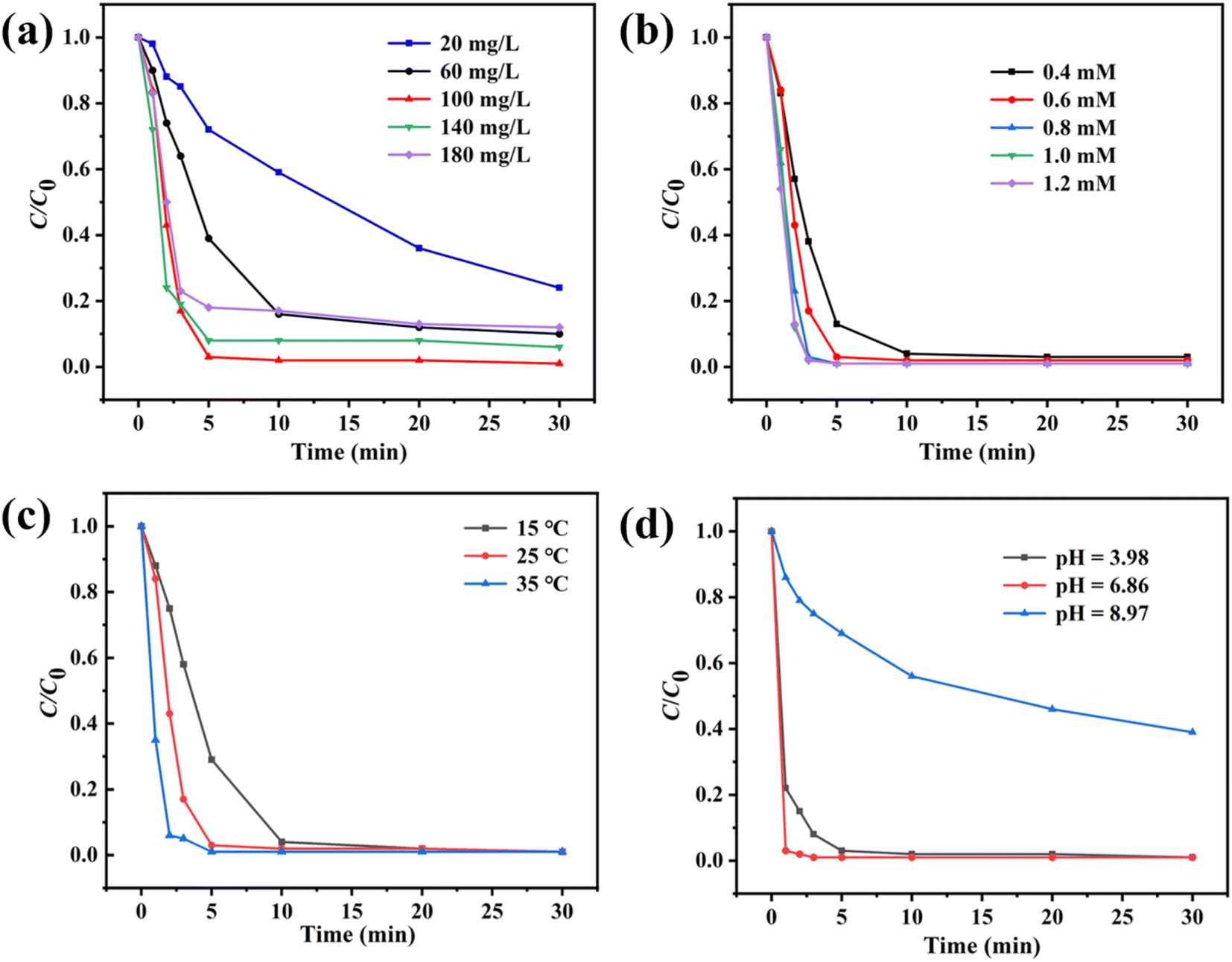 | ||
| Fig. 4 Influence of (a) catalyst usage, (b) PMS usage, (c) temperature, and (d) pH on Co3O4@NC-350/PMS system. | ||
Various anions and organic matter widely exist in the environmental water system. In this study, the influence of Cl−, HCO3−, HPO42−, and humic acid (HA) was evaluated. Fig. 5a and c showed that Cl− and HPO42− barely influenced the SMX removal in the Co3O4@NC-350/PMS system. In Fig. 5b, the introduction of HCO3− blocked the catalytic process. The degradation efficiency of SMX was decreased to 58% in 30 min in the presence of 50 mM HCO3−. This might result from the quenching effect ˙OH or SO4˙− between HCO3−. For organic HA, the influence of HA on SMX degradation was not obvious when the concentration of HA was lower than 20 mg L−1 (Fig. 5d). However, when the concentration of HA was increased to 50 mg L−1, the catalytic efficiency for Co3O4@NC-350 was significantly decreased with a degradation rate of 74% in 30 min, which could be ascribed to the degradation competition between SMX and HA. The above results demonstrated that the Co3O4@NC-350/PMS system had an acceptable resistance to co-existing anions and organic matters.
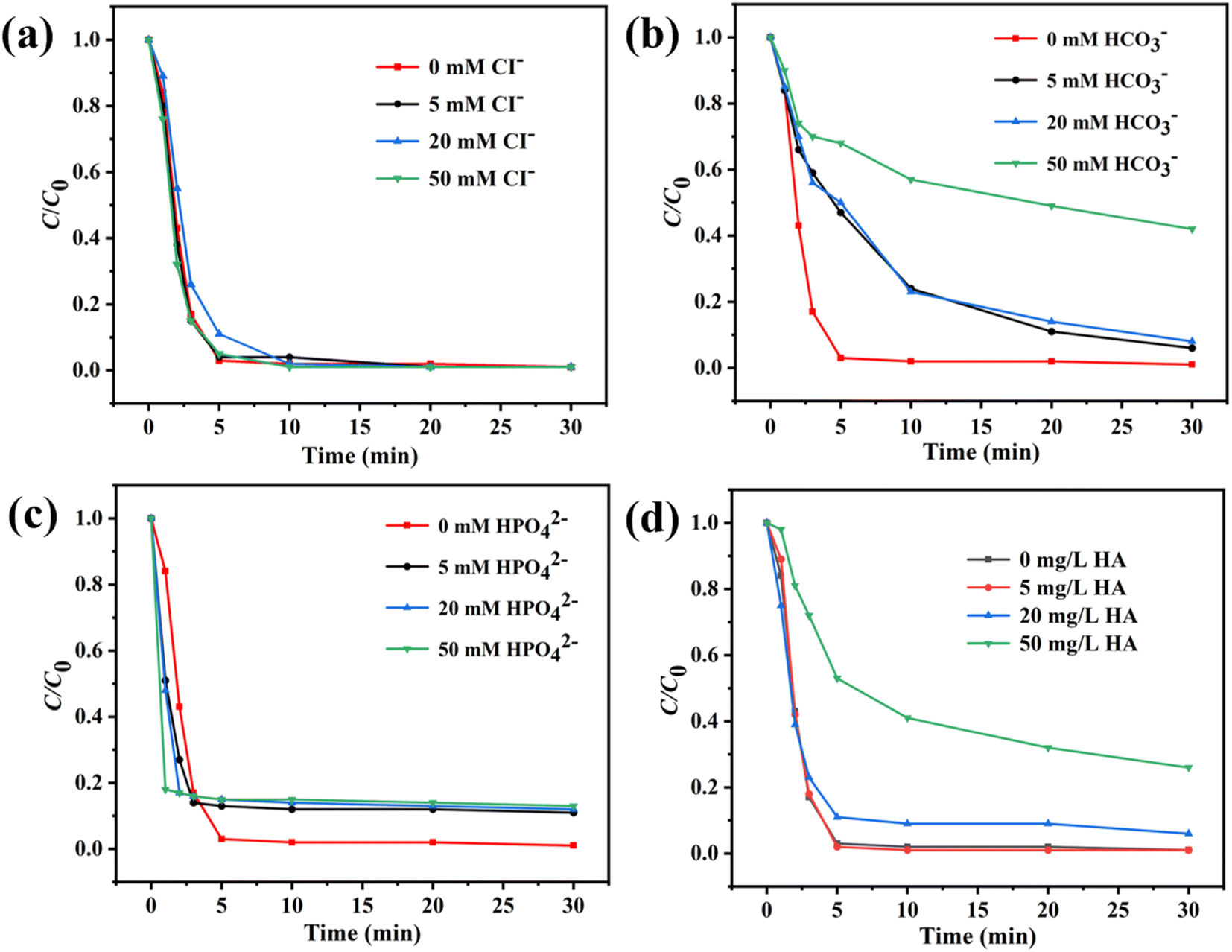 | ||
| Fig. 5 Influence of (a) Cl−, (b) HCO3−, (c) HPO42−, and (d) HA on Co3O4@NC-350/PMS system. | ||
3.3 Mechanism of Co3O4@NC-350/PMS system
In general, the ROS including ˙OH, SO4˙−, ˙O2− and 1O2 generated in transition metal oxide/PMS catalytic system are responsible for the efficient decomposition of organic pollutants. To identify the ROS that existed in the Co3O4@NC-350/PMS system, quenching experiments were carried out. Herein, methanol (ME) was used to quench both ˙OH and SO4˙−, while tertiary butanol (TBA) was employed to quench ˙OH. Besides, L-ascorbic acid (L-AA), and furfuryl alcohol (FFA) were utilized as scavengers of ˙O2− and 1O2, respectively. As shown in Fig. 6a, in comparison with the control experiment, the degradation efficiency of SMX was dramatically reduced in the presence of ROS scavengers, indicating ˙OH, SO4˙−, ˙O2− and 1O2 were formed through Co3O4@NC-350 activating PMS.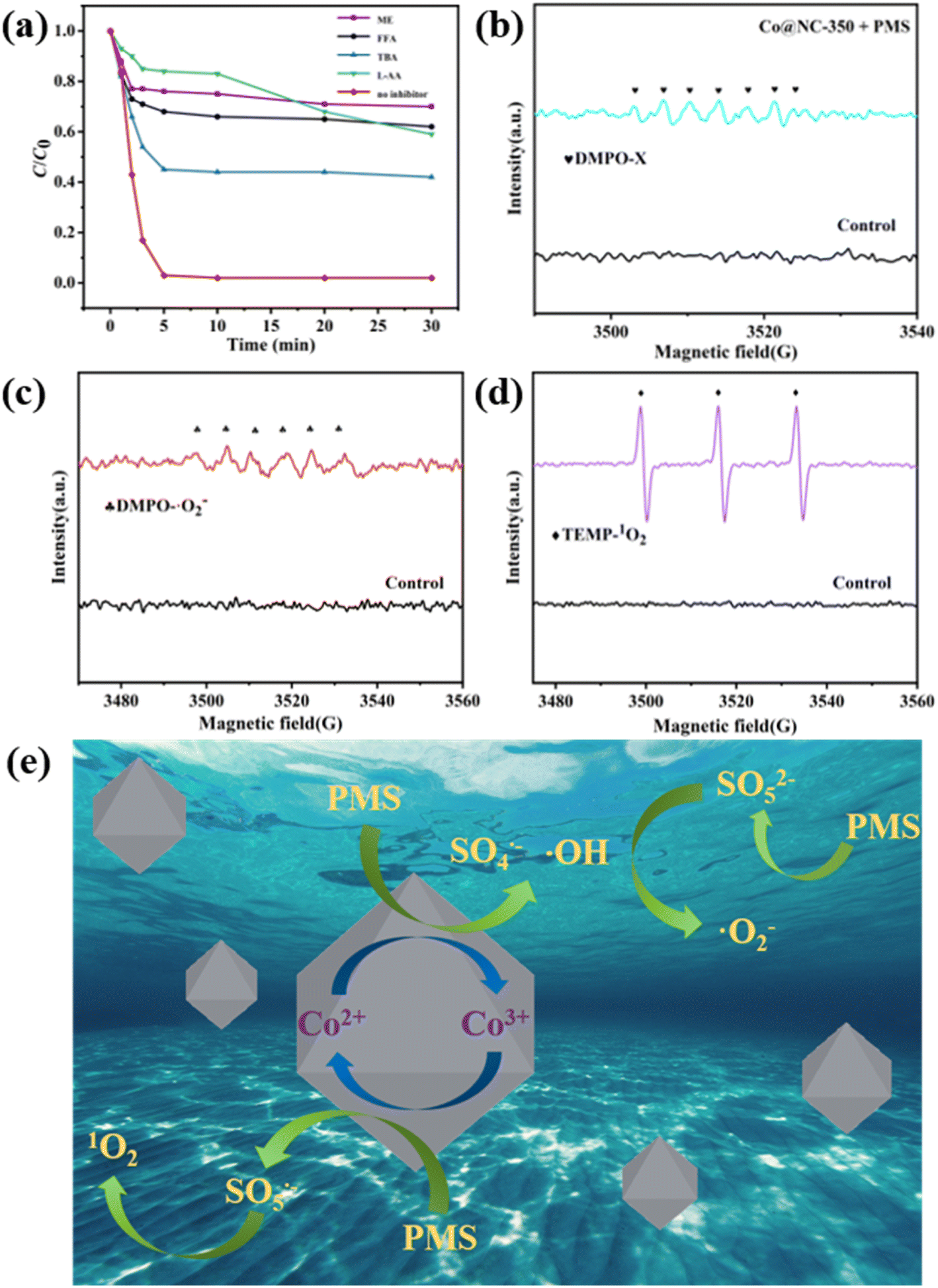 | ||
| Fig. 6 (a) Effects of different scavengers on SMX degradation. EPR spectra of (b) DMPO-X, (c) DMPO-O2−, and (d) TEMP-1O2 in Co3O4@NC-350/PMS system. (e) Proposed mechanism for SMX degradation in Co3O4@NC-350/PMS system. | ||
To directly identify the existence of the above ROS in the process of PMS activation, the electron paramagnetic resonance (EPR) technique was utilized with 5,5-dimethyl-1-pyrrolidone-N-oxide (DMPO) as the ˙OH, SO4˙− scavenger, DMPO in ME as the ˙O2− scavenger, and 2,2,6,6-tetramethylpiperidinooxy (TEMP) as the nonradical 1O2 scavenger. As illustrated in Fig. 6b, the typical seven-line signal of DMPO-X was observed in the Co3O4@NC-350/PMS/DMPO system, which can be ascribed to the oxidation of DMPO by ˙OH and SO4˙−.37 When the Co3O4@NC-350/PMS/DMPO system was in the ME medium, the typical DMPO-O2− peaks could be detected (Fig. 6c). Moreover, the characteristic triplet signal of TEMP-1O2 was clearly identified in Fig. 6d. The above EPR analyses demonstrated that the existence of ˙OH, SO4˙−, ˙O2− and 1O2 could be directly detected, which was consistent with the results of quenching experiments.
Based on the above experiments and previous reports, Co3O4 should be the main active site for PMS activation to generate ROS. Thus, a possible PMS activation mechanism with Co3O4@NC-350 was proposed and shown in Fig. 6e.38 Firstly, the PMS reacted with the Co2+ center of Co3O4@NC-350 to produce SO4˙− and ˙OH, and Co2+ was oxidized to Co3+ (eqn (1) and (2)). Meanwhile, the original and regenerated Co(III) center activated PMS to yield SO5˙− (eqn (3)). Furthermore, the SO5− decomposed into SO42− and 1O2 (eqn (4)). Besides, the PMS could undergo an ionization process to generate SO52−, which further reacted with ˙OH to produce ˙O2− (eqn (5) and (6)). The generated ˙OH, SO4˙−, ˙O2− and 1O2 were responsible for the decomposing SMX into inorganic species.
| Co2+ + HSO5− → Co3+ + SO4˙− + OH− | (1) |
| Co2+ + HSO5− → Co3+ + SO42− + ˙OH | (2) |
| Co3+ + HSO5− → Co2+ + SO5˙− + H+ | (3) |
| 2SO5˙− → SO42− + 1O2 | (4) |
| HSO5− → SO52− + H+ | (5) |
| SO52− + ˙OH → SO42− + ˙O2− + H+ | (6) |
3.4 Possible degradation pathways and toxicity prediction
The degradation pathway of SMX was proposed based on the analysis of high-performance liquid chromatography-mass spectrometry (HPLC-MS) as shown in Fig. 7 and S5–S15.† In pathway I, the isoxazole ring of SMX was cleaved to generate P1 (m/z 200). The –NH2 group in P1 could be further oxidized to –NO group (P4: m/z 212). Meanwhile, the S–C bond in P1 might be broken to form P5 (m/z 94). For pathway II, the benzene of SMX was hydroxylated to form mono hydroxylated P2 (m/z 270). Subsequently, P6 (m/z 110), P7 (m/z 99), and P8 (m/z 274) formed the cleavage of S–C bond or isoxazole ring of P2. P7 could be further oxidized into P10 (m/z 111) and P11 (m/z 119). In pathway III, the amino group of SMX was firstly oxidized to nitro group, forming intermediate P3 (m/z 284). Afterward, the breakage of S–C bond in P3 also occurred, resulting in generating P9 (m/z 123).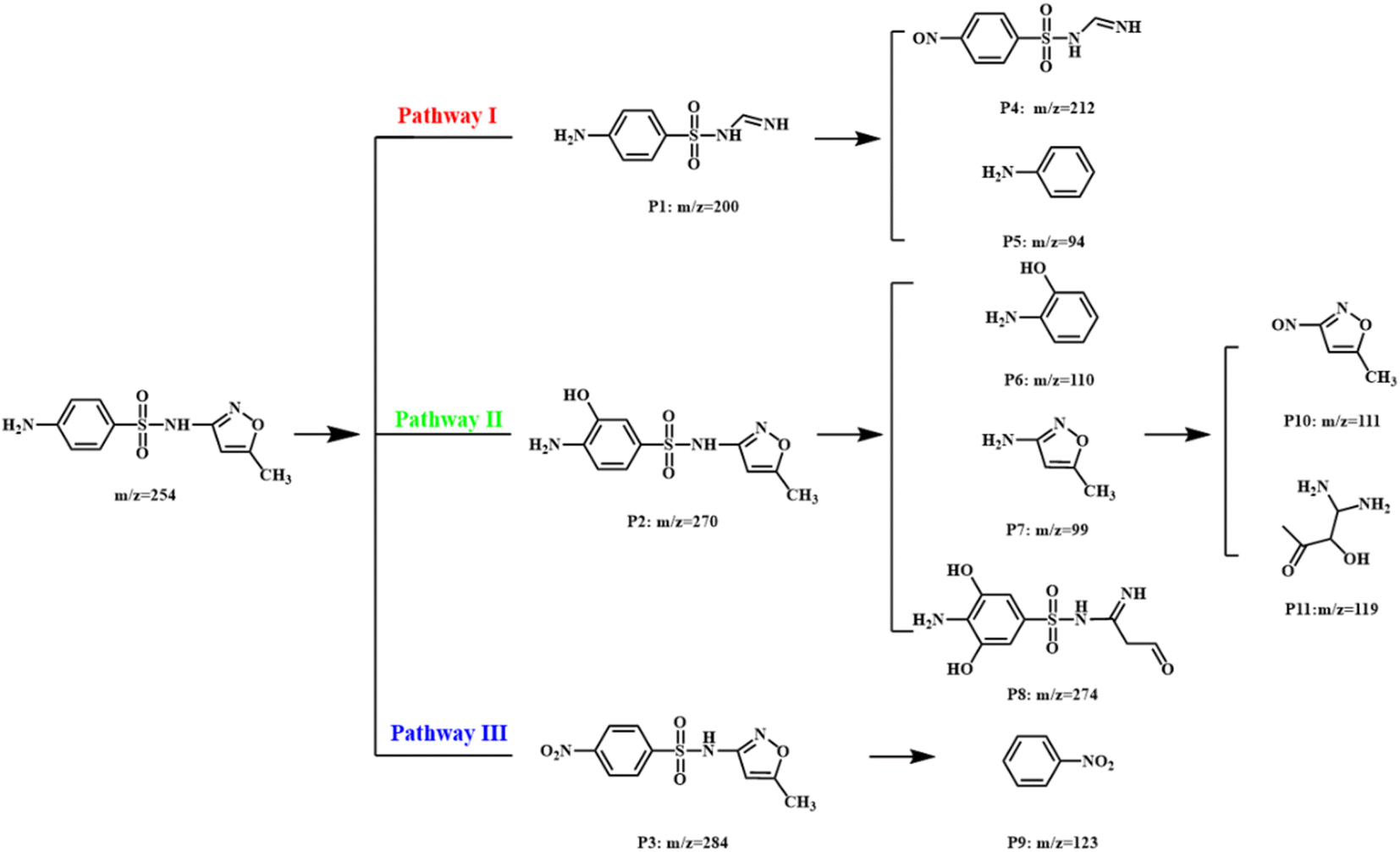 | ||
| Fig. 7 Degradation pathway of SMX in Co3O4@NC-350/PMS system. | ||
The toxicity of degradation intermediates including fathead minnow LC50, bioaccumulation factor, development toxicity, and mutagenicity was predicted with the Toxicity Estimation Software Tool (TEST).39–42 As shown in Fig. 8a, the fathead minnow lethal concentration of deeply oxidized intermediates was higher than that of SMX, except for P2 and P3. With the decomposition of SMX, the bioaccumulation factor of products decreased significantly (Fig. 8b). In Fig. 8c, the development toxicity of most of the intermediates was reduced, and P7, P9, and P10 were development non-toxicant. Fig. 8d showed the SMX and intermediates were mutagenicity negative except for P10. Thus, we could conclude that the SMX degradation process via Co3O4@NC-350 activating PMS was biological and environmentally friendly.
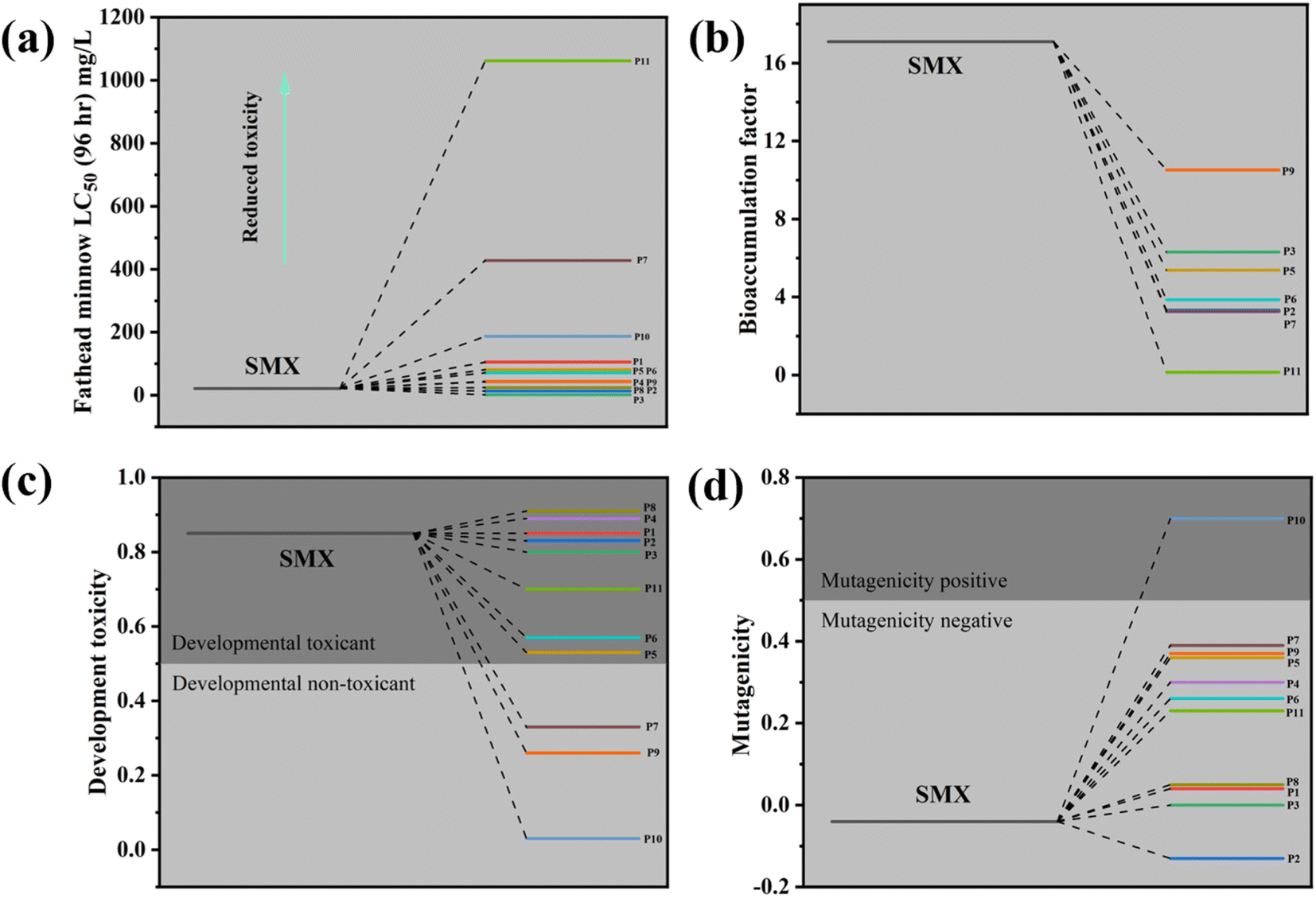 | ||
| Fig. 8 Toxicity prediction of intermediates via TEST, (a) fathead minnow LC50, (b) bioaccumulation factor, (c) development toxicity, and (d) mutagenicity. | ||
3.5 Stability of Co3O4@NC-350/PMS system
To evaluate the catalytic stability, the Co3O4@NC-350 was collected after each degradation reaction and subsequently used in successive runs. As shown in Fig. 9a, the catalytic performance of Co3O4@NC-350 decreased slightly in the continuous cycles, but 98% of SMX could still be removed in 10 min after five cycles. In the rapid oxidation process, the pores on the catalyst surface might be blocked by pollutants, which could be one of the reasons for the performance degradation. On the other hand, the leakage of cobalt ions in the catalyst might reduce the amounts of active species. The structural stability was evaluated by XRD and SEM. As shown in Fig. 9b, the XRD pattern of the used Co3O4@NC-350 was identical to the fresh one. In addition, the polyhedral morphology was maintained even after successive runs (Fig. 9c). These indicated that the Co3O4@NC-350 had satisfied catalytic and structural stability during PMS activation.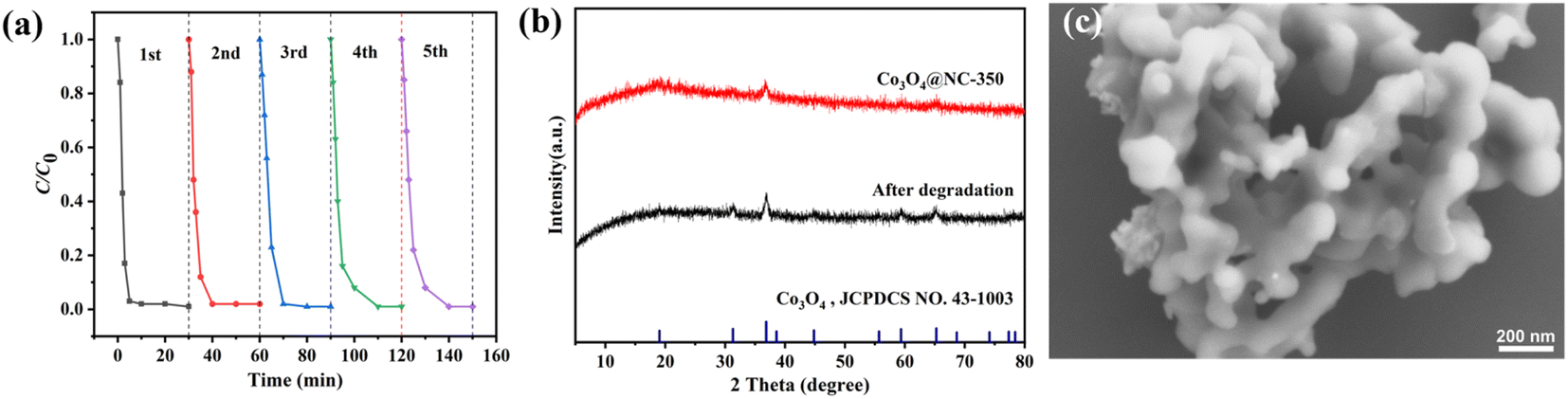 | ||
| Fig. 9 (a) Recycling performance of Co3O4@NC-350 for PMS activation. (b) XRD patterns of fresh and used Co3O4@NC-350. (c) SEM image of used Co3O4@NC-350. | ||
4. Conclusion
In summary, the Co3O4@NC-350 catalyst was constructed by half-pyrolyzing carbon-rich ZIF-9. In comparison with ZIF-9 and other derived materials, the obtained Co3O4@NC-350 showed the highest efficiency toward PMS activation to degrade SMX with a k value of 0.73364 min−1. Moreover, the Co3O4@NC-350 could be successively reused over five times without obvious structural collapse. According to various characterization analyses, the improved catalytic performance could be attributed to ultra-small and rich Co3O4 nanoparticles, uniform morphology, incomplete decomposed functional group, and enhanced surface area. In addition, the ˙OH, SO4˙−, ˙O2−, and 1O2 were identified during the PMS activation process. By verifying the structure and toxicity of intermediates, the SMX degradation reaction with Co3O4@NC-350/PMS system was environmentally and biologically friendly. This work provides new insight into the fabricating of efficient MOF-based heterogeneous catalysts for PMS activation.Author contributions
Bin Su: writing-original draft, investigation. Lu Zhang: investigation, data curation. Yifan Wang: investigation, data curation. Yuxin Li: formal analysis. Tianyu Zhou: data curation, formal analysis. Bo Liu: funding acquisition. Wei Jiang: funding acquisition, writing – review & editing. Linlin Liu: funding acquisition, formal analysis. Chunhong Ma: funding acquisition, project administration.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work is supported by the National Natural Science Foundation (21902060), the Natural Science Foundation Project of Jilin Province (20210203105SF, 20220201125GX, YDZJ202102CXJD049, 20210101117JC, YDZJ202101ZYTS078, 20210101113JC).References
- D. Pan and J. Tang, Sci. Total Environ., 2021, 751, 141550 CrossRef CAS PubMed.
- S. Giannakis, K.-Y. A. Lin and F. Ghanbari, Chem. Eng. J., 2021, 406, 127083 CrossRef CAS.
- Z. Li, Y. Xie, Y. Wang, Y. Peng, Z. Deng, B. Liu, G. Zhang, X. Wang, F. Zhang and L. Zhu, J. Solid State Chem., 2023, 317, 123707 CrossRef CAS.
- Y.-J. Zhang, G.-X. Huang, L. R. Winter, J.-J. Chen, L. Tian, S.-C. Mei, Z. Zhang, F. Chen, Z.-Y. Guo, R. Ji, Y.-Z. You, W.-W. Li, X.-W. Liu, H.-Q. Yu and M. Elimelech, Nat. Commun., 2022, 13, 3005 CrossRef CAS PubMed.
- L. Jiang, Z. Wei, Y. Ding, Y. Ma, X. Fu, J. Sun, M. Ma, W. Zhu and J. Wang, Appl. Catal., B, 2022, 307, 121184 CrossRef CAS.
- J. Li, Y. Mei, S. Ma, Q. Yang, B. Jiang, B. Xin, T. Yao and J. Wu, J. Colloid Interface Sci., 2022, 608, 2075–2087 CrossRef CAS PubMed.
- W. Jiang, Z. Li, C. Liu, D. Wang, G. Yan, B. Liu and G. Che, J. Alloys Compd., 2021, 854, 157166 CrossRef CAS.
- Y. Yan, H. Zhang, W. Wang, W. Li, Y. Ren and X. Li, J. Hazard. Mater., 2021, 411, 124952 CrossRef CAS PubMed.
- Y. Gao, Q. Zhao, Y. Li, Y. Li, J. Gou and X. Cheng, Chem. Eng. J., 2021, 405, 126719 CrossRef CAS.
- L. Li, Y. Zhang, S. Yang, S. Zhang, Q. Xu, P. Chen, Y. Du and Y. Xing, RSC Adv., 2022, 12, 7284–7294 RSC.
- M.-M. Chen, H.-Y. Niu, C.-G. Niu, H. Guo, S. Liang and Y.-Y. Yang, J. Hazard. Mater., 2022, 424, 127196 CrossRef CAS PubMed.
- H. Yang, J. Zhou, E. Yang, H. Li, S. Wu, W. Yang and H. Wang, Chemosphere, 2021, 263, 128011 CrossRef CAS PubMed.
- L. Wang, Q. Jin, Y. Xiang, L. Gan, L. Xu, Y. Wu, X. Fang, H. Lu, S. Han and J. Cui, Chem. Eng. J., 2022, 435, 134877 CrossRef CAS.
- J. Cao, S. Sun, X. Li, Z. Yang, W. Xiong, Y. Wu, M. Jia, Y. Zhou, C. Zhou and Y. Zhang, Chem. Eng. J., 2020, 382, 122802 CrossRef CAS.
- Y. Liu, H. Zou, H. Ma, J. Ko, W. Sun, K. A. Lin, S. Zhan and H. Wang, Chem. Eng. J., 2022, 430, 132816 CrossRef CAS.
- B. Li, L.-Y. Dai, W.-S. Wang and H.-Y. Xu, Mater. Today Commun., 2022, 33, 104388 CrossRef CAS.
- H. Zhang, Q. An, Y. Su, X. Quan and S. Chen, J. Hazard. Mater., 2023, 448, 130987 CrossRef CAS PubMed.
- Q. Wang, Z. Xu, Y. Cao, Y. Chen, X. Du, Y. Yang and Z. Wang, Chem. Eng. J., 2022, 427, 131953 CrossRef CAS.
- L.-Z. Dong, Y.-F. Lu, R. Wang, J. Zhou, Y. Zhang, L. Zhang, J. Liu, S.-L. Li and Y.-Q. Lan, Nano Res., 2022, 15, 10185–10193 CrossRef CAS.
- J. Wang, Y. Jiang, C. Liu, Y. Wu, B. Liu, W. Jiang, H. Li and G. Che, J. Colloid Interface Sci., 2022, 614, 532–537 CrossRef CAS PubMed.
- J. Hu, Q. Yi, Z. Xiao, F. Tian, T. Shu, X. Liu, Y. Wang, L. Li and J. Zhou, RSC Adv., 2022, 12, 35666–35675 RSC.
- M. Xie, J. Wang, X.-L. Du, N. Gao, T. Liu, Z. Li, G. Xiao, T. Li and J.-Q. Wang, RSC Adv., 2022, 12, 32518–32525 RSC.
- J. Hang, X.-H. Yi, C.-C. Wang, H. Fu, P. Wang and Y. Zhao, J. Hazard. Mater., 2022, 424, 127415 CrossRef CAS PubMed.
- S. Wang, W. Huo, F. Fang, Z. Xie, J. K. Shang and J. Jiang, Chem. Eng. J., 2022, 429, 132410 CrossRef CAS.
- C.-C. Wang, X.-H. Yi and P. Wang, Appl. Catal., B, 2019, 247, 24–48 CrossRef CAS.
- Y. Cao, X. Mi, X. Li and B. Wang, Front. Chem., 2021, 9, 673738 CrossRef CAS PubMed.
- J.-S. Wang, X.-H. Yi, X. Xu, H. Ji, A. M. Alanazi, C.-C. Wang, C. Zhao, Y. V. Kaneti, P. Wang, W. Liu and Y. Yamauchi, Chem. Eng. J., 2022, 431, 133213 CrossRef CAS.
- C. Zhao, L. Meng, H. Chu, J.-F. Wang, T. Wang, Y. Ma and C.-C. Wang, Appl. Catal., B, 2023, 321, 122034 CrossRef CAS.
- Q. Li, Z. Ren, Y. Liu, C. Zhang, J. Liu, R. Zhou, Y. Bu, F. Mao and H. Wu, Chem. Eng. J., 2023, 452, 139545 CrossRef CAS.
- Y. Hu, G. Luo, L. Wang, X. Liu, Y. Qu, Y. Zhou, F. Zhou, Z. Li, Y. Li, T. Yao, C. Xiong, B. Yang, Z. Yu and Y. Wu, Adv. Energy Mater., 2020, 11, 2002816 CrossRef.
- M. Khalid, A. M. B. Honorato, H. Varela and L. Dai, Nano Energy, 2018, 45, 127–135 CrossRef CAS.
- Y. Liang, L. Li, C. Yang, L. Ma, W. Mao and H. Yu, J. Alloys Compd., 2022, 907, 164504 CrossRef CAS.
- A. Elaouni, M. E. Ouardi, M. Zbair, A. BaQais, M. Saadi and H. A. Ahsaine, RSC Adv., 2022, 12, 31801–31817 RSC.
- J. Cong, F. Lei, T. Zhao, H. Liu, J. Wang, M. Lu, Y. Li, H. Xu and J. Gao, J. Solid State Chem., 2017, 256, 10–13 CrossRef CAS.
- K.-Y. A. Lin and H.-A. Chang, J. Taiwan Inst. Chem. Eng., 2015, 53, 40–45 CrossRef CAS.
- Y. Yu, Y. Ji, J. Lu, X. Yin and Q. Zhou, Chem. Eng. J., 2021, 406, 126759 CrossRef CAS.
- Y. Pang, J. Zhou, X. Yang, Y. Lan and C. Chen, Chem. Eng. J., 2021, 418, 129401 CrossRef CAS.
- D. Liu, X. Xue, X. Zhang, Y. Huang and P. Feng, Sep. Purif. Technol., 2023, 305, 122451 CrossRef CAS.
- J. Zhang, Y. Ma, Y. Sun, Y. Zhu, L. Wang, F. Lin, Y. Ma, W. Ji, Y. Li and L. Wang, J. Colloid Interface Sci., 2022, 628, 448–462 CrossRef CAS PubMed.
- H. Shi, Y. He, Y. Li, T. He and P. Luo, Sep. Purif. Technol., 2022, 280, 119864 CrossRef CAS.
- T. Zhou, J. Shi, G. Li, B. Liu, B. Hu, G. Che, C. Liu, L. Wang and L. Yan, J. Colloid Interface Sci., 2023, 632, 285–298 CrossRef CAS PubMed.
- W. Jiang, J. Li, Y. Jiang, S. Zhou, B. Liu, T. Zhou, C. Liu and G. Che, Polyhedron, 2022, 226, 116091 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3ra00323j |
| This journal is © The Royal Society of Chemistry 2023 |