
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceCarbon confined GeO2 hollow spheres for stable rechargeable Na ion batteries†
Dongyun Hana,
Lei Liangab,
Yongya Zhang b,
Lilan Yiab,
Xincheng Hub and
Wei Wei*b
b,
Lilan Yiab,
Xincheng Hub and
Wei Wei*b
aSchool of Petrochemical Engineering, Liaoning Shihua University, Fushun, Liaoning, P. R. China
bSchool of Chemistry and Chemical Engineering, Henan Engineering Center of New Energy Battery Materials, Shangqiu Normal University, Shangqiu 476000, P. R. China. E-mail: weiweizzuli@163.com
First published on 27th March 2023
Abstract
Germanium (Ge) based nanomaterials are regarded as promising high-capacity anode materials for Na ion batteries, but suffer fast capacity fading problems caused by the alloying/de-alloying reactions of Na–Ge. Herein, we report a new method for preparing highly dispersed GeO2 by using molecular-level ionic liquids (ILs) as carbon sources. In the obtained GeO2@C composite material, GeO2 exhibits hollow spherical morphology and is uniformly distributed in the carbon matrix. The as-prepared GeO2@C exhibits improved Na ion storage performances including high reversible capacity (577 mA h g−1 at 0.1C), rate property (270 mA h g−1 at 3C), and high capacity retention (82.3% after 500 cycles). The improved electrochemical performance could be attributed to the unique nanostructure of GeO2@C, the synergistic effect between GeO2 hollow spheres and the carbon matrix ensures the anode material effectively alleviates the volume expansion and the particle agglomeration problems.
1 Introduction
Recently, Ge-based anode materials have attracted much attention for their potential application as novel electrode materials for Na ion batteries (SIBs).1–4 Ge features an acceptable specific capacity (590 mA h g−1 for Na), a high electrical conductivity of ∼1 S m−1, as well as fast ion diffusivity (10−12 to 10−8 cm2 s−1 for Li ion).5–7 In addition, the price of Ge is expected to be significantly reduced with the development of exploration technology, because Ge has a high content in the crust, ranking 50th among all elements.8,9During the charge/discharge cycles, the alloying/de-alloying reactions of Na–Ge accompany huge volume variation, which leads to a rapid deterioration of the electrode performance.10–15 It is the aggregation of the primary particles during the sodiation processes that causes a degradation in the capacity of Ge-based electrode materials.15–20 To enhance Ge-based anode materials for Na+ storage, on the one hand, it is necessary to ensure that the primary particles are highly dispersed, and on the other hand, it is necessary to ensure that the particles do not agglomerate during the charge–discharge cycles.21–25 Achieving the above two requirements depends on material-controllable synthesis technology.26–32
GeO2 is a representative material for Ge-based materials.33 The widely used strategies to solve the problem of rapid capacity decay of GeO2 are: (i) hybridize GeO2 materials with carbonaceous materials (including graphene, carbon nanofibers, etc.);34–37 (ii) use elemental (N, F, S) doped carbon to encapsulate GeO2 and reasonably prepare porous and hollow nanostructures;38–41 (iii) fabricate nanoscale GeO2/Ge materials and use the catalytic properties of Ge to enhance the cycle stability.42,43 These synthetic methods are often limited by high temperatures, complex equipment, or expensive precursors and hazardous solvents. Therefore, it is urgent and challenging to develop simple and effective methods to synthesize high-performance GeO2 anode materials as used in SIBs.
In this work, by using molecular-level ionic liquids (ILs), we prepared a novel carbon/GeO2 composite nanostructure (denoted as GeO2@C). ILs are salts composed entirely of anions and cations in a liquid state at or near room temperature, and room temperature ILs are often accompanied by the presence of hydrogen bonds, so ILs are not only effective in stabilizing metal nanoparticles, but also have good solubility.16–18 In a typical process, Ge4+ initially dissolves in vinyl functionalized ILs to a uniform solution, followed by cross-linking and polymerization of the ionic liquids to form solid carriers with three-dimensional (3D) polymer networks so that Ge4+ can be fully dispersed within the ILs carrier. After hydrolysis of Ge4+ and carbonization, GeO2 confined by ILs-derived carbon was obtained. In this GeO2@C nanostructure, GeO2 shows hollow spherical morphology and is distributed homogeneously in the ILs-derived carbon matrix. The hollow structure of GeO2 is also filled with carbon materials. In the GeO2@C, GeO2 hollow spheres are uniformly distributed in a carbon matrix, which effectively prevents them from aggregation.
Taking the above advantages, the as-prepared GeO2@C as an anode material for SIBs exhibited long-term cycling stability, high capacity, and rate performance. Moreover, the in situ confined polymerization method utilizing ILs opens an avenue to prepare other high-performance electrode materials, such as SnO2, for SIBs.
2 Experimental section
2.1 Materials
Argon gas was supplied from the Longhai plant with a purity of 99.999%. Vinylimidazole, divinylbenzene (DVB), and dibromo butane were purchased from J&K. The starter 2,2-azobisisobutyronitrile (AIBN) from Aladdin. Additional chemicals and solvents were received from local providers. The water used in all experiments was ultrapure.2.2 The prepared ionic liquid monomer 1,4-butyl-3,3′-divinylimidazolium dibromide [BDVIm]Br2
Divide vinylimidazole (9.41 g, 100 mmol) and 1,4-dibromobutane (10.80 g, 50 mmol) in chloroform (100 mL) into a 500 mL flask. The mixture was stirred for 24 hours under reflux conditions. At the end of the reaction, remove the top phase, by washing the oily residue with chloroform three times and then drying at reduced pressure to eliminate the residual chloroform. A white powder (19.81 g) was attained in 98% yield.2.3 Preparation of GeO2@C
At room temperature, 1.50 g of [BDVIm]Br2 and 1.00 g of GeCl4 were dispersed in 10 mL of ethanol with uninterrupted stirring for 2 h. Then 0.25 g of AIBN was added to the above solution and the reaction was carried out 75 °C for 12 h. After washing three times with deionized water and drying overnight, PIL-supported GeO2 nanoparticles (GeO2@PIL) were obtained. Finally, GeO2@PIL was carbonized at 500 °C for 10 h under argon protection to obtain nitrogen-doped carbon-constrained GeO2 nanostructures (GeO2@C).2.4 Characterizations
The crystal phase structure of the samples was determined by powder X-ray diffractometry (Bruker D8, Cu Kα radiation, λ = 1.5406 Å, voltage: 40 kV, current: 40 mA). The X-ray photoelectron spectra (XPS) characterizations were conducted on an ESCALAB 250Xi (Thermo Fisher) with a monochrome Al Kα source. The microscopic morphology of the samples was investigated by using a field-emission scanning electron microscope (FESEM, Thermo Fisher, Varios G4 UC). The projected crystal structure morphology of the samples was examined on transmission electron microscopy (TEM, Tecnai G2 F20) and high-resolution transmission electron microscopy (HRTEM).2.5 Electrochemical measurements
The electrode slurry consists of GeO2@C sample powder with a binder (polyvinylidene fluoride, PVDF) and acetylene black dissolved in N-methyl-2-pyrrolidone (NMP) solution into (mass ratio 8![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1:1). Using a squeegee, the mixed slurry was uniformly applied to the copper foil (14 mm in diameter), and then the electrode foil was dried overnight at 80 °C under vacuum, with a mass loading of ∼10 mg after drying. In a glove box full of argon gas (O2 < 0.01 ppm, H2O < 0.01 ppm, Mikrouna), sodium foil with a circular diameter of about 12 mm was made from metallic sodium (Aladdin, 99.9%). The electrolyte for NIB was a 1.0 M NaClO4 solution (1:1) volume ratio of dimethyl carbonate (DMC), ethylene carbonate (EC), and 5% fluoroethylene carbonate (FEC), while the electrolyte for LIB was a 1.0 M LiPF6 solution (1:1:1 volume ratio of ethylene carbonate (EC), ethyl methyl carbonate (EMC), and dimethyl carbonate (DMC)). Glass fiber (Whatman GF/D) and polypropylene (PP) films (Celgard 2500) were used as separators for NIBs and LIBs, respectively. Coin cells (CR2032 for NIBs and CR2016 for LIBs) were assembled in an argon-filled glove box. Cyclic voltammetry (CV) curves were obtained in the voltage range of 0.01 to 3 V at a scan rate of 0.1 mV s−1 (Zennium, IM6, Germany). Electrochemical impedance spectroscopy (EIS) tests were performed with a frequency window between 100 kHz and 100 mHz (amplitude of 5 mV). Cell performance tests were performed at 25 °C using a LAND CT 2001A instrument (Wuhan, China) cycled with an applied voltage window ranging from 0.01–3.0 V (V vs. Na+/Na).
:1:1). Using a squeegee, the mixed slurry was uniformly applied to the copper foil (14 mm in diameter), and then the electrode foil was dried overnight at 80 °C under vacuum, with a mass loading of ∼10 mg after drying. In a glove box full of argon gas (O2 < 0.01 ppm, H2O < 0.01 ppm, Mikrouna), sodium foil with a circular diameter of about 12 mm was made from metallic sodium (Aladdin, 99.9%). The electrolyte for NIB was a 1.0 M NaClO4 solution (1:1) volume ratio of dimethyl carbonate (DMC), ethylene carbonate (EC), and 5% fluoroethylene carbonate (FEC), while the electrolyte for LIB was a 1.0 M LiPF6 solution (1:1:1 volume ratio of ethylene carbonate (EC), ethyl methyl carbonate (EMC), and dimethyl carbonate (DMC)). Glass fiber (Whatman GF/D) and polypropylene (PP) films (Celgard 2500) were used as separators for NIBs and LIBs, respectively. Coin cells (CR2032 for NIBs and CR2016 for LIBs) were assembled in an argon-filled glove box. Cyclic voltammetry (CV) curves were obtained in the voltage range of 0.01 to 3 V at a scan rate of 0.1 mV s−1 (Zennium, IM6, Germany). Electrochemical impedance spectroscopy (EIS) tests were performed with a frequency window between 100 kHz and 100 mHz (amplitude of 5 mV). Cell performance tests were performed at 25 °C using a LAND CT 2001A instrument (Wuhan, China) cycled with an applied voltage window ranging from 0.01–3.0 V (V vs. Na+/Na).
3 Results and discussions
The formation mechanisms of GeO2@C are speculated as follows. GeO2@C was prepared from a nanogels dispersion strategy with divinyl functionalized ionic liquid N,N′-methylene bisvinylimidazoliumbromide ([BDVIm]Br2) as the carbon sources. [BDVIm]Br2 was facilely synthesized from the reaction of vinyl imidazole with 1,2-dibromobutane. There are two fixed cations and two free anions in [BDVIm]Br2. One of the anions is distributed in the middle of the two cations, and the other one is partially free. [BDVIm]Br2 and GeCl4 were firstly dissolved in ethanol, then AIBN was added and [BDVIm]Br2 was polymerized to form the cross-linked polymeric nanogels (CLPNs), in which the imidazolium cations were linked together and arranged alternately with halogen anions. The free anions were distributed on the surface of spherical particles due to their mobility and electrostatic repulsion. Consequently, Ge4+ ions were uniformly adsorbed onto the surface of CLNPs, as illustrated in Scheme 1. After being immersed in water, Ge4+ were hydrolyzed in situ forming a core–shell structure with PILs serving as the core and nano-sized GeO2 well-dispersed on the surface as the shell. GeO2@C was obtained after carbonization under an inert atmosphere at 500 °C. | ||
| Scheme 1 Schematic diagram of GeO2@C formation process. | ||
As seen in Fig. 1a, the GeO2 nanoparticles show a circular distribution and are embedded in the carbon matrix, no aggregation or superposition occurred. Irregular sub-micro-pores with a diameter of 0.2–0.5 μm distributed in the carbon substrate. A magnified view of Fig. 1a is shown in Fig. 1b, from which we can estimate the GeO2 “rings” have a diameter of ∼500 nm. By carefully observing the GeO2 “ring” in the lower right corner of Fig. 1b, we can find that it shows hemispherical morphology. It can be speculated that these GeO2 “rings” may be cross-sections of GeO2 hollow spheres that are confined in a carbon matrix. A further enlarged view of Fig. 1b is shown in Fig. 1c, from which we can find that there are plenty of 20–30 nm sized pores around the GeO2 rings. The widely existing submicron pores and nanopores in the carbon matrix would facilitate the rapid infiltration of electrolytes and the rapid transport of sodium ions. Fig. 1d–f are the corresponding mixed energy dispersive spectrum (EDS) elemental mappings of Fig. 1a–c. In these colorful figures, green, red, and white represent C, Ge, and O elements, respectively. The uniform and orderly dispersion of GeO2 in the carbon matrix is more vividly shown in Fig. 1d–f. The transmission electron microscopy (TEM) image of GeO2@C shown in Fig. 1g directly confirms their hollow spherical nanostructure. Combined with the above scanning electron microscope (SEM) analysis, it is clear that GeO2@C is composed of GeO2 hollow spheres buried in carbon matrix and porous carbon matrix. Their corresponding O (Fig. 1h) and Ge (Fig. 1i) elemental mappings further verify the hollow spheres are GeO2.
 | ||
| Fig. 1 (a–c) SEM images of GeO2@C at different magnifications and (d–f) are their corresponding mixed EDS elemental mappings, green color represents the carbon matrix, white and red colors represent O and Ge respectively. (g–i) TEM image (g) and the corresponding EDS elemental mapping images of GeO2@C, orange (h), and purple (i) represent O and Ge elements, respectively. | ||
Next, we analyzed the crystal structure of the GeO2@C sample by X-ray diffraction (XRD). As shown in Fig. 2a, three diffraction peaks centered at 20.5°, 26.3°, and 38.2° appear, corresponding to the (100), (101), and (102) planes of crystalline GeO2, respectively, which can be perfectly classified as hexagonal phase GeO2 (JCPDS # 36-1463).14 Three diffraction peaks centered at 37°, 45°, and 65° appear, corresponding to the (012), (021) and (203) planes of the crystalline phase GeO2, which can be fully classified as hexagonal phase GeO2 (JCPDS # 65-8052).44 No diffraction peak belonging to the (002) plane of the carbon crystal was detected near 26.6°, indicating that the carbon matrix is an amorphous structure. Then the GeO2@C samples were characterized by Raman spectroscopy (Fig. 2b). A weak peak at 443 cm−1 was observed, in correspondence with the characteristic peak of GeO2.45 The reason for the weak GeO2 peak may be that most of the GeO2 hollow spheres are hidden in the carbon matrix. The Raman peaks at 1356 and 1597 cm−1 belong to the D and G bands of carbon elements, respectively.37,46,47 The D-band reflects the sp3 defects in carbon, while the G-band reflects the E2g vibrations of the sp2 hybridized graphitized carbon atoms. The parameters of these two peaks (position, width, and intensity ratio) are used to characterize the carbon material. For example, an increase in the ID/IG ratio is due to an increase in the number and/or size of sp2 clusters. A ratio of the integration region (IG/ID) > 1 at the same laser wavelength test conditions indicates high graphitization (or conductivity) of the carbon material in GeO2@C.48,49 It is favorable to enhance the electrical conductivity of GeO2@C.The GeO2 content in GeO2@C was determined by thermogravimetric analysis (TG). From the TG curve of GeO2@C, it can be directly calculated that the weight ratio of GeO2 is 42.1% (Fig. 2c).
 | ||
| Fig. 2 (a) XRD patterns of GeO2@C, (b) Raman spectra of GeO2@C. (c) TG analysis of GeO2@C. (d) Nitrogen adsorption and desorption isotherms of GeO2@C, inset images are the corresponding pore size distribution plots. | ||
Fig. 2d plots the nitrogen adsorption/desorption isotherms of the GeO2@C sample. The BET surface area of the GeO2@C sample is 41.1 m2 g−1. The GeO2@C sample shows a wide pore distribution, the pores could be generally divided into two types: one is 20 nm in diameter, and the other is between 100 and 300 nm. This result is consistent with the SEM observations.
The surface chemistry and element bonding configurations of the GeO2@C sample are identified by X-ray photoelectron spectroscopy (XPS) measurements. The overall XPS spectrum confirms the existence of C, N, Ge, and O elements (Fig. 3a). For the GeO2@C sample, the main 32.5 eV peak in the Ge 3d spectrum (Fig. 3b) was refined into three separate peaks at 31.5, 32.4, and 33.8 eV, corresponding to Ge2+, Ge3+, and Ge4+, with none at 29.3 eV, indicating the absence of elementary Ge metals in the GeO2@C sample.50,51 The stoichiometric ratio of Ge:O is 1:1.6 as shown by the XPS results of Ge 3d. The N 1s spectrum shows two main peaks of 398.4 eV and 400.8 eV, corresponding to the Ge–N bond and pyridine N, respectively, indicating the formation of Ge–N bond between GeO2 nanoparticles and carbon matrix, which is beneficial for improving the electrochemical properties (Fig. 3c).52 The high-resolution XPS spectrum of C 1s has two peaks, the peak at 284.7 eV is typical of C–C bonding and the other peak at 286.4 eV is the fitted counterpart of C–N bonding (Fig. 3d).53
 | ||
| Fig. 3 XPS spectra of GeO2@C: (a) complete XPS spectra of GeO2@C; (b–d) high-resolution spectra Ge 3d, N 1s, and C 1s, respectively. | ||
Cyclic voltammetry (CV) was used to detect the sodiation/desodiation behavior of GeO2@C. A spike of 0.1 V and a peak of 0.7 V at the first cathodic scan can be attributed to the alloying reaction between Na and Ge and the formation of an irreversible solid electrolyte interphase (SEI) (Fig. 4a).54 In the following cycles, the 0.7 V peak disappears. The two anodic peaks at around 0.4 and 1.2 V are the desaturation behavior of the GeO2@C electrode.55 The near-overlapping CV curves in the next four cycles indicate that the GeO2@C electrode has good cycling stability. Moreover, according to the CV curves and charge/discharge distributions (Fig. 4e), as well as the similarity of LIBs and SIBs, it can be speculated that at the beginning of the reduction process, GeO2 reacts with sodium ions and irreversibly transforms into germanium nanoparticles and Na2O matrix, and then the germanium nanoparticles reversibly react with sodium ions through the alloying mechanism for the subsequent sodiation/desodiation process,56 and the possible reaction mechanism is as follows:
| GeO2 + 4Na → Ge + 2Na2O | (I) |
| Ge + xNa → NaxGe (x ∼ 1) | (II) |
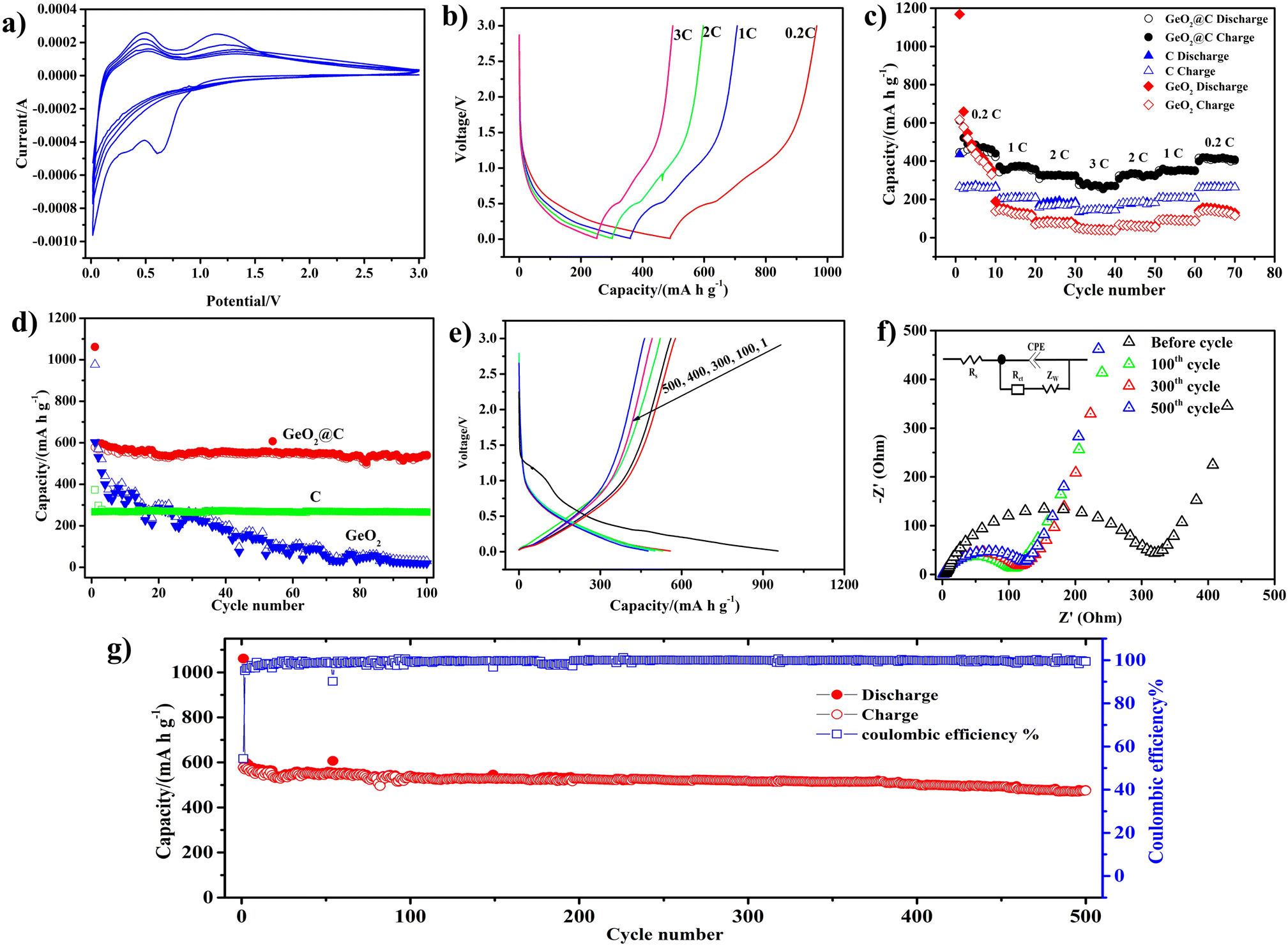 | ||
| Fig. 4 Electrochemical performance of the GeO2@C electrode. (a) Cyclic voltammograms of the GeO2@C electrode for the first five cycles at a scan rate of 0.1 mV s−1; (b) discharge–charge curves of the cycles at different current rates; (c) rate performance of the GeO2@C, GeO2, and C electrode; (d) comparison of the cycling performance of the GeO2@C, pure C, and GeO2 powder electrodes (current density: 0.1C); (e) discharge–charge profiles of GeO2@C electrode at the 1st, 100th, 300th, 400th and 500th cycles; (f) EIS spectra of GeO2@C electrode before cycling and the 100th, 300th and 500th cycles, the inset presents the equivalent circuit model; (g) long cycling performance under the current density of 0.1C. | ||
To further evaluate the electrochemical performance of the GeO2@C electrode, we tested the GeO2@C electrode at different current densities (0.2C to 3C, 1C = 500 mA g−1). The discharge–charge profiles of GeO2@C electrode show that, at 0.2, 0.5, 2, and 3C the electrode could deliver reversible capacities of about 460, 365, 305, and 270 mA h g−1, respectively (Fig. 4b). The capacity is calculated based on the total mass of GeO2@C. When the current density is restored to 0.2C, the rated capacity can be almost restored to the initial value, which indicates that the GeO2@C electrode has good electrochemical reversibility (Fig. 4c).
To testify the improved cycling stability of GeO2@C electrode, pure C (obtained in the absence of Ge precursor) and commercial GeO2 electrodes were fabricated for comparison (their corresponding SEM images are provided in Fig. S1a–f, ESI†). As shown in Fig. 4d and S3 (ESI†), all three electrodes were tested for 100 cycles at a current density of 0.1C. The capacity of GeO2@C electrode was maintained above 500.0 mA h g−1 with a flat capacity graph and no significant capacity decay after 100 cycles, indicating that it can combine high capacity and cycling performance. The initial reversible charging capacity of the GeO2@C electrode was 577.5 mA h g−1, and after 100 cycles, the reversible charging capacity was 537.3 mA h g−1, corresponding to a capacity retention rate of 93.0%. The pure C electrode showed only 260–270 mA h g−1. The pure GeO2 electrode exhibited an initial capacity comparable to that of the GeO2@C electrode (602.2 mA h g−1), but the capacity decayed rapidly. After 100 cycles, the GeO2 electrode capacity contribution was minimal (19.0 mA h g−1).
The GeO2@C electrode provided a discharge capacity of 955.1 mA h g−1 and a charging capacity of 577.5 mA h g−1 with an initial Coulomb efficiency of 60.4% in a current density of 0.1C initially (Fig. 4e). The reversible capacity of the GeO2@C electrode is enhanced by the presence of abundant mesopores in the active material, which not only promote the diffusion of Na+ but also serve as storage sites for more Na+.57 Generally, the doping of N elements improves the electrochemical reactivity and electron conduction of the material, which in turn leads to higher charge/discharge capacity.58 The capacity loss in the first cycle (425 mA h g−1) may be caused by the formation of an irreversible SEI layer in the initial cycle.
The ion migration rate and charge transfer kinetics of GeO2@C electrodes are usually analyzed by electrochemical impedance spectroscopy (EIS). The Nyquist plot contains a high-frequency region (a semicircle) and a low-frequency region (an inclined line). The semicircle is caused by the charge transfer resistance (Rct) and the SEI film impedance (Rs).59 The ion diffusion within the anode material resulted in an inclined line in the low-frequency region.60,61 The EIS spectra of GeO2@C electrode at 100th, 300th and 500th cycles were displayed in Fig. 4f, with the inset showing the corresponding equivalent circuits. The semicircle diameters of GeO2@C electrode at 100th, 300th, and 500th cycles slightly increase, suggesting that the Rs and Rct of the electrode did not obviously increase. Results also indicate that the Na+ migration rate and charge-transfer kinetics remain largely the same even after a long-term cycle.
A long-cycle test was performed for the GeO2@C electrode at a current density of 0.1C (Fig. 4g and S4 ESI†). The initial reversible charging capacity of the GeO2@C electrode was 577.2 mA h g−1, and the capacity remained at 475.1 mA h g−1 after 500 cycles, with a capacity retention rate of 82.3%, indicating that the GeO2@C electrode has good long-term cycling stability.
To broaden the application of the GeO2@C electrode, the lithium storage performance of the GeO2@C electrode was further tested as shown in Fig. S2 (Fig. S2, ESI†). The high overlap of the CV curves (2–5 cycles) indicates that the GeO2@C electrode has good cycling stability (Fig. S2a, ESI†), and at a current density of 100 mA g−1, the electrode can provide a high reversible capacity of 1197.3 mA h g−1 (Fig. S2b, ESI†). After 100 cycles, the capacity retention was 89.9% (Fig. S2c, ESI†). At current densities of 0.2, 1, 2, 3, and 5 A g−1, the GeO2@C electrodes delivered capacities of 900, 755, 630, and 505 mA h g−1, respectively (Fig. S2d, ESI†). Therefore, GeO2@C is also a lithium-ion battery anode material with excellent performance.
4 Conclusions
In summary, a novel carbon-confined GeO2 hollow spheres nanocomposite has been synthesized by using ILs as the carbon source and the template assistant agent. The GeO2@C anode material shows excellent electrochemical performance in SIBs with a combination of high reversible discharge capacity and cycling stability (577 mA h g−1 at 0.1C; 500 cycles at 0.1C with 82.3% capacity retention) and excellent rate performance (270 mA h g−1 at 3C). The unique nanostructure of GeO2@C anode is beneficial to the improvement of its electrochemical performance. (1) The uniformly dispersed GeO2 hollow spheres are confined in the carbon matrix, which can effectively alleviate the volume expansion of Ge during the sodiation process, and in addition, the carbon framework can effectively avoid the agglomeration of Ge nanoparticles. (2) The multi-level pore structure of the carbon matrix facilitates the electron conduction and rapid movement of Na+ in the GeO2@C electrode, which can enhance the electron and Na+ transport kinetics and thus improve the capacity, cycling, and rate performance. In addition, this is also closely related to the high Na+ storage capacity of GeO2 itself.Conflicts of interest
The authors declare no competing financial interest.Acknowledgements
This study was supported by the National Natural Science Foundation of China (22104079) and the Science and Technology Innovation Talents in Universities of Henan Province (No. 22HASTIT028).Notes and references
- N. Khossossi, A. Banerjee, I. Essaoudi, A. Ainane, P. Jena and R. Ahuja, J. Power Sources, 2021, 485, 229318 CrossRef CAS
.
- M. Li, Z. Wang, J. Fu, K. Ma and E. Detsi, Scr. Mater., 2019, 164, 52–56 CrossRef CAS
- X. Wang, L. Fan, D. Gong, J. Zhu, Q. Zhang and B. Lu, Adv. Funct. Mater., 2016, 26, 1104–1111 CrossRef CAS
- W. Wei, Y. Zhang, L. Liang, K. Wang, Q. Zhou, Y. Zhou and H. Wang, Mater. Chem. Front., 2021, 5, 7778–7786 RSC
- L. Li, Y. Zheng, S. Zhang, J. Yang, Z. Shao and Z. Guo, Energy Environ. Sci., 2018, 11, 2310–2340 RSC
- X. Zhong, H. Huan, X. Liu and Y. Yu, Nano Res., 2018, 11, 3702–3709 CrossRef CAS
- S. C. Jung, H.-J. Kim, Y.-J. Kang and Y.-K. Han, J. Alloys Compd., 2016, 688, 158–163 CrossRef CAS
- R. Amatya and R. J. Ram, J. Electron. Mater., 2011, 41, 1011–1019 CrossRef
- K. H. Seng, M. H. Park, Z. P. Guo, H. K. Liu and J. Cho, Angew. Chem., Int. Ed., 2012, 51, 5657–5661 CrossRef CAS PubMed
- Z. Yi, N. Lin, T. Li, Y. Han, Y. Li and Y. Qian, Nano Res., 2019, 12, 1824–1830 CrossRef CAS
- X. Xiao, X. Li, S. Zheng, J. Shao, H. Xue and H. Pang, Adv. Mater. Interfaces, 2017, 4, 1600798 CrossRef
- H. Wu, L. Zheng, W. Liu, X. Xia, C. Xiao, J. Xie, L. Su, L. Wang and N. Du, J. Alloys Compd., 2019, 771, 169–175 CrossRef CAS
- H. Wu, W. Liu, L. Zheng, D. Zhu, N. Du, C. Xiao, L. Su and L. Wang, ChemistryOpen, 2019, 8, 298–303 CrossRef CAS PubMed
- C. Shang, L. Hu, D. Luo, K. Kempa, Y. Zhang, G. Zhou, X. Wang and Z. Chen, Adv. Sci., 2020, 7, 2002358 CrossRef CAS PubMed
- F. Li, Z. Wei, A. Manthiram, Y. Feng, J. Ma and L. Mai, J. Mater. Chem. A, 2019, 7, 9406–9431 RSC
- A. K. Thakur, M. S. Ahmed, G. Oh, H. Kang, Y. Jeong, R. Prabakaran, M. P. Vikram, S. W. Sharshir, J. Kim and J.-Y. Hwang, J. Mater. Chem. A, 2021, 9, 2628–2661 RSC
- H. Kang, Y. Liu, K. Cao, Y. Zhao, L. Jiao, Y. Wang and H. Yuan, J. Mater. Chem. A, 2015, 3, 17899–17913 RSC
- Y. Yan, Y. Liu, Y. Zhang, C. Qin, Z. Bakenov and Z. Wang, J. Colloid Interface Sci., 2021, 592, 103–115 CrossRef CAS PubMed
- F. Liu, Y. Wang, J. Shi, J. Lin, W. Zhou and A. Pan, Electrochim. Acta, 2019, 318, 314–321 CrossRef CAS
- Y. Wei, J. He, Q. Zhang, C. Liu, A. Wang, H. Li and T. Zhai, Mater. Chem. Front., 2017, 1, 1607–1614 RSC
- W. Wei, H. Wang, A. Tian, K. Wang, J. Wang, P. Qu, S. Zhang and L. Guo, Nanoscale, 2018, 10, 6872–6877 RSC
- J. Zhou, Y. Zhou, X. Zhang, L. Cheng, M. Qian, W. Wei and H. Wang, Nanoscale, 2020, 12, 79–84 RSC
- W. Li, L. Ke, Y. Wei, S. Guo, L. Gan, H. Li, T. Zhai and H. Zhou, J. Mater. Chem. A, 2017, 5, 4413–4420 RSC
- Q.-L. Zhu and Q. Xu, Chem, 2016, 1, 220–245 CAS
- M. Zhou, C. Li and J. Fang, Chem. Rev., 2021, 121, 736–795 CrossRef CAS PubMed
- S. Mathur, H. Shen, N. Donia, T. Rügamer, V. Sivakov and U. Werner, J. Am. Chem. Soc., 2007, 129, 9746–9752 CrossRef CAS PubMed
- M. Javadi, Z. Yang and J. G. Veinot, Chem. Commun., 2014, 50, 6101–6104 RSC
- D. V. Averyanov, I. S. Sokolov, I. A. Karateev, A. N. Taldenkov, O. A. Kondratev, O. E. Parfenov, A. M. Tokmachev and V. G. Storchak, J. Mater. Chem. C, 2021, 9, 17012–17018 RSC
- K. Ma and N. Lin, Inorg. Chem. Front., 2019, 6, 1897–1903 RSC
- K. von Fieandt, F. O. L. Johansson, O. Balmes, R. Lindblad, L. Riekehr, A. Lindblad and E. Lewin, Inorg. Chem., 2019, 58, 11100–11109 CrossRef CAS PubMed
- D. Carolan and H. Doyle, J. Nanomater., 2015, 2015, 1–9 CrossRef
- A. M. Ahadi, K. I. Hunter, N. J. Kramer, T. Strunskus, H. Kersten, F. Faupel and U. R. Kortshagen, Appl. Phys. Lett., 2016, 108, 093105 CrossRef
- H. Nhung Thi Nguyen, P. Nguyen Ngoc, H. Tran Huu, T. Thuy Trang Phan, D. Nhan Nguyen, T. Huong Thi Nguyen, T. Nguyen Van, L. Nguyen Thi, M. Kha Le, V. Man Tran, M. Loan Phung Le and V. Vo, Chem. Phys. Lett., 2022, 801, 139747 CrossRef CAS
- W. Xie, W. Wang, L. Duan, W. Zheng, S. Liang, S. Liu, F. Liu, X. Wang, H. Sun and X. Sun, J. Alloys Compd., 2022, 918, 165687 CrossRef CAS
- A. Jahel, A. Darwiche, C. Matei Ghimbeu, C. Vix-Guterl and L. Monconduit, J. Power Sources, 2014, 269, 755–759 CrossRef CAS
- F. Pantò, Y. Fan, S. Stelitano, E. Fazio, S. Patanè, P. Frontera, P. Antonucci, N. Pinna and S. Santangelo, Int. J. Hydrogen Energy, 2017, 42, 28102–28112 CrossRef
- J. Liang, W. Wei, D. Zhong, Q. Yang, L. Li and L. Guo, ACS Appl. Mater. Interfaces, 2012, 4, 454–459 CrossRef CAS PubMed
- Y. Lin, K. Zhong, J. Zheng, M. Liang, G. Xu, Q. Feng, J. Li and Z. Huang, ACS Appl. Energy Mater., 2021, 4, 9848–9857 CrossRef CAS
- L. Han, J. Tang, Q. Wei, C. Chen and M. Wei, Chem. Commun., 2019, 55, 14319–14322 RSC
- Y. Xue, T. Yu, J. Chen, X. Wan, X. Cai, X. Guo, F. Zhang, W. Xiong, Y. Liu, Q. Kong, A. Yuan and J. Zhang, J. Solid State Chem., 2020, 286, 121303 CrossRef CAS
- X. Han, P. Gao, S. Deng, P. Mei, Q. Zhang and Y. Yang, Mater. Today Chem., 2020, 17, 100293 CrossRef CAS
- D. McNulty, H. Geaney, D. Buckley and C. O'Dwyer, Nano Energy, 2018, 43, 11–21 CrossRef CAS
- S. Yan, H. Song, S. Lin, H. Wu, Y. Shi and J. Yao, Adv. Funct. Mater., 2019, 29, 1807946 CrossRef
- Y. Pan, L. Li, J. Lu, R. Pang, L. Wan and S. Huang, Dalton Trans., 2016, 45, 9506–9512 RSC
- H.-S. Choe, S.-J. Kim, M.-C. Kim, D.-M. Kim, G.-H. Lee, S.-B. Han, D.-H. Kwak and K.-W. Park, RSC Adv., 2016, 6, 72926–72932 RSC
- Q. Ma, W. Wang, P. Zeng and Z. Fang, Langmuir, 2017, 33, 2141–2147 CrossRef CAS PubMed
- J. Xu, N. Yang, S. Heuser, S. Yu, A. Schulte, H. Schönherr and X. Jiang, Adv. Energy Mater., 2019, 9, 1803623 CrossRef
- L. G. Cancado, A. Jorio, E. H. Ferreira, F. Stavale, C. A. Achete, R. B. Capaz, M. V. Moutinho, A. Lombardo, T. S. Kulmala and A. C. Ferrari, Nano Lett., 2011, 11, 3190–3196 CrossRef CAS PubMed
- E. H. Martins Ferreira, M. V. O. Moutinho, F. Stavale, M. M. Lucchese, R. B. Capaz, C. A. Achete and A. Jorio, Phys. Rev. B: Condens. Matter Mater. Phys., 2010, 82, 125429 CrossRef
- K. Prabhakaran, F. Maeda, Y. Watanabe and T. Ogino, Appl. Phys. Lett., 2000, 76, 2244–2246 CrossRef CAS
- X. Ma, Y. Zhou, M. Chen and L. Wu, Small, 2017, 13, 1700403 CrossRef PubMed
- C. Zhang, R. Hao, H. Liao and Y. Hou, Nano Energy, 2013, 2, 88–97 CrossRef CAS
- R. Hao, H. Lan, C. Kuang, H. Wang and L. Guo, Carbon, 2018, 128, 224–230 CrossRef CAS
- R. Liu, F. Luo, L. Zeng, J. Liu, L. Xu, X. He, Q. Xu, B. Huang, Q. Qian, M. Wei and Q. Chen, J. Colloid Interface Sci., 2021, 584, 372–381 CrossRef CAS PubMed
- Q. Li, Z. Zhang, S. Dong, C. Li, X. Ge, Z. Li, J. Ma and L. Yin, Part. Part. Syst. Charact., 2017, 34, 1600115 CrossRef
- W. Qin, T. Chen, B. Hu, Z. Sun and L. Pan, Electrochim. Acta, 2015, 173, 193–199 CrossRef CAS
- C. Xu, Y. Xu, C. Tang, Q. Wei, J. Meng, L. Huang, L. Zhou, G. Zhang, L. He and L. Mai, Nano Energy, 2016, 28, 224–231 CrossRef CAS
- G. Hu, A. Chen, R. Yu, K. Zhong, Y. Zhang, J. Wu, L. Zhou and L. Mai, ACS Appl. Energy Mater., 2021, 4, 6277–6283 CrossRef CAS
- C. Zhang, Z. Lin, Z. Yang, D. Xiao, P. Hu, H. Xu, Y. Duan, S. Pang, L. Gu and G. Cui, Chem. Mater., 2015, 27, 2189–2194 CrossRef CAS
- J. Yang, H. Wang, P. Hu, J. Qi, L. Guo and L. Wang, Small, 2015, 11, 3744–3749 CrossRef CAS PubMed
- T.-T. Wei, X. Liu, S.-J. Yang, P.-F. Wang and T.-F. Yi, J. Energy Chem., 2023, 80, 603–613 CrossRef CAS
Footnote |
| † Electronic supplementary information (ESI) available: Experimental section, SEM images of pure C obtained at the absence of Ge precursor and commercial GeO2 powders at different magnification, and Li storage performance of the Ge@C electrode. See DOI: https://doi.org/10.1039/d3ra00460k |
| This journal is © The Royal Society of Chemistry 2023 |