
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceDegradable Fe3O4-based nanocomposite for cascade reaction-enhanced anti-tumor therapy†
Yang Wang *a,
Xun Li‡
b,
Yuan Fanga,
Jianhua Wanga,
Danhong Yana and
Baisong Chang*b
*a,
Xun Li‡
b,
Yuan Fanga,
Jianhua Wanga,
Danhong Yana and
Baisong Chang*b
aDepartment of Medical Technology, Suzhou Chien-shiung Institute of Technology, Taicang 215411, Jiangsu Province, P.R. China. E-mail: wangy0070@csit.edu.cn
bState Key Laboratory of Advanced Technology for Materials Synthesis and Processing, Wuhan University of Technology, Wuhan, 430070, P.R. China
First published on 9th March 2023
Abstract
Cascade catalytic therapy has been recognized as a promising cancer treatment strategy, which is due in part to the induced tumor apoptosis when converting intratumoral hydrogen peroxide (H2O2) into highly toxic hydroxyl radicals (˙OH) based on the Fenton or Fenton-like reactions. Moreover this is driven by the efficient catalysis of glucose oxidization associated with starving therapy. The natural glucose oxidase (GOx), recognized as a “star” enzyme catalyst involved in cancer treatment, can specially and efficiently catalyze the glucose oxidization into gluconic acid and H2O2. Herein, pH-responsive biodegradable cascade therapeutic nanocomposites (Fe3O4/GOx–PLGA) with dual enzymatic catalytic features were designed to respond to the tumor microenvironment (TME) and to catalyze the cascade reaction (glucose oxidation and Fenton-like reaction) for inducing oxidase stress. The GOx-motivated oxidation reaction could effectively consume intratumoral glucose to produce H2O2 for starvation therapy and the enriched H2O2 was subsequently converted into highly toxic ˙OH by a Fe3O4-mediated Fenton-like reaction for chemodynamic therapy (CDT). In addition, the acidity amplification owing to the generation of gluconic acid will in turn accelerate the degradation of the nanocomposite and initiate the Fe3O4–H2O2 reaction for enhancing CDT. The resultant cooperative cancer therapy was proven to provide highly efficient tumor inhibition on HeLa cells with minimal systemic toxicity. This cascade catalytic Fenton nanocomposite might provide a promising strategy for efficient cancer therapy.
1 Introduction
Cancer, one of the most difficult diseases to overcome over the past decades, has seriously threatened human health.1 As an emerging strategy of nanocatalytic medicine, chemodynamic therapy (CDT) can transform H2O2 into highly reactive oxygen species (ROS) in a tumor microenvironment (TME) by employing the Fenton reaction or Fenton-like reaction with metal ion catalysts (e.g., Fe2+, Cu+, Mn2+ and V2+).2–6 The hydroxyl radical (·OH), the most toxic reactive oxygen species (ROS), can cause apoptosis of tumor cells owing to their high oxidation capability.7 Compared with chemotherapy, radiotherapy, photothermal therapy and photodynamic therapy, CDT holds unique advantages including low side effects, high selectivity, activation by endogenous stimulus, and low treatment cost.8 Most importantly, this approach ensures normal tissue safety to a certain extent, because the Fenton reaction will be substantially suppressed under slight alkaline conditions or in a normal microenvironment with insufficient H2O2 levels.9 Compared with other nano-Fenton catalysts, ferroferric oxide nanoparticles (Fe3O4 NPs) have attracted tremendous attention for biomedical applications due to their superparamagnetism, biodegradability, low toxicity, and cost-effectiveness.10–12 Mazuel et al. evidenced a near-complete intracellular degradation of Fe3O4 NPs by using stem cell spheroids as a tissue model and global spheroid magnetism as a fingerprint of the degradation process.13 However, because of the relatively low drug loading and iron leakage, Fe3O4 NPs presents the weak antitumor efficacy when used as drug carriers and iron source for CDT.14 In addition, the CDT efficacy and clinical translation are restricted by the low conversion efficiency from Fe2+ to Fe3+, consumption of hydroxyl radicals by glutathione (GSH),15 the limited endogenous supply of H2O2.16,17Several unique characteristics including mild acidity, rich glucose, low catalase activity and overproduced H2O2 owing to the complex biological microstructure of TME open the “gate” for selective and efficient tumor treatments.18–23 Clinical practice and in-depth exploration indicates that monotherapy is incapable of eliminating tumor cells completely, thus, recent studies have gradually gained a focus on the synergistic therapy. Numerous new TME-responsive strategies have acquired the gratifying achievements in nanomedicine, such as hypoxia-responsive controlled release drug delivery systems,24 starvation therapy for tumor growth inhibition by glucose depletion,25 and so on. Glucose oxidase (GOx), inherently biocompatible and degradable, can lead to the consumption of glucose in tumors for starvation therapy by efficient catalytic oxidization.26 Furthermore, the generated H2O2 in cancer cells can be effectively conversed by Fenton reagents into toxic reactive oxygen species for CDT. Thus, CDT is expected to combine with GOx-mediated starvation therapy to arouse more efficient tumor suppression.
Different from inorganic theranostics, nanomedicines, fabricated from biodegradable polymer, demonstrate their unparalleled advantages such as biocompatibility, high drug loading, controlled drug release and stimuli-responsiveness.27 For instance, Zhang et al. constructed a dual-catalytic nanoreactor for synergistic chemodynamic-starvation therapy by encapsulating O2 carrier perfluorohexane into the hole of HMSNs, simultaneously loading GOx and Fe3O4 nanoparticles on the surface of HMSNs and then coating the obtained NPs with the cancer cell membrane.25 As the dual-catalytic nanoreactor promoted cascade catalytic reactions, the sequential glucose depletion and ·OH aggregation synergistically suppressed tumor metastasis with negligible side effects. In addition, Ke et al. fabricated a degradable polymersome nanoreactor containing polyprodrug, ultrasmall Fe3O4 NPs, and GOx was loaded to integrate starvation therapy, CDT, and camptothecin-induced chemotherapy together for cooperative cancer therapy.28 The nanocomposite design inducing tumor-activable cascade reactions represents an insightful paradigm for precise cooperative cancer therapy. More importantly, the modification of Fe3O4 NPs with biodegradable polymer could improve the biocompatibility and effectively slow down the ion leakage in normal tissues during the delivery.
Toward the issues, herein, a biodegradable cascade catalytic nanocomposite (Fe3O4/GOx–PLGA) for cancer therapy was developed by encapsulating oleic acid (OA)-modified Fe3O4 NPs and GOx into poly(lactic-co-glycolic acid) (PLGA) microspheres with emulsification-solvent evaporation technology (Scheme 1). Owing to better biocompatibility, higher drug loading and prolonged retention time, PLGA, approved by the U.S. Food and Drug Administration (FDA) as anticancer drug carrier, can be hydrolyzed into lactic acid and glycolic acid and subsequently eliminated from body by metabolic pathways.29 By co-loading GOx and Fe3O4 NPs into biodegradable PLGA polymer NPs, GOx reacts with glucose and oxygen in the tumor cells to produce gluconic acid along with inexhaustible H2O2, which sustains Fe3O4 NPs-mediated Fenton-like reaction to generate highly toxic ·OH. The excessive consumption of glucose in tumor cells, resulted in nutritional deficiencies and suppression of tumor growth. Moreover, because of the gluconic acid generation, the pH decrease would in turn accelerate the degradation of PLGA and promote the Fe3O4–H2O2 reaction for an increasing generation of ·OH. Simultaneously, the intracellular oxygen was consumed during GOx-catalyzed oxidation of glucose, exacerbating the hypoxic state of tumors. Such orchestrated designed cascade catalytic nanocomposite integrates advantages of CDT, starvation therapy and hypoxia therapy. Therefore, the cascade reaction will dramatically improve the efficacy of tumor treatment and significantly reduce the side effects on normal tissues or organs.
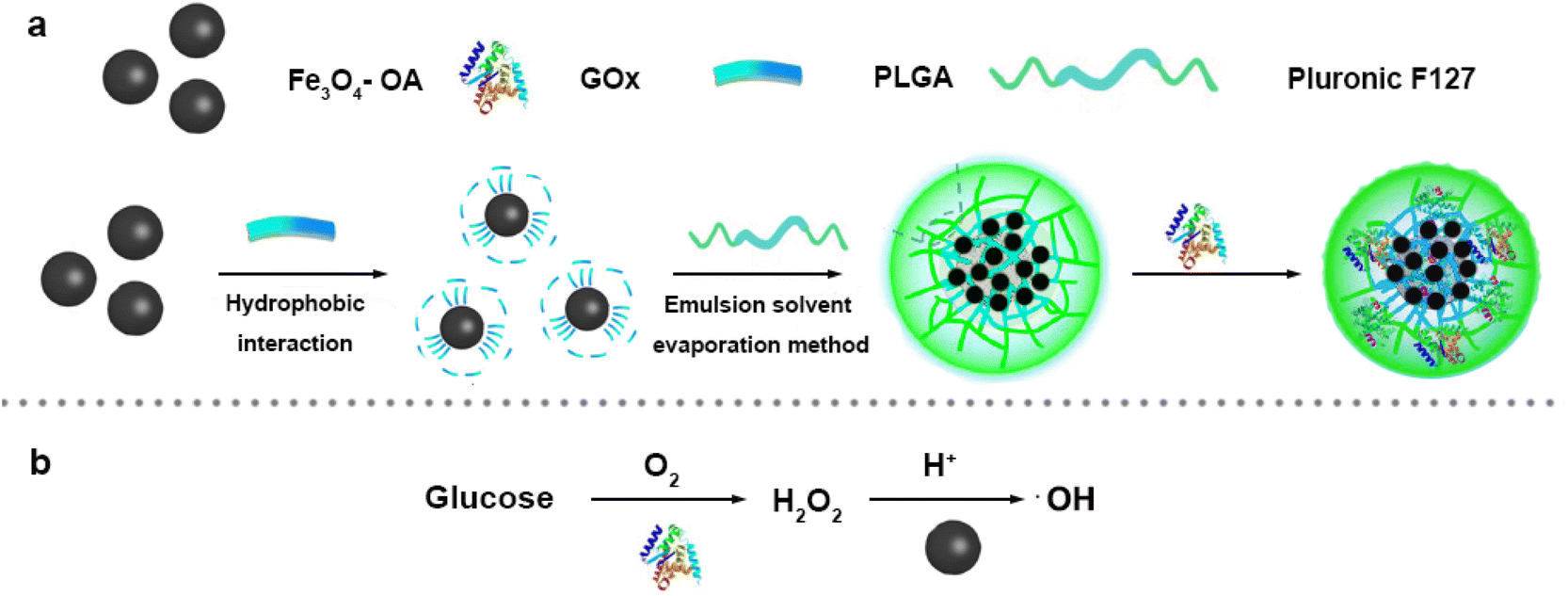 | ||
| Scheme 1 (a) Synthetic scheme of Fe3O4/GOx–PLGA. (b) The mechanism schematic of ·OH generation by cascade catalytic reaction. | ||
2 Experimental section
2.1 Materials
3,3′,5,5′-Tetramethylbenzidine (TMB, 99%, CAS 54827-17-7) was purchased from Alfa Company. β-D-Glucose (β-D-Glu, 99%, CAS 28905-12-6) was purchased from TCI Company. Phosphate-buffered saline (PBS), trypsin (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :250, CAS 9002-07-7), Dulbecco’s modified eagle medium (DMEM), high-glucose DMEM (4500 mg L−1) and fetal bovine serum albumin (FBSA, 98%, CAS 9048-46-8) were purchased from Gibco, USA. HeLa cells were purchased from Beyotime Biotechnology Co. Ltd and Cell Counting Kit-8 (CCK8) was obtained from Beyotime Biotechnology Co. Ltd. Calcein acetoxymethyl ester (Calcein-AM, 90%, CAS 148504-34-1)/Propidium iodide (PI, 95%, CAS 25535-16-4) staining reagents were purchased from Yeasen Biotechnology (Shanghai) Co. Ltd. 5,5-Dimethyl-1-pyrroline N-oxide (DMPO, 97%, CAS 3317-61-1) was bought from J&K Scientific Company. Ferric chloride hexahydrate (FeCl3·6H2O, 98%, CAS 7705-08-0), ferrous chloride tetrahydrate (FeCl2·4H2O, 99%, CAS 13478-10-9), oleic acid (OA, 90%, CAS 112-80-1), polyoxyethylene oxypropylene ether block copolymer (Pluronic F127, MW 12600, CAS 9003-11-6), poly(D,L-lactic acid-co-glycolide) (PLGA (50:50), MW 7000–17000, CAS 26780-50-7) were purchased from Sigma-Aldrich Company. Glucose oxidase (GOx, 100–250 U mg−1, CAS 9001-37-0) was purchased from Shanghai Yuanye Biotechnology Co. Ltd. Ammonia (NH3·H2O, 25%, CAS 1336-21-6), acetone (C3H6O, 97%, CAS 67-64-1), n-octane (n-C8H18, 96%, CAS 111-65-9), chloroform (CHCl3, 97%, CAS 67-66-3), hydrogen peroxide (H2O2, 30%, CAS 7722-84-1), terephthalic acid (TA, 99%, CAS 100-21-0), citric acid monohydrate (CA·H2O, 99%, CAS 5949-29-1), potassium dihydrogen phosphate (KH2PO4, 99%, CAS 7778-77-0), disodium hydrogen phosphate (Na2HPO4, 99%, CAS 7558-79-4), ethanol (C2H5OH, 99.5%, CAS 64-17-5) and methanol (CH3OH, 99.5%, CAS 67-56-1), were analytically pure and obtained from Sinopharm reagent company. All reagents were analytical grade and were used directly without further purification. Throughout, Milli-Q ultrapure water was used in all needed experiments.
:250, CAS 9002-07-7), Dulbecco’s modified eagle medium (DMEM), high-glucose DMEM (4500 mg L−1) and fetal bovine serum albumin (FBSA, 98%, CAS 9048-46-8) were purchased from Gibco, USA. HeLa cells were purchased from Beyotime Biotechnology Co. Ltd and Cell Counting Kit-8 (CCK8) was obtained from Beyotime Biotechnology Co. Ltd. Calcein acetoxymethyl ester (Calcein-AM, 90%, CAS 148504-34-1)/Propidium iodide (PI, 95%, CAS 25535-16-4) staining reagents were purchased from Yeasen Biotechnology (Shanghai) Co. Ltd. 5,5-Dimethyl-1-pyrroline N-oxide (DMPO, 97%, CAS 3317-61-1) was bought from J&K Scientific Company. Ferric chloride hexahydrate (FeCl3·6H2O, 98%, CAS 7705-08-0), ferrous chloride tetrahydrate (FeCl2·4H2O, 99%, CAS 13478-10-9), oleic acid (OA, 90%, CAS 112-80-1), polyoxyethylene oxypropylene ether block copolymer (Pluronic F127, MW 12600, CAS 9003-11-6), poly(D,L-lactic acid-co-glycolide) (PLGA (50:50), MW 7000–17000, CAS 26780-50-7) were purchased from Sigma-Aldrich Company. Glucose oxidase (GOx, 100–250 U mg−1, CAS 9001-37-0) was purchased from Shanghai Yuanye Biotechnology Co. Ltd. Ammonia (NH3·H2O, 25%, CAS 1336-21-6), acetone (C3H6O, 97%, CAS 67-64-1), n-octane (n-C8H18, 96%, CAS 111-65-9), chloroform (CHCl3, 97%, CAS 67-66-3), hydrogen peroxide (H2O2, 30%, CAS 7722-84-1), terephthalic acid (TA, 99%, CAS 100-21-0), citric acid monohydrate (CA·H2O, 99%, CAS 5949-29-1), potassium dihydrogen phosphate (KH2PO4, 99%, CAS 7778-77-0), disodium hydrogen phosphate (Na2HPO4, 99%, CAS 7558-79-4), ethanol (C2H5OH, 99.5%, CAS 64-17-5) and methanol (CH3OH, 99.5%, CAS 67-56-1), were analytically pure and obtained from Sinopharm reagent company. All reagents were analytical grade and were used directly without further purification. Throughout, Milli-Q ultrapure water was used in all needed experiments.
2.2 Characterization
Hydrodynamic diameter and zeta potential of the particles were determined by a dynamic light scattering (DLS) particle size analyzer (Malvern Nano-ZS90). Fourier transform infrared (FT-IR) spectra were measured on a FT-IR spectrometer (Vertex 80V, Bruker Germany) in the range of 4000–400 cm−1 to confirm the chemical composition and structure information. Crystal structure of Fe3O4–PLGA was analyzed using X-ray diffraction (XRD, D&ADVANCE, Bruker Germany). The morphology microstructure of the nanocomposites was observed by transmission electron microscopy (TEM) (JEM-2100F STEM, JEOL Japan) and atomic force microscope (AFM) (Multimode 8, Bruker Germany). UV-vis spectra were obtained using a PerkinElmer Lambda 35 spectrophotometer (UV-2550, SHIMADZU, Japan) to analyze the catalytic oxidation activity. The hysteresis loops (300 K) of different Fe3O4 NPs were tested on a vibrating sample magnetometer (VSM) (model 7404, LakeShore USA) to characterize their saturation magnetization. Fluorescence spectra were obtained using a fluorescence spectrum analyzer (LS55, PerkinElmer USA). Electron paramagnetic resonance (EPR) spectra were measured by JEOL JES-FA200 spectrometer (JEOL, Japan). The cytotoxicity of HeLa cell was observed by a fluorescence microplate reader (Synergy TM MX, Berton Corporation USA), and the ROS was observed on fluorescence microscope (Olympus IX71) and confocal laser scanning microscope (Leica TCS SP5 II).2.3 Synthesis of Fe3O4/GOx–PLGA
2.4 Fenton-like reaction activity of Fe3O4–PLGA
Terephthalic acid (TA), adopted as a fluorescent probe, can react with·OH to form highly fluorescent 2-hydroxyterephthalic acid with an emission peak at 430 nm upon exposure to the excitation wavelength of 325 nm. Thus, TA was used to detect the ·OH production to verify Fe3O4–PLGA-induced Fenton reaction. Typically, 0.4 mL of Fe3O4–PLGA aqueous, disodium hydrogen phosphate–citrate buffer (pH 3.5, 5 mL), TA solution (5 mM, 1 mL), and different concentrations (0, 2, 4, 6, 8, 10 mM) of H2O2 solution were mixed uniformly. After incubation for 30 min in the dark at room temperature, NaOH (1 M, 1 mL) was added to terminate the reaction, and the remained Fe3O4–PLGA nanocomposites were removed by magnetic separation, then 3 mL of the supernatant was taken for fluorescence spectrum detection (λex = 325 nm, λem = 430 nm).The catalytic activity of Fe3O4–PLGA was evaluated by chromogenic substrate 3,3′,5,5′-tetramethylbenzidine (TMB). Under acidic conditions, Fenton-like reaction catalyzed H2O2 to generate ·OH, and further oxidized colorless TMB to form a blue mixture with maximum absorbance at 652 nm. Typically, TMB (5 mM), H2O2 (0–10 mM) and Fe3O4–PLGA (0.05–0.20 mg mL−1) were mixed evenly at different pH (3.0–7.4). After incubation for 30 min in the dark at room temperature, the reaction was terminated by removing the catalyst Fe3O4–PLGA with magnetic separation, and the UV spectra were detected on the microplate reader. Steady-state kinetics of Fe3O4–PLGA was investigated at pH 4.0 with 5 mM TMB and 0.20 mg mL−1 Fe3O4–PLGA while changing the concentration of H2O2. The apparent kinetic parameters were calculated in the light of Lineweaver–Burk plots derived from Michaelis–Menten equation. The molar absorptivity of TMB was 39000 M−1 cm−1.30
2.5 Cascade catalytic activity of Fe3O4/GOx–PLGA
TA (0.5 mM), β-D-glucose solution (0–5 mM) and Fe3O4/GOx–PLGA (0–5 mg mL−1) were mixed uniformly in the buffer solution of pH 4.0. After incubation for 1 h in the dark at room temperature, 1 M NaOH solution was added to terminate reaction. The Fe3O4/GOx–PLGA were removed with the assistance of a magnet, and 3 mL of the supernatant was collected for fluorescence spectroscopic detection (λex = 325 nm, λem = 430 nm).TMB was used to evaluate the cascade catalytic performance of Fe3O4/GOx–PLGA in the presence of glucose. The typical procedure was as follows: Fe3O4/GOx–PLGA dispersion (0.4 mL, 5 mg mL−1), buffer solutions at different pH (5 mL, pH 3.0–7.4), TMB solution (1 mL, 5 mM) and β-D-glucose solution (1 mL, 5 mM) were added in turn into a PE tube, then ultrapure water was added to make the total volume reach 10 mL. After 1 h incubation, the Fe3O4/GOx–PLGA was removed to terminate reaction with the aid of a magnet.
2.6 Evaluation of ·OH generation ability of Fe3O4–PLGA and Fe3O4/GOx–PLGA
Electron paramagnetic resonance (EPR) was used to measure the production of ·OH by using 50 mM of DMPO as the spin trapper.31 The samples were prepared as following: Fe3O4–PLGA (20 μg mL−1) with 10 mM H2O2 in pH 5.0 buffer, Fe3O4/GOx–PLGA (20 μg mL−1) in pH 5.0 buffer with 5 mM β-D-glucose, 10 mM H2O2 in pH 5.0 buffer for control. The spectra of DMPO/·OH were collected with an interval of 10 min.2.7 Cytotoxicity experiment
2.8 Fe3O4/GOx–PLGA-induced oxidative stress in HeLa cells
HeLa Cells were placed into 96-well plate with 8 × 103 cells per well and cultured for 24 h in DMEM medium to allow the attachment of cells. The medium of the 96-well plate was discarded followed by rinsing with PBS twice. Subsequently, Fe3O4–PLGA and Fe3O4/GOx–PLGA were separately dispersed into the 10% FBS containing high-glucose DMEM medium at the concentration of 4500 mg L−1, and then incubated into the 96-well plate. The pH value was respectively adjusted to 6.0 and 7.4 by the addition of HCl. After incubation for 24 h, the medium was then treated with 100 μL of fresh medium containing 10 μL of CCK-8 solution, then incubated at 37 °C for another 4 h. The absorbance of each well OD 490 was determined on the microplate reader.For ROS observations by fluorescence microscope, 5 × 104 of HeLa cancer cells were digested and resuspended into 1 mL 10% FBS containing high-glucose DMEM medium and subcultured into culture disk for another 6 h incubation. Subsequently, the medium was discarded and the disks were rinsed by PBS twice before 1 mL of high-glucose DMEM (pH 7.4 and 6.0) containing 1.0–10 μg mL−1 of Fe3O4–PLGA or Fe3O4/GOx–PLGA was replaced. Finally, the above medium was removed completely by PBS rinsing followed by the fluorescence probe addition.
For viable and dead cells observations after 6 h cytotoxicity, the Calcein acetoxymethyl ester (Calcein-AM)/Propidium iodide (PI) staining reagents were respectively applied to stain the viable cells as green fluorescence and dead cells as red fluorescence under 490 nm excitation. Specifically, 100 μL of 5 μM Calcein-AM solution and 100 μL of 10 μM PI solution were added after the removal of the culture medium and rinsing of the disks. After 15 min of incubation, staining solution were removed and rinsed by PBS twice and the samples were subsequently visualized on fluorescence microscope and confocal laser scanning microscope.
3 Results and discussion
3.1 Preparation and characterization of Fe3O4–PLGA and Fe3O4/GOx–PLGA
Fe3O4–PLGA were prepared by a two-step chemical synthesis process. First, oleic acid was used to modify Fe3O4 NPs synthesized by coprecipitation method32 to form hydrophobic Fe3O4–OA NPs with excellent stability and monodispersity. Next, the hydrophobic Fe3O4–OA NPs were encapsulated into biodegradable PLGA with emulsification-solvent evaporation method by using Pluronic F127 as an emulsifier to generate hydrophilic Fe3O4–PLGA nanocomposite.33 Fe3O4/GOx–PLGA were synthesized by further loading GOx into Fe3O4–PLGA to selectively convert intratumoral abundant glucose into highly oxidative ·OH.![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O stretching of PLGA,5 further indicating that the absence of chemical changes in PLGA during the encapsulation process. The peaks at 1270 cm−1 and 1344 cm−1 from Fe3O4–PLGA attributed to the bending vibrations of C–H, and the peaks at 840 cm−1 and 1087 cm−1 ascribed to stretching vibrations of C–O–C all indicated the presence of PEO on the surface of Fe3O4–PLGA,39 proving the successful modification of Pluronic F127 (Fig. S1†).
O stretching of PLGA,5 further indicating that the absence of chemical changes in PLGA during the encapsulation process. The peaks at 1270 cm−1 and 1344 cm−1 from Fe3O4–PLGA attributed to the bending vibrations of C–H, and the peaks at 840 cm−1 and 1087 cm−1 ascribed to stretching vibrations of C–O–C all indicated the presence of PEO on the surface of Fe3O4–PLGA,39 proving the successful modification of Pluronic F127 (Fig. S1†).
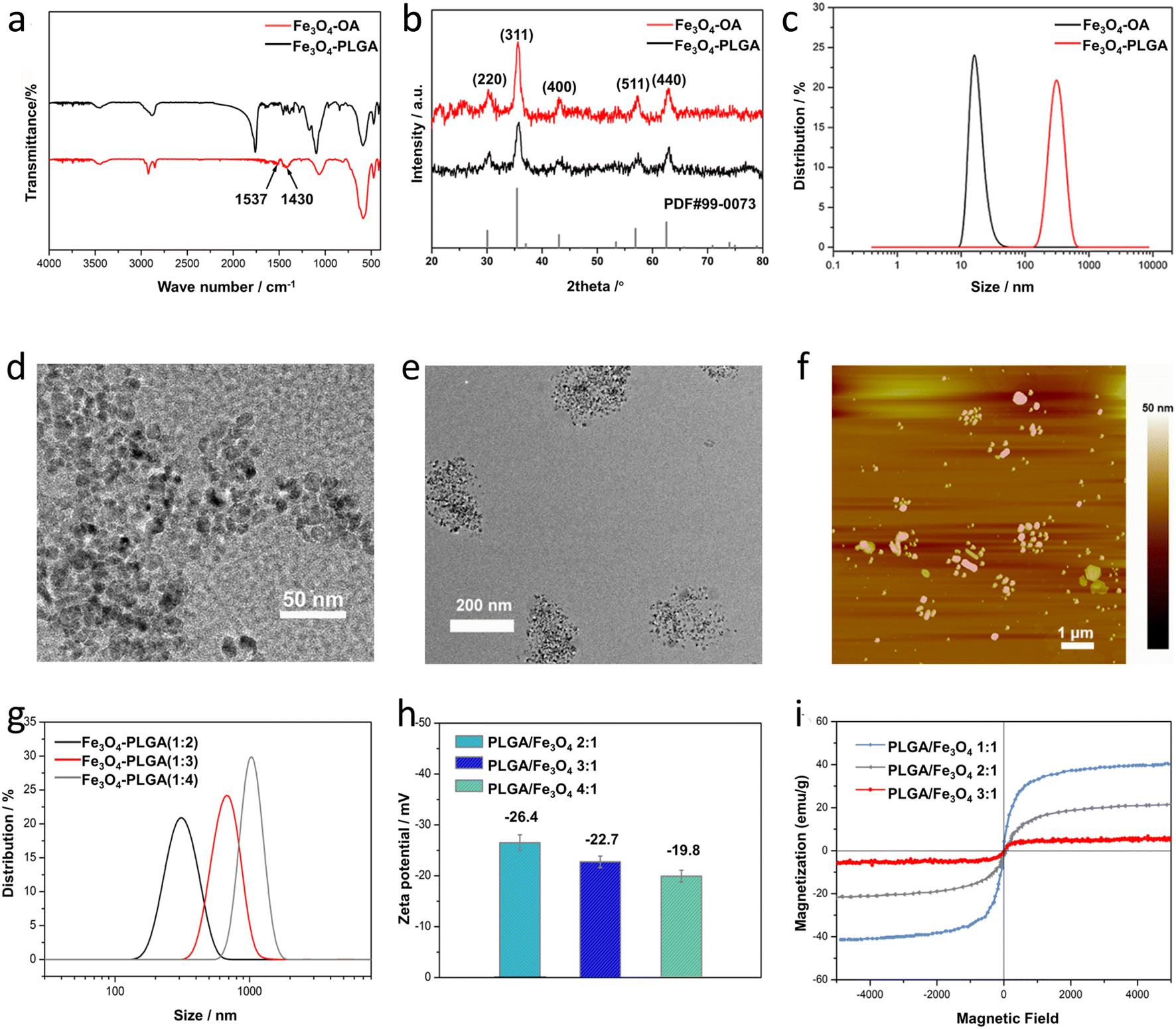 | ||
| Fig. 1 (a) FT-IR spectra of Fe3O4–OA NPs and Fe3O4–PLGA. (b) XRD spectra of Fe3O4–OA NPs and Fe3O4–PLGA. (c) Hydrodynamic diameter distribution of OA-capped Fe3O4 NPs and Fe3O4–PLGA nanocomposite. (d) TEM images of Fe3O4–OA NPs. (e) TEM images of Fe3O4–PLGA nanocomposite. (f) AFM images of Fe3O4–PLGA nanocomposite. (g) Hydrodynamic diameter distribution with different Fe3O4/PLGA mass ratio. (h) Zeta potential of Fe3O4–PLGA with different PLGA/Fe3O4 mass ratio. (i) VSM for Fe3O4–PLGA with different PLGA/Fe3O4 mass ratio. | ||
The influence of the PLGA doping amount on the particle size and morphology of Fe3O4–PLGA was investigated by changing the PLGA/Fe3O4 mass ratio from 2:1 to 4:1. Obviously, with the continuous increase of PLGA content, Fe3O4–PLGA gradually became larger, and the number of Fe3O4 NPs encapsulated in a single polymer nanoparticle increased (Fig. S2†). The particle size of the synthesized Fe3O4–PLGA with the PLGA/Fe3O4 mass ratio of 2:1 was about 270 nm (Fig. S2 a and d†) and rose to about 550 nm when the mass ratio increased to 3:1 (Fig. S2b and e†) with unchanged spherical morphology. And as the mass ratio of PLGA/Fe3O4 reached 4:1, Fe3O4–PLGA nanocomposites were nearly spherical and the size was close to 1 μm (Fig. S2c and f†). The larger size of the Fe3O4–PLGA nanocomposite with the increased PLGA/Fe3O4 mass ratio suggested more Fe3O4 NPs encapsulated in single PLGA NP. The more PLGA content brought about higher viscosity of the reaction system, resulting in a larger emulsion droplet where more Fe3O4 NPs were encapsulated owing to the decreased diffusion rate of the Fe3O4 NPs during emulsification.40 Furthermore, the higher PLGA concentration facilitated aggregation of the hydrophobic Fe3O4 NPs and PLGA inside the large emulsion droplets, also bringing about a larger particle size.
Similarly, the hydrodynamic diameters of Fe3O4–PLGA nanocomposites prepared with different PLGA/Fe3O4 mass ratio (2:1, 3:1, and 4:1) were respectively 324 ± 15 nm, 679 ± 21 nm and 1045 ± 45 nm (Fig. 1g), further proving that the increased amount of PLGA could enlarge the size of the Fe3O4–PLGA. In addition, the surface zeta potential of the Fe3O4–PLGA presented a downward trend and changed from −26.4 eV to −22.7 eV and −19.8 eV (Fig. 1h). The PLGA shell became thicker with the increase of the PLGA amount, bringing about the part screening of the negative charges on the surface of Fe3O4–OA NPs. The increase of the PLGA/Fe3O4 mass ratio lead to larger size and poorer stability of the Fe3O4–PLGA nanocomposite, also demonstrating that it was difficult to maintain spherical morphology and better stability for the prepared Fe3O4–PLGA if the PLGA/Fe3O4 mass ratio exceeded 4:1. Therefore, considering the encapsulated number of Fe3O4–OA NPs in the nanocomposites and better biocompatibility, the optimal PLGA/Fe3O4 mass ratio was chosen as 4:1 in the following experiments.
:1) to 21.9 emu g−1 (2:1) and 5.1 emu g−1 (3:1). In general, superparamagnetism is mainly relative to the particle size and surface-modified nonmagnetic materials.41 The smaller grain size favours the higher value of Ms, while the presence of non-magnetic coating materials can lead to a decrease in magnetic saturation due to the reduction of the effective weight fraction of the magnetic cores encapsulated in PLGA NPs.423.2 Fenton-like reaction of Fe3O4–PLGA
Terephthalic acid (TA) was severed as a probe to quantitatively analyze the content of ·OH, further verifying the catalyzing mechanism of Fe3O4–PLGA for Fenton-like reaction. It was based that in the presence of ·OH, TA would be oxidized into hydroxyterephthalic acid (TAOH) with strong fluorescence emission at 430 nm. And the fluorescence intensity of TAOH would enhance with the increased ·OH in the system. As shown in Fig. 2a, only when H2O2 was introduced in the system of Fe3O4–PLGA–TA, the strong fluorescence emission signal appeared at λem 430 nm. This result indicated that Fe3O4–PLGA catalytically oxidized H2O2 to generate ·OH. As H2O2 concentration increased from 0 mM to 10 mM, the fluorescence intensity of TAOH at λem 430 nm increased almost linearly, indicating the increasing generation of ·OH by Fe3O4–PLGA-induced Fenton catalysis was associated with the growing H2O2 concentration within a certain concentration range.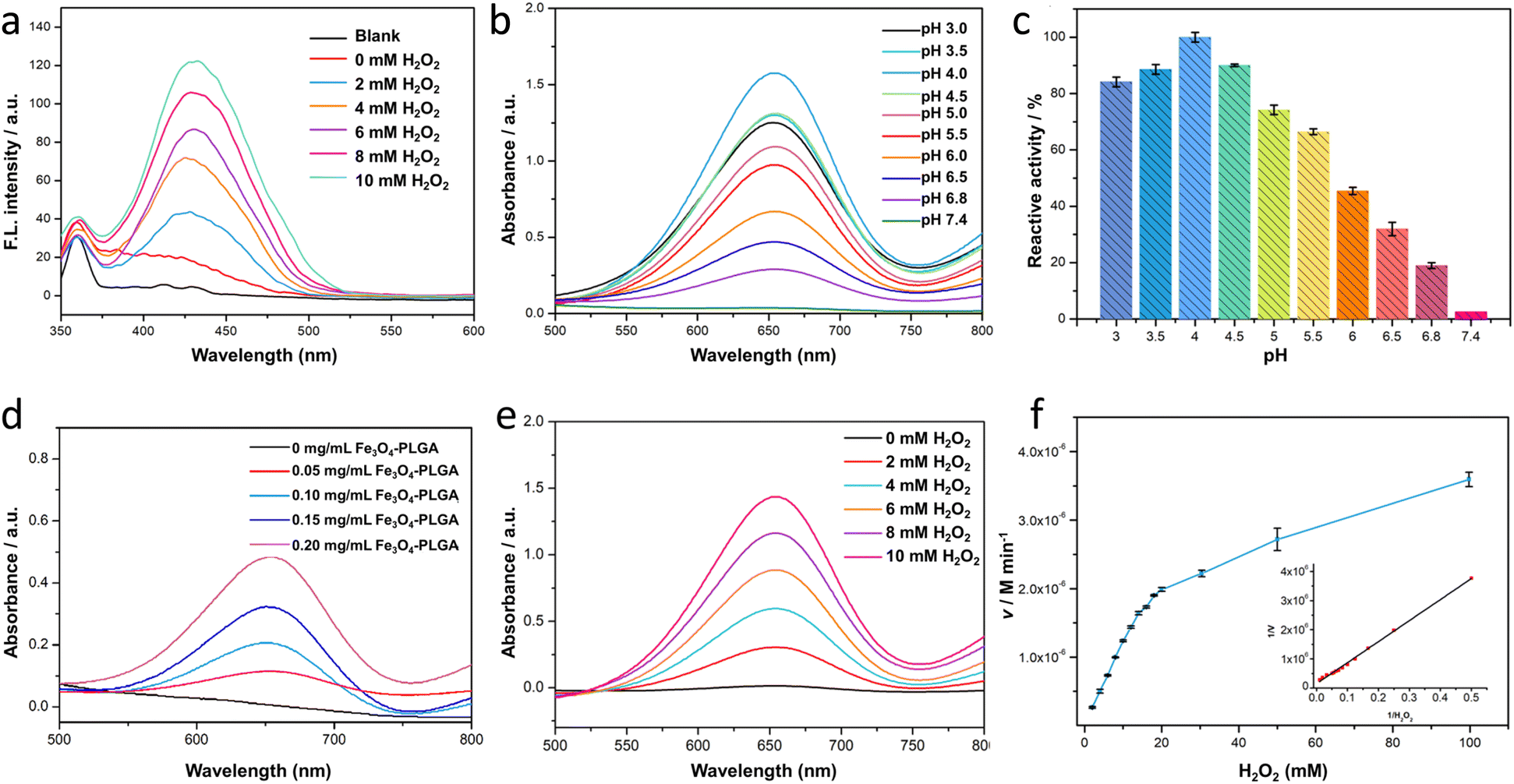 | ||
| Fig. 2 (a) Fluorescence spectra of Fe3O4–PLGA–H2O2–TA system. (b) The absorption curves of Fe3O4–PLGA–H2O2–TMB systems at different pH. (c) The influence of pH on the catalytic relative activity of Fe3O4–PLGA nanocomposite. (d) The absorption curves of Fe3O4–PLGA–H2O2–TMB systems with different concentrations of Fe3O4–PLGA nanocomposite. (e) The absorption curves of Fe3O4–PLGA–H2O2–TMB systems upon varied concentrations of H2O2. (f) Michaelis–Menten kinetics and Lineweaver–Burk plotting (insert figure) of Fe3O4–PLGA nanocomposite. | ||
3,3′,5,5′-Tetramethylbenzidine (TMB), one of the most sensitive dyes,30 was used to optimize reaction conditions (pH, nanocomposite concentration) of Fenton-like reaction catalyzed by Fe3O4–PLGA. The produced ·OH from the disproportionation of H2O2 under the catalysis by Fe3O4–PLGA in acidic environment would oxidize colorless TMB to chromogenic TMB cation-free radicals, which could be assayed at 652 nm (E = 3.9 × 104 M−1 cm−1) in UV-Vis spectrometer. In Fig. 2b, the UV absorption intensity at 652 nm from TMB oxidate has a strong dependence on pH of the reaction system (Fig. 2b).11 The results suggested that Fe3O4–PLGA showed the higher catalytic activity from pH 4.0 to 6.0 (Fig. 2c), which was related to high catalytic activity of Fe3O4 under acidic condition and looser state of the nanocomposites due to the PLGA degradation under acidic condition. Moreover, the acid environment could prevent dissolved iron from precipitation, and higher concentration of dissolved iron contributed to an increased yield of ·OH generation. The highest catalytic relative activity at pH 4.0 was defined as 100%, and the catalytic relative activity remained about 30% even the system pH was adjusted to 6.5. The tumor acidity boosted the degradation of Fe3O4–PLGA and the released Fe3O4 NPs further mediated the Fenton-like reactions for CDT use. Only 2.6% of relative activity at pH 7.4 was attributed to the slower degradation rate of PLGA and lower catalytic activity of Fe3O4 NPs under neutral condition. Then, it was found that the absorbance of Fe3O4–PLGA–TMB–H2O2 increased gradually in the acidic environment when more Fe3O4–PLGA nanocomposites were added into the system within a certain concentration range (Fig. 2d), while no change was observed without the addition of Fe3O4–PLGA, further confirming excellent catalytic activity of nanocomposites for Fenton-like reactions. In the last, with the increased H2O2 concentration, the absorbance intensity of Fe3O4–PLGA–H2O2–TMB system increased linearly at 652 nm, consistent with the results of TA fluorescence reaction. It further proved the intrinsic biodegradability and catalytic capacity of Fe3O4–PLGA at acidic pH, and also indicated that the increased H2O2 concentration contributed to the improvement of catalytic efficiency (Fig. 2e).
Based on the above optimal conditions, Fenton-like reaction kinetics of Fe3O4–PLGA was investigated by changing H2O2 concentration, so as to further evaluate the enzymatic catalytic activity as peroxidase. It was found that Fe3O4–PLGA followed the typical Michaelis–Menten model toward H2O2 by tracking change curves of absorbance (652 nm) in a real-time manner (Fig. 2f and S4†). The catalytic kinetic constant (Km) value of Fe3O4–PLGA with H2O2 as the substrate was 48.76 mM, much lower than that of Fe3O4 magnetic nanoparticles (154 mM)43 and Fe2O3 magnetic nanoparticles (324 mM),44 revealing strong affinity to H2O2. The Km of the catalyst, closely related with catalytic rate and substrate concentration, reflected the catalyst specificity to catalytic substrate.39 The lower the Km, the higher the physical affinity to substrate.45 Subsequently, the Vmax was calculated to be 1.12 × 10−7 M s−1 from Lineweaver–Burk plot. Interestingly, the initial fast (burst) increase of absorbance (Fig. S4†) might be associated with a small amount of Fe3O4 NPs through the external side of the Fe3O4–PLGA NPs. Then the gradually increased absorbance implied looser nanocomposites due to the acid degradation of PLGA and subsequent rapid release of Fe3O4 to generate ·OH.
3.3 Cascade catalytic activity of Fe3O4/GOx–PLGA
The cascade reaction of Fe3O4/GOx–PLGA was expected in the order that the loading GOx converted endogenous glucose into gluconic acid and H2O2 with the help of intracellular oxygen, and subsequently Fe3O4–PLGA catalyzed the oxidation of H2O2 into toxic ·OH via Fenton-like reaction under TME. The process of catalytic cascade reaction was verified by measuring the fluorescence spectra of the Fe3O4/GOx–PLGA–glucose–TA system without addition of H2O2. It was found in Fig. 3a and b that the TA-containing system showed a strong fluorescence emission peak at 435 nm in the presence of both Fe3O4/GOx–PLGA and β-D-glucose, indicating the cascade catalytic activity of Fe3O4/GOx–PLGA to β-D-glucose. It confirmed that Fe3O4/GOx–PLGA can efficiently boot the yield of ·OH in the presence of β-D-glucose. Moreover, it was also found that the increased concentrations of Fe3O4/GOx–PLGA led to a gradient ascent in the fluorescence intensity of Fe3O4/GOx–PLGA–glucose–TA system (Fig. 3a), which indicated that the cascade catalytic capacity of Fe3O4/GOx–PLGA was positively correlated with the concentration of Fe3O4/GOx–PLGA in a certain range. Similarly, the fluorescence intensity of Fe3O4/GOx–PLGA–glucose–TA system grew almost linearly with the ascent of the glucose concentration (0–5 mM) under the conditions of Fe3O4/GOx–PLGA (0.2 mg mL−1), TA (0.5 mM), and pH 4.0. The cascade catalytic activity of Fe3O4/GOx–PLGA was related to the concentration of the substrate β-D-glucose, which conformed to the catalytic principle (Fig. 3b). The concentration dependence of the Fe3O4/GOx–PLGA NPs' cascade catalytic activity on Fe3O4/GOx–PLGA NPs and glucose was similar to that of Fe3O4–PLGA NPs' catalytic activity on Fe3O4–PLGA and H2O2. The above results indicated that the Fe3O4/GOx–PLGA will provide superior therapeutic efficiency for cancer by simultaneously depleting glucose and boosting cascade reaction.46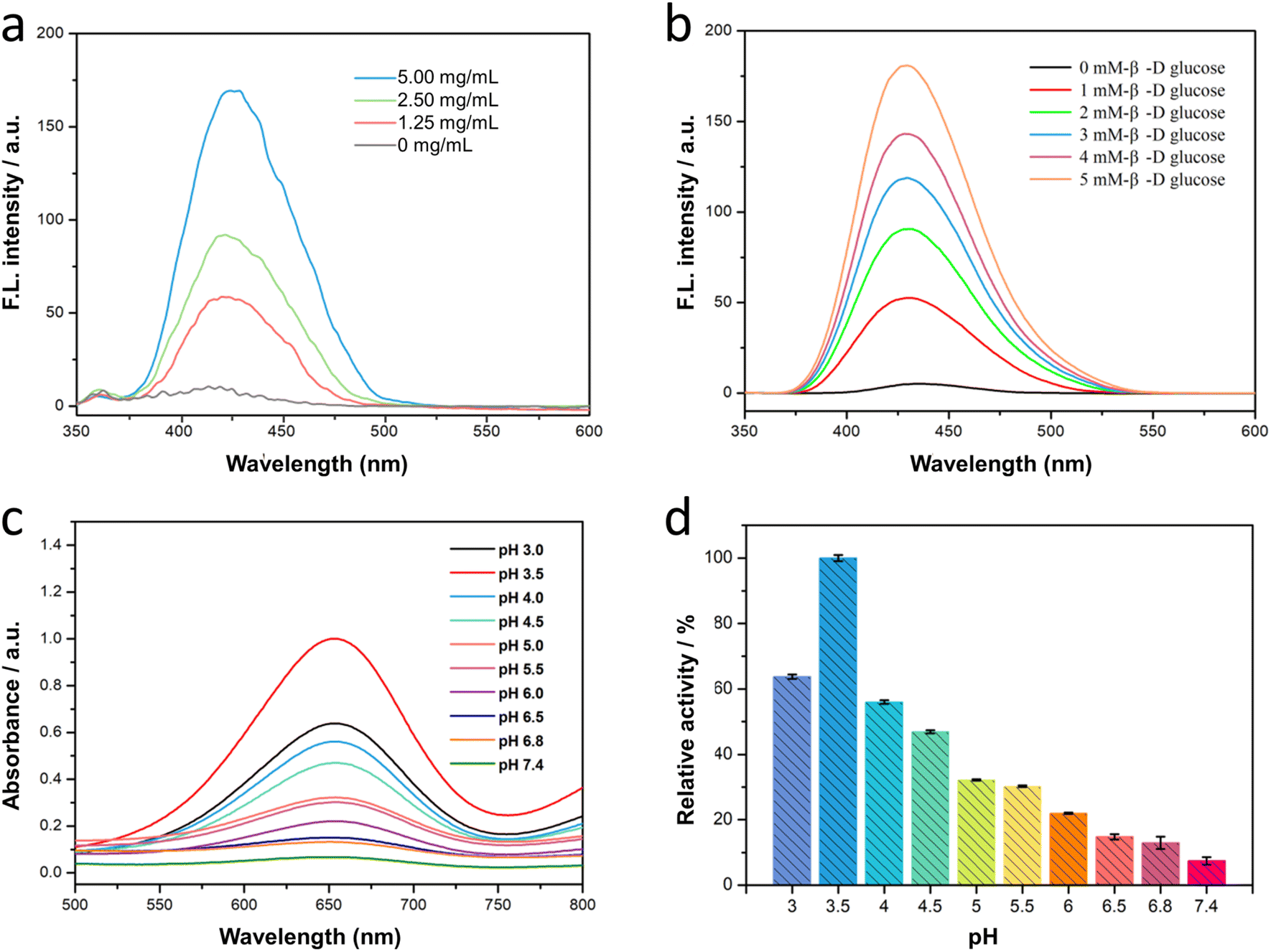 | ||
| Fig. 3 (a) Effects of different Fe3O4/GOx–PLGA nanocomposite concentration and (b) different β-D-glucose concentration on the fluorescence intensity of Fe3O4/GOx–PLGA–glucose–TA system. (c) The absorption curves of Fe3O4/GOx–PLGA–glucose–TMB system under different pH conditions. (d) The influence of pH on the catalyzed activity of Fe3O4/GOx–PLGA nanocomposite. | ||
Furthermore, to clarify the influence of pH on the cascade catalytic activity of Fe3O4/GOx–PLGA nanocomposite, TMB was applied to monitor the radical production by colorimetric reaction. The absorbance of Fe3O4/GOx–PLGA–glucose–TMB system was the highest at pH 3.5 (Fig. 3c), slightly different from the optimal pH of Fe3O4–PLGA. It might be due to the influence of GOx catalytic reaction. It was also found that Fe3O4/GOx–PLGA–glucose–TMB system presented different absorbance at 652 nm from pH 3 to pH 7.4. The relative activity of cascade catalysis at pH 3.5 was the highest and defined as 100% to quantify the influence on pH. Interestingly, Fe3O4/GOx–PLGA still remained more than 20% of reactive activity in a certain pH range from 3.0 to 6.0, while only 5.3% of reactive activity at pH 7.4 (Fig. 3d). The pH dependence of the cascade catalytic activity of Fe3O4/GOx–PLGA nanocomposite was associated with the acid degradation of PLGA and high catalytic activity of Fe2+ for Fenton reaction at acidic pH. The above results further confirmed the huge potential of Fe3O4/GOx–PLGA for TME-specific CDT.
Electron paramagnetic resonance (EPR) spectroscopy using 5,5-dimethyl-1-pyrroline-N-oxide (DMPO) as the radical trapper also clearly validated the generation of ·OH. As shown in Fig. S5,† typical EPR spectrum with 1:2:2:1 intensity characteristic for ·OH,47 were obtained for Fe3O4/GOx–PLGA and Fe3O4–PLGA nanocomposites, while no signal was found for control. Moreover, the intensity of the Fe3O4/GOx–PLGA was higher than that of Fe3O4–PLGA, which further evidenced that GOx consumed glucose into H2O2 and the enriched H2O2 was subsequently converted into abundant highly toxic ·OH by a Fe3O4-mediated Fenton-like reaction. Together, these results demonstrated that the Fe3O4/GOx–PLGA nanocomposite afforded a H2O2 self-supplying CDT platform under acidic condition.
3.4 Cytotoxicity analysis
The excellent Fenton-like activity of Fe3O4–PLGA and Fe3O4/GOx–PLGA further inspired us to explore its potential to induce oxidative stress. To this end, the cytotoxicity of Fe3O4–PLGA and Fe3O4/GOx–PLGA on PC12 cells was evaluated by using CCK-8 colorimetric assay. PC12 cells were respectively incubated with Fe3O4–PLGA and Fe3O4/GOx–PLGA for 24 h, and cell viability was evaluated by CCK-8 colorimetric assay. As shown in Fig. 4a, no obvious cytotoxicity was observed after 24 h incubation in the concentration range of 0.1–20 μg mL−1, and the cell viability was over 90% even at high concentration (20 μg mL−1), which validated that Fe3O4–PLGA and Fe3O4/GOx–PLGA nanocomposites presented low cytotoxicity. No significant decreases of cell viabilities in both cases may be ascribed to excellent biocompatibility of nanocomposites based on PLGA and Fe3O4. It also provided a basis for the dose design of nanocomposites for subsequent oxidative stress experiments.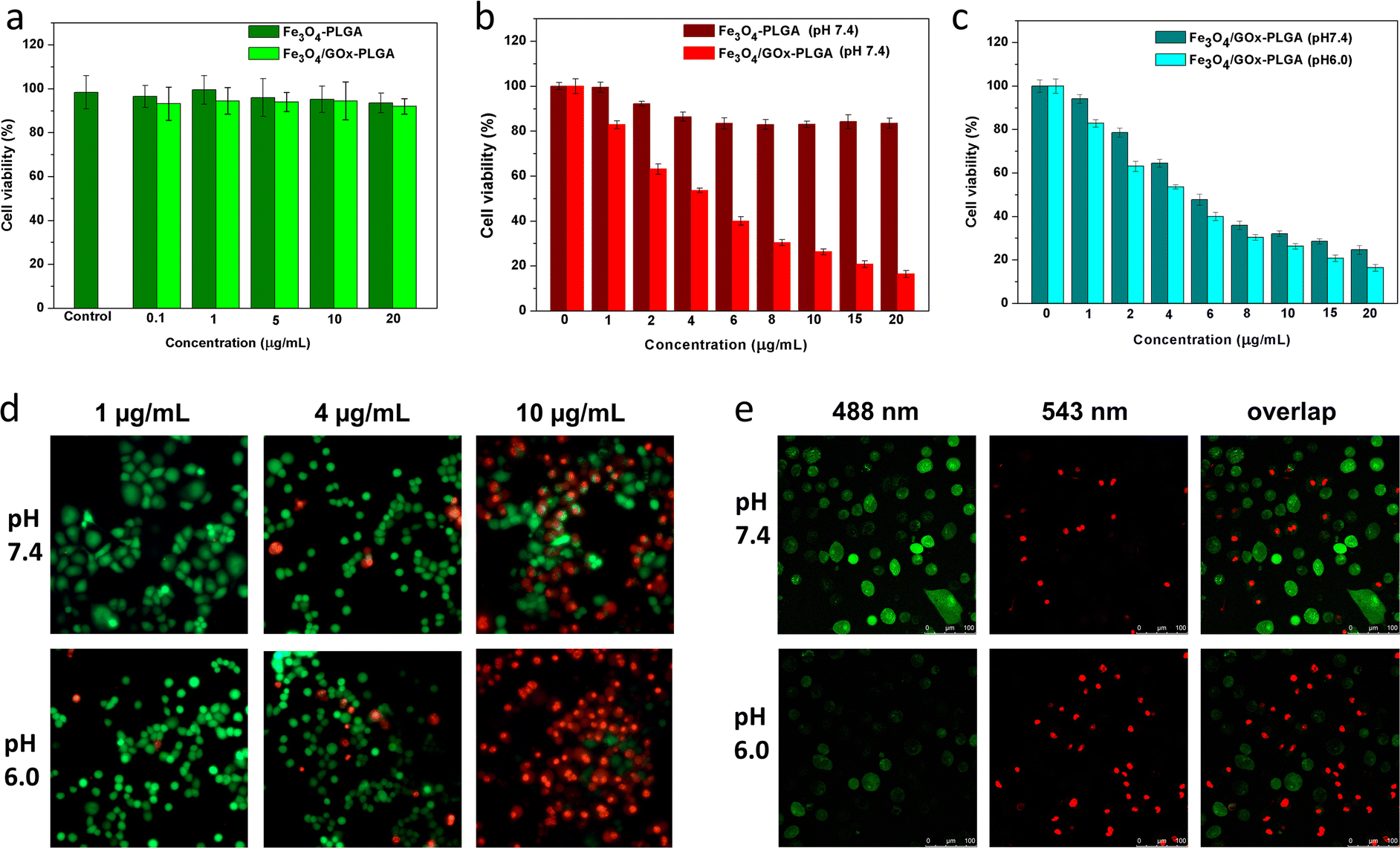 | ||
| Fig. 4 (a) PC12 cell viability of Fe3O4–PLGA and Fe3O4/GOx–PLGA with different concentrations from 0.1 to 20 μg mL−1 for 24 h, respectively. (b) Concentration effects of Fe3O4–PLGA and Fe3O4/GOx–PLGA under neutral (pH = 7.4) condition on the viability of HeLa cells. (c) Concentration effects of Fe3O4/GOx–PLGA under neutral (pH = 7.4) and acidic (pH = 6.0) conditions on the viability of HeLa cells. (d) Fluorescence images of viable and dead cell distributions after co-incubation with Fe3O4/GOx–PLGA under neutral (pH = 7.4) and acidic (pH = 6.0) conditions at varied concentrations for 6 h. (e) CLSM images of HeLa cells after co-incubation with Fe3O4/GOx–PLGA under neutral (pH = 7.4) and acidic (pH = 6.0) conditions at 10 μg mL−1 for 6 h. | ||
3.5 Fe3O4/GOx–PLGA NCs-induced oxidative stress in HeLa cells
The inhibitory effect of oxidative stress from Fe3O4/GOx–PLGA on HeLa cells were evaluated at the concentration of 1–20 μg mL−1 based on the results of cytotoxicity analysis on PC12 cells. HeLa cells were used as a model of tumor cells to verify the ability of Fe3O4–PLGA and Fe3O4/GOx–PLGA to induce oxidative stress. Under the condition with high concentration of glucose at pH 7.4, Fe3O4–PLGA showed no obvious cytotoxicity at the concentration of 1–20 μg mL−1 for HeLa cells (Fig. 4b). Compared with Fe3O4–PLGA, the higher inhibition rate from Fe3O4/GOx–PLGA meant more efficient oxidative stress and apoptosis inducement caused by a large amount of ·OH generated from glucose by cascade catalysis of GOx and Fe3O4 in glucose-rich TME. The cytotoxicity of Fe3O4/GOx–PLGA at pH 6.0 and 7.4 was respectively evaluated within the concentration range of 1–20 μg mL−1. In Fig. 4c, it was found that the Fe3O4/GOx–PLGA showed obvious inhibition on HeLa cells in a dose-dependent manner both at pH 6.0 and 7.4, but higher inhibition rate at pH 6.0.To visually observe distributions of the viable and dead cells, HeLa cancer cells were stained with calcein-AM and PI solution after separate incubation with Fe3O4–PLGA and Fe3O4/GOx–PLGA at varied concentrations (2, 6, and 10 μg mL−1) under neutral conditions for 6 h. Likewise, HeLa cells were respectively incubated with Fe3O4–PLGA and Fe3O4/GOx–PLGA nanocomposite at varied concentrations(1, 4, and 10 μg mL−1) in both acidic (pH = 6.0) and neutral (pH = 7.4) culture mediums for 6 h. The viable and dead cells were respectively stained with green and red fluorescence before fluorescence and CLSM observation. The fluorescence and CLSM images showed that no significant damage on HeLa cells for Fe3O4–PLGA at pH 7.4 (Fig. S6 and S7†), and influence of cell apoptosis on the varied concentration of Fe3O4–PLGA and Fe3O4/GOx–PLGA (1, 4, 10 μg mL−1) were assayed under neutral and acidic conditions, respectively. More dead cells were observed when incubated with Fe3O4/GOx–PLGA at varied concentration (1, 4, 10 μg mL−1) under acid condition (Fig. 4d and S8†). Obviously, under acidic condition, almost all HeLa cancer cells were dead at 10 μg mL−1 of Fe3O4/GOx–PLGA (Fig. 4e). The improved ability of killing cancer cells for Fe3O4/GOx–PLGA at pH 6.0 might be related to the GOx-initiating and Fe3O4-mediated generation of ·OH and cell starvation resulting from consumption of intracellular glucose. The above results confirmed cooperative enhancement interactions between starvation therapy and cascade cancer therapy by self-sufficient H2O2 in situ and the continuous generation of ·OH inducing oxidase stress and apoptosis.
4 Conclusions
In summary, a new strategy responding to TME was developed by intelligently integrate GOx and Fe3O4 NPs into PLGA NPs with emulsification-solvent evaporation method, realizing a tumor-selected combination of chemodynamic and starving therapy. The GOx effectively converted glucose into H2O2, accompanied by decline of localized O2 content and pH. The pH decline in turn promoted the degradation of PLGA and sequent Fe3O4 release, to give rise to Fenton reaction between Fe ions and H2O2 for generation of ·OH. The synthesized Fe3O4–PLGA nanocomposite was proved to show remarkable Fenton catalytic activity and better affinity to substrate H2O2 in tumor microenvironment, indicating excellent capability of producing ·OH to kill cancer cells. After loading GOx, intracellular oxidative stress suggested that Fe3O4/GOx–PLGA nanocomposite could enhance the ability to kill cancer cells through continuous conversion of intratumoral glucose into H2O2 for Fenton reaction and cell starvation due to glucose consumption. Thus, Fe3O4/GOx–PLGA nanocomposite as an excellent cascade reaction-based nanoplatform will provide a new perspective for cancer therapy.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was financially supported by the Young Top-notch Talent Cultivation Program of Hubei Province, China, Leading Talents in specialty construction of colleges and universities in Taicang of China, Natural Science Foundation of Jiangsu Province (No. BK20220301) and Science Projects of Taicang City, China (TC2022JC27).Notes and references
- C. R. Leach, S. V. Hudson and M. A. Diefenbach, Cancer, 2022, 128, 597–605 CrossRef PubMed.
- L. Z. Zhang, C. Y. Zhu, R. T. Huang, Y. W. Ding, C. P. Ruan and X. C. Shen, Front. Chem., 2021, 18, 630969 CrossRef PubMed.
- M. Patel and A. Prabhu, Int. J. Pharm., 2022, 618, 121697 CrossRef CAS PubMed.
- Y. B. Peng, Y. Ren, H. Zhu, Y. An, B. S. Chang and T. L. Sun, RSC Adv., 2021, 11, 14517–14526 RSC.
- Y. Zhang, A. R. Khan, X. Y. Yang, M. F. Fu, R. J. Wang, L. Q. Chi and G. X. Zhai, J. Drug Delivery Sci. Technol., 2021, 61, 102266 CrossRef CAS.
- C. Y. Jia, Y. X. Guo and F. G. Wu, Small, 2021, 02, 2103868 Search PubMed.
- H. Simon, A. Haj-Yehia and F. Levi-Schaffer, Apoptosis, 2000, 05, 415–418 CrossRef CAS PubMed.
- Z. M. Tang, Y. Y. Liu, M. Y. He and W. B. Bu, Angew. Chem., Int. Ed., 2019, 58, 946–956 CrossRef CAS PubMed.
- S. D. Zhai, X. L. Hu, Y. J. Hu, B. Y. Wu and D. Xing, Biomaterials, 2017, 121, 41–54 CrossRef CAS PubMed.
- Z. Shen, T. Liu, Y. Li, J. Lau, Z. Yang, W. Fan, Z. Zhou, C. Shi, C. Ke, V. I. Bregadze, S. K. Mandal, Y. Liu, Z. Li, T. Xue, G. Zhu, J. Munasinghe, G. Niu, A. Wu and X. Chen, ACS Nano, 2018, 12, 11355–11365 CrossRef CAS PubMed.
- Y. P. Wang, Y. T. Liao, C. H. Liu, J. Yu, H. R. Alamri, Z. A. Alothman, M. S. A. Hossain, Y. Yamauchi and K. C. W. Wu, ACS Biomater. Sci. Eng., 2017, 3, 2366–2374 CrossRef CAS PubMed.
- W. H. Wang, Z. W. Huang, Y. Huang, X. Pan and C. B. Wu, Int. J. Pharm., 2020, 589, 119815 CrossRef CAS PubMed.
- F. Mazuel, A. Espinosa, N. Luciani, M. Reffay, R. L. Borgne, L. Motte, K. Desboeufs, A. Michel, T. Pellegrino, Y. Lalatonne and C. Wilhelm, ACS Nano, 2016, 10, 7627–7638 CrossRef CAS PubMed.
- P. Ma, H. Xiao, C. Yu, J. Liu, Z. Cheng, H. Song, X. Zhang, C. Li, J. Wang, Z. Gu and J. Lin, Nano Lett., 2017, 17, 928–937 CrossRef CAS PubMed.
- L. S. Lin, J. Song, L. Song, K. Ke, Y. Liu, Z. Zhou, Z. Shen, J. Li, Z. Yang, W. Tang, G. Niu, H. H. Yang and X. Chen, Angew. Chem., Int. Ed., 2018, 57, 4902–4906 CrossRef CAS PubMed.
- L. Yu, Y. Chen, M. Wu, X. Cai, H. Yao, L. Zhang, H. Chen and J. Shi, J. Am. Chem. Soc., 2016, 138, 9881–9894 CrossRef CAS PubMed.
- G. Chen, I. Roy, C. Yang and P. N. Prasad, Chem. Rev., 2016, 116, 2826–2885 CrossRef CAS PubMed.
- X. Y. Chen, H. L. Zhang, M. Zhang, P. R. Zhao, R. X. Song, T. Gong, Y. Y. Liu, X. H. He, K. Zhao and W. B. Bu, Adv. Funct. Mater., 2019, 29, 1908365 Search PubMed.
- L. H. Fu, C. Qi, Y. R. Hu, J. Lin and P. Huang, Adv. Mater., 2019, 31, 1808325–1808339 CrossRef PubMed.
- L. H. Fu, Y. R. Hu, C. Qi, T. He, S. S. Jiang, C. Jiang, J. He, J. L. Qu, J. Lin and P. Huang, ACS Nano, 2019, 13, 13985–13994 CrossRef CAS PubMed.
- N. Mauro, C. Scialabba, R. Puleio, P. Varvarà, M. Licciardi, G. Cavallaro and G. Giammon, Int. J. Pharm., 2019, 555, 207–219 CrossRef CAS PubMed.
- Y. L. Dai, C. Xu, X. L. Sun and X. Y. Chen, Chem. Soc. Rev., 2017, 46, 3830–3852 RSC.
- N. N. Zheng, Y. Fu, X. J. Liu, Z. W. Zhang, J. X. Wang, Q. X. Mei, X. Y. Wang, G. Y. Deng, J. Lu and J. Q. Hu, J. Mater. Chem. B, 2022, 10, 637–645 RSC.
- R. Kumari, D. Sunil and R. S. Ningthoujam, J. Controlled Release, 2020, 319, 135–156 CrossRef CAS PubMed.
- H. W. Zhang, F. Lu, W. Pan, Y. G. Ge, B. J. Cui, S. H. Gong, N. Li and B. Tang, Biomater. Sci., 2021, 9, 3814–3820 RSC.
- W. P. Fan, N. Lu and P. Huang, Angew. Chem., Int. Ed., 2016, 55, 1–6 CrossRef.
- K. Fu, G. Kai, L. Hsieh, A. M. Klibanov and R. Langera, J. Controlled Release, 1999, 3, 357–366 CrossRef PubMed.
- W. Ke, J. Li, F. Mohammed, Y. Wang, K. Tou, X. Liu, P. Wen, H. Kinoh, Y. Anraku, H. Chenk, K. Kataoka and Z. Ge, ACS Nano, 2019, 13, 2357–2369 CAS.
- J. M. Anderson and M. S. Shive, Adv. Drug Delivery Rev., 2012, 64, 72–82 CrossRef.
- B. Jiang, D. M. Duan, M. J. Zhou, K. L. Fan, Y. Tang and J. Q. Xi, Nat. Protoc., 2018, 13, 1506–1520 CrossRef CAS PubMed.
- C. Rota, C. F. Chignell and R. P. Mason, Free Radical Biol. Med., 1999, 27, 873–881 CrossRef CAS PubMed.
- W. C. Elmore, Phys. Rev., 1938, 54, 309–310 CrossRef CAS.
- S. Doppalapudi, A. Jain, A. J. Domb and W. Khan, Expert Opin. Drug Delivery, 2016, 13, 891–909 CrossRef CAS PubMed.
- Q. Gao, F. H. Chen, J. L. Zhang, G. Y. Hong, J. Z. Ni, X. Wei and D. J. Wang, J. Magn. Magn. Mater., 2009, 321, 1052–1057 CrossRef CAS.
- V. V. Korolev, A. G. Ramazanova and A. V. Blinov, Russ. Chem. Bull. Int. Ed., 2002, 51, 2044–2049 CrossRef CAS.
- M. Khalil, A. Fahmi, N. M. Nizardo, Z. Amir and B. M. Jan, Langmuir, 2021, 37, 8855–8865 CrossRef CAS PubMed.
- L. Zhang, R. He and H. C. Gu, Appl. Surf. Sci., 2006, 253, 2611–2617 CrossRef CAS.
- R. D. Palma, S. Peeters, M. J. Van Bael, V. D. R. Heidik, K. Bonroyk, W. Laureynk, J. Mullens, G. Borghs and G. Maes, Chem. Mater., 2007, 19, 1821–1831 CrossRef.
- Y. Wang, J. S. Nie, B. S. Chang, Y. F. Sun and W. L. Yang, Biomacromolecules, 2013, 14, 3034–3046 CrossRef CAS PubMed.
- F. Yan, J. Li, J. J. Zhang, F. Q. Liu and W. S. Yang, J. Nanopart. Res., 2009, 11, 289–296 CrossRef CAS.
- Y. W. Jun, J. S. Choia and J. W. Cheon, Chem. Commun., 2007, 12, 1203–1214 RSC.
- K. Nejati-Koshk, M. Mesgari, E. Ebrahimi, F. Abbasalizadeh and A. Akbarzadeh, Microencapsul, 2014, 06, 1464–5246 Search PubMed.
- L. Z. Gao, L. Zhuang, J. B. Nie, Y. Zhang, N. Zhang, T. H. Gu, J. Wang, D. L. Feng, S. P. Yang and X. Yan, Nat. Nanotechnol., 2007, 2, 577–583 CrossRef CAS PubMed.
- X. Q. Zhang, S. W. Gong, Y. Zhang, T. Yang, C. Y. Wang and N. Gu, J. Mater. Chem., 2010, 20, 5110–5116 RSC.
- A. Asati, S. Santra, C. Kaittanis, S. Nath and J. M. Perez, Angew. Chem., Int. Ed., 2009, 48, 2308–2312 CrossRef CAS PubMed.
- L. H. Fu, C. Qi, J. Lin and P. Huang, Chem. Soc. Rev., 2018, 47, 6454–6472 RSC.
- Y. J. Han, J. Ouyang, Y. Z. Li, F. L. Wang and J. H. Jiang, ACS Appl. Mater. Interfaces, 2020, 12, 288–297 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3ra00527e |
| ‡ Co-first author. |
| This journal is © The Royal Society of Chemistry 2023 |