DOI:
10.1039/D3RA00546A
(Paper)
RSC Adv., 2023,
13, 5762-5769
Herbal molecule-mediated dual network hydrogels with adhesive and antibacterial properties for strain and pressure sensing†
Received
26th January 2023
, Accepted 10th February 2023
First published on 15th February 2023
Abstract
Multifunctional integration is the focus of hydrogel-based flexible sensors, and formation of a dual network (DN) could shed light on the fabrication of hydrogels with multifunctionality and enhanced properties. In this study, a DN hydrogel was fabricated by the self-assembly of herbal molecule glycyrrhizic acid (GA) as the first hydrogel network and subsequent photocrosslinking of methacrylated sodium alginate (SA-MA) to form the second network. Profiting from the good compatibility between the two hydrogel networks, the obtained DN hydrogels with a homogeneous porous microstructure were endowed with remarkably enlarged stretching (114.5%) and compression (74.4%) strains. In addition, they were demonstrated to display excellent bacteriostatic activity (>99.9%) against Escherichia coli and Staphylococcus aureus owing to the synergetic antibacterial effect of GA and SA-MA. The DN hydrogels as strain sensors possessed high sensitivity (GF = 1.39), linear sensing (R2 > 0.99), rapid response (180 ms), and good stability (1300 times) for human motion detection. Besides, the DN hydrogels could also be used to conduct pressure sensing such as application of heavy weights and even human pulses. All results suggest that the developed DN hydrogels have great potential in serving as epidermal and implantable flexible sensors for human health monitoring.
1. Introduction
Flexible and wearable sensors that possess the capability to convert a mechanical stimulus into electrical signals have been extensively developed in the fields of human motion monitoring, physiological monitoring, soft robots, electronic skin, and human–machine interfaces.1–3 Compared with traditional sensors made of metals or elastomers, flexible hydrogels are superior in terms of adjustable stretchability, skin-like modulus, biocompatibility, and biodegradability.4,5 These intrinsic advantages make them promising candidates for flexible sensors. Nowadays, ionic hydrogels containing free ions and conductive hydrogels loaded with electrical materials (e.g., carbon materials, metal nanoparticles, conductive polymers, MXenes, and liquid metals) are undergoing further multifunctional integration, for example with self-healing ability, anti-freezing properties, stimulus responsiveness, and antibacterial properties.6–9 The multifunctional hydrogel sensors can be used in more scenarios with better sensing performance, stronger environmental tolerance, and longer service life.
Polymer-based hydrogels have been widely developed for the preparation of flexible sensors. Among them, the copolymer hydrogels could bring about adaptable mechanical properties and other appealing properties such as hydrophobicity and stimulus responsiveness,10–13 which enable the sensors to be robust and smart. Recently, dual network (DN) hydrogels, first established in 2003 by Gong et al.,14 have been recently demonstrated to be effective in constructing flexible sensors possessing enhanced physicochemical properties and integrated multifunctionality.15–18 For example, Li et al. fabricated a tough DN hydrogel through combining thermoreversible κ-carrageenan hydrogel with polyacrylamide network, where the former rendered good recoverability and self-healing capability to the DN hydrogel while the latter enabled 3D printing by means of UV light-triggered photopolymerization.19 Gu et al. employed the host–guest interaction and dynamic covalent bond to prepare DN hydrogels, acquiring satisfactory comprehensive performance including stretchability, strength, remodeling ability, self-healing property, self-adhesiveness, and biocompatibility.20 When the DN hydrogels were used as flexible strain sensors, the human motions and respiratory movement could be detected with good stability. Unfortunately, most DN hydrogels still lack antibacterial property required in practical applications. Indeed, the wearable sensors are inevitable to contact with biological surfaces (e.g., human skin and even organs). Besides, the bacterial growth within and/or surrounding the hydrogel sensors may cause inestimable health hazards to users and severely decrease the service life of hydrogel sensors.21,22
Recently, many kinds of antibacterial agents were introduced into the DN hydrogels with the purpose of integrating antibacterial property.23–25 Hu et al. developed a DN hydrogel where gradient silver nanoparticles were in situ formed on the hydrogel surface.26 It was found that this hydrogel exhibited excellent antibacterial activity against both Gram-negative bacteria and Gram-positive bacteria. Wu et al. constructed a flexible nanocellulose DN hydrogel through the combination with polyvinyl alcohol network. After loading a typical antibiotic amoxicillin, antibacterial ratios of about 99.0% for Escherichia coli (E. coli) and 99.6% for Staphylococcus aureus (S. aureus) were obtained.27 Though significant advances have been made in the improving antibacterial DN hydrogel sensors, the common antibacterial materials have potential drawbacks. For instance, silver nanoparticles were demonstrated to possess cytotoxicity to human cells and the antibiotics could lead to the problem of bacterial resistance.28,29 Therefore, pursuing new approaches to prepare multifunctional DN hydrogel sensors with antibacterial property presents a challenging task.
Glycyrrhizic acids (GAs), small herbal molecules of triterpenic saponin extracted from licorice,30 intrinsically consist of di-glucuronic acid (hydrophilic part) and 18b-glycyrrhetinic acid (hydrophobic part). Due to their unique structural amphiphilicity, the GA molecules are prone to self-assemble into nanofibrils in aqueous solution and further entangle to form supramolecular hydrogels.31 Besides, GA and its derivatives have been demonstrated to possess efficient antibacterial effect,32 and poly(methacrylic acid) and methacrylic copolymer display a broad spectrum of antibacterial properties.33,34 It is envisioned that elaborate combination of GA hydrogel and methacrylated biopolymers may provide a promising solution to intrinsically antibacterial and multifunctional DN hydrogels. Herein, in this work, a flexible DN hydrogel was constructed through the self-assembly of GA molecules and in situ photopolymerization of SA-MA. Benefiting from their good compatibility and synergetic antibacterial effect, the obtained DN hydrogels exhibited enhanced mechanical property, good transparency, self-adhesion to human skin, and excellent antibacterial property. Furthermore, the hydrogel could be used as strain and pressure dual-modal sensors to detect the human motions and pulse signals with high sensitivity, fast response, and acceptable reproducibility. This versatile DN hydrogel as a flexible sensor owns the great potential for applications in health monitoring and soft electronic skin.
2. Materials and methods
2.1 Materials
Glycyrrhizic acid ammonium salt (98%, Yuanye Biotechnology Co., Ltd.) was used as hydrogel precursor. Methacrylic anhydride (94%, Aladdin) was used to modify sodium alginate (Mw = 388![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000, Haizhilin Biotechnology Co., Ltd.), and the obtained product was used as macromonomers. Triethanolamine (99%, Aladdin), eosin Y (AR, Aladdin) and N-vinylpyrrolidone (99%, Aladdin) were used as photoinitiator system for photopolymerization. Fresh porcine skin was bought from local market. All reagents were used as received without further purification, and ultrapure water (18.2 MΩ) was used throughout the experiments.
000, Haizhilin Biotechnology Co., Ltd.), and the obtained product was used as macromonomers. Triethanolamine (99%, Aladdin), eosin Y (AR, Aladdin) and N-vinylpyrrolidone (99%, Aladdin) were used as photoinitiator system for photopolymerization. Fresh porcine skin was bought from local market. All reagents were used as received without further purification, and ultrapure water (18.2 MΩ) was used throughout the experiments.
2.2 Preparation of hydrogels
2.2.1 Preparation of SA hydrogels. The SA-MA precursors were synthesized according to the published process35 and confirmed by 1H NMR. Briefly, 2.0 g SA was placed into 100 mL deionized water and stirred until completely dissolved. Excess methacrylate (10 mL) was added dropwise in an ice bath, and the solution was stirred for 24 h at 4 °C, during which 5 M NaOH solution was used to control the solution pH value at constantly 8. White precipitates were obtained and washed through adding ethanol.Typically, 30 mg SA-MA was initially dissolved in 1 mL deionized water, and then 5 μL of 0.5 wt% eosin Y solution in 1-vinyl-2-pyrrolidinone and 5 μL of 2 M triethanolamine in water were added. The above photoinitiator solution was transferred into a mold and finally irradiated by green LED (100 W, 525 nm, 10 cm distance to sample) for 30 s to obtain SA3 hydrogel.
2.2.2 Preparation of GA hydrogels. 20 mg GA were placed in 1 mL deionized water and heated to 80 °C until completely dissolved. After cooling to room temperature, the GA self-assembled to produce hydrogel rapidly.
2.2.3 Preparation of GA/SA DN hydrogels. Taking GA/SA3 DN hydrogel as an example, 30 mg SA-MA and 20 mg GA were mixed in 1 mL deionized water, and 5 μL of 0.5 wt% eosin Y solution in 1-vinyl-2-pyrrolidinone and 5 μL of 2 M triethanolamine in water were added. The above mixture was heated to 90 °C until completely dissolved, and then injected into a mold to stand at room temperature. After the emergence of GA hydrogel, green LED (100 W, 525 nm, 10 cm distance to sample) was utilized to trigger the formation of GA/SA DN hydrogel. For GA/SA2, GA/SA4, GA/SA5 DN hydrogels, the preparation procedures were similar except that the amounts of SA-MA were 20 mg, 40 mg, and 50 mg, respectively.
2.3 Characterization
2.3.1 General measurements. 1H NMR spectra were recorded on a Bruker spectrometer (AVANCE III HD 600 MHz) using tetramethylsilane as internal standard (δ = 0 ppm). The morphologies of hydrogels were examined by a field emission scanning electron microscope (Quanta PEG-450, FEI, America) after sprayed with a thin gold layer. Before SEM observation, the freeze drying of hydrogels was used for sample preparation.
2.3.2 Tensile tests. Mechanical tensile stress–strain tests were conducted by a TA-XTplusC mechanical testing instrument (Stable Micro Systems, England) equipped with a 500 N tension sensor at room temperature at a stretching rate of 6 mm min−1 until the sample broke. All hydrogels were cast using polytetrafluoroethylene molds into dumbbell-shaped samples (1.5 mm thickness, 5.0 mm width at center).
2.3.3 Compression tests. The compression tests of hydrogel samples were performed on TA-XTplusC mechanical testing instrument (Stable Micro Systems, England) with a 500 N load cell. Each hydrogel disk with a diameter of 15 mm (about 8 mm thickness) was compressed until failure at a rate of 6 mm min−1. All samples were measured at room temperature.
2.3.4 Lap-shear tests. The adhesive strength of the hydrogel samples to diverse substrates was characterized by lap-shear tests on a TA-XTplusC mechanical testing instrument (Stable Micro Systems, England) at ambient temperature. The samples were cut into a rectangular shape (15 mm (length) × 15 mm (width) × 1.5 mm (thickness)), and then adhered between two pieces of substrates. Each end of the substrates was clamped with a tensile tester, and the shear velocity was 10 mm min−1. The adhesion strength was calculated by the measured maximum tensile force (N) divided by the contact area (m2).
2.3.5 Rheological measurements. The testing was conducted on a hybrid rheometer (DHR-1, TA, America) at 25 °C using a 20 mm diameter flat plate with the gap set to 1.0 mm. Excess sample was trimmed off the lower plate. The storage modulus (G′) and loss modulus (G′′) were measured and recorded by performing a shear strain-sweep test using a 10 rad s−1 angular frequency and a strain sweeping from 0.1% to 1000%.
2.3.6 Electrical measurements. The electrical conductivity of the hydrogels (30 mm (length) × 10 mm (width) × 2 mm (thickness)) was measured using a CHI760E electrochemical workstation with a 4-probe alternating current impedance method. The electrical conductivity of the hydrogels was calculated using the following formula: s = L/RS, where s is the conductivity (S m−1), L is the length of the hydrogel strip (m), R is the hydrogel resistance (Ω), and S is the cross-sectional area of the hydrogel (m2).
2.3.7 Tests of GA/SA DN hydrogel as a strain and pressure sensor. The sensing performance tests were recorded using a CHI760E electrochemical workstation at a constant voltage of 2 V. For all the strain sensor tests, the hydrogel samples were controlled to be cuboid in shape with the length, width and thickness being 30 mm, 10 mm, and 2 mm, respectively. The relative resistance change can be calculated using the following formula ΔR/R0 = (R − R0)/R0 × 100%, where R0 and R represent the resistance before and after the applied strain. For all the pressure sensor tests, the hydrogel samples were GA/SA3 DN hydrogel disks with a diameter of 15 mm and about 8 mm thickness. To investigate the performance as a strain sensor, the GA/SA DN hydrogel was directly adhered to the human skin due to the good self-adhesive properties of the hydrogels, and written consent was obtained from the human subject taking part in the experiments. All the measurements were conducted at room temperature.
2.3.8 In vitro antibacterial assay. The antibacterial properties of SA3 and GA/SA3 hydrogels were evaluated by the flat colony counting method.36,37 E. coli (ATCC 25922) and S. aureus (ATCC 29213) were chosen as Gram-negative and Gram-positive bacteria, respectively. The detailed procedure is as follows: first, the hydrogel sample was placed in a 6 well plate and exposed under a UV lamp for 30 min; then 10 μL of the 1.0 × 106 CFU per mL bacterial stock solution in liquid Luria–Bertani medium was introduced to the surface of the given hydrogel and incubated at 37 °C for 18 h. 990 μL sterile PBS solution was added to each well and bacteria on hydrogel surface were ultrasonically detached for 3 min. After serial 10-fold dilutions of the previous bacterial solution (10−2, 10−3, and 10−4 times for E. coli and 10−3, 10−4, and 10−5 times for S. aureus), the bacterial suspension (100 μL) was spread evenly on a solid Luria–Bertani medium and incubated at 37 °C for 18 h. Last, the number of bacterial colonies was recorded and counted by digital camera. For comparison, identical bacterial suspensions without the addition of hydrogel were used as the control group. The bacteriostatic ratio is calculated using the reported equation.38
3. Results and discussion
3.1 Construction of GA/SA DN hydrogels
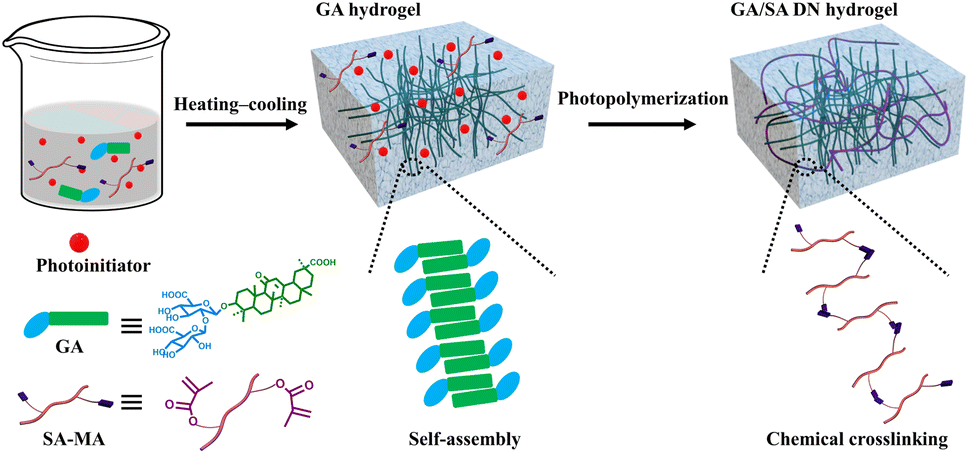
To lay the foundation for the preparation of GA/SA DN hydrogels, the pure GA and SA hydrogels were investigated preliminarily. SA-MA by reacting sodium alginate with methacrylic anhydride was prepared and confirmed via 1H NMR spectrum, where the characteristic peaks assigned to methacrylic anhydride could be clearly observed (Fig. S1†). Then SA-MA (2 wt%, 3 wt%, 4 wt%, 5 wt% in aqueous solution) and photoinitiator were homogeneously mixed, light yellow SA hydrogels could be obtained after a facile photopolymerization process (Fig. S2a†). In addition, herbal GA molecules have been reported to self-assemble into hydrogels through a simple heating–cooling process.31 The GA molecules could yield a head-to-head configuration due to lateral hydrophobic interactions between the hydrophobic moieties and form nanofibrils at low concentration. The increase of GA concentration enabled strong interfibrillar interactions triggered by hydrogen bonds, leading to further entanglement of GA nanofibrils to form supramolecular hydrogel network. In our work, 1 wt%, 2 wt%, and 3 wt% GA solutions were tested to clarify their gelation behaviors. As shown in Fig. S3a,† 1 wt% GA remained a solution state while the others generated stable transparent hydrogels. In addition, several experiments were conducted to evaluate the influence of SA-MA macromonomers and photoinitiators (e.g., eosin Y, 1-vinyl-2-pyrrolidinone and triethanolamine) on the self-assembly behavior of GA (Table S1†). It could be seen from the Fig. S4† that hydrogels formed under all circumstances with variable gelation times. The rheological measurement revealed a lower stiffness of GA hydrogel after the addition of above compounds (Fig. S5†). In general, the self-assembly of GA was well retained in the presence of SA-MA and photoinitiators. In terms of the stability of SA-MA during the heating–cooling process, the as-prepared SA-MA and SA-MA sample after heating–cooling for three cycles were tested by 1H NMR. Fig. S6† depicts almost identical characteristic peaks, revealing that no change of SA-MA molecule structure occurred. On this basis, we designed a one-pot two-step fabrication process for the GA/SA DN hydrogels (Fig. 1). Briefly, GA, SA-MA, and the photoinitiator were all added into water, heated at 80 °C until fully dissolved, and then allowed to stand 15 minutes. In this way, GA hydrogel containing uniformly distributed SA-MA and photoinitiator was obtained (Fig. S3b†). After that, the GA hydrogel further underwent a photopolymerization process to achieve GA/SA DN hydrogel (Fig. S2b†). As shown in Fig. 2a, the as-prepared GA/SA DN hydrogel could be twisted and bended to form a circular ring, suggesting the excellent flexibility of hydrogels. A thin GA/SA DN hydrogel could withstand a weight of 50 g without cracking (Fig. 2b). In addition, owing to the good compatibility between GA and SA hydrogel, the GA/SA DN hydrogel remained highly transparent (Fig. 2c).
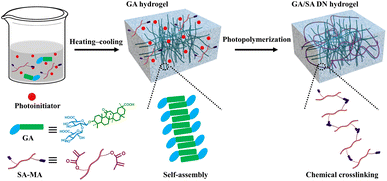 |
| | Fig. 1 Schematic illustration of the preparation process of the GA/SA DN hydrogels. | |
 |
| | Fig. 2 Photos showing (a) the GA/SA DN hydrogels during twisting and bending, (b) the lifting weight of GA/SA DN hydrogel, and (c) the transparency of GA/SA DN hydrogel and SA hydrogel. | |
SEM characterizations were conducted to investigate the micromorphologies of GA, SA, and GA/SA DN hydrogels. Fig. 3a–d displays SEM images of SA hydrogels showing variable porous structures. With the increase of SA-MA concentration from 2 wt% to 5 wt%, the porous structure gradually became inconspicuous, corresponding to the denser cross-linking network. As depicted in Fig. 3e, the 2 wt% GA hydrogel exhibited discrete thin layers, which was consistent with previous report that high concentration led to the formation of thin layers composed of highly aligned GA fibrils.31 In the circumstance of GA/SA DN hydrogels, the second SA hydrogel network should be affected by GA hydrogel to a large extent. In detail, a low concentration of SA-MA could not produce regular second network due to the steric hindrance effect of GA hydrogel (Fig. 3f). Particularly, for the critical content of 3 wt% SA-MA, the SA and GA hydrogels were comparable to each other. With this balance, GA/SA3 DN hydrogel network characterized by both distinguishable GA layers and homogeneous porous structure formed (Fig. 3g). A slight increase of SA-MA content in the GA/SA4 endowed similar inner structures with that of GA/SA3 hydrogel except that the pore size remarkably decreased (Fig. 3h). On the contrary, high loading amount of SA-MA made it dominate in the competition for space, resulting in the embedment of GA network into SA hydrogel (Fig. 3i).
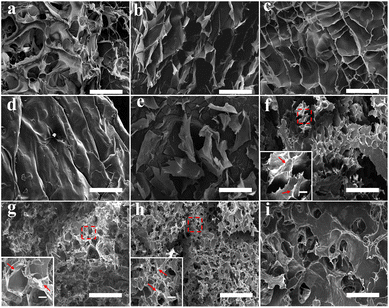 |
| | Fig. 3 SEM images of the hydrogel micromorphologies: (a) SA2 hydrogel, (b) SA3 hydrogel, (c) SA4 hydrogel, (d) SA5 hydrogel, (e) GA hydrogel, (f) GA/SA2 DN hydrogel, (g) GA/SA3 DN hydrogel, (h) GA/SA4 DN hydrogel, and (i) GA/SA5 DN hydrogel. The scale bars were 200 μm while that of the magnified images were 20 μm. | |
3.2 Mechanical properties of GA/SA DN hydrogels
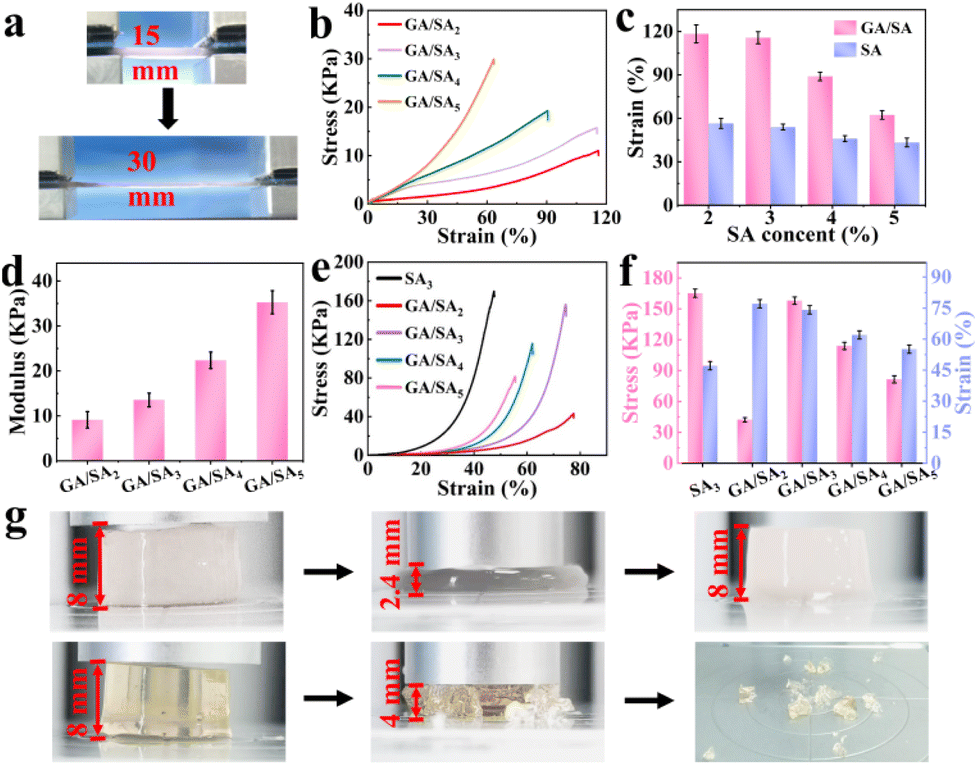
As shown in Fig. 4a, the GA/SA3 DN hydrogel could be stretched up to two times of its original length. Then the tensile stress–strain curves of SA and GA/SA hydrogels with different SA contents were measured to quantitatively evaluate their mechanical properties (Video S1†). With the increase of SA-MA content, the strength and modulus of the SA hydrogels increased continuously while the breaking elongation decreased from 56.7% to 44.5% (Fig. S7†). Not surprisingly, similar tendency was found in the GA/SA DN hydrogels (Fig. 4b). Meanwhile, the corresponding fracture strains of DN hydrogels all enlarged compared to their analogues without the addition of GA (Fig. 4c). For example, the GA/SA2 and GA/SA3 hydrogels exhibited the highest breaking elongation of 114.5%, which was two times that of the original SA hydrogels. Fig. 4d shows that the elastic modulus of DN hydrogels were in an acceptable level ranging from 9.1 kPa to 35.3 kPa. In contrast, the G′ and G′′ values of 2 wt% GA hydrogels were 958 Pa and 208 Pa respectively (Fig. S5a†), and its critical strain value was 7.6% in the oscillatory strain sweep test. Thus, the formation of DN hydrogel not only brought about superior stretchability to pure SA hydrogel, but also endowed significantly enhanced robustness compared with GA hydrogel. Apart from stretchability, Fig. 4e depicts the typical compressive stress–strain curves of SA3 and GA/SA DN hydrogels. With the content of SA increased from 2 to 3 wt%, their critical compressive strains were similar, being 77.5% and 74.4% respectively. Surprisingly, the compression stress of GA/SA3 DN hydrogel reached 156.1 kPa while the value of GA/SA2 hydrogel was just 42.5 kPa (Fig. 4f). The further addition of SA caused a marked decline in both compressive stress and compressive strain for GA/SA4 and GA/SA5 hydrogels. In comparison with SA3 hydrogel, the GA/SA3 DN hydrogel exhibited a much higher compressive strain while its compressive stress was slightly lower (Fig. 4g, Videos S2 and S3†). Hence, the GA/SA3 DN hydrogel owned the highest compression stress and near maximum compression strain, which could be attributed to its homogeneous DN microstructures observed in SEM image. All above results showed that the GA/SA3 DN hydrogel had excellent tensile and compression properties, beneficial for the strain and pressure sensing. Thus, it was chosen as the matrix material in the following study unless otherwise indicated.
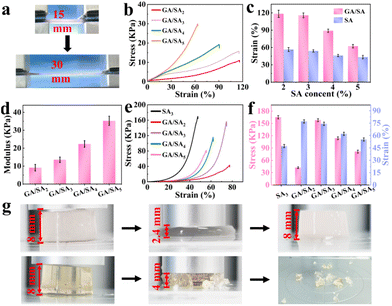 |
| | Fig. 4 (a) Photos showing the stretching of GA/SA DN hydrogel, (b) tensile curves of the GA/SA DN hydrogels, (c) tensile strain of the GA/SA DN hydrogels and SA hydrogels as a function of SA content, (d) elastic modulus of the GA/SA DN hydrogels, (e) compressive curves of the GA/SA DN hydrogels and SA3 hydrogel, (f) the corresponding compression stress and strain values, and (g) photos showing the compression behaviors of GA/SA3 DN hydrogel (top) and SA3 hydrogel (bottom). | |
3.3 Adhesive properties of GA/SA DN hydrogels
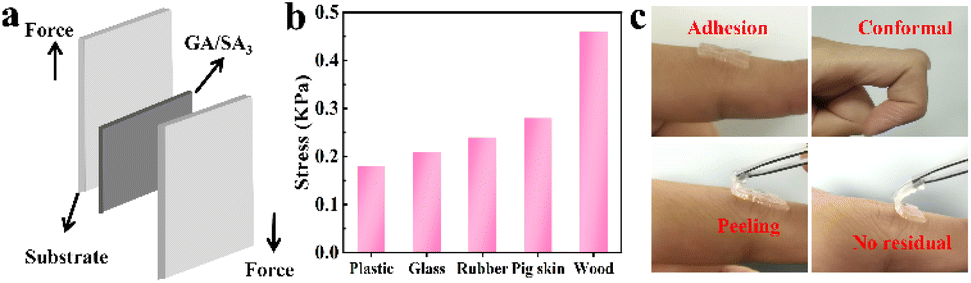
First, qualitative test was performed to figure out the adhesive properties of GA/SA3 DN hydrogel on different materials. As illustrated in Fig. S8,† the hydrogel could not only adhere to hydrophilic materials like glass and paper, also exhibited self-adhesion to hydrophobic substances including rubber, plastic, and polytetrafluoroethylene. In addition to showing adhesion to flat surfaces, the hydrogel also adhered to cambered surface. The adhesion of GA/SA3 DN to various materials is beneficial in practical applications. Fig. 5a displays a schematic diagram of 180° lap-shear test, aiming to figure out the adhesion strengths of GA/SA3 DN hydrogel to typical substrates. As shown in Fig. 5b, the adhesion strengths to plastic, glass, rubber, pig skin, and wood were 0.18, 0.21, 0.24, 0.28, and 0.46 kPa, respectively. These results indicated that the GA/SA3 DN hydrogels possessed moderate adhesion capability on different substrates, especially the skin surface. This level of adhesion strength enabled the hydrogel to self-adhere to the human finger joint and even persist conformal adhesion when bending the finger at 90° (Fig. 5c). Meanwhile, the peeling off from human skin without any residual left could be realized.
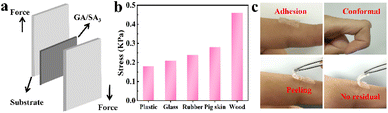 |
| | Fig. 5 (a) Schematic illustration of the lap-shear test, (b) corresponding adhesion strengths of GA/SA3 DN hydrogel toward different substrates, and (c) photos showing the conformal adhesion and peeling off demonstration of the GA/SA3 DN hydrogel on human finger skin. | |
3.4 Strain sensing performance
The presence of large amount of Na+ rendered ionic conductivity to the SA hydrogel, which was 0.06 S m−1 at a SA-MA content of 2 wt% (Fig. S9†). However, with the increase of SA-MA, the much denser hydrogel network significantly inhibited the ion migration, leading to gradually decreased conductivities. In the GA/SA DN hydrogels, GA could provide NH4+ as free ions to further enhance the ionic conductivity. For GA/SA2 DN hydrogel, its value reached 0.12 S m−1, more than two times that of SA2 hydrogel. However, the GA/SA3 DN hydrogel achieved the highest ionic conductivity of 0.14 S m−1, resulting from the homogeneous porous structure that could promote ion transport.
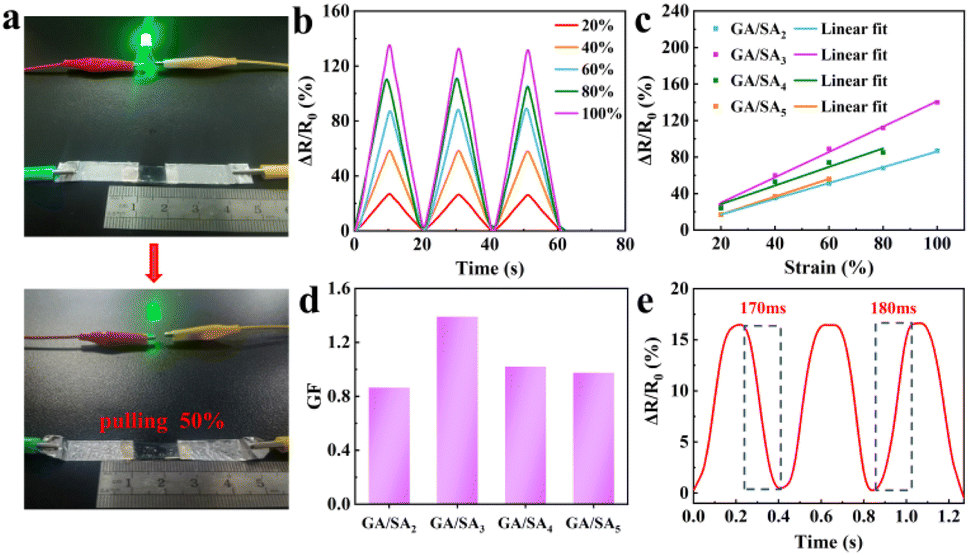
As shown in Fig. 6a, a GA/SA3 DN hydrogel film was connected to complete circuit and found to light up a light-emitting diode bulb. When the stretching strain reached 50%, the brightness of the bulb became obviously darkened, suggesting a strain-dependent sensing property. Fig. 6b and S10† showed the relative resistances under different strain states for GA/SA DN hydrogels. Noticeably, quasi-linear relationship of ΔR/R0 values as a function of the applied strain was found for all DN hydrogels (Fig. 6c). According to the linear fitting, the gauge factors (GFs) of GA/SA2, GA/SA3, GA/SA4, and GA/SA5 hydrogels were 0.87 (R2 = 0.998), 1.39 (R2 = 0.995), 1.01 (R2 = 0.943) and 0.98 (R2 = 0.944), respectively (Fig. 6d). Clearly, the strain sensing property of GA/SA3 DN hydrogel was superior to others, corresponding to its outstanding microstructures, mechanical characteristics, and ionic conductivity. The sensing linearity can be beneficial for obtaining precise detection results in real strain sensing applications. Besides, the GA/SA3 DN hydrogel owned the relaxation times of 170 and 180 ms respectively (Fig. 6e), indicating a rapid response in strain sensing.
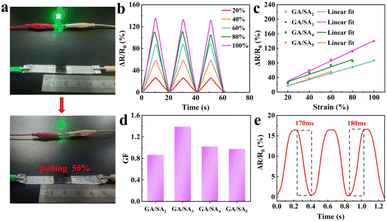 |
| | Fig. 6 (a) Digital photographs of the brightness of a LED bulb in response to the 50% strain change of the GA/SA3 DN hydrogel, (b) relative resistance change of the GA/SA3 DN hydrogel under different strains, (c) the relative resistance changes of GA/SA DN hydrogels as a function of strain, (d) their corresponding GF values, and (e) the response and relaxation times of GA/SA3 DN hydrogel. | |
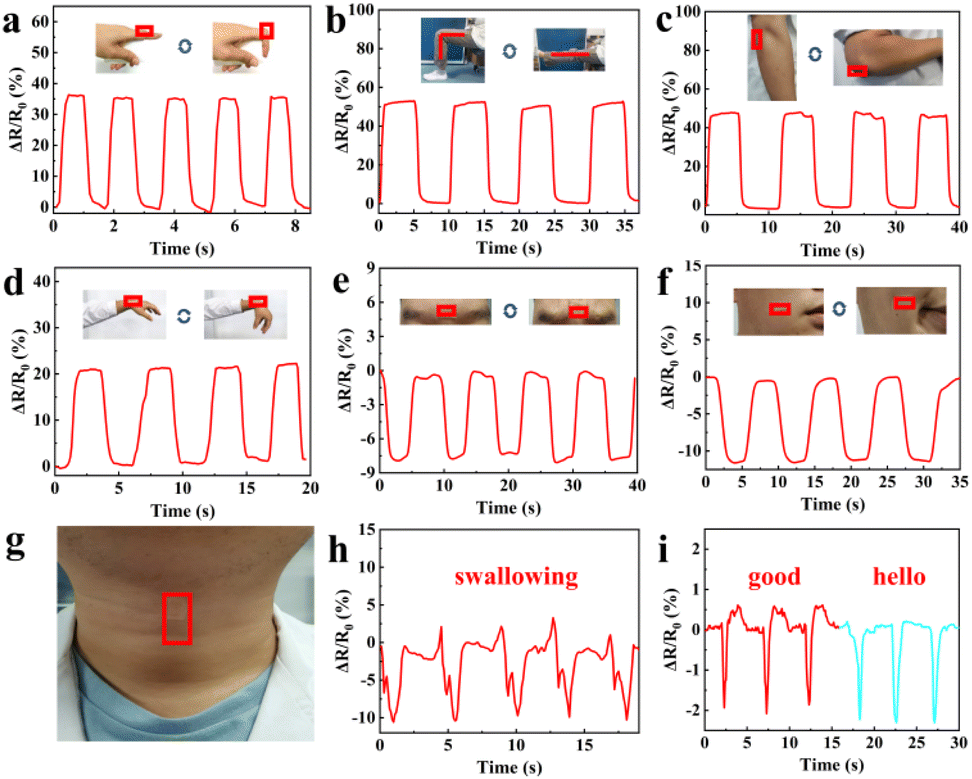
Since the GA/SA3 DN hydrogel displayed superior mechanical property, conformal adhesion to the human skin, and good strain sensing sensitivity, it was suitable candidate for detecting human motions. Therefore, the GA/SA3 DN hydrogel film as a sensor was directly attached to different body joints without the assistance of adhesive tape, and the corresponding resistance changes were recorded. As shown in Fig. 7a–d, the repetitive bending movements of forefinger, knee, elbow, and wrist could yield stable signals with distinguishable pattern and amplitude. For instance, the hydrogel that adhered on finger joint was repeatedly stretched and relaxed during its cyclic bending at the angle of 0 and 90°. Thus, the hydrogel sensor in stretched state generated a ΔR/R0 value of 35%, which could be restored to 0% when the finger became straightened (Fig. 7a). In addition to large scale human joint movements, the GA/SA3 DN hydrogel also held the capability to monitor the subtle motions. When the volunteer frowned and smiled, the sensor could respond sensitively to such facial expressions, endowing stable and repeatable electrical signals (Fig. 7e and f). Moreover, swallowing and speaking as typical complicated and tiny throat movements were tested (Fig. 7g). In the scene of swallowing, complex but discernible monitoring curve was obtained with acceptable amplitude (Fig. 7h). Saying the words of “good” and “hello” could give different curve features though the similar syllable and pronunciation (Fig. 7i), further confirming that the GA/SA3 DN hydrogel possessed high sensitivity in monitoring subtle human motions.
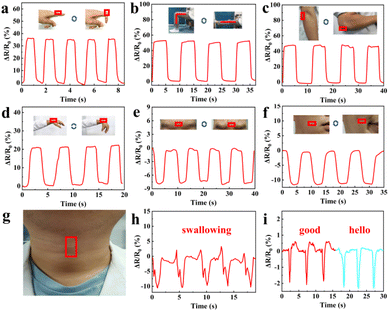 |
| | Fig. 7 Resistance changes of the GA/SA3 DN hydrogel responding to (a) finger bending, (b) knee bending, (c) elbow bending between 0 and 90°, (d) wrist bending and releasing, and monitoring of facial expressions of (e) frowning and (f) smiling. (g) Photo showing the GA/SA3 DN hydrogel film adhered on the throat without assistance, and corresponding detection of (h) swallowing and (i) speaking. | |
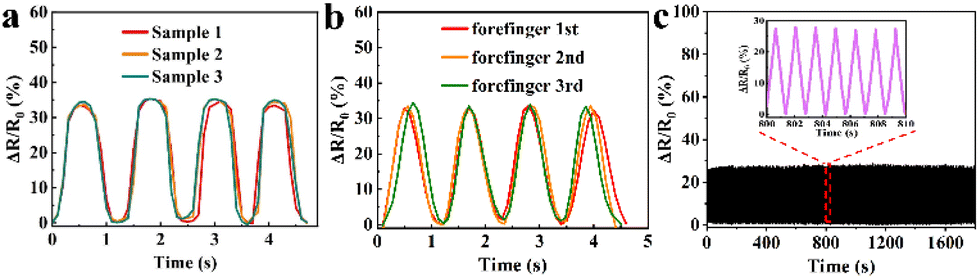
To validate the reproducibility of GA/SA3 DN hydrogel sensor, three hydrogel samples were prepared independently and tested in the finger movements. In addition, one hydrogel sensor was successively tested on the forefinger, middle finger, and forefinger again under same finger bending states. The GA/SA3 DN hydrogel sensors in both scenarios yielded almost identical relative resistance values (Fig. 8a and b), demonstrating their good reproducibility. In addition, the GA/SA3 DN hydrogel was adhered onto the surface of a balloon to perform cyclic stability measurement by utilizing a stepping machine to periodically extrude the balloon. Fig. 8c represents the monitoring result of the loading/unloading strain for about 1300 times, where the stable signals proved the excellent reproducibility of GA/SA3 DN hydrogel sensor.
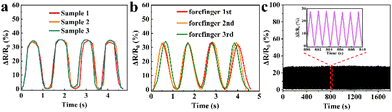 |
| | Fig. 8 The reproducibility demonstration of GA/SA3 DN hydrogel sensors by (a) three samples of the same batch, (b) three independent tests on human forefinger, and (c) cyclic stability measurement. | |
3.5 Pressure sensing performance
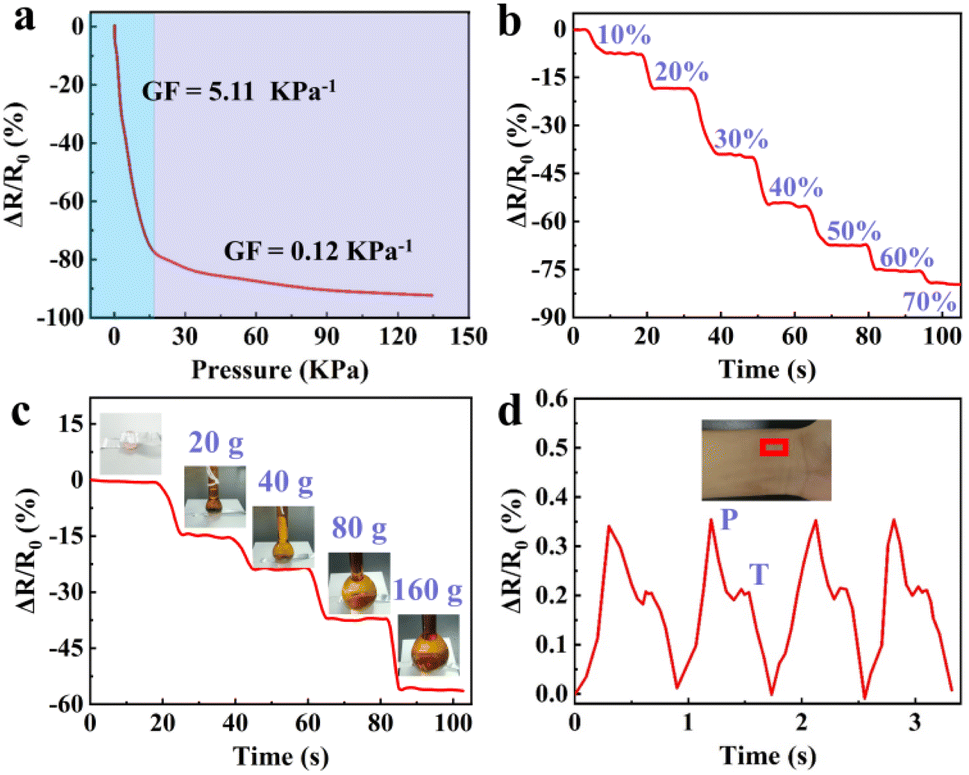
Besides the strain sensing property, the GA/SA3 DN hydrogels could also serve as pressure sensors owing to their excellent compression behavior. The real-time resistance changes of the GA/SA3 DN hydrogel to successive compressive pressure were recorded, as shown in Fig. 9a, where two regions with different slopes emerged. The GF value was up to 5.11 kPa−1 in the 0–16 pressure range while it decreased to 0.12 kPa−1 when the compressive pressure varied from 16 to 135 kPa. According to the compressive stress–strain curve (Fig. 4f), the continuously increasing pressure could not enable the GA/SA3 DN hydrogel to deform linearly, thus leading to the nonlinear relative resistance change under pressure. Besides, the relaxation times of 190 and 170 ms of GA/SA3 DN hydrogel in pressure sensing revealed a quick response as demonstrated in strain sensing (Fig. S11†). Furthermore, there was a stepwise change in the relative resistance as the increasing compressive strain from 0 to 70% (Fig. 9b). On this basis, we tested the pressure sensing performance of the GA/SA3 DN hydrogel by adding heavy weights. Fig. 9c illustrates that the increase of applied weight on the hydrogel from 0 to 160 g resulted in a corresponding gradual resistance change to 56.2%. As a health indicator for cardiovascular conditions, the pulse wave signals were also investigated, aiming to justify the potential practical application of hydrogel pressure sensor. Distinguishable and stable pulse peaks, about 70 times per min, could be observed in Fig. 9d, which were assigned to percussion wave (P) and tidal wave (T) respectively.39,40 The precise detection of pulse waves with good stability suggested the potential of the GA/SA3 DN hydrogels as pulse monitoring device.
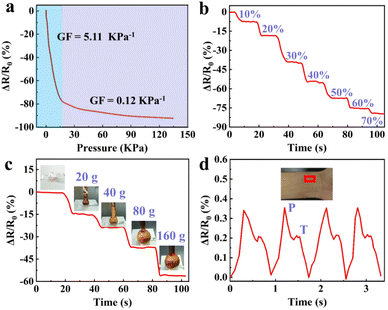 |
| | Fig. 9 (a) GF factors of the GA/SA3 DN hydrogel under different compressive pressures, and its relative resistance change (b) under stepped compression to 70% strain, (c) response to different weights, and (d) during the detection of pulse waves. | |
3.6 Antibacterial property
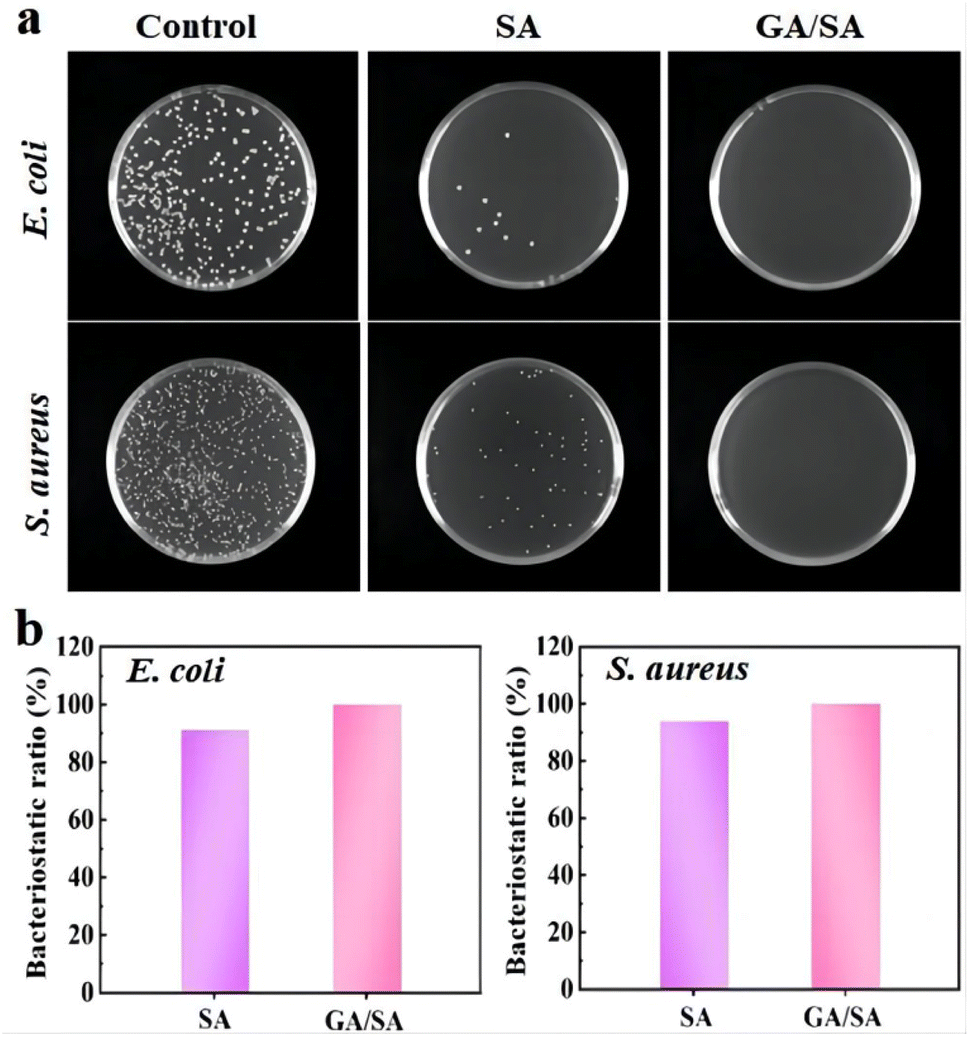
The antibacterial ability is a crucial factor for the hydrogel-based sensors, which should necessarily contact with human skin and even the surface of organs. Therefore, the antibacterial activities of the SA3 and GA/SA3 DN hydrogels were studied against Gram-negative E. coli and Gram-positive S. aureus. In this study, bacterial stock solution was inoculated onto the surfaces of SA3 and GA/SA3 DN hydrogels and incubated at 37 °C for 18 h. The survived bacteria on the hydrogel sample were washed out and then cultured on solid Luria–Bertani medium at 37 °C for another 18 h. It can be observed from Fig. 10a that the amounts of colonies of E. coli and S. aureus both obviously decreased in the case of SA3 hydrogel while no visible colonies were observed for the GA/SA3 DN hydrogel. The bacteriostatic ratios of two hydrogels against E. coli and S. aureus were calculated and displayed in Fig. 10b. The bacteriostatic ratios of SA3 hydrogel reached 91.2% for E. coli and 94.0% for S. aureus, respectively. Considering that the contact with bacteria has been reported to be pivotal for the hydrogels to show antibacterial activity, this decent antibacterial property of SA3 hydrogel can be mainly attributed to the presence of methacrylic acid groups and its direct contact with bacteria in this work.33,41,42 Significantly, the GA/SA3 DN hydrogel exhibited high bacteriostatic ratios beyond 99.9% against E. coli and S. aureus bacteria benefiting from the synergetic antibacterial effect of SA and GA hydrogels. These results demonstrated excellent antibacterial ability of GA/SA3 DN hydrogels.
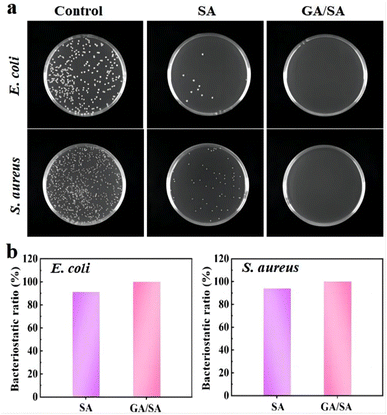 |
| | Fig. 10 (a) Photograph of E. coli and S. aureus on the Luria–Bertani agar plates of the control group, SA3 hydrogel and GA/SA3 DN hydrogel, and (b) bacteriostatic ratios of the SA3 and GA/SA3 DN hydrogels against E. coli and S. aureus. | |
4. Conclusion
In summary, a DN hydrogel was constructed by self-assembly of herbal GA molecules and photopolymerization of SA-MA, which possessed flexibility, transparency, and conformal adhesion on human skin. Specially, the GA/SA DN hydrogel exhibited superior mechanical property and ionic conductivity compared to either GA or SA hydrogel. Thanks to these merits, the DN hydrogel could serve as strain sensor to detect human motions with high sensitivity, fast response, and good reproducibility. When used as pressure sensor, it was demonstrated to be effective in monitoring applied heavy weights and even subtle pulse signals. In addition, benefiting from the synergy of GA and SA hydrogels, the GA/SA DN hydrogel exhibited excellent antibacterial property toward E. coli and S. aureus. Overall, this study could provide new ideas to develop multifunctional herbal molecules-based DN hydrogels for fabricating flexible sensors.
Author contributions
Conceptualization: Lei Shi and Hao Sun, methodology: Qiaoqiao Zhao, data curation: Hao Sun, writing—original draft preparation: Lei Shi, writing—review and editing: Xinyu Kou and Zhibin Dong, project administration: Yuning Ma, funding acquisition: Yuxia Ma. All authors have read and agreed to the published version of the manuscript.
Conflicts of interest
The authors declare no conflict of interest.
Acknowledgements
This research was funded by the general program of Shandong Natural Science Foundation (No. ZR2021MH373), the Joint Fund of Natural Science Foundation of Shandong Province (No. ZR2021LZY044), Jinan “GaoXiao 20 Tiao” Funding Project Contract (No. 2020GXRC005), and Qilu Health Leading Talent Project.
Notes and references
- K. C. Xu, Y. Y. Lu and K. Takei, Adv. Mater. Technol., 2019, 4, 1800628 CrossRef.
- B. Zazoum, K. M. Batoo and M. A. A. Khan, Sensors, 2022, 22, 4653 CrossRef PubMed.
- T. Qin, W. C. Liao, L. Yu, J. H. Zhu, M. Wu, Q. Y. Peng, L. B. Han and H. B. Zeng, J. Polym. Sci., 2022, 60, 2607–2634 CrossRef CAS.
- S. H. Lin, C. M. Papadakis, J. J. Kang, J. M. Lin and S. H. Hsu, Chem. Mater., 2021, 33, 3945–3958 CrossRef CAS.
- H. Liu, M. X. Li, O. Y. Chen, T. J. Lu, F. Li and F. Xu, Small, 2018, 14, 1801711 CrossRef PubMed.
- Z. Chen, Y. J. Chen, M. S. Hedenqvist, C. Chen, C. Cai, H. Li, H. Z. Liu and J. Fu, J. Mater. Chem. B, 2021, 9, 2561–2583 RSC.
- X. Y. Li, J. Wang, Y. K. Lin, Y. Q. Cheng, W. J. Han, G. L. Yuan and H. B. Jia, Colloids Surf., A, 2022, 635, 128091 CrossRef CAS.
- X. B. Li, L. Z. He, Y. F. Li, M. Y. Chao, M. K. Li, P. B. Wan and L. Q. Zhang, ACS Nano, 2021, 15, 7765–7773 CrossRef CAS PubMed.
- W. S. Zhang, L. X. Xu, M. J. Zhao, Y. N. Ma, T. Zheng and L. Shi, Soft Matter, 2022, 18, 1644–1652 RSC.
- A. Haleem, S. B. Syaal, M. Ajmal, J. Ambreen, S. Rauf, N. Ali, S. Muhammad, A. Shah, A. B. Zia and M. Siddiq, Korean J. Chem. Eng., 2020, 37, 614–622 CrossRef CAS.
- S. Zahid, A. K. Alzahrani, N. Kizilbash, J. Ambreen, M. Ajmal, Z. H. Farooqi and M. Siddiq, RSC Adv., 2022, 51, 33215–33228 RSC.
- J. Ambreen, F. F. A. Harbi, H. Sakhawat, M. Ajmal, H. Naeem, Z. H. Farooqi, N. Batool and M. Siddiq, J. Mol. Liq., 2022, 355, 118931 CrossRef CAS.
- K. Naseem, Z. H. Farooqi, R. Begum, W. Wu, A. Irfand and M. Ajmal, Colloids Surf., A, 2020, 594, 124646 CrossRef CAS.
- J. P. Gong, Y. Katsuyama, T. Kurokawa and Y. Osada, Adv. Mater., 2003, 15, 1155–1158 CrossRef CAS.
- X. J. Ning, J. N. Huang, Y. A, N. N. Yuan, C. Chen and D. H. Lin, Int. J. Mol. Sci., 2022, 23, 15757 CrossRef CAS PubMed.
- X. X. Huang, J. C. Li, L. Luo, Q. Gao, A. Mao and J. Z. Li, Mater. Today Commun., 2021, 29, 102757 CrossRef CAS.
- H. D. Zhang, H. Shen, J. N. Lan, H. Wu, L. J. Wang and J. P. Zhou, Carbohydr. Polym., 2022, 295, 119848 CrossRef CAS PubMed.
- Q. Diao, H. Y. Liu and Y. Y. Yang, Gels, 2022, 8, 424 CrossRef CAS PubMed.
- S. J. Liu and L. Li, ACS Appl. Mater. Interfaces, 2017, 9, 26429–26437 CrossRef CAS PubMed.
- X. Liu, Z. J. Ren, F. F. Liu, L. Zhao, Q. J. Ling and H. B. Gu, ACS Appl. Mater. Interfaces, 2021, 13, 14612–14622 CrossRef CAS PubMed.
- P. Rahmani and A. A. Shojaei, Adv. Colloid Interface Sci., 2021, 298, 102553 CrossRef CAS PubMed.
- T. X. Zhu, Y. M. Ni, G. M. Biesold, Y. Cheng, M. Z. Ge, H. Q. Li, J. Y. Huang, Z. Q. Lin and Y. K. Lai, Chem. Soc. Rev., 2023, 52, 473–509 RSC.
- H. Lei, J. Zhao, X. X. Ma, H. Li and D. D. Fan, Adv. Healthcare Mater., 2021, 10, 2101089 CrossRef CAS PubMed.
- Z. H. Li, W. L. Xu, X. H. Wang, W. Q. Jiang, X. L. Ma, F. J. C. Wang, C. L. Zhang and C. G. Ren, Eur. Polym. J., 2021, 146, 110253 CrossRef CAS.
- J. H. Hwang, Y. L. Cha, L. Ramos, T. Y. Zhu, L. B. Kurnaz and C. B. Tang, Chem. Mater., 2022, 34, 5663–5672 CrossRef CAS.
- H. Zhang, N. Tang, X. Yu, Z. K. Guo, Z. Liu, X. M. Sun, M. H. Li and J. Hu, Chem. Eng. J., 2022, 430, 132779 CrossRef CAS.
- L. Y. Zhang, C. C. Wan, J. H. Su, C. H. Zhang, S. Wei, W. Y. Tian, X. Y. Liu, W. J. Cheng, X. G. Li, X. J. Li, X. Guo, K. T. Yong and Y. Q. Wu, Mater. Des., 2022, 215, 110464 CrossRef CAS.
- P. V. AshaRani, G. L. K. Mun, M. P. Hande and S. Valiyaveettil, ACS Nano, 2009, 3, 279–290 CrossRef CAS PubMed.
- X. H. Zhang, J. J. Wei, S. C. Lu, H. Xiao, Q. X. Miao, M. Zhang, K. Liu, L. H. Chen, L. L. Huang and H. Wu, ACS Appl. Polym. Mater., 2021, 3, 5798–5807 CrossRef CAS.
- L. A. Stecanella, A. P. R. Bitencourt, G. R. Vaz, E. Quarta, J. O. C. S. Júnior and A. Rossi, Pharmaceutics, 2021, 13, 1792 CrossRef CAS PubMed.
- A. Saha, J. Adamcik, S. Bolisetty, S. Handschin and R. Mezzenga, Angew. Chem., 2015, 127, 5498–5502 CrossRef.
- E. A. H. Mohammed, Y. Peng, Z. Wang, X. Qiang and Q. Zhao, Russ. J. Bioorg. Chem., 2022, 48, 906–918 CrossRef CAS PubMed.
- K. Sharma, V. Kumar, C. Swart-Pistor, B. Chaudhary and H. C. Swart, J. Bioact. Compat. Polym., 2017, 32, 74–91 CrossRef CAS.
- S. Mageswari and K. Subramanian, Polym.-Plast. Technol. Eng., 2012, 51, 1296–1302 CrossRef CAS.
- A. Kirillova, R. Maxson, G. Stoychev, C. T. Gomillion and L. Ionov, Adv. Mater., 2017, 29, 1703443 CrossRef PubMed.
- D. J. Chen, X. L. Zhao, H. Gao, G. L. Ren, J. N. Luo, H. X. Wang, C. Y. Zha, K. W. Yang and P. X. Jia, ACS Biomater. Sci. Eng., 2022, 8, 2624–2635 CrossRef CAS PubMed.
- A. Song, A. A. Rane and K. L. Christman, Acta Biomater., 2012, 8, 41–50 CrossRef CAS PubMed.
- L. Fan, J. L. Xie, Y. P. Zheng, D. X. Wei, D. D. Yao, J. Zhang and T. D. Zhang, ACS Appl. Mater. Interfaces, 2020, 12, 22225–22236 CrossRef CAS PubMed.
- B. Shen, J. Li, Y. T. Tang, H. H. Xu and F. Y. Li, Micromachines, 2021, 12, 789 CrossRef PubMed.
- M. Wu, J. S. Chen, Y. H. Ma, B. Yan, M. F. Pan, Q. Y. Peng, W. D. Wang, L. B. Han, J. F. Liu and H. B. Zeng, J. Mater. Chem. A, 2020, 8, 24718–24733 RSC.
- S. Rahman, F. F. A. Harbi, M. Ajmal, A. Naseem, Z. H. Farooqi and M. Siddiq, J. Mater. Sci., 2022, 57, 6763–6779 CrossRef CAS.
- M. Ajmal, S. Anwar, H. Naeem, M. A. Zia and M. Siddiq, Polym. Eng. Sci., 2020, 60, 2918–2929 CrossRef CAS.
|
| This journal is © The Royal Society of Chemistry 2023 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence *a,
Yuning Ma
*a,
Yuning Ma