
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSpirocitrinols A and B, citrinin derivatives with a spiro[chromane-2,3′-isochromane] skeleton from Penicillium citrinum†
Junjie Tong‡
ab,
Yang Zhang‡ c,
Yang Xub,
Yangyang Hanb,
Chuan Li*b,
Wenying Zhuangd and
Yongsheng Che*ab
c,
Yang Xub,
Yangyang Hanb,
Chuan Li*b,
Wenying Zhuangd and
Yongsheng Che*ab
aTianjin University of Traditional Chinese Medicine, Tianjin, 300193, People's Republic of China
bNHC Key Laboratory of Biotechnology of Antibiotics, Institute of Medicinal Biotechnology, Chinese Academy of Medical Sciences & Peking Union Medical College, Beijing, 100050, People's Republic of China. E-mail: ysche@imb.cams.cn; lichuan@imb.cams.cn
cState Key Laboratory of Toxicology & Medical Countermeasures, Beijing Institute of Pharmacology & Toxicology, Beijing, 100850, People's Republic of China
dState Key Laboratory of Mycology, Institute of Microbiology, Chinese Academy of Sciences, Beijing, 100101, People's Republic of China
First published on 20th February 2023
Abstract
Spirocitrinols A (1) and B (2), two new citrinin-derived metabolites possessing a spiro[chromane-2,3′-isochromane] skeleton, were isolated from cultures of Penicillium citrinum. Their structures were elucidated primarily by NMR experiments. The absolute configurations of 1 and 2 were assigned by electronic circular dichroism calculations. Compound 2 is the first naturally occurring trimeric citrinin derivative with a spiro[chromane-2,3′-isochromane] core. Compound 1 showed modest cytotoxicity against A549 human tumor cells.
Introduction
Citrinin is a typical polyketide mycotoxin first discovered from a Penicillium citrinum strain in 1931.1 Citrinin derivatives could be dimerized or trimerized to form highly complex polycyclic natural products with a variety of biological activities.2,3 This class of natural products are encountered as the secondary metabolites of fungi, especially among species of Penicillium sp. and Monascus sp. but also sporadically in Aspergillus sp. from both terrestrial and marine environments.4Benzannulated spiroketals are privileged scaffolds commonly encountered in natural products and drugs with the benzannulated 5,5-, 6,5-, and 6,6-spiroketal cores.5 While naturally occurring bisbenzannulated 6,6-spiroketals are categorized into two type of skeletons (Fig. 1), 2,2′-spirobi[chromane] and spiro[chromane-2,3′-isochromane]. To our knowledge, only a few precedents with a spiro[chromane-2,3′-isochromane] core have been reported as dimeric citrinin derivatives to date,6,7 two of which are xerucitrinic acids A and B isolated from the fungus Xerula sp. BCC56836,6 and xerucitrinins A–C identified from Penicillium citrinum.7
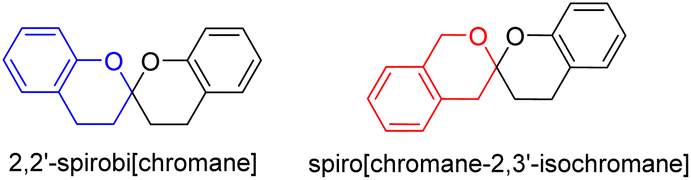 | ||
| Fig. 1 Two types of skeletons in naturally occurring bisbenzannulated 6,6-spiroketals. | ||
Penicillium citrinum has attracted much attention due to frequent discovery of structurally diverse and biologically active natural products from this fungal species.8 In efforts to identify new cytotoxic metabolites from the fungi of various habitats,9 a strain of P. citrinum isolated from a soil sample collected from the Yongxing Island, Hainan province, People's Republic of China, was subjected to a chemical investigation. Fractionation of an EtOAc extract prepared from the solid-substrate fermentation cultures led to the isolation of spirocitrinols A (1, Fig. 2) and B (2), two new citrinin-derived metabolites possessing a rare spiro[chromane-2,3′-isochromane] skeleton. Details of the structure elucidation, cytotoxicity evaluation, and plausible biogenesis of 1 and 2 are reported herein.
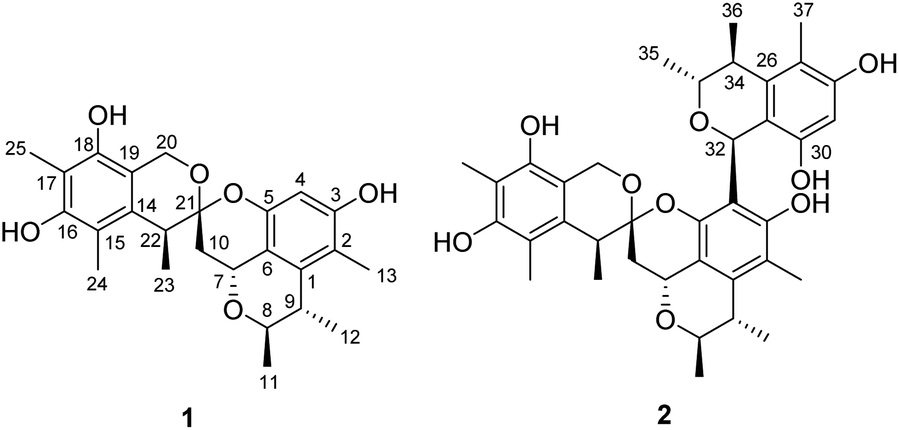 | ||
| Fig. 2 Structures of compounds 1 and 2. | ||
Results and discussion
Spirocitrinol A (1) had a molecular formula of C25H30O6 (11 degrees of unsaturation) as deduced from its HRESIMS and the NMR spectroscopic data (Table 1). Analysis of its NMR data revealed the presence of three exchangeable protons (δH 9.05, 7.83, and 7.88, respectively), six methyl groups, two methylene units (one oxygenated), four methines (two oxygenated), one doubly oxygenated sp3 quaternary carbon (δC 101.2), and 12 aromatic carbons with one protonated. These data accounted for all of the NMR resonances, suggesting that 1 was a pentacyclic compound. The 1H–1H COSY NMR data of 1 (Fig. 3) showed three isolated spin-systems, which were C-7–C-10, C-11–C-8–C-9–C-12, and C-22–C-23. The HMBC correlations from H-4 to C-2, C-3, C-5, and C-6, H-7 to C-1 and C-6, H-8 to C-7, and from H-9 to C-1 established an isochromane moiety, which was partially supported by the downfield chemical shifts of C-7 (δC 59.3) and C-8 (δC 74.2). The H3-11, H3-12, and H3-13 methyl groups were located at C-8, C-9, and C-2, respectively, by relevant HMBC cross peaks. While the exchangeable proton at 9.05 ppm was assigned to the free hydroxy group attached to C-3 based on the HMBC correlations from this proton (OH-3) to C-2, C-3, and C-4. Further correlations from H-7 to C-10 and C-21 led to the connection of C-10 to C-21, and those of H2-10 and H-22 with the doubly oxygenated tertiary carbon at 101.2 ppm enabled connection of C-21 to C-10 and C-22. In turn, the HMBC correlations from H2-20 and H-22 to C-14 and C-19, H3-24 to C-14, C-15, and C-16, and from H3-25 to C-16, C-17, and C-18 established the second polysubstituted aryl ring in 1. The remaining two exchangeable protons at 7.83 and 7.88 ppm were assigned to the free hydroxy groups located at C-16 and C-18, respectively, on the basis of relevant HMBC correlations. HMBC correlation from H2-10 to C-21 revealed the presence of the second isochromane unit in 1. Considering the unsaturation requirement of 1, the doubly oxygenated nature of C-21 (δC 101.2), and the chemical shift values for C-5 (δC 149.2) and C-20 (δC 60.8), the two C-21 bonded oxygen atoms were individually attached to C-5 and C-20 by default to complete the gross structure of 1 as shown. Compound 1 was found to be a stereoisomer of the known fungal metabolite xerucitrinin A,7 when comparison of its NMR data acquired in deuterated MeOH (Table S1†) with those of the known precedent.| No. | 1 | 2 | ||
|---|---|---|---|---|
| δCa, type | δHb (J in Hz) | δCa, type | δHb (J in Hz) | |
| a Recorded in Recorded in DMSO-d6 at 150 MHz.b Recorded in DMSO-d6 at 600 MHz.c These assignments are interchangeable. | ||||
| 1 | 136.5, qC | 134.0, qC | ||
| 2 | 114.5, qC | 113.8, qC | ||
| 3 | 155.6, qC | 153.1, qC | ||
| 4 | 99.9, qC | 5.91, s | 113.8, qC | |
| 5 | 149.2, qC | 147.6, qC | ||
| 6 | 110.7, qC | 112.7, qC | ||
| 7 | 59.3, CH | 4.77, dd (4.2, 12.0) | 60.4, CH | 4.72, brd (12.0) |
| 8 | 74.2, CH | 4.02, m | 74.2, CH | 4.02, m |
| 9 | 34.6, CH | 2.62, m | 34.3, CH | 2.58, m |
| 10a | 35.9, CH2 | 2.31, dd (4.8, 12.0) | 38.2, CH2 | 2.38, dd (4.2, 12.0) |
| 10b | 1.68, t (12.0) | 1.61, t (12.0) | ||
| 11 | 18.7, CH3 | 1.28, d (6.6) | 18.5, CH3 | 1.28, d (6.6) |
| 12 | 22.7, CH3 | 1.11, d (7.2) | 22.5, CH3 | 1.12, d (6.6) |
| 13 | 10.5, CH3 | 1.94, s | 10.3, CH3 | 1.85, s |
| 14 | 133.6, qC | 133.8, qC | ||
| 15 | 113.7, qC | 113.3, qC | ||
| 16 | 152.6, qC | 152.0, qC | ||
| 17 | 110.6, qC | 110.0, qC | ||
| 18 | 148.3, qC | 148.3, qC | ||
| 19 | 111.9, qC | 111.7, qC | ||
| 20a | 60.8, CH2 | 4.45, d (15.0) | 61.3, CH2 | 4.71, d (14.4) |
| 20b | 4.71, d (15.0) | 4.77, d (13.8) | ||
| 21 | 101.2, qC | 101.4, qC | ||
| 22 | 38.0, CH | 2.85, brd (7.2) | 38.6, CH | 2.92, t (6.6) |
| 23 | 18.6, CH3 | 1.05, d (6.6) | 17.9, CH3 | 1.07, d (6.6) |
| 24 | 11.0, CH3 | 2.01, s | 10.6, CH3 | 1.89, s |
| 25 | 9.9, CH3 | 2.04, s | 9.7, CH3 | 1.95, s |
| 26 | 138.5, qC | |||
| 27 | 112.9, qC | |||
| 28 | 155.4, qC | |||
| 29 | 100.8, CH | 6.11, s | ||
| 30 | 152.3, qC | |||
| 31 | 111.5, qC | |||
| 32 | 61.9, CH | 5.45, s | ||
| 33 | 73.0, CH | 3.67, m | ||
| 34 | 35.0, CH | 2.40, m | ||
| 35 | 16.1, CH3 | 0.50, d (6.6) | ||
| 36 | 20.3, CH3 | 1.21, d (6.6) | ||
| 37 | 10.4, CH3 | 1.92, s | ||
| 3-OH | 9.05, s | 5.90, s | ||
| 16-OH | 7.83, s | 7.59 c, s | ||
| 18-OH | 7.88, s | 7.80 c, s | ||
| 28-OH | 8.99 c, s | |||
| 30-OH | 8.75, s | |||
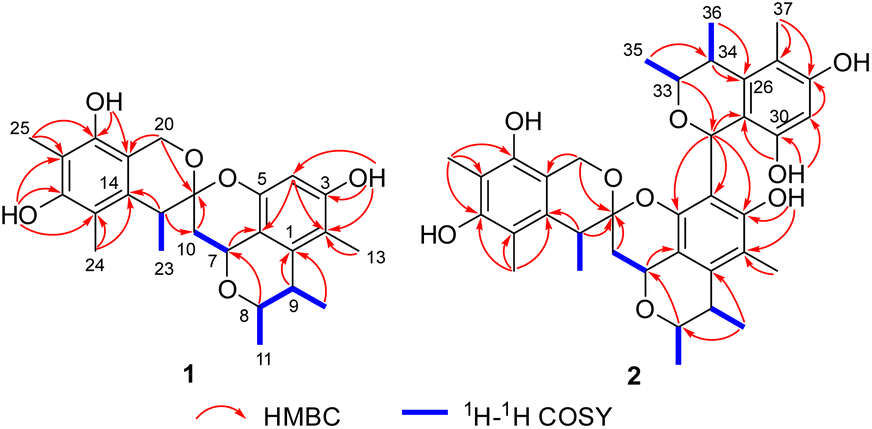 | ||
| Fig. 3 Key 1H–1H COSY and HMBC correlations of 1 and 2. | ||
The relative configuration of 1 was deduced by analysis of the 1H–1H coupling constants and NOESY data (Fig. 4). A coupling constant of 4.8 Hz observed between H-10a and H-7 suggested their cis relationship.6 The NOESY cross peaks of H3-11 with H-7 and H-9, and of H-10a with H-7 and H3-23 placed these protons on the same face of 3,3a,5,6-tetrahydro-2H-pyrano[2,3,4-de]chromene moiety, whereas the correlations of H-10b and H-22, and of H-8 with H3-12 revealed their spatial proximity, and suggested that they were on the opposite face of the ring system. On the basis of these data, the relative configuration of 1 was proposed as shown. The absolute configuration of 1 was deduced by comparison of the experimental and simulated electronic circular dichroism (ECD) spectra generated by time-dependent density functional theory (TDDFT), for enantiomers 7R,8R,9S,21R,22S-1 (1a) and 7S,8S,9R,21S,22R-1 (1b).10 The MMFF94 conformational search followed by reoptimaization using TDDFT at the B3LYP/6-31G(d) basis set level afforded the lowest-energy conformers for 1a (Fig. S14†). The overall calculated ECD spectra of 1a was then generated by Boltzmann weighting of their lowest-energy conformers by their relative free energies. The absolute configuration of 1 was then extrapolated by comparison of the experimental and calculated ECD spectra of 1a and 1b (Fig. 5). The experimental ECD spectrum of 1 was nearly identical to the calculated ECD curve of 7R,8R,9S,21R,22S-1 (1a), both showing two negative Cotton effects (CEs) in the ranges of 210–250 and 250–300 nm, respectively, suggesting that 1 has the 7R,8R,9S,21R,22S absolute configuration.
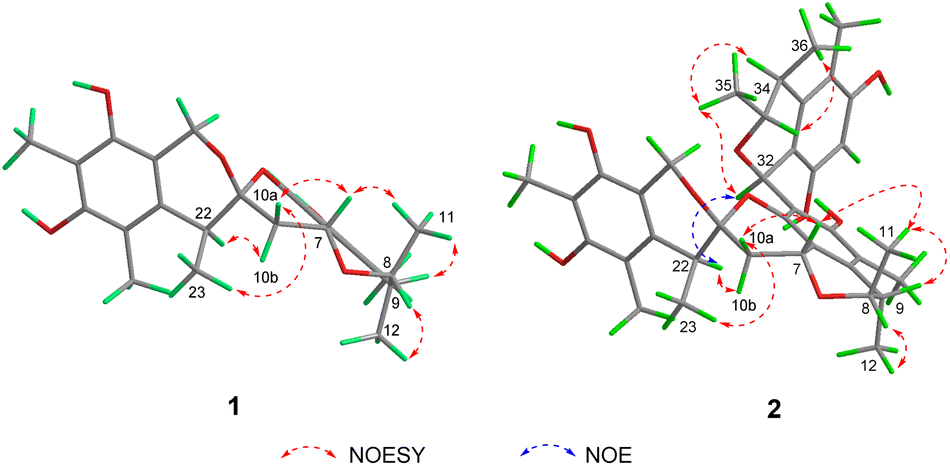 | ||
| Fig. 4 Key NOESY correlations of 1 and 2 and NOE correlation of 2. | ||
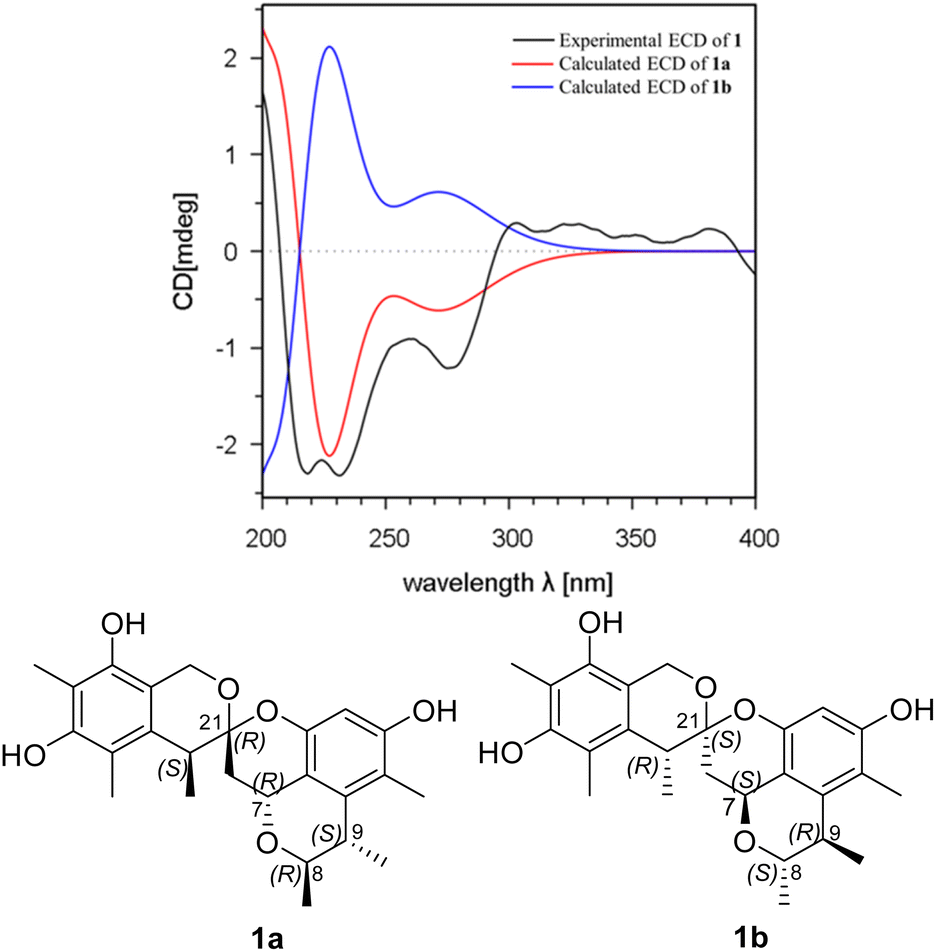 | ||
| Fig. 5 Experimental ECD spectrum of 1 in MeOH and the calculated ECD spectra of 1a and 1b. | ||
Spirocitrinol B (2) was determined to have a molecular formula of C37H44O9 (16 degrees of unsaturation) based on HRESIMS and NMR data (Table 1). Comparison of 1H and 13C NMR data with those of 1 indicated that there was a 4-substituted-1 substructure in 2. The remaining resonances for 2 indicated the presence of a known citrinin derivative, decarboxydihydrocitrinin,2a which was confirmed by interpretation of the 2D NMR data. The HMBC correlations from H-29 to C-27, C-28 and C-30, from H-32 to C-26 and C-31, from H-33 to C-32, and from H-34 to C-26 and C-31, plus the downfield chemical shifts of C-32 (δC 61.9) and C-33 (δC 73.0), established the third isochromane unit in 2. The H3-35, H3-36, and H3-37 methyl groups were located at C-33, C-34, and C-27, respectively, by relevant HMBC cross peaks. The exchangeable proton at 8.75 ppm was assigned to the free hydroxy group attached to C-30 based on the HMBC correlations from this proton (OH-30) to C-29, C-30, and C-31. Considering the chemical shift of C-28 (δC 155.4) and by comparison to the similar chemical shift of C-3 (δC 155.6) in 1, another free hydroxy group is required to attach to C-28, although no additional evidence was provided by the HMBC data. In addition, the HMBC correlations from H-32 to C-3, C-4, and C-5, established the C-4–C-32 linkage between the two units. Collectively, the planar structure of a novel trimeric citrinin derivative for 2 was tentatively assigned as shown. The relative configuration of the 4-substituted-1 substructure in 2 was determined to be the same as that of 1 by analysis of its coupling constant and NOESY data. The NOESY correlations of H3-35 with H-32 and H-34 indicated that these protons adopt the same orientation for the remaining 3,4,5-trimethylisochromane-6,8-diol unit. In addition, NOE correlation of H-22 with H-32 revealed their proximity in space, thereby allowing deduction of the relative configuration for 2. The absolute configuration of 2 was proposed by comparison of the experimental and calculated ECD spectra generated for enantiomers 2a and 2b (Fig. 6). MMFF94 conformational search followed by B3LYP/6-31G(d)DFT reoptimization afforded lowest-energy conformers (Fig. S15†). The calculated ECD spectra of 2a and 2b were then generated by Boltzmann weighting of their lowest-energy conformers. The experimental ECD spectrum of 2 matches the calculated ECD curve of 2a but is opposite to that of 2b, suggesting the 7R,8R,9S,21R,22S,32S,33R,34S configuration.
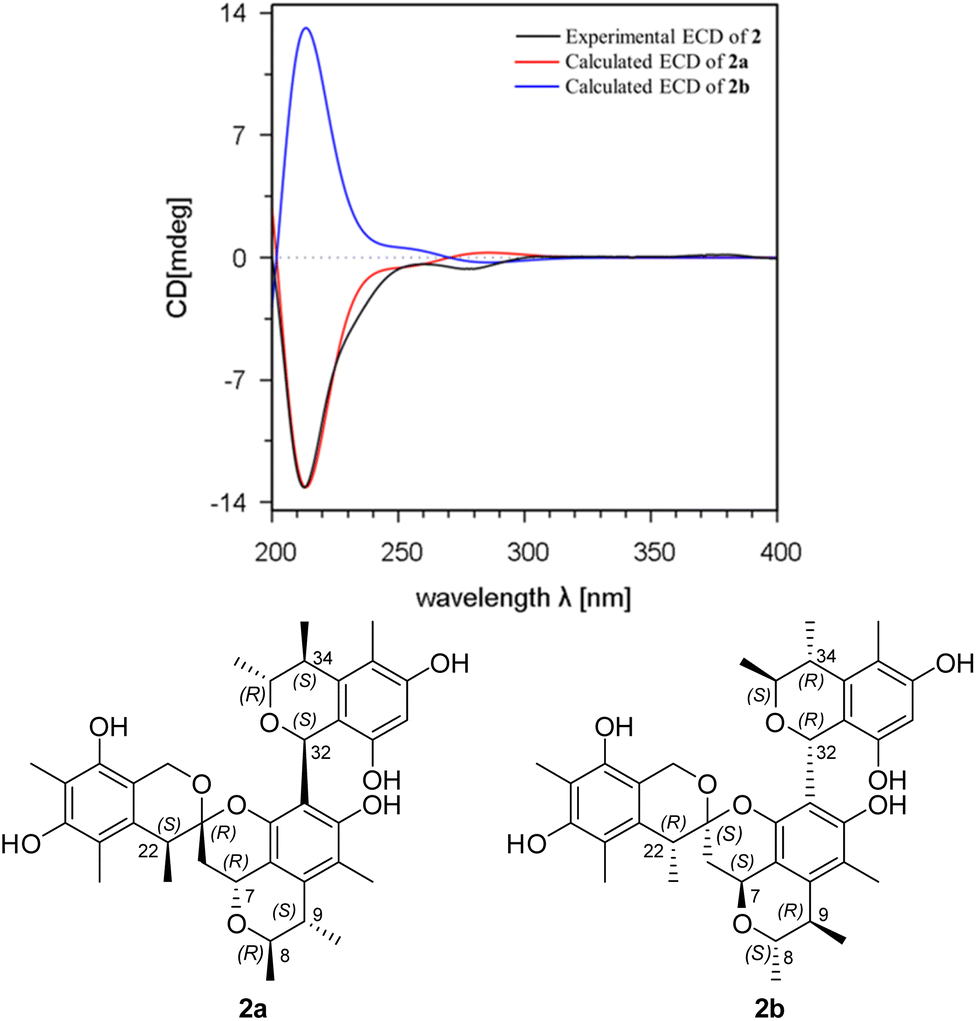 | ||
| Fig. 6 Experimental ECD spectrum of 2 in MeOH and the calculated ECD spectra of 2a and 2b. | ||
In vitro cytotoxic activity of compounds 1 and 2 was evaluate against seven tumor cell lines including SKBR3 (human breast cancer), HepG2 (human hepatoma), A549 (human lung cancer), J82 (human bladder carcinoma), MB49 (STR) (cisplatin resistant mouse bladder cancer), Huh7 (human hepatoma) cell lines, and MB49 (WT) (cisplatin sensitive mouse bladder cancer) (Table 2). Compound 1 showed modest cytotoxicity against A549 cells, with IC50 value of 4.3 ± 0.4 μM, compound 2 was cytotoxic to A549 cells, with IC50 value of 11.8 ± 2.8 μM, while the positive control cisplatin showed IC50 of 6.5 ± 1.0 μM.11,12
| Compounds | IC50 a(μM) | ||||||
|---|---|---|---|---|---|---|---|
| SKBR3 | HepG2 | A549 | J82 | MB49 (STR) | Huh7 | MB49 (WT) | |
| a IC50 values were averaged from at least three independent experiments.b Positive control. | |||||||
| 1 | 49.2 ± 3.3 | 41.6 ± 8.1 | 4.3 ± 0.4 | 45.6 ± 2.5 | 49.3 ± 7.8 | 48.2 ± 5.6 | 43.3 ± 4.5 |
| 2 | 48.1 ± 6.5 | 41.2 ± 6.3 | 11.8 ± 2.8 | 48.7 ± 2.8 | 55.7 ± 10.8 | 44.1 ± 7.9 | 64.5 ± 6.7 |
| Cisplatinb | 3.6 ± 0.6 | 4.8 ± 0.9 | 6.5 ± 1.0 | 0.61 ± 0.1 | 9.6 ± 1.9 | 5.1 ± 1.6 | 5.1 ± 1.6 |
Experimental
General experimental procedures
The culture of Penicillium citrinum was isolated from a soil sample that was collected in Yongxing Island, Hainan province, People's Republic of China, in November 2016. The isolate was identified based on morphology and sequence (Genbank Accession No. ON307318) analysis of the ITS region of the rDNA. The strain was cultured on slants of potato dextrose agar at 25 °C for 10 days. Agar plugs were cut into small pieces (about 0.5 × 0.5 × 0.5 cm3) under aseptic conditions, and 25 pieces were used to inoculate in five 250 mL Erlenmeyer flasks, each containing 50 mL of media (0.4% glucose, 1% malt extract, and 0.4% yeast extract; pH adjusted to 6.5 and sterilized by autoclave). Five flasks of the inoculated media were incubated at 25 °C on a rotary shaker at 170 rpm for 5 days to prepare the seed culture. Fermentation was carried out in 20 Fernbach flasks (500 mL), each containing 80 g of rice. Distilled H2O (120 mL) was added to each flask, and the contents were soaked overnight before autoclaving at 15 psi for 30 min. After cooling to room temperature, each flask was inoculated with 5.0 mL of the spore inoculum and incubated at 25 °C for 40 days.Extraction and isolation
The fermentation material was extracted repeatedly with EtOAc (2 × 3.0 L), and the organic solvent was evaporated to dryness under vacuum to afford 5.2 g of extract. The extract was fractionated by silica gel vacuum liquid chromatography (VLC) using petroleum ether–EtOAc–MeOH gradient elution. The fraction (80.2 mg) eluted with 100![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :30 petroleum ether–EtOAc was subjected to Sephadex LH-20 column chromatography (CC) eluting with 1:1 CH2Cl2–MeOH and the resulting subfractions were combined and purified using sempreparative HPLC (45% MeCN in H2O, Agilent Zorbax SB-C18 column; 5 μm; 9.4 × 250 mm, 3 mL min−1) to give 1 (12.3 mg, tR 18.2 min), the resulting subfractions were combined and purified using sempreparative HPLC (50% MeCN in H2O, Agilent Zorbax SB-C18 column; 5 μm; 9.4 × 250 mm, 3 mL min−1) to give 2 (4.0 mg, tR 30.0 min).
:30 petroleum ether–EtOAc was subjected to Sephadex LH-20 column chromatography (CC) eluting with 1:1 CH2Cl2–MeOH and the resulting subfractions were combined and purified using sempreparative HPLC (45% MeCN in H2O, Agilent Zorbax SB-C18 column; 5 μm; 9.4 × 250 mm, 3 mL min−1) to give 1 (12.3 mg, tR 18.2 min), the resulting subfractions were combined and purified using sempreparative HPLC (50% MeCN in H2O, Agilent Zorbax SB-C18 column; 5 μm; 9.4 × 250 mm, 3 mL min−1) to give 2 (4.0 mg, tR 30.0 min).
Spirocitrinol A (1): colorless oil; [α]25D +18.0 (c 0.1, MeOH); UV (MeOH) λmax (log ε) 204 (0.88), 284 (0.03); IR (KBr) νmax 3412, 2922, 1631, 1105, 908 cm−1; ECD (c 2.0 × 10−5 M, MeOH) λmax (Δε) 231 (−2.30), 275 (−1.21) nm; 1H and 13C NMR data see Table 1; HMBC data (DMSO-d6, 600 MHz) H-4 → C-2, 3, 5, 6; H-7 → C-1, 6, 10, 21; H-8 → C-7, 9; H-9 → C-1, 8; H-10a → C-6, 7, 21; H-10b → C-6, 7, 21; H-11 → C-8, 9; H-12 → C-1, 8, 9; H-13 → C-1, 2, 3; H-20a → C-14, 19, 21; H-20b → C-14, 19, 21; H-22 → C-10, 14, 19, 21, 23; H-23 → C-14, 21, 22; H-24 → C-14, 15, 16; H-25 → C-16, 17, 18; OH-3 → C-2, 3, 4; OH-16 → C-15, 16, 17; OH-18 → C-18, 19; HRESIMS m/z 425.1973 (calcd. for C25H29O6, 425.1969).
Spirocitrinol B (2): colorless oil; [α]25D −16.6 (c 0.1, MeOH); UV (MeOH) λmax (logε) 205 (0.40), 281 (0.08); IR (KBr) νmax 3421, 2931, 1606, 1105, 964 cm−1; ECD (c 1.2 × 10−4 M, MeOH) λmax (Δε) 212 (−13.15), 278 (−0.65) nm; 1H and 13C NMR data see Table 1; HMBC data (DMSO-d6, 600 MHz) H-7 → C-1, 6; H-8 → C-7, 9; H-9 → C-1, 8; H-10a → C-7, 21; H-10b → C-7, 21; H-11 → C-8, 9; H-12 → C-1, 8, 9; H-13 → C-1, 2, 3; H-20a → C-14, 19, 21; H-20b → C-10, 14, 19, 21, 23; H-22 → C-10, 14, 19, 21, 23; H-23 → C-14, 21, 22; H-24 → C-14, 15, 16; H-25 → C-16, 17, 18; H-29 → C-27, 28, 30; H-32 → C-3, 4, 5, H-33 → C-32, 34; H-34 → C-26, 31, 33; H-35 → C-33, 34; H-36 → C-26, 33, 34; H-37 → C-26, 27, 28; OH-3 → C-2, 3; OH-30 → C-29, 30, 31; HRESI-MS m/z 631.2917 (calcd. for C37H43O9 631.2912).
Computational details10
Conformational analysis within an energy window of 3.0 kcal mol−1 was performed by using the OPLS3 molecular mechanics force field via the MacroModel panel of Maestro 10. The conformers were then further optimized with the software package Gaussian 09 at the B3LYP/6-31G(d) level, and the harmonic vibrational frequencies were also calculated to confirm their stability. Then the 60 lowest electronic transitions for the obtained conformers in vacuum were calculated using time-dependent density functional theory (TD-DFT) methods at the CAM-B3LYP/6-31G(d) level. ECD spectra of the conformers were simulated using a Gaussian function. The overall theoretical ECD spectra were obtained according to the Boltzmann weighting of each conformer.Cytotoxicity assays
MTT assays were performed as previously described.11,12 Briefly, cells were seeded into 96-well plates at a density of 5 × 103 cells per well for 24 h, and were the exposed to different concentrations of test compounds. After incubation for 72 h, cells were stained with 25 μL of MTT solution (5 mg mL−1) for 25 min. Finally, the mixture of medium and MTT solution was removed and 75 μL DMSO was added to dissolve formazan crystals. Absorbance of each well was measured at 544 nm (test wavelength) and 690 nm (background) using the Multi-Mode Microplate Reader. Background was subtracted from the absorbance of each well. A549 (human lung cancer) and Huh7 (human hepatoma), and HepG2 (human hepatoma) was purchased from the American Type Culture Collection (ATCC). SK-BR-3 (human breast cancer) was purchased from the Procell Life Science&Technology Co., Ltd J82 (human bladder cancer cell line) and MB49 (WT) (mouse bladder carcinoma cell line) was obtained from the Merck Millipore. MB49 (STR), the cisplatin resistant subclone of MB49 cells, was established in our lab by weekly exposure to cisplatin.Conclusions
In summary, spirocitrinols A (1) and B (2) are new citrinin derivatives with a spiro[chromane-2,3′-isochromane] skeleton. Compound 1 shares the same plane structure as the known fungal metabolites xerucitrinin A and B, but differs in having different configurations at C-7/C-21. Compound 2 possesses the same spiro[chromane-2,3′-isochromane] skeleton as 1, but differs in having a 3,4,5-trimethylisochromane-6,8-diol unit at C-4 via C–C linkage between C-4 and C-32. Although citrinin dimers have been documented in many P. citrinum strains,2 only three trimeric citrinin derivatives, tricitrinols A–C have been reported.3 To our knowledge, spirocitrinol B (2) is the fourth example of trimeric citrinin metabolite, and is the first example possessing the spiro[chromane-2,3′-isochromane] skeleton. Dimeric and trimeric citrinin are described to be derived from the same or different citrinin monomers through Diels–Alder, Michael addition, or nucleophilic reactions.2,3 Natural occurring citrinin-anthraquinone, and citrinin-alkaloid adducts also have been documented from P. citrinum strains.8h,i From a biosynthetic aspect, 1 and 2 could be derived from a hypothetical precursor e, which is formed via an intermolecular hetero Diels–Alder reaction between c and d.2a,6 Although the known compounds a and b and the intermediates c and d were not coisolated in the current work, they could be the hypothetical biosynthetic precursors leading to the formation of 1 and 2 through a series of reactions including hetero Diels–Alder cycloaddition and Michael addition as illustrated in the proposed plausible biosynthetic pathways shown in Scheme S1.†2,3,13–16Conflicts of interest
The authors declare no conflict interest.Acknowledgements
This work was financially supported by the CAMS Innovation Fund for Medical Sciences (2021-1-I2M-030 and 2021-1-I2M-1-028).Notes and references
- (a) J. P. Brown, N. J. Cartwright, A. Robertson and W. B. Whalley, Nature, 1948, 162, 72–73 CAS; (b) S. Kovac, P. Nemec and J. Balan, Nature, 1961, 190, 1104–1105 CrossRef CAS; (c) R. K. Hill and L. A. Gardella, J. Org. Chem., 1964, 29, 766–767 CrossRef CAS.
- (a) D. Wakana, T. Hosoe, T. Itabashi, K. Okada, G. M. D. C. Takaki, T. Yaguchi, K. Fukushima and K. I. Kawai, J. Nat. Med., 2006, 60, 279–284 CrossRef CAS; (b) Z. Y. Lu, Z. J. Lin, W. L. Wang, L. Du, T. J. Zhu, Y. C. Fang, Q. Q. Gu and W. M. Zhu, J. Nat. Prod., 2008, 71, 543–546 CrossRef CAS PubMed; (c) L. Du, D. H. Li, G. J. Zhang, T. J. Zhu, J. Ai and Q. Q. Gu, Tetrahedron, 2010, 66, 9286–9290 CrossRef CAS; (d) W. Q. Guo, D. Li, J. X. Pen, T. J. Zhu, Q. Q. Gu and D. H. Li, J. Nat. Prod., 2015, 78, 306–310 CrossRef CAS PubMed; (e) L. Chen, Y. Y. Zhao, R. F. Lan, L. Du, B. S. Wang, T. Zhou, Y. P. Li, Q. Q. Zhang, M. G. Ying, Q. H. Zheng and Q. Y. Liu, Tetrahedron, 2017, 73, 5900–5911 CrossRef CAS; (f) L. Wang, C. L. Li, G. H. Yu, Z. C. Sun, G. J. Zhang, Q. Q. Gu, T. J. Zhu, Q. Che, H. S. Guan and D. H. Li, Tetrahedron Lett., 2019, 60, 151182 CrossRef CAS; (g) W. Y. Wang, Y. Y. Liao, B. B. Zhang, M. L. Gao, W. Q. Ke, F. Li and Z. Z. Shao, Mar. Drugs, 2019, 17, 46 CrossRef CAS PubMed; (h) A. Sabdaningsih, Y. Liu, U. Mettal, J. Heep, Riyanti, L. Wang, O. Cristianawati, H. Nuryadi, M. T. Sibero, M. Marner, O. K. Radjasa, A. Sabdono, A. Trianto and T. F. Schäberle, Mar. Drugs, 2020, 18, 227 CrossRef CAS PubMed; (i) H. T. Li, R. T. Duan, T. Liu, R. N. Yang, J. P. Wang, S. X. Liu, Y. B. Yang, H. Zhou and Z. T. Ding, Fitoterapia, 2020, 146, 104711 CrossRef CAS PubMed; (j) M. M. Cheng, P. L. Li, Y. Jiang, X. L. Tang, W. J. Zhang, Q. Wang and G. Q. Li, J. Nat. Prod., 2021, 84, 1345–1352 CrossRef CAS PubMed; (k) H. Fan, Z. M. Shi, Y. H. Lei, M. X. Si-Tu, F. G. Zhou, C. Feng, X. Wei, X. H. Shao, Y. Chen and C. X. Zhang, Mar. Drugs, 2022, 20, 443 CrossRef CAS PubMed.
- (a) L. Du, H. C. Liu, W. Fu, D. H. Li, Q. M. Pan, T. J. Zhu, M. Y. Geng and Q. Q. Gu, J. Med. Chem., 2011, 54, 5796–5810 CrossRef CAS PubMed; (b) J. H. Wei, X. X. Chen, Y. C. Ge, Q. Z. Yin, X. D. Wu, J. S. Tang, Z. J. Zhang and B. Wu, J. Org. Chem., 2022, 87, 13270–13279 CrossRef CAS PubMed.
- J. M. Gao, S. X. Yang and J. C. Qin, Chem. Rev., 2013, 113, 4755–4811 CrossRef CAS PubMed.
- (a) J. Sperry, Z. E. Wilson, D. C. K. Rathwell and M. A. Brimble, Nat. Prod. Rep., 2010, 27, 1117–1137 RSC; (b) D. J. Atkinson and M. A. Brimble, Nat. Prod. Rep., 2015, 32, 811–840 RSC; (c) K. Hiesinger, D. Dar’in, E. Proschak and M. Krasavin, J. Med. Chem., 2021, 64, 150–183 CrossRef CAS PubMed.
- K. Sadorn, S. Saepua, N. Boonyuen, P. Laksanacharoen, P. Rachtawee and P. Pittayakhajonwut, RSC Adv., 2016, 6, 94510–94523 RSC.
- (a) L. Salendra, X. P. Lin, W. H. Chen, X. Y. Pang, X. W. Luo, J. Y. Long, S. R. Liao, J. F. Wang, X. F. Zhou, Y. H. Liu and B. Yang, Nat. Prod. Res., 2021, 35, 900–908 CrossRef CAS PubMed; (b) S. J. Chen, D. M. Tian, J. H. Wei, C. Li, Y. H. Ma, X. S. Gou, Y. R. Shen, M. Chen, S. H. Zhang, J. Li, B. Wu and J. S. Tang, Front. Mar. Sci., 2022, 961356 CrossRef.
- (a) C. H. Chen, C. Y. Shaw, C. C. Chen and Y. C. Tsai, J. Nat. Prod., 2002, 65, 740–741 CrossRef CAS PubMed; (b) T. Amagata, A. Amagata, K. Tenney, F. A. Valeriote, E. Lobkovsky, J. Clardy and P. Crews, Org. Lett., 2003, 5, 4393–4396 CrossRef CAS PubMed; (c) M. Tsuda, Y. Kasai, K. Komatsu, T. Sone, M. Tanaka, Y. Mikam and J. Kobayashi, Org. Lett., 2004, 6, 3087–3089 CrossRef CAS PubMed; (d) M. Tsuda, M. Sasaki, T. Mugishima, K. Komatsu, T. Sone, M. Tanaka, Y. Mikami and J. Kobayashi, J. Nat. Prod., 2005, 68, 273–276 CrossRef CAS PubMed; (e) M. Sasaki, M. Tsuda, M. Sekiguchi, Y. Mikami and J. Kobayashi, Org. Lett., 2005, 7, 4261–4264 CrossRef CAS PubMed; (f) T. Mugishima, M. Tsuda, Y. Kasai, H. Ishiyama, E. Fukushi, J. Kawabata, M. Watanabe, K. Akao and J. Kobayashi, J. Org. Chem., 2005, 70, 9430–9435 CrossRef CAS PubMed; (g) L. Du, T. J. Zhu, Y. C. Fang, Q. Q. Gu and W. M. Zhu, J. Nat. Prod., 2008, 71, 1343–1351 CrossRef CAS PubMed; (h) N. Khamthong, V. Rukachaisirikul, S. Phongpaichit, S. Preedanon and J. Sakayaroj, Tetrahedron, 2012, 68, 8245–8250 CrossRef CAS; (i) M. El-Neketi, W. Ebrahim, W. Lin, S. Gedara, F. Badria, H. E. A. Saad, D. Lai and P. Proksch, J. Nat. Prod., 2013, 76, 1099–1104 CrossRef CAS PubMed; (j) K. Trisuwan, V. Rukachaisirikul, K. Borwornwiriyapan, S. Phongpaichi and J. Sakayaroj, Tetrahedron Lett., 2014, 55, 1336–1338 CrossRef CAS; (k) L. H. Meng, Y. Liu, X. M. Li, G. M. Xu, N. Y. Ji and B. G. Wang, J. Nat. Prod., 2015, 78, 2301–2305 CrossRef CAS PubMed.
- (a) L. Liu, Y. Li, S. Liu, Z. Zheng, X. Chen, H. Zhang, L. Guo and Y. Che, Org. Lett., 2009, 11, 2836–2839 CrossRef CAS PubMed; (b) C. Ma, Y. Li, S. Niu, H. Zhang, X. Liu and Y. Che, J. Nat. Prod., 2011, 74, 32–37 CrossRef CAS PubMed; (c) J. Ren, F. Zhang, X. Liu, L. Li, G. Liu, X. Liu and Y. Che, Org. Lett., 2012, 14, 6226–6229 CrossRef CAS PubMed; (d) E. Li, F. Zhang, S. Niu, X. Liu, G. Liu and Y. Che, Org. Lett., 2012, 14, 3320–3323 CrossRef CAS PubMed; (e) J. Zhang, L. Liu, B. Wang, Y. Zhang, L. Wang, X. Liu and Y. Che, J. Nat. Prod., 2015, 78, 3058–3066 CrossRef CAS PubMed; (f) F. Ren, S. Chen, Y. Zhang, S. Zhu, J. Xiao, X. Liu, R. Su and Y. Che, J. Nat. Prod., 2018, 81, 1752–1759 CrossRef CAS PubMed; (g) J. Zhang, Y. Li, F. Ren, Y. Zhang, X. Liu and Y. Che, J. Nat. Prod., 2019, 82, 1678–1685 CrossRef CAS PubMed.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, B. Mennucci, G. A. Petersson, H. Nakatsuji, M. Caricato, X. Li, H. P. Hratchian, A. F. Izmaylov, J. Bloino, G. Zheng, J. L. Sonnenberg, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, J. A. Montgomery Jr, J. E. Peralta, F. Ogliaro, M. Bearpark, J. J. Heyd, E. Brothers, K. N. Kudin, V. N. Staroverov, R. Kobayashi, J. Normand, K. Raghavachari, A. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, N. Rega, J. M. Millam, M. Klene, J. E. Knox, J. B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, R. L. Martin, K. Morokuma, V. G. Zakrzewski, G. A. Voth, P. Salvador, J. J. Dannenberg, S. Dapprich, A. D. Daniels, O. Farkas, J. B. Foresman, J. V. Ortiz, J. Cioslowski and D. J. Fox, Gaussian 09, Rev D.01, Gaussian, Inc., Wallingford, CT, 2009 Search PubMed.
- C. Y. Wang, L. Engelke, D. Bickel, A. Hamacher, M. Frank, P. Proksch, H. Gohlke and U. M. Kassack, Bioorg. Med. Chem., 2019, 27, 115044 CrossRef CAS PubMed.
- N. Zhang, Y. Chen, R. Jiang, E. Li, X. Chen, Z. Xi, Y. Guo, X. Liu, Y. Zhou, Y. Che and X. Jiang, Autophagy, 2011, 7, 598–612 CrossRef CAS PubMed.
- (a) H. Hajjaj, A. Klaebe, M. O. Loret, G. Goma, P. J. Blanc and J. Francois, Appl. Environ. Microbiol., 1999, 65, 311–314 CrossRef CAS PubMed; (b) B. R. Clark, R. J. Capon, E. Lacey, S. Tennant and J. H. Gill, Org. Biomol. Chem., 2006, 4, 1520–1528 RSC.
- D. Zhang, X. Li, J. S. Kang, H. D. Choi, J. H. Jung and B. W. Son, J. Microbiol. Biotechnol., 2007, 17, 865–867 CAS.
- Q. Chen, J. Gao, C. Jamieson, J. Liu, M. Ohashi, J. Bai, D. Yan, B. Liu, Y. Che, Y. Wang, K. N. Houk and Y. Hu, J. Am. Chem. Soc., 2019, 141, 14052–14056 CrossRef CAS PubMed.
- Y. Zhai, Y. Li, J. Zhang, Y. Zhang, F. Ren, X. Zhang, G. Liu, X. Liu and Y. Che, Fungal Genet. Biol., 2019, 129, 7–15 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: HRESIMS, NMR spectra, and ECD calculations of compounds 1 and 2. See DOI: https://doi.org/10.1039/d3ra00665d |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2023 |