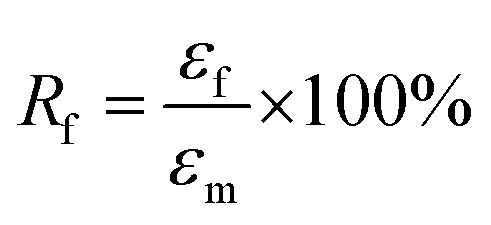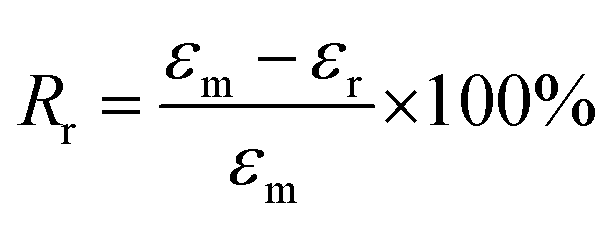DOI:
10.1039/D3RA00670K
(Paper)
RSC Adv., 2023,
13, 13120-13127
Biocompatible shape-memory poly(propylene carbonate)/silk fibroin blend with body temperature responsiveness†
Received
31st January 2023
, Accepted 21st April 2023
First published on 27th April 2023
Abstract
The high value-added medical applications surely represent the leading edge of the shape-memory materials (SMPs) field. Herein, the biomedical SMPs were easily prepared via incorporating silk fibroin (SF) into poly(propylene carbonate) (PPC) through directly melt blending. Based on the intrinsic glass transition of PPC at ∼37 °C, the blends showed a body temperature responsiveness without a complex procedure for adjusting the switching temperature. By varying the SF content, the blend exhibited tunable shape-memory effects (SME), with a first enhancing but then worsening shape recoverability and a stable and excellent shape fixity. And the blend with 3 wt% SF achieved the best SME, enabling an efficient shape reconfiguration under a 37 °C water bath. It was revealed that SF acted as physical cross-links to connect the PPC chains forming a shape-memory network, thus can well retard irreversible the chain slipping of PPC, leading to the improvement of recoverability. Moreover, the results obtained from cell compatibility testing showed the huge application potential of this material in the biomedical field. This work proposed a facile preparation strategy for developing biocompatible body heat actuated shape-memory materials.
1 Introduction
Shape-memory polymeric materials (SMPs) can encode geometry information into a temporary structure, then utilize that information to regain the initial shape when exposed to an environmental change.1–5 Their ability to create diverse stimuli-responsive movements such as contracting, expanding, twisting, bending, or jumping, has rendered them a promising material for biomedical devices, artificial muscles, micro-robots, actuators, among others.6–8 Particularly, the high value-added medical applications with the elaboration of blood stents, surgical sutures, and bone fixators surely represent the leading edge of the SMP field.9–12
Generally, SMPs contain two functional regions of switching unit and permanent unit. The former with a well-defined phase/thermal transition determines the switching temperature (Ts) to enable the fixation and activation of the temporary shape formed by thermal-mechanical processing, while the latter memorizes the permanent shape via restraining irreversible deformation or/and providing recovery force.13,14 Regarding to the limitation of human physiological environment, one of the most important design considerations of biomedical SMPs is locating Ts at around body temperature. In addition, the shape-memory effect (SME) featuring the performances of temporary shape fixation and permanent shape recovery is governed by the synergy between switching and permanent units.15 Thus, when trying to develop a high-performance body heat-triggered biomedical SMP, the intrinsic property and mass ratio as well as molecular interaction of the two shape-memory components must be taken into account.
With the advantage of tailoring macroscopic properties by the control of molecular parameters, blockcopolymer as the structural concept is usually applied to tune both the Ts and SME. As an example, Xue and coworkers synthesized a series of biodegradable shape-memory copolymers comprising hyperbranched three-arm poly(ε-caprolactone) (h-PCL) as the switching unit and poly(3-hydroxybutyrate-co-3-hydroxyvalerate) (PHBV) as the permanent domain.16 The melting temperature (Tm) from h-PCL, which directly determined the Ts, was adjusted in the range of 38–47 °C through changing the block ratio. And a biomedical stent made from the copolymer containing 25 wt% PHBV completed an excellent self-expansion upon 37 °C water bath within 25 s. Besides, the simple melt blending has been used to fabricate SMPs with tunable Ts because of its ease in material integration and composition control. Ajili et al. blended the biocompatible polyurethane (PU) and PCL with the elastic PU serving as the permanent unit and the crystalline PCL serving as the switching unit.17 By manipulating the crystallization behavior of PCL, the Ts related to the Tm of PCL decreased to the body temperature range, enabling the blend to realize shape recovery at 37 °C. However, the preparation of biomedical SMPs by either chemical synthesis or melt processing requires a complex regulation of Ts due to the limitation in material choice of switching unit. Regarding to that, finding a biocompatible polymer with a thermal/phase transition temperature around body temperature would greatly simplify the produce procedure.
In recent years, the growing interest in green chemistry has spawned many sustainable polymers derived from natural resource such as starch,18 cellulose,19 polylactide (PLA),20 bacterial polyesters,21 etc. Among them, the amorphous poly(propylene carbonate) (PPC) created by copolymerization of carbon dioxide (CO2) and propylene oxide has attracted concerns from both industrial and academic fields because of its efficiency in harnessing CO2.22 Particularly, PPC has a low glass transition temperature (Tg) located between 20 and 40 °C, which is a perfect candidate to serve as the body temperature switch. However, for the amorphous PPC molecular chains featuring high flexibility and weak small inter-chain interaction,23 it is easy to generate irreversible slipping, which are regarded as the major contributor to the deterioration of shape recoverability.24 One effective way to address this issue is introducing stiff particles such as graphene nanoplatelets and carbon nanotubes as permanent component to enhance the mechanical strength and retard chain slipping of the matrix, and through that way some amorphous polymers (e.g., PLA,25 poly(vinyl acetate) (PVAc),26 polyvinyl alcohol (PVA),27 etc.) successfully obtained favorable SME. Also, Qi et al. constructed a strong nanoconfinement effect via hydrogen bonding between PPC and graphene oxide, leading to an enhancement of SME.28
Silk fibroin (hereafter called “SF”), as a structural protein from the Bombyx mori silkworm, has gained extensive attention owing to its advantageous properties including biocompatibility, biodegradability, and unique mechanical performance, showing good suitability for versatile applications ranging from biocompatible reinforcing fillers to textiles, to biomedical devices and moisture-responsive actuator.29–31 Moreover, SF possesses abundant amide groups enabling to provide active centers for blending with polyester,32 benefiting the formation of efficient physical crosslinking shape-memory network. Hence, in this work, the shape-memory PPC was fabricated via incorporating different content of SF through simple melt blending. Aiming at the biomedical applications, the body temperature triggered SME of PPC/SF blends was explored. The interaction of PPC with SF was discussed for revealing the operative shape-memory mechanism of this blending system. Besides, the cell contacting experiment was performed to further characterize the biocompatibility of this material.
2 Experimental
2.1 Materials
PPC was kindly provided by the State Key Laboratory of Polymer Physics and Chemistry of China, with a weight-average molecular weight of 2.5 × 105 g mol−1. Silkworm cocoon (Bombyx mori) was bought from local farms. Anhydrous sodium carbonate (Na2CO3) was supplied by Kelong reagent Co. Ltd (China).
2.2 Degumming of silk fibers
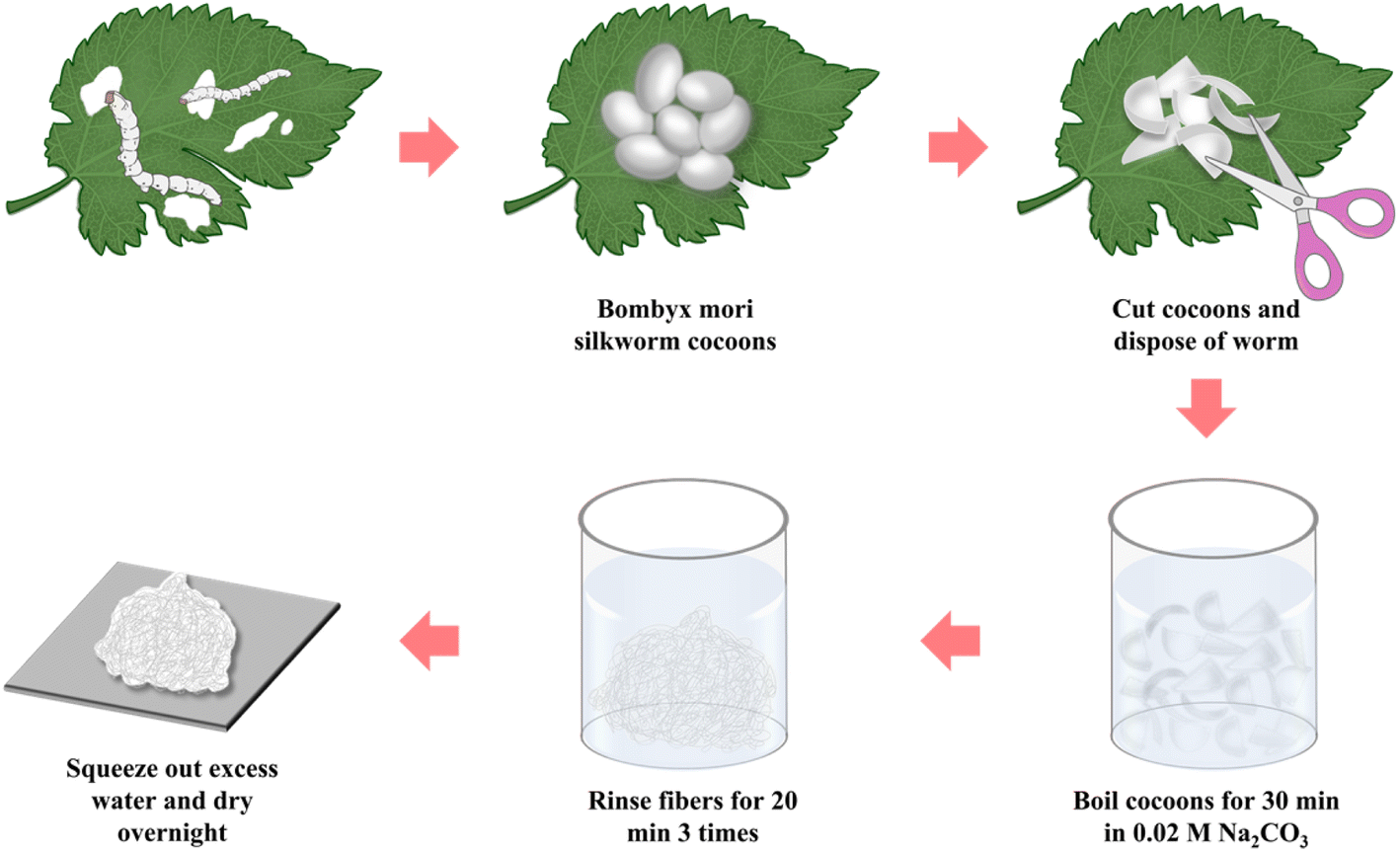
First, the sericin was removed from Bombyx mori silkworm cocoons in accordance with the protocol previously established in the published literature.33 As shown in Fig. 1, after cutting the cocoon shells into pieces, 10 g cocoons were immersed in a 0.02 M Na2CO3 solution and boiled for 30 min, then followed by thoroughly washing with deionized water three times. The excess water was squeezed out and then the degummed silk fiber was air-dried overnight in a convection oven at a room temperature. Finally, the dried SF was cut gently into short fibers.
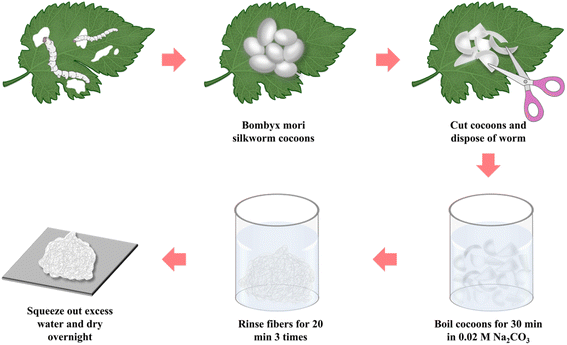 |
| | Fig. 1 Schematic of the silk fibroin extraction procedure. | |
2.3 Preparation of PPC/SF blends
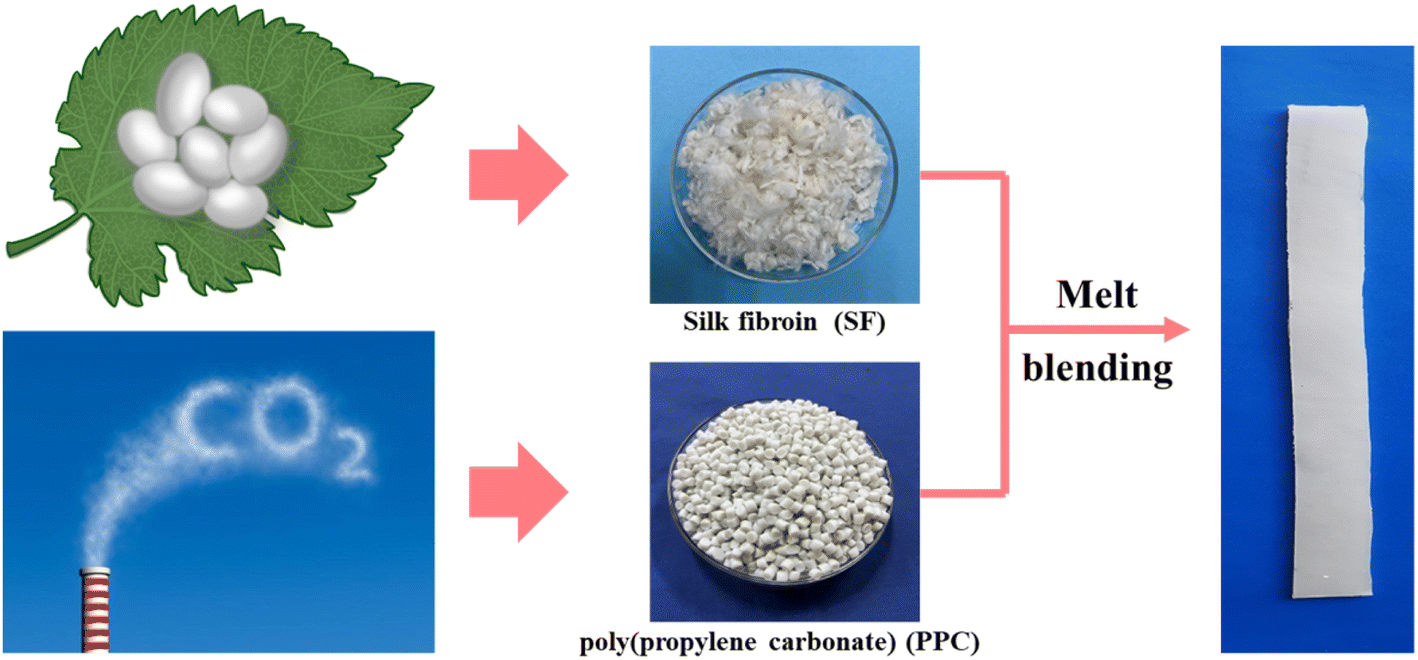
Prior to the melt blending process, PPC was dried at 40 °C and SF was dried at 80 °C for 24 h. PPC was melted in a HAAKE torque rheometer (Thermo Fisher Scientific, USA) at 50 rpm for 5 min, and the temperature was set at 150 °C considering its poor thermostability.34 Then, SF at different weight ratios (1–5 wt%) was added and blending with PPC for an extra 5 min. The prepared PPC/SF blends (respectively denoted as PPC-1, PPC-2, PPC-3, PPC-4, and PPC-5 where the number refers to the weight ratio of SF) as well as neat PPC, were molded into 1.0 mm film using a hot-press at 150 °C by applying 10 MPa for 8 minutes, Fig. 2.
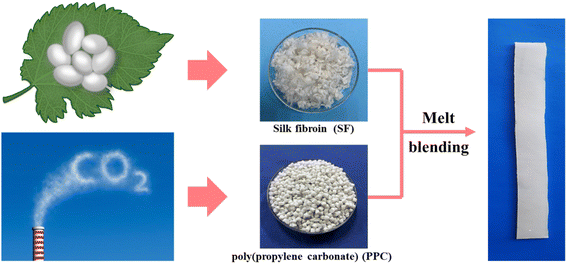 |
| | Fig. 2 Facile fabrication of shape-memory PPC/SF blend. | |
2.4 Observation of SF dispersion
An approximately 10 μm think slice was cut from the sample using a microtome. Then, the optical microscope (BX51, OLYMPUS) was used to directly observe the dispersion of SF in PPC without using the polarizer.
2.5 Differential scanning calorimetry (DSC)
DSC experiments were performed on TA Q2000 DSC (TA Instruments, New Castle, DE) in nitrogen atmosphere, with a scanning rate of 10 °C min−1 from 0 to 80 °C.
2.6 Dynamic mechanical analysis (DMA)
A TA Q800 DMA in multifrequency strain mode was used to measure the moduli of the blends. Dynamic tensile scans from 0 to 80 °C with a heating rate of 3 °C min−1, an amplitude of 10 μm, and a frequency of 1 Hz.
2.7 Dynamic rheological analysis
The rheological properties of each sample were characterized using dynamic oscillatory rheometer (AR1500ex, TA Instrument) with a parallel plate geometry (25 mm in diameter). The test was conducted at 150 °C with the frequency ranging from 0.1 to 100 rad s−1, and the strain amplitude was set at 2% that was within the linear viscoelastic regime.
2.8 Fourier transform infrared spectroscopy (FTIR)
FTIR spectra of each blend were obtained with a Nicolet-IS10 spectrometer (Thermo Electron Co.) working in attenuated total reflection mode. The spectrum was acquired from 4000 to 400 cm−1 in a resolution of 4 cm−1.
2.9 Shape-memory characterizations
The samples for the shape-memory characterizations were cut from the films into rectangular specimens with a dimension of 8.0 mm (length) × 2.0 mm (width) × 1.0 mm (thickness). The SME was explored using the controlled force mode of the DMA Q800. Samples were heated to 37 °C and held for 5 min, followed by loading a constant force to stretch the sample. Once the maximum strain (εm) equilibrated, the deformed sample suffered a rapid quenching to room temperature under the stress. Subsequently, the force was removed to obtain the temporary shape (εf). Upon reheating to 37 °C, the sample recovered to the original shape, and the recovering progress went go for 20 min to allow the strain to equilibrate (εr). Based on the shape-memory cycle, the shape-fixing and -recovering ratios (Rf and Rr) were determined by following eqs:35| |
 | (1) |
| |
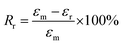 | (2) |
2.10 Evaluation of cytocompatibility
The HEK 293T cells were cultured in Dulbecco's Modified Eagle's Medium (DMEM), which has been supplemented with 10% fetal bovine serum (GIBCO) and 1% penicillin–streptomycin solution (GIBCO), under a humidified atmosphere containing 5% CO2 at 37 °C. The cytocompatibility of the composites was evaluated by live staining assay. Prior the tests, the rectangle specimens with a length of 15 mm and a width of 10 mm were cut from the blend films, and then sterilized by 75% ethanol for 24 h and subsequently washed with phosphate buffer saline (PBS). Then, the specimens were put in a 12-well plate, and 1 mL 293T cell suspension (5 × 104 cells per mL) was seeded on them. The control group was treated with DMEM. The cells were incubated for 1 and 3 days in a 37 °C biochemical incubator. After carefully removing the culture media, the cells were stained with 1 mL calcein-AM (1 μg mL−1) for 30 min, and then gently washed twice with PBS. The survival of the cells was visualized under an Olympus BX51 fluorescence microscope (Olympus America Inc., USA).
3 Results and discussion
3.1 Thermal behaviors
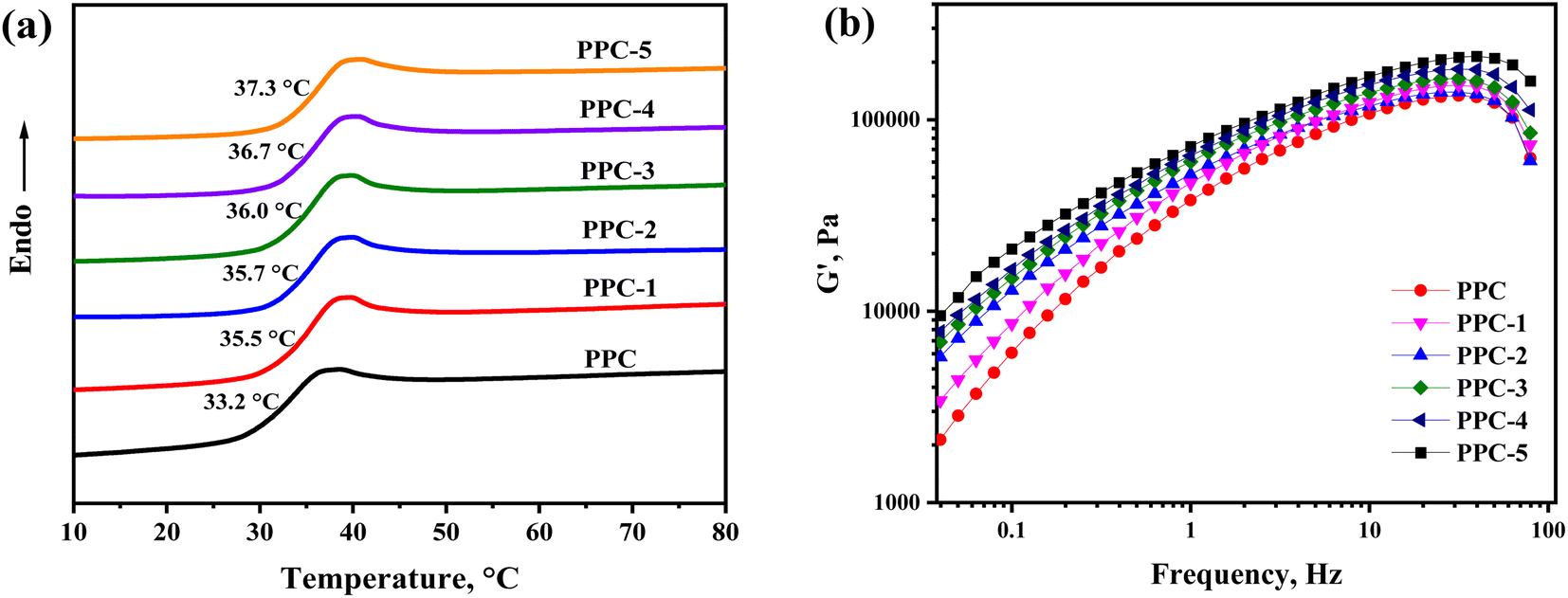
The thermal transitions of PPC and the PPC/SF blends were explored by DSC, and in Fig. 3a the heating thermograms of each sample are shown. The amorphous PPC showed a glass transition at 33.2 °C, which gradually increased with the addition of SF and reached 37.3 °C of PPC-5. It indicated that SF limited the chain motion of PPC, which could be further evidenced by the rheological curves in Fig. 3b showing the strengthened elastic response of blend as more SF introduced. In accordance with the shape-memory mechanism,36 Ts of the as-prepared blends was supposed to be slightly higher than the Tg of PPC. Hence, Ts could be set at 37 °C for characterizing the body heat-triggered SME of each specimen.
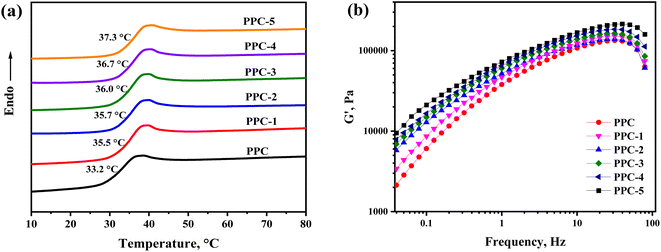 |
| | Fig. 3 (a) DSC heating curves for the blend systems and (b) the frequency dependence of storage modulus (G′) of the PPC/SF blending systems at 150 °C. | |
3.2 Shape-memory performance
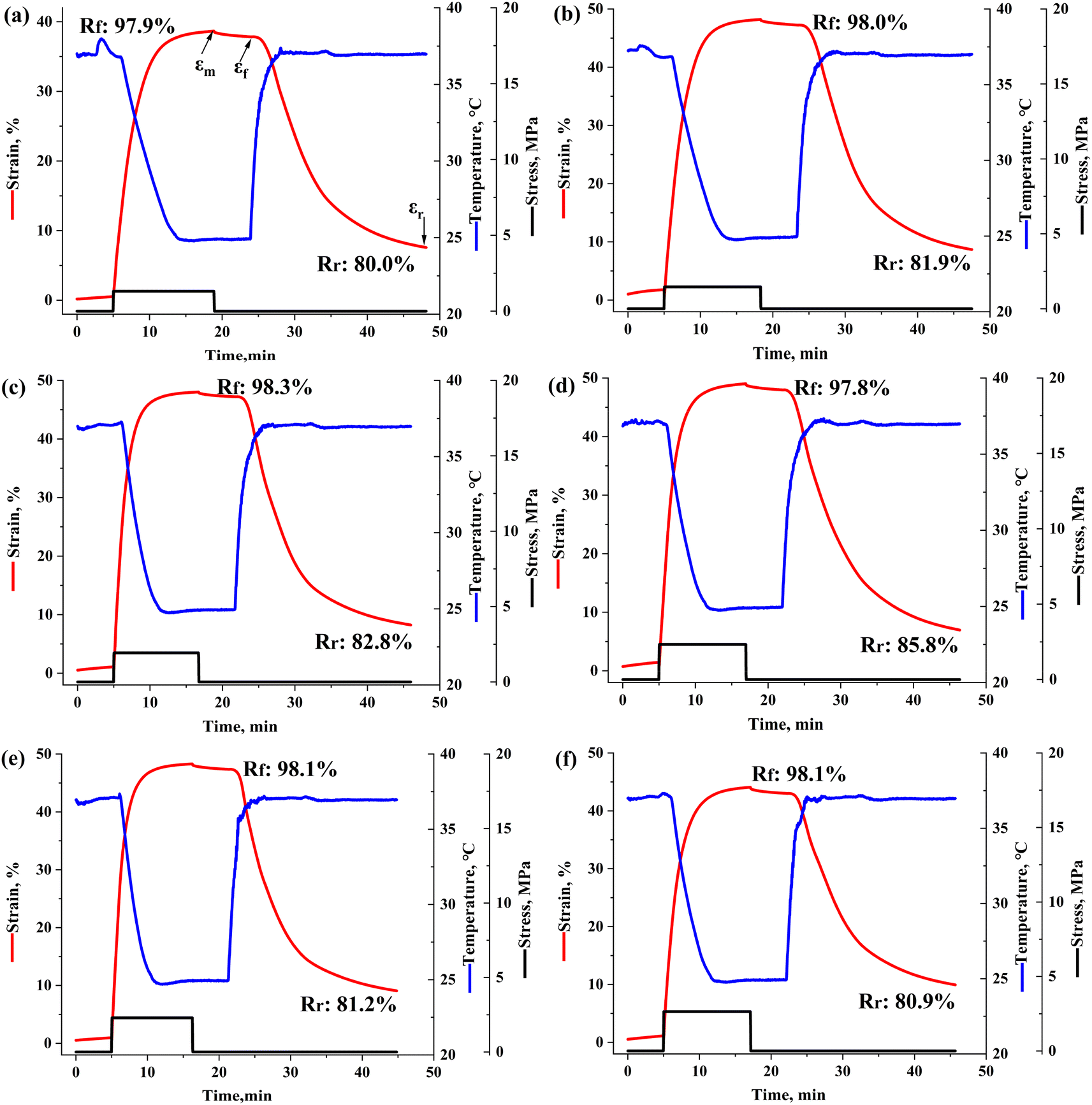
The SME of each sample was researched by thermos-mechanical cycling test, and the shape-memory curves are exhibited in Fig. 4, with the calculated Rf and Rr marked beside. The Rf of the samples stayed at ∼98.0%, meaning that the pure PPC has an excellent intrinsic shape-fixing performance and the incorporation of SF had little effect on Rf. In contrast, the Rr increased from 80.0% to 85.8% as 3 wt% SF introduced, but then decreased to 80.9% with a more addition of SF up to 5 wt%. In comparison, the PPC-3 sample showed the best body heat-triggered shape-memory performance.
 |
| | Fig. 4 Thermo-mechanical cycles of PPC and the PPC/SF blends: (a) PPC, (b) PPC-1, (c) PPC-2, (d) PPC-3, (e) PPC-4, (f) PPC-5. | |
Afterwards, a thermo-mechanical cycle was performed in water for intuitively demonstrating the SME of PPC and PPC-3. The strip sample was thermally reshaped into a spiral at 37 °C and fixed via quenching. Then, reheating recovered to the original shape upon water bathing at 37 °C. From Video S1,† a fast and good recovery was observed in PPC-3 against a slow and poor recovery in PPC, indicating that blending PPC with SF could not only optimize the Rr but also accelerate the recovering speed. The geometries of PPC-3 at different shape-memory stages were recorded in Fig. 5, further illustrating its good SME. In this way, the great potential for medical applications of the PPC/SF blend was outlined.
 |
| | Fig. 5 Macroscopic shape-memory progress of PPC-3. | |
3.3 Shape-memory mechanism
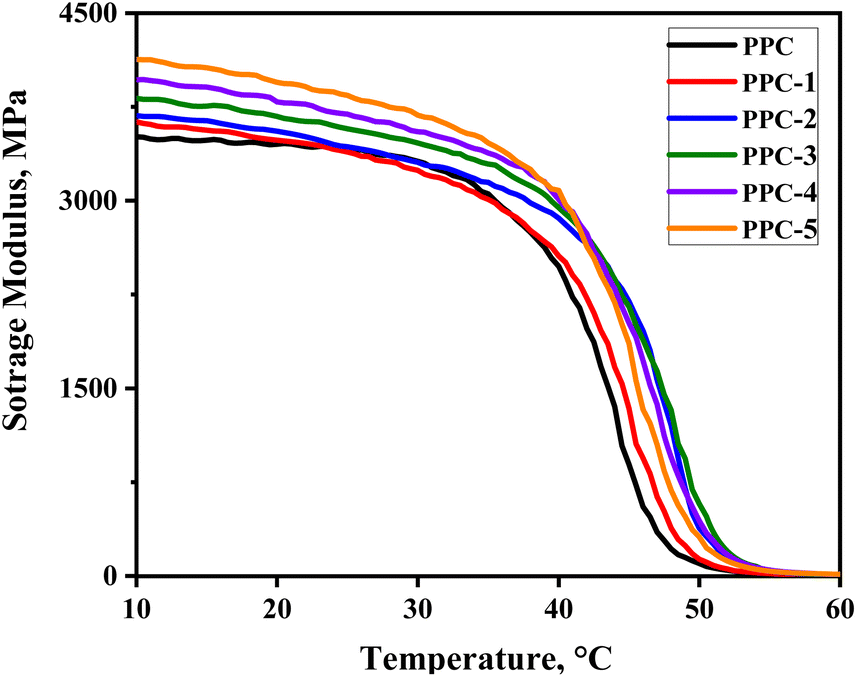
3.3.1 Fixation of temporary shape. The operative mechanism of PPC/SF shape-memory blend composites was discussed in terms of shape-fixing and -recovering progress. As the thermally deformed sample was cooled to room temperature, the PPC chains became glassy to maintain the temporary deformation and store the strain energy. From Fig. 6, PPC exhibited high stiffness at room temperature with a storage modulus above 3400 MPa, thus could provide the enough mechanical support to maintain the deformation and achieve an excellent Rf of 97.9%. As a result, although the addition of SF strengthened PPC, it had little effect on Rf.
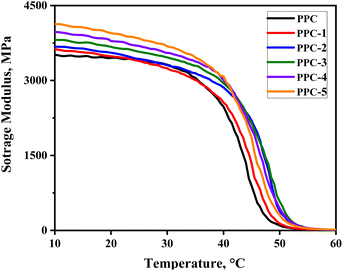 |
| | Fig. 6 The evolution of storage modulus of each sample with temperature. | |
3.3.2 Recovery of permanent shape. Upon heating to Ts, the activated PPC chains released the elastic energy and returned to the original location. However, the pure PPC showed a poor Rr of 80.0% because of the irreversible chain sliding induced by thermal stretching. With the addition of SF, Rr first increased but then decreased as the filler network became more denser, Fig. 7a. The filler–polymer interaction was explored via infrared analysis. As shown in Fig. S1a,† SF had three absorptions at 1650 cm−1 (amide I), 1540 cm−1 (amide II) and 1235 cm−1 (amide III), which were assigned to α-helix or random coil conformation, whereas the peak at 1265 cm−1 was attributed to β-sheet structure.37 On the other hand, the strong absorption peaks centered at 1749 cm−1 and 1233 cm−1 belonged to the stretching vibrations of the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O group and C–O–C band in PPC, respectively, Fig. S1b.† For the blends, the amide I bond of SF disappeared and the characterization peaks in amide III region of SF were overlapped with the absorption peaks of PPC, Fig. 7b and c. Moreover, the amide II band at 1540 cm−1 shifted to 1530 cm−1 assigned to β-sheet, and its relative intensity was slightly increased with more SF added, indicating a conformation transition of SF after blending with PPC. According to the previous reports,38,39 these changes were ascribed to the hydrogen bonding between the carbonyl groups of PPC and the esters hydroxyl groups of amino acids of SF. Hence, the SF fillers could act as physical crosslinks to retard the slipping of PPC chains, leading to the optimization of Rr. However, Rr was deteriorated when the filler content was over 3 wt%. This is due to the excess SF, especially which failed to interact with PPC chain, showed an aggregative distribution and would significantly inhibit chain motion, turning to impede the elasticity of the blend. Afterwards, the creep-recovery test was performed to further support the above view. As shown in Fig. 7d, the addition of SF could continuously modify the creep resistance of PPC. Besides, the PPC-3 sample showed a lower residual strain thus a better recoverability, while the PPC-5 sample presented a recovering performance similar with PPC.
O group and C–O–C band in PPC, respectively, Fig. S1b.† For the blends, the amide I bond of SF disappeared and the characterization peaks in amide III region of SF were overlapped with the absorption peaks of PPC, Fig. 7b and c. Moreover, the amide II band at 1540 cm−1 shifted to 1530 cm−1 assigned to β-sheet, and its relative intensity was slightly increased with more SF added, indicating a conformation transition of SF after blending with PPC. According to the previous reports,38,39 these changes were ascribed to the hydrogen bonding between the carbonyl groups of PPC and the esters hydroxyl groups of amino acids of SF. Hence, the SF fillers could act as physical crosslinks to retard the slipping of PPC chains, leading to the optimization of Rr. However, Rr was deteriorated when the filler content was over 3 wt%. This is due to the excess SF, especially which failed to interact with PPC chain, showed an aggregative distribution and would significantly inhibit chain motion, turning to impede the elasticity of the blend. Afterwards, the creep-recovery test was performed to further support the above view. As shown in Fig. 7d, the addition of SF could continuously modify the creep resistance of PPC. Besides, the PPC-3 sample showed a lower residual strain thus a better recoverability, while the PPC-5 sample presented a recovering performance similar with PPC.
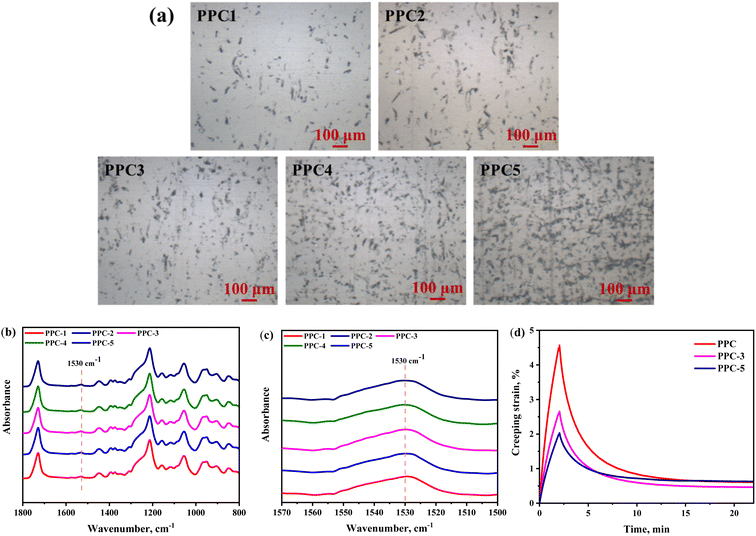 |
| | Fig. 7 (a) The optical microscope images and (b) FTIR spectroscopy of the PPC/SF blends; (c) zooming of the 1530 cm−1 region; (d) the creep-recovery curves of PPC, PPC-3, and PPC-5. | |
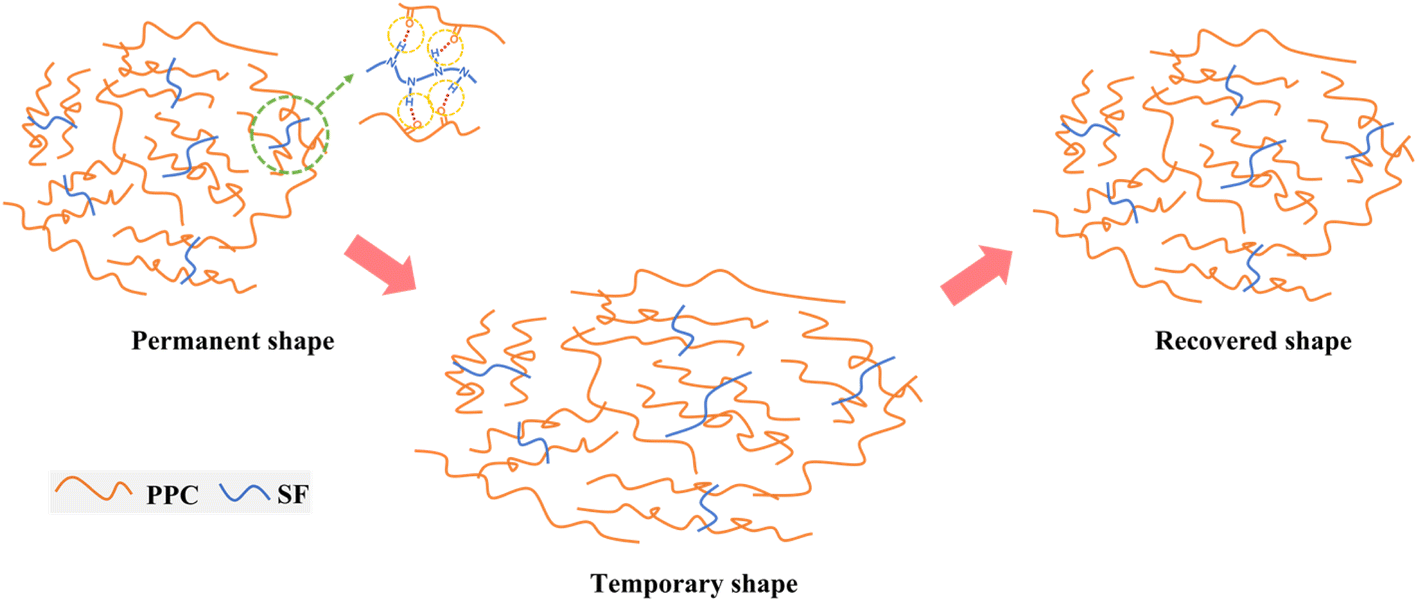
Here, a scheme of the shape-memory mechanism was proposed in Fig. 8. The SF fillers act as physical cross-links to connect the PPC chains forming a shape-memory network via the N–H⋯OC interactions. During the progress of configuration of temporary shape, the PPC chains were elongated upon thermal stretching and then frozen after quenching to room temperature, thus can well fix the deformation and exhibit a high Rf. Meanwhile, SF could effectively restrict the irreversible chain slipping of PPC. Thus, once reheated to Ts, the defrosted PPC chains could efficiently recover back via releasing the strain energy.
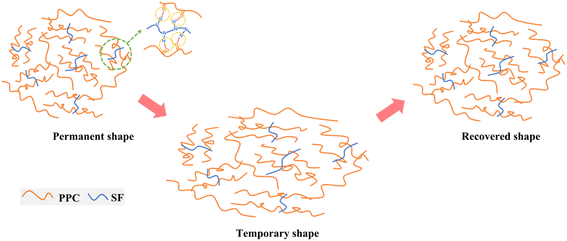 |
| | Fig. 8 Schematic illustration of the interactions between PPC and SF and the shape-memory progress. | |
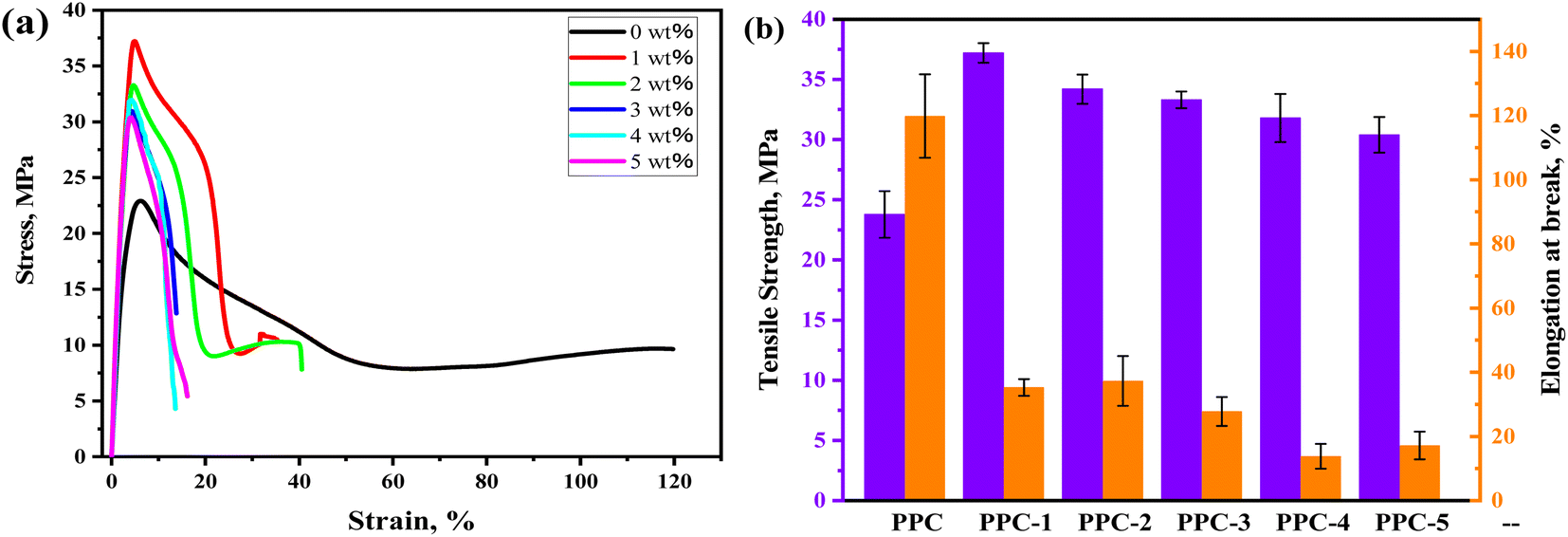
3.4 Mechanical properties
Mechanical properties of the blends are characterized, and the stress–strain curves and the summarizing results of tensile strength (σ) and the elongation at break (ε) are displayed in Fig. 9. PPC has the σ of 23.8 MPa and ε of 119.8% due to its high chain flexibility and weak inter-chain interaction. With SF adding, the σ of PPC increased but accompanied by the sacrifice of ductility. The PPC-1 blend achieved the best σ of 37.2 MPa, which was enhanced 56.3% compared to that of PPC. A further adding of SF led to agglomeration thus the deterioration of σ, however, which remained above 30 MPa.
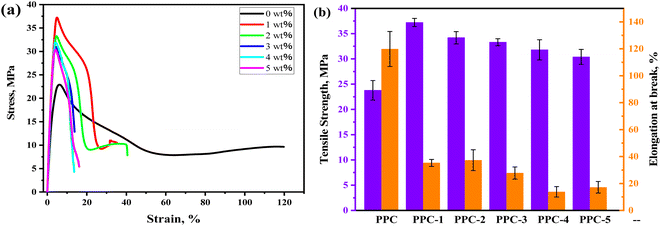 |
| | Fig. 9 Mechanical properties of the PPC/SF blends. | |
3.5 Cytocompatibility
Cell compatibility is vital for the clinical application of polymeric materials. Herein, the 393T cells were treated with the PPC-3 specimens for 1 and 3 days, respectively. Then, the cytocompatibility was evaluated by the living staining assay, in accordance with the principle that the normal cells stained with calcein-AM would show a green morphology. As shown in Fig. 10, the cells spread more and formed tighter intercellular after cultured with the composite by 1 day, as compared to the control group. After 3 days, the cell proliferation could be observed, and there were no distinct differences of cells amounts between the treated and control ones. These results well verified the good cytocompatibility of PPC/SF composite, which demonstrated a guarantee for further biomedical application.
 |
| | Fig. 10 The fluorescence images of cells after cultured on the specimens. | |
4 Conclusions
In this work, the body temperature-responsive shape-memory effect was easily achieved via directly melt blending PPC with SF. It was demonstrated that SF could connect the PPC chains to form an efficient physical crosslinking network, well restricting the irreversible chain slipping of PPC. Hence, the incorporation of SF could tune the shape recoverability of PPC, meanwhile had little effect on its inherent excellent shape fixity. By comparison, the blend with 3 wt% SF not only exhibited the best experimental shape-memory performance but also enabled a favorable shape recovery upon a water bath at 37 °C. Moreover, this material was proved biocompatible through a cell treating test, which conveys a potential application for development of smart biomedical devices.
Conflicts of interest
There are no conflicts of interest to declare.
Acknowledgements
This work was financially supported by the National Natural Science Foundation of China (Grant No. 51903165) and the State Key Laboratory of Polymer Materials Engineering (sklpme2020-3-05).
References
- H. Chen, F. Zhang, Y. Sun, B. Sun, B. Gu, J. Leng and W. Zhang, Smart Mater. Struct., 2021, 30, 025040 CrossRef CAS.
- X. Gong, L. Liu, Y. Liu and J. Leng, Smart Mater. Struct., 2016, 25, 035036 CrossRef.
- A. Asar, M. S. Irfan, K. A. Khan, W. Zaki and R. Umer, Compos. Sci. Technol., 2022, 221, 109219 CrossRef CAS.
- W. Peng, G. Zhang, J. Liu, S. Nie, Y. Wu, S. Deng, G. Fang, J. Zhou, J. Song, J. Qian, P. Pan, Q. Zhao and T. Xie, Adv. Funct. Mater., 2020, 30, 2000522 CrossRef CAS.
- Y. Deng, F. Zhang, M. Jiang, Y. Liu, H. Yuan and J. Leng, ACS Appl. Mater. Interfaces, 2022, 14, 42568–42577 CrossRef CAS PubMed.
- Y. Zhang, N. Zhang, H. Hingorani, N. Ding, D. Wang, C. Yuan, B. Zhang, G. Gu and Q. Ge, Adv. Funct. Mater., 2019, 29, 1806698 CrossRef.
- Q. Zhao, H. Qi and T. Xie, Prog. Polym. Sci., 2015, 49–50, 79–120 CrossRef CAS.
- Y. Xia, Y. He, F. Zhang, Y. Liu and J. Leng, Adv. Mater., 2020, 33, 2000713 CrossRef PubMed.
- J. G. Hardy, M. Palma, S. J. Wind and M. J. Biggs, Adv. Mater., 2016, 28, 5717–5724 CrossRef CAS PubMed.
- W. Zhao, L. Liu, F. Zhang, J. Leng and Y. Liu, Mater. Sci. Eng., C, 2019, 97, 864–883 CrossRef CAS PubMed.
- Y. Deng, B. Yang, F. Zhang, Y. Liu, J. Sun, S. Zhang, Y. Zhao, H. Yuan and J. Leng, Biomaterials, 2022, 291, 121886 CrossRef CAS PubMed.
- F. Zhang, L. Wang, Z. Zheng, Y. Liu and J. Leng, Composites, Part A, 2019, 125, 105571 CrossRef CAS.
- J. Huang, J. Fan, S. Yin and Y. Chen, Compos. Sci. Technol., 2019, 182, 107732 CrossRef CAS.
- X. Xu, P. Fan, J. Ren, Y. Cheng, J. Ren, J. Zhao and R. Song, Compos. Sci. Technol., 2018, 168, 255–262 CrossRef CAS.
- M. Balk, M. Behl, U. Nöchel and A. Lendlein, Macromol. Mater. Eng., 2012, 297, 1184 CrossRef CAS.
- L. Xue, S. Y. Dai and Z. Li, Biomaterials, 2010, 31, 8132–8140 CrossRef CAS PubMed.
- S. H. Ajili, N. G. Ebrahimi and M. Soleimani, Acta Biomater., 2009, 5, 1519–1530 CrossRef CAS PubMed.
- A. Regazzi, M. Teil, P. J. J. Dumont, B. Harthong, D. Imbault, R. Peyroux and J. L. Putaux, Compos. Sci. Technol., 2019, 182, 107755 CrossRef CAS.
- Z. Zhang, S. Cai, Y. Li, Z. Wang, Y. Long, T. Yu and Y. Shen, Compos. Sci. Technol., 2020, 194, 108151 CrossRef CAS.
- J. M. Raquez, Y. Habibi, M. Murariu and P. Dubois, Prog. Polym. Sci., 2013, 38, 1504–1542 CrossRef CAS.
- R. W. Lenz and R. H. Marchessault, Biomacromolecules, 2005, 6, 1–8 CrossRef CAS PubMed.
- Y. Xu, L. Lin, M. Xiao, S. Wang, A. T. Smith, L. Sun and Y. Meng, Prog. Polym. Sci., 2018, 80, 163–182 CrossRef CAS.
- X. D. Shi and Z. H. Gan, Eur. Polym. J., 2007, 43, 4852–4858 CrossRef CAS.
- D. H. Jung, H. M. Jeong and B. K. Kim, J. Mater. Chem., 2010, 20, 3458 RSC.
- M. Keramati, I. Ghasemi, M. Karrabi, H. Azizi and M. Sabzi, Fibers Polym., 2016, 17, 1062–1068 CrossRef CAS.
- M. Babaahmadi, M. Sabzi, G. R. Mahdavinia and M. Keramati, Polymer, 2017, 112, 26–34 CrossRef CAS.
- P. Miaudet, A. Derre, M. Maugey, C. Zakri, P. M. Piccione, R. Inoubli and P. Poulin, Science, 2007, 318, 1294 CrossRef CAS PubMed.
- X. Qi, Y. Guo, Y. Wei, P. Dong and Q. Fu, J. Phys. Chem. B, 2016, 120, 11064–11073 CrossRef CAS PubMed.
- H. Y. Cheung, K. T. Lau, X. M. Tao and D. Hui, Composites, Part B, 2008, 39, 1026–1033 CrossRef.
- M. K. DeBari, C. I. King III, T. A. Altgold and R. D. Abbott, ACS Biomater. Sci. Eng., 2021, 7, 3530–3544 CrossRef CAS PubMed.
- G. Manikandan, A. Murali, R. Kumar and D. K. Satapathy, ACS Appl. Mater. Interfaces, 2021, 13, 8880–8888 CrossRef CAS PubMed.
- H. Lee and G. Kim, J. Biomater. Sci., Polym. Ed., 2010, 21, 1687–1699 CrossRef CAS PubMed.
- D. N. Rockwood, R. C. Preda, T. Yücel, X. Wang, M. L. Lovett and D. L. Kaplan, Nat. Protoc., 2011, 6, 1612–1631 CrossRef CAS PubMed.
- X. Qi, P. Dong, Z. Liu, T. Liu and Q. Fu, Compos. Sci. Technol., 2016, 125, 38–46 CrossRef CAS.
- Y. Zheng, B. Zeng, L. Yang, J. Shen and S. Guo, Ind. Eng. Chem. Res., 2020, 59, 2977–2987 CrossRef CAS.
- T. Xie, Polymer, 2011, 52, 4985–5000 CrossRef CAS.
- H. Zhu, X. Feng, H. Zhang, Y. Guo, J. Zhang and J. Chen, J. Biomater. Sci., Polym. Ed., 2009, 20, 1259–1274 CrossRef CAS PubMed.
- J. He, Y. Qin, S. Cui, Y. Gao and S. Wang, J. Mater. Sci., 2011, 46, 2938–2946 CrossRef CAS.
- S. Balali, S. M. Davachi, R. Sahraeian, B. S. Heidari, J. Seyfi and I. Hejazi, Biomacromolecules, 2018, 19, 4358–4369 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: The FTIR spectroscopy of the SF and PPC (Fig. S1). Video S1 showing the comparative shape recovery efficiency of PPC and the PPC-3 blend in a water bath at 37 °C (MP4). |
| ‡ These authors contributed equally to this work. |
|
| This journal is © The Royal Society of Chemistry 2023 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence *abc and
Shaoyun Guoab
*abc and
Shaoyun Guoab