DOI:
10.1039/D3RA00693J
(Paper)
RSC Adv., 2023,
13, 11160-11170
Hyaluronidase-trigger nanocarriers for targeted delivery of anti-liver cancer compound†
Received
1st February 2023
, Accepted 30th March 2023
First published on 11th April 2023
Abstract
Chemotherapy is recognized as one of the significant treatment methods for liver cancer. The compound celastrol (CSL) could effectively inhibit the proliferation, migration, and invasion of liver cancer cells, which is regarded as a promising candidate to become a mainstream anti-liver cancer drug. However, the application of CSL in liver cancer chemotherapy is limited due to its systemic toxicity, poor water solubility, multidrug resistance, premature degradation, and lack of tumor targeting. Meanwhile, in order to comply with the current concept of precision medicine, precisely targeted delivery of the anti-liver compound CSL was desired. This paper takes into account that liver cancer cells were equipped with hyaluronic acid (HA) receptors (CD44) on their surface and overexpressed. Hyaluronidase (HAase) capable of degrading HA, HAase-responsive nanocarriers (NCs), named HA/(MI)7-β-CD NCs, were prepared based on the electrostatic interaction between HA and imidazole moieties modified β-cyclodextrin (MI)7-β-CD. HA/(MI)7-β-CD NCs showed disassembly properties under HAase stimuli, which was utilized to trap, deliver, and the controllable release of the anti-liver cancer compound CSL. Furthermore, cytotoxicity assay experiments revealed that CSL-trapped HA/(MI)7-β-CD NCs not only reduced cytotoxicity for normal cells but also effectively inhibited the survival for five tumor cells, and even the apoptotic effect of CSL-trapped NCs with a concentration of 5 μg mL−1 on tumor cells (SMMC-7721) was consistent with free CSL. Cell uptake experiments demonstrated HA/(MI)7-β-CD NCs possessed the capability of targeted drug delivery to cancerous cells. HA/(MI)7-β-CD NCs exhibited site-specific and controllable release performance, which is anticipated to proceed further in precision-targeted drug delivery systems.
1 Introduction
Liver cancer is one of the most frequently diagnosed solid tumors worldwide and the third leading cause of cancer-related deaths.1 In addition, liver cancer is the only one of the top five most lethal cancers to show an annual percentage increase in occurrence.2 In recent decades, the effective treatment of liver cancer has become one of the clinical challenges arising from the growing number of liver cancer cases. Chemotherapy is considered to be one of the most effective treatments for liver cancer.3 Traditional Chinese medicine (TCM) has become a research hotspot in the field of chemotherapy drugs because it can act on preventing tumorigenesis attenuating toxicity, enhancing the treatment effect, and reducing tumor recurrence and metastasis. Among them, celastrol (CSL) is a natural active product extracted from the root bark of Tripterygium wilfordii Hook, which displays strong antioxidant, anti-cancer, anti-rheumatoid and other effects.4–7 In clinical practice, CSL has been successfully used to treat autoimmune diseases such as rheumatoid arthritis and leprosy reactions.8,9 It is worth mentioning that recent research has demonstrated that CSL can effectively inhibit the proliferation, metastasis, and invasion of liver cancer cells.10 However, CSL is limited use in hepatocellular carcinoma chemotherapy due to its systemic toxicity, poor water solubility, multidrug resistance, premature degradation, and lack of tumor targeting.11 Therefore, the development of adjuvants that can reduce systemic toxicity and improve tumor targeting of CSL is expected to advance the clinical application of CSL.
Stimulus-responsive drug delivery system (DDS) is a functional nanocarrier that can release drugs rapidly or at a certain rate after entering the body by changing the structure and configuration of the carrier under the stimulation of a specific environment including light, heat, ultrasound, enzyme, and pH changes.12–18 Especially, enzyme-responsive nanocarriers have been investigated extensively due to their high sensitivity, catalytic activity, and mild reaction.19–24 As we all know, abnormal expression of enzymes is one of the markers of the occurrence and development of tumors.25 For example, hyaluronidase (HAase) is demonstrated with high expression levels in larynx cancer, bladder cancer, neck squamous cell, carcinoma cells, breast tumor, and liver cancer cells.26–31 Thus, HAase overexpressed in cancer cells provide the basis for the targeted delivery of anticancer drugs. Besides, HAase plays an important role in the degradation of hyaluronic acid (HA), which is a glycosaminoglycan with the advantages of good biocompatibility and biodegradability, low toxicity, and non-immunogenicity.32–34 HA receptors, CD44 and RHAMM, are highly expressed on the surface of many cancer cells, such as breast, lung, colon, and liver cancers.35–38 Accordingly, it is reasonable to assume that the HAase-based enzyme-responsive DDS is capable of specifically depolymerizing HA-containing nanoparticles and is expected to be a model for precisely targeted delivery antitumor drugs. For example, Kim et al. designed HA stimuli-responsive nanoparticles by grafted 3-diethylaminopropyl (DEAP) to the carboxylic acid groups of HA.39 The nanoparticles not only are destabilized at an acidic pH but also can efficiently degrade in HAase-rich endosomes. Raju et al. prepared HAase-responsive nanoparticles by coating the surface of PEI-Au/Fe nanoparticles with HA.40 The nanoparticles as intracellular transport carriers of ADAM10 siRNA can effectively inhibit the growth of prostate cancer cells.
Supramolecular nanocarriers are constructed based on non-covalent interactions such as electrostatic forces, hydrogen bonds, van der Waals forces, etc.41,42 They have the advantages of a simple construction process, easy functionalization, and do not depend on too many organic reagents, which is one of the important branches of stimulus-responsive DDS.43,44 Cyclodextrins (CDs) is a naturally occurring, non-toxic, biodegradable cyclic oligosaccharide that is used to form supramolecular nanocarriers for various drug delivery systems.45–48 For example, Bai et al. prepared supramolecular self-assemblies by grafting β-CD onto the HA backbone to achieve drug-targeted delivery.49 Singh et al. grafted β-CD onto HA to generate a supramolecular polymer (HA-β-CD) and explored its targeted drug delivery applications.50 Although those nanocarriers exhibit good biocompatibility, high drug loading capacity, targeted drug delivery, and controlled release properties, the construction of HAase-responsive nanocarriers is generally a tedious and time-consuming process, i.e., the nanocarriers are not easily destroyed due to HA was grafted onto the nanocarriers by covalent interaction, resulting to limit the development of such nanocarriers for tumor therapy. Therefore, a simple combination of HA with nanocarriers would be an ideal vector for precision-targeted delivery antitumor drugs.
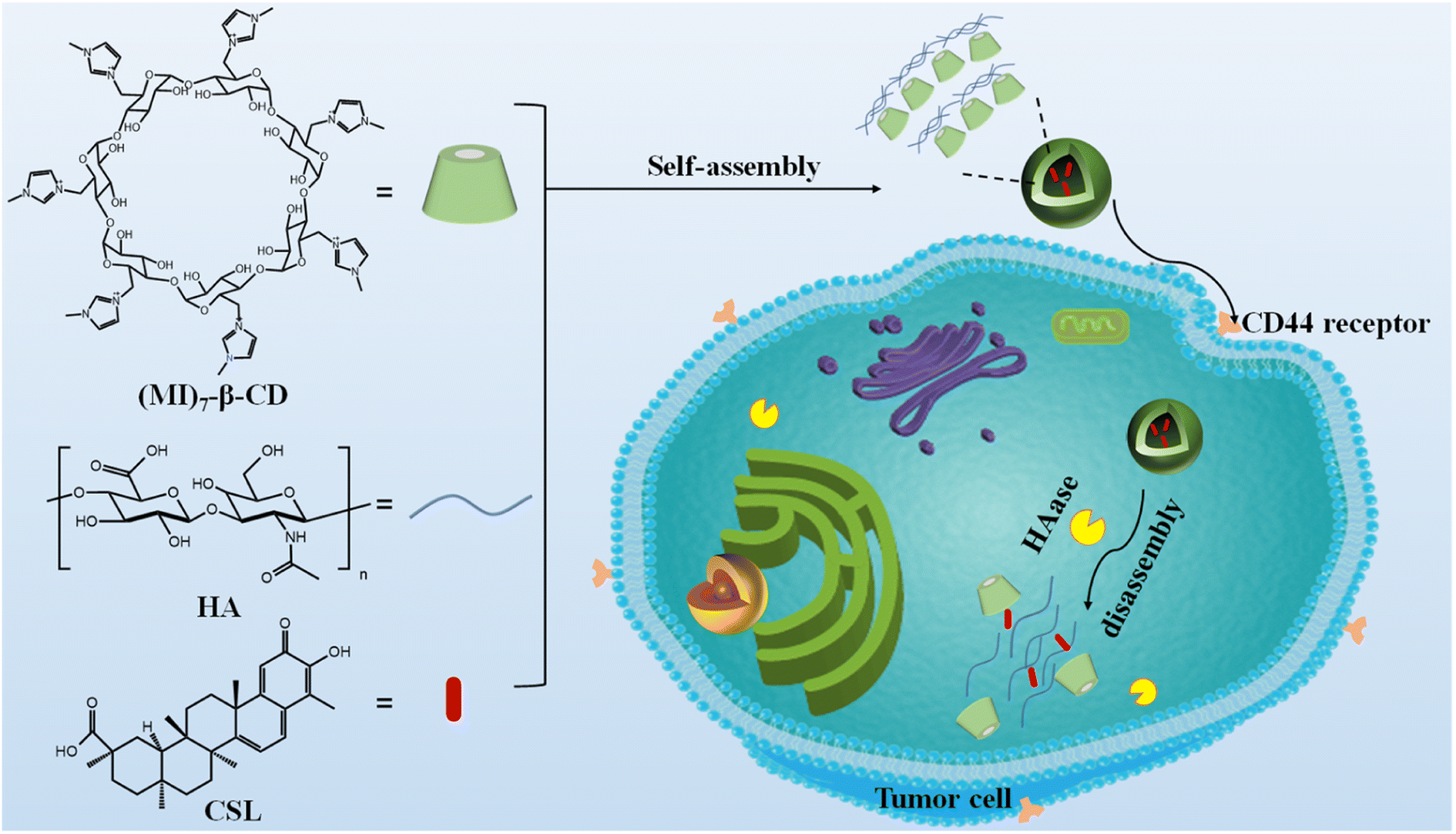
In this study, HA and per-6-deoxy-6-(1-methylimidazol-3-ium-3-yl)-β-CD ((MI)7-β-CD) were chosen as the targeted agent and building block, respectively. The HAase-responsive supramolecular nanocarriers, named HA/(MI)7-β-CD NCs, were prepared based on the electrostatic interaction between negatively charged HA and positively charged (MI)7-β-CD as shown in Fig. 1. The preparation method possessed some advantages including easy preparation, cheap preparation cost, and short preparation time. HA/(MI)7-β-CD NCs showed disassembly properties under HAase stimuli, which was utilized to trap, deliver, and controllable the release of anti-liver cancer compound CSL. Furthermore, cytotoxicity assay experiments revealed that CSL-trapped HA/(MI)7-β-CD NCs not only reduced cytotoxicity for normal cells but also presented an effective therapeutic effect for five tumor cells, and even the apoptotic effect of CSL-trapped NCs with a concentration of 5 μg mL−1 on tumor cells (SMMC-7721) was consistent with free CSL. Cell uptake experiments demonstrated that CSL-trapped NCs were more easily taken up by cancer cells than free drugs, which greatly increased the concentration of drugs in cancer cells. The HAase-responsive nanocarrier exhibited the site-specific and controllable release of the anti-liver cancer drug, which makes it have potential applications in liver cancer therapy.
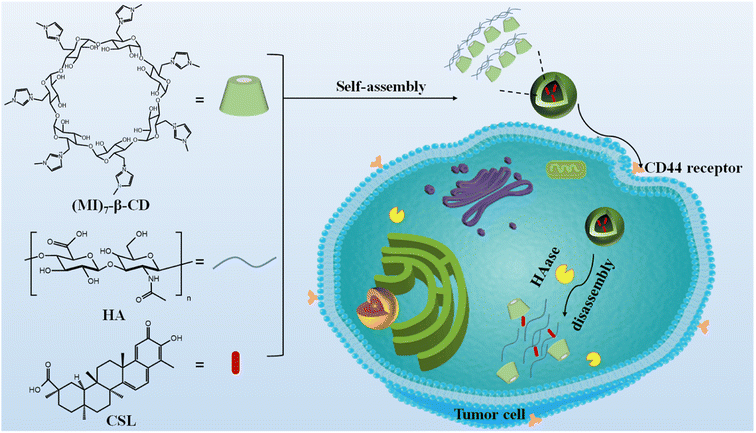 |
| | Fig. 1 Schematic diagram of the formation and the enzyme-responsive behavior of CSL-trapped HA/(MI)7-β-CD NCs. | |
2 Experimental sections
2.1 Materials
β-CD, iodine, 1-methylimidazole, triphenylphosphine (PPh3), methanol, and sodium methoxide were obtained from Adamas company. Hyaluronidase was obtained from TCL corporation. Lysozyme (LZM) was purchased from Sigma-Aldrich. β-CD purchased from International Group Chemical Reagents. Celastrol (CSL) was acquired from Chengdu Manster Biotechnology Co. Hyaluronan was provided from Shanghai Yuanye Biotechnology Co., LTD.
2.1.1 Synthesis of per-6-deoxy-6-iodo-β-CD. Per-6-deoxy-6-iodo-β-CD was synthesized according to previous ref. 51. Firstly, triphenylphosphine (PPh3) (24.0 g) and solid iodine (24.5 g) were resolved into the anhydrous DMF. The iodine solution was then slowly dropped into PPh3. The mixture solutions were kept stirred and increased to 50 °C for 10 minutes. Then β-CD (5.8 g) was added to the mixture under nitrogen protection. The temperature of the reaction was kept for 18 h. Finally, 6-deoxy-6-iodine-β-CD with a yield of 31.95% was obtained after cooling, rotary evaporation, methanol washing, and vacuum suction filter. 1H NMR (DMSO-d6, 500 MHz, δppm): 3.23–3.47 (m, 21H), 3.54–3.69 (m, 14H), 3.72–3.89 (d, 7H), 4.92–5.04 (d, 7H), 5.86–5.98 (d, 7H), 6.01–6.12 (d, 7H).
2.1.2 Synthesis of per-6-deoxy-6-(1-methylimidazol-3-ium-3-yl)-β-CD. Per-6-deoxy-6-(1-methylimidazol-3-ium-3-yl)-β-CD ((MI)7-β-CD) was synthesized based on previous report,51 and the resultant roadmap is provided in Fig. S1.† Per-6-deoxy-6-iodo-β-CD (2.0 g) was dissolved in methylimidazole (10.0 mL) under nitrogen gas protection at a temperature of 80 °C. The mixture was stirred continuously for 48 hours and then poured into acetone (100.0 mL) to obtain a white precipitate. The precipitate was filtered, washed with acetone, recrystallized in deionized water, and dried under vacuum to obtain (MI)7-β-CD with a yield of 32.1%. The 1H NMR is shown in Fig. S2.† 1H NMR (D2O-d2, 500 MHz, δppm): 3.29–3.41 (t, 1H), 3.49–3.58 (d, 1H), 3.73–3.84 (s, 3H), 3.90–4.02 (t, 1H), 4.07–4.55 (m, 3H), 5.01–5.09 (s, 1H), 7.40–7.55 (s, 2H).
2.1.3 Preparation of HA/(MI)7-β-CD NCs. Supramolecular nanoparticles were prepared by the homogeneous method. HA and (MI)7-β-CD aqueous solutions were mixed and stood for 1 h at room temperature. The final concentrations of HA and (MI)7-β-CD were 20.00 μg mL−1 and 14.73 μg mL−1, respectively.
2.1.4 Preparation of CSL-trapped HA/(MI)7-β-CD NCs. A certain amount of HA, (MI)7-β-CD and CSL solution were mixed, and the volume was fixed to 25.0 mL in distilled water to obtain CSL-trapped NCs. The final concentrations of CSL, HA, and (MI)7-β-CD were 45.06 μg mL−1, 20.00 μg mL−1, and 14.73 μg mL−1, respectively. CSL not trapped by HA/(MI)7-β-CD NCs was removed by a 3500 Da dialysis bag.
2.2 Characterization of HA/(MI)7-β-CD NCs
2.2.1 UV-vis spectroscopy. The solution to be tested was poured into a quartz cell with a 10 mm optical path and the wavelength range was controlled from 190 to 825 nm at room temperature. The absorbance and transmittance were recorded by Shimadzu UV-3600.
2.2.2 X-ray diffraction (XRD). HA, (MI)7-β-CD and HA/(MI)7-β-CD NCs were tested by Dandong DX-2700 diffractometer at a scan rate of 5° min−1, with a scan step of 2θ = 0.02° and a scan range of 5–60°.
2.2.3 Transmission electron microscopy (TEM). The transmission electron microscope images of nanocarriers, drug-trapped nanocarriers, and depolymerized nanocarriers were measured by JEM-1400 Flash transmission electron microscope at an accelerating voltage of 120 keV.
2.2.4 Dynamic light scattering (DLS). The average particle size of nanocarriers and drug-trapped nanocarriers was obtained by using a laser scattering spectrometer with a scattering angle of 90° and the average values were measured for three tests.
2.2.5 Zeta potential measurements. The zeta potentials of nanocarriers and drug-trapped nanocarriers were measured by the Malvern-Zetasizer Nano ZS 90 instrument. During the test, deionized water was used as the background and the average value of three tests was taken as the zeta potential value of the sample.
2.2.6 Fourier transform infrared spectrometry (FT-IR). HA, (MI)7-β-CD and HA/(MI)7-β-CD were mixed with KBr and pressed respectively to obtain the samples to be tested. The transmittance of each sample was measured by FT-IR in the range of 4000–400 cm−1 (tensor 27; Bruker, Germany).
2.3 Calculation of loading efficiency (LE) and encapsulation efficiency (EE)
Standard curves were drawn after the CSL was dissolved in ethanol and measured with UV-vis at 425 nm.
Loading efficiency and encapsulation efficiency were calculated by the following eqn (1) and (2), respectively.52
| | |
LE% = {mo/(mc + mo)} × 100%
| (1) |
mo is the mass after encapsulating drugs in the nanocarriers;
mD is the total mass of drugs;
mc is the mass of the nanocarriers.
2.4 Release studies of CSL from HA/(MI)7-β-CD NCs
The drug release behavior of CSL-trapped HA/(MI)7-β-CD NCs in response to enzyme stimulation was studied by adding different enzyme solutions. Dialysis bags with molecular weight truncated 3500 Da were immersed in deionized water for 72 h in advance. To simulate the tumor environment, the buffer solutions (pH = 7.4 and 5) were used as drug release medium, and kept stirred for 1 h at 37 °C. 4.0 mL solution was removed from the dialysis bags and refilled with the same volume of buffer at regular intervals. The absorbance curve of the removed solution was measured by UV-vis spectra and the cumulative release rate of CSL was calculated by eqn (3).14mt is drug amount of the CSL-trapped NCs after dialysis; mo is drug amount of initial solution before dialysis.
2.5 Cytotoxicity assay of CSL-trapped HA/(MI)7-β-CD NCs
The cytotoxicity of nanocarriers, CSL-trapped NCs, and free CSL was detected by MTS assay. In short, five strains of tumor cells (liver cancer SMMC-7721, colon cancer SW480, leukemia HL-60, breast cancer MCF-7, and lung cancer A-549 cell) and human normal BEAS-2B cells were added to DMEM medium containing 10% FBS, and various cells lines were seeded into 96-well plates (3000 ∼ per well). The prepared cell solution was incubated at 37 °C with 5% CO2 for 48 h. The drug-trapped nanocarriers, nanocarriers, and free CSL were dissolved in DMSO to make different concentration solutions (0.10, 1.00, and 5.00 μg mL−1). Different concentrations of solution were added to each cell culture medium and incubated for 48 h. Then 20.0 μL MTS solution and 100 μL culture medium were added to each well and incubate for another 4 h. The absorbance of each sample was recorded at 492 nm with a Multiskan FC microplate reader. The cell growth curve was drawn with a cell survival rate as ordinate and concentration as abscissa, and the IC50 value of the compound was calculated by the two-point method.
2.6 Cell apoptosis analysis
PE Annexin V/7-AAD labeled with fluorescein was used as the fluorescent probe and flow cytometry was used to detect the apoptosis-inducing effects of CSL-trapped NCs and CSL on tumor cells (SMMC-7721). Liver cancer cells were inoculated and cultured in DMEM for 24 h. CSL-trapped NCs and CSL were dissolved in DMSO to make samples of different concentrations. Tumor cells in the logarithmic growth stage were seeded into 6-well plates (3 × 105 cells per well), and a certain amount of sample was added to each well. 1 μM DOX was used as the positive control group. After 24 h of incubation, the cells were collected and centrifuged for 5 minutes (300 RPM per min), and the supernatant was discarded. Then 5.0 μL of PE Annexin V and 5 μL of 7-AAD were added to the solution, mixed well, and incubated for 15 minutes at room temperature in the dark. The stained cell samples were immediately analyzed by flow cytometry.
2.7 Cellular uptake
The cell uptake of CSL-trapped NCs was studied in SMMC-7721 cells. Rhodamine-B was used instead of non-fluorescent CSL. The preparation of Rhodamine B-trapped NCs was similar to that of CSL-trapped NCs described above. After incubation with free Rhodamine-B and Rhodamine B-trapped NCs for 4 h, the SMMC-7721 cells were photographed by laser scanning confocal microscope (LSM880, ZEISS).
3 Results and discussion
3.1 Preparation of HA/(MI)7-β-CD NCs
The preparation process of the designed nanocarriers was performed by adding (MI)7-β-CD in HA aqueous solution. As shown in Fig. S3,† the mixture solution containing cationic (MI)7-β-CD and anionic HA showed a noticeable Tyndall effect and reduced transmittance, indicating aggregates formed between (MI)7-β-CD and HA. Note that no Tyndall effects were observed after replacing (MI)7-β-CD with neutral β-CD, which suggested that the electrostatic interaction may play a dominant role in the formation of the aggregates. To study the critical aggregation concentration (CAC) between (MI)7-β-CD and HA, the various concentration of (MI)7-β-CD were added into HA solution (30.00 μg mL−1). The optical transmittance at 450 nm of HA showed mildly change when the concentration of (MI)7-β-CD rose from 0 to 14.25 μg mL−1, while having a rapid downward trend with (MI)7-β-CD concentration increasing from 14.25 to 18.21 μg mL−1, indicating the CAC of (MI)7-β-CD was 14.25 μg mL−1 (see in Fig. S4a and b†). Furthermore, the optimal concentration between HA and (MI)7-β-CD was tested by adding the different concentrations of HA into (MI)7-β-CD solution (14.73 μg mL−1). The optical transmittance at 450 nm of the mixed solution showed a downward trend with the concentration of HA increasing from 0.00 to 20.00 μg mL−1, then rising, and eventually reaching a plateau with the concentration of HA increasing from 20.00 to 110.00 μg mL−1, suggesting the optimum mixing concentration between (MI)7-β-CD and HA w 14.73 μg mL−1 and 20.00 μg mL−1, respectively (see in Fig. S4c and d†). The possible causes of decreased transmittance can be illustrated in that the aggregates were formed based on electrostatic interaction when HA and (MI)7-β-CD were reaching the optimum mixing concentration. The reason that the optical transmittance enhanced with adding extra HA in (MI)7-β-CD solution could be summed up as that HA enters into the cavity of (MI)7-β-CD, which leads to the disassembly of the polymer.
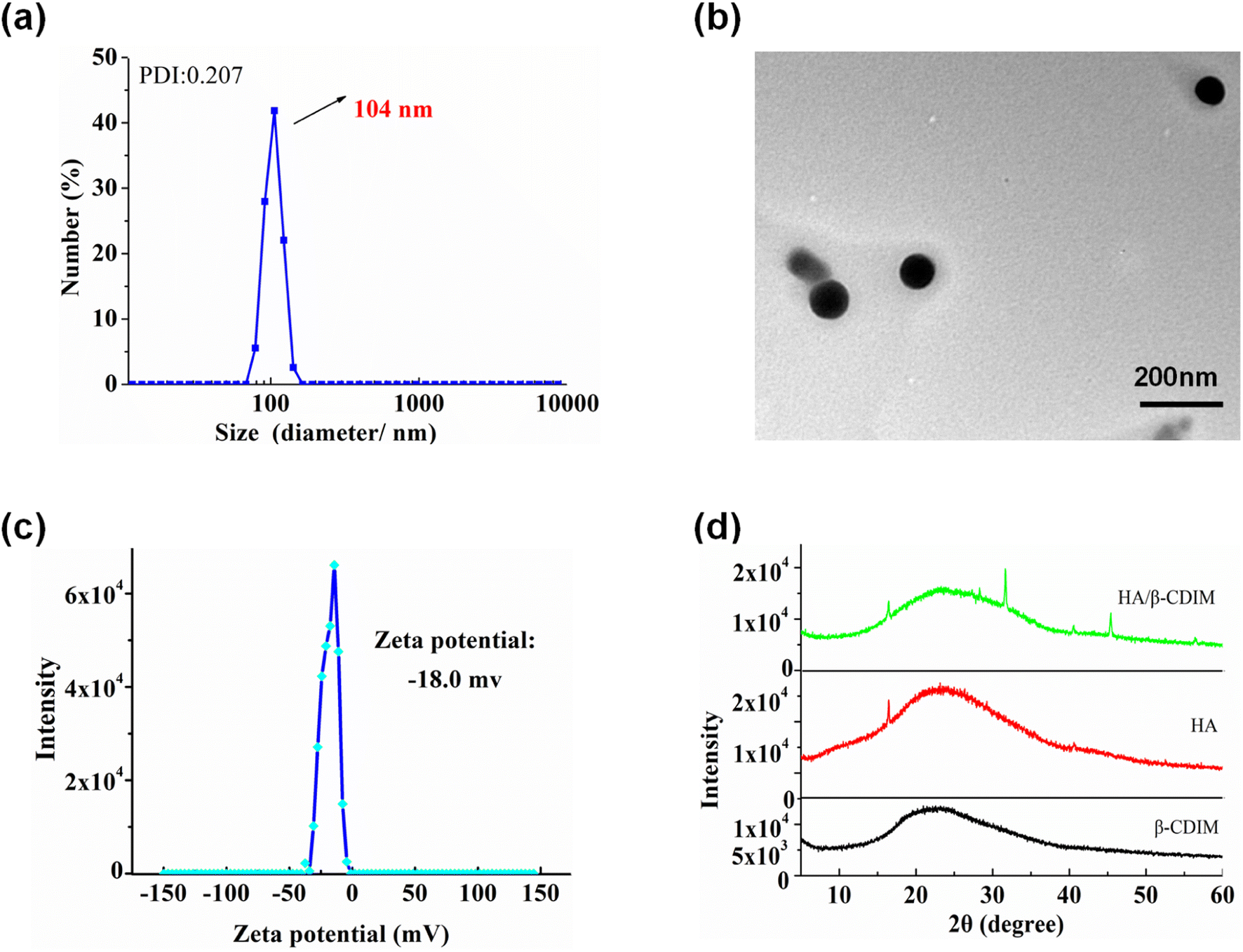
The structure and phase morphology of the nanocarriers were characterized by DLS, TEM, zeta potential, XRD, and FT-IR respectively. The DLS results illustrated that the HA/(MI)7-β-CD NCs exhibited a narrow particle size distribution with an average hydrodynamic diameter of ∼104 nm, and the PDI index was 0.207 (see Fig. 2a). TEM images showed a spherical shape with a diameter of about 100 nm, which was approximately identical to the DLS (see Fig. 2b). As is shown in Fig. 2c, the zeta potential of the HA/(MI)7-β-CD NCs was close to −18.0 mV, suggesting the anionic charge HA was exposed on the surface of the assembly. As is shown in Fig. 2d, XRD patterns of the nanocarriers exhibited sharp and enhanced diffraction peaks at 2θ = 28.35°, 31.69°, 45.40°, and 56.46° compared with the individual (MI)7-β-CD and HA. In the analysis of FT-IR, HA at 3403, 2902, 1614, 1411, and 1041 cm−1 showed strong characteristic absorption peaks from stretching vibration of –OH, –C–H, –COO− and –C–O, respectively. (MI)7-β-CD in 3393 (hydroxyl stretching vibration of CD), 3083 (C–H stretching vibration in imidazole ring), 1634 (C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N stretching vibration), 1572 and 1421 (CC stretching vibration), 1045 cm−1 (C–O stretching vibration of CD). Compared with the FT-IR spectrum of HA and (MI)7-β-CD, the absorption peak at 1411 and 1634 cm−1 moved to 1417 and 1644 cm−1, possibly due to the electrostatic interaction between HA and (MI)7-β-CD (see Fig. S5†). The above results indicated that HA/(MI)7-β-CD NCs have been successfully prepared. Moreover, the optical transmittance of the nanocarriers with the various time and temperatures showed unobvious changes, suggesting HA/(MI)7-β-CD NCs possessed good stability (see Fig. S6a and b†).
N stretching vibration), 1572 and 1421 (CC stretching vibration), 1045 cm−1 (C–O stretching vibration of CD). Compared with the FT-IR spectrum of HA and (MI)7-β-CD, the absorption peak at 1411 and 1634 cm−1 moved to 1417 and 1644 cm−1, possibly due to the electrostatic interaction between HA and (MI)7-β-CD (see Fig. S5†). The above results indicated that HA/(MI)7-β-CD NCs have been successfully prepared. Moreover, the optical transmittance of the nanocarriers with the various time and temperatures showed unobvious changes, suggesting HA/(MI)7-β-CD NCs possessed good stability (see Fig. S6a and b†).
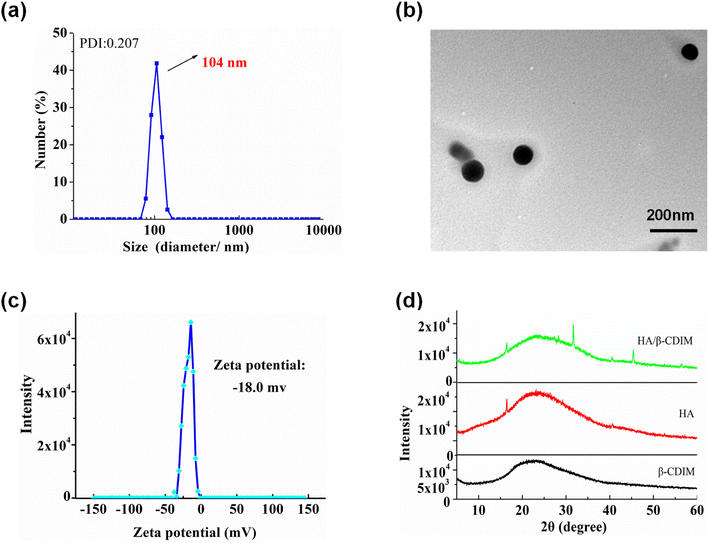 |
| | Fig. 2 The DLS diagram (a), TEM image (b), zeta potential (c), XRD patterns (d) of HA/(MI)7-β-CD NCs were measured at 25 °C, [HA] = 20 μg mL−1, [(MI)7-β-CD] = 14.73 μg mL−1. | |
3.2 Enzyme-responsive property of HA/(MI)7-β-CD NCs
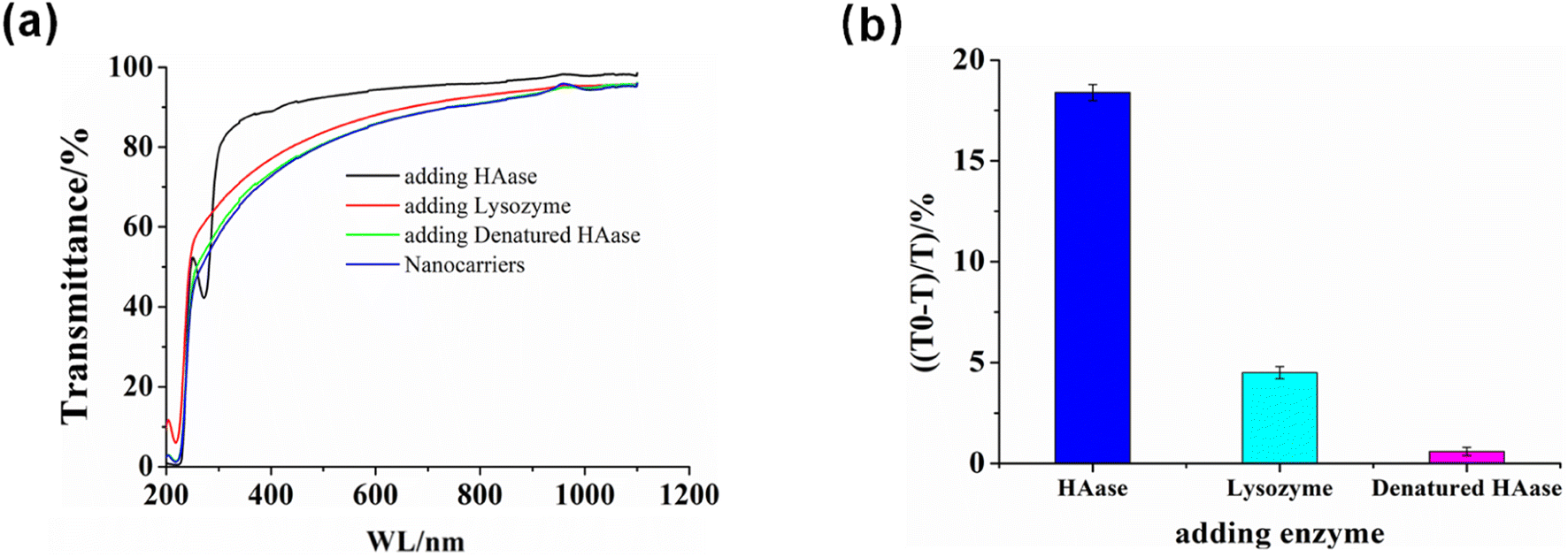
Due to HA being the building block of HA/(MI)7-β-CD NCs, we expected HA/(MI)7-β-CD NCs could respond to hyaluronidase (HAase) specifically. The enzyme-responsive of HA/(MI)7-β-CD NCs was investigated by recording the optical transmittance of HA/(MI)7-β-CD NCs solution after adding HAase, lysozyme (LZM), and denatured HAase. This is shown in Fig. 3, the optical transmittance of HA/(MI)7-β-CD NCs enhanced from 77.25% to 91.46% with adding HAase for 1 h at 25 °C, while showed negligible changes after adding LZM and denatured HAase, indicated the HAase-induced specific disassemble ability of HA/(MI)7-β-CD NCs. TEM images were used to trace the disassembly processes. As shown in Fig. S7a,† no spherical particles were observed after adding HAase for 1 h at 25 °C. The reason that HA/(MI)7-β-CD NCs disassembled could be attributed to the HA chains being cleaved into small fragments by HAase, leading to the increase of water solubility. Moreover, the disassembly rate of HA/(MI)7-β-CD NCs was studied by recording the optical transmittance by adding HAase into HA/(MI)7-β-CD NCs solution and maintaining the various culture time (Fig. S7b†). The optical transmittance of HA/(MI)7-β-CD NCs solution increased from 79% to 87% and 93% in 0.5 and 1 h, respectively, suggesting HA/(MI)7-β-CD NCs could respond to HAase rapidly. Thus, the HAase-responsive properties of HA/(MI)7-β-CD NCs were the basis for achieving controlled drug release.
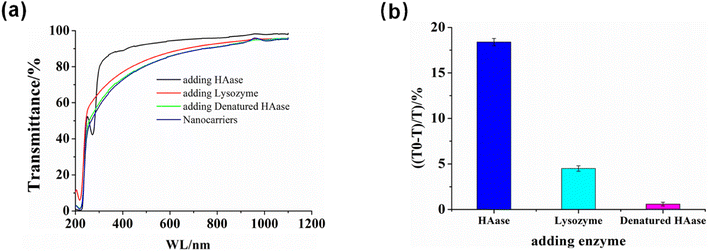 |
| | Fig. 3 The enzymes-responsive performance of HA/(MI)7-β-CD NCs. (a) Optical transmittance curves of HA/(MI)7-β-CD NCs solution were obtained by adding HAase, denatured HAase, and LZM at 25 °C for 1 h. (b) The relationship of the transmittance changes at 450 nm in (a) versus the enzyme type. [HA] = 20 μg mL−1, [(MI)7-β-CD] = 14.73 μg mL−1, [HAase] = 5 mg mL−1 and [LZM] = 5 mg mL−1. | |
3.3 Drug loaded and HAase-induced release behavior studies
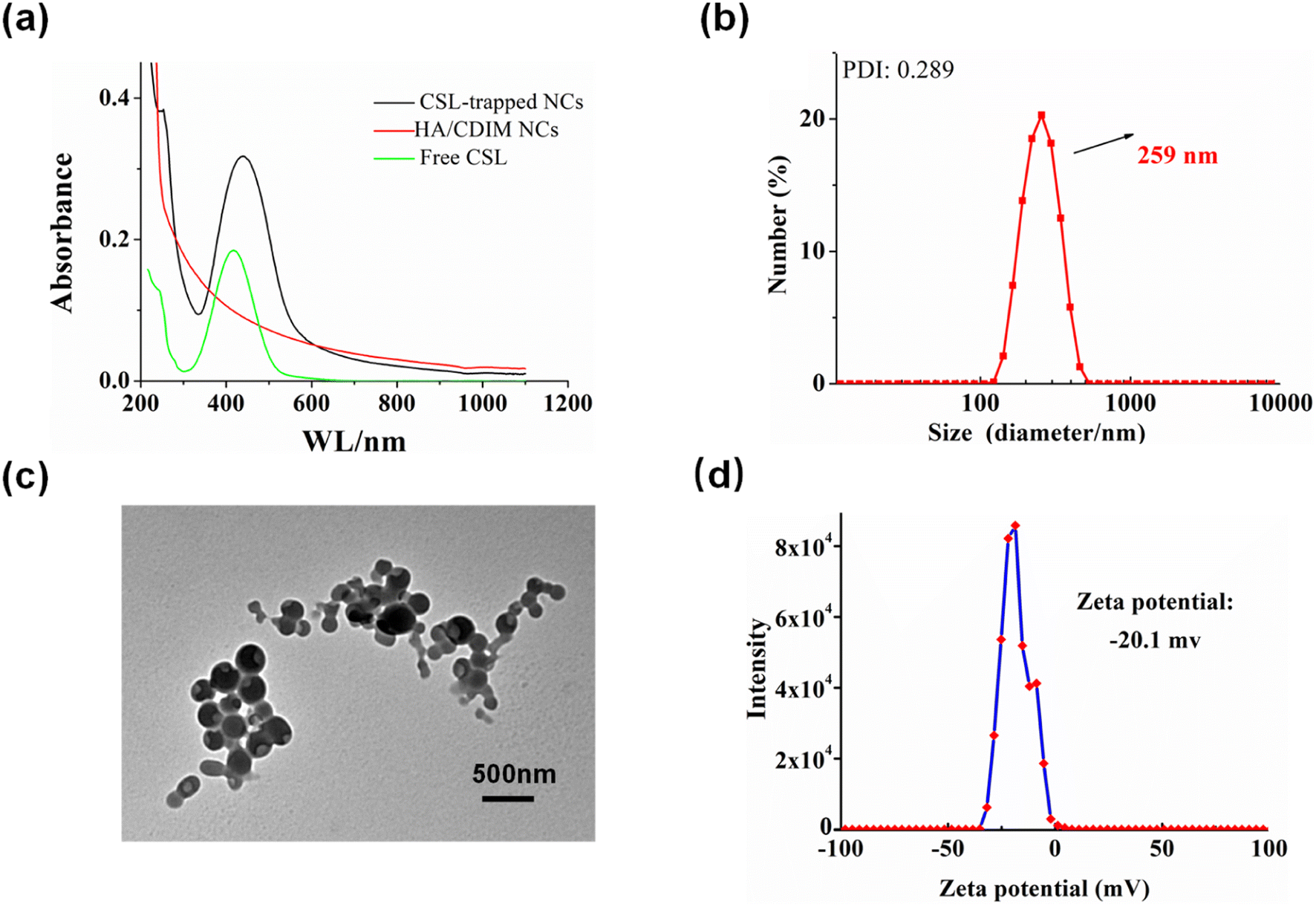
The HAase-induced disassembly of HA/(MI)7-β-CD NCs provided a potential pathway for the release of the antitumor drug CSL. The loading and release properties of HA/(MI)7-β-CD NCs were investigated by recording UV-vis curves. As shown in Fig. 4a, CSL-trapped HA/(MI)7-β-CD NCs showed the enhanced characteristic absorption peak, revealing that CSL was successfully loaded into HA/(MI)7-β-CD NCs. Meanwhile, the dynamic light scattering (DLS) and transmission electron microscopy (TEM) indicated that the average diameter of the CSL-trapped NCs was significantly larger than those of CSL-untrapped NCs (see Fig. 4b and c). DLS showed that the particle size of CSL-trapped NCs was about 259 nm, and the PDI index was 0.289. TEM showed that the particle size was consistent with that of DLS. The reason can be attributed to the fact that CSL was captured inside HA/(MI)7-β-CD NCs, which in turn occupies part of the space of HA/(MI)7-β-CD NCs, resulting in an increased diameter. The zeta potential of CSL-trapped HA/(MI)7-β-CD NCs decreased from −18.0 to −20.1 mV contrasting with HA/(MI)7-β-CD NCs, which may be due to the partially negatively charged CSL located on the surface of HA/(MI)7-β-CD NCs. The stability of CSL-trapped NCs was characterized by continuously recording the UV-vis absorbance curves for 16 h. As shown in Fig. S8,† no significant absorbance changes were observed showing that CSL-trapped NCs can exist stably at room temperature. In short, the above results proved that CSL was successfully trapped in HA/(MI)7-β-CD NCs.
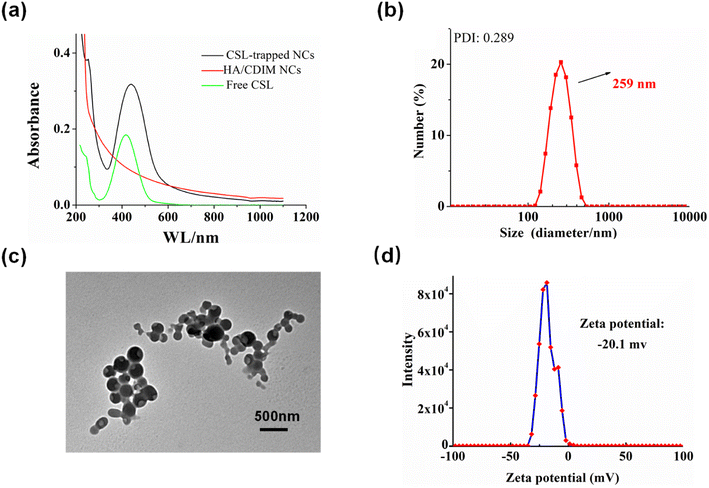 |
| | Fig. 4 The characterization of CSL-trapped HA/(MI)7-β-CD NCs, UV-vis absorption curves (a), DLS results (b), TEM image (c), and zeta potential (d). | |
The encapsulation efficiency (EE%) and loading efficiency (LE%) of HA/(MI)7-β-CD NCs on CSL were calculated according to formulas (1) and (2) and the standard curve of drugs. The LE% and EE% of HA/(MI)7-β-CD NCs on CSL were 25% and 33.42%, respectively, which indicate HA/(MI)7-β-CD NCs could be used as a drug delivery system.
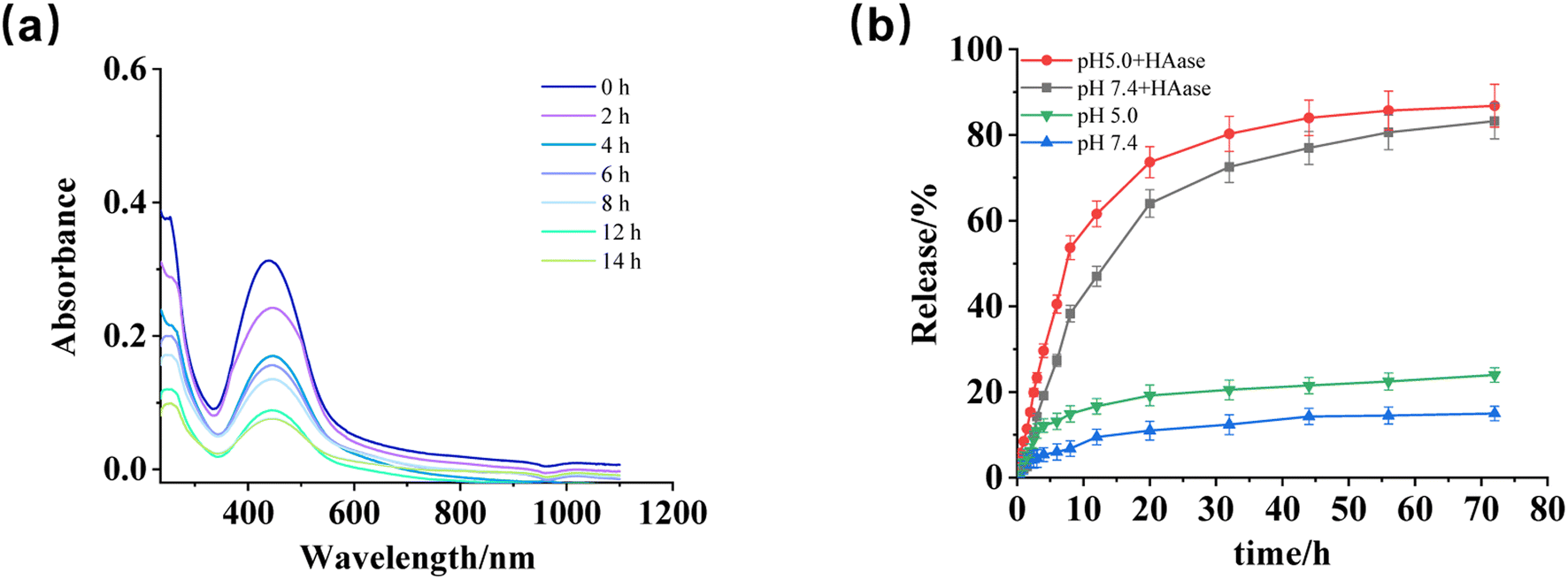
The release behavior of CSL-trapped HA/(MI)7-β-CD NCs was studied by recording the absorbance changes within 72 h under different conditions (pH, enzyme). As shown in Fig. 5a, when pH was 5 and HAase was added, the absorbance curves decreased gradually with treatment time, indicating pH and HAase triggered the depolymerization of CSL-trapped NCs, as well as led to the release of CSL. Furthermore, the above results were compared with the absorbance of CSL-trapped NCs in different environments (pH 7.4 with HAase, pH 5, and pH 7.4). The pictures were displayed in Fig. S9a–c,† and the drug release rate were calculated. As depicted in Fig. 5b. In addition, in order to better simulate the tumor environment, it was found that the release rate of CSL increased to 86.82% when pH was set at 5.0 and HAase was present. Compared with pH 7.4, the release rate was slightly increased, indicating that the drug release behavior of the nanocarriers was mainly controlled by HAase. The results indicated that CSL-trapped NCs possessed HAase-triggered release properties, which enabled the release of CSL at the HAase overexpression site.
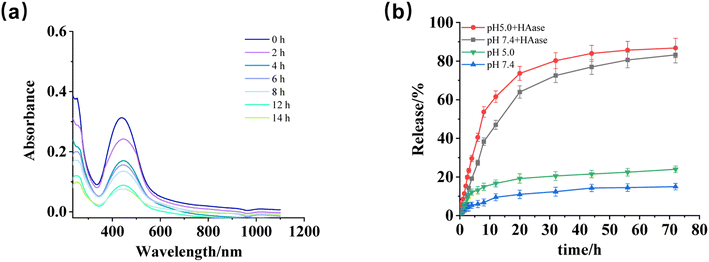 |
| | Fig. 5 (a) UV-vis absorbance curves of CSL-trapped HA/(MI)7-β-CD NCs at various times after adding HAase under pH5. (b) The release rate was obtained after treating CSL-trapped NCs solution with HAase at pH of 5.0 and 7.4 for 72 h. | |
3.4 Cytotoxicity assay
The cytotoxicity activity and antitumor efficiency of CSL-trapped HA/(MI)7-β-CD NCs against five human cancer cell lines (SMMC-7721, SW480, HL-60, MCF-7, and A-549 cells) and normal lung human epithelial cells (BEAS-2B) were researched by MTT assay.53 Cisplatin and Taxol were used as control groups. As shown in Table 1, HA/(MI)7-β-CD NCs showed IC50 of ∼41–65 μg L−1 for five tumor cells and ∼45 μg L−1 for normal cells, proving that the NCs showed no obvious toxicity to normal cells and cancer cells. IC50 value of some cancer cells (MCF-7, SW480) was slightly higher than that of normal cells, which might be caused by the intrinsic drug resistance of cancer cells.54,55 CSL-trapped NCs and free CSL displayed higher cytotoxicity to the five tumor cells compared to cisplatin, suggesting that both CSL-trapped NCs and free CSL can effectively inhibit the growth of tumor cells. The overall antitumor activity of CSL-trapped NCs against tumor cells was lower than that of free CSL. The possible reason is that the content of CSL trapped in HA/(MI)7-β-CD NCs is lower than free CSL in the same mass of the sample. The above results demonstrated that CSL-trapped HA/(MI)7-β-CD NCs showed favorable performance in inhibiting cancer cells, as well as low toxicity to normal cells, which exhibited great promise in the treatment of cancer.
Table 1 The cytotoxicity of CSL, HA/(MI)7-β-CD NCs and CSL-trapped NCs, cisplatin and Taxol against five human cancer cell lines and the normal cells BEAS-2B
| Compounds |
IC50 ± SD (μg L−1) |
| HL-60 |
A549 |
SMMC-7721 |
MCF-7 |
SW480 |
BEAS-2B |
| HA/(MI)7-β-CD NCs |
41.850 ± 0.780 |
45.360 ± 0.901 |
45.077 ± 0.823 |
65.493 ± 0.785 |
64.636 ± 0.898 |
44.340 ± 0.820 |
| CSL-trapped NCs |
0.851 ± 0.090 |
0.833 ± 0.068 |
0.359 ± 0.005 |
3.040 ± 0.037 |
1.973 ± 0.037 |
1.864 ± 0.043 |
| CSL |
0.201 ± 0.011 |
0.387 ± 0.012 |
0.072 ± 0.006 |
1.071 ± 0.093 |
0.435 ± 0.038 |
0.379 ± 0.004 |
| Cisplatin |
8.245 ± 0.306 |
25.630 ± 0.530 |
16.830 ± 0.660 |
20.000 ± 1.140 |
22.250 ± 0.190 |
>40 |
| Taxol |
<0.008 |
<0.008 |
0.558 ± 0.030 |
<0.008 |
<0.008 |
1.833 ± 0.090 |
3.5 Cell apoptosis analysis
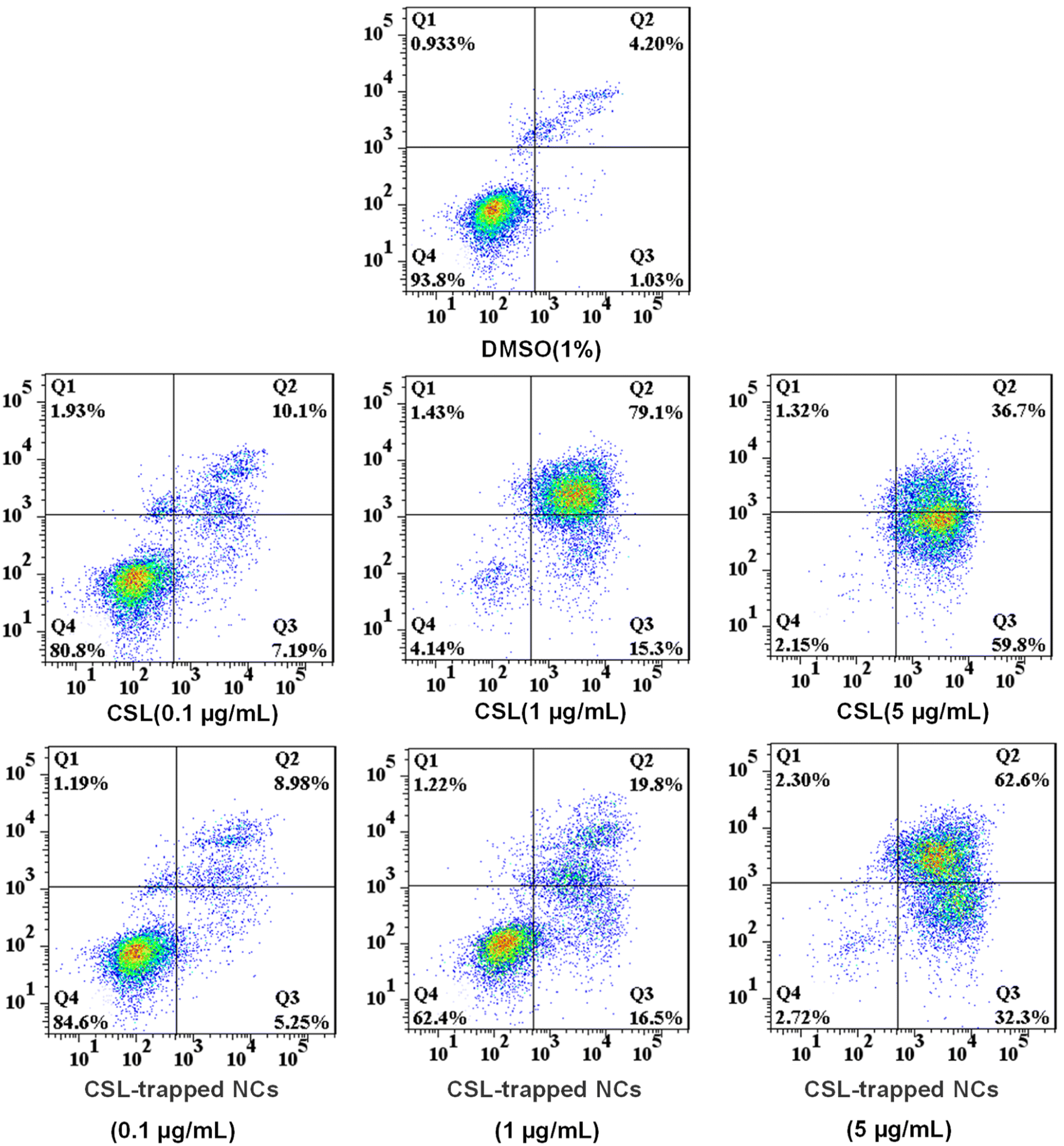
Since CSL-trapped NCs exhibited good toxicity to liver cancer cells SMMC-7721, we chose SMMC-7721 as model cells to investigate the apoptosis rate of CSL-trapped NCs. Apoptosis files of SMMC-7721 were investigated by PE Annexin V-APC/7-AAD double staining assay with incubating SMMC-7721 with various concentrations of free CSL and CSL-trapped NCs. As shown in Fig. 6, the apoptosis rate showed a concentration-dependent relationship, i.e., it increased from 17.29% to 94.40% and 96.50% for free CSL, and from 14.23% to 36.30% and 94.90% for CSL-trapped NCs with the concentration of free CSL and CSL-trapped NCs increasing from 0.1 μg mL−1 to 5 μg mL−1. Notably, when the concentration of CSL-trapped NCs was 5 μg mL−1, the apoptosis rate was similar to that of free CSL even though the CSL content in the CSL-trapped NCs was lower than that of free CSL. The reason could be attributable to the ability of nanocarriers to targeted delivery CSL compared with free CSL, resulting in an increased cumulative concentration of CSL in SMMC-7721 cells.
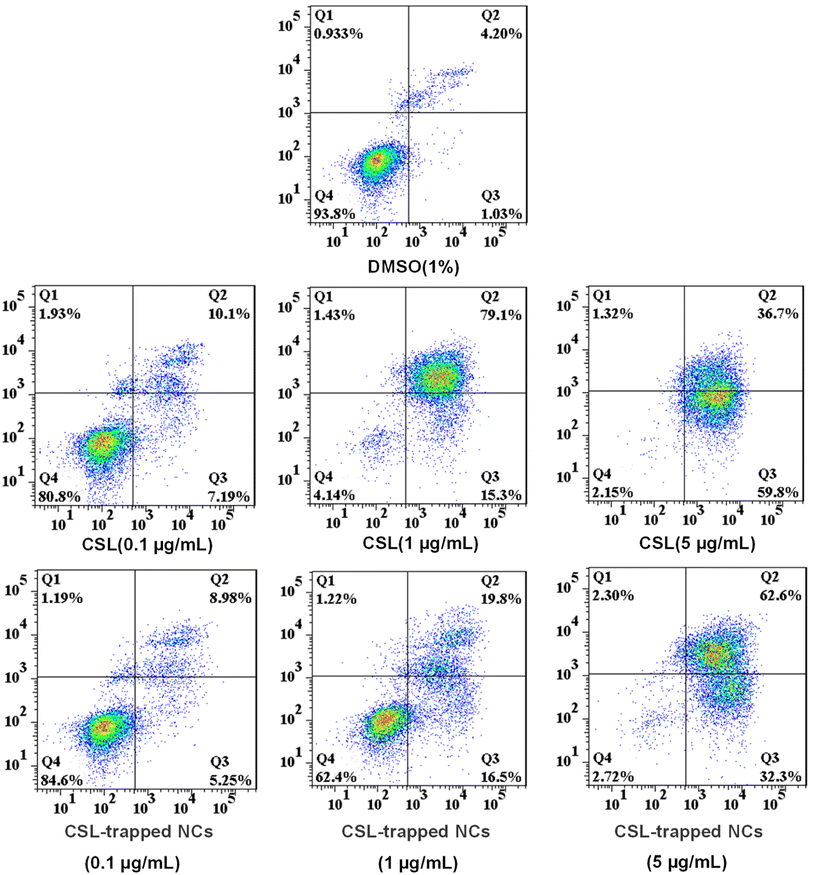 |
| | Fig. 6 The apoptotic activity for treating SMMC-7721 cells with different concentrations of free CSL and CSL-trapped HA/(MI)7-β-CD NCs (0.1 μg mL−1, 1 μg mL−1, and 5 μg mL−1) for 24 h. Graphics are divided into four quadrants: (Q1) necrosis cells, (Q2) late apoptotic cells, (Q3) early apoptotic cells, and (Q4) live cells. | |
3.6 Cellular uptake
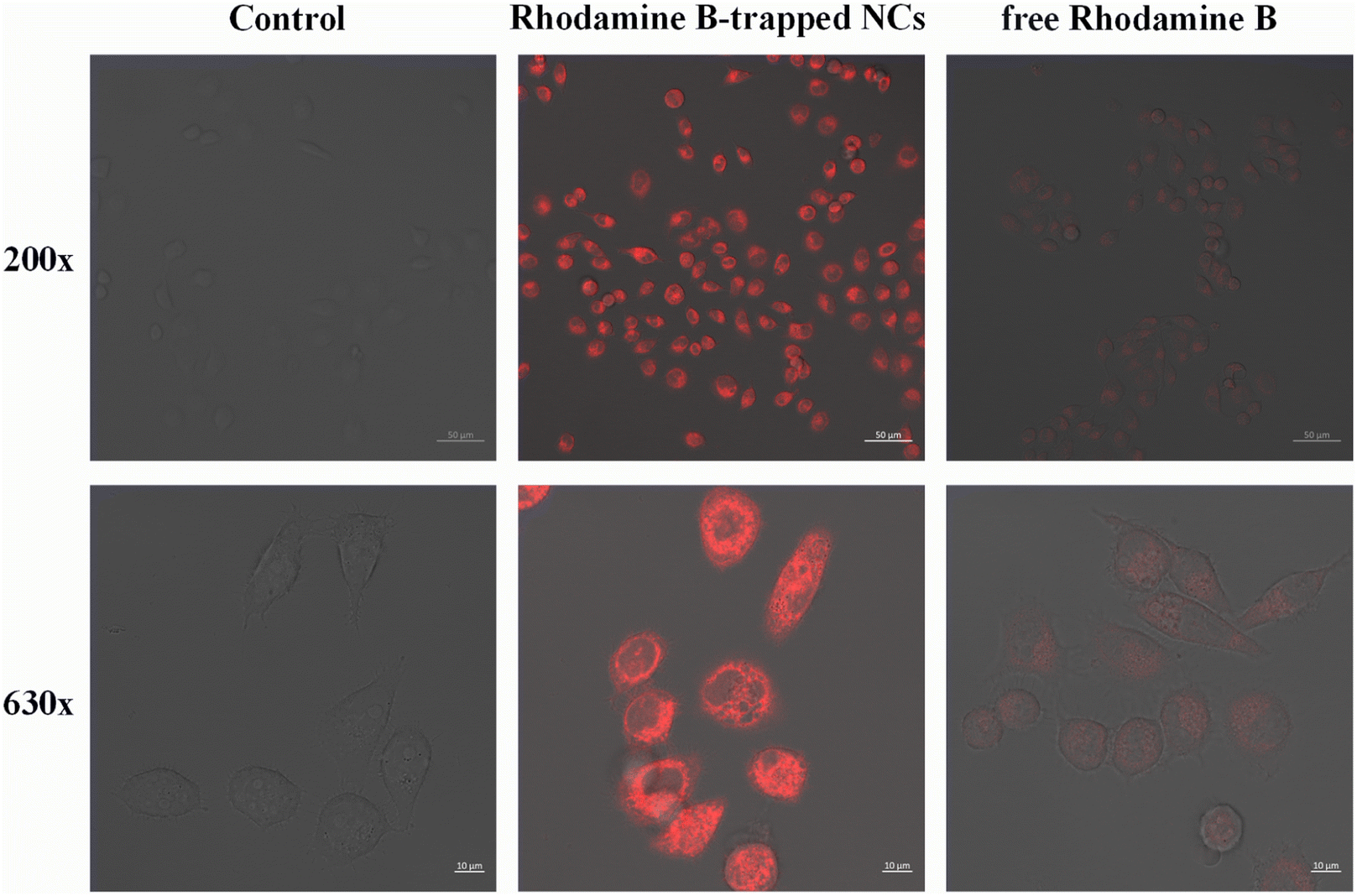
To evaluate the feasibility of targeted delivery of HA/(MI)7-β-CD NCs, we replaced non-fluorescent CSL with Rhodamine-B and selected CD44 receptor overexpressed tumor cells SMMC-7721 for cell uptake studies. As shown in Fig. 7, after the sample was incubated with the SMMC-7721 cells for 4 hours, red fluorescence appeared in the cells, and the fluorescence intensity of Rhodamine B-trapped NCs was significantly higher than that of free Rhodamine B, which indicated that the NCs had a highly specific binding ability to CD44 receptors and were internalized through receptor-mediated endocytosis. Therefore, the HA/(MI)7-β-CD NCs could accurately transport drugs into SMMC-7721 cells and effectively increase the cumulative concentration of drugs in the cells.
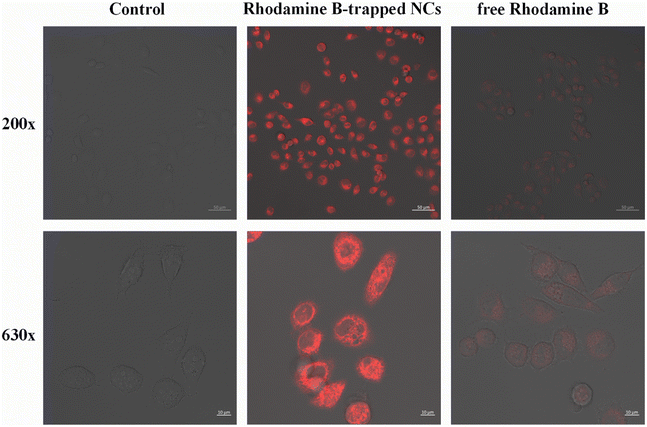 |
| | Fig. 7 Laser scanning confocal microscope images of SMMC-7721 cells at 200× and 630× magnification after incubating with Rhodamine-B-trapped NCs and free Rhodamine-B for 4 h, respectively (the scale is 50 μm and 10 μm, and [Rhodamine-B-trapped NCs] = 5 μg mL−1, [Rhodamine-B] = 5 μg mL−1). | |
4 Conclusion
In conclusion, we designed and synthesized HAase-responsive supramolecular nanocarriers, HA/(MI)7-β-CD NCs, based on electrostatic interaction between negatively charged HA and positively charged (MI)7-β-CD. HA/(MI)7-β-CD NCs exhibit rapid depolymerization behavior in the presence of HAase, which was used as a nanocarrier for the capture, delivery, and release of the anti-hepatocellular carcinoma drug CSL. In cellular experiments, CSL-trapped NCs not only reduced cytotoxicity for normal cells but also showed good performance in therapeutic effect for five tumor cells. Furthermore, when SMMC-7721 cells were treated with CSL-trapped NCs and free CSL at a concentration of 5 μg mL−1, respectively, CSL-trapped NCs exhibited apoptotic rates similar to those of free CSL. The above results suggested that CSL-trapped HA/(MI)7-β-CD NCs may be potentially useful in the field of anti-liver cancer drug targeting delivery.
Data availability
The data that support the findings of this study are available from the corresponding author upon reasonable request.
Author contributions
Junxin Xu: conceptualization, methodology, investigation, formal analysis, writing-original draft preparation. Siling Chen: investigation, formal analysis. Jianmei Yang: investigation, visualization. Zhengquan Nie: investigation, formal analysis. Junnan He: software, validation. Yong Zhao: investigation, visualization. Jin Zhang: writing-reviewing and editing, supervision, funding acquisition. Yan Zhao: writing-reviewing and editing, supervision, funding acquisition.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
This work was supported by the National Natural Science Foundations (no. 21961046, 21362046, and 21062030) and Yunnan Normal University Doctoral Research Initiation Program (no. 01100205020503180), which are gratefully acknowledged.
References
- P. Rawla, T. Sunkara, P. Muralidharan and J. P. Raj, Contemp. Oncol., 2018, 22, 141–150 CAS.
- K. D. Miller, L. Nogueira, A. B. Mariotto, J. H. Rowland, K. R. Yabroff, C. M. Alfano, A. Jemal, J. L. Kramer and R. L. Siegel, Ca-Cancer J. Clin., 2019, 69, 363–385 CrossRef PubMed.
- S. Y. Qin, A. Q. Zhang, S. X. Cheng, L. Rong and X. Z. Zhang, Biomaterials, 2017, 112, 234–247 CrossRef CAS PubMed.
- M. H. Abu Bakar, M. R. Sarmidi, J. S. Tan and M. N. Mohamad Rosdi, Eur. J. Pharmacol., 2017, 799, 73–83 CrossRef CAS PubMed.
- W. Zhang, Z. Wu, H. Qi, L. Chen, T. Wang, X. Mao, H. Shi, H. Chen, M. Zhong, X. Shi, X. Wang and Q. Li, Phytomedicine, 2022, 104, 154280 CrossRef CAS PubMed.
- Y. Wang, Inflammation Res., 2021, 70, 323–341 CrossRef CAS PubMed.
- D. Fan, S. Parhira, G. Y. Zhu, Z. H. Jiang and L. P. Bai, Fitoterapia, 2016, 113, 69–73 CrossRef CAS PubMed.
- S. H. Venkatesha, S. Dudics, B. Astry and K. D. Moudgil, Pathog. Dis., 2016, 74, 6 Search PubMed.
- R. Cascao, J. E. Fonseca and L. F. Moita, Front. Med., 2017, 4, 69 CrossRef PubMed.
- Z. T. Jiang, Y. Han, X. Y. Liu, L. Y. Lv, J. H. Pan and C. D. Liu, OncoTargets Ther., 2020, 13, 2973–2985 CrossRef CAS PubMed.
- X. Du, X. He, Y. Huang, B. Fu, B. Liang, C. Lv, Y. Li, X. Gao and Y. Wang, J. Chromatogr. B: Biomed. Sci. Appl., 2019, 1113, 1–13 CrossRef CAS PubMed.
- X. Xiao, Z. Xu, W. Wang, S. Sun, Y. Qiao, L. Jiang, Y. Yan and J. Huang, Langmuir, 2021, 37, 8348–8355 CrossRef CAS PubMed.
- T. Liu, Q. Wan, C. Zou, M. Chen, G. Wan, X. Liu and H. Chen, Chem. Eng. J., 2021, 417, 128004 CrossRef CAS.
- B.-L. Li, J. Zhang, W. Jin, X.-Y. Chen, J.-M. Yang, S.-M. Chi, Q. Ruan and Y. Zhao, React. Funct. Polym., 2022, 172, 105175 CrossRef CAS.
- C. Ji, Y. Deng, H. Yuan, W. Yuan, Y. Song and Z. Li, Macromol. Mater. Eng., 2021, 306, 610533 CrossRef.
- M. A. Tovar, A. Parkhurst, E. Matuczinski, S. Balenger and L. C. Giancarlo, Nanotechnology, 2019, 30, 465101 CrossRef CAS PubMed.
- M. P Daryasari, M. R. Akhgar, F. Mamashli, B. Bigdeli and M. Khoobi, RSC Adv., 2016, 107, 104949–106045 Search PubMed.
- R. Deng, H. Yi, F. Fan, L. Fu, Y. Zeng, Y. Wang, Y. Li, Y. Liu, S. Ji and Y. Su, RSC Adv., 2016, 6, 77083–77092 RSC.
- J. Zhang, S. Chen, J. Teng, B. Li, L. Wang, J. Yang and Y. Zhao, Macromol. Chem. Phys., 2022, 223, 2200028 CrossRef CAS.
- H. Li, Q. Li, W. Hou, J. Zhang, C. Yu, D. Zeng, G. Liu and F. Li, ACS Appl. Mater. Interfaces, 2019, 11, 43581–43587 CrossRef CAS PubMed.
- G. Iaccarino, M. Profeta, R. Vecchione and P. A. Netti, Acta Biomater., 2019, 89, 265–278 CrossRef CAS PubMed.
- Z. T. Zhang, M. Y. Huang-Fu, W. H. Xu and M. Han, Eur. J. Pharm. Biopharm., 2019, 137, 122–130 CrossRef CAS PubMed.
- Y. Li, H. Hu, Q. Zhou, Y. Ao, C. Xiao, J. Wan, Y. Wan, H. Xu, Z. Li and X. Yang, ACS Appl. Mater. Interfaces, 2017, 9, 19215–19230 CrossRef CAS PubMed.
- S. Li, L. Chen, K. Huang, N. Chen, Q. Zhan, K. Yi, H. Qi, C. Liu, Y. Tan, X. Hou, Y. Lu, J. Zhao, X. Yuan and C. Kang, Adv. Funct. Mater., 2019, 29, 1903296 CrossRef CAS.
- H. Zhang, J. Fei, X. Yan, A. Wang and J. Li, Adv. Funct. Mater., 2015, 25, 1193–1204 CrossRef CAS.
- A. Kaul, W. D. Short, X. Wang and S. G. Keswani, Int. J. Mol. Sci., 2021, 22, 3204 CrossRef CAS PubMed.
- H. C. Inan, M. Yener, N. Buyru, A. Celebi, M. Yilmaz and N. Comunoglu, Clin. Otolaryngol., 2019, 44, 914–918 CrossRef PubMed.
- Z. Liang, Q. Zhang, C. Wang, F. Shi, H. Cao, Y. Yu, M. Zhang and X. Liu, Neoplasma, 2017, 64, 901–908 CrossRef CAS PubMed.
- E. J. Franzmann, G. L. Schroeder, W. J. Goodwin, D. T. Weed, P. Fisher and V. B. Lokeshwar, Int. J. Cancer, 2003, 106, 438–445 CrossRef CAS PubMed.
- Z. Mazidi, S. Javanmardi, S. M. Naghib and Z. Mohammadpour, Chem. Eng. J., 2022, 433, 134569 CrossRef CAS.
- N. Schatz-Siemers, Y.-T. Chen, Z. Chen, D. Wang, L. H. Ellenson and Y.-C. N. Du, Appl. Immunohistochem. Mol. Morphol., 2020, 28, 453–459 CrossRef CAS PubMed.
- E. Chen, S. Han, B. Song, L. Xu, H. Yuan, M. Liang and Y. Sun, Int. J. Nanomed., 2020, 15, 6311–6324 CrossRef CAS PubMed.
- M. Grandoch, P. L. Bollyky and J. W. Fischer, Circ. Res., 2018, 122, 1341–1343 CrossRef CAS PubMed.
- F. M. Almutairi, A. A. Abd-Rabou and M. S. Mohamed, Bioorg. Med. Chem., 2019, 27, 1629–1638 CrossRef CAS PubMed.
- Z. Luo, Y. Dai and H. Gao, Acta Pharm. Sin. B, 2019, 9, 1099–1112 CrossRef PubMed.
- V. Quagliariello, A. Gennari, S. A. Jain, F. Rosso, R. V. Iaffaioli, A. Barbarisi, M. Barbarisi and N. Tirelli, Mater. Sci. Eng. C, 2021, 131, 112475 CrossRef CAS PubMed.
- P. Heldin, C. Kolliopoulos, C. Y. Lin and C. H. Heldin, Cell. Signalling, 2020, 65, 109427 CrossRef CAS PubMed.
- S. Bhattacharya, D. Singh, J. Aich, Ajazuddin and M. B. Shete, Mater. Today Commun., 2022, 31, 103757 CrossRef CAS.
- S. W. Kim, K. T. Oh, Y. S. Youn and E. S. Lee, Colloids Surf., B, 2014, 116, 359–364 CrossRef CAS PubMed.
- R. Panday, A. M. E. Abdalla, Y. Miao, X. Li, M. Neupane, C. Ouyang and G. Yang, J. Bioact. Compat. Polym., 2020, 35, 504–516 CrossRef CAS.
- Z. Wu, Y. Liu, J. Cao, L. Huang and X. Luo, Macromol. Mater. Eng., 2018, 303, 1800330 CrossRef.
- S. Sargazi, U. Laraib, M. Barani, A. Rahdar, I. Fatima, M. Bilal, S. Pandey, R. K. Sharma and G. Z. Kyzas, J. Mol. Struct., 2022, 1261, 132922 CrossRef CAS.
- Y. Zhou, Y. Zhao, B. Niu, Q. Luo, Y. Zhang, G. Quan, X. Pan and C. Wu, Int. J. Pharm., 2020, 588, 119777 CrossRef CAS PubMed.
- A. Rajan, B. Kaczmarek-Szczepańskac and N. K. Sahu, Mater. Today Commun., 2021, 28, 102583 CrossRef CAS.
- B. Tian, S. Hua and J. Liu, Carbohydr. Polym., 2020, 232, 115805 CrossRef CAS PubMed.
- J. Rincón-López, Y. C. Almanza-Arjona, A. P. Riascos and Y. Rojas-Aguirre, J. Drug Delivery Sci. Technol., 2021, 61, 102156 CrossRef PubMed.
- Y. Chen, H. Zhang, X. Cai, J. Ji, S. He and G. Zhai, RSC Adv., 2016, 6, 92073–92091 RSC.
- G. Mohammadi Ziarani, M. Malmir, N. Lashgari and A. Badiei, RSC Adv., 2019, 9, 25094–25106 RSC.
- Y. Bai, C. P. Liu, D. Chen, C. F. Liu, L. H. Zhuo, H. Li, C. Wang, H. T. Bu and W. Tian, Carbohydr. Polym., 2020, 246, 116654 CrossRef CAS PubMed.
- P. Singh, Y. Chen, D. Tyagi, L. Wu, X. Ren, J. Feng, A. Carrier, T. Luan, Y. Tang, J. Zhang and X. Zhang, Int. J. Pharm., 2021, 602, 120602 CrossRef CAS PubMed.
- D. Zhao, Y. Chen and Y. Liu, Chin. Chem. Lett., 2015, 26, 829–833 CrossRef CAS.
- S. Chen, F. D. Zhu, B. L. Li, J. M. Yang, T. Yang, X. Q. Liu, J. Zhang and Y. Zhao, J. Appl. Polym. Sci., 2022, 139, 53163 CrossRef.
- A. Dos Santos Correa, L. A. Contreras, W. J. Keijok, D. H. F. Barcelos, A. C. H. Pereira, R. R. Kitagawa, R. Scherer, D. C. de Oliveira Gomes, A. R. da Silva, D. C. Endringer, J. P. de Oliveira and M. C. C. Guimaraes, Mater. Sci. Eng. C, 2018, 91, 853–858 CrossRef CAS PubMed.
- J. Khaled, M. Kopsida, H. Lennernas and F. Heindryckx, Cells, 2022, 11, 632 CrossRef CAS PubMed.
- Y. Liu, Q. Bao, Z. Chen, L. Yao, Z. Ci, X. Wei, Y. Wu, J. Zhu, K. Sun, G. Zhou, S. Li, W. Ma and K. Tao, Nano Lett., 2021, 21, 9115–9123 CrossRef CAS PubMed.
|
| This journal is © The Royal Society of Chemistry 2023 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence *a
*a