
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Pyrazole-based lamellarin O analogues: synthesis, biological evaluation and structure–activity relationships†
Karolina Dzedulionytė a,
Nina Fuxreiterb,
Ekaterina Schreiber-Brynzakb,
Asta Žukauskaitėc,
Algirdas Šačkusad,
Verena Pichler*b and
Eglė Arbačiauskienė*a
a,
Nina Fuxreiterb,
Ekaterina Schreiber-Brynzakb,
Asta Žukauskaitėc,
Algirdas Šačkusad,
Verena Pichler*b and
Eglė Arbačiauskienė*a
aDepartment of Organic Chemistry, Faculty of Chemical Technology, Kaunas University of Technology, Radvilėnų pl. 19, LT-50254 Kaunas, Lithuania. E-mail: egle.arbaciauskiene@ktu.lt
bDepartment of Pharmaceutical Sciences, Division of Pharmaceutical Chemistry, Faculty of Life Sciences, University of Vienna, Althanstraße 14, 1090 Vienna, Austria. E-mail: verena.pichler@univie.ac.at
cDepartment of Chemical Biology, Faculty of Science, Palacký University, Šlechtitelů 27, CZ-78371 Olomouc, Czech Republic
dInstitute of Synthetic Chemistry, Faculty of Chemical Technology, Kaunas University of Technology, K. Baršausko g. 59, LT-51423 Kaunas, Lithuania
First published on 10th March 2023
Abstract
A library of pyrazole-based lamellarin O analogues was synthesized from easily accessible 3(5)-aryl-1H-pyrazole-5(3)-carboxylates which were subsequently modified by bromination, N-alkylation and Pd-catalysed Suzuki cross-coupling reactions. Synthesized ethyl and methyl 3,4-diaryl-1-(2-aryl-2-oxoethyl)-1H-pyrazole-5-carboxylates were evaluated for their physicochemical property profiles and in vitro cytotoxicity against three human colorectal cancer cell lines HCT116, HT29, and SW480. The most active compounds inhibited cell proliferation in a low micromolar range. Selected ethyl 3,4-diaryl-1-(2-aryl-2-oxoethyl)-1H-pyrazole-5-carboxylates were further investigated for their mode of action. Results of combined viability staining via Calcein AM/Hoechst/PI and fluorescence-activated cell sorting data indicated that cell death was triggered in a non-necrotic manner mediated by mainly G2/M-phase arrest.
Introduction
Lamellarins are a group of natural marine-derived alkaloids with a characteristic central pyrrole moiety and widely reported biological activity. Since their discovery in 1985, more than 50 compounds of this family have been isolated from various marine organisms, mainly, but not exclusively, sponges and ascidians.1–3 Most lamellarins contain 6H[1]benzo-pyrano[4′,3′:4,5]pyrrolo[2,1-a]isoquinolinone chromophore and fall into two subtypes – 5,6-saturated and 5,6-unsaturated lamellarins, designated as type Ia and type Ib, respectively. Structurally less complex type II lamellarins form a smaller class of compounds bearing 3,4-diarylpyrrole-2-carboxylate fragment (Fig. 1).4,5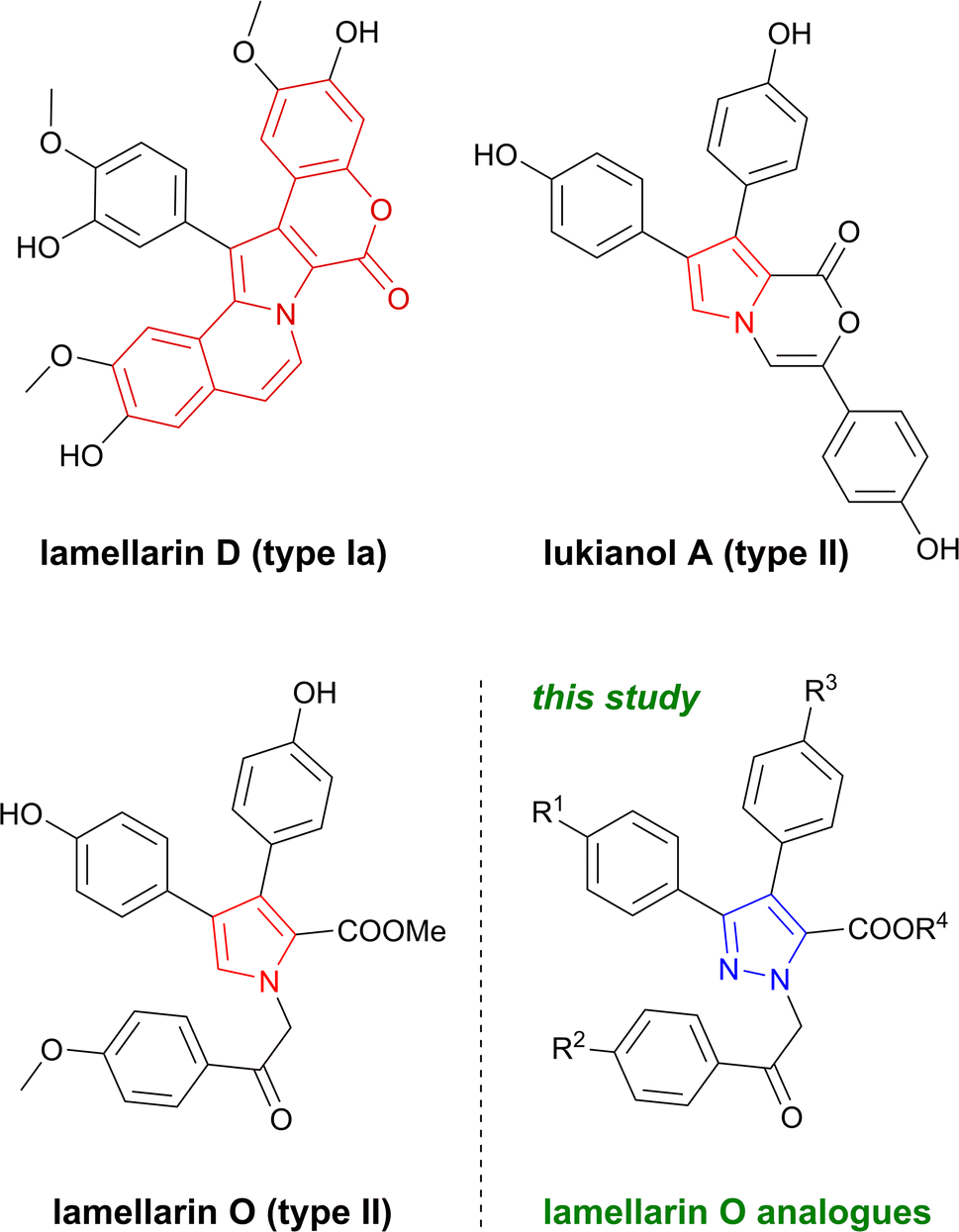 | ||
| Fig. 1 Relevant lamellarin alkaloid representatives of type I and type II bearing a characteristic central pyrrole core and novel pyrazole containing analogues. | ||
The biological spectrum of lamellarins is manifold, with pronounced cytotoxic activity.6–11 In detail, lamellarin D (Fig. 1), a leading compound in the family of type I, induces its anticancer activity through topoisomerase I inhibition and mitochondrial targeting that triggers cell death.12–15 Lamellarin O was investigated by Huang et al.16 As reported, cytotoxicity effects of some natural lamellarins were assessed towards colorectal cancer SW620 and its multi-drug resistant daughter cell line SW620/Ad300. Lamellarin O exhibited moderate cytotoxicity against aforementioned cells at IC50 of 20.0 μM and 22.3 μM respectively. Whereas lukianol A, a natural cyclized lactone of lamellarin O, is known to exhibit cytotoxicity against human epidermoid carcinoma cell lines.17 Other lamellarins can reverse multidrug resistance and consequently promote therapeutic activity of conventional cytotoxic drugs towards chemoresistant tumours.5,18
The unbridled key goal is, besides appropriate strategies to synthetically approach these natural compounds,17,19–21 to create new analogues as innovative drug-like compounds with potent antitumor activities.22–24 Zheng et al. reported synthesis and investigation of novel glycosylated lamellarin D compounds wherein glycosyl moieties improve important physicochemical properties of active compounds, especially the solubility in water.25 Another study revealed A-ring modified lamellarin N analogues as potent noncovalent inhibitors of EGFR T790M/L858R mutant, which is responsible for non-small cell lung cancer resistance.26 More recent investigation on A-ring modified azalamellarins revealed that synthetic analogues selectively inhibit the proliferation of EGFR T790M/L858R mutant cells over EGFR WT cells.27 Moreover, Klumthong et al. presented a diversity-oriented synthesis of azalamellarins, where lactone-to-lactam modification resulted in increased cytotoxicity against HeLa cervical cancer cells.28
Following these significant discoveries, interest in lamellarin based research has been growing and remains highly relevant. The structure of lamellarin O is easily accessible for replacement of the central pyrrole ring to design new derivatives with structural similarities like shape and electronic configuration by other five membered ring systems. Pyrazole is a versatile moiety taking place in various biologically active compounds as well as in-use pharmaceuticals.29–31 Replacement of the central pyrrole to pyrazole is expected to change e.g., the energy of the highest occupied molecular orbital (HOMO) which is associated with increased metabolic stability.32
In continuation of our previous works devoted to synthesis and investigation of pyrazole derivatives,33–41 in this study we ought to synthesize and investigate various functionalized pyrazole derivatives of lamellarin O. The goal was based on the scaffold hopping of the pyrrole ring in natural lamellarin O to its pyrazole counterpart. Synthetic strategy involves 3,5-substituted pyrazole formation and pyrazole functionalization at 4-position by Pd-catalysed Suzuki cross-coupling reaction. Obtained compounds were evaluated for their physicochemical properties and further investigated as potent agents against human colon cancer cell lines HCT116, HT29 and SW480. Moreover, after structure–activity relationship determination, the most cytotoxic compounds were used to investigate their mode of action in the before mentioned cell lines.
Results and discussion
Chemistry
As outlined in Scheme 1, Claisen condensation of commercially available acetophenones 1a–c with diethyl oxalate and subsequent cyclocondensation with hydrazine hydrate afforded 3(5)-aryl-1H-pyrazole-5(3)-carboxylates 2a–c in good yields as previously described by Wu et al.42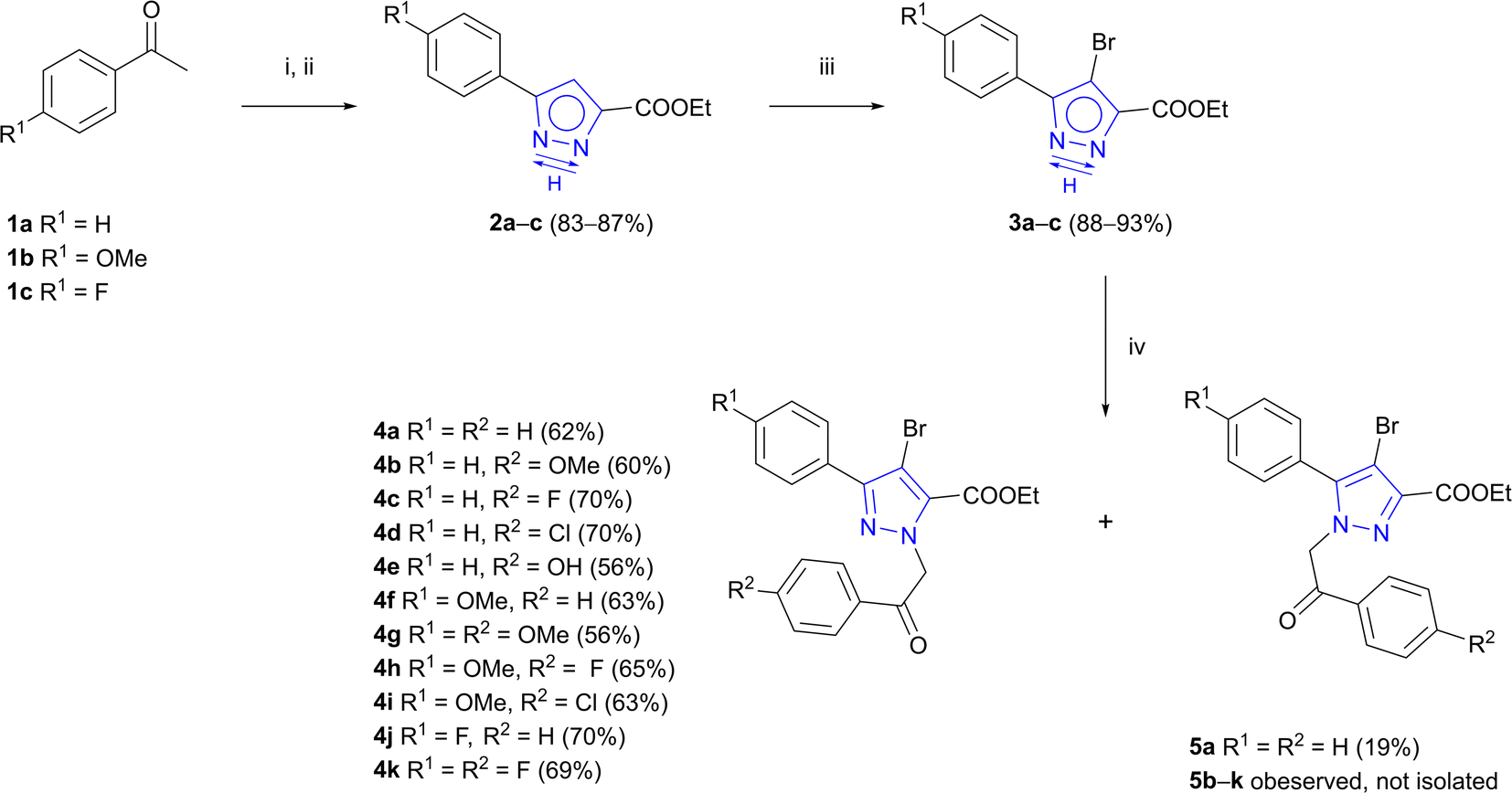 | ||
| Scheme 1 Synthesis of intermediates 4a–k. Reagents and conditions: (i) diethyl oxalate, NaOEt, EtOH, rt, 16 h;42 (ii) NH2NH2·H2O, AcOH, EtOH, rt, 16 h;42 (iii) NBS, DCM, 35 °C, 16 h;43 (iv) appropriate 2-bromoacetophenone, Na2CO3, DMF, 60–70 °C, 5–8 h. | ||
For introduction of phenyl substituents at C-4 of pyrazole ring, a halogen atom at the indicated position was introduced beforehand. 3(5)-Aryl-1H-pyrazole-5(3)-carboxylates 2a–c underwent bromination reaction using NBS in DCM and products 3a–c were obtained in 88–93% yields.43 Thereafter, two alternative pathways could be employed – either by first forming C–C bond via cross-coupling reaction and subsequently conducting N-alkylation, or vice versa. Multiple experiments were carried out towards Suzuki cross-coupling with pyrazole 3a bearing free –NH group (see ESI, Table S1†). Unfortunately, most C–C bond formation reactions resulted in complex mixtures and product yield did not exceed 22%. To tackle this problem, functionalization of free –NH group had to be performed in the first place.44
It is known that NH-pyrazoles usually exhibit annular N,N-prototropy.45 Typically, N-alkylation of asymmetrically ring-substituted 1H-pyrazoles results in the formation of a mixture of regioisomeric N-substituted products,46–49 therefore regioselective N-alkylation requires optimisation of reaction conditions. In this work, the goal was to carry out alkylation of 3(5)-aryl-4-bromo-1H-pyrazole-5(3)-carboxylates 3a–c in a regioselective manner to obtain desired isomers 4a–k as major products. Experiments were carried out with 3(5)-aryl-1H-pyrazole-5(3)-carboxylate 3a and the influence of the solvent and/or base on the regiochemical outcome of the reaction was evaluated. Comparison of Na2CO3, K2CO3 and NaH bases using DMF or ACN as a solvent revealed that combination of Na2CO3 and less polar DMF gave the best regioselectivity ratio of isomers 4a and 5a. Therefore, reaction conditions with Na2CO3-DMF system were applied for the synthesis of intermediates 4a–k.
N-Alkylated products 4a and 5a were fully characterized based on 1H, 1H-COSY, 1H, 1H-NOESY, 1H, 13C-HSQC, 1H, 13C- and 1H, 15N-HMBC experimental data (Fig. 2). To assign the regiochemistry of isomers, 1H, 13C-HMBC experiment was fundamental. Obtained data of regioisomer 4a revealed strong heteronuclear three-bond correlation between NCH2 protons at δ 6.23 ppm and annular C-5 of pyrazole at δ 132.1 ppm. Another correlation for distinguishing regioisomers is a long-range coupling between the same NCH2 protons at δ 6.23 ppm and carboxylate ester carbon at δ 158.1 ppm. Minor isomer 5a can be easily identified in a similar manner. A strong three-bond coupling was observed between NCH2 protons at δ 5.94 ppm and pyrazole C-5 at δ 144.5 ppm. Distinct NOE was observed between NCH2 protons at δ 5.94 and 5-phenyl ring 2(6)-H protons at δ 7.35–7.41 ppm, which confirms their proximity in space. The absence of heteronuclear correlation between NCH2 protons and carbon atom from carboxylate ester indicates the structure of 5-phenyl-1H-pyrazole-3-carboxylate 5a.
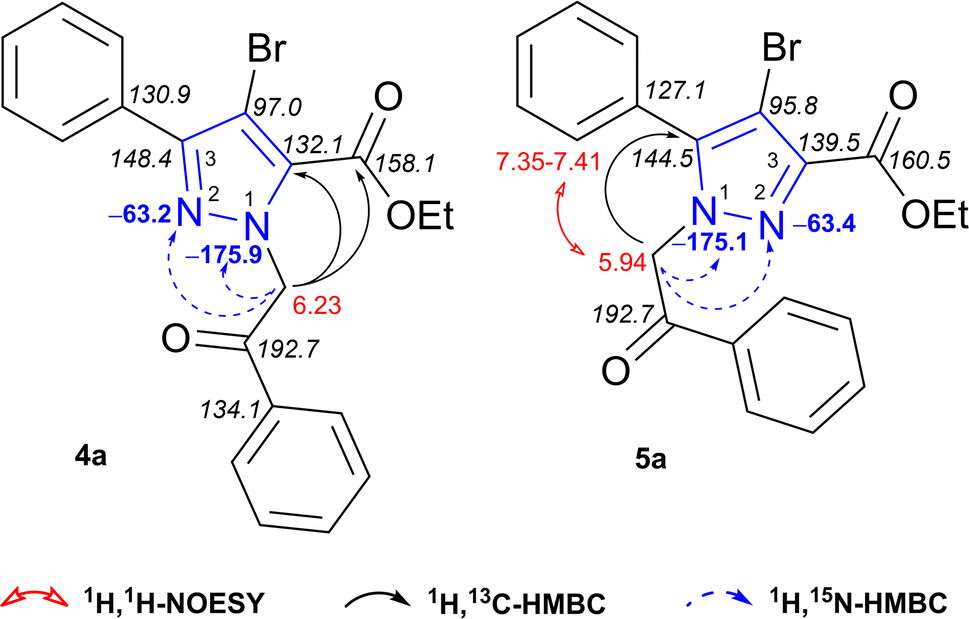 | ||
| Fig. 2 1H (red), 13C (italic), 15N (blue) NMR chemical shifts and relevant 1H, 13C-HMBC, 1H, 15N-HMBC, 1H, 1H-NOESY correlations of regioisomers 4a and 5a. | ||
Synthesized 3-aryl-4-bromo-1H-pyrazole-5-carboxylate in-termediates 4a–k were used for the final derivatization step i.e., construction of C–C bond via Pd-catalysed Suzuki cross-coupling reaction. As the efficiency and yield of transition metal catalysed reactions are influenced by various factors such as catalysts, solvents, bases, ligands, or other additives,50–53 optimization was carried out beforehand using carboxylate 4a as a model compound (Table 1). For the C–C coupling Pd(PPh3)4 was employed as a catalyst. Investigation started with the use of K3PO4 in DMF and water as a co-solvent for the dissolution of inorganic base. In the initial attempt, reaction mixture was stirred at 100 °C for 16 h affording hydrolysed product 1-(2-oxo-2-phenylethyl)-3,4-diphenyl-1H-pyrazole-5-carboxylic acid (6a′) in 53% yield (Table 1, entry 1). As reaction with K3PO4 in DMF/H2O resulted in successful C–C bond formation, it was repeated using MW-assisted heating (Table 1, entry 2). In the latter experiment hydrolysed product was once again isolated as a sole reaction product, however, the outcome of the reaction has improved and reached 77% yield.
|
||||||||
|---|---|---|---|---|---|---|---|---|
| Entry | Catalyst | Base | Solvent | Heating | Temperature (°C) | Time (h) | Yielda (%) | |
| 6a | 6a′![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) b b |
|||||||
| a Isolated yield.b Hydrolysed product. | ||||||||
| 1 | Pd(PPh3)4 | K3PO4 | DMF/H2O | Conventional | 100 | 16 | — | 53 |
| 2 | Pd(PPh3)4 | K3PO4 | DMF/H2O | MW-assisted | 140 | 1 | — | 77 |
| 3 | Pd(PPh3)4 | K3PO4 | Dioxane/H2O | Conventional | 100 | 24 | 51 | 38 |
| 4 | Pd(PPh3)4 | K3PO4 | Dioxane/H2O | MW-assisted | 100 | 1 | 78 | — |
| 5 | Pd(PPh3)4 | Na2CO3 (sat.) | Toluene/EtOH | Conventional | 80 | 24 | 16 | — |
| 6 | Pd(PPh3)4 | Cs2CO3 | DMF/H2O | MW-assisted | 140 | 1 | — | 73 |
| 7 | Pd(PPh3)4 | Cs2CO3 | Dioxane/H2O | MW-assisted | 100 | 1 | 84 | — |
| 8 | Pd(OAc)2 | Cs2CO3 | Dioxane/H2O | MW-assisted | 100 | 1 | 50 | — |
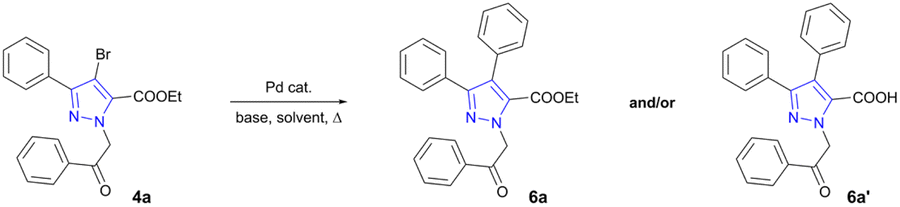
Further investigations involved multiple experiments using K3PO4 in less polar dioxane or dioxane/water solvent systems. Using conventional heating (Table 1, entry 3) both desired carboxylate ester 6a and hydrolysed product 6a′ were isolated in 51% and 38% yields, respectively. To our satisfaction, reaction with K3PO4 in dioxane/water system under MW irradiation proceeded without the undesired hydrolysis, giving rise to 6a in 78% yield (Table 1, entry 4). Additional experiment using saturated Na2CO3 solution in toluene/EtOH mixture54 was carried out giving very low 16% yield of 6a (Table 1, entry 5). Few more reactions were investigated using Cs2CO3 as a base (Table 1, entries 6 and 7) and Pd(OAc)2 as a catalyst (Table 1, entry 8), revealing Pd(PPh3)4 and Cs2CO3–dioxane–H2O system as the most suitable approach for C–C bond formation. In contrast to a cross-coupling under conventional heating, MW-assisted heating dramatically shortened reaction times, formed relatively pure products, and increased overall yields. Therefore, MW irradiation took a significant role in successful formation of the target compounds.
Optimized Suzuki cross-coupling reaction conditions were applied to evaluate the scope of the reaction and to couple phenyl-, 4-methoxyphenyl- and 4-fluorophenylboronic acids with N-alkylated pyrazole-5-carboxylates 4a–k (Scheme 2). Microwave-assisted Suzuki cross-coupling was successfully exploited to synthesize a library of ethyl 3,4-diaryl-1-(2-aryl-2-oxoethyl)-1H-pyrazole-5-carboxylates 6a–o in 73–97% yields. Additionally, three 3,4-diaryl-1H-pyrazole-5-carboxylates 6a–c underwent transesterification in the presence of K2CO3 in refluxing methanol to derive methyl carboxylates 7a–c in 92–96% yields.
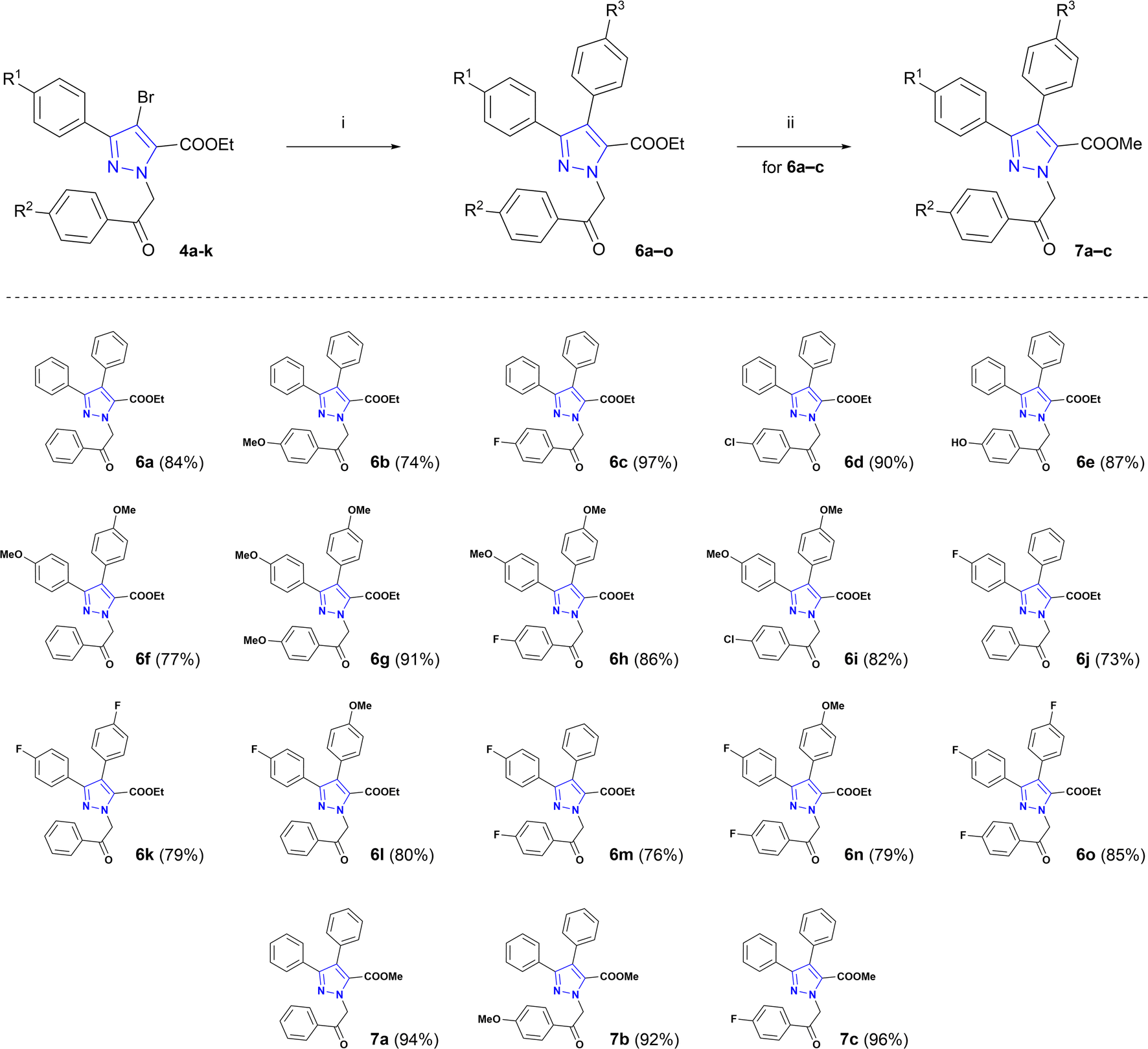 | ||
| Scheme 2 Synthesis of target pyrazole core-bearing lamellarin O analogues. Reagents and conditions: (i) appropriate phenylboronic acid, Pd(PPh3)4, Cs2CO3, dioxane, H2O, MW, 100 °C, 1 h; (ii) K2CO3, MeOH, reflux, 3 h. | ||
Physicochemical properties
Estimation of the drug-likeness is facilitated by the evaluation of physicochemical properties and their causal relationships to predict pharmacokinetics. Despite the development of the rule of five reported by Lipinski et al.55 to identify key properties for potential bioavailability in drug design, natural products and drugs based on naturally occurring compounds were not explicitly included in this systematization.56The lipophilicity of compounds 6a–o and 7a–c was estimated using both, high throughput chromatographic method employing an octadecyl–poly(vinyl alcohol) stationary phase57 and computational methods (Table 2). Besides, topological polar surface area (tPSA), pKa of strongest acid as well as hydrogen bond donors (HBD) and acceptors (HBA) were calculated (see ESI, Table S2†). The calculated logP (clogP) and measured HPLC–logP were in a narrow range of 4.81–5.84 and 3.803–4.784, respectively, with the hydroxy-substituted derivative 6e being the least lipophilic one. All natural products, lamellarin O, lamellarin D, lamellarin I and lukianol A had a lower lipophilicity in the range of 3.79–4.57 for clogP. A similar trend was observed for the calculated tPSA values, where the pyrazole-based derivatives presented a reduced polarity. The N-substitution pattern on the pyrazole ring, based on the structural similarity to lamellarin O, was the main reason for the increase of lipophilicity. Once the nitrogen in position 1 is substituted, the pyrazole loses the amphoteric properties58 in parallel to the lower HOMO energy of the pyrazole versus pyrrole ring.32
P and clogP data of ethyl and methyl 3,4-diaryl-1-(2-aryl-2-oxoethyl)-1H-pyrazole-5-carboxylates 6a–o and 7a–c
| Compound | HPLC–logPa |
clogPb |
|---|---|---|
| a Data is provided as a mean value ± standard deviation (SD) of at least three independent experiments.b Calculated using ChemDraw 13.0. | ||
| Lamellarin O | — | 4.00 |
| Lukianol A | — | 4.57 |
| Lamellarin D | — | 3.79 |
| Lamellarin I | — | 4.17 |
| 6a | 4.381 ± 0.002 | 5.28 |
| 6b | 4.468 ± 0.006 | 5.15 |
| 6c | 4.453 ± 0.005 | 5.44 |
| 6d | 4.711 ± 0.006 | 5.84 |
| 6e | 3.803 ± 0.012 | 4.89 |
| 6f | 4.462 ± 0.005 | 5.02 |
| 6g | 4.531 ± 0.004 | 4.90 |
| 6h | 4.531 ± 0.006 | 5.18 |
| 6i | 4.784 ± 0.005 | 5.58 |
| 6j | 4.490 ± 0.006 | 5.44 |
| 6k | 4.546 ± 0.005 | 5.59 |
| 6l | 4.499 ± 0.006 | 5.31 |
| 6m | 4.545 ± 0.006 | 5.59 |
| 6n | 4.564 ± 0.004 | 5.47 |
| 6o | 4.598 ± 0.004 | 5.75 |
| 7a | 4.279 ± 0.005 | 4.94 |
| 7b | 4.347 ± 0.007 | 4.81 |
| 7c | 4.327 ± 0.007 | 5.10 |
Taken together, being as close as possible to the structure of lamellarin O, all pyrazole-based derivatives are outliers of Lipinski's rule of five. However, it is reported that natural products and compounds derived from natural products are among the most favourable exceptions from the Lipinski's rule of five.59
Biological evaluation
Synthesized ethyl and methyl 3,4-diaryl-1-(2-aryl-2-oxoethyl)-1H-pyrazole-5-carboxylates 6a–o and 7a–c were first evaluated for their cytotoxicity against three human colorectal cancer cell lines HCT116, HT29 and SW480 (Table 3). Six out of eighteen compounds did not reach GI50 in the given 20 μM concentration range because of their wide therapeutic window and low solubility in cell culture medium. Ethyl 3,4-diaryl-1-(2-aryl-2-oxoethyl)-1H-pyrazole-5-carboxylates 6c, h, j, k, m–o, and methyl 1-[2-(4-fluorophenyl)-2-oxoethyl]-3,4-diphenyl-1H-py-razole-5-carboxylate (7c) have shown activity in the low micromolar range towards all tested cell lines. In most of the cases compounds were more active towards HCT116 cells, except for ethyl 3,4-diaryl-1-(2-aryl-2-oxoethyl)-1H-pyrazole-5-carboxylates 6m–o which have shown slightly higher cytotoxicity against SW480 cells. As expected, compounds bearing fluorine substituent demonstrated the best results.60 Fluorine substitution at R2 had the highest impact on advantageous cytotoxicity, whereas fluorine substitution on R1 and R3 had only minor or no effects. Among them, compound 6m proved to be the most active and reached lowest GI50 concentrations of 1.456 μM, 2.688 μM, and 1.441 μM towards HCT116, HT29 and SW480 cells, respectively.| Compound | General structure | R1 | R2 | R3 | R4 | GI50 ± SDa (μM) | ||
|---|---|---|---|---|---|---|---|---|
| HCT116 | HT29 | SW480 | ||||||
| a Data is provided as a mean value ± standard deviation (SD) of at least three independent experiments. | ||||||||
| 6a | 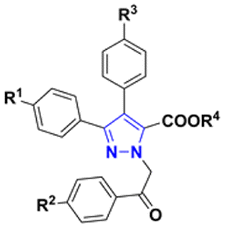 |
H | H | H | Et | 5.462 ± 0.393 | >20 | >20 |
| 6b | H | OMe | H | Et | >20 | >20 | >20 | |
| 6c | H | F | H | Et | 1.964 ± 0.266 | 4.979 ± 2.620 | 5.523 ± 3.719 | |
| 6d | H | Cl | H | Et | 14.459 ± 3.915 | >20 | >20 | |
| 6e | H | OH | H | Et | >20 | >20 | >20 | |
| 6f | OMe | H | OMe | Et | 2.293 ± 0.218 | >20 | >20 | |
| 6g | OMe | OMe | OMe | Et | >20 | >20 | >20 | |
| 6h | OMe | F | OMe | Et | 2.072 ± 1.450 | 4.759 ± 2.331 | 4.759 ± 2.331 | |
| 6i | OMe | Cl | OMe | Et | >20 | >20 | >20 | |
| 6j | F | H | H | Et | 3.751 ± 1.056 | 13.757 ± 0.861 | 9.243 ± 2.147 | |
| 6k | F | H | F | Et | 3.386 ± 0.620 | 11.125 ± 2.619 | 7.807 ± 2.017 | |
| 6l | F | H | OMe | Et | 5.051 ± 1.164 | >20 | 8.275 ± 2.339 | |
| 6m | F | F | H | Et | 1.456 ± 0.247 | 2.688 ± 0.110 | 1.441 ± 0.173 | |
| 6n | F | F | OMe | Et | 2.097 ± 0.198 | 6.783 ± 0.463 | 1.641 ± 0.022 | |
| 6o | F | F | F | Et | 2.362 ± 0.039 | 6.666 ± 2.121 | 2.088 ± 0.032 | |
| 7a | H | H | H | Me | >20 | >20 | >20 | |
| 7b | H | OMe | H | Me | >20 | >20 | >20 | |
| 7c | H | F | H | Me | 2.699 ± 0.689 | 9.725 ± 3.711 | >20 | |
Comparing synthesized ethyl carboxylates 6a–c with their methyl esters 7a–c it was noticed that ethyl group positively affects activity of the compounds. Interestingly, compound 6a appeared to be selective towards HCT116, and 6c, bearing fluorine atom at 4′-position of acetophenone, was active against all tested cell lines. In comparison, compounds 7a, b showed no activity at all, whereas methyl carboxylate 7c was less active than its counterpart 6c.
Influence of the substituent at 4′-position (Fig. 3) was evaluated in pyrazoles having identical substituents at 3- and 4-position. Interestingly, ethyl 3,4-diaryl-1-(2-aryl-2-oxoethyl)-1H-pyrazole-5-carboxylates 6a and 6f with no substituents at 4′-position were more active compared to those possessing Cl, OH or OMe groups and, as anticipated, fluorine substituent improved cytotoxicity of both 6c and 6h towards all cell lines. Additionally, presence of OMe group at both 4- and 4′′-positions of compound 6f improved activity against HCT116 cells two-fold compared to carboxylate 6a. In case of ethyl 3,4-diaryl-1-(2-aryl-2-oxoethyl)-1H-pyrazole-5-carboxylates 6j, l, m, n containing fluorine atom, it was noticed that compounds with different substituents at 3,4-positions do not lose their activity. On the contrary, most active compound 6m possessed 4-fluorophenyl and phenyl substituents in the 3- and 4-positions of the pyrazole ring respectively.
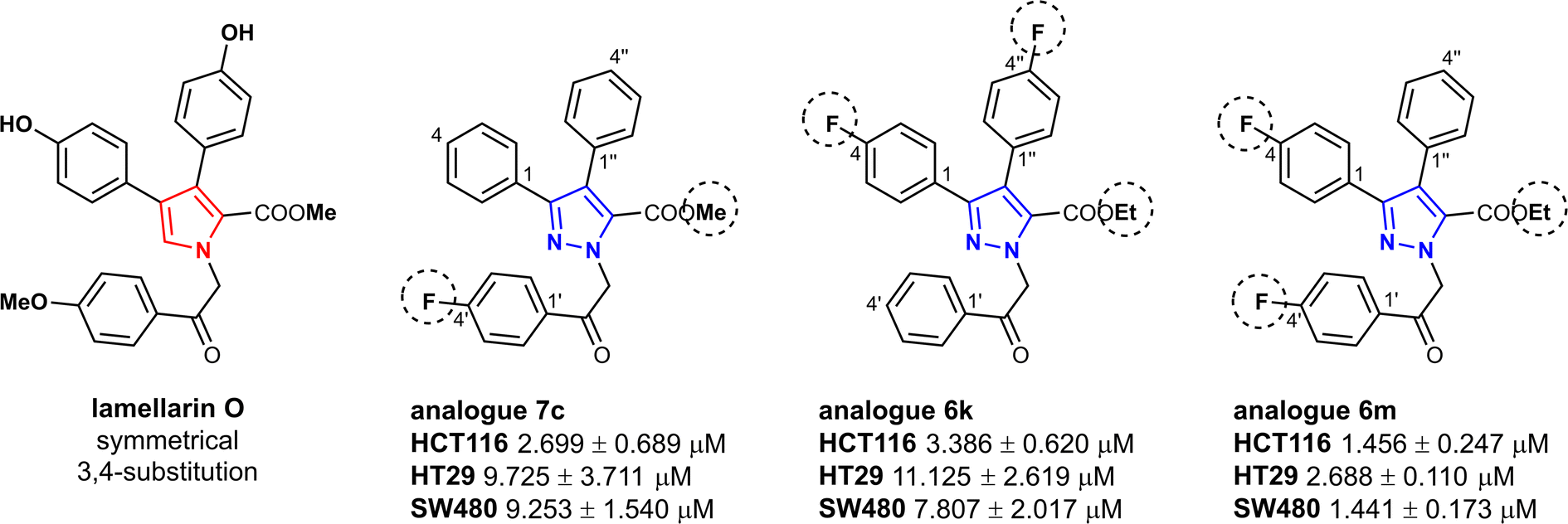 | ||
| Fig. 3 Structure and activity comparison of compounds 6k, 6m, and 7c. | ||
The significant cytotoxicity of pyrazole-based derivatives motivated us to investigate the mode of action of the most potent compounds. First, we evaluated the compounds by a conventional live/dead counterstain including propidium iodide (PI), Hoechst 33258 and Calcein AM.61 Calcein AM is a well-described cell-permeable dye staining living cells, whereas PI can only bind to DNA when the cellular membrane is disrupted and allows the visualization of necrotic cells. Hoechst 33258 stain intensity increases during apoptosis due to nuclear condensation indicating apoptosis. The absence of any PI staining of the cells indicated the absence of necrotic and late-apoptotic cells for all tested compounds in all three colorectal cancer cells (ESI Fig. S134–S136†).
Cell cycle distribution was determined using flow cytometry in order to verify if chosen ethyl 3,4-diaryl-1-(2-aryl-2-oxoethyl)-1H-pyrazole-5-carboxylates 6c and 6m could induce cell cycle arrest. HCT116 cells were treated for 72 h with the respective compounds in a dose-dependent manner. As shown in Fig. 4, treatment of HCT116 cells with target compounds resulted in an induction of G1 and predominantly G2/M cell cycle arrest for both compounds already for the lowest concentration of 5 μM. Compared to negative control cells, the G1 phase increased significantly from 49.5 ± 1.8% to 63.2 ± 1.4% and 61.0 ± 1.8% for 6c and 6m respectively. In parallel, the G2/M phase increased from 14.2 ± 1.1% to 25.0 ± 0.5% and 26.9 ± 0.8%, respectively. No significant increase of cell debris was observed over the whole concentration range. Cell cycle arrest in the G2 phase was also described for lamellarin D in P388 leukaemia cells,62 whereas to the best of our knowledge no mode of action studies were published for lamellarin O. However, it seems likely that our newly synthesized isosteric analogues of lamellarin O and lamellarin D exhibit comparable cytotoxicity and cellular effects.
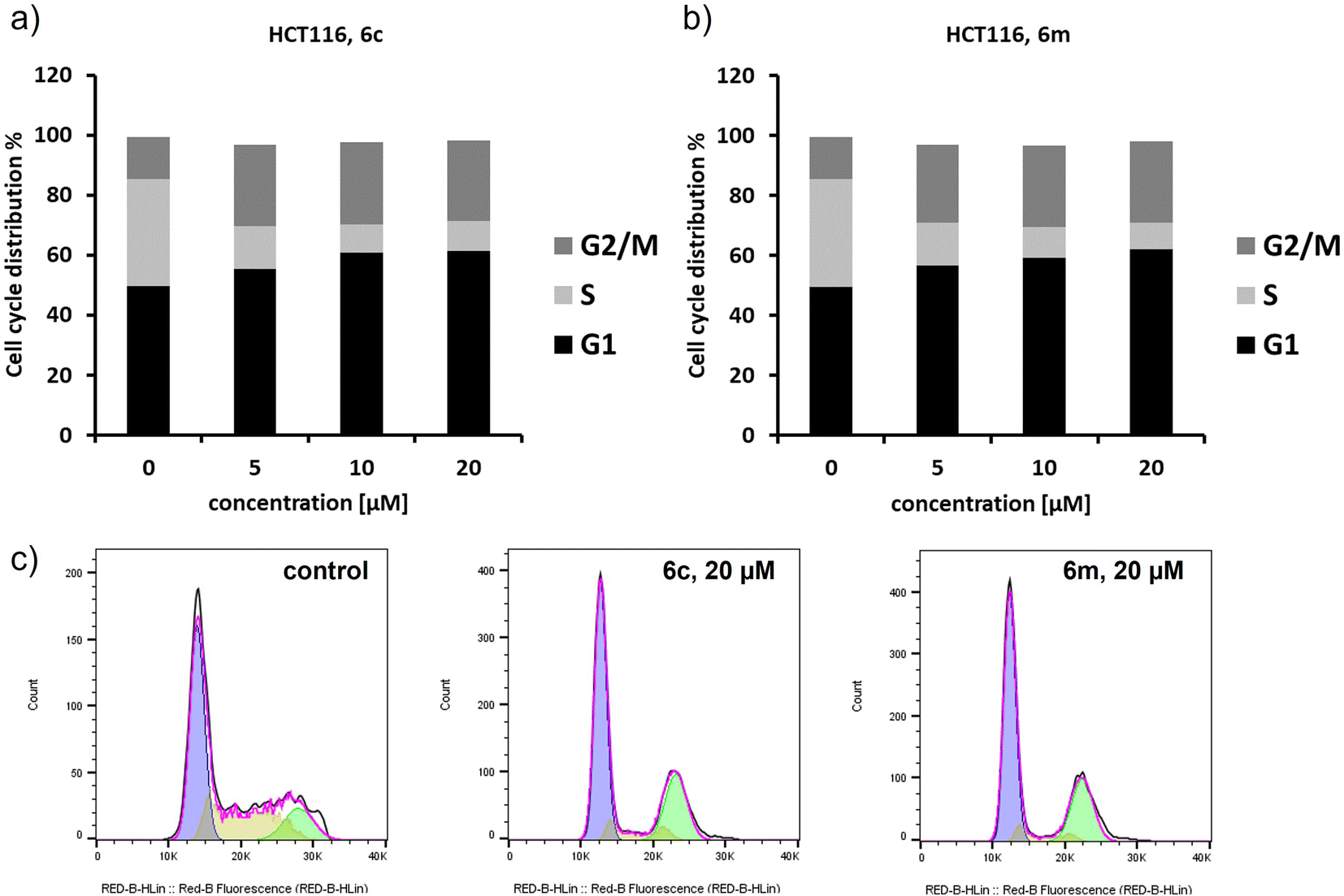 | ||
| Fig. 4 Cell cycle analysis in HCT116 cells after treatment with 6c and 6m: (a) dose-dependent increase of G1 and G2/M phase for 6c; (b) dose-dependent increase of G1 and G2/M phase for 6m; (c) representative histograms for the negative control, and at 20 μM of 6c and 6m. Data is provided as mean value of at least three independent experiments. | ||
Additionally, plasma protein binding (PPB) experiments by means of a high-throughput HPLC method applying albumin-bound stationary phase were performed with selected ethyl 3,4-diaryl-1-(2-aryl-2-oxoethyl)-1H-pyrazole-5-carboxylates 6a, c, f, h, m–o. Compounds 6a, c, m–o showed very strong (above 95%) binding to human serum albumin (see ESI, Table S3†). The relationship between %PPB and calculated logP values correlated – increased compound lipophilicity results in stronger binding to albumin. However, studied compounds bind differently than would have been expected only from their lipophilicity, as the HPLC–logP values did not show linear correlation with %PPB data. High PPB decreases the free plasma fraction of the drug being part of the free drug hypothesis stating that only the unbound fraction of a drug can unfold their biological efficacy.63 This hypothesis is opposing the evidence of albumin being a versatile drug carrier via the enhanced permeability and retention (EPR) effect for oncological issues and that 45% of newly approved drugs between 2003 and 2013 possessed PPB of >95%.64
Materials and methods
Chemistry
Ethyl 4-bromo-1-(2-oxo-2-phenylethyl)-3-phenyl-1H-pyrazole-5-carboxylate (4a). White solid, yield 62% (256 mg). Rf = 0.69 (n-hexane/ethyl acetate 7/3, v/v), mp 93–94 °C. IR (KBr) νmax, cm−1: 2982, 2952, 1704 (C
![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O), 1448, 1269, 1091, 688. 1H NMR (400 MHz, DMSO-d6) δH ppm: 1.15 (t, J = 7.1 Hz, 3H, CH3), 4.23 (q, J = 7.1 Hz, 2H, OCH2), 6.23 (s, 2H, NCH2), 7.43–7.54 (m, 3H, 3-Ph 3,4,5-H), 7.58–7.64 (m, 2H, C(O)Ph 3,5-H), 7.71–7.77 (m, 1H, C(O)Ph 4-H), 7.79–7.84 (m, 2H, 3-Ph 2, 6-H), 8.06–8.10 (m, 2H, C(O)Ph 2,6-H). 13C NMR (101 MHz, DMSO-d6) δC ppm: 13.7 (CH3), 60.3 (NCH2), 61.5 (OCH2), 97.0 (C-4), 127.8 (3-Ph C-2,6), 128.1 (C(O)Ph C-2,6), 128.6 (3-Ph C-3,5), 128.7 (3-Ph C-4), 129.0 (C(O)Ph C-3,5), 130.9 (3-Ph C-1), 132.1 (C-5), 134.1 (C(O)Ph C-1), 134.2 (C(O)Ph C-4), 148.4 (C-3), 158.1 (COO), 192.7 (CO). 15N NMR (40 MHz, DMSO-d6) δN ppm: −175.9 (N-1), −63.2 (N-2). HRMS (ESI) for C20H17BrN2NaO3 ([M + Na]+): calcd m/z 435.0315, found m/z 435.0316.
O), 1448, 1269, 1091, 688. 1H NMR (400 MHz, DMSO-d6) δH ppm: 1.15 (t, J = 7.1 Hz, 3H, CH3), 4.23 (q, J = 7.1 Hz, 2H, OCH2), 6.23 (s, 2H, NCH2), 7.43–7.54 (m, 3H, 3-Ph 3,4,5-H), 7.58–7.64 (m, 2H, C(O)Ph 3,5-H), 7.71–7.77 (m, 1H, C(O)Ph 4-H), 7.79–7.84 (m, 2H, 3-Ph 2, 6-H), 8.06–8.10 (m, 2H, C(O)Ph 2,6-H). 13C NMR (101 MHz, DMSO-d6) δC ppm: 13.7 (CH3), 60.3 (NCH2), 61.5 (OCH2), 97.0 (C-4), 127.8 (3-Ph C-2,6), 128.1 (C(O)Ph C-2,6), 128.6 (3-Ph C-3,5), 128.7 (3-Ph C-4), 129.0 (C(O)Ph C-3,5), 130.9 (3-Ph C-1), 132.1 (C-5), 134.1 (C(O)Ph C-1), 134.2 (C(O)Ph C-4), 148.4 (C-3), 158.1 (COO), 192.7 (CO). 15N NMR (40 MHz, DMSO-d6) δN ppm: −175.9 (N-1), −63.2 (N-2). HRMS (ESI) for C20H17BrN2NaO3 ([M + Na]+): calcd m/z 435.0315, found m/z 435.0316.
Ethyl 4-bromo-1-[2-(4-methoxyphenyl)-2-oxoethyl]-3-phenyl-1H-pyrazole-5-carboxylate (4b). White solid, yield 60% (266 mg). Rf = 0.61 (n-hexane/ethyl acetate 7/3, v/v), mp 131–132 °C. IR (KBr) νmax, cm−1: 3035, 3006, 2977, 2947, 1714 (C
O), 1601, 1449, 1174, 694. 1H NMR (400 MHz, CDCl3) δH ppm: 1.26 (t, J = 7.1 Hz, 3H, CH3), 3.82 (s, 3H, OCH3), 4.26 (q, J = 7.1 Hz, 2H, OCH2), 5.95 (s, 2H, NCH2), 6.87–6.95 (m, 2H, C(O)Ph 3,5-H), 7.28–7.41 (m, 3H, 3-Ph 3,4,5-H), 7.75–7.82 (m, 2H, 3-Ph 2,6-H), 7.86–7.94 (m, 2H, C(O)Ph 2,6-H). 13C NMR (101 MHz, CDCl3) δC ppm: 14.1 (CH3), 55.7 (OCH3), 59.7 (NCH2), 61.8 (OCH2), 98.3 (C-4), 114.3 (C(O)Ph C-3,5), 127.6 (C(O)Ph C-1), 128.44 (3-Ph C-3,5), 128.46 (3-Ph C-2,6), 128.7 (3-Ph C-4), 130.4 (C(O)Ph C-2,6), 131.5 (3-Ph C-1), 132.7 (C-5), 150.0 (C-3), 159.4 (COO), 164.3 (C(O)Ph C-4), 190.1 (CO). 15N NMR (40 MHz, CDCl3) δN ppm: −179.4 (N-1), −65.7 (N-5). HRMS (ESI) for C21H19BrN2NaO4 ([M + Na]+): calcd m/z 465.0420, found m/z 465.0423.
Ethyl 4-bromo-1-[2-(4-fluorophenyl)-2-oxoethyl]-3-phenyl-1H-pyrazole-5-carboxylate (4c). White solid, yield 70% (302 mg). Rf = 0.64 (n-hexane/ethyl acetate 7/1, v/v), mp 96–97 °C. IR (KBr) νmax, cm−1: 3071, 2985, 2946, 1704 (C
O), 1595, 1090, 836, 699. 1H NMR (400 MHz, CDCl3) δH ppm: 1.35 (t, J = 7.1 Hz, 3H, CH3), 4.34 (q, J = 7.1 Hz, 2H, OCH2), 6.03 (s, 2H, NCH2), 7.16–7.24 (m, 2H, C(O)Ph 3,5-H), 7.37–7.49 (m, 3H, 3-Ph 3,4,5-H), 7.82–7.89 (m, 2H, 3-Ph 2,6-H), 7.99–8.06 (m, 2H, C(O)Ph 2,6-H). 13C NMR (101 MHz, CDCl3) δC ppm: 14.1 (CH3), 59.8 (NCH2), 61.9 (OCH2), 98.4 (C-4), 116.38 (d, 2JCF = 22.1 Hz, C(O)Ph C-3,5), 128.44 (3-Ph C-2,6), 128.47 (3-Ph C-3,5), 128.8 (3-Ph C-4), 130.84 (d, 3JCF = 9.5 Hz, C(O)Ph C-2,6), 131.05 (d, 4JCF = 3.1 Hz, C(O)Ph C-1), 131.41 (3-Ph C-1), 132.5 (C-5), 150.2 (C-3), 159.4 (COO), 166.40 (d, JCF = 256.4 Hz, C(O)Ph C-4), 190.3 (CO). 15N NMR (40 MHz, CDCl3) δN ppm: −180.0 (N-1), −65.7 (N-2). HRMS (ESI) for C20H16BrFN2NaO3 ([M + Na]+): calcd m/z 453.0221, found m/z 453.0218.
Ethyl 4-bromo-1-[2-(4-chlorophenyl)-2-oxoethyl]-3-phenyl-1H-pyrazole-5-carboxylate (4d). White solid, yield 70% (313 mg). Rf = 0.77 (n-hexane/ethyl acetate 7/3, v/v), mp 87–88 °C. IR (KBr) νmax, cm−1: 3061, 2983, 2941, 1707 (C
O), 1580, 1226, 1092, 697. 1H NMR (400 MHz, CDCl3) δH ppm: 1.35 (t, J = 7.1 Hz, 3H, CH3), 4.33 (q, J = 7.1 Hz, 2H, OCH2), 6.01 (s, 2H, NCH2), 7.38–7.52 (m, 5H, 3-Ph 3,4,5-H; C(O)Ph 3,5-H), 7.84–7.89 (m, 2H, 3-Ph 2,6-H), 7.90–7.95 (m, 2H, C(O)Ph 2,6-H). 13C NMR (101 MHz, CDCl3) δC ppm: 14.1 (CH3), 59.8 (NCH2), 61.9 (OCH2), 98.4 (C-4), 128.41 (3-Ph C-2,6), 128.46 (3-Ph C-3,5), 128.8 (3-Ph C-4), 129.47 (C(O)Ph C-3,5), 129.48 (C(O)Ph C-2,6), 131.4 (3-Ph C-1), 132.5 (C-5), 132.9 (C(O)Ph C-1), 140.7 (C(O)Ph C-4), 150.1 (C-3), 159.4 (COO), 190.7 (CO). 15N NMR (40 MHz, CDCl3) δN ppm: −180.1 (N-1), −65.6 (N-2). HRMS (ESI) for C20H16BrClN2NaO3 ([M + Na]+): calcd m/z 468.9925, found m/z 468.9923.
Ethyl 4-bromo-1-[2-(4-hydroxyphenyl)-2-oxoethyl]-3-phenyl-1H-pyrazole-5-carboxylate (4e). White solid, yield 56% (240 mg). Rf = 0.63 (n-hexane/ethyl acetate 1/1, v/v), mp 191–192 °C. IR (KBr) νmax, cm−1: 3377 (OH), 2984, 2945, 1707 (C
O), 1580, 1176, 1090, 837. 1H NMR (400 MHz, DMSO-d6) δH ppm: 1.15 (t, J = 7.1 Hz, 3H, CH3), 4.22 (q, J = 7.1 Hz, 2H, OCH2), 6.10 (s, 2H, NCH2), 6.88–6.96 (m, 2H, C(O)Ph 3,5-H), 7.41–7.54 (m, 3H, 3-Ph 3,4,5-H), 7.76–7.83 (m, 2H, 3-Ph 2,6-H), 7.91–7.99 (m, 2H, C(O)Ph 2,6-H), 10.60 (s, 1H, OH). 13C NMR (101 MHz, DMSO-d6) δC ppm: 13.7 (CH3), 60.0 (NCH2), 61.5 (OCH2), 96.84 (C-4), 115.6 (C(O)Ph C-3,5), 125.6 (C(O)Ph C-1), 127.8 (3-Ph C-2,6), 128.6 (3-Ph C-3,5), 128.7 (3-Ph C-4), 130.8 (C(O)Ph C-2,6), 131.0 (3-Ph C-1), 132.3 (C-5), 148.3 (C-3), 158.1 (COO), 163.0 (C(O)Ph C-4), 190.5 (CO). 15N NMR (40 MHz, DMSO-d6) δN ppm: −175.1 (N-1), −63.4 (N-2). HRMS (ESI) for C20H17BrN2NaO4 ([M + Na]+): calcd m/z 451.0264, found m/z 451.0265.
Ethyl 4-bromo-3-(4-methoxyphenyl)-1-(2-oxo-2-phenylethyl)-1H-pyrazole-5-carboxylate (4f). White solid, yield 63% (279 mg). Rf = 0.79 (n-hexane/ethyl acetate 7/3, v/v), mp 152–153 °C. IR (KBr) νmax, cm−1: 3068, 2955, 2835, 1704 (C
O), 1455, 1251, 1177, 1093, 1029, 841. 1H NMR (400 MHz, DMSO-d6) δH ppm: 1.14 (t, J = 7.1 Hz, 3H, CH3), 3.81 (s, 3H, OCH3), 4.22 (q, J = 7.0 Hz, 2H, OCH2), 6.19 (s, 2H, NCH2), 7.01–7.11 (m, 2H, 3-Ph 3,5-H), 7.56–7.65 (m, 2H, C(O)Ph 3,5-H), 7.70–7.79 (m, 3H, C(O)Ph 4-H; 3-Ph 2,6-H), 8.03–8.11 (m, 2H, C(O)Ph 2,6-H). 13C NMR (101 MHz, DMSO-d6) δC ppm: 13.7 (CH3), 55.2 (OCH3), 60.3 (NCH2), 61.5 (OCH2), 96.7 (C-4), 114.0 (3-Ph C-3,5), 123.3 (3-Ph C-1), 128.1 (C(O)Ph C-2,6), 129.0 (C(O)Ph C-3,5), 129.2 (3-Ph C-2,6), 132.0 (C-5), 134.15 (C(O)Ph C-1), 134.23 (C(O)Ph C-4), 148.3 (C-3), 158.1 (COO), 159.6 (3-Ph C-4), 192.8 (CO). 15N NMR (40 MHz, DMSO-d6) δN ppm: −177.1 (N-1), −64.3 (N-2). HRMS (ESI) for C21H19BrN2NaO4 ([M + Na]+): calcd m/z 465.0420, found m/z 465.0418.
Ethyl 4-bromo-3-(4-methoxyphenyl)-1-[2-(4-methoxyphenyl)-2-oxoethyl]-1H-pyrazole-5-carboxylate (4g). White solid, yield 56% (265 mg). Rf = 0.59 (n-hexane/ethyl acetate 7/3, v/v), mp 109–110 °C. IR (KBr) νmax, cm−1: 2991, 2945, 2839, 1709 (C
O), 1689, 1234, 1176, 1030, 840. 1H NMR (400 MHz, CDCl3) δH ppm: 1.33 (t, J = 7.1 Hz, 3H, CH3), 3.85 (s, 3H, Ph 4-OCH3), 3.89 (s, 3H, C(O)Ph 4-OCH3), 4.32 (q, J = 7.0 Hz, 2H, OCH2), 6.00 (s, 2H, NCH2), 6.92–7.04 (m, 4H, C(O)Ph 3,5-H; 3-Ph 3,5-H), 7.74–7.85 (m, 2H, 3-Ph 2,6-H), 7.93–8.00 (m, 2H, C(O)Ph 2,6-H). 13C NMR (101 MHz, CDCl3) δC ppm: 14.1 (CH3), 55.4 (3-Ph 4-OCH3), 55.7 (C(O)Ph 4-OCH3), 59.6 (NCH2), 61.8 (OCH2), 98.0 (C-4), 113.9 (3-Ph C-3,5), 114.3 (C(O)Ph C-3,5), 124.1 (3-Ph C-1), 127.6 (C(O)Ph C-1), 129.8 (3-Ph C-2,6), 130.4 (C(O)Ph C-2,6), 132.5 (C-5), 149.8 (C-3), 159.4 (COO), 160.0 (3-Ph C-4), 164.3 (C(O)Ph C-4), 190.2 (CO). 15N NMR (40 MHz, CDCl3) δN ppm: −180.3 (N-1), −67.0 (N-2). HRMS (ESI) for C22H21BrN2NaO5 ([M + Na]+): calcd m/z 495.0526, found m/z 495.0523.
Ethyl 4-bromo-1-[2-(4-fluorophenyl)-2-oxoethyl]-3-(4-methoxy-phenyl)-1H-pyrazole-5-carboxylate (4h). White solid, yield 65% (230 mg). Rf = 0.73 (n-hexane/ethyl acetate 3/2, v/v), mp 157–158 °C. IR (KBr) νmax, cm−1: 3083 and 3073 (doublet), 2951, 2934, 2837, 1704 (C
O), 1598, 1268, 839, 611 and 591 (doublet). 1H NMR (400 MHz, CDCl3) δH ppm: 1.37 (t, J = 7.1 Hz, 3H, CH3), 3.87 (s, 3H, OCH3), 4.35 (q, J = 7.0 Hz, 2H, OCH2), 6.04 (s, 2H, NCH2), 6.96–7.03 (m, 2H, 3-Ph 3,5-H), 7.18–7.26 (m, 2H, C(O)Ph 3,5-H), 7.78–7.85 (m, 2H, 3-Ph 2,6-H), 8.01–8.08 (m, 2H, C(O)Ph 2,6-H). 13C NMR (101 MHz, CDCl3) δC ppm: 14.1 (CH3), 55.4 (OCH3), 59.8 (NCH2), 61.9 (OCH2), 98.2 (C-4), 113.9 (3-Ph C-3,5), 116.37 (d, 2JCF = 22.1 Hz, C(O)Ph C-3,5), 124.0 (3-Ph C-1), 129.8 (3-Ph C-2,6), 130.83 (d, 3JCF = 9.5 Hz, C(O)Ph C-2,6), 131.09 (d, 4JCF = 3.0 Hz, C(O)Ph C-1), 132.4 (C-5), 150.0 (C-3), 159.5 (COO), 160.1 (3-Ph C-4), 166.39 (d, JCF = 256.3 Hz, C(O)Ph C-4), 190.4 (CO). 15N NMR (40 MHz, CDCl3) δN ppm: −180.9 (N-1). −67.1 (N-2). HRMS (ESI) for C21H18BrFN2NaO4 ([M + Na]+): calcd m/z 483.0326, found m/z 483.0332.
Ethyl 4-bromo-1-[2-(4-chlorophenyl)-2-oxoethyl]-3-(4-methoxy-phenyl)-1H-pyrazole-5-carboxylate (4i). White solid, yield 63% (301 mg). Rf = 0.70 (n-hexane/ethyl acetate 7/3, v/v), mp 119–120 °C. IR (KBr) νmax, cm−1: 2993, 2952, 1705 (C
O), 1252, 1177, 1092, 1025, 842. 1H NMR (400 MHz, DMSO-d6) δH ppm: 1.15 (t, J = 7.1 Hz, 3H, CH3), 3.81 (s, 3H, OCH3), 4.22 (q, J = 7.1 Hz, 2H, OCH2), 6.19 (s, 2H, NCH2), 7.01–7.10 (m, 2H, 3-Ph 3,5-H), 7.66–7.77 (m, 4H, C(O)Ph 3,5-H; 3-Ph 2,6-H), 8.05–8.11 (m, 2H, C(O)Ph 2,6-H). 13C NMR (101 MHz, DMSO-d6) δC ppm: 13.7 (CH3), 55.2 (OCH3), 60.2 (NCH2), 61.5 (OCH2), 96.7 (C-4), 114.0 (3-Ph C-3,5), 123.3 (3-Ph C-1), 129.14 (3-Ph C-2,6), 129.17 (C(O)Ph C-3,5), 130.0 (C(O)Ph C-2,6), 131.9 (C-5), 132.9 (C(O)Ph C-1), 139.2 (C(O)Ph C-4), 148.3 (C-3), 158.1 (COO), 159.6 (3-Ph C-4), 192.0 (CO). 15N NMR (40 MHz, DMSO-d6) δN ppm: −177.2 (N-1), −64.5 (N-2). HRMS (ESI) for C21H18BrClN2NaO4 ([M + Na]+): calcd m/z 499.0031, found m/z 499.0029.
Ethyl 4-bromo-3-(4-fluorophenyl)-1-(2-oxo-2-phenylethyl)-1H-pyrazole-5-carboxylate (4j). White solid, yield 70% (302 mg). Rf = 0.70 (n-hexane/ethyl acetate 7/3, v/v), mp 97–98 °C. IR (KBr) νmax, cm−1: 3001 and 2982 (doublet), 2938, 2360, 2342, 1708 (C
O), 1445, 1255, 1226, 1165, 844, 756, 688. 1H NMR (400 MHz, CDCl3) δH ppm: 1.34 (t, J = 7.1 Hz, 3H, CH3), 4.33 (q, J = 7.1 Hz, 2H, OCH2), 6.06 (s, 2H, NCH2), 7.09–7.18 (m, 2H, 3-Ph 3,5-H), 7.49–7.57 (m, 2H, C(O)Ph 3,5-H), 7.61–7.69 (m, 1H, C(O)Ph 4-H), 7.81–7.89 (m, 2H, 3-Ph 2,6-H), 7.97–8.03 (m, 2H, C(O)Ph 2,6-H). 13C NMR (101 MHz, CDCl3) δC ppm: 14.1 (CH3), 60.0 (NCH2), 61.9 (COO), 98.2 (C-4), 115.49 (d, 2JCF = 21.6 Hz, 3-Ph C-3,5), 127.60 (d, 4JCF = 3.3 Hz, 3-Ph C-1), 128.1 (C(O)Ph C-2,6), 129.2 (C(O)Ph C-3,5), 130.31 (d, 3JCF = 8.4 Hz, 3-Ph C-2,6), 132.7 (C-5), 134.3 (C(O)Ph C-4), 134.6 (C(O)Ph C-1), 149.2 (C-3), 159.30 (COO), 163.12 (d, JCF = 248.1 Hz, 3-Ph C-4), 191.7 (CO). 15N NMR (40 MHz, CDCl3) δN ppm: −179.8 (N-1), −66.0 (N-2). HRMS (ESI) for C20H16BrFN2NaO3 ([M + Na]+): calcd m/z 453.0221, found m/z 453.0225.
Ethyl 4-bromo-3-(4-fluorophenyl)-1-[2-(4-fluorophenyl)-2-oxo-ethyl]-1H-pyrazole-5-carboxylate (4k). White solid, yield 69% (310 mg). Rf = 0.77 (n-hexane/ethyl acetate 3/2, v/v), mp 99–100 °C. IR (KBr) νmax, cm−1: 3084, 2989, 2943, 1708 (C
O), 1598, 1443, 1233, 841. 1H NMR (400 MHz, DMSO-d6) δH ppm: 1.15 (t, J = 7.1 Hz, 3H, CH3), 4.23 (q, J = 7.0 Hz, 2H, OCH2), 6.21 (s, 2H, NCH2), 7.31–7.39 (m, 2H, 3-Ph 3,5-H), 7.41–7.49 (m, 2H, C(O)Ph 3,5-H), 7.81–7.88 (m, 2H, 3-Ph 2,6-H), 8.13–8.20 (m, 2H, C(O)Ph 2,6-H). 13C NMR (101 MHz, DMSO-d6) δC ppm: 13.7 (CH3), 60.3 (NCH2), 61.6 (OCH2), 96.9 (C-4), 115.59 (d, 2JCF = 21.7 Hz, 3-Ph C-3,5), 116.15 (d, 2JCF = 22.0 Hz, C(O)Ph C-3,5), 127.38 (d, 4JCF = 3.2 Hz, 3-Ph C-1), 129.99 (d, 3JCF = 8.5 Hz, 3-Ph C-2,6), 130.89 (d, 4JCF = 2.8 Hz, C(O)Ph C-1), 131.24 (d, 3JCF = 9.7 Hz, C(O)Ph C-2,6), 132.2 (C-5), 147.6 (C-3), 158.0 (COO), 162.28 (d, JCF = 246.0 Hz, 3-Ph C-4), 165.59 (d, JCF = 253.0 Hz, C(O)Ph C-4), 191.39 (CO). 15N NMR (40 MHz, DMSO-d6) δN ppm: −175.9 (N-1), −63.7 (N-2). HRMS (ESI) for C20H15BrF2N2NaO3 ([M + Na]+): calcd m/z 471.0126, found m/z 471.0130.
Ethyl 4-bromo-1-(2-oxo-2-phenylethyl)-5-phenyl-1H-pyrazole-3-carboxylate (5a). Pale yellow solid, yield 19% (79 mg). Rf = 0.35 (n-hexane/ethyl acetate 7/3, v/v), mp 148–149 °C. IR (KBr) νmax, cm−1: 2981, 2952, 1714 (C
O), 1460 and 1451 (doublet), 1226, 1054. 1H NMR (400 MHz, DMSO-d6) δH ppm: 1.31 (t, J = 7.1 Hz, 3H, CH3), 4.33 (q, J = 7.1 Hz, 2H, OCH2), 5.94 (s, 2H, NCH2), 7.35–7.41 (m, 2H, 3-Ph 2,6-H), 7.45–7.56 (m, 5H, 3-Ph 3,4,5-H; C(O)Ph 3,5-H), 7.65–7.71 (m, 1H, C(O)Ph 4-H), 7.89–7.95 (m, 2H, C(O)Ph 2,6-H). 13C NMR (101 MHz, DMSO-d6) δC ppm: 14.1 (CH3), 57.8 (NCH2), 60.7 (OCH2), 95.8 (C-4), 127.1 (3-Ph C-1), 128.1 (C(O)Ph C-2,6), 128.90 (3-Ph C-3,5), 128.94 (C(O)Ph C-3-5), 129.6 (3-Ph C-2,6), 129.9 (3-Ph C-4), 133.8 (C(O)Ph C-1), 134.4 (C(O)Ph C-4), 139.5 (C-3), 144.5 (C-5), 160.5 (COO), 192.7 (CO). 15N NMR (40 MHz, DMSO-d6) δN ppm: −175.1 (N-1), −63.4 (N-2). HRMS (ESI) for C20H17BrN2NaO3 ([M + Na]+): calcd m/z 435.0315, found m/z 435.0313.
1-(2-Oxo-2-phenylethyl)-3,4-diphenyl-1H-pyrazole-5-carboxylic acid (6a′). White solid, yield 77% (147 mg). Rf = 0.46 (ethyl acetate), mp 209–210 °C. IR (KBr) νmax, cm−1: 3058, 3029, 2989, 2940, 2556, 1716 (C
O), 1702 (CO), 1449, 1228, 1089, 700. 1H NMR (400 MHz, DMSO-d6) δH ppm: 6.21 (s, 2H, CH2), 7.19–7.42 (m, 10H, 3-Ph 2,3,4,5,6-H; 4-Ph 2,3,4,5,6-H), 7.56–7.65 (m, 2H, C(O)Ph 3,5-H), 7.68–7.78 (m, 1H, C(O)Ph 4-H), 8.03–8.13 (m, 2H, C(O)Ph 2,6-H), 13.16 (s, 1H, COOH). 13C NMR (101 MHz, DMSO-d6) δC ppm: 59.3 (CH2), 123.8 (C-4), 127.3 (4-Ph C-4), 127.5 (4-Ph C-2,6), 127.7 (3-Ph C-4), 128.0 (3-Ph C-2,6), 128.1 (3-Ph C-3,5), 128.2 (C(O)Ph C-2,6), 129.0 (C(O)Ph C-3,5), 130.4 (4-Ph C-3,5), 132.2 (3-Ph C-1), 132.4 (C-5), 133.0 (4-Ph C-1), 134.0 (C(O)Ph C-4), 134.5 (C(O)Ph C-1), 147.8 (C-3), 160.8 (COOH), 193.2 (CO). 15N NMR (40 MHz, DMSO-d6) δN ppm: −177.9 (N-1), −65.8 (N-2). HRMS (ESI) for C24H19N2O3 ([M + H]+): calcd m/z 383.1390, found m/z 383.1389.
Ethyl 1-(2-oxo-2-phenylethyl)-3,4-diphenyl-1H-pyrazole-5-carboxylate (6a). White solid, yield 84% (172 mg). Rf = 0.63 (n-hexane/ethyl acetate 7/3, v/v), mp 112–113 °C. IR (KBr) νmax, cm−1: 3063, 2987, 2932, 1701 (C
O), 1449, 1226, 1096, 763, 708. 1H NMR (400 MHz, CDCl3) δH ppm: 0.92 (t, J = 7.1 Hz, 3H, CH3), 4.03 (q, J = 7.1 Hz, 2H, OCH2), 6.13 (s, 2H, NCH2), 7.20–7.24 (m, 3H, 3-Ph 3,4,5-H), 7.28–7.37 (m, 5H, 4-Ph 2,3,4,5,6-H), 7.38–7.43 (m, 2H, 3-Ph 2,6-H), 7.50–7.56 (m, 2H, C(O)Ph 3,5-H), 7.61–7.67 (m, 1H, C(O)Ph 4-H), 8.01–8.06 (m, 2H, C(O)Ph 2,6-H). 13C NMR (101 MHz, CDCl3) δC ppm: 13.5 (CH3), 59.2 (NCH2), 61.0 (OCH2), 125.2 (C-4), 127.4 (4-Ph C-4), 127.8 (3-Ph C-4), 127.9 (4-Ph C-2,6), 128.12 (3-Ph C-2,6), 128.14 (C(O)Ph C-2,6), 128.3 (3-Ph C-3,5), 129.1 (C(O)Ph C-3,4), 130.8 (4-Ph C-3,5), 131.8 (C-5), 132.5 (3-Ph C-1), 133.3 (4-Ph C-1), 134.1 (C(O)Ph C-4), 134.9 (C(O)Ph C-1), 149.5 (C-3), 160.4 (COO), 192.5 (CO). 15N NMR (40 MHz, CDCl3) δN ppm: −181.7 (N-1), −67.6 (N-2). HRMS (ESI) for C26H22N2NaO3 ([M + Na]+): calcd m/z 433.1523, found m/z 433.1523.
Ethyl 1-[2-(4-methoxyphenyl)-2-oxoethyl]-3,4-diphenyl-1H-pyrazole-5-carboxylate (6b). White solid, yield 74% (163 mg). Rf = 0.28 (n-hexane/ethyl acetate 4/1, v/v), mp 144–145 °C. IR (KBr) νmax, cm−1: 3053, 2989, 2962, 2837, 1708 (C
O), 1601, 1235, 1176, 1095, 838, 699. 1H NMR (400 MHz, CDCl3) δH ppm: 0.91 (t, J = 6.8 Hz, 3H, CH3), 3.90 (s, 3H, OCH3), 3.98–4.08 (m, 2H, OCH2), 6.09 (s, 2H, NCH2), 6.95–7.06 (m, 2H, C(O)Ph 3,5-H), 7.18–7.25 (m, 3H, 3-Ph 3,4,5-H), 7.28–7.47 (m, 7H, 3-Ph 2,6-H; 4-Ph 2,3,4,5,6-H), 7.95–8.08 (m, 2H, C(O)Ph 2,6-H). 13C NMR (101 MHz, CDCl3) δC ppm: 13.6 (CH3), 55.7 (OCH3), 58.9 (CH2), 61.0 (OCH2), 114.3 (C(O)Ph C-3,5), 125.1 (C-4), 127.4 (4-Ph C-4), 127.78 (3-Ph C-4), 127.86 (C(O)Ph C-1), 127.91 (4-Ph C-2,6), 128.1 (3-Ph C-2,6), 128.3 (3-Ph C-3,5), 130.5 (C(O)Ph C-2,6), 130.8 (4-Ph C-3,5), 132.0 (C-5), 132.5 (3-Ph C-1), 133.3 (4-Ph C-1), 149.3 (C-3), 160.4 (COO), 164.2 (C(O)Ph C-4), 190.8 (CO). 15N NMR (40 MHz, CDCl3) δN ppm: −181.0 (N-1), −68.5 (N-2). HRMS (ESI) for C27H24N2NaO4 ([M + Na]+): calcd m/z 463.1628, found m/z 463.1631.
Ethyl 1-[2-(4-fluorophenyl)-2-oxoethyl]-3,4-diphenyl-1H-pyrazole-5-carboxylate (6c). White solid, yield 97% (208 mg). Rf = 0.71 (n-hexane/ethyl acetate 7/3, v/v), mp 114–115 °C. IR (KBr) νmax, cm−1: 3060, 2984, 2941, 1707 (C
O), 1598, 1231, 1095, 837, 699. 1H NMR (400 MHz, CDCl3) δH ppm: 0.92 (t, J = 7.1 Hz, 3H, CH3), 4.03 (q, J = 7.1 Hz, 2H, OCH2), 6.10 (s, 2H, NCH2), 7.17–7.25 (m, 5H, C(O)Ph 3,5-H; 3-Ph 3,4,5-H), 7.28–7.43 (m, 7H, 3-Ph 2,6-H; 4-Ph 2,3,4,5,6-H), 8.02–8.10 (m, 2H, C(O)Ph 2,6-H). 13C NMR (101 MHz, CDCl3) δC ppm: 13.5 (CH3), 59.0 (NCH2), 61.1 (OCH2), 116.32 (d, 2JCF = 22.1 Hz, C(O)Ph C-3,5), 125.2 (C-4), 127.5 (4-Ph C-4), 127.9 (3-Ph C-4), 128.0 (4-Ph C-2,6), 128.1 (3-Ph C-2,6), 128.3 (3-Ph C-3,5), 130.8 (4-Ph C-3,5), 130.85 (d, 3JCF = 9.7 Hz, C(O)Ph C-2,6), 131.31 (d, 4JCF = 3.0 Hz, C(O)Ph C-1), 131.8 (C-5), 132.4 (3-Ph C-1), 133.2 (4-Ph C-1), 149.5 (C-4), 160.5 (COO), 166.33 (d, J = 256.1 Hz, C(O)Ph C-4), 190.9 (CO). 15N NMR (40 MHz, CDCl3) δN ppm: −181.8 (N-1), −67.6 (N-2). 19F NMR (376 MHz, CDCl3) δF ppm: −103.4 (C(O)Ph 4-F). HRMS (ESI) for C26H21FN2NaO3 ([M + Na]+): calcd m/z 451.1428, found m/z 451.1431.
Ethyl 1-[2-(4-chlorophenyl)-2-oxoethyl]-3,4-diphenyl-1H-pyrazole-5-carboxylate (6d). White solid, yield 90% (200 mg). Rf = 0.74 (n-hexane/ethyl acetate 7/3, v/v), mp 135–136 °C. IR (KBr) νmax, cm−1: 3062, 2999 and 2992 (doublet), 2939, 1702 (C
O), 1224, 1094, 696. 1H NMR (400 MHz, CDCl3) δH ppm: 0.91 (t, J = 7.1 Hz, 3H, CH3), 4.03 (q, J = 7.1 Hz, 2H, OCH2), 6.08 (s, 2H, NCH2), 7.19–7.24 (m, 3H, 3-Ph 3,4,5-H), 7.28–7.44 (m, 7H, 3-Ph 2,6-H; 4-Ph 2,3,4,5,6-H), 7.47–7.55 (m, 2H, C(O)Ph 3,5-H), 7.92–8.00 (m, 2H, C(O)Ph 2,6-H). 13C NMR (101 MHz, CDCl3) δC ppm: 13.4 (CH3), 58.9 (NCH2), 60.9 (OCH2), 125.2 (C-4), 127.3 (4-Ph C-4), 127.7 (3-Ph C-4), 127.8 (4-Ph C-2,6), 128.0 (3-Ph C-2,6), 128.2 (3-Ph C-3,5), 129.3 (C(O)Ph C-3,5), 129.4 (C(O)Ph C-2,6), 130.7 (4-Ph C-3,5), 131.6 (C-5), 132.3 (3-Ph C-1), 133.04 (C(O)Ph C-1), 133.07 (4-Ph C-1), 140.4 (C(O)Ph C-4), 149.4 (C-3), 160.3 (COO), 191.3 (CO). 15N NMR (40 MHz, CDCl3) δN ppm: −182.0 (N-1), −67.7 (N-2). HRMS (ESI) for C26H21ClN2NaO3 ([M + Na]+): calcd m/z 467.1133, found m/z 467.1134.
Ethyl 1-[2-(4-hydroxyphenyl)-2-oxoethyl]-3,4-diphenyl-1H-pyrazole-5-carboxylate (6e). White solid, yield 87% (186 mg). Rf = 0.68 (n-hexane/ethyl acetate 1/1, v/v), mp 143–144 °C. IR (KBr) νmax, cm−1: 3300 (OH), 3059, 2984, 2949, 1709 (C
O), 1603, 1234, 1170, 700. 1H NMR (400 MHz, DMSO-d6) δH ppm: 0.81 (t, J = 7.1 Hz, 3H, CH3), 3.95 (q, J = 7.0 Hz, 2H, OCH2), 6.11 (s, 2H, NCH2), 6.88–6.99 (m, 2H, C(O)Ph 3,5-H), 7.20–7.43 (m, 10H, 3-Ph 2,3,4,5,6-H; 4-Ph 2,3,4,5,6-H), 7.94–8.01 (m, 2H, C(O)Ph 2,6-H), 10.56 (s, 1H, OH). 13C NMR (101 MHz, DMSO-d6) δC ppm: 13.2 (CH3), 58.9 (NCH2), 60.6 (OCH2), 115.5 (C(O)Ph C-3,5), 124.1 (C-4), 125.8 (C(O)Ph C-1), 127.42 (3-Ph C-2,6; 4-Ph C-4), 127.7 (3-Ph C-4), 128.0 (4-Ph C-2,6), 128.3 (3-Ph C-3,5), 130.3 (4-Ph C-3,5), 130.7 (C(O)Ph C-2,6), 131.5 (3-Ph C-1), 132.2 (C-5), 132.8 (4-Ph C-1), 147.6 (C-3), 159.2 (COO), 162.8 (C(O)Ph C-4), 190.8 (CO). 15N NMR (40 MHz, DMSO-d6) δN ppm: −177.7 (N-1), −64.9 (N-2). HRMS (ESI) for C26H22N2NaO4 ([M + Na]+): calcd m/z 449.1472, found m/z 449.1469.
Ethyl 3,4-bis(4-methoxyphenyl)-1-(2-oxo-2-phenylethyl)-1H-pyrazole-5-carboxylate (6f). White solid, yield 77% (181 mg). Rf = 0.40 (n-hexane/ethyl acetate 4/1, v/v), mp 62–63 °C. IR (KBr) νmax, cm−1: 2982, 2939, 2836, 1706 (C
O), 1450, 1248, 1176, 1093, 1032, 834. 1H NMR (400 MHz, CDCl3) δH ppm: 0.97 (t, J = 7.1 Hz, 3H, CH3), 3.77 (s, 3H, 3-Ph 4-OCH3), 3.84 (s, 3H, 4-Ph 4-OCH3), 4.05 (q, J = 7.1 Hz, 2H, OCH2), 6.11 (s, 2H, NCH2), 6.72–6.80 (m, 2H, 3-Ph 3,5-H), 6.84–6.94 (m, 2H, 4-Ph 3,5-H), 7.19–7.25 (m, 2H, 4-Ph 2,6-H), 7.30–7.37 (m, 2H, 3-Ph 2,6-H), 7.48–7.57 (m, 2H, C(O)Ph 3,5-H), 7.60–7.68 (m, 1H, C(O)Ph 4-H), 7.99–8.07 (m, 2H, C(O)Ph 2,6-H). 13C NMR (101 MHz, CDCl3) δC ppm: 13.7 (CH3), 55.3 (3-Ph 4-OCH3), 55.4 (4-Ph 4-OCH3), 59.4 (NCH2), 61.0 (OCH2), 113.4 (4-Ph C-3,5), 113.7 (3-Ph C-3,5), 124.5 (C-4), 125.1 (3-Ph C-1), 125.4 (4-Ph C-1), 128.1 (C(O)Ph C-2,6), 129.1 (C(O)Ph C-3,5), 129.4 (3-Ph C-2,6), 131.8 (C-5), 132.0 (4-Ph C-2,6), 134.0 (C(O)Ph C-4), 134.9 (C(O)Ph C-1), 149.4 (C-3), 159.0 (3-Ph C-4), 159.3 (4-Ph C-4), 160.5 (COO), 192.5 (CO). 15N NMR (40 MHz, CDCl3) δN ppm: −183.6 (N-1), −71.6 (N-2). HRMS (ESI) for C28H26N2NaO5 ([M + Na]+): calcd m/z 493.1734, found m/z 493.1732.
Ethyl 3,4-bis(4-methoxyphenyl)-1-[2-(4-methoxyphenyl)-2-oxoethyl]-1H-pyrazole-5-carboxylate (6g). White solid, yield 91% (228 mg). Rf = 0.29 (n-hexane/ethyl acetate 4/1, v/v), mp 67–68 °C. IR (KBr) νmax, cm−1: 2938, 2837, 1707 (C
O), 1699, 1601, 1320, 1245, 1174, 1092, 1031, 835. 1H NMR (400 MHz, CDCl3) δH ppm: 0.96 (t, J = 7.1 Hz, 3H, CH3), 3.76 (s, 3H, 4-Ph 4-OCH3), 3.84 (s, 3H, 3-Ph 4-OCH3), 3.90 (s, 3H, C(O)Ph 4-OCH3), 4.05 (q, J = 7.1 Hz, 2H, OCH2), 6.05 (s, 2H, NCH2), 6.73–6.79 (m, 2H, 3-Ph 3,5-H), 6.85–6.91 (m, 2H, 4-Ph 3,5-H), 6.97–7.02 (m, 2H, C(O)Ph 3,5-H), 7.19–7.25 (m, 2H, 4-Ph 2,6-H), 7.30–7.36 (m, 2H, 3-Ph 2,6-H), 7.98–8.03 (m, 2H, C(O)Ph 2,6-H). 13C NMR (101 MHz, CDCl3) δC ppm: 13.6 (CH3), 55.16 (3-Ph 4-OCH3), 55.23 (4-Ph 4-OCH3), 55.6 (C(O)Ph 4-OCH3), 58.7 (NCH2), 60.8 (OCH2), 113.2 (4-Ph C-3,5), 113.6 (3-Ph C-3,5), 114.1 (C(O)Ph C-3,5), 124.3 (C-4), 125.2 (3-Ph C-1), 125.4 (4-Ph C-1), 127.8 (C(O)Ph C-1), 129.2 (3-Ph C-2,6), 130.3 (C(O)Ph C-2,6), 131.6 (C-5), 131.9 (4-Ph C-2,6), 149.2 (C-3), 158.8 (3-Ph C-4), 159.1 (4-Ph C-4), 160.4 (COO), 164.1 (C(O)Ph C-4), 190.8 (CO). 15N NMR (40 MHz, CDCl3) δN ppm: −182.9 (N-1), −69.1 (N-2). HRMS (ESI) for C29H28N2NaO6 ([M + Na]+): calcd m/z 523.1840, found m/z 523.1840.
Ethyl 1-[2-(4-fluorophenyl)-2-oxoethyl]-3,4-bis(4-methoxy-phenyl)-1H-pyrazole-5-carboxylate (6h). White solid, yield 66% (161 mg). Rf = 0.67 (n-hexane/ethyl acetate 3/2, v/v), mp 104–105 °C. IR (KBr) νmax, cm−1: 2998, 2940, 2836, 1705 (C
O), 1597, 1436, 1249, 1178, 1091, 1033, 839. 1H NMR (400 MHz, DMSO-d6) δH ppm: 0.84 (t, J = 7.1 Hz, 3H, CH3), 3.71 (s, 3H, 3-Ph 4-OCH3), 3.78 (s, 3H, 4-Ph 4-OCH3), 3.96 (q, J = 7.1 Hz, 2H, OCH2), 6.17 (s, 2H, NCH2), 6.80–6.87 (m, 2H, 3-Ph 3,5-H), 6.91–6.97 (m, 2H, 4-Ph 3,5-H), 7.13–7.19 (m, 2H, 4-Ph 2,6-H), 7.23–7.30 (m, 2H, 3-Ph 2,6-H), 7.40–7.48 (m, 2H, C(O)Ph 3,5-H), 8.15–8.22 (m, 2H, C(O)Ph 2,6-H). 13C NMR (101 MHz, DMSO-d6) δC ppm: 13.4 (CH3), 55.1 (2 × OCH3), 59.2 (NCH2), 60.6 (OCH2), 113.5 (4-Ph C-3,5), 113.8 (3-Ph C-3,5), 116.14 (d, 2JCF = 22.0 Hz, C(O)Ph C-3,5), 123.5 (C-4), 124.71 (3-Ph C-1), 124.73 (4-Ph C-1), 128.7 (3-Ph C-2,6), 131.16, 131.20 and 131.26 (m, C-5, C(O)Ph C-1,2,6), 131.5 (4-Ph C-2,6), 147.9 (C-3), 158.6 (4-Ph 4-OCH3), 158.9 (3-Ph 4-OCH3), 159.4 (COO), 165.56 (d, JCF = 252.9 Hz, C(O)Ph C-4), 191.9 (CO). 15N NMR (40 MHz, DMSO-d6) δN ppm: −180.0 (N-1), −66.2 (N-1). 19F NMR (376 MHz, DMSO-d6) δF ppm: −104.5 (C(O)Ph 4-F). HRMS (ESI) for C28H25FN2NaO5 ([M + Na]+): calcd m/z 511.1640, found m/z 511.1639.
Ethyl 1-[2-(4-chlorophenyl)-2-oxoethyl]-3,4-bis(4-methoxy-phenyl)-1H-pyrazole-5-carboxylate (6i). White solid, yield 82% (207 mg). Rf = 0.35 (n-hexane/ethyl acetate 4/1, v/v), mp 132–133 °C. IR (KBr) νmax, cm−1: 2982, 2939, 2836, 1706 (C
O), 1450, 1248, 1176, 1092, 1032, 834. 1H NMR (400 MHz, CDCl3) δH ppm: 0.96 (t, J = 7.1 Hz, 3H, CH3), 3.76 (s, 3H, 3-Ph 4-OCH3), 3.84 (s, 3H, 4-Ph 4-OCH3), 4.04 (q, J = 7.1 Hz, 2H, OCH2), 6.04 (s, 2H, NCH2), 6.72–6.81 (m, 2H, 3-Ph 3,5-H), 6.84–6.93 (m, 2H, 4-Ph 3,5-H), 7.17–7.24 (m, 2H, 4-Ph 2,6-H), 7.29–7.37 (m, 2H, 3-Ph 2,6-H), 7.45–7.53 (m, 2H, C(O)Ph 3,5-H), 7.90–8.00 (m, 2H, C(O)Ph 2,6-H). 13C NMR (101 MHz, CDCl3) δC ppm: 13.6 (CH3), 55.16 (3-Ph 4-OCH3), 55.22 (4-Ph 4-OCH3), 58.9 (NCH2), 60.9 (OCH2), 113.3 (4-Ph C-3,5), 113.6 (3-Ph C-3,5), 124.4 (C-4), 125.0 (3-Ph C-1), 125.3 (4-Ph C-1), 129.2 (3-Ph C-2,6), 129.3 (C(O)Ph C-3,5), 129.4 (C(O)Ph C-2,6), 131.4 (C-5), 131.8 (4-Ph C-2,6), 133.1 (C(O)Ph C-1), 140.4 (C(O)Ph C-4), 149.4 (C-3), 158.9 (4-Ph C-4), 159.17 (3-Ph C-4), 160.4 (COO), 191.4 (CO). 15N NMR (40 MHz, CDCl3) δN ppm: −183.5 (N-1), −69.1 (N-2). HRMS (ESI) for C28H25ClN2NaO5 ([M + Na]+): calcd m/z 527.1344, found m/z 527.1346.
Ethyl 3-(4-fluorophenyl)-1-(2-oxo-2-phenylethyl)-4-phenyl-1H-pyrazole-5-carboxylate (6j). White solid, yield 73% (156 mg). Rf = 0.74 (n-hexane/ethyl acetate 7/3, v/v), mp 135–136 °C. IR (KBr) νmax, cm−1: 3063, 2978, 2955, 1716 (C
O), 1523, 1449, 1305, 1222, 1096, 846, 768. 1H NMR (400 MHz, DMSO-d6) δH ppm: 0.80 (t, J = 7.1 Hz, 3H, CH3), 3.95 (q, J = 7.0 Hz, 2H, OCH2), 6.22 (s, 2H, NCH2), 7.06–7.18 (m, 2H, 3-Ph 3,5-H), 7.22–7.45 (m, 7H, 3-Ph 2,6-H; 4-Ph 2,3,4,5,6-H), 7.58–7.67 (m, 2H, C(O)Ph 3,5-H), 7.70–7.79 (m, 1H, C(O)Ph 4-H), 8.05–8.16 (m, 2H, C(O)Ph 2,6-H). 13C NMR (101 MHz, DMSO-d6) δC ppm: 13.2 (CH3), 59.3 (NCH2), 60.7 (OCH2), 115.29 (d, 2JCF = 21.5 Hz, 3-Ph C-3,5), 124.0 (C-4), 127.5 (4-Ph C-4), 128.1 (4-Ph C-3,5; C(O)Ph C-2,6), 128.62 (d, 4JCF = 3.1 Hz, 3-Ph C-4), 129.0 (C(O)Ph C-3,5), 129.43 (d, 3JCF = 8.2 Hz, 3-Ph C-2,6), 130.3 (4-Ph C-2,6), 131.4 (C-5), 132.5 (4-Ph C-1), 134.1 (C(O)Ph C-4), 134.3 (C(O)Ph C-1), 146.9 (C-3), 159.2 (COO), 161.72 (d, JCF = 245.2 Hz, 3-Ph C-4), 193.0 (CO). 15N NMR (40 MHz, DMSO-d6) δN ppm: −178.1 (N-1), −65.2 (N-2). 19F NMR (376 MHz, DMSO-d6) δF ppm: −114.0 (3-Ph 4-F). HRMS (ESI) for C26H21FN2NaO3 ([M + Na]+): calcd m/z 451.1428, found m/z 451.1427.
Ethyl 3,4-bis(4-fluorophenyl)-1-(2-oxo-2-phenylethyl)-1H-pyrazole-5-carboxylate (6k). White solid, yield 79% (176 mg). Rf = 0.71 (n-hexane/ethyl acetate 7/3, v/v), mp 103–104 °C. IR (KBr) νmax, cm−1: 3063, 2976, 2949, 1704 (C
O), 1524, 1442, 1223, 1095, 856, 602. 1H NMR (400 MHz, DMSO-d6) δH ppm: 0.84 (t, J = 7.0 Hz, 3H, CH3), 3.97 (q, J = 7.0 Hz, 2H, OCH2), 6.22 (s, 2H, NCH2), 7.10–7.38 (m, 8H, 3-Ph 2,3,5,6-H; 4-Ph 2,3,5,6-H), 7.58–7.66 (m, 2H, C(O)Ph 3,5-H), 7.71–7.78 (m, 1H, C(O)Ph 4-H), 8.06–8.13 (m, 2H, C(O)Ph 2,6-H). 13C NMR (101 MHz, DMSO-d6) δC ppm: 13.2 (CH3), 59.4 (NCH2), 60.7 (NCH2), 115.01 (d, 2JCF = 21.4 Hz, 4-Ph C-3,5), 115.38 (d, 2JCF = 21.6 Hz, 3-Ph C-3,5), 123.0 (C-4), 128.1 (C(O)Ph C-2,6), 128.47 (d, 4JCF = 3.2 Hz, 3-Ph C-1), 128.77 (d, 4JCF = 3.2 Hz, 4-Ph C-1), 129.0 (C(O)Ph C-3,5), 129.50 (d, 3JCF = 8.3 Hz, 3-Ph C-2,6), 131.5 (C-5), 132.40 (d, 3JCF = 8.2 Hz, 4-Ph C-2,6) 134.1 (C(O)Ph C-1), 134.3 (C(O)Ph C-4), 147.1 (C-3), 159.1 (COO), 161.67 (d, JCF = 244.1 Hz, 4-Ph C-4), 161.76 (d, JCF = 245.2 Hz, 3-Ph C-4), 193.0 (CO). 15N NMR (40 MHz, DMSO-d6) δN ppm: −177.8 (N-1), −65.1 (N-2). 19F NMR (376 MHz, DMSO-d6) δF ppm: −114.7 (4-F), −113.9 (4-F). HRMS (ESI) for C26H20F2N2NaO3 ([M + Na]+): calcd m/z 469.1334, found m/z 469.1334.
Ethyl 3-(4-fluorophenyl)-4-(4-methoxyphenyl)-1-(2-oxo-2-phenylethyl)-1H-pyrazole-5-carboxylate (6l). White solid, yield 80% (183 mg). Rf = 0.68 (n-hexane/ethyl acetate 3/2, v/v), mp 87–88 °C. IR (KBr) νmax, cm−1: 2984, 2941, 2837, 1724, 1702 (C
O), 1440, 1223, 1176, 1087, 832. 1H NMR (400 MHz, CDCl3) δH ppm: 0.97 (t, J = 7.1 Hz, 3H, CH3), 3.84 (s, 3H, OCH3), 4.06 (q, J = 7.1 Hz, 2H, OCH2), 6.11 (s, 2H, NCH2), 6.86–6.96 (m, 4H, 3-Ph 3,5-H; 4-Ph 3,5-H), 7.18–7.24 (m, 2H, 4-Ph 2,6-H), 7.35–7.41 (m, 2H, 3-Ph 2,6-H), 7.50–7.57 (m, 2H, C(O)Ph 3,5-H), 7.61–7.68 (m, 1H, C(O)Ph 4-H), 7.98–8.08 (m, 2H, C(O)Ph 2,6-H). 13C NMR (101 MHz, CDCl3) δC ppm: 13.7 (CH3), 55.4 (OCH3), 59.2 (NCH2), 61.1 (OCH2), 113.5 (4-Pc C-3,5), 115.26 (d, 2JCF = 21.5 Hz, 3-Ph C-3,5), 124.7 (C-4), 125.0 (4-Ph C-1), 128.1 (C(O)Ph C-2,6), 128.67 (d, 4JCF = 3.2 Hz, 3-Ph C-1), 129.1 (C(O)Ph C-3,5), 129.88 (d, 3JCF = 8.1 Hz, 3-Ph C-2,6), 131.88 (C-5), 131.91 (4-Ph C-2,6), 134.1 (C(O)Ph C-4), 134.8 (C(O)Ph C-1), 148.7 (C-3), 159.1 (4-Ph C-4), 160.4 (COO), 162.55 (d, JCF = 247.1 Hz, 3-Ph C-4), 192.4 (CO). 15N NMR (40 MHz, CDCl3) δN ppm: −182.2 (N-1), −70.0 (N-2). 19F NMR (376 MHz, CDCl3) δF ppm: −114.2 (3-Ph 4-F). HRMS (ESI) for C27H23FN2NaO4 ([M + Na]+): calcd m/z 481.1534, found m/z 481.1535.
Ethyl 3-(4-fluorophenyl)-1-[2-(4-fluorophenyl)-2-oxoethyl]-4-phenyl-1H-pyrazole-5-carboxylate (6m). White solid, yield 76% (170 mg). Rf = 0.64 (n-hexane/ethyl acetate 3/2, v/v), mp 104–105 °C. IR (KBr) νmax, cm−1: 3059, 2999, 2962, 1711 (C
O), 1599, 1232, 1156, 1095, 840. 1H NMR (400 MHz, DMSO-d6) δH ppm: 0.80 (t, J = 7.1 Hz, 3H, CH3), 3.95 (q, J = 7.1 Hz, 2H, OCH2), 6.21 (s, 2H, NCH2), 7.07–7.17 (m, 2H, 3-Ph 3,5-H), 7.21–7.51 (m, 9H, 3-Ph 2,6-H; 4-Ph 2,3,4,5,6-H; C(O)Ph 3,5-H), 8.16–8.22 (m, 2H, C(O)Ph 2,6-H). 13C NMR (101 MHz, DMSO-d6) δC ppm: 13.2 (CH3), 59.3 (NCH2), 60.7 (OCH2), 115.29 (d, 2JCF = 21.6 Hz, 3-Ph C-3,5), 116.12 (d, 2JCF = 22.0 Hz, C(O)Ph C-3,5), 124.0 (C-4), 127.5 (4-Ph C-4), 128.1 (4-Ph C-3,5), 128.60 (d, 4JCF = 3.1 Hz, 3-Ph C-1), 129.42 (d, 4JCF = 8.3 Hz, 3-Ph C-2,6), 130.3 (4-Ph C-2,6), 131.10 (d, 4JCF = 2.8 Hz, C(O)Ph C-1), 131.21 (d, 3JCF = 9.6 Hz, C(O)Ph C-2,6), 131.4 (C-5), 132.5 (4-Ph C-1), 147.0 (C-3), 159.2 (COO), 161.72 (d, JCF = 245.1 Hz, 3-Ph C-4), 165.54 (d, JCF = 252.9 Hz, C(O)Ph C-4), 191.7 (CO). 15N NMR (40 MHz, DMSO-d6) δN ppm: −177.7 (N-1), −65.2 (N-2). 19F NMR (376 MHz, DMSO-d6) δF ppm: −114.0 (4-F), −104.4 (4-F). HRMS (ESI) for C26H20F2N2NaO3 ([M + Na]+): calcd m/z 469.1334, found m/z 469.1332.
Ethyl 3-(4-fluorophenyl)-1-[2-(4-fluorophenyl)-2-oxoethyl]-4-(4-methoxyphenyl)-1H-pyrazole-5-carboxylate (6n). White solid, yield 79% (188 mg). Rf = 0.69 (n-hexane/ethyl acetate x/x, v/v), mp 81–82 °C. IR (KBr) νmax, cm−1: 2993, 2956, 2840, 1706 (C
O), 1597, 1440, 1232, 1093, 844. 1H NMR (400 MHz, CDCl3) δH ppm: 0.97 (t, J = 7.1 Hz, 3H, CH3), 3.85 (s, 3H, OCH3), 4.05 (q, J = 7.1 Hz, 2H, OCH2), 6.07 (s, 2H, NCH2), 6.86–6.96 (m, 4H, 4-Ph 3,5-H; C(O)Ph 3,5-H), 7.16–7.24 (m, 4H, 3-Ph 3,5-H; 4-Ph 2,6-H), 7.33–7.42 (m, 2H, 3-Ph 2,6-H), 8.01–8.11 (m, 2H, C(O)Ph 2,6-H). 13C NMR (101 MHz, CDCl3) δC ppm: 13.6 (CH3), 55.3 (OCH3), 58.9 (NCH2), 61.0 (OCH2), 113.4 (4-Ph C-3,5), 115.16 (d, 2JCF = 21.5 Hz, C(O)Ph C-3,5), 116.23 (d, 2JCF = 22.0 Hz, 3-Ph C-3,5), 124.6 (C-4), 124.9 (4-Ph C-1), 128.51 (d, 4JCF = 3.3 Hz, 3-Ph C-1), 129.75 (d, 3JCF = 8.1 Hz, 3-Ph C-2,6), 130.73 (d, 3JCF = 9.5 Hz, C(O)Ph C-2,6), 131.15 (d, 4JCF = 3.1 Hz, C(O)Ph C-1), 131.7 (C-5), 131.8 (4-Ph C-2,6), 148.7 (C-3), 159.0 (4-Ph C-4), 160.3 (COO), 162.45 (d, JCF = 247.1 Hz, 3-Ph C-4), 166.24 (d, JCF = 256.1 Hz, C(O)Ph C-4), 190.8 (CO). 15N NMR (40 MHz, CDCl3) δN ppm: −182.8 (N-1), −69.2 (N-2). 19F NMR (376 MHz, CDCl3) δF ppm: −114.2 (4-F), −103.3 (4-F). HRMS (ESI) for C27H22F2N2NaO4 ([M + Na]+): calcd m/z 499.1440, found m/z 499.1439.
Ethyl 3,4-bis(4-fluorophenyl)-1-[2-(4-fluorophenyl)-2-oxoethyl]-1H-pyrazole-5-carboxylate (6o). White solid, yield 85% (197 mg). Rf = 0.62 (n-hexane/ethyl acetate 7/3, v/v), mp 105–106 °C. IR (KBr) νmax, cm−1: 3000, 2984, 2946, 1710 (C
O), 1599, 1444, 1231, 840. 1H NMR (400 MHz, DMSO-d6) δH ppm: 0.84 (t, J = 7.1 Hz, 3H, CH3), 3.97 (q, J = 7.0 Hz, 2H, OCH2), 6.22 (s, 2H, NCH2), 7.09–7.38 (m, 8H, 3-Ph 2,3,5,6-H; 3-Ph 2,3,5,6-H), 7.40–7.51 (m, 2H, C(O)Ph 3,5-H), 8.12–8.25 (m, 2H, C(O)Ph 2,6-H). 13C NMR (101 MHz, DMSO-d6) δC ppm: 13.2 (CH3), 59.3 (NCH2), 60.8 (OCH2), 115.02 (d, 2JCF = 21.4 Hz, 4-Ph C-3,5), 115.39 (d, 2JCF = 21.5 Hz, 3-Ph C-3,5), 116.13 (d, 2JCF = 22.0 Hz, C(O)Ph C-3,5), 123.0 (C-4), 128.45 (d, 4JCF = 3.2 Hz, 3-Ph C-1), 128.75 (d, 4JCF = 3.3 Hz, 4-Ph C-1), 129.50 (d, 3JCF = 8.3 Hz, 3-Ph C-2,6), 131.09 (d, 4JCF = 2.7 Hz, C(O)Ph C-1), 131.21 (d, 3JCF = 9.6 Hz, C(O)Ph C-2,6), 131.5 (C-5), 132.40 (d, 3JCF = 8.3 Hz, 4-Ph C-2,6), 147.1 (C-3), 159.1 (COO), 161.67 (d, JCF = 244.1 Hz, 4-Ph C-4), 161.77 (d, JCF = 245.3 Hz, 3-Ph C-4), 165.55 (d, JCF = 253.0 Hz, C(O)Ph C-4), 191.69 (CO). 15N NMR (40 MHz, DMSO-d6) δN ppm: −177.7 (N-1), −65.1 (N-2). 19F NMR (376 MHz, DMSO-d6) δF ppm: −114.7 (4-F), −113.9 (4-F), −104.4 (4-F). HRMS (ESI) for C26H19F3N2NaO3 ([M + Na]+): calcd m/z 487.1240, found m/z 487.1238.
Methyl 1-(2-oxo-2-phenylethyl)-3,4-diphenyl-1H-pyrazole-5-carboxylate (7a). White solid, yield 94% (93 mg). Rf = 0.63 (n-hexane/ethyl acetate 7/3, v/v), mp 138–139 °C. IR (KBr) νmax, cm−1: 3059, 2976, 2939, 1716 (C
O), 1451, 1355, 1319, 1231, 1100, 774 and 757 (doublet), 705 and 691 (doublet). 1H NMR (400 MHz, acetone-d6) δH ppm: 3.53 (s, 3H, CH3), 6.24 (s, 2H, CH2), 7.19–7.45 (m, 10H, 3-Ph 2,3,4,5,6-H; 4-Ph 2,3,4,5,6-H), 7.59–7.68 (m, 2H, C(O)Ph 3,5-H), 7.70–7.78 (m, 1H, C(O)Ph 4-H), 8.12–8.19 (m, 2H, C(O)Ph 2,6-H). 13C NMR (101 MHz, acetone-d6) δC ppm: 52.0 (NCH2), 60.1 (OCH2), 125.4 (C-4), 128.2 (4-Ph C-4), 128.5 (3-Ph C-4), 128.7 (4-Ph C-2,6), 128.8 (3-Ph 2,6-H), 128.9 (3-Ph 3,5-H; C(O)Ph C-2,6), 129.8 (C(O)Ph C-3,5), 131.4 (4-Ph C-3,5), 132.4 (C-5), 133.7 (3-Ph C-1), 134.1 (4-Ph C-1), 134.8 (C(O)Ph C-4), 135.9 (C(O)Ph C-1), 149.5 (C-3), 161.2 (COO), 193.4 (CO). 15N NMR (40 MHz, acetone-d6) δN ppm: −178.7 (N-1), −63.3 (N-2). HRMS (ESI) for C25H20N2NaO3 ([M + Na]+): calcd m/z 419.1366, found m/z 419.1367.
Methyl 1-[2-(4-methoxyphenyl)-2-oxoethyl]-3,4-diphenyl-1H-pyrazole-5-carboxylate (7b). White solid, yield 92% (98 mg). Rf = 0.49 (n-hexane/ethyl acetate 7/3, v/v), mp 163–164 °C. IR (KBr) νmax, cm−1: 3025, 2962, 2836, 1712 (C
O), 1601, 1237, 1176, 1096, 698. 1H NMR (400 MHz, acetone-d6) δH ppm: 3.53 (s, 3H, COOCH3), 3.94 (s, 3H, OCH3), 6.17 (s, 2H, CH2), 7.09–7.17 (m, 2H, C(O)Ph 3,5-H), 7.20–7.43 (m, 10H, 3-Ph 2,3,4,5,6-H; 4-Ph 2,3,4,5,6-H), 8.09–8.17 (m, 2H, C(O)Ph 2,6-H). 13C NMR (101 MHz, acetone-d6) δC ppm: 51.9 (COO![[C with combining low line]](https://www.rsc.org/images/entities/char_0043_0332.gif) H3), 56.1 (OCH3), 59.8 (CH2), 115.0 (C(O)Ph C-3,5), 125.3 (C-4), 128.2 (4-Ph C-4), 128.4 (3-Ph C-4), 128.68 (4-Ph C-2,6), 128.72 (C(O)Ph C-1), 128.8 (3-Ph C-2,6), 128.9 (3-Ph C-3,5), 131.2 (C(O)Ph C-2,6), 131.4 (4-Ph C-3,5), 132.5 (C-5), 133.7 (3-Ph C-1), 134.2 (4-Ph C-1), 149.4 (C-3), 161.2 (COO), 165.2 (C(O)Ph C-4), 191.5 (CO). 15N NMR (40 MHz, acetone-d6) δN ppm: −178.2 (N-1), −63.5 (N-2). HRMS (ESI) for C26H22N2NaO4 ([M + Na]+): calcd m/z 449.1472, found m/z 449.1470.
H3), 56.1 (OCH3), 59.8 (CH2), 115.0 (C(O)Ph C-3,5), 125.3 (C-4), 128.2 (4-Ph C-4), 128.4 (3-Ph C-4), 128.68 (4-Ph C-2,6), 128.72 (C(O)Ph C-1), 128.8 (3-Ph C-2,6), 128.9 (3-Ph C-3,5), 131.2 (C(O)Ph C-2,6), 131.4 (4-Ph C-3,5), 132.5 (C-5), 133.7 (3-Ph C-1), 134.2 (4-Ph C-1), 149.4 (C-3), 161.2 (COO), 165.2 (C(O)Ph C-4), 191.5 (CO). 15N NMR (40 MHz, acetone-d6) δN ppm: −178.2 (N-1), −63.5 (N-2). HRMS (ESI) for C26H22N2NaO4 ([M + Na]+): calcd m/z 449.1472, found m/z 449.1470.
Methyl 1-[2-(4-fluorophenyl)-2-oxoethyl]-3,4-diphenyl-1H-pyrazole-5-carboxylate (7c). White solid, yield 96% (100 mg). Rf = 0.66 (n-hexane/ethyl acetate 7/3, v/v), mp 114–115 °C. IR (KBr) νmax, cm−1: 3061, 2929, 1720 (C
O), 1596, 1232, 1086, 841, 700. 1H NMR (400 MHz, acetone-d6) δH ppm: 3.56 (s, 3H, CH3), 6.23 (s, 2H, CH2), 7.13–7.20 (m, 2H, C(O)Ph 3,5-H), 7.23–7.29 (m, 3H, 3-Ph 3,4,5-H), 7.31–7.41 (m, 4H, 3-Ph 2,6-H; 4-Ph 2,6-H), 7.59–7.66 (m, 2H, 4-Ph 3, 5H), 7.70–7.77 (m, 1H, 4-Ph 4-H), 8.12–8.18 (m, 2H, C(O)Ph 2,6-H). 13C NMR (101 MHz, acetone-d6) δC ppm: 52.0 (CH3), 60.2 (CH2), 115.64 (d, 2JCF = 21.6 Hz, C(O)Ph C-3,5), 124.3 (C-4), 128.6 (4-Ph C-4), 128.7 (3-Ph C-4), 128.9 (3-Ph C-3,5), 129.0 (4-Ph C-2,6), 129.8 (3-Ph C-2,6), 130.29 (d, 4JCF = 3.4 Hz, C(O)Ph C-1), 132.4 (4-Ph C-3,5), 133.39 (d, 3JCF = 8.2 Hz, C(O)Ph C-2,6), 133.5 (3-Ph C-1), 134.8 (4-Ph C-1), 135.9 (C-5), 149.6 (C-3), 161.1 (COO), 163.13 (d, JCF = 244.5 Hz, C(O)Ph C-4), 193.3 (CO). 15N NMR (40 MHz, acetone-d6) δN ppm: −178.3 (N-1), −63.2 (N-2). 19F NMR (376 MHz, acetone-d6) δF ppm: −116.5 (C(O)Ph 4-F). HRMS (ESI) for C25H19FN2NaO3 ([M + Na]+): calcd m/z 437.1272, found m/z 437.1269.
Determination of HPLC–log![[thin space (1/6-em)]](https://www.rsc.org/images/entities/h3_char_2009.gif) P
P
HPLC–logP values were determined using Shimadzu HPLC system equipped with apHera C18 column (10 × 6 mm, 5 μm, Supelco, Bellefonte, PA, USA). Each sample was dissolved in the internal standard mixture, consisting of triphenylene (99.9%, Carl Roth) and toluene (≥98%, Sigma-Aldrich). Analysis was performed at 1.5 mL min−1 flow rate in the linear gradient where mobile phase consisted of methanol and 0.01 M phosphate buffer (pH 7.4). The HPLC–logP values were calculated from the measured retention times using previously published equation.57
Plasma protein binding (PPB)
Retention times of the analytes were measured with Shimadzu HPLC system on the CHIRALPAK®HAS stationary phase (50 × 3 mm, 5 μm, Chiral Technologies, DAICEL Group, Europe SAS, France). The mobile phase A consisted of 50 mM aqueous ammonium acetate buffer (pH 7.4) and phase B of 2-propanol according to Valko et al.65 Analysis was performed at prolonged 1 mL min−1 flow rate in the linear gradient. Retention capacity factors (k′) were calculated by using DMSO or a substance with 0% HAS binding for systems' dead time (Rt0). The system was calibrated by injecting the reference compounds: acetylsalicylic acid (CAS 69-72-7), betamethasone (CAS 378-44-9), budesonide (CAS 5133-22-3), carbamazepine (CAS 298-46-4), cimetidine (CAS 51481-61-9), ciprofloxacin (CAS 85721-33-1), indomethacin (CAS 53-86-1), isoniazid (CAS 54-85-3), metronidazole (CAS 443-48-1), nicardipine (CAS 55985-32-5), nizatidine (CAS 76963-41-2) and warfarin (CAS 81-81-2) obtained from Sigma-Aldrich, diclofenac (CAS 15307-86-5) from EMD Chemicals Inc., flumazenil (CAS 78755-81-4) from ABX and ketoprofen (CAS 22071-15-4) from LKT Labs. The logarithmic capacity factors of the references' Rt (log(k′)) on the HSA column were plotted against the %PPB values from literature. The slope and the intercept were used to convert the log(k′) of the compounds (6a, c, f, h, m–o) to %PPB using the regression equation.66Biological evaluation
000 units penicillin and 10 mg streptomycin per mL in 0.9% NaCl) and L-glutamine (Sigma; 200 mM solution). Cell cultures were cultivated at 37 °C, maintaining a humidified atmosphere consisting of 5% CO2. Gibco™ trypsin–EDTA (0.05%) was used for cell passage.:1 ratio in non-supplemented RPMI-1640 medium and were additionally incubated for 2 h. After incubation, the medium was removed, and formazan product was dissolved in DMSO. Optical densities were measured at 550 nm with TECAN Infinite® M200 PRO microplate reader using a reference wavelength of 690 nm to correct unspecific absorption. The quantity of viable cells was normalized to untreated controls. The GI50 values were calculated from dose–response curves.000 cells per well) were treated with the test compounds (6c, 6m) for 72 h in the concentration range of 5, 10 and 20 μM. After treatment cells were washed with DPBS (Sigma; modified, without calcium chloride and magnesium chloride) and trypsinized using a trypsin–EDTA solution (Gibco™, 0.05%). After additional washing with DPBS, cells were stained at +4 °C overnight using PI/HFS solution (50 μg mL−1).67 Samples were then analysed using Guava® easyCyte™ 8HT (Merck Millipore) flow cytometer with Guava Clean 3.1 software. The amount of debris was in the range of 9.2 ± 2.0%.Conclusions
Proceeding from the natural marine alkaloid lamellarin O, a scaffold hopping from a central pyrrole to pyrazole resulted in 18 fully characterized derivatives. Structure–activity relationships revealed the importance of a fluorine in the para-position of the phenyl substituent in phenyl-2-oxoethyl scaffold. The most cytotoxic compounds inhibited cell proliferation in the low micromolar range in three colorectal cancer cell lines, namely HCT116, HT29, and SW480. The similarity of the investigated GI50 values demonstrated the absence of conventional resistance mechanism between the cell lines. Pronounced effects on the cell cycle were observed resulting in G1 and predominantly G2/M phase arrest. This set of lamellarin O inspired pyrazole derivatives is an important entry for natural product-derived drug candidates.Author contributions
Conceptualization, E. A., A. Ž. and V.·P.; methodology, E. A., A. Ž. and V. P.; formal analysis, K. D., N. F. E. S.-B.; investigation, K. D., N. F., E. S.-B., V. P.; resources, A. Š., E. A. and V. P.; data curation, E. A. and V. P.; writing—original draft preparation, K. D.; writing—review and editing, E. A., A. Ž., A. Š. and V. P.; visualization, K. D. and V. P.; supervision, E. A., A. Ž. and V. P.; funding acquisition, V. P. and E. A. All authors have read and agreed to the published version of the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
Open access funding provided by University of Vienna. Authors are thankful to the Institute of Cancer Research, Department of Medicine, Medical University of Vienna for provided cell lines. Furthermore, we thank K. Cseh for the support of FACS measurement and the Institute of Inorganic Chemistry, University of Vienna for the provision of the FACS device.References
- J. Bracegirdle, L. P. Robertson, P. A. Hume, M. J. Page, A. v. Sharrock, D. F. Ackerley, A. R. Carroll and R. A. Keyzers, J. Nat. Prod., 2019, 82, 2000–2008 CrossRef CAS PubMed.
- H. Zhang, M. M. Conte, X. C. Huang, Z. Khalil and R. J. Capon, Org. Biomol. Chem., 2012, 10, 2656–2663 RSC.
- H. Fan, J. Peng, M. T. Hamann and J. F. Hu, Chem. Rev., 2008, 108, 264–287 CrossRef CAS PubMed.
- T. Fukuda, F. Ishibashi and M. Iwao, Alkaloids: Chemistry and Biology, Elsevier Inc., 1st edn, 2020, vol. 83, pp. 1–112, ISSN: 1099-4831 Search PubMed.
- C. Bailly, Curr. Med. Chem.: Anti-Cancer Agents, 2004, 4, 363–378 CrossRef CAS PubMed.
- C. Bailly, Mar. Drugs, 2015, 13, 1105–1123 CrossRef CAS PubMed.
- D. Pla, F. Albericio and M. Álvarez, MedChemComm, 2011, 2, 689–697 RSC.
- P. Krishnaiah, V. L. N. Reddy, G. Venkataramana, K. Ravinder, M. Srinivasulu, T. V. Raju, K. Ravikumar, D. Chandrasekar, S. Ramakrishna and Y. Venkateswarlu, J. Nat. Prod., 2004, 67, 1168–1171 CrossRef CAS PubMed.
- D. Matulja, F. Vranješević, M. Kolympadi Markovic, S. K. Pavelić and D. Marković, Molecules, 2022, 27, 1449, DOI:10.3390/molecules27041449.
- F. Plisson, X. Huang, H. Zhang, Z. Khalil and R. J. Capon, Chem.–Asian J., 2012, 7, 1616–1623 CrossRef CAS PubMed.
- P. Sopha, N. Phutubtim, B. Chantrathonkul, P. Ploypradith, S. Ruchirawat and M. Chittchang, Toxicology, 2021, 462, 152963, DOI:10.1016/j.tox.2021.152963.
- S. Khiati, Y. Seol, K. Agama, I. D. Rosa, S. Agrawal, K. Fesen, H. Zhang, K. C. Neuman and Y. Pommier, Mol. Pharmacol., 2014, 86, 193–199 CrossRef PubMed.
- C. Ballot, A. Martoriati, M. Jendoubi, S. Buche, P. Formstecher, L. Mortier, J. Kluza and P. Marchetti, Marine Drugs, 2014, 12, 779–798 CrossRef CAS PubMed.
- C. Ballot, J. Kluza, S. Lancel, A. Martoriati, S. M. Hassoun, L. Mortier, J. C. Vienne, G. Briand, P. Formstecher, C. Bailly, R. Nevière and P. Marchetti, Apoptosis, 2010, 15, 769–781 CrossRef CAS PubMed.
- C. Ballot, J. Kluza, A. Martoriati, U. Nyman, P. Formstecher, B. Joseph, C. Bailly and P. Marchetti, Mol. Cancer Ther., 2009, 8, 3307–3317 CrossRef CAS PubMed.
- X. C. Huang, X. Xiao, Y. K. Zhang, T. Talele, A. Salim, Z. S. Chen and R. Capon, Mar. Drugs, 2014, 12, 3818–3837 CrossRef CAS PubMed.
- I. Satyanarayana, D. Y. Yang and T. J. Liou, RSC Adv., 2020, 10, 43168–43174 RSC.
- Q. Zhang, Y. Feng and D. Kennedy, Cell. Mol. Life Sci., 2017, 74, 777–801 CrossRef CAS PubMed.
- V. Kumar, A. Awasthi, A. Salam and T. Khan, J. Org. Chem., 2019, 84, 11596–11603 CrossRef CAS PubMed.
- D. Imbri, J. Tauber and T. Opatz, Mar. Drugs, 2014, 12, 6142–6177 CrossRef CAS PubMed.
- D. Morikawa, K. Morii, Y. Yasuda, A. Mori and K. Okano, J. Org. Chem., 2020, 85, 8603–8617 CrossRef CAS PubMed.
- V. Colligs, S. P. Hansen, D. Imbri, E. J. Seo, O. Kadioglu, T. Efferth and T. Opatz, Bioorg. Med. Chem., 2017, 25, 6137–6148 CrossRef CAS PubMed.
- K. Klumthong, P. Chalermsub, P. Sopha, S. Ruchirawat and P. Ploypradith, J. Org. Chem., 2021, 86, 14883–14902 CrossRef CAS PubMed.
- T. Fukuda, Y. Nanjo, M. Fujimoto, K. Yoshida, Y. Natsui, F. Ishibashi, F. Okazaki, H. To and M. Iwao, Bioorg. Med. Chem., 2019, 27, 265–277 CrossRef CAS PubMed.
- L. Zheng, T. Gao, Z. Ge, Z. Ma, J. Xu, W. Ding and L. Shen, Eur. J. Med. Chem., 2021, 214, 113226, DOI:10.1016/j.ejmech.2021.113226.
- T. Fukuda, T. Umeki, K. Tokushima, G. Xiang, Y. Yoshida, F. Ishibashi, Y. Oku, N. Nishiya, Y. Uehara and M. Iwao, Bioorg. Med. Chem., 2017, 25, 6563–6580 CrossRef CAS PubMed.
- T. Fukuda, M. Anzai, A. Nakahara, K. Yamashita, K. Matsukura, F. Ishibashi, Y. Oku, N. Nishiya, Y. Uehara and M. Iwao, Bioorg. Med. Chem., 2021, 34, 116039, DOI:10.1016/j.bmc.2021.116039.
- K. Klumthong, P. Chalermsub, P. Sopha, S. Ruchirawat and P. Ploypradith, J. Org. Chem., 2021, 86, 14883–14902 CrossRef CAS PubMed.
- P. K. Mykhailiuk, Chem. Rev., 2021, 121, 1670–1715 CrossRef CAS PubMed.
- W. Byon, S. Garonzik, R. A. Boyd and C. E. Frost, Clin. Pharmacokinet., 2019, 58, 1265–1279 CrossRef PubMed.
- X. Li, Y. Yu and Z. Tu, Molecules, 2021, 26, 1202, DOI:10.3390/molecules26051202.
- P. R. Lazzara and T. W. Moore, RSC Med. Chem., 2020, 11, 18–29 RSC.
- V. Milišiūnaitė, A. Kadlecová, A. Žukauskaitė, K. Doležal, M. Strnad, J. Voller, E. Arbačiauskienė, W. Holzer and A. Šačkus, Mol. Diversity, 2020, 24, 1025–1042 CrossRef PubMed.
- V. Milišiūnaitė, R. Paulavičiūtė, E. Arbačiauskienė, V. Martynaitis, W. Holzer and A. Šačkus, Beilstein J. Org. Chem., 2019, 15, 679–684 CrossRef PubMed.
- V. Milišiūnaitė, E. Plytninkienė, R. Bakšienė, A. Bieliauskas, S. Krikštolaitytė, G. Račkauskienė, E. Arbačiauskienė and A. Šačkus, Molecules, 2021, 26, 5604, DOI:10.3390/molecules26185604.
- B. Razmienė, E. Řezníčková, V. Dambrauskienė, R. Ostruszka, M. Kubala, A. Žukauskaitė, V. Kryštof, A. Šačkus and E. Arbačiauskienė, Molecules, 2021, 26, 6747, DOI:10.3390/molecules26216747.
- G. Matulevičiūtė, E. Arbačiauskienė, N. Kleizienė, V. Kederienė, G. Ragaitė, M. Dagilienė, A. Bieliauskas, V. Milišiūnaitė, F. A. Sløk and A. Šačkus, Molecules, 2021, 26, 3808, DOI:10.3390/molecules26133808.
- V. Milišiūnaitė, E. Arbačiauskienė, E. Řezníčková, R. Jorda, V. Malínková, A. Žukauskaitė, W. Holzer, A. Šačkus and V. Kryštof, Eur. J. Med. Chem., 2018, 150, 908–919 CrossRef PubMed.
- B. Razmienė, V. Vojáčková, E. Řezníčková, L. Malina, V. Dambrauskienė, M. Kubala, R. Bajgar, H. Kolářová, A. Žukauskaitė, E. Arbačiauskienė, A. Šačkus and V. Kryštof, Bioorg. Chem., 2022, 119, 105570, DOI:10.1016/j.bioorg.2021.105570.
- G. Varvuolytė, L. Malina, A. Bieliauskas, B. Hošíková, H. Simerská, H. Kolářová, N. Kleizienė, V. Kryštof, A. Šačkus and A. Žukauskaitė, Dyes Pigm., 2020, 183, 108666, DOI:10.1016/j.dyepig.2020.108666.
- E. Arbačiauskienė, V. Laukaitytė, W. Holzer and A. Šačkus, Eur. J. Org. Chem., 2015, 2015, 5663–5670 CrossRef.
- Y. Wu, C. Tang, R. Rui, L. Yang, W. Ding, J. Wang, Y. Li, C. C. Lai, Y. Wang, R. Luo, W. Xiao, H. Zhang, Y. Zheng and Y. He, Acta Pharm. Sin. B, 2020, 10, 512–528 CrossRef CAS PubMed.
- B. Xiong, S. Chen, P. Zhu, M. Huang, W. Gao, R. Zhu, J. Qian, Y. Peng, Y. Zhang, H. Dai and Y. Ling, Med. Chem., 2019, 15, 743–754 CrossRef CAS PubMed.
- M. A. Düfert, K. L. Billingsley and S. L. Buchwald, J. Am. Chem. Soc., 2013, 135, 12877–12885 CrossRef PubMed.
- A. Secrieru, P. M. O'Neill and M. L. S. Cristiano, Molecules, 2019, 25, 42, DOI:10.3390/molecules25010042.
- R. Lin, G. Chiu, Y. Yu, P. J. Connolly, S. Li, Y. Lu, M. Adams, A. R. Fuentes-Pesquera, S. L. Emanuel and L. M. Greenberger, Bioorg. Med. Chem. Lett., 2007, 17, 4557–4561 CrossRef CAS PubMed.
- M. Guerrero, J. Pérez, J. Ros, V. Branchadell, E. Pellicer, J. Sort and J. Pons, Curr. Org. Synth., 2013, 11, 149–155 CrossRef.
- M. Iškauskienė, G. Ragaitė, F. A. Sløk and A. Šačkus, Mol. Diversity, 2020, 24, 1235–1251 CrossRef PubMed.
- K. Dzedulionytė, M. Veikšaitė, V. Morávek, V. Malinauskienė, G. Račkauskienė, A. Šačkus, A. Žukauskaitė and E. Arbačiauskienė, Molecules, 2022, 27, 8666, DOI:10.3390/molecules27248666.
- C. F. R. A. C. Lima, A. S. M. C. Rodrigues, V. L. M. Silva, A. M. S. Silva and L. M. N. B. F. Santos, ChemCatChem, 2014, 6, 1291–1302 CrossRef CAS.
- J. Sherwood, J. H. Clark, I. J. S. Fairlamb and J. M. Slattery, Green Chem., 2019, 21, 2164–2213 RSC.
- S. Bhat K, V. Lanke, J. D. Prasad and K. R. Prabhu, Appl. Catal., A, 2020, 596, 117516, DOI:10.1016/j.apcata.2020.117516.
- K. S. M. Salih and Y. Baqi, Catalysts, 2020, 10, 4, DOI:10.3390/catal10010004.
- Z. Li, C. Liu, W. Shi, X. Cai, Y. Dai, C. Liao, W. Huang and H. Qian, Bioorg. Med. Chem. Lett., 2018, 26, 703–711 CrossRef CAS PubMed.
- C. A. Lipinski, F. Lombardo, B. W. Dominy and P. J. Feeney, Adv. Drug Delivery Rev., 2012, 64, 4–17 CrossRef.
- D. Camp, A. Garavelas and M. Campitelli, J. Nat. Prod., 2015, 78, 1370–1382 CrossRef CAS PubMed.
- C. Vraka, L. Nics, K. H. Wagner, M. Hacker, W. Wadsak and M. Mitterhauser, Nucl. Med. Biol., 2017, 50, 1–10 CrossRef CAS PubMed.
- G. M. Nitulescu, Molecules, 2022, 27, 3300, DOI:10.3390/molecules27103300.
- C. A. Lipinski, Adv. Drug Delivery Rev., 2016, 101, 34–41 CrossRef CAS PubMed.
- B. M. Johnson, Y.-Z. Shu, X. Zhuo and N. A. Meanwell, J. Med. Chem., 2020, 63, 6315–6386 CrossRef CAS PubMed.
- L. C. Crowley, B. J. Marfell and N. J. Waterhouse, Cold Spring Harbor Protocols, 2016, 2016, 778–781 Search PubMed.
- C. Ballot, A. Martoriati, M. Jendoubi, S. Buche, P. Formstecher, L. Mortier, J. Kluza and P. Marchetti, Mar. Drugs, 2014, 12, 779–798 CrossRef CAS PubMed.
- S. G. Summerfield, J. W. T. Yates and D. A. Fairman, Pharm. Res., 2022, 39, 213–222 CrossRef CAS PubMed.
- X. Liu, M. Wright and C. E. C. A. Hop, J. Med. Chem., 2014, 57, 8238–8248 CrossRef CAS PubMed.
- K. Valko, S. Nunhuck, C. Bevan, M. H. Abraham and D. P. Reynolds, J. Pharm. Sci., 2003, 92, 2236–2248 CrossRef CAS PubMed.
- C. Vraka, S. Mijailovic, V. Fröhlich, M. Zeilinger, E. M. Klebermass, W. Wadsak, K. H. Wagner, M. Hacker and M. Mitterhauser, Nucl. Med. Biol., 2018, 58, 20–32 CrossRef CAS PubMed.
- I. Nicoletti, G. Migliorati, M. C. Pagliacci, F. Grignani and C. Riccardi, J. Immunol. Methods, 1991, 139, 271–279 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: 1H, 13C, 1H–15N HMBC, 19F NMR, HRMS, physicochemical parameters and Calcein AM/Hoechst/PI assay results. See DOI: https://doi.org/10.1039/d3ra00972f |
| This journal is © The Royal Society of Chemistry 2023 |