
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceMulti-enzyme catalysed processes using purified and whole-cell biocatalysts towards a 1,3,4-substituted tetrahydroisoquinoline†‡
Douglas Weber ab,
Lucas de Souza Bastosa,
Margit Winklercd,
Yeke Nie,
Abil E. Alieve,
Helen C. Hailese and
Doerte Rother*ab
ab,
Lucas de Souza Bastosa,
Margit Winklercd,
Yeke Nie,
Abil E. Alieve,
Helen C. Hailese and
Doerte Rother*ab
aInstitute of Bio- and Geosciences (IBG-1): Biotechnology, Forschungszentrum Juelich GmbH, 52425 Juelich, Germany. E-mail: do.rother@fz-juelich.de
bAachen Biology and Biotechnology (ABBt), RWTH Aachen University, Worringer Weg 1, 52062 Aachen, Germany
cacib GmbH, Krenngasse 37, A-8010 Graz, Austria
dInstitute of Molecular Biotechnology, Graz University of Technology, Petersgasse 14, 8010 Graz, Austria
eDepartment of Chemistry, University College London, London, WC1H 0AJ, UK
First published on 29th March 2023
Abstract
In this work, two multi-enzyme catalysed processes to access a 1,3,4-substituted tetrahydroisoquinoline (THIQ), using either purified enzymes or lyophilised whole-cell catalysts, are presented. A key focus was the first step in which the reduction of 3-hydroxybenzoic acid (3-OH-BZ) into 3-hydroxybenzaldehyde (3-OH-BA) was catalysed by a carboxylate reductase (CAR) enzyme. Incorporation of the CAR-catalysed step enables substituted benzoic acids as the aromatic components, which can potentially be obtained from renewable resources by microbial cell factories. In this reduction, the implementation of an efficient cofactor regeneration system of both ATP and NADPH was crucial. Two different recycling approaches, either using purified enzymes or lyophilised whole-cells, were established and compared. Both of them showed high conversions of the acid into 3-OH-BA (>80%). However, the whole-cell system showed superior performance because it allowed the combination of the first and second steps into a one-pot cascade with excellent HPLC yields (>99%, enantiomeric excess (ee) ≥ 95%) producing the intermediate 3-hydroxyphenylacetylcarbinol. Moreover, enhanced substrate loads could be achieved compared to the system employing only purified enzymes. The third and fourth steps were performed in a sequential mode to avoid cross-reactivities and the formation of several side products. Thus, (1R,2S)-metaraminol could be formed with high HPLC yields (>90%, isomeric content (ic) ≥ 95%) applying either purified or whole-cell transaminases from Bacillus megaterium (BmTA) or Chromobacterium violaceum (Cv2025). Finally, the cyclisation step was performed using either a purified or lyophilised whole-cell norcoclaurine synthase variant from Thalictrum flavum (ΔTfNCS-A79I), leading to the formation of the target THIQ product with high HPLC yields (>90%, ic > 90%). As many of the educts applied are from renewable resources and a complex product with three chiral centers can be gained by only four highly selective steps, a very step- and atom efficient approach to stereoisomerically pure THIQ is shown.
Introduction
For the synthesis of biologically active, stereoisomerically enriched molecules, such as pharmaceuticals and fine chemicals, high reaction selectivity is extremely important.1–3 Among the key properties of biocatalysts, their high stereoselectivity is very significant in asymmetric synthesis.1,4–7 Biocatalysis is therefore an enabling technology that can also be combined with traditional synthetic methods for the preparation of single-isomer compounds.7,8The demand for non-racemic chiral compounds is continuing to increase.9 Active pharmaceutical ingredients (APIs) were traditionally produced as racemates but the preference now is for single enantiomer synthesis.9,10 The switch from a racemic to a single-enantiomer API (also known as the chiral switch) is crucial to both the life cycle management and effectiveness of the application as a drug.9,11,12 As a result, biocatalysis over the past few years has matured into an essential tool for modern, cost effective, and sustainable pharmaceutical manufacturing.2
Among the plentiful APIs which can be produced by means of biocatalysis,8,9,11,13–15 there are some classes of compounds that play a relevant role. For example, in this work a chiral 2-hydroxy ketone intermediate is formed in a carboligation reaction. In general, 2-hydroxy ketones are highly valuable building blocks for many applications since they are precursors of several fine chemicals and pharmaceuticals,16 such as a broad range of amino alcohols (e.g., norephedrines)17 and tetrahydroisoquinolines (THIQs). The latter class of compound show relevant biological activities, including use as antidepressants, antitumor, anti-HIV, anti-inflammatory, and antimalarial drugs.17–19 Therefore, producing them in high enantiomeric purity is important in both academia and industry.
Many THIQ-containing compounds are found in bioactive natural products but most of them are present in very low concentrations, making THIQ extraction challenging.20 Nevertheless, the diversity of accessible THIQs can be significantly expanded in the laboratory. Chemical syntheses of these compounds are feasible, but they usually require multiple steps and might not give high stereoselectivities. Moreover, they often depend on the use of toxic or environmentally harmful chemicals.19,22 Thus, novel synthetic approaches towards THIQs are of significant interest.
Biocatalysis provides viable methods of producing complex THIQs in high stereoselectivities and under mild conditions.23 For instance, a three-step enzymatic cascade to access a 1,3,4-substituted THIQ has been shown.21 In this approach, the commercially available 3-hydroxybenzaldehyde (3-OH-BA) serves as starting material. Although this aldehyde is inexpensive, it is still predominantly obtained from petroleum resources. From a sustainability and circular bioeconomy standpoint, the use of renewable materials is preferable. Therefore, a four-step enzymatic cascade towards this trisubstituted THIQ starting with 3-hydroxybenzoic acid (3-OH-BZ), which can ultimately be obtained by microbial cell factories utilizing renewable resources, such as D-glucose, D-xylose or glycerol as the C-source,24,25 is proposed (Scheme 1).
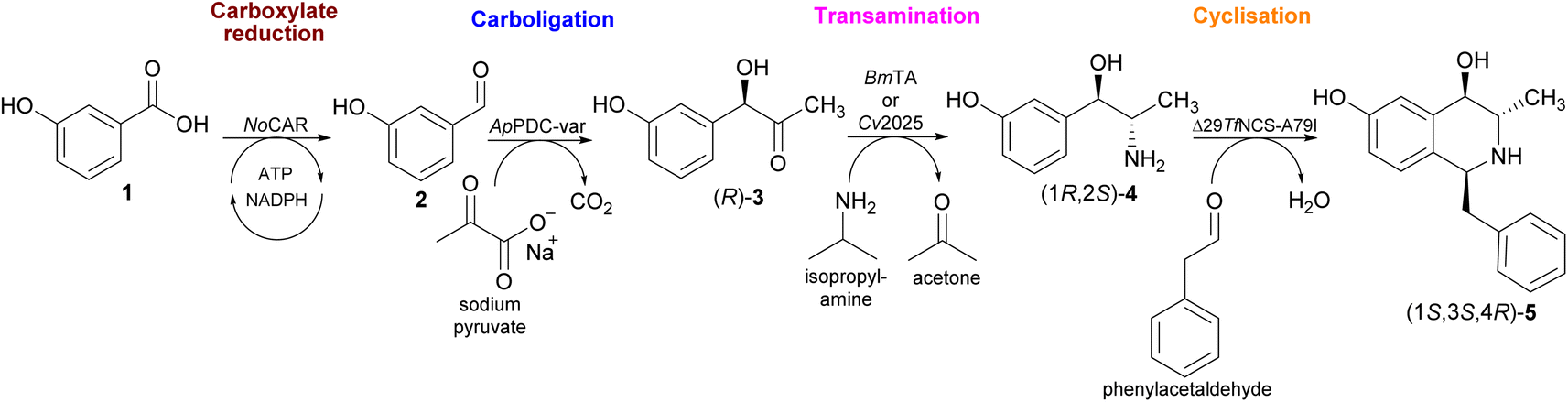 | ||
| Scheme 1 In this work, an extended multi-enzyme catalysed process starting from potentially renewable 3-hydroxybenzoic acid 1 is shown. In STEP 1, the carboxylate reductase from Nocardia otitidiscaviarum (NoCAR) catalyses the selective reduction of 1 into the aldehyde 2. In STEP 2, the carboligation reaction forms the 2-hydroxy ketone (R)-3 catalysed by a (R)-selective pyruvate decarboxylase variant from Acetobacter pasteurianus (ApPDC-var). In STEP 3, the carbonyl transamination towards (1R,2S)-4 is catalysed by either Cv2025 or the amine transaminase from Bacillus megaterium (BmTA). Lastly, in STEP 4, the cyclisation step towards the final product (1S,3S,4R)-5 is catalysed by a norcoclaurine synthase variant from Thalictrum flavum (ΔTfNCS-A79I).21 | ||
As shown in Scheme 1, the novelty of this cascade is the integration of the reduction of 3-OH-BZ into 3-OH-BA by a carboxylic acid reductase (CAR) enzyme. In general, the chemical reduction of carboxylic acids into their corresponding aldehydes is a challenging reaction as it relies on harsh reaction conditions and lacks selectivity.26 On the other hand, CARs (E.C.1.2.1.30) are a group of enzymes capable of carrying out the reduction reaction of carboxylic acids in only one-step with little or no over-reduction into the alcohol product. CARs activate the carboxylate substrate with the aid of adenosine 5′-triphosphate (ATP) and catalyse the reduction step using nicotinamide adenine dinucleotide phosphate (NADPH) as hydride donor.27,28 Therefore, to employ CARs in large-scale applications, continuous regeneration of the cofactors ATP and NADPH is required.29,30 This therefore creates the challenge of ensuring that the incorporation of CARs is feasible economically.
The implementation of a cofactor regeneration system can reduce the costs of synthesis by driving the reaction to completion, simplifying product isolation, and preventing the accumulation of inhibitory cofactor by-products.31,32 Various methodologies of cofactor regeneration have been developed to allow their use in catalytic amounts.31–33 Evidently, one of the simplest ways to regenerate ATP and NADPH in CAR-catalysed reactions is to apply them as whole-cell catalysts to leverage the cofactor regeneration machinery of the cell and supply glucose for cell metabolic function.29 This greatly simplifies cofactor regeneration and makes the addition of expensive external cofactors unnecessary in most cases.34
Whole-cell biotransformations can however show several potential limitations.29,34 One of the major drawbacks is that endogenous enzymes present can reduce the desired aldehyde product of the CAR reaction, leading to significant quantities of alcohol by-product. Efforts to overcome this issue have included expressing CARs in strains (like the E. coli K-12 MG1655 strain)35 which have inherently reduced reductase activity and/or to delete genes for endogenous reductase enzymes to minimize the amount of background over-reduction.29,36 An additional requirement is that the substrate must first enter the cell, which can present a mass transfer limitation.29 Moreover, in the case of CAR-catalysed transformations, high aldehyde concentrations can cause cell toxicity when no other measures are taken to protect them.28,34,35,37
Alternatively, in vitro regeneration of ATP and NADPH can overcome some of these limitations and prevent undesired reactions but impact the economic efficiency due to the addition of several coenzymes and cosubstrates.29,30,38 Nevertheless, cell-free CAR-mediated synthesis of aldehydes as end products or intermediates in cascade reactions is a promising approach.
In this study, the aim was to establish multi-step enzymatic processes starting with commercially available 3-OH-BZ towards a 1,3,4-substituted tetrahydroisoquinoline (Scheme 1) with a view to ultimately using microbially produced 3-OH-BZ. Two approaches were considered: (i) a four-step process using only purified enzymes, including an in vitro regeneration of both ATP and NADPH, and (ii) a two-step one-pot cascade followed by a two-step sequential process using whole-cell catalysts.
Results and discussion
Screening of CARs and substrate scope investigation
An important feature of CAR enzymes is their broad substrate tolerance, making them valuable biocatalysts to be applied in (chemo)enzymatic cascades targeting high-value compounds of synthetic interest.29 The substrate specificity of CARs is described to be determined by the adenylating core domain (A-domain), where the carboxylate moiety is first activated by ATP for subsequent reduction in the reducing domain (R-domain), forming the aldehyde product.39 As sequence similarities between different subtypes and between fungal and bacterial origins are below 25%, substrate spectra of CARs are expected to vary substantially.28,40Herewith, a small toolbox of CARs was setup and is now available for the enzymatic reduction of a variety of (hydroxy)benzoates to their corresponding aldehydes (Table S2 in the ESI‡). The first approach used to screen CARs was the determination of their initial rate towards each substrate and evaluation of the rate with which NADPH was depleted. The results obtained are given in the Fig. S13 in the ESI.‡
The photometric assay to determine CAR activity rapidly gave insights into which CARs would be promising catalysts for further applications. However, the assay does not provide data on the overall catalytic performance of CARs, especially when high overall conversions are sought. A more detailed analysis is given by analytical methods, which directly detect the products of the reaction (both the aldehyde and the alcohol, the potential side product due to over-reduction). Therefore, each CAR was assembled with an in vitro cofactor regeneration system (Fig. 1A and Table S14 in the ESI‡) for quantification of the unreacted acid, desired aldehyde and undesired alcohol. A similar reaction setup was tested with the CAR from Segniliparus rotundus (SrCAR) but under considerably different conditions.41
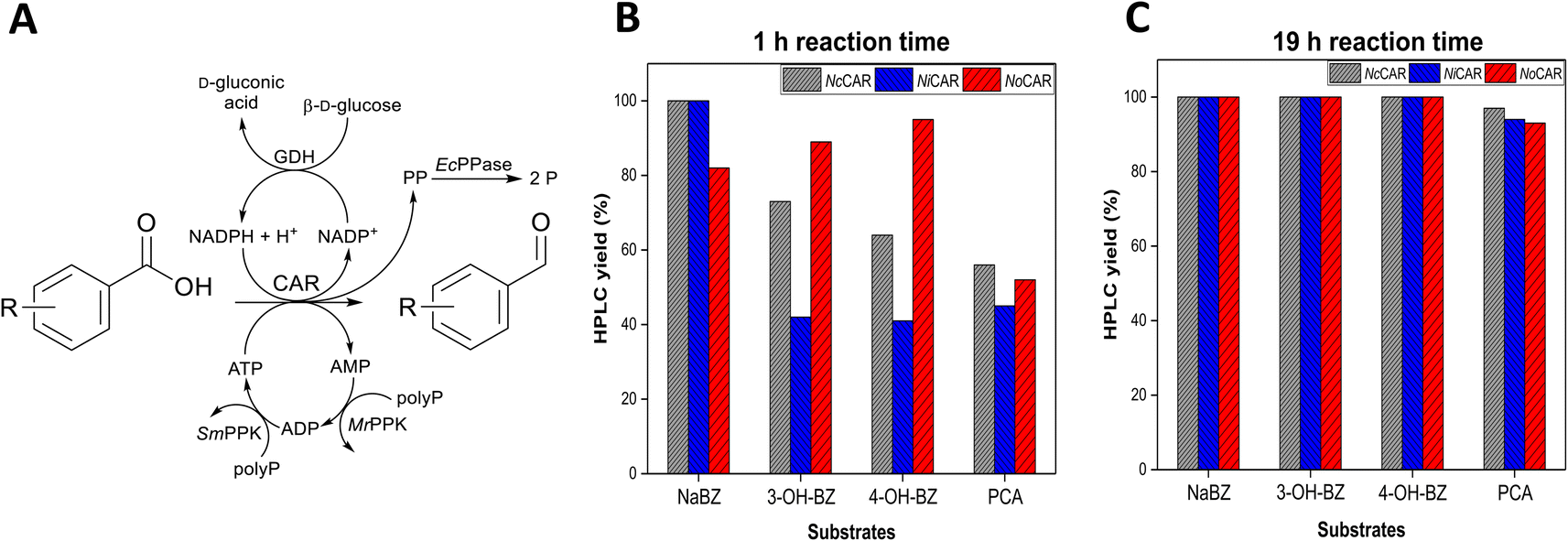 | ||
| Fig. 1 (A) Reaction scheme for the in vitro reduction of several (hydroxy)benzoates including full regeneration of all cofactors. Abbreviations – polyP: sodium polyphosphate, PP: pyrophosphate, P: ortho-phosphate, MrPPK and SmPPK are the polyphosphate kinases from Meiothermus ruber and Sinorhizobium meliloti, respectively.42,43 The reaction scheme was adapted from Strohmeier et al.30 Plots show aldehyde formation (HPLC yield) catalysed by NcCAR, NiCAR, or NoCAR after (B) 1 h and (C) 19 h of reaction. Substrates (5 mM) and CARs were assembled with the in vitro cofactor regeneration system applying the following reaction conditions: 100 mM MOPS buffer (pH 7.5), 5 mM substrate, 6.25 mM MgCl2, 100 mM β-D-glucose, 4 mg mL−1 sodium polyphosphate, 0.5 mM NADPH, 1 mM ATP, 100 μg mL−1 purified CAR, 100 μg mL−1 purified MrPPK, 40 μg mL−1 purified SmPPK, 25 μg mL−1 purified EcPPase, 50 μg mL−1 purified GDH, 30 °C, 800 rpm. Overall reaction volume of 250 μL. Data points show the product yield (HPLC yield) in percentage. | ||
All CARs, except NoCAR, NiCAR and NcCAR, gave poor levels of reduction of the substrates sodium benzoate (NABZ), 3-hydroxybenzoic acid (3-OH-BZ), 4-hydroxybenzoic acid (4-OH-BZ), and protocatechuic acid (PCA) into their corresponding aldehydes. These CARs either did not convert any of the substrate or showed conversions ≤16% even after 19 h of reaction (Table S14 in the ESI‡). On the other hand, it was confirmed that NoCAR, NcCAR, and NiCAR were able to convert each substrate with very good yields under the tested conditions and in the first hour of reaction. The HPLC yields (amount of product determined by HPLC, according to the process metrics available in the ESI‡) for NoCAR, NcCAR, and NiCAR towards the four substrates after 1 h and 19 h of reaction are shown in Fig. 1B and C.
According to Fig. 1B, in the first hour of reaction NoCAR converted >80% of NaBZ, 3-OH-BZ, and 4-OH-BZ into the corresponding aldehydes and about 50% of PCA was converted into 3,4-dihydroxybenzaldehyde under applied reaction conditions. NiCAR also had an outstanding performance, showing HPLC yields of >99% for benzaldehyde and about 40% for the aldehydes derived from 3-OH-BZ, 4-OH-BZ and PCA. Importantly, no alcohol product was detected in any case.
Finally, NcCAR was able to convert >99% of NaBZ, >70% of 3-OH-BZ, >60% of 4-OH-BZ, and >50% PCA into the corresponding aldehyde in the first hour. Similarly, when NiCAR and NcCAR were employed as catalysts, no over-reduction was observed. These observations highlighted that these CAR enzymes are highly selective under the applied reaction conditions. In addition, almost a full conversion of all substrates was obtained after 19 h of reaction (Fig. 1C). Overall, the excellent performance of NoCAR, NcCAR, and NiCAR when assembled with the in vitro cofactor regeneration system made them ideal candidates for applications in the targeted cascade. As NoCAR had been explored in our group previously,44 if not stated otherwise, it was the preferred enzyme used in this reaction step.
Multi-enzyme catalysed processes towards (1S,3S,4R)-THIQ
As mentioned previously, the main goal was to combine CAR-catalysed reactions with other biocatalysts in multistep biocatalytic reactions to access two pharmaceutical ingredients, metaraminol and a 1,3,4-substituted THIQ (Scheme 1) to close the gap between microbial cell factories and modular enzyme cascades. Herein, two biocatalytic approaches were setup to access this THIQ product, differing mainly on the formulation of the biocatalysts employed to generate as much product as possible.In the first multi-enzyme catalysed process, only purified enzymes were employed, which included an in vitro system for the regeneration of both ATP and NADPH (as shown in Fig. 2A). In the second process, lyophilised whole-cell catalysts were applied. In the latter approach, no additional coenzymes for the regeneration of ATP and NADPH were added. Challenges and limitations of each biocatalytic approach will be discussed in more details below.
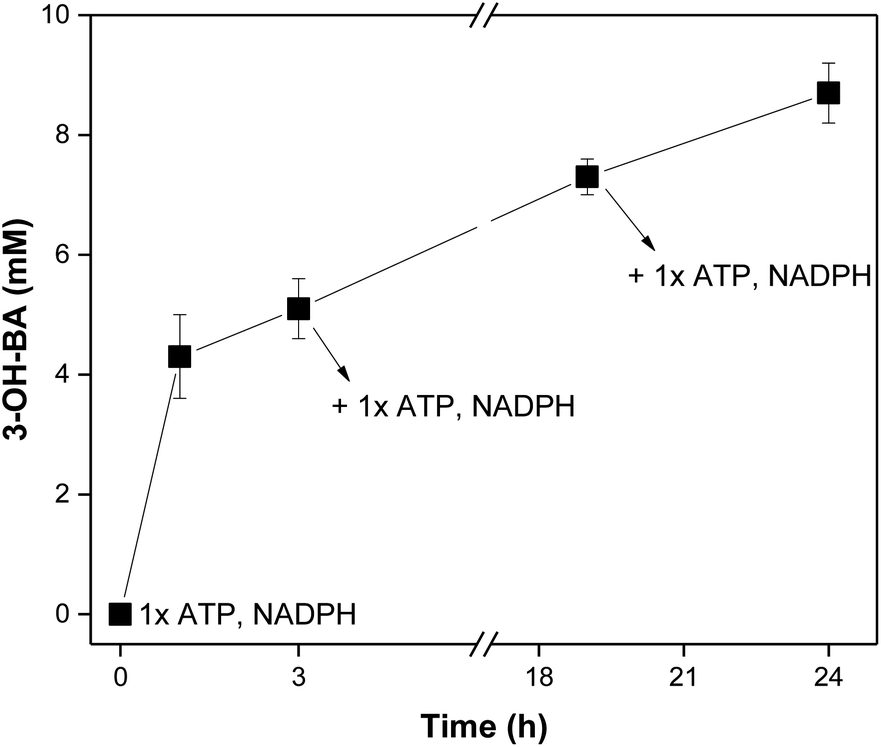 | ||
| Fig. 2 In vitro reduction of 3-hydroxybenzoic acid (3-OH-BZ, 10 mM) using a purified NoCAR preparation assembled with the in vitro regeneration of cofactors, including a cofactor feeding. ATP and NADPH were added in three different time points within 24 h of reaction (at t = 0, 3, and 20 h). Reaction conditions: 200 mM MOPS buffer (pH 7.5), 12.5 mM MgCl2, 100 mM β-D-glucose, 8 mg mL−1 sodium polyphosphate, 100 μg mL−1 purified NoCAR, 100 μg mL−1 purified MrPPK, 40 μg mL−1 purified SmPPK, 25 μg mL−1 purified EcPPase, 50 μg mL−1 purified GDH, 30 °C, 800 rpm. (1× ATP = 1 mM; 1× NADPH = 0.5 mM). Data points are the average of three technical replicates. Error bars represent the standard deviation. | ||
Multi-step biocatalytic process using purified enzymes
In a previous report,44 a highly active, purified form of NoCAR was obtained and its ability to produce benzaldehyde from sodium benzoate with excellent conversions was established. Although this enzyme showed outstanding performance by accepting high substrate loads (up to 75 mM sodium benzoate), it was rather unstable under the storage methods developed. Experiments performed with a purified formulation of NoCAR showed that it loses activity rapidly depending on what protocol is adopted for enzyme storage. Therefore, for in vitro cascades, the continuous production and purification of the enzyme might be required to maintain its high catalytic efficiency. Extending this approach into an industrial setting, the need for continuous production and purification of the biocatalyst(s) can be a limiting factor, leading to additional and undesired costs. On the other hand, robust and stable catalysts are ideal.When freshly produced and purified NoCAR was employed in the reduction of 3-OH-BZ into 3-OH-BA, up to 25 mM substrate was converted into aldehyde with high product yields (>90%) (Fig. S14, ESI‡). However, reduced catalytic efficiencies were noted when NoCAR enzyme preparations older than a month (CAR in 10 mM Tris–HCl buffer, pH 7, and stored at −20 °C) were used. Increasing the amount of biocatalyst in the reaction did not show major improvements. Therefore, additional efforts to address this issue was considered and it seemed reasonable that the amount of cofactors in the reaction might influence the performance of the enzyme (Fig. S15 in the ESI‡).
Knowing that increasing the concentration of cofactor would probably benefit the production of aldehyde, a cofactor feeding was performed. Here, instead of adding 2-fold the concentration of the cofactors in one portion, they were sequentially added at different time-points and the conversion was monitored. The obtained results are presented in Fig. 2.
At the start of the reaction, 0.5 mM NADPH and 1 mM ATP were added. After 3 h of reaction, about 5 mM of 3-OH-BA had been formed (50% conversion). At this point, a second portion of cofactors was added (0.5 mM NADPH and 1 mM ATP). After an additional 17 h, conversions increased up to 75% (7.5 mM 3-OH-BA) and a third portion of cofactor was made (0.5 mM NADPH and 1 mM ATP). The reaction was carried for 24 h in total, producing ∼9 mM of 3-OH-BA (87 ± 5% HPLC yield). The results obtained showed the applicability of CARs even after a long storage period if fresh cofactors were added. However, it has to be decided if it is more efficient to continuously generate fresh CAR preparations, or if extra quantities of cofactors in combination with a more extended enzyme storage is more appropriate. Nevertheless, based on the obtained results, we can conclude that CARs assembled with the in vitro approach to regenerate both ATP and NADPH have potential applications in cascades. Here, a sequential cascade mode should be the preferred choice since many components and enzymes are required, which could affect the next steps.
The following reaction steps (STEPs 2–4) were optimised previously. In particular, the carboligation reaction using the pyruvate decarboxylase ApPDC wild-type as well as other variants to produce a library of 2-hydroxy ketones was previously studied for acetohydroxy acid synthase I from Escherichia coli (EcAHAS-I).17,45 The optimised conditions were applied here, but now using a (R)-selective pyruvate decarboxylase variant (ApPDC-var) instead of EcAHAS-I as it was more recently established that this enzyme might have a better performance. Other authors have also obtained analogous results46–48 using the same ApPDC-var in related cascades.
Purified form of ApPDC-var was employed in this study. Using the reaction mixture of the previous step containing 3-OH-BA as starting material (about 9 mM, Fig. 2), the carboligation step worked well under the applied reaction conditions and (R)-3 was produced with very good conversions (96 ± 1% HPLC yield). The carboligation between 3-OH-BA and sodium pyruvate catalysed by ApPDC-var was rapid since 2 h of reaction were sufficient to achieve almost full conversion of the substrate and excellent product yields. The stereoselectivity of the carboligase was confirmed by chiral 2D-UHPLC analysis (Fig. S16, ESI‡). The enantiomeric excess (ee) was estimated to be >95%, confirming that this ApPDC-var is highly selective towards the formation of (R)-enantiomers.
Subsequently, the transamination reaction (STEP 3) was performed. It is important to highlight that herein a sequential reaction mode is crucial to avoid any cross-reactivity between the transaminase and any remaining aldehyde from STEP 1. In addition, elimination of the transaminase after the completion of this step has to be assured since this enzyme can also accept phenylacetaldehyde (co-substrate of the last step of the cascade) as substrate, leading to the formation of unwanted side products (for more details, see Scheme S5 in the ESI‡).
First attempts to perform the transamination reaction using the reaction mixture of step 2 containing (R)-3 was quite challenging. Applying previously determined reaction conditions,21 optimised for the unsubstituted hydroxy ketone, including the well described transaminase Cv2025, did not lead to satisfactory conversions of (R)-3 into (1R,2S)-4 (<20% conversion, data not shown) even after long reaction times. Cv2025 is known to perform at lower reaction rates compared to other TAs, such as the amine transaminase from Bacillus megaterium (BmTA), although previous reports showed outstanding performance of Cv2025 for catalysing similar reactions.17,21 In this study, due to the better reaction rates achieved with BmTA, this enzyme was selected for transamination optimisation experiments starting with (R)-3 to give (1R,2S)-4. Next, the best conditions when using BmTA were exploited in the reactions catalysed by Cv2025 to have a final overview on the performance of both TAs before deciding on the most suitable catalyst to proceed in the cascade. Thus, reaction parameters such as the amount of enzyme, molar ratio of substrates, reaction mode (opened or closed lid), and selection of amine donor to shift the equilibrium of the reaction were investigated (see the ESI‡).
After the optimisation, BmTA and IPA were selected as the best performing enzyme and amine donor, respectively. Under the optimised reaction conditions (Fig. S21 in the ESI‡), product (1R,2S)-4 was obtained with high conversions (91 ± 7% HPLC yield). The carboligase and transaminase employed in this cascade have previously been shown to be valuable catalysts for cascade reactions with unsubstituted aromatic aldehydes and 2-hydroxy ketones. Now, they also proved to be suitable catalysts for a substituted benzaldehyde, yielding biocatalytic access to (1R,2S)-4.
The final fourth step towards the production of (1S,3S,4R)-5 was catalysed by the norcoclaurine synthase Δ29TfNCS-A79I. Similarly, the cyclisation reaction was also optimised to determine the best reaction conditions (see the ESI‡).
By employing the best reaction conditions (section 5.6, ESI‡), the reaction mixture of the third step containing (1R,2S)-4 served as a substrate for the cyclisation catalysed by Δ29TfNCS-A79I. This reaction, when catalysed by the selected enzyme, yielded mainly the (1S,3S,4R)-product. When non-enzymatically catalysed (by KPi buffer), the stereocomplementary stereoisomer is obtained in significant amounts.21 The product (1S,3S,4R)-5 was formed with 93 ± 5% HPLC yield after 2 h of reaction.
After the completion of the last step, the overall product yield (overall amount determined by HPLC, not isolated yield) of the four-step sequential cascade was 71 ± 8%. This is a promising result considering the complexity of the process and starting with low-cost substrates, which can ultimately be obtained from renewables. In addition, it is important to note that the success of this in vitro multi-enzyme catalysed process is also due to the fact it was performed in a sequential mode. As discussed previously, cross-reactivity can take place and could have negatively influenced the formation of the final product. A complete overview of the four-step sequential process applying purified enzymes starting with 3-OH-BZ, including the product yields of each step, is shown in Scheme 2A.
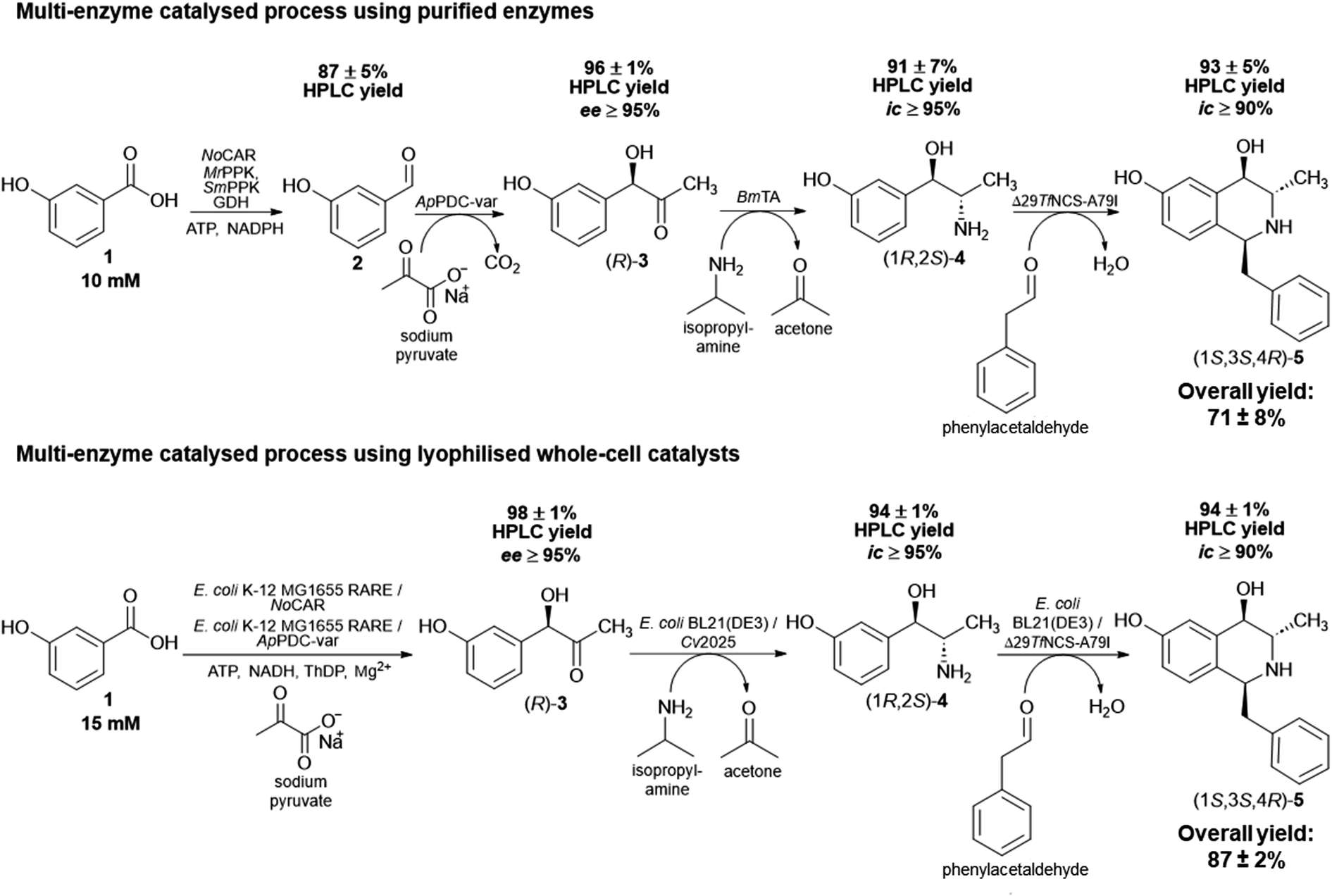 | ||
| Scheme 2 Overview of the multi-enzyme catalysed processes performed in this work. (A) Four-step sequential reactions using only purified enzymes towards (1S,3S,4R)-5 starting with 3-OH-BZ (10 mM). (B) Two-step one pot cascade using whole-cell catalysts starting with 1 (15 mM) towards (R)-3 followed by a two-step sequential reaction towards (1S,3S,4R)-5. The complete multi-enzyme catalysed process procedures are described in the Experimental section. Product yields were determined by HPLC and are the average of three technical replicates ± standard deviation. Abbreviations: ee: enantiomeric excess; ic: isomeric content (see ESI‡ for more details on how these parameters were determined). | ||
Multi-step biocatalytic process using whole-cell catalysts
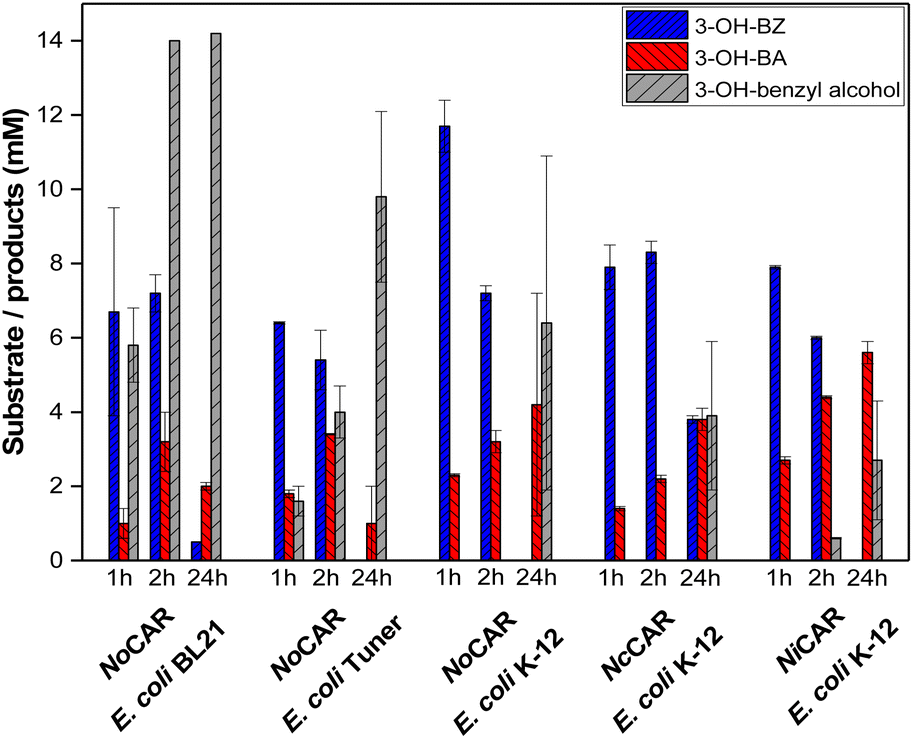 | ||
| Fig. 3 Reduction of 3-hydroxybenzoic acid (3-OH-BZ, 20 mM) using CARs as lyophilised whole-cell catalysts. Here, E. coli BL21, E. coli Tuner, and E. coli K-12 MG1655 RARE were used as host strains. Reaction conditions: 200 mM MOPS buffer (pH 7.5), 20 mM 3-OH-BZ, 25 mM MgCl2, 48 mM β-D-glucose, 24 mM sodium citrate, 0.5 mM NADPH, 1 mM ATP, 10 mg mL−1 lyophilised cells co-expressing EcPPTase and NoCAR, NcCAR or NiCAR, 30 °C, 1000 rpm. Reaction volume of 1 mL. Reactions were performed in technical triplicates. Error bars represent the standard deviation. | ||
According to Fig. 3, E. coli BL21 and E. coli Tuner cells expressing NoCAR and EcPPTase showed a pronounced endogenous aldehyde reduction activity. In both cases, 3-OH-benzyl alcohol was formed in the first hour of the reaction and the aldehyde barely accumulated in the cells. By the end of the reaction (24 h), almost full conversion of 3-OH-BZ was observed but the major product obtained was the alcohol (>10 mM of 20 mM substrate in both cases). Our results confirm that these E. coli strains are not suitable for CAR applications as whole-cell catalysts when the aldehyde product is targeted. However, the overall aim was not to obtain the aldehyde 2 as final product but to convert it further in a follow-up reaction into the 2-hydroxy ketone 3.
In addition, the amount of biocatalyst in the whole-cell catalysed carboxylate reduction was addressed. High amounts of biocatalyst introduce more CAR in the reaction, although less biomass in the biotransformation should decrease endogenous cofactor pools and enzymes with aldehyde reductase activity. On the other hand, overloading with biocatalyst makes the overall process more expensive and the separation of substrate/product more difficult in some cases. Thus, the optimal amount of lyophilised cells in the CAR-catalysed bioreduction of 3-OH-BZ was investigated.
The results (Fig. S25 in the ESI‡) indicated that 10 mg mL−1 lyophilised cells was the optimal amount of bicatalyst, in which 3.3 mM of 3-OH-BA was formed after 2 h of reaction. Doubling the concentration of biocatalyst did not enhance the product yields (2.5 mM 3-OH-BA) any further; therefore, 10 mg mL−1 lyophilised cells was the concentration chosen for further experiments using CARs as whole-cell catalysts. It is important to mention that no alcohol was formed in any of the reactions performed here, probably because the reactions were carried out only for 2 h.
In order to investigate whether lyophilised E. coli K-12 MG1655 cells co-expressing NoCAR and EcPPTase would already contain sufficient amount of cofactors or if they needed to be externally added to the bioreductions, the reduction of 3-OH-BZ was investigated in the presence or absence of additional cofactors. In addition, NADH was also tested as a possible substitute for NADPH. Overall, this experiment should investigate whether lyophilised cells co-expressing NoCAR and EcPPTase would still be metabolically active and regenerate both cofactors or not. The results obtained in this study are shown in Fig. 4.
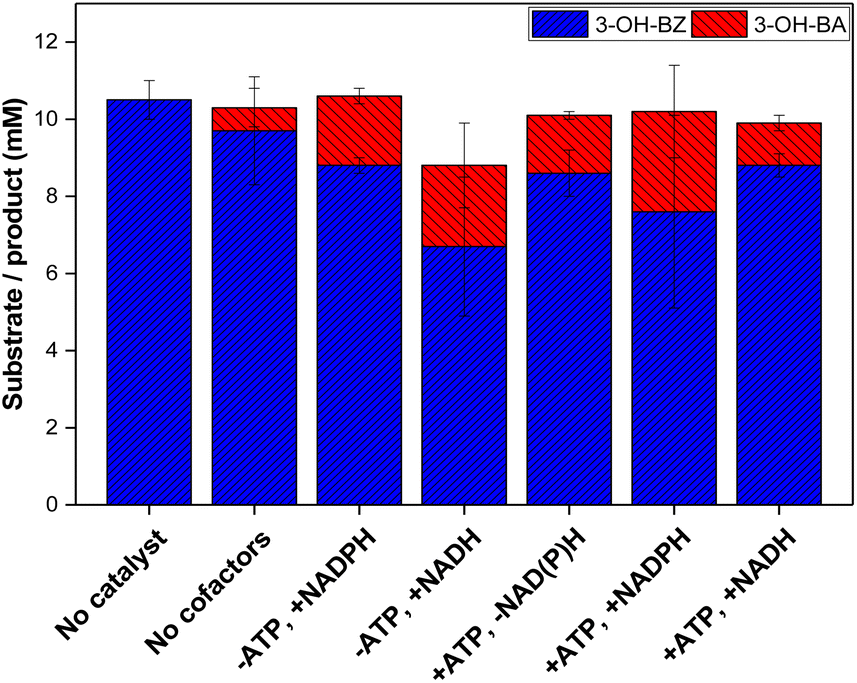 | ||
| Fig. 4 Reduction of 3-hydroxybenzoic acid (3-OH-BZ, 10 mM) using E. coli K-12 MG1655 RARE lyophilised cells co-expressing NoCAR and EcPPTase. At the x-axes, (−) represents absence and (+) the presence of the cofactor(s) in the reaction. When present, 0.5 mM NAD(P)H and 1 mM ATP were used. Reaction conditions: 200 mM MOPS buffer (pH 7.5), 10 mM 3-OH-BZ, 25 mM MgCl2, 48 mM β-D-glucose, 24 mM sodium citrate, 10 mg mL−1 lyophilised cells co-expressing NoCAR and EcPPTase, 30 °C, 1000 rpm, 2 h reaction. Reaction volume of 1 mL. Reactions were performed in technical triplicates. Error bars represent the standard deviation of three technical replicates. | ||
In respect to the cofactor demand, a trend of increased product yield by cofactor addition could be observed. Feed of both ATP and NAD(P)H increased product yield the most (about 2.6 mM 3-OH-BA was formed). Addition of ATP or NAD(P)H alone had a similar effect in both cases in respect to the formation of aldehyde (1.8 and 2.1 mM 3-OH-BA, respectively). In addition, substituting NADH to NADPH did not affect the reaction much, although less product was formed (2.1 and 2.6 mM 3-OH-BA, respectively). As mentioned before, NADPH can be generated in situ in whole-cell mediated transformations from NADH, which has been already described in the literature.52–54 Therefore, it is possible to conclude that NADH promotes CAR activity in whole-cell bioreductions.
Based on these results, it can be concluded that the NoCAR-catalysed reduction of 3-OH-BZ was limited in cofactor supply and could not achieve its full potential. A similar trend was recently reported by Horvat and Winkler28 who concluded that the bioreduction of octanoic acid into octanal was limited by ATP supply even when living cells were applied as the biocatalyst. This limitation could be circumvented by using the whole-cell biocatalyst in combination with in vitro regeneration of ATP.
The conclusion was that the intrinsic pool of both cofactors present within lyophilised cells is not sufficient to supply NoCAR to enable the full catalytic performance of CARs. Thus, the addition of both cofactors is still crucial.
Increasing loads of 3-OH-BZ were tested with lyophilised E. coli K-12 MG1655 RARE cells co-expressing NoCAR and EcPPTase. In general, the bioreductions starting with substrate concentrations higher than 10 mM did not perform very well. For example, when 50 mM substrate was used, overall conversions of the substrate were lower than 10% even after 24 h of reaction and, in some cases, a mixture of aldehyde and alcohol was formed (Fig. S26, ESI‡). As a solution, we combined the CAR-catalysed biotransformation with the carboligation reaction (STEP 2) in a two-step one-pot cascade towards (R)-3 (Fig. 5A). In this approach, all the components required for the carboxylate reduction and carboligation reactions were combined in a one-pot system to yield (R)-3. This strategy should give a two-fold benefit: to pull aldehyde from the CAR to avoid inhibition and at the same time consume it in situ before it can undergo over-reduction. Here, not only the CAR had to be expressed in E. coli K-12 MG1655 RARE but also the ApPDC-var. However, it is important to note that the ApPDC-var was not co-expressed with NoCAR. They were heterologously produced and lyophilised separately, and subsequently mixed in the reaction mixture (as shown in Fig. 5A).
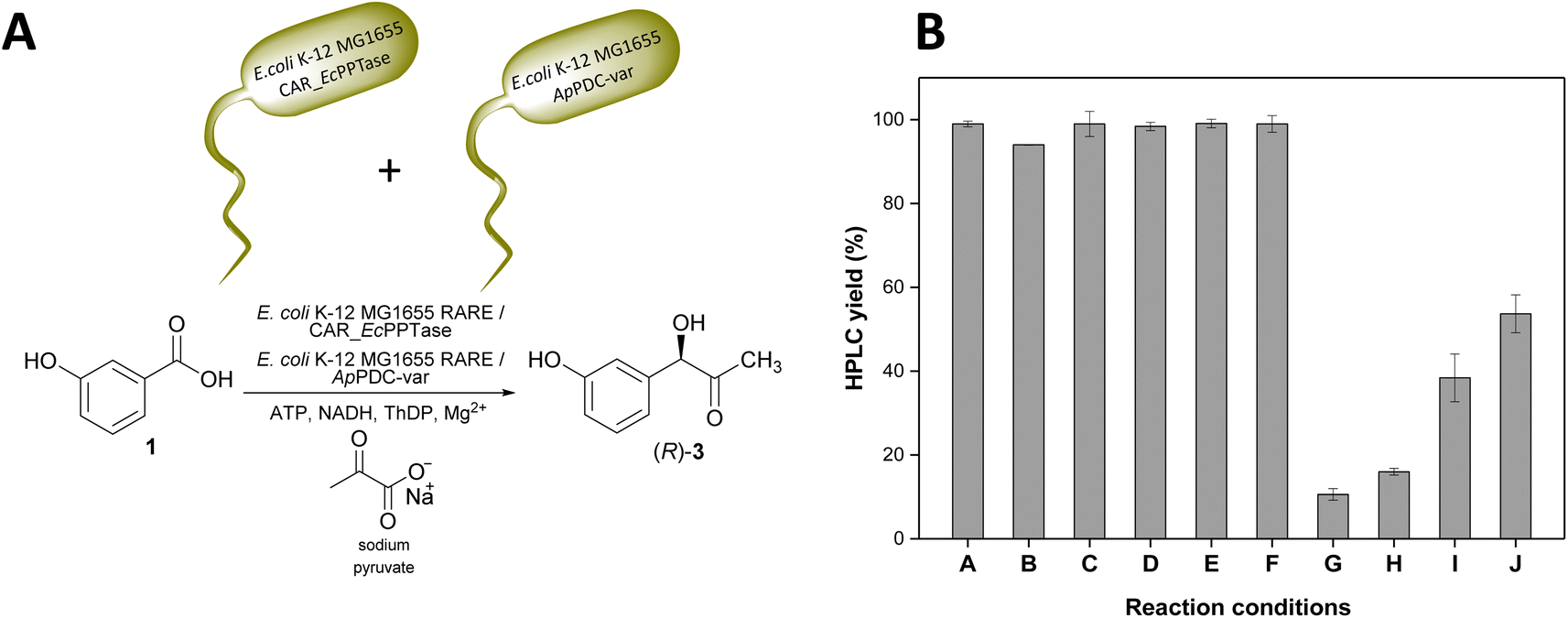 | ||
| Fig. 5 (A) Reaction scheme of the one-pot two-step cascade towards (R)-3-OH-PAC ((R)-3) starting with 3-OH-BZ. E. coli K-12 MG1655 RARE lyophilised cells co-expressing a CAR enzyme and EcPPTase were added together with E. coli K-12 MG1655 RARE lyophilised cells expressing ApPDC-var. In this cascade, 3-OH-BZ is first reduced to 3-OH-BA (in situ formation) by 10 mg mL−1 E. coli K-12 MG1655 RARE lyophilised cells expressing a CAR enzyme. Next, 3-OH-BA reacts with sodium pyruvate to produce (R)-3 catalysed by 10 mg mL−1 E. coli K-12 MG1655 RARE lyophilised cells expressing ApPDC-var. (B) HPLC yields (% of product detected) obtained when different reaction conditions were applied. Reaction conditions – A: 10 mM 3-OH-BZ, 1 mM ATP and 0.5 mM NADH, 6 h reaction, catalysed by cells expressing NoCAR; B: 10 mM 3-OH-BZ, 1 mM ATP and 0.5 mM NADH, 6 h reaction, catalysed by cells expressing NiCAR; C: 10 mM 3-OH-BZ, 1 mM ATP and 0.5 mM NADH, 6 h reaction, catalysed by cells expressing NcCAR; D: 15 mM 3-OH-BZ, 1 mM ATP and 0.5 mM NADH, 6 h reaction, catalysed by cells expressing NoCAR; E: 20 mM 3-OH-BZ, 1 mM ATP and 0.5 mM NADH, 6 h reaction, catalysed by cells expressing NoCAR; F: 20 mM 3-OH-BZ, 2 mM ATP and 1 mM NADH, 6 h reaction, catalysed by cells expressing NoCAR; G: 50 mM 3-OH-BZ, 1 mM ATP and 0.5 mM NADH, 6 h reaction, catalysed by cells expressing NoCAR; H: 50 mM 3-OH-BZ, 1 mM ATP and 0.5 mM NADH, 24 h reaction, catalysed by cells expressing NoCAR; I: 50 mM 3-OH-BZ, 5 mM ATP and 2.5 mM NADH, 6 h reaction, catalysed by cells expressing NoCAR; J: 50 mM 3-OH-BZ, 5 mM ATP and 2.5 mM NADH, 24 h reaction, catalysed by cells expressing NoCAR; for A–J: 200 mM MOPS buffer (pH 7.5), 25 mM MgCl2, 1 mM THDP, 48 mM β-D-glucose, 24 mM sodium citrate, 3% (v v−1) DMSO, 30 °C, 850 rpm. Reaction volume of 1 mL. Product yields are the amount of product determined by HPLC and are the average of three technical replicates ± standard deviation. | ||
In this cascade, 3-OH-BA would be first formed in situ and should then be consumed by the carboligase, avoiding its accumulation in the cells. Furthermore, no alcohol should be formed since the carboligation step is much faster and 3-OH-BA would not accumulate long enough to be reduced. In other words, this one-pot two-step cascade is the best strategy possible because it cannot only solve the problem of over-reduction but also can shorten the time to perform the overall cascade since two steps are sequential. Fig. 5B shows the results obtained for the one-pot two-step cascade under different conditions.
According to the data shown in Fig. 5B, the combination of the reduction with the carboligation reactions in a one-pot system showed encouraging results. E. coli K-12 MG1655 RARE cells expressing either NoCAR, NiCAR or NcCAR showed a similar performance on the reactions starting with 10 mM 3-OH-BZ (entries A–C). For all these biocatalysts, product yields of >94% were obtained in 6 h of reaction time.
Next, increasing substrate concentrations were evaluated using E. coli K-12 MG1655 RARE cells expressing NoCAR. Thus, concentrations of 15 mM (entry D), 20 mM (entries E and F), and 50 mM (entries G–J) of 3-OH-BZ were tested in this setup. Substrate tolerance in this one-pot system was much higher compared to single reaction steps. By combining two steps in one system, up to 20 mM of 3-OH-BZ was almost fully consumed and (R)-3 could be obtained with excellent yields. In contrast, with purified CAR, only 10 mM 3-OH-BZ was fully converted. For instance, reactions starting with 15 mM 3-OH-BZ yielded about 98% (R)-3 in 6 h (entry D). When the substrate was increased to 20 mM, ≥99% product yields were obtained in the same period (entry E). Here, doubling the concentration of ATP and NADH was confirmed not to be necessary since excellent yields were already obtained with the standard cofactor concentrations (entries E and F).
The most significant difference was observed for the reactions starting with 50 mM 3-OH-BZ. In this case, increasing the concentration of ATP and NADH up to 5 times enhanced product yields up to 54% in the reactions performed up to 24 h. In this case, increasing the concentration of the cofactors and the time of reaction were decisive to enhance the product yields from 11% (1-fold concentration of ATP and NADH, 6 h of reaction) to 54% (5-fold concentration of ATP and NADH, 24 h of reaction). To summarise, with higher substrate concentrations the limiting factor was the cofactor regeneration rather than inhibiting effects of the substrate acid or the aldehyde.
The results here obtained were very pleasing because (i) diverging flux to 3-OH-benzyl alcohol was fully suppressed; (ii) the substrate tolerance was higher compared to single reaction steps and up to nearly 20 mM 3-OH-BZ could be fully converted into (R)-3 (product yields ≥98%, ee ≥ 95%); and (iii) less need for manipulation/work-up in simultaneous cascades as compared to sequential cascades.
After the promising results obtained in the one-pot two-step cascade towards (R)-3, the next step was to perform the subsequent steps to give (1S,3S,4R)-5. Again, the third and fourth steps were performed in a sequential mode to avoid the formation of any by-product due to cross-reactivity. In these steps, lyophilised E. coli BL21 cells expressing Cv2025 and Δ29TfNCS-A79I were employed in the transamination and cyclisation reactions, respectively. BmTA as whole-cell catalyst was also used but, in contrast to the results obtained for the in vitro biocatalytic process, it did not outperform Cv2025 (data not shown). Therefore, Cv2025 was chosen to be used in the cascade with whole-cell catalysts.
The product yields obtained by HPLC analysis for each step are given in Scheme 2B. Starting with 15 mM 3-OH-BZ, (R)-3 was obtained with yields of >98% after 6 h of reaction (ee > 95%). Then, the supernatant served as the substrate for the transamination catalysed by lyophilised E. coli BL21 expressing Cv2025, in which (1R,2S)-4 was obtained in excellent yields of 94 ± 1% (ic > 95%) product yield after 4 h of reaction. This result is certainly promising since this step has been shown to be challenging due to both low enzyme activity and equilibrium issues. It is important to mention that the increase in the concentration of IPA (80 mM in the reaction, which corresponds to more than 5-fold excess) was decisive to obtain high product yields after 4 h. Again, IPA outperformed α-MBA as amine donor (data not shown), which was also observed for the in vitro process. Heating of the reaction mixture (80 °C for 15 min) followed by centrifugation of the supernatant after the completion of the third step was crucial to eliminate any remaining transaminase activity and the formation of side products. Just as importantly, the cyclisation reaction catalysed by lyophilised E. coli BL21 expressing Δ29TfNCS-A79I also gave (1S,3S,4R)-5 in an excellent yield (94 ± 1%) after 3 h of reaction (ic > 90%). In short, the cascade employing whole-cell catalysts showed outstanding performance and excellent product yields. The overall product yield obtained in this biocatalytic process was 87 ± 2% and the ee and ic values for the intermediates and final product were similar to the ones obtained in vitro. This observation was important to confirm that applying enzymes as whole cell catalysts did not affect their stereoselectivities.
In the future, 3-OH-BZ obtained from microbial cell factories could be applied in biocatalytic processes as a more sustainable alternative to oil-based starting materials. As a proof-of-concept, 3-OH-BZ microbially produced and present in fermentation broths were directly applied in a one-pot two-step cascade combining NoCAR and ApPDC-var as whole-cell catalysts. The hydroxy-ketone (R)-3 was obtained with very high HPLC yields (≥99%, for more details, see ESI‡). This is certainly a great example of a hybrid process that could fill the gap between aromatic carboxylic acids obtained from microbial cell factories and enzyme cascades targeting important pharmaceutical precursors.
Conclusions
In summary, we have reported two multi-enzyme catalysed processes to access two relevant molecules, namely metaraminol and a THIQ-containing molecule, starting from 3-hydroxybenzoic acid. These processes differed mainly on the formulation of the biocatalysts employed.Firstly, the implementation of a toolbox of CAR enzymes and investigation of their substrate scope (towards sodium benzoate, 3-hydroxybenzoic acid, 4-hydroxybenzoic acid, and protocatechuic acid) highlighted that NoCAR, NcCAR, and NiCAR were the most promising enzymes for the production of aldehydes and in enzymatic cascades targeting more complex molecules.
Biocatalytic processes using only purified enzymes and starting with 3-OH-BZ showed encouraging results. The success of the NoCAR-catalysed reduction step to produce 3-OH-BA proved to be highly dependent on the enzyme stability upon storage. In general, fresh enzyme preparations were preferable. Enzyme preparations that had been stored for long time could still be used but in this case the concentration of ATP and NADPH needed to be increased (up to 3-fold) to guarantee maximal performance of the biocatalyst. Cofactor feed turned out to be beneficial for reactions at a mL scale; however, substrate loads became limiting in vitro. The subsequent three steps towards the THIQ product also gave good results and high product yields were obtained. The overall product yield of the four-step sequential process was 71% when starting with 10 mM 3-OH-BZ.
Multi-enzyme catalysed processes using lyophilised whole-cell catalysts however outperformed the processes with purified catalysts. Splitting the overall process into two parts (first, two-step one pot cascade to obtain (R)-3 followed by two-step sequential process towards the THIQ product) was a successful strategy to: (i) reduce the over-reduction of 3-OH-BA into the alcohol within the cells; (ii) maximize the formation of (R)-3 by combining CARs and ApPDC-var in one-pot system; (iii) reduce reaction steps, generating less waste and avoiding the loss of products due to liquid transference and/or implicit dilutions (e.g., due to the addition of components of the subsequent step); and (iv) enhance the substrate loading (up to 50 mM 3-OH-BZ could be converted with good product yields) without deactivating the biocatalyst. Whole-cell transamination and cyclisation to obtain (1S,3S,4R)-5 succeeded with an overall yield of 87% when starting with 15 mM 3-OH-BZ. Moreover, lyophilised whole-cell biocatalysts could be used without loss of activity even after many months of storage, showing that this formulation is ideal to keep the enzymes active for long-term usage.
In conclusion, we were able to demonstrate the applicability of multi-enzyme catalysed processes and enzyme cascades for the formation of a complex chiral compound bearing three chiral centres from low-cost starting material. In a future work, scaling of the reaction into preparative scale will enable determination of minor impurities.
Experimental details
Materials and chemicals
Chemicals were purchased from Sigma-Aldrich (Steinheim, Germany), Roth (Karlsruhe, Germany), Fluka (Buchs, Germany), AlfaAesar (Kandel, Germany), or Merck (Darmstadt, Germany) and used without further purification. NADH was purchased from Biomol GmbH (Hamburg, Germany) and NADPH from PanReac AppliChem (Darmstadt, Germany). Glucose dehydrogenase from Pseudomonas sp. was purchased from Sigma-Aldrich. (1R,2S)-3-(2-Amino-1-hydroxypropyl)phenol (metaraminol bitartrate) was bought from Toronto Research Chemicals (TRC, Canada), thiamine pyrophosphate (ThDP) was purchased from AppliChem (Darmstadt, Germany). HPLC-MS grade acetonitrile was obtained from Biosolve Chimie (Dieuze, France). The compound standard (R)-3 was enzymatically synthesised as described elsewhere.48 The compound standards of (1S,3S,4R)-5 and its (1S,3S,4S)-isomer were enzymatically synthesised as described in the ESI.‡Gene expression and protein production
Competent E. coli BL21 (DE3) cells were purchased from Thermo Fisher Scientific (Waltham, MA, USA). Competent E. coli Tuner (DE3) cells were donated by Prof. Dr Thomas Drepper (Heinrich-Heine-University Duesseldorf). Competent E. coli K-12 MG1655 RARE (DE3) cells were donated by Prof. Dr Kristala L. J. Prather (Massachusetts Institute of Technology). Plasmid vectors encoding for CAR enzymes used in this study are listed in Table S2 in the ESI.‡ In general, cells were transformed with the respective plasmid vector by adding 1 μL plasmid solution (100 ng μL−1) to 100 μL bacterial solution (OD600 = ∼12, in 80 mM CaCl2, 20% v v−1 glycerol, stored at −80 °C). After incubation on ice for 30 min, a heat shock was performed at 42 °C for 45 s and the cells were stored on ice for 2 min. Subsequently, 900 μL S.O.C. medium (Thermo Fisher Scientific, Waltham, MA, USA) was added and the cells were incubated for 1 h at 37 °C and 600 rpm in a thermomixer (Eppendorf, Hamburg, Germany). The transformed cells were plated (50–100 μL) on lysogeny broth (LB) agar containing 50 μg mL−1 kanamycin or 100 μg mL−1 ampicillin and incubated overnight at 37 °C.Depending on the target protein, distinct cultivation and gene expression conditions were applied. The complete overview of the cultivation methods applied are shown in Table S4 in the ESI.‡ Proteins were produced in baffled shaking flasks with a filling volume up to 20%. A single colony from the respective overnight plates was transferred to 50 mL LB medium with the addition of the appropriate antibiotic (50 μg mL−1 kanamycin or 100 μg mL−1 ampicillin) and the pre-cultures were cultivated overnight at 37 °C and 150 rpm (Multitron shaker, Infors HT, Bottmingen, Switzerland). The main cultivation was conducted for 48 h at 80 rpm (Multitron shaker, Infors HT, Bottmingen, Switzerland). After 3–4 h cultivation time, the temperature was reduced to 15–20 °C (depending on the cultivation method) and IPTG was added. Cells were harvested after the cultivation by centrifugation at 4 °C and 8000 rpm for 45 min in a Beckman centrifuge (rotor JA 8.1000, Brea, USA) and stored at −20 °C until further processing.
Protein purification and formulation
Depending on the target protein, different buffer compositions for the purification were used. The complete overview of the composition of the purification buffers are shown in Tables S5–S10 in the ESI.‡After thawing, cells were re-suspended in equilibration buffer containing lysozyme (1 mg mL−1). The resulting suspension (∼15% w v−1) was disrupted by sonication (Hielscher Ultrasonics Sonotrode S14, 70% amplitude, 0.5 s cycle). The crude cell extract was centrifuged (Beckman JA-20 rotor, 18![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 rpm, 45 min, 4 °C) and purified by nickel affinity chromatography (Ni-NTA) on an Äkta purification system. For this, the column was first equilibrated with equilibration buffer. Next, the crude cell extract was loaded into the column and washed with equilibration buffer. Subsequently, the column was washed with washing buffer and the target protein was eluted with elution buffer. The protein containing fractions were pooled and loaded into a column for size-exclusion chromatography (Sephadex®G-25). The sample was desalted with desalting buffer. The protein content of each purification step was estimated by Bradford assay, as described elsewhere.44 The desalted protein was either freeze-dried (Martin Christ Gefriertrocknungsanlagen, Osterode am Harz, Germany; conditions: 0.46 mbar, −46 °C, 72 h) or stored as liquid stocks. Both purified formulations were stored at −20 °C or −80 °C until further use.
000 rpm, 45 min, 4 °C) and purified by nickel affinity chromatography (Ni-NTA) on an Äkta purification system. For this, the column was first equilibrated with equilibration buffer. Next, the crude cell extract was loaded into the column and washed with equilibration buffer. Subsequently, the column was washed with washing buffer and the target protein was eluted with elution buffer. The protein containing fractions were pooled and loaded into a column for size-exclusion chromatography (Sephadex®G-25). The sample was desalted with desalting buffer. The protein content of each purification step was estimated by Bradford assay, as described elsewhere.44 The desalted protein was either freeze-dried (Martin Christ Gefriertrocknungsanlagen, Osterode am Harz, Germany; conditions: 0.46 mbar, −46 °C, 72 h) or stored as liquid stocks. Both purified formulations were stored at −20 °C or −80 °C until further use.
Lyophilisation of whole-cell catalysts
Directly after harvesting, fresh cells were transferred to a crystallisation dish, spread evenly keeping a high surface area, and frozen at atmospheric pressure (usually overnight at −20 °C). The lyophilisation process (0.46 mbar, −46 °C) lasted for 48 h or until the samples were visibly dry, depending on the surface and total volume of sample. Subsequently, lyophilised cells were removed from the crystallisation dish with a spatula and crushed in a mortar or directly in a Falcon tube until a uniformly fine powder was obtained. Then, lyophilised whole cells were stored at −20 °C until further use.Biocatalytic procedures towards (1S,3S,4R)-THIQ
STEP 2 – Carboligation: in this step, 800 μL of the reaction mixture from the previous step were transferred into a 2 mL glass vial. Next, 100 mM HEPES buffer at pH 6.5 (1 M in water), 2.5 mM MgSO4 (250 mM in water), 0.1 mM thiamine diphosphate (ThDP) (10 mM in water), 2.5% (v v−1) DMSO, and a 5-fold sodium pyruvate (2 M in water) in respect to the concentration of 3-OH-BA were added to the reaction mixture. The reaction started by adding purified, lyophilised ApPDC-var (0.8 mg protein per mL final concentration).44 Reactions took place in a total volume of 1 mL, at 30 °C and 850 rpm for 3–6 h. Reactions were stopped by ultrafiltration to remove the biocatalyst (Merck Millipore Ltd: Microcon-10, centrifugal filters).
STEP 3 – Transamination: in this step, a determined volume of the reaction mixture from the previous step (ranging from 800–900 μL) was transferred into a 2 mL glass vial. Next, 100 mM HEPES buffer at pH 8.0 (1 M in water), 0.3 mM PLP (10 mM in water), 3% (v v−1) DMSO, and a 4-fold amine donor (2 M in 100 mM HEPES buffer, final pH 8) in respect to the concentration of (R)-3 were added to the reaction mixture. The reaction started by adding purified, lyophilised Cv2025 or BmTA (2.5 mg mL−1 estimated by Bradford assay). Reactions took place in an overall volume of 1.2 mL, at 30 °C and 1000 rpm for 6 h. Reactions were terminated by ultrafiltration (Merck Millipore Ltd: Microcon-10, centrifugal filters). Alternatively, the enzyme was deactivated by heat (at 80 °C for 15 min), precipitated, and removed by centrifugation. The heating step was incorporated into this procedure to make sure that no residual activity of the transaminase would be still present in the supernatant before proceeding to the cyclisation reaction to avoid undesired amination of the co-substrate added in the fourth step.
STEP 4 – Cyclisation: in this step, 900 μL of the reaction mixture from the STEP 3 was transferred into a 2 mL glass vial. Next, 100 mM HEPES buffer at pH 7.5 (1 M in water), 5% (v v−1) DMSO, and a 4-fold phenylacetaldehyde (300 mM in DMSO) in respect to the concentration of (1R,2S)-4 were added to the reaction mixture. The reaction started by adding purified and lyophilised Δ29TfNCS-A79I (1 mg mL−1 protein final concentration). Reactions took place in an overall volume of 1 mL, at 37 °C and 850 rpm for up to 3 h.
STEP 1 and 2 – One-pot two-step cascade towards (R)-3: the production of (R)-3 in a one-pot system starting with 3-OH-BZ was performed by combining the components of the first two steps of the cascade shown in Scheme 1B. E. coli K-12 MG1655 RARE cells co-expressing a CAR enzyme (NcCAR, NiCAR or NoCAR) and EcPPTase as well as E. coli K-12 MG1655 RARE cells expressing the ApPDC-var were used as whole-cell catalysts. In a 2 mL glass vial, 200 mM HEPES buffer at pH 7.5 (1 M in water), 48 mM β-D-(+)-glucose (480 mM in water), and 24 mM sodium citrate (480 mM in water) were mixed. In addition, 10–100 mM 3-OH-BZ (250 mM in 250 mM KOH), 5 equivalents of sodium pyruvate (2 M in water) in respect to the concentration of 3-OH-BZ, 3% (v v−1) DMSO, 6.5 mM MgSO4 (250 mM in water), 1 mM ThDP (10 mM in water), 0.5 mM NAD(P)H (10 mM in water), and 1 mM ATP (20 mM in water) were added to the reaction mixture. NcCAR, NiCAR or NoCAR as lyophilised whole-cell catalysts were used at a concentration of 10 mg mL−1. ApPDC-var as lyophilised whole-cell catalyst was also used at a concentration of 10 mg mL−1. The total reaction volume was 1 mL and reactions were carried out for up to 3 h, at 30 °C and 850 rpm. After the formation of (R)-3, the reaction mixture was centrifuged (15000 rpm, 10 min) to precipitate the cells and prepare the supernatant for STEP 3.
STEP 3 – Transamination: in this step, 800 μL of the supernatant from the previous step was transferred into a 2 mL glass vial. In addition, 8 equivalents of IPA (2 M in 100 mM HEPES, final pH 8) in respect to the concentration of (R)-3 was added together with 0.3 mM PLP (10 mM in water) and 3% (v v−1) DMSO. The overall reaction volume was 1 mL. The reaction was started by the addition of lyophilised E. coli BL21 cells expressing Cv2025 or BmTA to a final concentration of 10 mg lyophilised cells per mL. Reactions were carried out for 3–6 h, at 30 °C and 1000 rpm. After complete formation of (1R,2S)-4, the reaction mixture was heated (80 °C, 15 min) to inactivate the transaminase and then centrifuged (15000 rpm, 10 min) to precipitate and remove the cells.
STEP 4 – Cyclisation: in this step, 900 μL of the supernatant from the previous step was transferred into a 2 mL glass vial. In addition, 15 mM phenylacetaldehyde (300 mM in DMSO) was added to the reaction mixture. The total reaction volume was 1 mL. The reaction was started by the addition of lyophilised E. coli BL21 cells expressing ΔTf29NCS-A79I to a final concentration of 10 mg lyophilised cells per mL. Reactions were carried out for 3–24 h, at 30 or 37 °C and 850 rpm.
Monitoring of the biocatalytic reactions
The detection and quantification of substrates and products in the cascade was accomplished by HPLC analytics. The complete overview of the analytical methods employed is given in the ESI.‡Determination of process metrics
Equations used to determine the main process metrics, such as enzymatic conversion, HPLC yields, enantiomeric excess (ee), and isomeric content (ic) are described in the ESI.‡Author contributions
DW and DR: conceptualization; DW and LSB: investigation, methodology, and formal analysis; DW, LSB, and DR: visualization and data curation; YN, AEA: data curation; DR: supervision and funding acquisition; DW, DR: writing – first draft. DW, MW, LSB, HCH, and DR: writing – review & editing.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We thank Prof. Dr Jennifer Andexer (University of Freiburg, Germany) and Prof. Dr Bernd Nidetzky (Graz University of Technology, Austria) for the donation of the genes encoding for the ATP-regenerating enzymes and EcPPase, respectively. In addition, we thank Prof. Dr Nicholas J. Turner (University of Manchester) for donating the gene of TpCAR and Prof. Dr Wolfgang Kroutil (University of Graz) for the donation of the plasmid encoding for the transaminase BmTA. We are also grateful for Kristala L. J. Prather (Massachusetts Institute of Technology) for providing the E. coli K-12 MG1655 RARE strain. We thank Prof. Dr John M. Ward (University College London) for providing the plasmids encoding for the norcoclaurine synthase variant ΔTf29NCS-A79I. We also thank the UCL Mass Spectrometry Facility and UCL NMR Facility in the Department of Chemistry UCL and 700 MHz NMR equipment support by EPSRC (EP/P020410/1). We thank Dr Stephan Noack and Dr Mohamed Labib (IBG-1, FZ Juelich) for donating fermentation broths containing 3-OH-BZ. This work received funding in frame of the Bioeconomy Science Center (BioSC) FocusLab “HyImPAct” (Hybrid processes for Important Precursor and Active pharmaceutical ingredients) by the Federal State of North-Rhine Westphalia (Germany) within the framework of the NRW Strategieprojekt BioSC (No. 313/323-400-002 13).References
- S. Wu, R. Snajdrova, J. C. Moore, K. Baldenius and U. T. Bornscheuer, Angew. Chem., Int. Ed., 2021, 60, 88–119 CrossRef CAS PubMed.
- J. P. Adams, M. J. B. Brown, A. Rodriguez-Diaz, R. C. Lloyd and G.-D. Roiban, Adv. Synth. Catal., 2019, 361, 2421–2432 CAS.
- H. Sun, H. Zhang, E. L. Ang and H. Zhao, Bioorg. Med. Chem., 2017, 26, 1275–1284 CrossRef PubMed.
- N. Ran, L. Zhao, Z. Chen and J. Tao, Green Chem., 2008, 10, 361–372 RSC.
- R. A. Sheldon and P. C. Pereira, Chem. Soc. Rev., 2017, 46, 2678–2691 RSC.
- A. Schmid, J. S. Dordick, B. Hauer, A. Kiener, M. Wubbolts and B. Witholt, Nature, 2001, 409, 258–268 CrossRef CAS PubMed.
- R. Wohlgemuth, New Biotechnol., 2021, 60, 113–123 CrossRef CAS PubMed.
- C. K. Winkler, J. H. Schrittwieser and W. Kroutil, ACS Cent. Sci., 2021, 7, 55–71 CrossRef CAS PubMed.
- R. N. Patel, Bioorg. Med. Chem., 2018, 26, 1252–1274 CrossRef CAS PubMed.
- W. H. Brooks, W. C. Guida and K. G. Daniel, Curr. Top. Med. Chem., 2011, 11, 760–770 CrossRef CAS PubMed.
- T. Classen and J. Pietruszka, Bioorg. Med. Chem., 2018, 26, 1285–1303 CrossRef CAS PubMed.
- L. A. Nguyen, H. He and C. Pham-Huy, Int. J. Biomed. Sci, 2006, 2, 85–100 CAS.
- M. T. Reetz, J. Am. Chem. Soc., 2013, 135, 12480–12496 CrossRef CAS PubMed.
- B. M. Nestl, S. C. Hammer, B. A. Nebel and B. Hauer, Angew. Chem., Int. Ed., 2014, 53, 3070–3095 CrossRef CAS PubMed.
- G. Li, J.-B. Wang and M. T. Reetz, Bioorg. Med. Chem., 2017, 26, 1241–1251 CrossRef PubMed.
- P. Hoyos, J. V. Sinisterra, F. Molinari, A. R. Alcántara and P. D. De María, Acc. Chem. Res., 2010, 43, 288–299 CrossRef CAS PubMed.
- T. Sehl, H. C. Hailes, J. M. Ward, U. Menyes, M. Pohl and D. Rother, Green Chem., 2014, 16, 3341–3348 RSC.
- J. D. Scott and R. M. Williams, Chem. Rev., 2002, 102, 1669–1730 CrossRef CAS PubMed.
- Faheem, B. K. Kumar, K. Venkata Gowri Chandra Sekhar, S. Chander, S. Kunjiappan and S. Murugesan, Expert Opin. Drug Discovery, 2021, 16, 1119–1147 CrossRef CAS PubMed.
- M. E. Pyne and V. J. J. Martin, Curr. Opin. Green Sustainable Chem., 2022, 33, 100561 CrossRef CAS.
- V. Erdmann, B. R. Lichman, J. Zhao, R. C. Simon, W. Kroutil, J. M. Ward, H. C. Hailes and D. Rother, Angew. Chem., Int. Ed., 2017, 56, 12503–12507 CrossRef CAS PubMed.
- L. Zeng, B. Huang, Y. Shen and S. Cui, Org. Lett., 2018, 20, 3460–3464 CrossRef CAS PubMed.
- R. Roddan, J. M. Ward, N. H. Keep and H. C. Hailes, Curr. Opin. Chem. Biol., 2020, 55, 69–76 CrossRef CAS PubMed.
- N. Kallscheuer and J. Marienhagen, Microb. Cell Fact., 2018, 17, 1–13 CrossRef PubMed.
- Y. Zhou, Z. Li, X. Wang and H. Zhang, Eng. Life Sci., 2019, 19, 389–395 CrossRef CAS PubMed.
- K. Napora-Wijata, G. A. Strohmeier and M. Winkler, Biotechnol. J., 2014, 9, 822–843 CrossRef CAS PubMed.
- M. Winkler, Curr. Opin. Chem. Biol., 2018, 43, 23–29 CrossRef CAS PubMed.
- M. Horvat and M. Winkler, ChemCatChem, 2020, 12, 5076–5090 CrossRef CAS.
- S. R. Derrington, N. J. Turner and S. P. France, J. Biotechnol., 2019, 304, 78–88 CrossRef CAS PubMed.
- G. A. Strohmeier, I. C. Eiteljörg, A. Schwarz and M. Winkler, Chem.–Eur. J., 2019, 25, 6119–6123 CrossRef CAS PubMed.
- H. Zhao and W. A. Van Der Donk, Curr. Opin. Biotechnol., 2003, 14, 583–589 CrossRef CAS PubMed.
- R. D. Woodyer, T. W. Johannes and H. Zhao, Enzyme Technology, Springer, New York, 2006, pp. 85–103 Search PubMed.
- S. Kara, J. H. Schrittwieser, F. Hollmann and M. B. Ansorge-Schumacher, Appl. Microbiol. Biotechnol., 2014, 98, 1517–1529 CrossRef CAS PubMed.
- J. Wachtmeister and D. Rother, Curr. Opin. Biotechnol., 2016, 42, 169–177 CrossRef CAS PubMed.
- A. M. Kunjapur and K. L. J. Prather, Appl. Environ. Microbiol., 2015, 81, 1892–1901 CrossRef CAS PubMed.
- A. M. Kunjapur, Y. Tarasova and K. L. J. J. Prather, J. Am. Chem. Soc., 2014, 136, 11644–11654 CrossRef CAS PubMed.
- M. Z. Xie, M. I. Shoulkamy, A. M. H. Salem, S. Oba, M. Goda, T. Nakano and H. Ide, Mutat. Res., Fundam. Mol. Mech. Mutagen., 2016, 786, 41–51 CrossRef CAS PubMed.
- W. Finnigan, R. Cutlan, R. Snajdrova, J. P. Adams, J. A. Littlechild and N. J. Harmer, ChemCatChem, 2019, 11, 3474–3489 CrossRef CAS PubMed.
- D. Gahloth, G. A. Aleku and D. Leys, J. Biotechnol., 2020, 307, 107–113 CrossRef CAS PubMed.
- H. Stolterfoht, D. Schwendenwein, C. W. Sensen, F. Rudroff and M. Winkler, J. Biotechnol., 2017, 257, 222–232 CrossRef CAS PubMed.
- Y. Duan, P. Yao, X. Chen, X. Liu, R. Zhang, J. Feng, Q. Wu and D. Zhu, J. Mol. Catal. B: Enzym., 2015, 115, 1–7 CrossRef CAS.
- A. E. Parnell, S. Mordhorst, F. Kemper, M. Giurrandino, J. P. Prince, N. J. Schwarzer, A. Hofer, D. Wohlwend, H. J. Jessen, S. Gerhardt, O. Einsle, P. C. F. Oyston, J. N. Andexer and P. L. Roach, Proc. Natl. Acad. Sci. U. S. A., 2018, 115, 3350–3355 CrossRef CAS PubMed.
- S. Mordhorst, J. Siegrist, M. Müller, M. Richter and J. N. Andexer, Angew. Chem., Int. Ed., 2017, 56, 4037–4041 CrossRef CAS PubMed.
- D. Weber, D. Patsch, A. Neumann, M. Winkler and D. Rother, ChemBioChem, 2021, 22, 1823–1832 CrossRef CAS PubMed.
- T. Sehl, S. Bock, L. Marx, Z. Maugeri, L. Walter, R. Westphal, C. Vogel, U. Menyes, M. Erhardt, M. Müller, M. Pohl and D. Rother, Green Chem., 2017, 19, 380–384 RSC.
- K. Mack, M. Doeker, L. Grabowski, A. Jupke and D. Rother, Green Chem., 2021, 23, 4892–4901 RSC.
- M. Labib, L. Grabowski, C. Brüsseler, N. Kallscheuer, L. Wachtendonk, T. Fuchs, A. Jupke, W. Wiechert, J. Marienhagen, D. Rother and S. Noack, ACS Sustainable Chem. Eng., 2022, 10, 5117–5128 CrossRef CAS.
- M. Doeker, L. Grabowski, D. Rother and A. Jupke, Green Chem., 2022, 24, 295–304 RSC.
- P. Loi, D. Iuso, M. Czernik, F. Zacchini and G. Ptak, Trends Biotechnol., 2013, 31, 688–695 CrossRef CAS PubMed.
- A. Merivaara, J. Zini, E. Koivunotko, S. Valkonen, O. Korhonen, F. M. Fernandes and M. Yliperttula, J. Controlled Release, 2021, 336, 480–498 CrossRef CAS PubMed.
- J. Wachtmeister and D. Rother, Curr. Opin. Biotechnol., 2016, 42, 169–177 CrossRef CAS PubMed.
- S. K. Spaans, R. A. Weusthuis, J. van der Oost and S. W. M. Kengen, Front. Microbiol., 2015, 6, 1–27 Search PubMed.
- W. Xiao, R. S. Wang, D. E. Handy and J. Loscalzo, Antioxid. Redox Signaling, 2018, 28, 251–272 CrossRef CAS PubMed.
- R. Singh, J. Lemire, R. J. Mailloux and V. D. Appanna, PLoS One, 2008, 3, e2682 CrossRef PubMed.
Footnotes |
| † We like to dedicate this article to our colleague Prof. Karl-Erich Jaeger, who is about to retire after many successful years in science. |
| ‡ Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3ra01210g |
| This journal is © The Royal Society of Chemistry 2023 |