
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSynthesis of 3-substituted 2,3-dihydropyrazino[1,2-a]indol-4(1H)-ones by sequential reactions of 2-indolylmethyl acetates with α-amino acids†
Antonella Goggiamani *a,
Antonio Arcadib,
Alessia Cioglia,
Martina De Angelisa,
Stefano Dessalvia,
Giancarlo Fabrizia,
Federica Iavaronecd,
Antonia Iazzetticd,
Alessio Sferrazza*ae and
Roberta Zoppolia
*a,
Antonio Arcadib,
Alessia Cioglia,
Martina De Angelisa,
Stefano Dessalvia,
Giancarlo Fabrizia,
Federica Iavaronecd,
Antonia Iazzetticd,
Alessio Sferrazza*ae and
Roberta Zoppolia
aDipartimento di Chimica e Tecnologie del Farmaco, Sapienza, Università di Roma, P.le A. Moro 5, 00185 Rome, Italy. E-mail: alessia.ciogli@uniroma1.it; m.deangelis@uniroma1.it; stefano.dessalvi@uniroma1.it; giancarlo.fabrizi@uniroms1.it; antonella.goggiamani@uniroma1.it; a.sferrazza@irbm.com; roberta.zoppoli@uniroma1.it
bDipartimento di Dipartimento di Scienze Fisiche e Chimiche, Università degli Studi di L'Aquila, Via Vetoio, 67100 Coppito (AQ), Italy. E-mail: antonio.arcadi@univaq.it
cDipartimento di Scienze Biotecnologiche di base, Cliniche Intensivologiche e Perioperatorie, Università Cattolica del Sacro Cuore, L.go Francesco Vito 1, 00168 Rome, Italy. E-mail: federica.iavarone@unicatt.it; antonia.iazzetti@unicatt.it
dPoliclinico Universitario ‘A. Gemelli’ Foundation-IRCCS, Rome, 00168, Italy
eAlessio Sferrazza is currently a research scientist in IRBM S.p.A., Medicinal Chemistry Department, Pomezia, Roma, Italy
First published on 29th March 2023
Abstract
The synthesis of 2,3-dihydropyrazino[1,2-a]indol-4(1H)-ones from the sequential reaction of amino acid methyl esters with readily available indole-2-ylmethyl acetates is described. The reaction proceeds in situ under basic conditions of highly unstable and reactive 2-alkylideneindolenines followed by Michael-type addition of α-amino acid methyl esters/intramolecular cyclization.
Introduction
The indole nucleus has a central position in organic and medicinal chemistry because it is an important structural motif of many natural and synthetic biologically active compounds.1 Moreover, N-fused indole derivatives display various pharmacological properties including antitumor2 and tubulin polymerization inhibitory activities.3 Amongst N-fused indole heterocycles, pyrazinoindoles and their congeners attract a great deal of attention due to their application as efficient pharmacophores (Fig. 1).4 Consequentially, the pyrazinoindole system still triggers organic chemists to develop efficient methodologies for diversity-oriented preparations.5 In this field, the biological properties of pyrazino[1,2-a]indol-1-ones have been explored and a variety of synthetic approaches have been developed.6 Conversely, the pyrazino[1,2-a]indol-4-ones have been less studied. To the best of our knowledge, only one process has been reported via stereospecific N-acylation of indoles with L-amino acids followed by intramolecular cyclization via the Pictet–Spengler reaction.7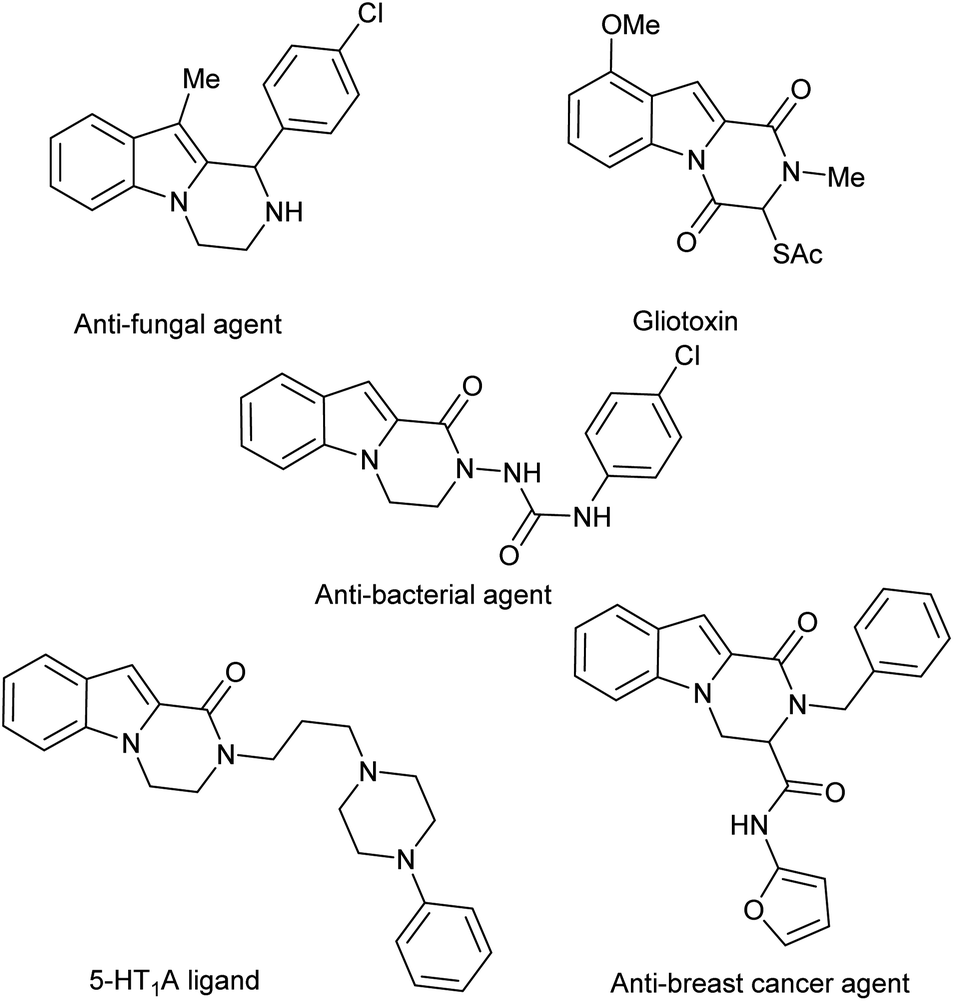 | ||
| Fig. 1 Biologically active pyrazinoindoles. | ||
Then, the synthesis of polysubstituted pyrazino[1,2-a]indol-4-one derivatives through straightforward one-pot approaches from easily available building blocks would be particularly significant considering their great potential as molecular scaffolds in drug discovery.
During our studies in the synthesis of heterocyclic compounds,8 great interest has been devoted to constructing/functionalizing indole and indole-fused polycyclic systems through simple domino processes.9 Recently, we reported the metal-free synthesis of 2-(aminomethyl), (tosylmethyl), and (aryloxymethyl) indoles 2 starting from readily available 2-indolylmethylacetates 1 and N, O, and S soft nucleophiles. The reaction proceeds through in situ generation of highly reactive 2-alkylideneindolenines I under basic conditions as provided through ESI-MS and IRMPD spectroscopy analyses.10 Furthermore, we developed a straightforward assembly of polysubstituted 1,2-dihydro-3H-pyrrolo[1,2-a]indol-3-ones 3 through a domino palladium-catalyzed reaction of 2-indolylmethylacetates 1 with 1,3-dicarbonyl compounds (Scheme 1).11
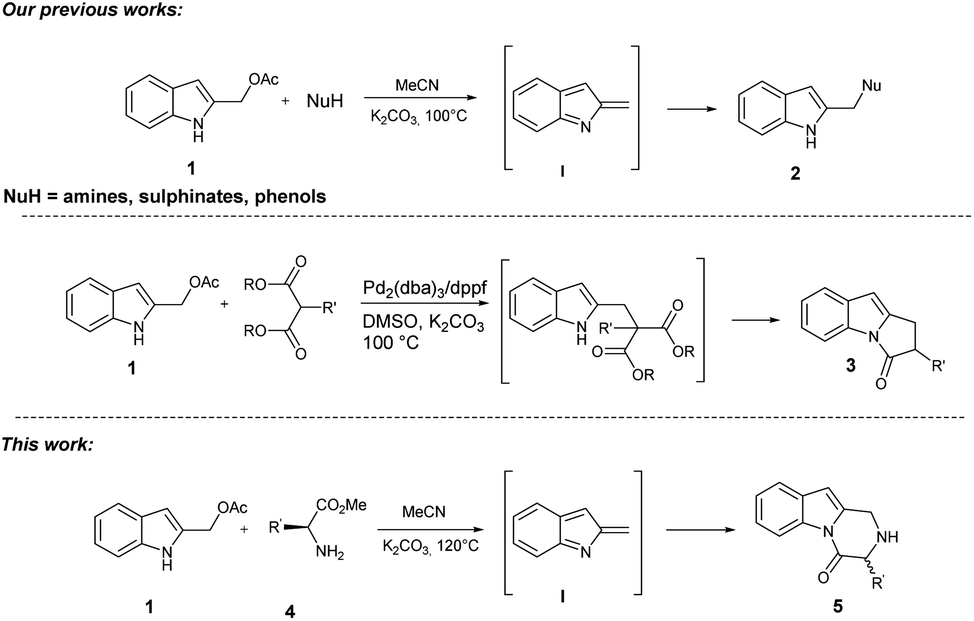 | ||
| Scheme 1 Our background and this work. | ||
Based on this background, we envisaged that the reaction of 2-indolylmethyl acetates 1 with α-amino acids 4 should achieve a general entry into the title target 5 through the in situ generation of 2-methide-2H-indole intermediate I/nucleophile Michael addition/cyclization cascade reaction according to the retrosynthetic analysis of Scheme 2.
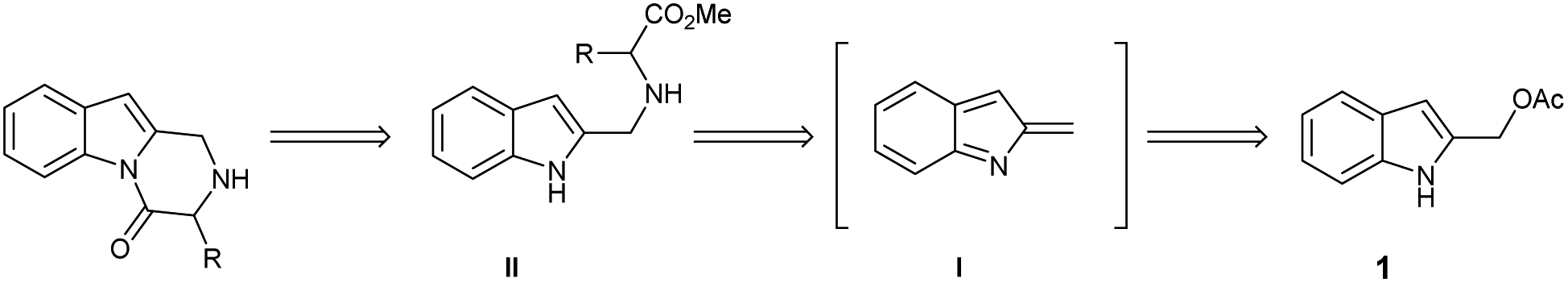 | ||
| Scheme 2 Retrosynthetic approach to the 2,3-dihydropyrazino[1,2-a]indol-4(1H)-one scaffold. | ||
Herein, we describe the scope and limitations of this approach to the synthesis of multi-substituted 2,3-dihydropyrazino[1,2-a]indol-4(1H)-one 5.
Results and discussion
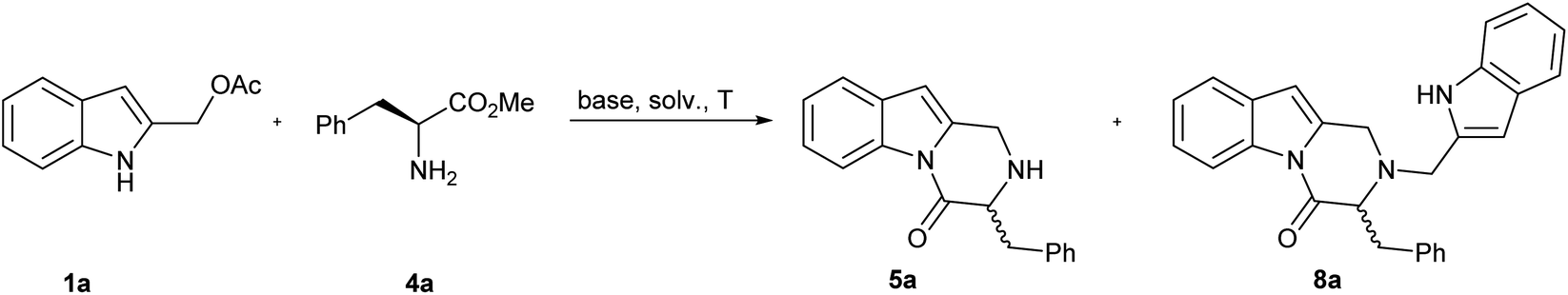
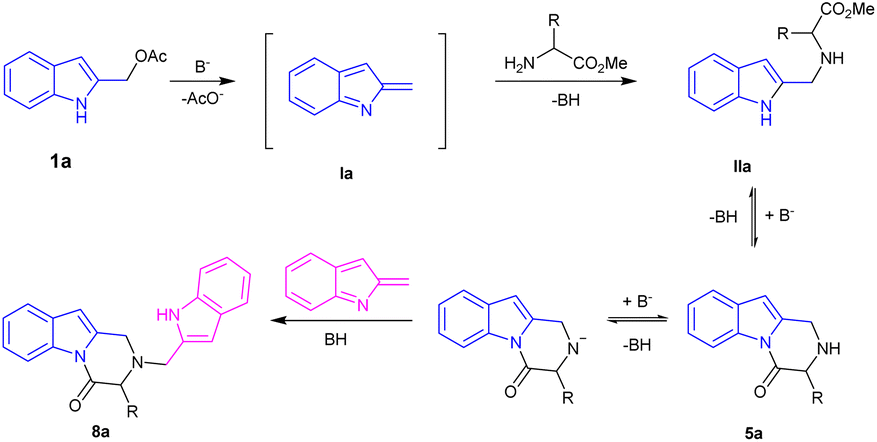
Indole-2-ylmethyl acetates 1 were obtained with excellent overall yield through the reduction of commercially available ethyl 1H-indole-2-carboxylates 6 followed by acetylation: both synthetic steps did not require further purification.10–12For our initial investigations, we examined the reaction of the indole-2-ylmethyl acetates 1a with two equivalents of methyl L-phenylalaninate 4a using K2CO3 in MeCN at 120 °C (Table 1, entry 1). The reaction proceeded to give the desired 3-benzyl-2,3-dihydropyrazino[1,2-a]indol-4(1H)-one 5a in 51% yield. A significant side reaction led to the formation of 2-((1H-indol-2-yl)methyl)-3-benzyl-2,3-dihydropyrazino[1,2-a]indol-4(1H)-one 8a in 26% yield. Very likely, the target product 5a is, also, prone to competitively give an aza-Michael addition on 2-alkylideneindolenine Ia to afford 8a (Scheme 3).
|
||||||||
|---|---|---|---|---|---|---|---|---|
| Entryb | Solvent | T (°C) | Base | 4a (equiv.) | t (h) | 5a (%) | 8a (%) | 1a (%) |
| a Unless otherwise stated, reactions were carried out on a 0.404 mmol scale using 2 equiv. of base in 3.0 mL of solvent.b Yields are given for isolated products.c Under microwave irradiation.d The reaction was carried out in 9.0 mL of solvent. | ||||||||
| 1 | MeCN | 120 | K2CO3 | 2 | 23 | 51 | 26 | — |
| 2c | MeCN | 170, 150 W | K2CO3 | 2 | 0.25 | 54 | 25 | — |
| 3d | MeCN | 120 | K2CO3 | 2 | 32 | 34 | 16 | 27 |
| 4 | DMSO | 120 | K2CO3 | 2 | 2 | 30 | 6 | — |
| 5 | DMSO | 100 | K2CO3 | 2 | 2 | 37 | 17 | — |
| 6 | DMF | 120 | K2CO3 | 2 | 0.75 | 39 | 20 | — |
| 8 | MeCN | 120 | K3PO4 | 2 | 24 | 9 | 5 | 53 |
| 9 | MeCN | 120 | Cs2CO3 | 2 | 2 | 36 | 21 | — |
| 10 | MeCN | 120 | Na2CO3 | 2 | 22 | — | — | 90 |
| 11 | MeCN | 120 | K2CO3 | 3 | 24 | 54 | 20 | — |
| 12 | MeCN | 120 | K2CO3 | 4 | 22 | 58 | 14 | — |
| 13 | MeCN | 120 | K2CO3 | 5 | 18 | 73 | 8 | — |
 | ||
| Scheme 3 Competitive formation of 8a. | ||
The features of solvent (Table 1, entries 4–6), base (Table 1, entries 8–10), dilution of the reaction mixture (Table 1, entry 3), and microwave irradiation (Table 1, entry 2) didn't influence the ratio 5a/8a. However, the wished 5a was isolated in 73% yield by carrying out the reaction in MeCN at 120 °C under the presence K2CO3 as the base and 5 equivalents of 4a. The excess of 4a was found to play a key role to achieve better product selectivity control (Table 1, entries 11–13).
This protocol was then used when the process was extended to include other indoles 1 and α-amino acid methyl esters 4. Our preparative results are summarized in Table 2.
|
||||||||
|---|---|---|---|---|---|---|---|---|
| Entryb | R1 | R2 | 1 | R3 | 4 | t (h) | 5 (%) | 8 (%) |
| a Unless otherwise stated, reactions were carried out on a 0.404 mmol scale at 120 °C using 5 equiv. of 4, 2 equiv. of K2CO3 in 3.0 mL of MeCN.b Yields are given for isolated products.c The reaction was carried out in presence of 2 equiv. of 4.d Yield was calculated from the 1H NMR analyses.e 1d was recovered in 15% of yield. | ||||||||
| 1c | H | H | 1a | CH2CH(Me)2 | 4b | 22 | 36 5b | 32 8b |
| 2 | H | H | 1a | CH2CH(Me)2 | 4b | 15 | 59 5b | 32 8b |
| 3c | H | H | 1a | CH(Me)2 | 4c | 19 | 59 5c | 29 8c |
| 4 | H | H | 1a | CH(Me)2 | 4c | 19 | 71 5c | 13 8c |
| 5 | Me | H | 1b | CH(Me)2 | 4c | 17 | 76 5d | 19 8d |
| 6 | Me | H | 1b | CH2Ph | 4a | 21 | 44 5e | 20 8e |
| 7 | OMe | H | 1c | CH2Ph | 4a | 6 | 67 5f | 15 8f |
| 8 | Cl | H | 1d | CH2Ph | 4a | 4.5 | 58 5g | 6 8gd,e |
| 9 | H | C6H5- | 1e | CH2Ph | 4a | 6.5 | 68 5h | 8 8h |
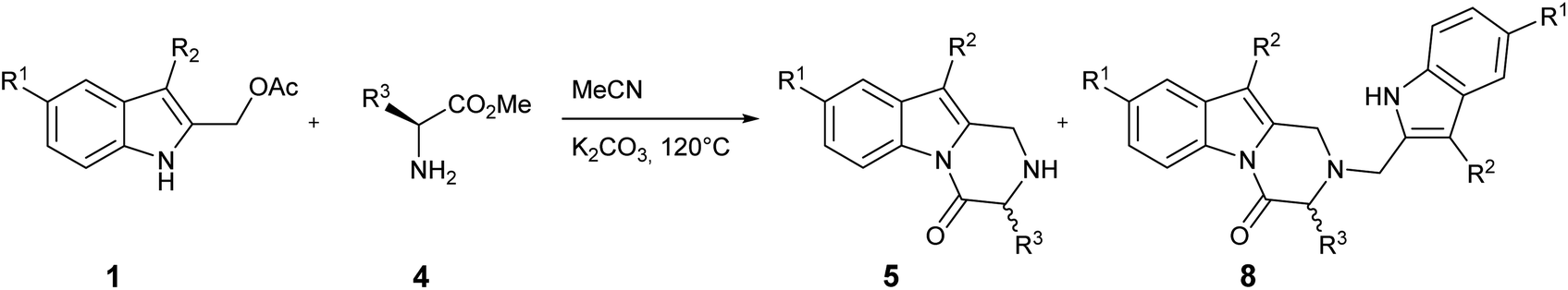
Several 2,3-dihydropyrazino[1,2-a]indol-4(1H)-one derivatives bearing a variety of useful functional groups have been prepared in good yields: in particular, the 5-chloro derivative (Table 2, entry 8) indicates that this protocol is a useful tool for obtaining more complex derivatives through subsequent transition metal-catalyzed cross-coupling reactions.
Indoles bearing electron-releasing and electron-donating groups at the C5 position show a comparable reactivity for the same α-amino acid 4 (Table 2, entries 6–8) in terms of overall yield and 5/8 ratio; this result suggests that the electrophilicity of 5-substituted 2-methide-2H-indole intermediates I, even modulated by the electronic effects of the substituents, do not affect the reaction outcome in presence of the strong neutral nitrogen nucleophiles 4 and 5.
Interestingly, the size of the R3 chain of the α-amino acid 4 seems to play a role in the achievement of the reaction: in fact, while a satisfactory yield was obtained with L-phenylalanine and L-valine methyl ester (Table 1, entry 13 and Table 2, entry 4), a significant lowering in the yield was observed with L-leucine (Table 2, entry 2).
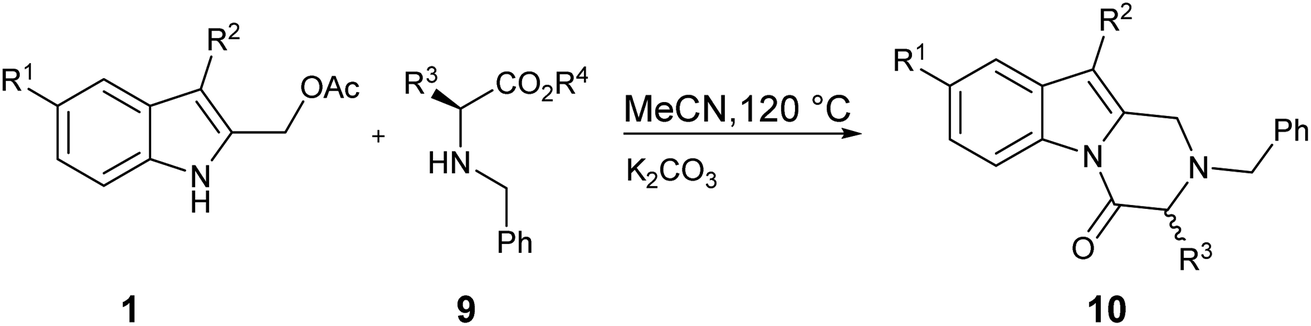
In order to avoid the side formation of 8 using a lower molar excess of nucleophile, we decided to utilize the α-amino acids methyl esters as N-benzyl derivatives 9. The brief investigation done showed that compounds 10 were obtained in good yield (Table 3, entry 1–5) without the isolation of side products. As reported before, the correlation between the steric hindrance of the alkyl substituent R3 in 9 and the yield of the reaction was observed (Table 3, entry 1 vs. entry 2–3).
|
||||||||
|---|---|---|---|---|---|---|---|---|
| Entryb | R1 | R2 | 1 | R3 | R4 | 9 | t (h) | 10 (%) |
| a Unless otherwise stated, reactions were carried out on a 0.290 mmol scale at 120 °C using 1.5 equiv. of 9, 2 equiv. of K2CO3 in 3.0 mL of MeCN.b Yields are given for isolated products. | ||||||||
| 1 | H | H | 1a | CH2Ph | Me | 9a | 24 | 84 10a |
| 2 | H | H | 1a | CH(CH3)2 | Et | 9b | 48 | 71 10b |
| 3 | H | H | 1a | CH(CH3)(C2H5) | Me | 9c | 16 | 67 10c |
| 4 | Cl | H | 1d | CH(CH3)(C2H5) | Me | 9c | 17 | 54 10d |
| 5 | OMe | H | 1c | CH2Ph | Me | 9a | 16 | 68 10e |
Finally, we briefly investigate the outcome of the reaction using L-proline 4d, to extend the scope of the process to the construction of the pirrolopirazinoindol-12-one tetracyclic core 11 (Table 4).
|
||||||
|---|---|---|---|---|---|---|
| Entryb | R1 | 1 | t (h) | 11 (%) | 12 (%) | 13 (%) |
| a Unless otherwise stated, reactions were carried out on a 0.404 mmol scale at 120 °C using 2 equiv. of 4d, 2 equiv. of K2CO3 in 3.0 mL of MeCN.b Yields are given for isolated products.c (1H-indol-2-yl)methanol was isolated in 14% yield. | ||||||
| 1 | H | 1a | 20 | 76 11a | 8 12a | — |
| 2c | OMe | 1c | 16 | Traces | 39 12c | 12 13c |
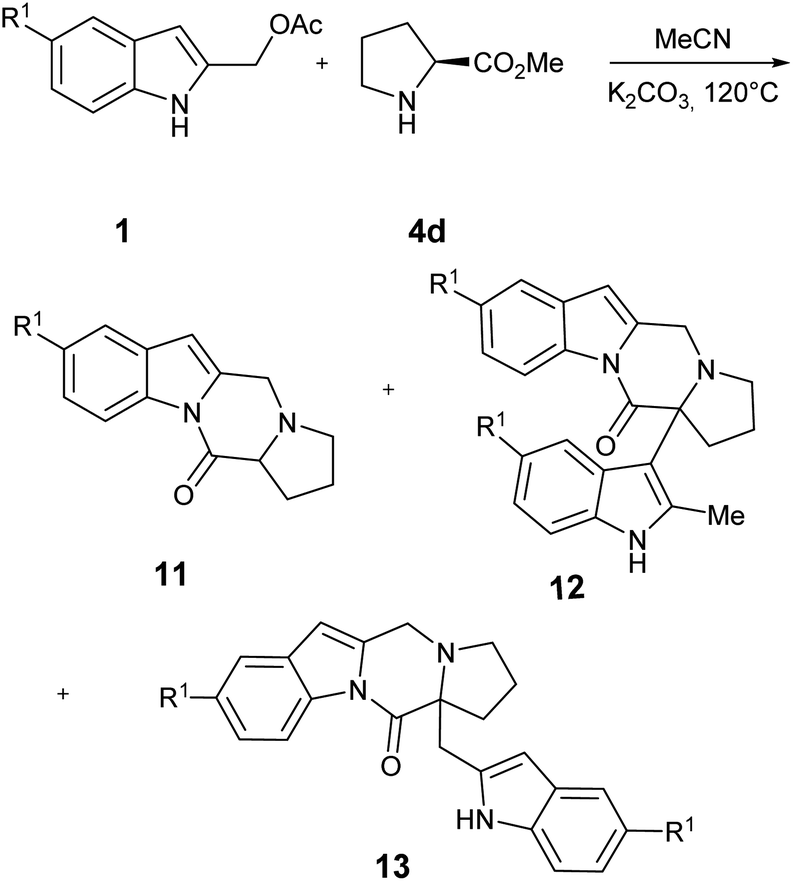
Table 4 shows the preliminary results obtained using two different indole precursors 1a and 1c. Interestingly, while the reaction between 1a and L-proline gave the target compound 11a in large excess over the side products 12a and 13a (Table 4, entry 1), using indole derivative 2b the outcome of the reaction is reversed, being the 12b and 13b products the principal, while 1b was isolated in traces (Table 4, point 2). A tentative explanation of these results could be made by taking into account the electronic effect of 5- substituent on the intermediate I: (Fig. 2) to this end, the HF calculations at 631G** level on Ia and Ib showed a significant difference in the partial positive charge on the methylidene carbon atom (see ESI† for more details).13 Since the carbanion derived from 11 may be considered highly delocalized, it is reasonable to believe that the greater electrophilicity of Ic prevents an accumulation of the product 11b but, on the contrary, favors its rapid transformation into 12c and 13c.
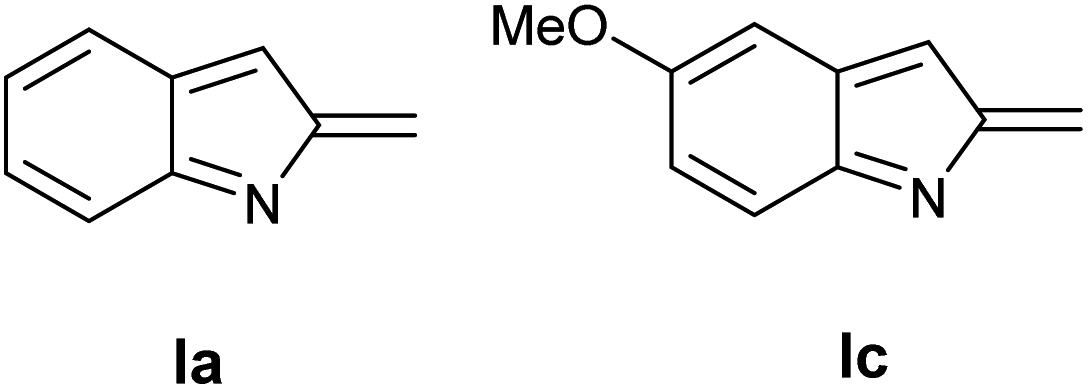 | ||
| Fig. 2 2-Methide-2H-indole I. | ||
Because of the crucial role of chiral nitrogen heterocycles in medicinal chemistry, we also analyzed the stereochemical outcome of our synthesis (Table 5).
|
|||||
|---|---|---|---|---|---|
| Entryc | T °(C) | t (h) | 5f (%) | e.r.d | 8f (%) |
| a Unless otherwise stated, reactions were carried out on a 0.404 mmol scale using 5 equiv. of 4a, 2 equiv. of K2CO3 in 3.0 mL of MeCN.b The major enantiomer L is shown.c Yields are given for isolated products.d e.r was calculated from enantioselective HPLC analysis (see ESI for more details).e The starting indole 1c was recovered in 50% of yield.f The starting indole 1c was recovered in 60% of yield.g The starting indole 1c was recovered almost quantitatively. | |||||
| 1 | 120 | 6 | 67 | 68![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :32 :32 |
15 |
| 2 | 90 | 6 | 69 | 85:15 |
— |
| 3e | 70 | 18 | 31 | 92:8 |
— |
| 4f | 70 | 6 | 27 | 95:5 |
— |
| 5g | 50 | 24 | — | — | |
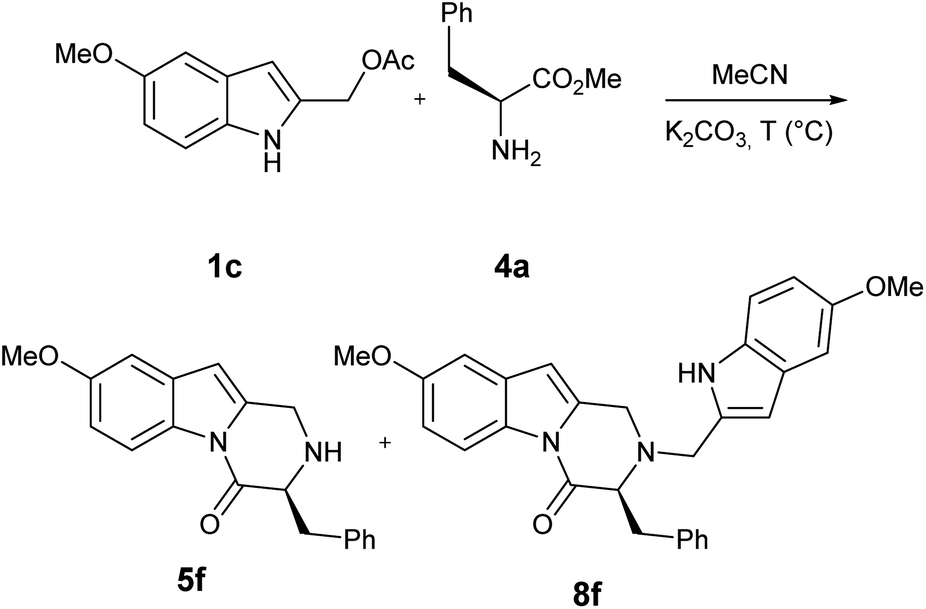
It has long been noted that amino acids rapidly undergo racemization process when heated in acidic or basic conditions and subsequently our target pyrazoindole could undergo racemization at C3 via enolate.14
Indeed, when the reaction of 1c with methyl L-phenylalaninate 4a was carried out at 120 °C, HPLC analysis showed the formation of 5f as enantioenriched mixture (68:32, Table 5, entry 1), while 8f was obtained as racemate. The enantiomeric ratio and its dependence on reaction temperature were established by HPLC on Chiralpak IA column of purified pyrazinoindoles. When the reaction was performed at lower temperatures (Table 5, entry 2–4) the loss of enantiomeric purity was limited (up to 95:5, Table 5, entry 4).
Experimental
A list of chemicals and instrumentation is provided in the ESI.†Typical procedure for the preparation of substituted (1H-indol-2-yl)methyl acetate 1(a–e)
The (1H-indol-2-yl)methyl acetates (1a–d) and 3-aryl-2-(1H-indol-2-yl)methyl acetates (1e) were previously synthesized in our laboratory10–12 according to the procedures reported in ESI.†Typical procedure for the preparation of 3-benzyl-2,3-dihydropyrazino[1,2-a]indol-4(1H)-one 5a
In a 50 mL Carousel Tube Reactor (Radely Discovery Technology) containing a magnetic stirring bar (1H-indol-2-yl)methyl acetate 1a (76.4 mg, 0.404 mmol, 1.0 equiv.) was dissolved at room temperature with 2.0 mL of anhydrous MeCN. Then, methyl L-phenylalaninate 4a (357.5 mg, 2.020 mmol, 5.0 equiv.), K2CO3 (112.0 mg, 0.808 mmol, 2.0 equiv), and 1.0 mL of solvent were added. The mixture was stirred for 18 h at 120 °C. After this time, the reaction mixture was cooled to room temperature, diluted with Et2O, washed with a saturated NaHCO3 solution and with brine. The organic extract was dried over Na2SO4, filtered, and concentrated under reduced pressure. The residue was purified by chromatography on SiO2 (25–40 μm), eluting with a 80/20 (v/v) n-hexane/AcOEt mixture (Rf = 0.21) to obtain 81.0 mg (73% yield) of 3-benzyl-2,3-dihydropyrazino[1,2-a]indol-4(1H)-one 5a and 7.0 mg (8% yield) of 2-((1H-indol-2-yl)methyl)-3-benzyl-2,3-dihydropyrazino[1,2-a]indol-4(1H)-one 8a.:20); IR (neat): 2989, 2796, 1444, 1272, 1183, 941, 734 cm−1; 1H NMR (400.13 MHz) (CDCl3): δ 8.36 (d, J = 8.0 Hz, 1H), 7.39 (d, J = 7.6 Hz, 1H), 7.27–7.16 (m, 7H), 6.17 (s, 1H), 4.11 (d, J = 16 Hz, 1H), 3.88 (d, J = 16 Hz, 1H), 3.79 (dd, J1 = 8.7 Hz, J2 = 4.0 Hz, 1H), 3.44 (dd, J1 = 14 Hz, J2 = 4.0 Hz, 1H), 3.06 (dd, J1 = 14.0 Hz, J2 = 8.7 Hz, 1H), 1.79 (s, 1H); 13C NMR (100.6 MHz) (CDCl3): δ 169.1 (C), 137.5 (C), 136.6 (C), 134.7 (C), 129.5 (CH), 128.8 (CH), 127.0 (CH), 124.5 (CH), 124.2 (CH), 120.2 (CH), 116.3 (CH), 103.1 (CH), 61.3 (CH), 41.9 (CH2), 36.3 (CH2). HRMS: m/z [M + H]+ calcd for C15H17NO2: 277.1335; found: 277.1336.:15); IR (neat): 3401, 2986, 2808, 1692, 1356, 1188, 691 cm−1; 1H NMR (400.13 MHz) (CDCl3): δ 8.40 (d, J = 7.9 Hz, 1H), 7.48 (d, J = 6.8 Hz, 1H), 7.41–7.18 (m, 10H), 7.06–7.02 (m, 1H), 7.00–6.94 (m, 2H), 6.36 (s, 1H), 6.11 (s, 1H), 4.43 (dd, J1 = 16.7, J2 = 1.8 Hz, 1H), 3.89–3.82 (m, 2H), 3.74–3.69 (m, 2H), 3.25 (dd, J1 = 14.1, J2 = 4.3 Hz, 1H), 3.09 (dd, J1 = 14.2, J2 = 11.5 Hz, 1H); 13C NMR (100.6 MHz) (CDCl3): δ 168.8 (C), 138.3 (C), 136.0 (C), 134.9 (C), 134.6 (C), 133.9 (C), 129.7 (C), 129.5 (CH), 128.8 (CH), 128.3 (C), 127.1 (CH), 124.9 (CH), 124.6 (CH), 121.7 (CH), 120.5 (CH), 120.2 (CH), 119.7 (CH), 116.5 (CH), 111.0 (CH), 105.8 (CH), 101.5 (CH), 64.6 (CH), 52.0 (CH2), 43.5 (CH2), 35.6 (CH2). HRMS: m/z [M + H]+ calcd for C27H24N3O: 406.1914; found: 406.1915.Typical procedure for the preparation of 2,3-dibenzyl-2,3-dihydropyrazino[1,2-a]indol-4(1H)-one 10a
In a 50 mL Carousel Tube Reactor (Radely Discovery Technology) containing a magnetic stirring bar (1H-indol-2-yl)methyl acetate (54.9 mg, 0.290 mmol, 1.0 equiv.) was dissolved at room temperature with 2.0 mL of anhydrous MeCN. Then, methyl benzyl L-phenylalaninate (369.8 mg, 1.45 mmol, 5.0 equiv.), K2CO3 (80.0 mg, 0.58 mmol, 2.0 equiv.), and 1.0 mL of solvent were added. The mixture was stirred for 24 h at 120 °C. After this time, the reaction mixture was cooled to room temperature, diluted with Et2O, washed with a saturated NaHCO3 solution, and with brine. The organic extract was dried over Na2SO4, filtered, and concentrated under reduced pressure. The residue was purified by chromatography on SiO2 (25–40 μm), eluting with a 96/4 (v/v) n-hexane/AcOEt mixture (Rf = 0.21) to obtain 89.2 mg (84% yield) of 2,3-dibenzyl-2,3-dihydropyrazino[1,2-a]indol-4(1H)-one.:4); IR (neat):3024, 1782, 1450, 1373, 695 cm−1; 1H NMR (400.13 MHz) (CDCl3): δ 8.54 (d, J = 7.9 Hz, 1H), 7.58 (d, 1H), 7.43–7.31 (m, 7H), 7.29–7.22 (m, 3H), 7.05–7.03 (m, 2H), 6.41 (s, 1H), 4.39 (dd, J1 = 16.9, J2 = 1.7 Hz, 1H), 3.93 (d, J = 4.4 Hz, 1H), 3.90 (d, 1H), 3.82 (d, J = 13.3 Hz, 1H), 3.66 (d, J = 13.3 Hz, 1H), 3.35 (dd, J1 = 14.4, J2 = 4.9 Hz, 1H), 3.25 (dd, J1 = 14.4, J2 = 10.1 Hz, 1H); 13C NMR (100.6 MHz) (CDCl3): δ 169.4 (C), 138.2 (C), 137.6 (C), 134.9 (C), 134.6 (C), 129.7 (C), 129.4 (CH), 128.8 (CH), 128.4 overlapping (CH), 127.5 (CH), 126.7 (CH), 124.6 (CH), 124.3 (CH), 120.3 (CH), 116.5 (CH), 105.4 (CH), 66.4 (CH), 58.1 (CH2), 43.1 (-CH2), 35.5 (CH2). HRMS: m/z [M + H]+ calcd for C25H23N2O: 367.1805; found: 367.1804.Conclusions
To sum up, this paper describes a general protocol for the synthesis of 2,3-dihydropyrazino[1,2-a]indol-4(1H)-ones from readily available indole-2-ylmethyl acetates. The reaction tolerates a variety of useful neutral, electron-rich, and electron-poor substituents and proceeds through in situ generation under basic conditions of highly unstable and reactive 2-alkylideneindolenines followed by Michael-type additions of α-amino acids methyl ester/intramolecular cyclization. The influence of the reaction conditions and the features of substrates on the stereochemical outcome of the procedure with the use of chiral α-amino esters has been explored.Author contributions
Conceptualization: G. F.; data curation: A. A. and A. S.; funding acquisition: G. F.; investigation: A. C., M. D. A., S. D., F. I., A. I., and R. Z.; methodology: A. G. and A. S.; project administration: G. F.; supervision: A. G.; writing—original draft: A. G.; writing—review and editing: A. I., A. A., and A. S. All authors have read and agreed to the published version of the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We gratefully acknowledge “Sapienza”, University of Rome, University of L′ Aquila, the Catholic University of Sacred Heart, and PRIN project 2017 “Targeting Hedgehog pathway: virtual screening identification and sustainable synthesis of novel Smo and Gli inhibitors and their pharmacological drug delivery strategies for improved therapeutic effects in tumors” (2017SXBSX4), for financial support.Notes and references
- For biological activities (a) T. P. Singh and O. M. Singh, Mini-Rev. Med. Chem., 2018, 18, 9 CrossRef CAS PubMed; (b) M.-Z. Zhang, Q. Chen and G.-F. Yang, Eur. J. Med. Chem., 2015, 89, 421 CrossRef CAS PubMed; (c) W. Zi, Z. Zuo and D. Ma, Acc. Chem. Res., 2015, 48, 702 CrossRef CAS PubMed; (d) A. J. Kochanowska-Karamyan and M. T. Hamann, Chem. Rev., 2010, 110, 4489 CrossRef CAS PubMed . For synthesis:; (e) M. G. Ciulla, S. Zimmermann and K. Kumar, Org. Biomol. Chem., 2019, 17, 413 RSC; (f) A. Kumari and R. K. Singh, Bioorg. Chem., 2019, 89, 103021 CrossRef CAS PubMed; (g) Z. Dong, X.-W. Zhang, W. Li, Z.-M. Li, W.-Y. Wang, Y. Zhang, W. Liu and W.-B. Liu, Org. Lett., 2019, 21, 1082 CrossRef CAS PubMed; (h) T.-T. Wang, L. Zhao, Y.-J. Zhang and W.-W. Liao, Org. Lett., 2016, 18, 5002–5005 CrossRef CAS PubMed; (i) S. Lancianesi, A. Palmieri and M. Petrini, Chem. Rev., 2014, 114, 7108 CrossRef CAS PubMed; (j) S. Cacchi, G. Fabrizi and A. Goggiamani, in Organic Reactions, HOBOKEN, NJ, Jonh Wiley & Sons, 2012, 76, p. 281 Search PubMed; (k) S. Cacchi, G. Fabrizi and A. Goggiamani, Org. Biomol. Chem., 2011, 9, 641 RSC; (l) S. Cacchi and G. Fabrizi, Chem. Rev., 2011, 111, PR215–PR283 CrossRef PubMed; (m) F. R. de Sa Alves, J. E. Barreiro and A. C. M. Fraga, Mini-Reviews in Med. Chem., 2009, 9, 782 CrossRef PubMed.
- N. Yoda and N. Hirayama, J. Med. Chem., 1993, 36, 1461 CrossRef CAS PubMed.
- M. Goldbrunner, G. Loidl, T. Polossek, A. Mannschreck and E. V. Angerer, J. Med. Chem., 1997, 40, 3524 CrossRef CAS PubMed.
- (a) K. Zhang, C. Tran, M. Alami, A. Hamze and O. Provot, Pharmaceuticals, 2021, 14, 779 CrossRef CAS PubMed; (b) E. A. Sokolova, Chem. Heterocycl. Compd., 2016, 52, 219e221 CrossRef; (c) A. R. Katritzky, A. K. Verma, H. Y. He and R. Chandra, Chem. Commun., 2011, 47, 3625e3627 Search PubMed; (d) R. K. Tiwari, A. K. Verma, A. K. Chhillar, D. Singh, J. Singh, V. K. Sankar, V. Yadav, G. L. Sharma and R. Chandra, Bioorg. Med. Chem., 2006, 14, 2747e2752 CrossRef PubMed; (e) R. K. Tiwari, D. Singh, J. Singh, V. Yadav, A. K. Pathak, R. Dabur, A. K. Chhillar, R. Singh, G. L. Sharma, R. Chandra and A. K. Verma, Bioorg. Med. Chem., 2006, 16, 413e416 Search PubMed; (f) J. Chang-Fong, J. Addo, M. Dukat, C. Smith, N. A. Mitchell, K. Herrick-Davis, M. Teitler and R. A. Glennon, Bioorg. Med. Chem. Lett., 2002, 12, 155e158 CrossRef PubMed; (g) F. Cafieri, E. Fattorusso and O. Taglialatela-Scafati, J. Nat. Prod., 1988, 61, 122e125 Search PubMed.
- (a) N. Thadem, M. Rajesh, H. Balaboina and S. Das, Org. Biomol. Chem., 2022, 20, 6368 RSC; (b) I. A. Wani, S. Das, S. Mondal and M. K. Ghorai, J. Org. Chem., 2018, 83, 14553 CrossRef CAS PubMed; (c) M. Nayak, G. Pandey and S. Batra, Tetrahedron, 2011, 67, 7563e7569 Search PubMed; (d) S. Laliberte, P. K. Dornan and A. Chen, Tetrahedron Lett., 2010, 51, 363e366 CrossRef; (e) R. K. Tiwari, J. Singh, D. Singh, A. K. Verma and R. Chandra, Tetrahedron, 2005, 61, 9513e9518 CrossRef; (f) G. Abbiati, A. Arcadi, E. Beccalli, E. Rossi and S. Zanzola, J. Org. Chem., 2005, 70, 4088 CrossRef CAS PubMed.
- (a) O. Sari, A. F. Seybek, S. Kaya, N. Menges, S. Erdem and M. Balci, Eur. J. Org. Chem., 2019, 5261 CrossRef CAS; (b) M. Palomba, L. Sancineto, F. Marini, C. Santi and L. Bagnoli, Tetrahedron, 2018, 74, 7156 CrossRef CAS; (c) G. Verniest and A. Padwa, Org. Lett., 2008, 10, 4379 CrossRef CAS PubMed; (d) J. B. Boothe, Y. Shen and J. P. Wolfe, J. Org. Chem., 2017, 82, 2777 CrossRef CAS PubMed; (e) M. Mahdavi, R. Hassanzadeh-Soureshjan, M. Saeedi, A. Ariafard, R. BabaAhmadi, P. R. Ranjbar and A. Shafiee, RSC Adv., 2015, 5, 101353 RSC; (f) G. Broggini, V. Barbera, E. M. Beccalli, E. Borsini, S. Galli, G. Lanza and G. Zecchi, Adv. Synth. Catal., 2012, 354, 159 CrossRef CAS; (g) J. An, N. J. Chang, L. D. Song, Y. Jin, Y. Q. Ma, J. R. Chen and W. J. Xiao, Chem. Commun., 2011, 47, 1869 RSC; (h) M. Bandini, A. Eichholzer, M. Monari, F. Piccinelli and A. Umani-Ronchi, Eur. J. Org. Chem., 2007, 2917 CrossRef CAS.
- A. Singh, S. Singh, S. Sewariya, N. Singh, P. Singh, A. Kumar, R. Bandichhor and R. Chandra, Tetrahedron, 2021, 84, 132017 CrossRef CAS.
- (a) V. Marsicano, A. Arcadi, M. Chiarini, G. Fabrizi, A. Goggiamani and A. Iazzetti, Org. Biomol. Chem., 2021, 19, 5177 RSC; (b) A. Arcadi, A. Ciogli, G. Fabrizi, A. Fochetti, R. Franzini, F. Ghirga, A. Goggiamani and A. Iazzetti, Org. Biomol. Chem., 2019, 17, 10065 RSC; (c) R. Alfonsi, B. Botta, S. Cacchi, L. Di Marcotullio, G. Fabrizi, R. Faedda, A. Goggiamani, A. Iazzetti and M. Mori, J. Med. Chem., 2017, 60, 1469 CrossRef CAS PubMed; (d) A. Arcadi, F. Blesi, S. Cacchi, G. Fabrizi, A. Goggiamani and F. Marinelli, Tetrahedron, 2013, 69, 1857 CrossRef CAS.
- (a) A. Arcadi, S. Cacchi, G. Fabrizi, F. Ghirga, A. Goggiamani, A. Iazzetti and F. Marinelli, Beilstein J. Org. Chem., 2018, 14, 2411 CrossRef CAS PubMed; (b) A. Arcadi, S. Cacchi, G. Fabrizi, F. Ghirga, A. Goggiamani, A. Iazzetti and F. Marinelli, Synthesis, 2018, 50, 1133 CrossRef CAS; (c) A. Arcadi, M. Chiarini, G. D'Anniballe, F. Marinelli and E. Pietropaolo, Org. Lett., 2014, 16, 1736 CrossRef CAS PubMed; (d) G. Cera, S. Piscitelli, M. Chiarucci, G. Fabrizi, A. Goggiamani, R. S. Ramón, S. P. Nolan and M. Bandini, Angew. Chem., Int. Ed., 2012, 51, 9891 CrossRef CAS PubMed; (e) G. Abbiati, A. Arcadi, M. Chiarini, F. Marinelli, E. Pietropaolo and E. Rossi, Org. Biomol. Chem., 2012, 10, 7801 RSC; (f) A. Arcadi, R. Cianci, G. Ferrara and F. Marinelli, Tetrahedron, 2010, 66, 2378 CrossRef CAS.
- A. Arcadi, G. Berden, A. Ciogli, D. Corinti, M. E. Crestoni, M. De Angelis, G. Fabrizi, A. Goggiamani, A. Iazzetti, F. Marrone, V. Marsicano, J. Oomens and A. Serraiocco, Eur. J. Org. Chem., 2022, e202201166 CAS.
- A. Iazzetti, A. Arcadi, S. Dessalvi, G. Fabrizi, A. Goggiamani, F. Marrone, A. Serraiocco, A. Sferrazza and K. Ullah, Catalysts, 2022, 12, 1516 CrossRef CAS.
- A. Arcadi, A. Calcaterra, M. Chiarini, G. Fabrizi, A. Fochetti, A. Goggiamani, A. Iazzetti, F. Marrone, V. Marsicano and A. Serraiocco, Synthesis, 2022, 54, 741 CrossRef CAS.
- Calculated by HF, 3-21G* in Titan 1.0.1 2000, Wavefunction inc. 18401, Von Karman Ave., Ste. 370, Irvine, CA, p. 92612.
- J. L. Bada, Racemization of Amino Acids, in Chemistry and Biochemistry of the Amino Acids, ed. G. C. Barrett, Springer, Dordrecht, 1985 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3ra01335a |
| This journal is © The Royal Society of Chemistry 2023 |