
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicencePhosphanyl-substituted tin half-sandwich complexes†
Carsten Müllera,
Justin Schua,
Bernd Morgensterna,
Michael Zimmera,
Marc Schmidtmannb and
André Schäfer *a
*a
aFaculty of Natural Sciences and Technology, Department of Chemistry, Saarland University, 66123 Saarbrücken, Germany. E-mail: andre.schaefer@uni-saarland.de
bSchool of Mathematics and Science, Institute of Chemistry, Carl von Ossietzky University of Oldenburg, 26129 Oldenburg, Germany
First published on 31st March 2023
Abstract
Phosphanyl-substituted tin(II) half sandwich complexes are reported. Due to the Lewis acidic tin center and Lewis basic phosphorous atom they form head-to-tail dimers. Their properties and reactivities were investigated both experimentally and theoretically. Furthermore, related transition metal complexes of these species are presented.
Introduction
Organometallic complexes exhibiting η5-bonded cyclopentadienyl (Cp) ligands are well-known species nowadays. The most common variants are complexes with two η5-Cp ligands, commonly referred to as metallocenes, and complexes with only one η5-Cp ligand, usually referred to as half-sandwich complexes.1 While many examples of such species are known for transitions metals, this structural motif can also be found in p-block chemistry.2 For instance, the first example of a group 14 half-sandwich complex was cyclopentadienyltin chloride, CpSnCl, which was reported by Noltes et al. in 1972,3 about 16 years after Fischer and Grubert had described the related metallocene parent compound stannocene, Cp2Sn,4 and a few years before Lappert et al. and Veith reported the first diorganostannylenes with σ-bonded substituents (Chart 1).5 While the latter are commonly regarded as the first examples of stable diorganostannylenes, stannocene, Cp2Sn, and cyclopentadienyltin chloride, CpSnCl, are also divalent tin(II) compounds of the general type R2Sn, but differ significantly from “typical” stannylenes in their properties and reactivities. Among other things, the central tin atoms in Cp2Sn and CpSnCl exhibit mostly electrophilic character but only minor nucleophilic character.6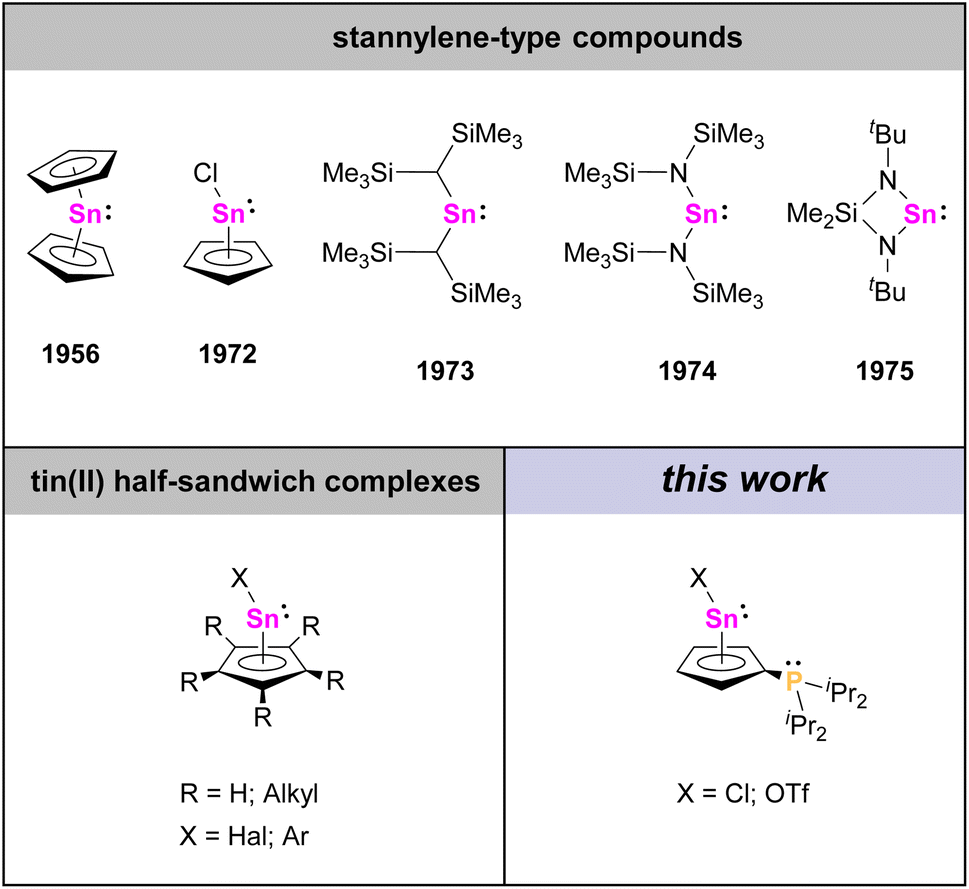 | ||
| Chart 1 Overview of selected R2Sn(II)-type compounds. | ||
Nowadays, different half-sandwich complexes of tin(II) with various cyclopentadienyl groups and different substituents bound to the tin atom are known, but noteworthy, the substitution patterns on the Cp ligands are almost exclusively limited to H atoms, or alkyl and aryl groups.2b,3,7,8 Donor groups at the Cp ligands of tin half-sandwich complexes on the other hand are unknown as of yet.
Since low valent tin species have attracted wide attention for their applications ranging from coordination chemistry to small molecule activation and homogeneous catalysis,9 and following our recent studies on phosphanyl-functionalized stannocenes and other main-group metallocenes,10 we herein report the related phosphanyl-functionalized tin half-sandwich complexes, which exhibit Lewis amphiphilic character due to a Lewis acidic tin atom and a Lewis basic phosphanyl group attached to the Cp ligand.
Results
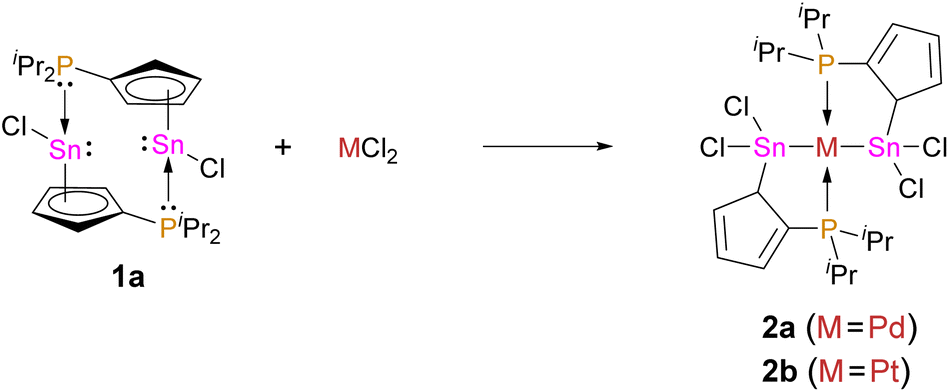
Recently, our group reported on diphosphanylstannocenes, which exhibit moderate Lewis amphiphilic character, due to Lewis basic phosphorus centres and mildly Lewis acidic tin atoms.10 Following established procedures for the synthesis of tin(II) half-sandwich complexes,2,7 we reacted bis(diisopropylphosphanyl)stannocene, dippSn, with tin(II) chloride and obtained the heteroleptic system 1a (Scheme 1). Furthermore, the corresponding triflate 1b could be obtained, by reacting 1a with trimethylsilyltriflate, in analogy to literature reports (Scheme 2).11 | ||
| Scheme 1 Synthesis of 1a by the reaction of dippSn with SnCl2. | ||
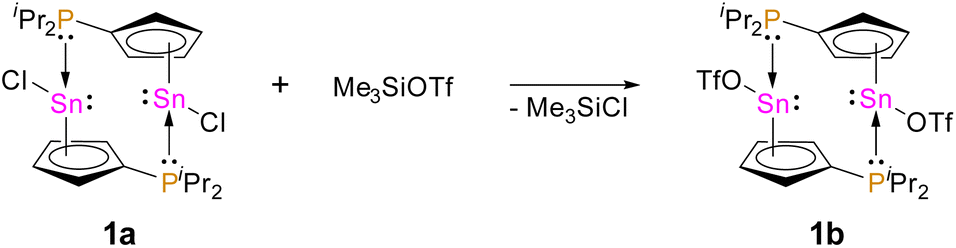 | ||
| Scheme 2 Synthesis of 1b via treatment of 1a with Me3SiOTf. | ||
To gain further insight into the electronic properties of phosphanyl-functionalized half-sandwich compound 1a,b and to assess whether the CpSnCl and 1a,b do indeed possess more Lewis acidic central atoms than the related homoleptic metallocenes, we performed DFT calculations to obtain fluoride ion affinities (FIA) and global electrophilicity indices (GEI) (Fig. 1).12
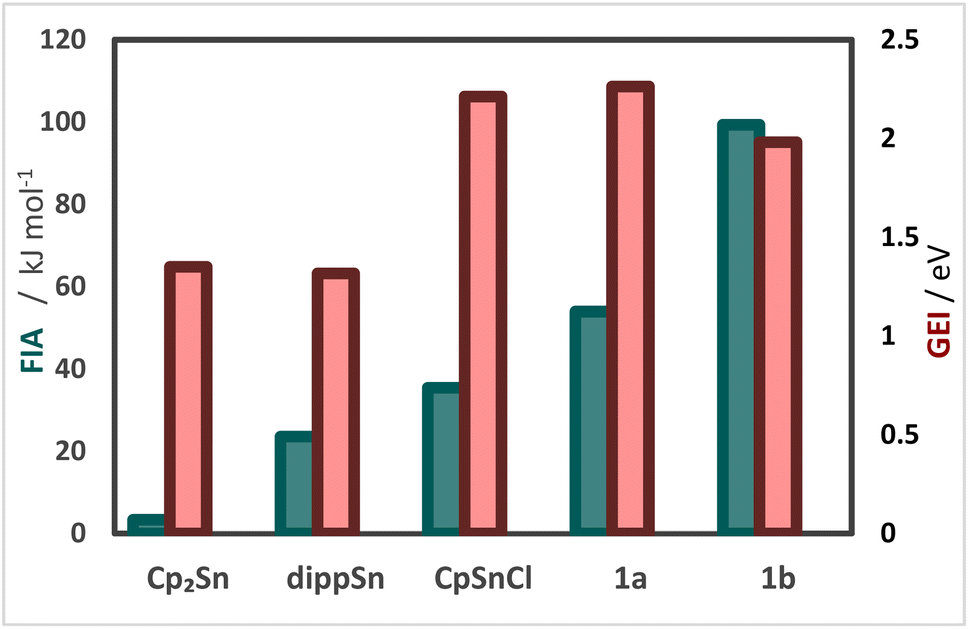 | ||
| Fig. 1 Fluoride ion affinities (FIA; green), and global electrophilicity indices (GEI; red), for Cp2Sn, dippSn, CpSnCl, 1a and 1b calculated at B3LYP-D3/def2-TZVP. | ||
The results suggest that the metallocene-type compounds Cp2Sn and dippSn possess GEIs of 1.32 to 1.35 eV, while GEIs of 1.98 to 2.26 eV are computed for CpSnCl and 1a,b. Similarly, lower FIAs are computed for Cp2Sn and dippSn than for CpSnCl and 1a,b. Thus, these results suggest that 1a,b do indeed possess a more pronounced Lewis acidity than their stannocene counterparts. To obtain experimental evidence for this, we conducted measurements following the Gutmann–Beckett method (Table 1).13 Indeed, significantly higher Δδ31P values of 14.9 to 17.8 ppm and acceptor numbers (ANs) of 32.9 to 39.3 are obtained for CpSnCl and 1a,b, than for their metallocene counterparts, Cp2Sn and dippSn, which is in qualitative agreement to the afore discussed DFT calculations.
1a and 1b exhibit 119Sn NMR chemical shifts of δ119Sn(1a)(CP/MAS) = −709 and δ119Sn(1b)(CP/MAS) = −928, which is rather unusual for a tin(II) half sandwich compound (e.g. δ119Sn(CpSnCl)(CP/MAS) = −1580).14 Moreover, the solid state 31P{1H} NMR spectra, exhibit signals at δ31P(1a)(CP/MAS) = 4.3 and δ31P(1b)(CP/MAS) = 19.2, accompanied by 117/119Sn satellites with coupling constants of 1JPSn = 950 Hz (1a), 1JPSn = 1460 Hz (1b), clearly reflecting an Sn–P bonding interaction. To gain further insight into the structures of 1a and 1b, we determined the crystal structures via X-ray diffraction. 1a crystallized in the monoclinic space group P21/c and 1b in the triclinic space group P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) , each with one formula unit ((iPr2PC5H4)SnX) per asymmetric unit. Interestingly, due to its Lewis amphiphilic character and inability for intramolecular quenching, 1a,b form head-to-tail dimers with Sn–P contacts of 279.08(13) pm (1a) and 279.58(4) pm (1b) (Fig. 2 and 3). Compared to related compounds, these Sn–P bonds are relatively long ([(TMS2N)2Sn(Cl)PPh]2: d(Sn–P) = 257.06(6) pm (ref. 15)). Furthermore, the Cp rings in 1a and 1b are bound in what may best be described as a heavily distorted η5 coordination. Although the Sn–CCp bonds are relatively unequal (1a: 243.56(44) to 315.68(43) pm; 1b: 245.73(18) to 300.45(19) pm), the C–C bonds within the Cp rings are fairly uniform (1a: 138.81(57) to 141.65(64) pm; 1b: 139.48(24) to 142.84(22) pm), indicating a certain degree of aromaticity.
, each with one formula unit ((iPr2PC5H4)SnX) per asymmetric unit. Interestingly, due to its Lewis amphiphilic character and inability for intramolecular quenching, 1a,b form head-to-tail dimers with Sn–P contacts of 279.08(13) pm (1a) and 279.58(4) pm (1b) (Fig. 2 and 3). Compared to related compounds, these Sn–P bonds are relatively long ([(TMS2N)2Sn(Cl)PPh]2: d(Sn–P) = 257.06(6) pm (ref. 15)). Furthermore, the Cp rings in 1a and 1b are bound in what may best be described as a heavily distorted η5 coordination. Although the Sn–CCp bonds are relatively unequal (1a: 243.56(44) to 315.68(43) pm; 1b: 245.73(18) to 300.45(19) pm), the C–C bonds within the Cp rings are fairly uniform (1a: 138.81(57) to 141.65(64) pm; 1b: 139.48(24) to 142.84(22) pm), indicating a certain degree of aromaticity.
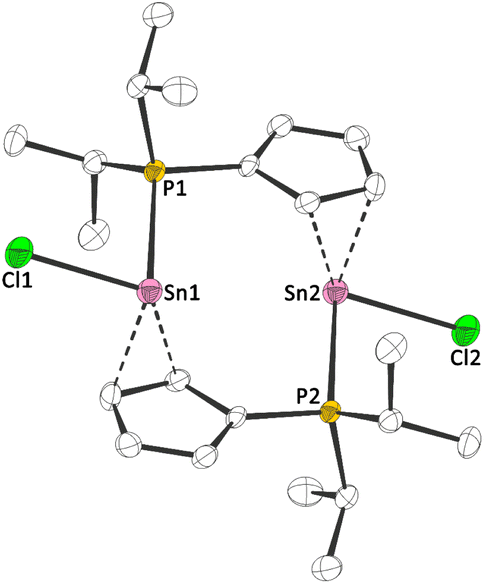 | ||
| Fig. 2 Molecular structure of 1a in the crystal (displacement ellipsoids at 50% probability level, hydrogen atoms omitted for clarity). Selected bond lengths [pm]: Sn–Cl: 247.54(2); Sn–P: 279.08(2). | ||
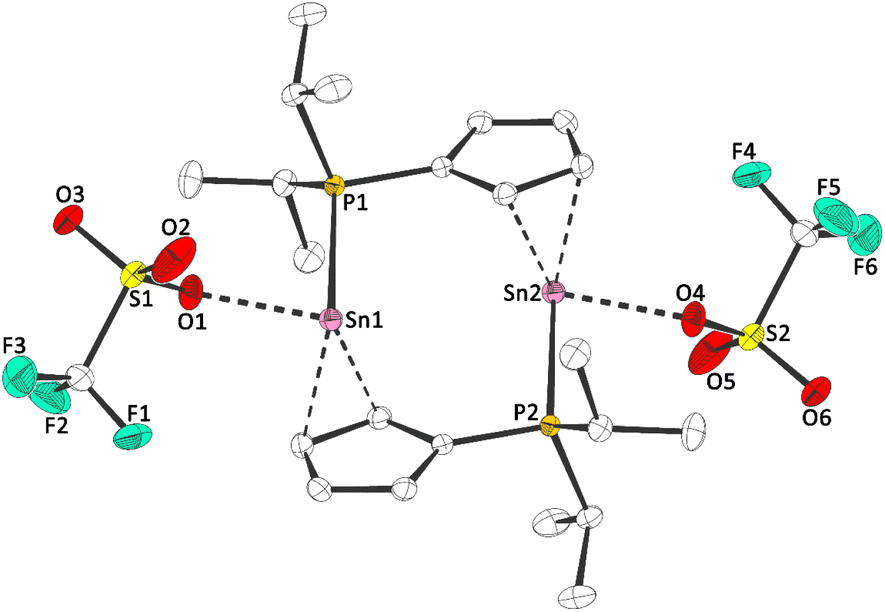 | ||
| Fig. 3 Molecular structure of 1b in the crystal (displacement ellipsoids at 50% probability level, hydrogen atoms omitted for clarity). Selected bond lengths [pm]: Sn–O: 233.48(14); Sn–P: 279.58(4). | ||
The Sn–Cl bond length in 1a is 247.54(12) pm, which is considerably shorter than in CpSnCl (324 to 326 pm16), but this is related to the different structure of CpSnCl in the solid-state. Noteworthy, the 31P NMR chemical shifts in solution are very similar to the ones observed in the solid-state (δ31P(1a)(CD2Cl2) = 3.6 and δ31P(1b)(CD2Cl2) = 19.8), suggesting the dimeric structures may be persistent in solution. Dimeric/oligomeric structures are not uncommon for tin(II) half-sandwich halides in the solid state, although CpSnCl forms a chloro-bridged polymer.16 To investigate the dimerization energy of 1a, we conducted DFT calculations (Fig. 4).
 | ||
| Fig. 4 Relative energy differences between monomeric 1a, related chloro-bridged dimer, and P–Sn dimer, 1a, calculated at B3LYP-D3/def2-SVP (energies in kJ mol−1). | ||
These calculations clearly suggest that a head-to-tail dimer with dative Sn–P bonds is preferred over a chloro-bridged structure and that the overall dimerization energy is 167.7 kJ mol−1. This is in line with the assumption that 1a and 1b exist predominantly as dimers in solution at room temperature.
Inspection of the frontier orbitals reveals that the LUMO has a large coefficient at the tin atom in the shape of a p orbital, which is typical for stannylene-type compounds. The HOMO corresponds to the lone-pair at the phosphorous atom, in-line with the compounds Lewis amphiphilicity and its head-to-tail dimerization, while the lone-pair at the tin atom is lower in energy and corresponds to the HOMO-3 (Fig. S25†).17
As many phosphines and tetrylenes are potent ligands for metal fragments and the latter can undergo oxidative addition reactions, we probed the reactivity of 1a towards metal halides and reacted it with palladium dichloride and platinum dichloride (Scheme 3). In both cases, an oxidative addition reaction at the low valent tin centres takes place and the corresponding stannylide complexes 2a,b were obtained. Storage of solutions of 2a,b at 253 K afforded red (2a) and orange (2b) crystals, suitable for single crystal X-ray diffraction analysis. Both compounds crystallize in the monoclinic space group C2/c with half a formula unit ((iPr2PC5H4)SnCl2M0.5) per asymmetric unit (Fig. 5). The structures show two stannylide ligands per metal centre coordinated in a bidentate square-planar fashion with Sn–[M] bond lengths of 256.34(4) pm ([M] = Pd)/257.02(4) pm ([M] = Pt) and P–[M] bond length of 233.48(4) pm ([M] = Pd)/232.58(10) pm ([M] = Pt). This is in-line with bond lengths found in related complexes ([ClPd(SnCl2Ph)(PPh2Py)2]:18 252.49(3) pm; [Pt(Sn(CH3)2(μ-C2H4)(PPh2))2]:19 259.73(5) to 260.47(5) pm). Two short and three long C–C bonds within the Cp rings in 2a,b indicate a diene-type situation, with η1/σ bound tin atoms. The coordination geometry of the group 10 metal is distorted square planar (2a: Sn–Pd–P: 82.2°, 97.7°; 2b: Sn–Pt–P: 82.1°, 97.8°).
 | ||
| Scheme 3 Synthesis of 2a,b by the reaction of 1a with (COD)PdCl2 and PtCl2. | ||
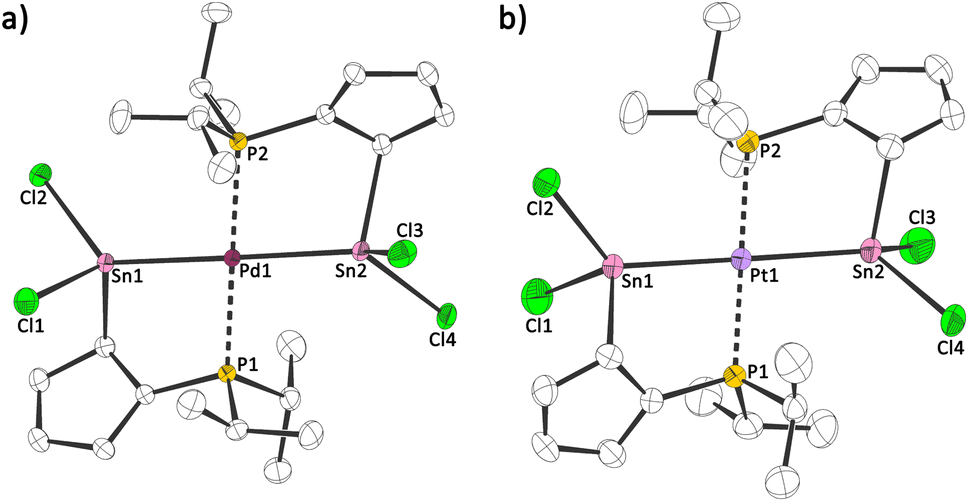 | ||
| Fig. 5 Molecular structures of (a) 2a and (b) 2b in the crystal (displacement ellipsoids at 50% probability level, hydrogen atoms omitted for clarity). Selected bond lengths [pm] and angles [°]: (a): Sn–Pd: 256.34(1); P–Pd: 233.48(1); Sn–Pd–P: 82.185(1); Sn–Pd–P: 97.735(1); (b): Sn–Pt: 257.02(1); P–Pt: 232.58(1); Sn–Pd–P: 82.131(1); Sn–Pd–P: 97.824(1). | ||
With the aim of increasing the Lewis acidity of the tin centre, we explored the possibility to generate a (diisopropylphosphanyl)-cyclopentadienyl tin cation. However, treatment of 1a with lithium and sodium salts of weakly coordinating anions (e.g. lithium hexafluorophosphate and sodium tetrakis-(pentafluorophenyl)borate) to abstract the chloride and obtain the corresponding stannyliumylidene were unsuccessful and led to complex mixtures of products. We therefore investigated the possibility of using a silver tetrakis(perfluoro-tert-butoxy)aluminate salt as chloride abstraction agent. This strategy was reported to be successful for the generation of a cyclopentadienyltin cation, by Krossing et al.20 Thus, we reacted 1a with silver tetrakis(perfluoro-tert-butoxy)aluminate in dichloromethane and indeed a chloride abstraction and precipitation of silver chloride was observed, along with the arise of a new resonance in the 119Sn{1H} NMR spectrum at δ119Sn NMR = −2122 (Scheme 4).
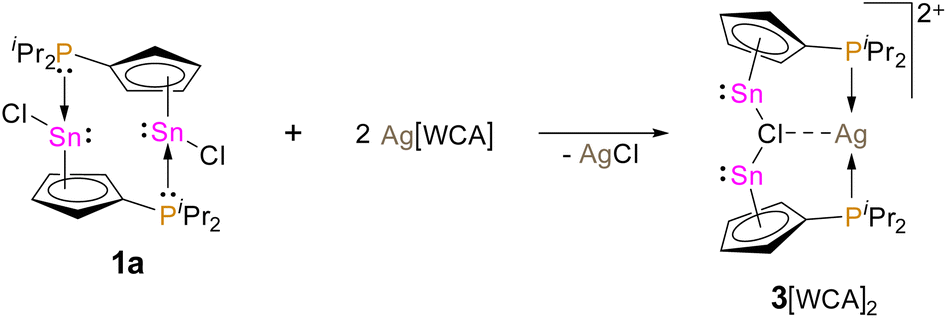 | ||
| Scheme 4 Synthesis of 3[WCA]2 by the reaction of 1a with Ag[WCA] (WCA = Al(OC(CF3)3)4). | ||
Storage of the reaction solution at 253 K resulted in the formation of colorless crystals suitable for single crystal X-ray diffraction. Most interesting, the solid-state structure did not reveal the targeted cation, but a complex with two (diisopropylphosphanyl)cyclopentadienyl tin cation units complexing a silver chloride moiety (Fig. 6). The Sn–Cp bonding in silver complex 3 can best be described as distorted η5, with relatively uniform C–C bond lengths within the Cp ring (138.81(13) to 143.69(74) pm), indicating a certain degree of aromaticity. The Sn–Cl bonds measure 283.66(18) and 285.26(19) pm which is significantly longer than in 1a (247.54(2) pm), in-line with the Sn–Cl–Sn multi-centre bond, but shorter than Sn–Cl contacts in CpSnCl (324 to 326 pm (ref. 7a)). The P–Ag bonds in 3 are 238.49(13) and 238.57(15) pm, which is slightly shorter than in comparable complexes ([(Ph3P)2AgCl]:16 246.7(2)/247.2(2) pm; [(Ph3P)3AgCl]:21 252.0(1)/255.2(1)/255.6(1) pm). Most interestingly, the Ag–Cl distance in 3 measures 285.03(17) pm, which is significantly longer than what is found in “traditional” silver chloride phosphine complexes ([(Ph3P)3AgCl]:21 255.2(1) pm; [(Ph3P)2AgCl]:16 259.6(2) pm), indicating a rather weak Ag–Cl bonding interaction. This elongation results from the unique bonding motive in 3, in which the chloride atom exhibits a formal coordination number of three, as it is bonded to the two tin atoms in addition to the silver centre, giving it a distorted trigonal planar geometry (Ag–Cl–Sn: 100.7°, 103.3°; Sn–Cl–Sn: 151.1°). Similarly, the silver atom also exhibits a distorted trigonal planar coordination environment (P–Ag–Cl: 98.8°, 96.3°; P–Ag–P: 164.8°). The structure of 3 is maintained in solution, as the 31P{1H} NMR spectrum shows two doublet which arises from coupling with the 107Ag and 109Ag nuclei (1J31P107Ag = 471 Hz; 1J31P109Ag = 544 Hz). However, prolong stirring of a solution of 3[WCA]2 in benzene-d6 led to decomposition.
 | ||
| Fig. 6 Molecular structure of 3 in the crystal (displacement ellipsoids at 50% probability level, hydrogen atoms and anions omitted for clarity). Selected bond lengths [pm]: Sn–Cl: 283.66(18)/285.26(19); P–Ag: 238.49(13)/238.57(15); Ag–Cl: 285.03(17); Sn–Cpcentriod: 223.51(5)/224.50(4). | ||
Conclusions
We were able to synthesize the first phosphanyl substituted tin(II) half-sandwich complexes, 1a,b, which properties differ significantly from previously reported CpSnCl. In the solid state, head-to-tail dimers with Sn–P bonds are observed, which are maintained in solution. Transition metal complexes 2a and 2b could be synthesized by oxidative addition reactions, which both exhibit a square planar geometry at the central transition metal atom in the solid state. By the reaction of 1a with silver tetrakis(perfluoro-tert-butoxy)aluminate, a cationic silver chloride complex, 3, with two tin half-sandwich moieties was obtained.Conflicts of interest
There are no conflicts to declare.Acknowledgements
Aylin Feuerstein is thanked for proof reading. Susanne Harling is thanked for elemental analysis. Linden ChroMasSpec GmbH is thanked for TOF MS LIFDI + measurements. Instrumentation and technical assistance for this work were provided by the Service Center X-ray Diffraction, with financial support from Saarland University and the Deutsche Forschungsgemeinschaft (INST 256/506-1). Funding by the Deutsche Forschungsgemeinschaft (Emmy Noether program, SCHA1915/3-1) is gratefully acknowledged. We acknowledge support by the Deutsche Forschungsgemeinschaft (DFG, German Research Foundation) and Saarland University within the ‘Open Access Publication Funding’ program.Notes and references
- A. Togni and R. L. Halterman, Metallocenes: Synthesis Reactivity Applications, WILEY-VCH Verlag GmbH, 1998, DOI:10.1002/9783527619542.
- (a) M. A. Beswick, J. S. Palmer and D. S. Wright, Chem. Soc. Rev., 1998, 27, 225–232 RSC; (b) P. Jutzi and N. Burford, Chem. Rev., 1999, 99, 969–990 CrossRef CAS PubMed.
- K. D. Bos, E. J. Bulten and J. G. Noltes, J. Organomet. Chem., 1972, 39, C52–C54 CrossRef CAS.
- E. O. Fischer and H. Grubert, Z. Naturforsch., 1956, 11b, 423 CAS.
- (a) P. J. Davidson and M. F. Lappert, J. Chem. Soc., Chem. Commun., 1973, 317a RSC; (b) D. H. Harris and M. F. Lappert, J. Chem. Soc., Chem. Commun., 1974, 895–896 RSC; (c) M. Veith, Angew. Chem., Int. Ed., 1975, 14, 263–264 CrossRef.
- (a) D. R. Armstrong, M. A. Beswick, N. L. Cromhout, C. N. Harmer, D. Moncrieff, C. A. Russell, P. R. Raithby, A. Steiner, A. E. H. Wheatley and D. S. Wright, Organometallics, 1998, 17, 3176–3181 CrossRef CAS; (b) C. Müller, A. Stahlich, L. Wirtz, C. Gretsch, V. Huch and A. Schäfer, Inorg. Chem., 2018, 57, 8050–8053 CrossRef PubMed; (c) C. Müller, D. M. Andrada, I.-A. Bischoff, M. Zimmer, V. Huch, N. Steinbrück and A. Schäfer, Organometallics, 2019, 38, 1052–1061 CrossRef.
- (a) K. D. Bos, E. J. Bulten, J. G. Noltes and A. L. Spek, J. Organomet. Chem., 1975, 99, 71–77 CrossRef CAS; (b) S. P. Constantine, G. M. De Lima, P. B. Hitchcock, J. M. Keates, G. A. Lawless and I. Marziano, Organometallics, 1997, 16, 793–795 CrossRef CAS; (c) K. D. Bos, E. J. Bulten and J. G. Noltes, J. Organomet. Chem., 1974, 67, C13–C15 CrossRef CAS; (d) M. C. Cassani, M. J. Davies, P. B. Hitchcock and M. F. Lappert, Inorg. Chim. Acta, 2005, 358, 1595–1604 CrossRef CAS; (e) M. Veith, C. Mathur and V. Huch, Organometallics, 1996, 15, 2858–2859 CrossRef CAS; (f) M. Veith, C. Mathur, S. Mathur and V. Huch, Organometallics, 1997, 16, 1292–1299 CrossRef CAS; (g) E. Y. Njua, A. Steiner and L. Stahl, J. Organomet. Chem., 2011, 696, 3301–3306 CrossRef CAS; (h) M. A. Paver, D. S. Wright and D. Stalke, Angew. Chem., Int. Ed., 1993, 32, 428–429 CrossRef; (i) T. Ochiai and S. Inoue, RSC Adv., 2017, 7, 801–804 RSC; (j) O. T. Summerscales, J. C. Fettinger and P. P. Power, J. Am. Chem. Soc., 2011, 133, 11960–11963 CrossRef CAS PubMed; (k) C. P. Sindlinger, A. Stasch, H. F. Bettinger and L. Wesemann, Chem. Sci., 2015, 6, 4737–4751 RSC; (l) P. Jutzi, B. Hampel, K. Stroppel, C. Krüger, K. Angermund and P. Hofmann, Chem. Ber., 1985, 118, 2789–2797 CrossRef CAS.
- (a) B. Rhodes, J. C. W. Chien and M. D. Rausch, Organometallics, 1998, 17, 1931–1933 CrossRef CAS; (b) G. M. de Lima, D. J. Duncalf and S. P. Constantine, Main Group Met. Chem., 2001, 24, 675–680 CAS; (c) G. M. de Lima, D. J. Duncalf and S. P. Constantine, Main Group Met. Chem., 2002, 25, 567–570 CAS; (d) J. N. Jones, J. A. Moore, A. H. Cowley and C. L. B. Macdonald, Dalton Trans., 2005, 3846–3851 RSC; (e) J. Beckmann, A. Duthie and M. Wiecko, Main Group Met. Chem., 2012, 35, 179–182 CAS; (f) M. Schleep, C. Hettich, J. Velázquez Rojas, D. Kratzert, T. Ludwig, K. Lieberth and I. Krossing, Angew. Chem., Int. Ed., 2017, 56, 2880–2884 CrossRef CAS PubMed; (g) Y. Ding, P. N. Ruth, R. Herbst-Irmer, D. Stalke, Z. Yang and H. W. Roesky, Dalton Trans., 2021, 50, 2067–2074 RSC; (h) P. Jutzi, F.-X. Kohl, E. Schlüter, M. B. Hursthouse and N. P. C. Walker, J. Organomet. Chem., 1984, 271, 393–402 CrossRef CAS; (i) P. Jutzi, F. Kohl, P. Hofmann, C. Krüger and Y.-H. Tsay, Chem. Ber., 1980, 113, 757–769 CrossRef CAS; (j) P. Jutzi, F. Kohl and C. Krüger, Angew. Chem., Int. Ed., 1979, 18, 59–60 CrossRef; (k) R. Hani and R. A. Geanangel, J. Organomet. Chem., 1985, 293, 197–205 CrossRef CAS; (l) P. Jutzi and R. Dickbreder, J. Organomet. Chem., 1989, 373, 301–306 CrossRef CAS; (m) F. X. Kohl, R. Dickbreder, P. Jutzi, G. Müller and B. Huber, Chem. Ber., 1989, 122, 871–878 CrossRef CAS.
- (a) Y. Mizuhata, T. Sasamori and N. Tokitoh, Chem. Rev., 2009, 109, 3479–3511 CrossRef CAS PubMed; (b) M. Asay, C. Jones and M. Driess, Chem. Rev., 2011, 111, 354–396 CrossRef CAS PubMed; (c) R. Dasgupta and S. Khan, in Advances in Organometallic Chemistry, ed. P. J. Pérez, Academic Press, 2020, vol. 74, pp. 105–152 Search PubMed; (d) W. P. Neumann, Chem. Rev., 1991, 91, 311–334 CrossRef CAS.
- C. Müller, J. Warken, V. Huch, B. Morgenstern, I.-A. Bischoff, M. Zimmer and A. Schäfer, Chem. – Eur. J., 2021, 27, 6500–6510 CrossRef PubMed.
- (a) F. X. Kohl and P. Jutzi, Chem. Ber., 1981, 114, 488–494 CrossRef CAS; (b) G. M. de Lima, D. J. Duncalf and S. P. Constantine, Main Group Met. Chem., 2002, 25, 567–570 CAS.
- (a) FIA were calculated following the procedure, in H. Großekappenberg, M. Reißmann, M. Schmidtmann and T. Müller, Organometallics, 2015, 34, 4952–4958 (Organometallics, 2015, 34, 5496) CrossRef; (b) GEI were calculated following method C in A. R. Jupp, T. C. Johnstone and D. W. Stephan, Inorg. Chem., 2018, 57, 14764–14771 CrossRef CAS PubMed.
- (a) V. Gutmann, Coord. Chem. Rev., 1976, 18, 225–255 CrossRef CAS; (b) G. C. Welch, L. Cabrera, P. A. Chase, E. Hollink, J. D. Masuda, P. Wei and D. W. Stephan, Dalton Trans., 2007, 3407–3414 RSC.
- In solution, no signals could be detected in the 119Sn NMR spectra in a range between +2000 and −2500 ppm and temperatures between 243 K and 323 K, which is however also the case for CpSnCl, presumably due to extreme signal broadening7b.
- J. K. West and L. Stahl, Organometallics, 2012, 31, 2042–2052 CrossRef CAS.
- A. Cassel, Acta Crystallogr., Sect. B: Struct. Crystallogr. Cryst. Chem., 1979, 35, 174–177 CrossRef.
- See ESI† for further details.
- S. Warsink, E. J. Derrah, C. A. Boon, Y. Cabon, J. J. M. de Pater, M. Lutz, R. J. M. Klein Gebbink and B.-J. Deelman, Chem. – Eur. J., 2015, 21, 1765–1779 CrossRef CAS PubMed.
- U. Baumeister, H. Hartung, T. Schulz and H. Weichmann, Acta Crystallogr., Sect. C: Cryst. Struct. Commun., 1998, 54, 333–335 CrossRef.
- M. Schleep, C. Hettich, J. Velázquez Rojas, D. Kratzert, T. Ludwig, K. Lieberth and I. Krossing, Angew. Chem., Int. Ed., 2017, 56, 2880–2884 CrossRef CAS PubMed.
- A. Cassel, Acta Crystallogr., Sect. B: Struct. Crystallogr. Cryst. Chem., 1981, 37, 229–231 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental details, NMR spectra, MS spectra, XRD data, DFT data, references. CCDC 2242130, 2242131, 2242132, 2242136 and 2242137. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d3ra01384g |
| This journal is © The Royal Society of Chemistry 2023 |