DOI:
10.1039/D3RA01589K
(Paper)
RSC Adv., 2023,
13, 10408-10413
Sinulariaone A: a novel diterpenoid with a 13-membered carbocyclic skeleton from an octocoral Sinularia species†
Received
10th March 2023
, Accepted 26th March 2023
First published on 3rd April 2023
Abstract
Chemical composition screening of an octocoral identified as Sinularia species led to the isolation of a novel diterpenoid, sinulariaone A (1), featuring a 13-membered carbocyclic skeleton. The structure of 1 was established by spectroscopic elucidation, computed calculation, and X-ray diffraction analysis. Moreover, a single-crystal X-ray diffraction analysis of chlorofurancembranoid B (2), obtained in our previous study from the same octocoral species, was reported for the first time to demonstrate the absolute configuration. Diterpenoid 1 showed cytotoxicity towards human promyelocytic leukemia HL-60 cells, with an IC50 value of 38.01 μM.
1 Introduction
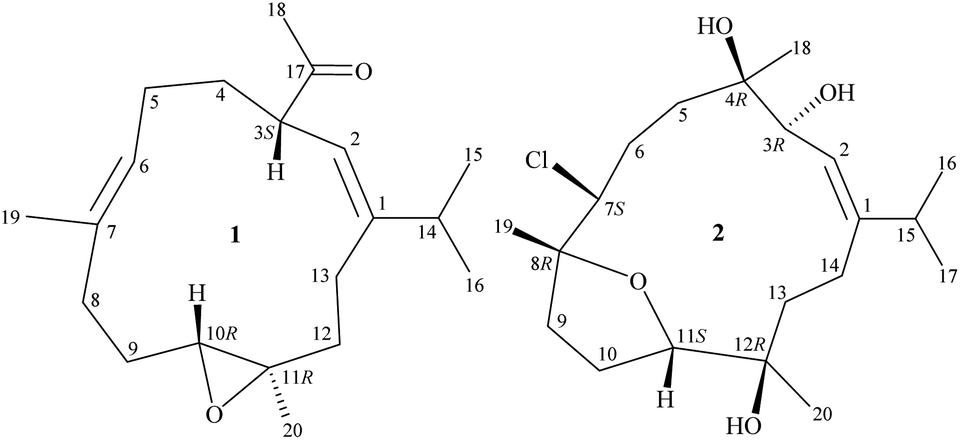
Octocorals of the genus Sinularia (phylum Cnidaria, sub-phylum Anthozoa, class Octocorallia, order Malacalcyonacea, family Sinulariidae)1 are one of the most common marine invertebrates natively distributed throughout tropical and subtropical regions of the Indo-Pacific Ocean. Despite their ecological importance, the secondary metabolites, in particular terpenoid derivatives from these organisms were proven to have potential for biomedical uses.2–4 In this research, we completed the preparation, structural identification, and cytotoxicity assessment of sinulariaone A (1), a diterpenoid featuring with a rare 13-membered carbocyclic skeleton and chlorofurancembranoid B (2)5 (Fig. 1), from an octocoral identified as Sinularia sp., collected from the waters of Taiwan, an area with high biodiversity at the intersection of the Kuroshio current, South China Sea surface current, and Mainland Coastal current.
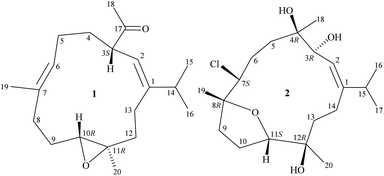 |
| | Fig. 1 Structures of sinulariaone A (1) and chlorofurancembranoid B (2). | |
2 Results and discussion
Sinulariaone A (1) was obtained as colorless prisms with the molecular formula determined to be C20H32O2 by (+)-HRESIMS at m/z 327.22928 (calcd for C20H32O2 + Na, 327.22945), corresponding to five double-bond equivalents (DBEs). The IR spectrum of 1 showed a strong absorption at νmax 1716 cm−1, consistent with a ketone moiety in the structure. The 13C spectrum (Table 1), in combination with the DEPT and HSQC spectrum, showed signals of 20 carbons, including a ketonic carbonyl (δC 210.0, C-17), four olefinic carbons (δC 149.8, C-1; 121.6, CH-2; 127.4, CH-6; 134.8, C-7), and two oxygenated carbons (δC 62.6, CH-10; 61.3, C-11), as well as five methyls, six aliphatic sp3 methylenes, and two aliphatic sp3 methines.
Table 1 1H and 13C NMR spectroscopic data of sinulariaone A (1)
| Position |
δHa (J in Hz) |
δCb, Mult.c |
| Spectra recorded at 400 MHz in CDCl3 at 25 °C. Spectra recorded at 100 MHz in CDCl3 at 25 °C. Multiplicity deduced by DEPT and HSQC spectrum and indicated by usual symbols. Signals overlapped. Signals overlapped. Signals overlapped. Signals overlapped. The coupling constants for H-9 were assigned by its geminal coupling with H-9′ and vicinal couplings with H-8 and H-10, respectively. |
| 1 |
|
149.8, C |
| 2 |
5.06 d (10.0) |
121.6, CH |
| 3 |
3.16 ddd (10.0, 10.0, 2.0) |
49.9, CH |
| 4 |
2.23 m |
31.5, CH2 |
| 4′ |
1.04 m |
|
| 5 |
1.97–2.05 md |
25.6, CH2 |
| 6 |
5.13 dd (6.8, 6.8) |
127.4, CH |
| 7 |
|
134.8, C |
| 8 |
2.36 ddd (12.4, 5.2, 4.4) |
36.7, CH2 |
| 8′ |
2.18 dd (12.4, 4.0) |
|
| 9 |
2.24 ddd (13.2, 5.2, 4.0)e,h |
24.4, CH2 |
| 9′ |
1.37 dddd (13.2, 10.0, 4.4, 4.0) |
|
| 10 |
2.74 dd (10.0, 4.0) |
62.6, CH |
| 11 |
|
61.3, C |
| 12α |
2.26 me |
40.7, CH2 |
| β |
1.07 m |
|
| 13 |
1.97–2.05 md |
25.2, CH2 |
| 14 |
2.25 me |
35.4, CH |
| 15 |
0.99 d (6.8)f |
21.9, CH3g |
| 16 |
0.99 d (6.8)f |
21.9, CH3g |
| 17 |
|
210.0, C |
| 18 |
2.06 s |
29.4, CH3 |
| 19 |
1.62 br s |
14.5, CH3 |
| 20 |
1.30 s |
16.0, CH3 |
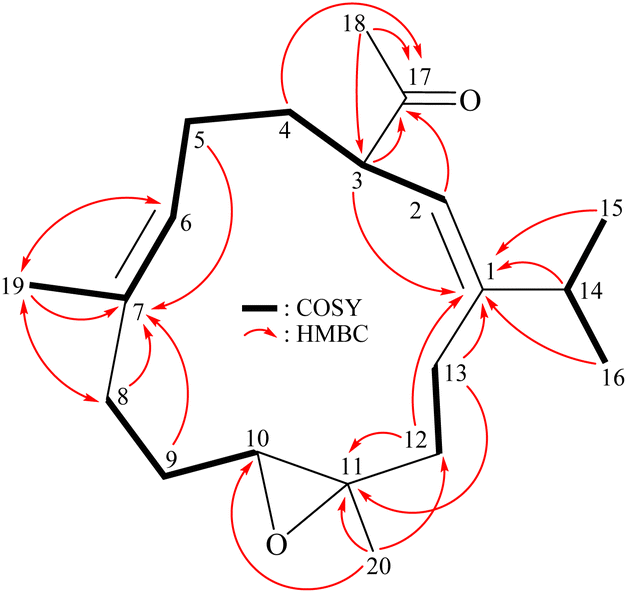
Analysis of 1H (Table 1), 13C, and HSQC spectra illustrated that 1 contained two trisubstituted carbon–carbon double bonds (δH 5.13, 1H, dd, J = 6.8, 6.8 Hz/δC 127.4, CH-6; δC 134.8, C-7; δH 5.06, 1H, d, J = 10.0 Hz/δC 121.6, CH-2; δC 149.8, C-1) and an acetyl group (δH 2.06, 3H, s/δC 29.4, CH3-18; δC 210.0, C-17). The 3J-proton-proton coupling information in the COSY spectrum led to the assignment of four continuous spin systems from H-2/H-3/H2-4/H2-5/H-6, H2-8/H2-9/H-10, H2-12/H2-13, and H-14/H3-15 (H3-16) (Fig. 2). The HMBC spectrum showed 2J- and 3J-heteronuclear correlations from neighbor protons to the non-protonated carbons such as H-3, H2-12, H2-13, H-14, H3-15, H3-16/C-1; H2-5, H2-8, H2-9, H3-19/C-7; H2-12, H2-13, H3-20/C-11; and H-2, H-3, H2-4, H3-18/C-17 (Fig. 2), confirming the presence of central 13-membered carbon macrocyclic ring system.6–10 The HMBC correlations from H3-20/C-10, C-11, and C-12 indicated that Me-20 was placed at C-11. The presence of a vinyl methyl (Me-19) at C-7 was substantiated by the HMBC correlations from H-6, H-8′ (δH 2.18)/C-19 and H3-19/C-6, C-7, C-8, and further confirmed by a long-range allylic coupling between H-6/H3-19 (Fig. 2). The presence of an isopropyl group at C-1 was substantiated by the HMBC correlations from H-14, H3-15, H3-16 to C-1. The above analysis enabled the establishment of the carbon skeleton of 1. A trisubstituted epoxide containing a methyl substituent in 1 was established from the signals of an oxygenated quaternary carbon at δC 61.3 (C-11) and an oxymethine (δH 2.74, 1H, dd, J = 10.0, 4.0 Hz/δC 62.6, CH-10), and from the proton signal of a methyl at δH 1.30 (3H, s, H3-20). An acetyl group at C-3 was confirmed by the HMBC correlations from the methine proton at δH 3.16 (H-3) to the ketonic carbonyl at δC 210.0 (C-17); the other HMBC correlations from the methyl protons resonating at δH 2.06 (H3-18) to C-17 ketonic carbonyl (δC 210.0) and C-3 methine (δC 49.9), further supporting that this group was positioned at C-3.
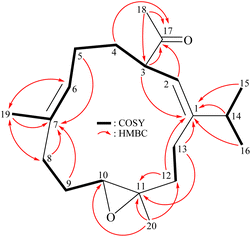 |
| | Fig. 2 Key COSY and HMBC correlations of 1. | |
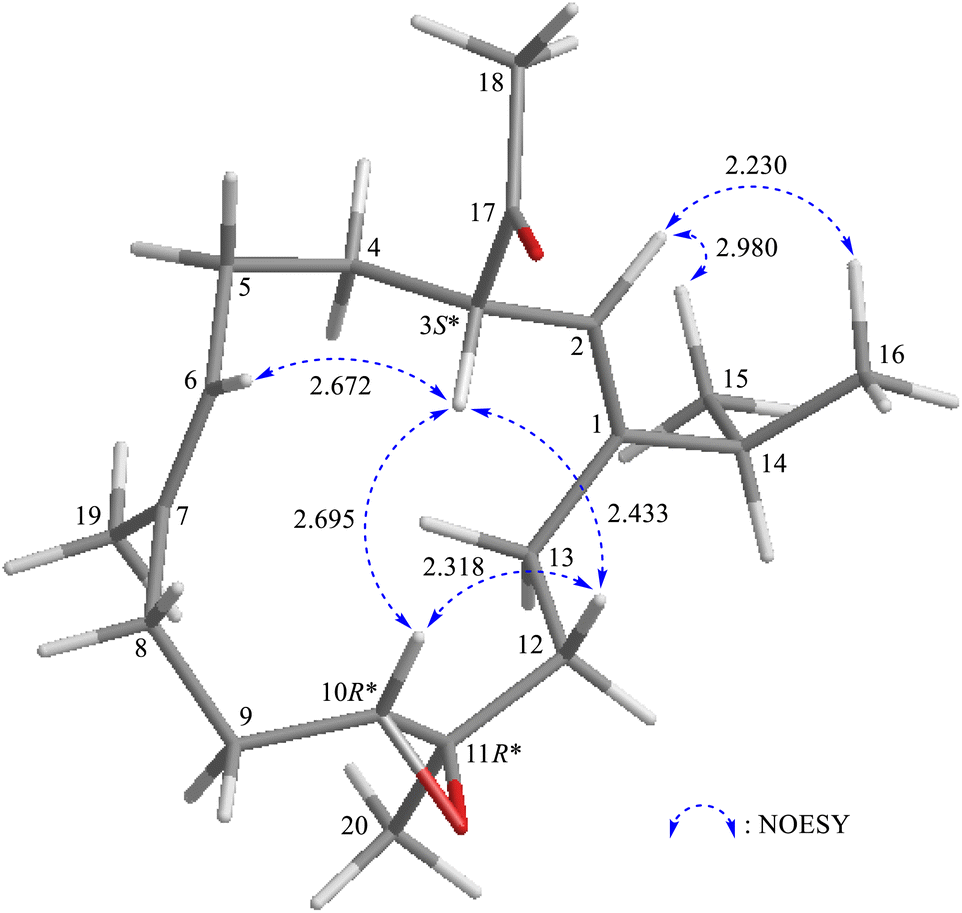
The relative stereochemistry of 1 was determined based on correlations obtained from NOESY experiments. In the NOESY spectrum (Fig. 3), H-10 exhibited cross-peaks with H-3 and one of the diastereotopic methylene protons at C-12 (δH 1.07, H-12β); and H-12β was correlated with H-3 but not with H3-20, which illustrated the β-orientations of H-3 and H-10, and the α-orientation of Me-20. The Z-form of Δ1 and Δ6 was confirmed by NOESY correlations between H-2 (olefin proton)/H3-15 (H3-16); and H-6 (olefin proton)/H-3, respectively, and there were no NOESY correlations were found between H-6/H3-19 (vinyl methyl) and H-2/H2-13.
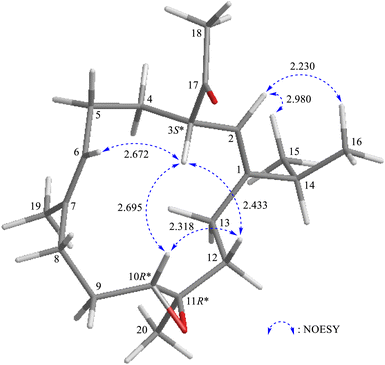 |
| | Fig. 3 Stereo-view of 1 (generated by computer modeling) and calculated distances (Å) between selected protons with key NOESY correlations. | |
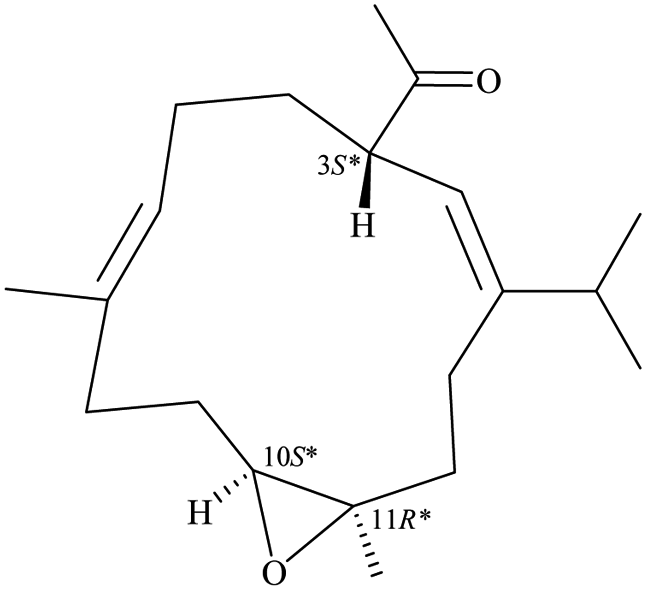
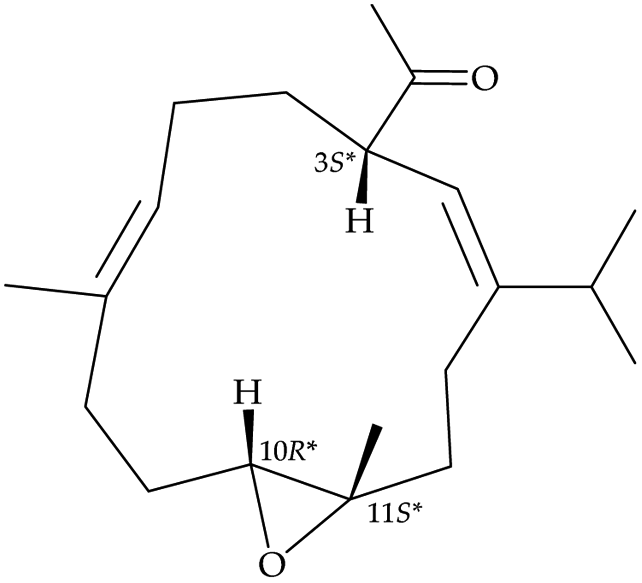
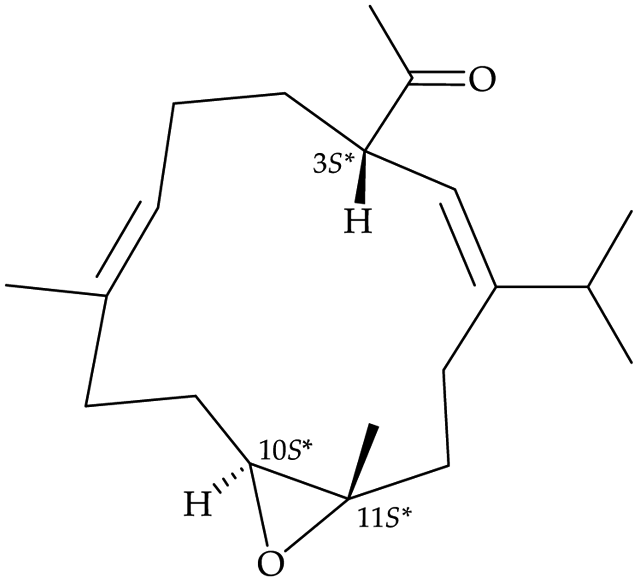
After the program of the above analysis, the gross structure of 1 displayed four possible relative configurations, including 1-3S*, 10R*, 11R*; 1-3S*, 10S*, 11R*; 1-3S*, 10R*, 11S*; and 1-3S*, 10S*,11S*. The four possible relative configurations were inputted into Spartan’16 and optimized at the MMFF94 level.11–13 The predicted distance of key NOESY of possible configurations is shown in Table 2. The 1-3S*, 10R*, 11R* displayed the best result matching the experimental key NOESY correlations, and the calculated single optical rotation (SOR) value of 1-3S, 10R, 11R and 1-3R, 10S, 11S were +226 and −226, respectively. Comparing the calculated with the experimental SOR value of 1 (+236), the absolute configuration of 1 could be assigned as 3S, 10R, 11R.
Table 2 The predicted distance (Å) of key NOESY of optimized top 3 possible relative configurations of 1
| |
3S*, 10R*, 11R* |
3S*, 10S*, 11R* |
| Structures |
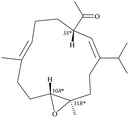 |
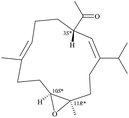 |
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) |
| |
Top 1 |
| H-3/H-6 |
2.775 |
2.688 |
| H-3/H-10 |
2.404 |
4.931 |
| H-3/H-12 |
2.468 |
2.479 |
|
| |
Top 2 |
| H-3/H-6 |
2.742 |
2.740 |
| H-3/H-10 |
2.430 |
4.857 |
| H-3/H-12 |
2.565 |
2.308 |
|
| |
Top 3 |
| H-3/H-6 |
2.636 |
2.517 |
| H-3/H-10 |
2.409 |
5.071 |
| H-3/H-12 |
2.411 |
2.479 |
| |
3S*, 10R*, 11S* |
3S*, 10S*, 11S* |
| Structures |
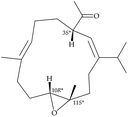 |
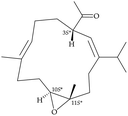 |
|
| |
Top 1 |
| H-3/H-6 |
2.597 |
3.197 |
| H-3/H-10 |
3.235 |
3.896 |
| H-3/H-12 |
4.123 |
2.346 |
|
| |
Top 2 |
| H-3/H-6 |
2.636 |
3.199 |
| H-3/H-10 |
3.138 |
3.839 |
| H-3/H-12 |
4.161 |
3.829 |
|
| |
Top 3 |
| H-3/H-6 |
2.604 |
3.269 |
| H-3/H-10 |
3.237 |
3.935 |
| H-3/H-12 |
4.229 |
3.220 |
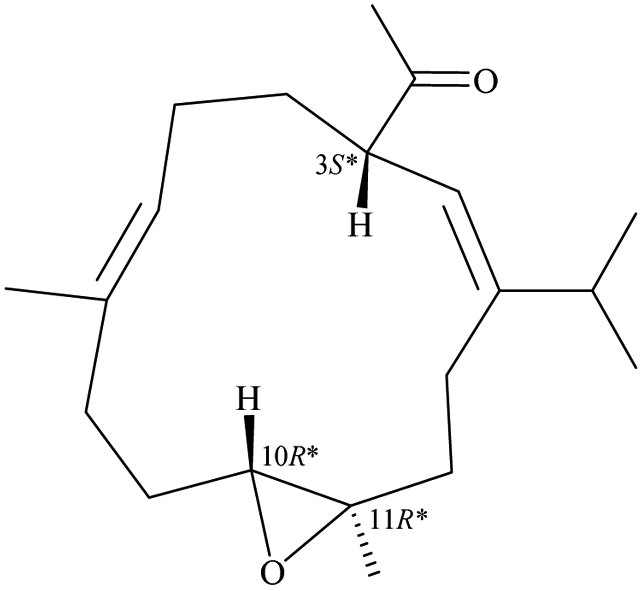
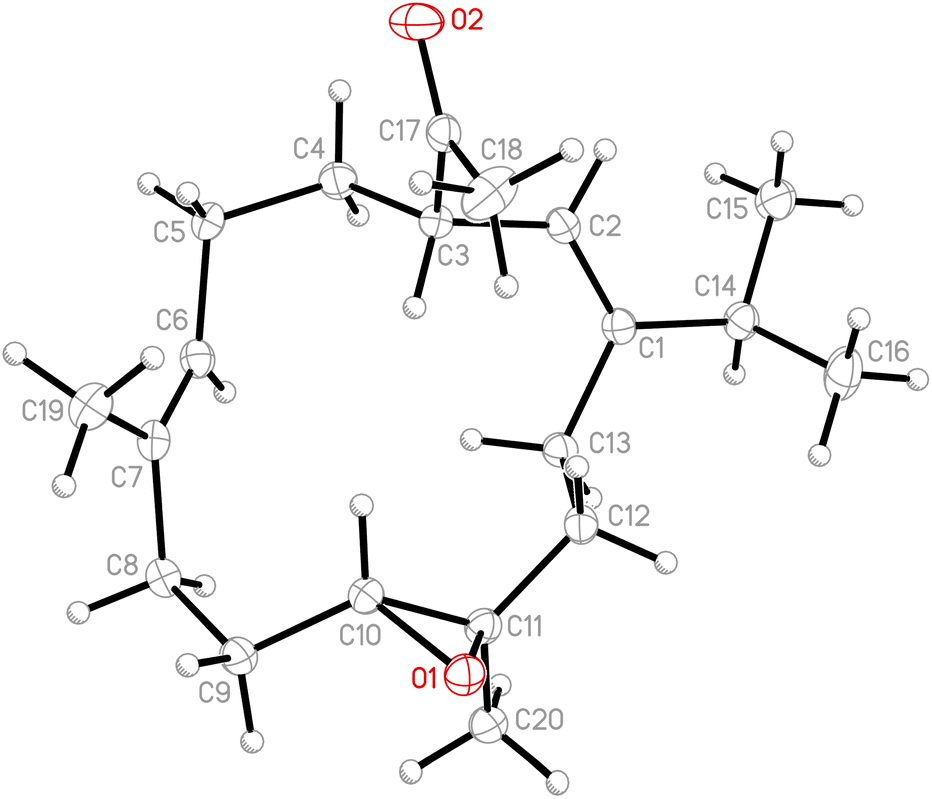
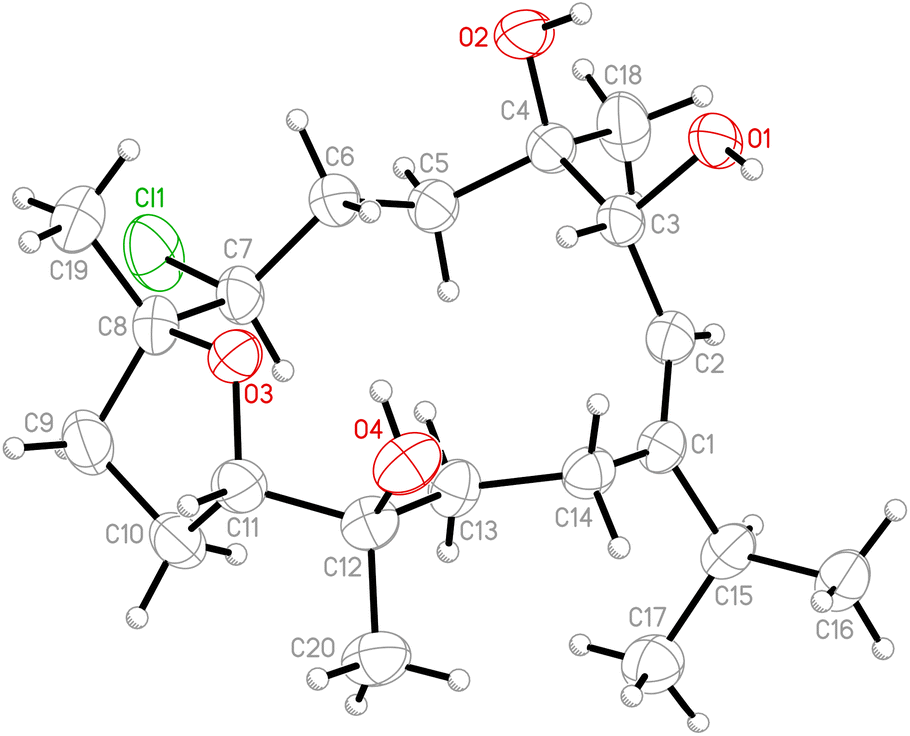
Due to the conformational mobility of the macrocycle, the stereochemistry of the stereogenic centers C-3, C-10, and C-11 of 1 would be further determined from an X-ray diffraction analysis. Regarding validation of the structure of 1, a single-crystal X-ray diffraction analysis was employed. The structure of 1 was fully established by X-ray crystallography, as observed by Cu Kα radiation (λ = 1.54178 Å) and the Flack parameter x = 0.0(3).14,15 The X-ray structure (Fig. 4) demonstrates the location of an acetyl group at C-3 and an epoxy group between C-10/11 in the 13-membered macrocycle ring. Based on the X-ray diffraction analysis, the stereogenic centers in 1 were assigned as 3S, 10R, 11R. From the above findings, the structure, including the absolute configuration, of 1 was therefore elucidated unambiguously.
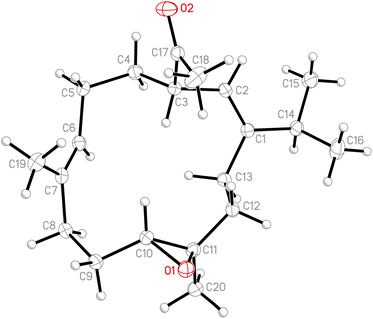 |
| | Fig. 4 The computer-generated ORTEP diagram of 1. | |
Chlorofurancembranoid B (2), a cytotoxic cembranoid toward human promyelocytic leukemia HL-60 cells, was reported in our previous publication, and its stereochemistry was established by combination of a NOESY experiment.5 Thus, in order to determine the absolute configuration. This compound has been crystallized, and the diffraction experiment was carried out with a diffractometer equipped with molybdenum radiation (Mo Kα, λ = 0.71073 Å) source. The ORTEP diagram (Fig. 5) showed the absolute configuration for all stereogenic centers were assigned as 3R, 4R, 7S, 8R, 11S, 12R.
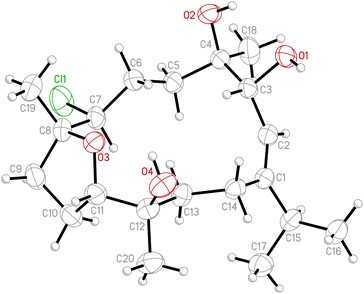 |
| | Fig. 5 The computer-generated ORTEP diagram of 2. | |
The cytotoxicity of 1 against cancer cells HL-60 and HepG2 (human hepatoma cell line) were investigated. The assay used in this study was performed as described in previous publications.16,17 The results are shown in Table 3. According to the outcomes of cytotoxic assays, diterpenoid 1 showed cytotoxicity towards human promyelocytic leukemia HL-60 cells, with an IC50 value of 38.01 μM.
Table 3 Effects of compound 1 on cell viability in HL-60 and HepG2 tumor cells
| Compound |
HL-60 |
HepG2 |
| Cell viabilitya (%) |
IC50b (μM) |
Cell viabilitya (%) |
IC50b (μM) |
| Cell viability at 50 μM for 48 h. Results are expressed as mean ± SEM (n = 3). ***p < 0.001 compared with DMSO alone. Concentration necessary for 50% inhibition (IC50). |
| 1 |
42.42 ± 0.70*** |
38.01 ± 1.21 |
71.76 ± 2.90 |
> 50 |
| DMSO |
100.00 ± 2.16 |
|
100.00 ± 0.72 |
|
3 Conclusions
In this study, the chemical composition of an octocoral identified as Sinularia sp. was screened, resulted in the isolation of a novel diterpenoid, sinulariaone A (1). It is to note that diterpenoid 1, involving an uncommon 13-membered carbocyclic carbon system, which was suggested biosynthesized from the common 14-membered carbocyclic cembrane analogues by ring contraction,6,7 however, to be one of a kind, this is the first time to obtain a 13-membered carbocyclic cembranolide analogue featuring with an acetyl group at C-3. The structure of 1, including the absolute configuration, was determined by spectroscopic methods and further confirmed by a single-crystal X-ray diffraction analysis and this compound showed cytotoxicity toward the HL-60 tumor cells. In addition, the absolute configuration of a known cytotoxic cembranoid, chlorofurancembranoid B (2), was determined using a single-crystal X-ray diffraction analysis with the molybdenum radiation source, with the material obtained in previous study.5
4 Experimental
4.1 General experimental procedures
Optical rotation values were measured using a JASCO P-1010 digital polarimeter. IR spectra were obtained with a Thermo Scientific Nicolet iS5 FT-IR spectrophotometer. NMR spectra were recorded on a 400 MHz Jeol ECZ NMR spectrometer using the residual CHCl3 (δH 7.26 ppm) and CDCl3 signals (δC 77.0 ppm) as internal standards for 1H and 13C NMR, respectively; coupling constants (J) are presented in Hz. ESIMS and HRESIMS were recorded using a Bruker 7 Tesla solariX FTMS system. Column chromatography was carried out with silica gel (230–400 mesh, Merck). TLC was performed on plates precoated with silica gel 60 F254 (Merck) and RP-18W/UV254 (0.15 mm-thick, Macherey-Nagel), then sprayed with 10% H2SO4 solution followed by heating to visualize the spots.
4.2 Animal material
Specimens of Sinularia sp. were collected on Turtle Island, Yilan County, Taiwan. The samples were stored in a freezer at −20 °C until extraction. A voucher specimen was deposited in the National Museum of Marine Biology & Aquarium, Taiwan (NMMBA-TW–SC–2018-0619). Identification of this organism was performed by comparison with previous descriptions.1,18
4.3 Extraction and isolation
Freeze-dried and sliced bodies (wet/dry weight = 510/172 g) of the coral specimens were extracted with a mixture of MeOH/CH2Cl2 (1:1) to give 17.8 g of crude extract, which was partitioned between EtOAc and H2O. The EtOAc extract (6.8 g) was subjected to silica gel column chromatography (Si C. C.) and eluted with gradients of n-hexane/EtOAc (100% n-hexane–100% EtOAc, stepwise) to furnish 14 sub-fractions A–N. Fraction D was chromatographed by Si C. C. and eluted with a mixture of CH2Cl2/EtOAc (20:1) to obtain 18 sub-fractions D1–D18. Fraction D13 was further separated by Si C. C. and eluted with CH2Cl2 to afford 1 (3.5 mg).
4.4 Structural characterization of undescribed compound
4.4.1 Sinulariaone A (1). Colorless prisms (MeOH); mp 102–104 °C; [α] + 236 (c 0.05, CHCl3); IR (KBr) νmax 1716 cm−1; 1H (400 MHz, CDCl3) and 13C (100 MHz, CDCl3) NMR data (see Table 1); ESIMS: m/z 327 [M + Na]+; HRESIMS m/z 327.22928 (calcd for C20H32O2 + Na, 327.22945).
4.5 Single-crystal X-ray crystallography of sinulariaone A (1)
Suitable colorless prisms of 1 were obtained from a solution of MeOH. The crystal (0.600 × 0.484 × 0.138 mm3) was identified as being of the orthorhombic system, space group P212121 (#19), with a = 9.0173(3) Å, b = 10.8659(3) Å, c = 18.6542(6) Å, V = 1827.76(10) Å3, Z = 4, Dcalcd = 1.106 Mg m−3 and λ (Cu Kα) = 1.54178 Å. Intensity data were obtained on a crystal diffractometer (Bruker, model: D8 Venture) up to a θmax of 69.999°. All measurement data of 18147 reflections were collected, of which 3472 were independent. The structure was solved by direct methods and refined by a full-matrix least-squares on F2 procedure.19,20 The refined structural model converged to a final R1 = 0.0601; wR2 = 0.1637 for 3095 observed reflections [I > 2σ(I)] and 199 variable parameters; and the absolute configuration was established from the Flack parameter x = 0.0(3).14,15 Crystallographic data for the structure of sinulariaone A (1) were submitted to the Cambridge Crystallographic Data Center (CCDC) with supplementary publication number CCDC 2226689 (data can be obtained from the CCDC website at https://www.ccdc.cam.ac.uk/conts/retrieving.html).
4.6 Single-crystal X-ray crystallography of chlorofurancembranoid B (2)
Suitable colorless prisms of 2 were obtained from a solution of MeOH. The crystal (0.388 × 0.145 × 0.028 mm3) was identified as being of the triclinic system, space group P1 (#1), with a = 10.1046(4) Å, b = 10.4180(3) Å, c = 10.9661(4) Å, V = 1066.93(7) Å3, Z = 2, Dcalcd = 1.167 Mg m−3 and λ (Mo Kα) = 0.71073 Å. Intensity data were obtained on a crystal diffractometer (Bruker, model: D8 Venture) up to a θmax of 29.998°. All measurement data of 38323 reflections were collected, of which 12414 were independent. The structure was solved by direct methods and refined by a full-matrix least-squares on F2 procedure.19,20 The refined structural model converged to a final R1 = 0.0561; wR2 = 0.1188 for 8865 observed reflections [I > 2σ(I)] and 468 variable parameters; and the absolute configuration was established from the Flack parameter x = −0.01(3).14,15 Crystallographic data for the structure of chlorofurancembranoid B (2) were deposited with the Cambridge Crystallographic Data Center (CCDC) as supplementary publication number CCDC 2208811 (data can be obtained from the CCDC website at https://www.ccdc.cam.ac.uk/conts/retrieving.html).
4.7 In silico calculations
The conformational search and calculated SOR results were carried out with the same method published as ref. 11–13. The brief procedure was described as follows. First, we optimized the minimized energy of the structure in the MM2 level and outputted an xyz file. Then, we submitted the file into spartan'16 software (Wavefunction Inc.; Irvine, CA, USA) at MMFF94 to generate conformational search results. The output data were imported into the Gaussian 09 software (Gaussian Inc.; Wallingford, CT, USA) and optimized using the time-dependent density functional theory (TDDFT) methodology at the B3LYP/6-31G* level in the gas phase and the B3LYP/6-31(d) levels in the solvent phase for SOR calculation, and the GIAO-DFT at the PCM/mpw1pw91/6-311 + g(d,p) level in the solvent phase for GIAO-NMR DP4+ analysis. The results were averaged by the proportion of each conformer.
4.8 In vitro cytotoxic assay
The cytotoxicity assay used in this study was performed as described in previous publications.16,17
Author contributions
Hsuan-Jung Tseng: investigation, analysis of results. Liang-Mou Kuo: investigation, analysis of results. Yu-Chi Tsai: investigation, software, modelling and simulation. Hao-Chun Hu: software, modelling and simulation. Po-Jen Chen: data curation, methodology. Su-Ying Chien: formal analysis, X-ray analysis. Jyh-Horng Sheu: conceptualization, supervision, visualization. Ping-Jyun Sung: analysis of results, conceptualization, visualization, supervision, writing-original draft, writing-reviewing and editing.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
The authors are grateful to Hsiao-Ching Yu and Chao-Lien Ho, of the High Valued Instrument Center, National Sun Yat-sen University, for obtaining the mass (MS 006500) and NMR (NMR 001100) spectra (NSTC 112-2740-M-110-002), and to the Instrumentation Center, National Taiwan University, for providing X-ray facilities (NSTC 112-2740-M-002-006, XRD 000200). This work was mainly funded by grants from the National Museum of Marine Biology & Aquarium, the National Science and Technology Council (MOST 109-2320-B-291-001-MY3, 111-2320-B-291-001, and 110-2314-B-242-003) and Chang Gung Memorial Hospital (CMRPG6L0311-3), Taiwan, awarded to L.-M. K. and P.-J. S. All funding is gratefully acknowledged.
Notes and references
- C. S. McFadden, L. P. van Ofwegen and A. M. Quattrini, Revisionary systematics of Octocorallia (Cnidaria: Anthozoa) guided by phylogenomics, Bull. Soc. Syst. Biol., 2022, 1, 8735 Search PubMed.
- H. V. N. Kamel and M. Slattery, Terpenoids of Sinularia: chemistry and biomedical applications, Pharm. Biol., 2005, 43, 253–269 CrossRef CAS.
- X. Yan, J. Liu, X. Leng and H. Ouyang, Chemical diversity and biological activity of secondary metabolites from soft coral genus Sinularia since 2013, Mar. Drugs, 2021, 19, 335 CrossRef CAS PubMed.
- N. B. A. Nguyen, L.-Y. Chen, M. El-Shazly, B.-R. Peng, J.-H. Su, H.-C. Wu, I.-T. Lee and K.-H. Lai, Towards sustainable medicinal resources through marine soft coral aquaculture: insights into the chemical diversity and the biological potential, Mar. Drugs, 2022, 20, 640 CrossRef CAS PubMed.
- H.-J. Tseng, L.-M. Kuo, P.-J. Chen, S.-H. Chen, C.-J. Liu, S.-Y. Chien, Y.-C. Tsai, Y.-J. Wu, T.-R. Su and P.-J. Sung, Chlorofurancembranoids A and B: novel cembranoids from octocoral Sinularia sp, Tetrahedron, 2022, 119, 132851 CrossRef CAS.
- B. F. Bowden, J. C. Coll, S. J. Mitchell and G. J. Stokie, Studies of Australian soft corals. VII Two new diterpenes from an unknown species of soft coral (genus Lobophytum), Aust. J. Chem., 1978, 31, 1303–1312 CrossRef CAS.
- Y. Yamada, S. Suzuki, K. Iguchi, K. Hosaka, H. Kikuchi, Y. Tsukitani, H. Horiai and F. Shibayama, Studies on marine natural products I. 13-Membered carbocyclic cembranolide diterpenes from the soft coral Lobophytum pauciflorum (Ehrenberg), Chem. Pharm. Bull., 1979, 27, 2394–2397 CrossRef CAS.
- K. Iguchi, M. Kitade, Y. Yamada, A. Ichikawa, I. Ohtani, T. Kusumi and H. Kakisawa, Stereostructures of unique 13-membered carbocyclic cembranolides from the soft coral Lobophytum pauciflorum, Chem. Lett., 1991, 20, 319–322 CrossRef.
- T. Iwagawa, Y. Shibata, H. Okamura, M. Nakatani and M. Shiro, Novel cembranoids with a 13-membered carbocyclic skeleton from a soft coral, Sarcophyton species, Tetrahedron Lett., 1994, 35, 8415–8416 CrossRef CAS.
- T. Iwagawa, S. Nakamura, T. Masuda, H. Okamura, M. Nakatani and M. Shiro, Irregular cembranoids containing a 13-membered carbocyclic skeleton isolated from a soft coral, Sarcophyton species, Tetrahedron, 1995, 51, 5291–5298 CrossRef CAS.
- G. H. Phan, H.-C. Hu, F.-R. Chang, Z.-H. Wen, J.-J. Chen, H.-M. Chung, Y.-C. Tsai and P.-J. Sung, Norsesquiterpenoids from the octocoral Paralemnalia thyrsoides (Ehrenberg 1834), RSC Adv., 2022, 12, 27970–27976 RSC.
- H.-C. Hu, S.-Y. Yu, X.-S. Hung, C.-H. Su, Y.-L. Yang, C.-K. Wei, Y.-B. Cheng, Y.-C. Wu, C.-H. Yen, T.-L. Hwang, S.-L. Chen, I. Szatmári, A. Hunyadi, Y.-H. Tsai and F.-R. Chang, Composition decipherment of Ficus pumila var. awkeotsang and its potential on COVID-19 symptom amelioration and in silico prediction of SARS-CoV-2 interference, J. Food Drug Anal., 2022, 30, 440–453 CrossRef.
- M. O. Marcarino, S. Cicetti, M. M. Zanardi and A. M. Sarotti, A critical review on the use of DP4+ in the structural elucidation of natural products: the good, the bad and the ugly. A practical guide, Nat. Prod. Rep., 2022, 39, 58–76 RSC.
- H. D. Flack, On enantiomorph-polarity estimation, Acta Crystallogr., 1983, A39, 876–881 CrossRef CAS.
- H. D. Flack and G. Bernardinelli, Absolute structure and absolute configuration, Acta Crystallogr., 1999, A55, 908–915 CrossRef CAS PubMed.
- L.-M. Kuo, P.-J. Chen, P.-J. Sung, Y.-C. Chang, C.-T. Ho, Y.-H. Wu and T.-L. Hwang, The bioactive extract of Pinnigorgia sp. induces apoptosis of hepatic stellate cells via ROS-ERK/JNK-caspase-3 signaling, Mar. Drugs, 2018, 16, 19 CrossRef PubMed.
- J. Yang, J. Mo, J. Dai, C. Ye, W. Cen, X. Zheng, L. Jiang and L. Ye, Cetuximab promotes RSL3-induced ferroptosis by suppressing the Nrf2/HO-1 signalling pathway in KRAS mutant colorectal cancer, Cell Death Dis., 2021, 12, 1079 CrossRef CAS PubMed.
- C.-F. Dai and C.-H. Chin, Octocoral Fauna of Dongsha Atoll, Marine National Park Headquarters, Kaohsiung, Taiwan, 2019, pp. 192–193 Search PubMed.
- G. M. Sheldrick, SHELXT-Integrated space-group and crystal-structure determination, Acta Crystallogr., 2015, A71, 3–8 CrossRef PubMed.
- G. M. Sheldrick, Crystal structure refinement with SHELXL, Acta Crystallogr., 2015, C71, 3–8 CrossRef PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: HRESI-MS, 1D and 2D NMR spectra of 1; experimental and calculated SOR values of 1; X-ray crystallo-graphic data of 1 and 2. CCDC 2226689 and 2208811. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d3ra01589k |
| ‡ These authors have contributed equally to this work. |
|
| This journal is © The Royal Society of Chemistry 2023 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence *behij
*behij