
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceThe influence of calcium on copper corrosion and its by-product release in drinking water
Ping Xu*,
Qiang Fu * and
Meihui Zhao*
* and
Meihui Zhao*
Key Laboratory of Urban Stormwater System and Water Environment, Ministry of Education, Beijing University of Civil Engineering and Architecture, Beijing 100044, China. E-mail: xuping@bucea.edu.cn
First published on 13th June 2023
Abstract
Copper is a high-quality material commonly used in drinking water supply pipes. Calcium is a prevalent cation found in drinking water. However, the effects of calcium on copper corrosion and its by-product release remain unclear. This study discusses the influences of Ca2+ on copper corrosion and its by-product release in drinking water under different conditions of Cl−, SO42−, and Cl−/SO42−, using electrochemical and scanning electron microscopy techniques. The results indicate that Ca2+ slows down the corrosion reaction of copper to some extent in comparison with Cl−, and the Ecorr shifts positively by 0.022 V, while Icorr decreases by 0.235 μA cm−2. However, the by-product release rate increases by 0.5 μg cm−2. The addition of Ca2+ causes the anodic process to become the controlling factor for corrosion, with an increase in resistance observed in both the inner and outer layers of the corrosion product film through SEM analysis. The corrosion product film becomes denser due to the reaction between Ca2+ and Cl−, forming a product that inhibits the entry of Cl− into the passive film on the copper surface. Adding Ca2+ promotes copper corrosion with the help of SO42− and the release of corrosion by-products. The anodic reaction resistance decreases while the cathodic reaction resistance increases, resulting in a small potential difference of only 10 mV between the anode and cathode. The resistance of the inner layer film decreases, while that of the outer layer film increases. SEM analysis shows that the surface becomes rougher with the addition of Ca2+, and 1–4 mm granular corrosion products are formed. This is due to the fact that Cu4(OH)6SO4 has low solubility and forms a relatively dense passive film that inhibits the corrosion reaction. The added Ca2+ also reacts with SO42− to form CaSO4, which reduces the amount of Cu4(OH)6SO4 generated at the interface, thus damaging the integrity of the passive film. Adding Ca2+ promotes the corrosion of copper by Cl− and SO42− and enhances the release of corrosion by-products, with the highest corrosion rate observed under the Cl−/SO42−/Ca2+ conditions. The resistance of the inner layer membrane decreases, while the mass transfer resistance of the outer layer membrane increases. Under the Cl−/SO42− conditions, the SEM surface of the Cu2O particles is uniform in size, arranged in an orderly and compact manner. After adding Ca2+, the size of the particles becomes uneven, and the surface becomes rough and uneven. This is because Ca2+ firstly combines with SO42−, thus promoting corrosion. And then the remaining Ca2+ combines with Cl−, which inhibits corrosion. Despite the amount of remaining Ca2+ being small, it still promotes corrosion. The amount of released corrosion by-products is mainly controlled by the redeposition reaction that occurs in the outer layer membrane, determining the amount of Cu2O to which the copper ions are converted. The increase in resistance of the outer layer membrane means that the charge transfer resistance of the redeposition reaction increases, and the reaction rate slows down. Consequently, the amount of Cu(II) converted to Cu2O decreases, leading to an increase in Cu(II) in the solution. Therefore, adding Ca2+ in all three conditions results in an increase in the release of corrosion by-products.
1 Introduction
Copper pipes have many advantages such as reliability, durability, and ease of installation, making them the preferred material for building water supply pipes.1 However, copper corrosion cannot be avoided, and extensive research has been conducted to study the factors that affect copper corrosion. The occurrence of corrosion in the copper pipes can lead to an increase in copper ion concentration in drinking water. Long-term excessive intake and contact with copper can be harmful to human health.2 Therefore, investigating copper corrosion and by-product release under drinking water quality conditions is an important prerequisite for studying the reasonable application of copper pipes and ensuring drinking water safety.3,4On the basis of a great deal of facts, it is evident that the mechanism behind the formation of copper's diverse corrosion products is strongly dependent upon the environment. Water constituents known to influence the nature of corrosion by-products include Cl−, SO42−, HCO3−, PO43−, and Ca2+.5,6 In general, the acidic or chloride-containing conditions are the most aggressive ones for copper. Cl− typically accelerates the corrosion process of copper.7 When Cl− ions are present, they can react with the copper oxide on the surface of copper, forming copper chloride precipitates. This may disrupts the protective layer of copper, making it more susceptible to corrosion by other corrosive substances.8 Additionally, Cl− can increase the electrochemical activity of the copper surface, leading to an elevated corrosion rate. In contrast, SO42− can generally exhibit inhibitory effects on copper corrosion. SO42− can react with the copper surface to form sulfate salt precipitates, which contribute to the formation of a protective film that hinders corrosion.9 This protective film can slow down the corrosion rate of copper and prevent further oxidation reactions. Therefore, SO42− can provide protection for copper when present at appropriate concentrations. HCO3− promotes the formation of pitting corrosion on copper at low concentrations (<1 mmol L−1), but it inhibits pitting corrosion at higher concentrations (>1 mmol L−1) by preventing the deterioration of the Cu2O film.10–12 The concentration of HCO3− in drinking water typically ranges from 0.5 to 10 mmol L−1, and its effect on copper pipe corrosion needs to be differentiated based on specific concentrations. In the short term, PO43− can effectively inhibit the corrosion of copper pipes. Adding 1–3 mg L−1 of PO43− to the water system can slow down the oxidation rate of copper metal and Cu2O film, leading to greater polarization of the copper surface and reducing the rate of electron transfer.13 As a result, the corrosion rate of copper is significantly reduced. However, in the long term, PO43− may hinder the natural formation of stable corrosion products, resulting in more Cu(II) entering the water in an ionized form.14–16 Precipitation–dissolution, complexation, and acid–base reactions govern copper speciation in the bulk water and in the metal–water interface. The formation reactions of copper scale are influenced by pH, DO, temperature, and the concentration and types of ions present in the water.17 Typically, when the pH is higher than 6, cupric ions precipitate forming scale.18,19 Pehkonen et al. reported that the stability, thickness, and hence mass transfer properties of a corrosion by-product film are dependent on pH and DO.20
According to literature research, there are currently limited research on the influence of Ca2+ as a single factor on copper corrosion, and it is usually considered together with alkalinity. Under low alkalinity water conditions, Ca2+ does not have a significant impact on the corrosion rate and copper leaching of copper.21 However, under higher alkalinity water conditions, increasing Ca2+ can effectively reduce the copper leaching compared to soft water.22 Ca2+ ions are common cations in drinking water. However, our current understanding of the effects of Ca2+ on copper corrosion under drinking water conditions is still limited. Additionally, the synergistic corrosion relationship between Ca2+ and common corrosive ions in drinking water, such as Cl− and SO42−, is not yet clearly understood. To ensure the safety of water quality, it is crucial to gain a deeper understanding of the correlation between copper corrosion behavior and the release of copper corrosion by-products. Therefore, the present work is aimed at studying the synergistic effects of Ca2+ and corrosive ions to explore their impact on the copper corrosion process and the release of corrosion by-products.
The investigation of different operating conditions was conducted using a combination of electrochemical methods, including the analysis of the open circuit corrosion potential (OCP), potentiodynamic (PD) polarization curves, and electrochemical impedance spectroscopy (EIS). Furthermore, the surface morphology was characterized by scanning electronic microscopy (SEM), and the attachment of the corrosive products was obtained by X-ray Photoelectron Spectroscopy (XPS). Such research is of significant importance for further understanding and controlling copper corrosion processes and the presence of corrosion by-products in drinking water.
2 Experimental
2.1 Material
In this experiment, deoxidized phosphorus copper that is commonly used in drinking water pipelines was selected as the research object. The copper specimen was a rectangular shape with a length of 5.0 cm, a width of 2.5 cm, and a height of 0.2 cm. Its elemental composition (wt) was 0.05 Fe, 0.013 P, 0.009 Sn, 0.006 Ni, 0.005 S, 0.005 Pb, and the remainder was Cu. Prior to the experiment, the specimen was pre-treated by following these specific steps: (1) soaking the specimen in acetone for a moment; (2) wiping it clean with medical degreased cotton; (3) immersing it in anhydrous ethanol for dehydration for about 1 minute; (4) wiping it clean again with medical degreased cotton; (5) air-drying it; (6) wrapping it in clean filter paper; (7) placing it in a drying dish for later use. Before the experiment, it needed to be taken out, recorded with a number, weighed, and sterilized under UV light for 30 minutes.The copper working electrode is a cylindrical shape with a bottom surface area of 1 cm2 and a height of 1 cm, one side of which served serving as the working face and the other side connected to a wire. Prior to the experiment, the copper electrode underwent a series of pre-treatment steps: (1) the area outside of the working face were covered with epoxy resin and left to solidify vertically; (2) the working face was polished using 180#, 360#, 600#, 1000#, 1500#, and 2000# water sandpaper until it was shiny, the work was followed by rinsing with distilled water; (3) the electrode was placed on chamois leather and was polished with alumina polishing powder (Al2O3), which was followed by rinsing with distilled water; (4) the electrode was dehydrated with anhydrous ethanol and was placed in a drying dish for later use.
The experimental water was prepared by using pure water as a basis and adjusting the ions concentrations to be within the range of drinking water quality. The compositions of the water used for the experiments are shown in Table 1. Ca2+ concentration was used as the control parameter to adjust the water quality, and a total of 6 sets of operating conditions were designed. The water quality parameters of each condition were shown in Table 2.
| Work condition | NaCl (mg L−1) | Na2SO4 (mg L−1) | CaCl2 (mg L−1) | CaSO4 (mg L−1) | Temperature (°C) |
|---|---|---|---|---|---|
| Cl− | 51.90 | — | — | — | 23 |
| Cl−/Ca2+ | — | — | 55.50 | — | 23 |
| SO42− | — | 217.88 | — | — | 23 |
| SO42−/Ca2+ | — | 150.88 | — | 68.00 | 23 |
| Cl−/SO42− | 51.90 | 217.88 | — | — | 23 |
| Cl−/SO42−/Ca2+ | 8.08 | 74.55 | 27.75 | 34.00 | 23 |
| Work condition | Cl− (mg L−1) | SO42− (mg L−1) | Ca2+ (mg L−1) | pH | Conductivity (μs cm−1) |
|---|---|---|---|---|---|
| Cl− | 30.0 | — | 0.0 | 6.68 | 120 |
| Cl−/Ca2+ | 30.0 | — | 50 | 6.67 | 126 |
| SO42− | — | 150 | — | 6.70 | 329 |
| SO42−/Ca2+ | — | 150 | 50 | 6.71 | 331 |
| Cl−/SO42− | 30.0 | 150.0 | — | 6.65 | 439 |
| Cl−/SO42−/Ca2+ | 30.0 | 150.0 | 50.0 | 6.65 | 436 |
2.2 Experimental device
Two parallel AR reactors were set up as shown in Fig. 1. The reactors included a stirring device that simulates hydraulic flow in actual pipeline networks, a temperature control system, and a pump that controls the inflow of water. The reactor had a volume of 2.5 L and was cylindrical in shape. The outer wall of the reactor, the internal rotating shaft, and the stirring blades that simulate water flow were made of organic glasses and equipped with organic glass slide racks. During the experiment, the slides were placed in the racks and remained relatively stationary. The stirring blades were driven by a motor to create water flow, generating shear forces between the water flow and the slides along with simulating the hydraulic conditions in actual water supply systems. The speed of the rotating shaft could be adjusted by a controller to simulate the water flow rate in copper pipe water supply systems. The reactor had a heating device inside to control water temperature.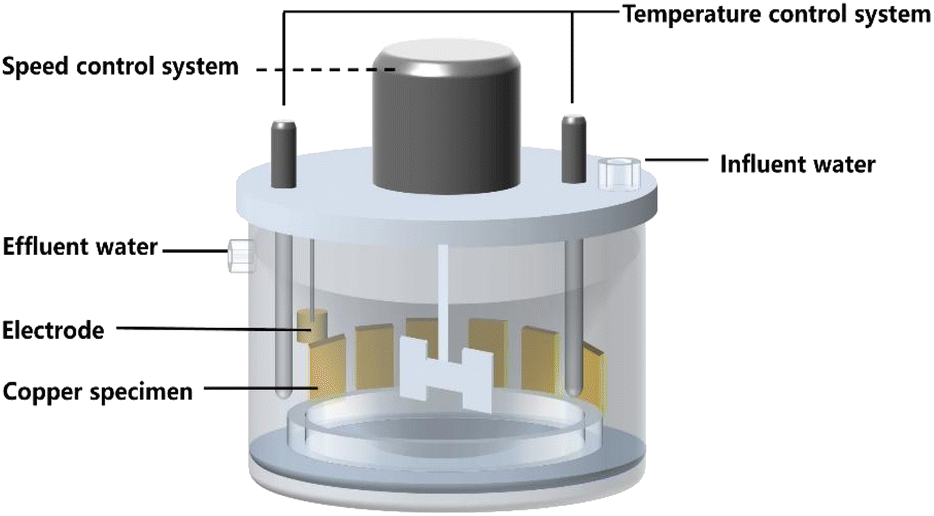 | ||
| Fig. 1 Schematic diagram of the experimental setup for the artificial reactor. | ||
Six sets of experimental conditions were established, and three parallel experiments were conducted in each set. Copper electrodes and copper test pieces were placed in each reaction vessel, and the experimental rotation speed was set at 60 rpm. The water was supplied intermittently, and the experiment was run for 15 days. Copper ion concentration in the water was measured using UV spectrophotometry at day 15 of each experimental set. At the same time, samples were taken to observe the surface of the copper under different experimental conditions using environmental scanning electron microscopy (ESEM). The elemental types and contents on the surface were analyzed using energy dispersive spectrometry (EDS). In addition, at day 15 of each experimental set, the corrosion products were qualitatively analyzed using X-ray photoelectron spectroscopy (XPS).
2.3 Analytical method
The experiment employed the DDTC-Na spectrophotometric method to determine the concentration of dissolved copper in water samples. Standard copper solutions were prepared and their absorbance was measured at a wavelength of 452 nm to establish a calibration curve. The curve was fitted with a requirement of a fitting error R2 value greater than 0.999. The concentration of copper in water was then converted to the amount of copper dissolved per unit area of the test specimen using the calculation formula is as follows.A: the amount of copper ions dissolved per unit area of coppers' test pieces, μg cm−2; c: copper ion concentration, mg L−1; V: reactor volume, L; S: copper specimen surface area, cm2; n: number of copper specimen, pieces.

The model number of the electrochemical workstation was CHI660C, which was a conventional three-electrode system. Prior to testing, hardware testing must be performed to ensure proper operation. The testing process included three parts, with the following parameters selected: open circuit potential, which must be within a fluctuation range of ±1 mV before testing can be performed. Polarization curve testing was conducted after the time when open circuit potential has stabilized, with a scan range of ±0.3 V set around the open circuit potential, one second per scan section, and a scan rate of 0.001 V s−1. Electrochemical impedance spectroscopy was conducted with the initial level set at the open circuit potential, with a scan frequency of 0.01–105 Hz, an amplitude of 0.005, and a settling time of 2 s.
The surface corrosion and morphological changes of copper were observed using scanning electron microscopy (SEM). The equipment used was a QUANTA 200F (Netherlands), with an accelerating voltage of 200 V to 30 kV and a sample chamber vacuum of <6 × 10−4 4000 Pa. EDS was used to determine the types and contents of some elements on the copper surface. Pre-treatment is required before testing.
In order to observe the corrosion status and the surface morphology changes of the copper sample, the copper test piece needed to undergo pre-treatment. The corrosion product was closely connected to the copper substrate and was difficult to scrape off from the substrate. Therefore, the copper sheet in the corroded area was placed on a conductive adhesive and was flattened for pressing before testing. The equipment used is the Quantera II X-ray photoelectron spectrometer from ULVAC PHI. Each XPS spectrum was calibrated using the C 1s outer standard method, with the C–C calibration position at 284.8 eV. The Avantage software was used for fitting and calibration.
3 Results
3.1 The impact of Ca2+ on copper corrosion and by-product release
 | ||
| Fig. 2 Polarization curves of copper electrode. | ||
| Work condition | Ecorr/V | Icorr/μA cm−2 | βa/mV dec−1 | −βc/mV dec−1 |
|---|---|---|---|---|
| Cl− | −0.051 | 1.456 | 126.887 | 188.573 |
| Cl−/Ca2+ | −0.029 | 1.221 | 246.003 | 181.488 |
| SO42− | −0.022 | 0.836 | 208.464 | 143.410 |
| SO42−/Ca2+ | −0.115 | 1.857 | 188.324 | 178.412 |
| Cl−/SO42− | −0.036 | 1.235 | 173.82 | 172.56 |
| Cl−/SO42−/Ca2+ | −0.090 | 2.420 | 349.90 | 187.58 |
Based on Fig. 2 and Table 3 and it could be observed that the polarization curve in the Cl−/Ca2+ system shifted positively compared to that in the Cl− system. Specifically, the corrosion potential (Ecorr) shifted from −0.051 V to −0.029 V, an increase of 43%, indicating that the addition of Ca2+ mitigated the corrosive tendency of the chloride ion system. Copper was less susceptible to corrosion in the Cl−/Ca2+ solution. The corrosion current density decreased from 1.456 μA cm−2 to 1.221 μA cm−2, a decrease of 0.235 μA cm−2, resulting in a corrosion inhibition rate of 16%. This suggested that Ca2+ inhibited the copper corrosion caused by Cl−.
Additionally, by examining βa and βc in Table 3 and it can be seen that βc for the Cl− system was 188.573 mV dec−1, which was greater than βa's 126.887 mV dec−1. This indicated that the cathodic reaction became the controlling factor, and the corrosion process was mainly controlled by the process of dissolved oxygen transfer. After adding Ca2+, βa increased by 119.116 mV dec−1, while βc remained relatively unchanged. At this point, the value of βa surpassed that of βc, indicating that the anodic reaction became the controlling factor, and the corrosion process was mainly controlled by the process of by-product release. This reflected that Ca2+ has an inhibitory effect on the anodic behavior of copper, similar to that of an anodic corrosion inhibitor.
Under the experimental conditions, the Icorr of the SO42− system was very small, at only 0.836 μA cm−2, indicating that corrosion was almost non-existent. This was due to the fact that the effect of SO42− on copper corrosion varied with concentration.23 When the concentration of SO42− was less than 0.01 mol L−1 within the range of drinking water quality, it accelerated the formation of stable corrosion products, rapidly forming a stable product film, thus inhibiting corrosion.24 However, when the concentration of SO42− exceeds 0.01 mol L−1, it increases the water's conductivity and the migration number of other anions, resulting in a relatively higher anodic current density. This, in turn, increases the overall corrosion susceptibility of copper, leading to pitting corrosion. In such cases, the Cu2O film formed on the copper surface contains numerous pores and is accompanied by a small amount of copper sulfate crystal precipitation, which promotes intergranular corrosion of the copper substrate.25 Within the range of concentrations that cause pitting corrosion, the tendency for pitting corrosion increases with the increasing concentration of SO42−. However, within the drinking water quality range, SO42− mainly played a role in inhibiting corrosion.
Compared with the SO42− system, the SO42−/Ca2+ system exhibited a significantly negative shift in the polarization curve. The Ecorr shifted negatively by 0.093 V and the Icorr increased from 0.836 μA cm−2 to 1.857 μA cm−2, an increase of 122%. This showed that the addition of Ca2+ significantly altered the inhibitory effect of 150 mg L−1 of SO42− on copper corrosion, promoting corrosion. In the SO42− system, the anodic reaction was the limiting factor. After adding Ca2+, βa decreased while βc increased, suggesting that Ca2+ has an effect on both the anodic and cathodic reactions. βa was slightly greater than βc, but the difference between the two was not significant. Compared with the Cl−/Ca2+ system, it can be concluded that the effect of Ca2+ on the anodic and cathodic reactions of Cl− and SO42− was different. Ca2+ mainly affected the anodic reaction of Cl−, slowing down its reaction rate, while Ca2+ had an effect on both the anodic and cathodic reactions of SO42−, mainly increasing the anodic reaction rate and decreasing the cathodic reaction rate.
It could be observed from Fig. 2 and Table 3 that when Cl−/SO42− coexisted, the addition of Ca2+ resulted in both the anodic and cathodic reactions of copper in the solution shifting towards a higher current. Ecorr negatively shifted with the addition of Ca2+ and Icorr increased by a factor of 2 compared to the Cl−/SO42− condition, indicating that the addition of Ca2+ significantly accelerated the corrosion of copper in the Cl−/SO42− solution.
From the Icorr values in Table 3 and it is evident that Cl− can promote copper corrosion, while SO42− can inhibit it. These results are in line with previous studies. Ghandehari et al. observed that the corrosion rate of copper in HCl solution is more intense than that in H2SO4 at the same pH value because of the catalysis of the chloride ion.26 On the other hand, the inhibiting film composed of adsorbed sulphate ions could mitigate the rate of oxygen reduction.
When they coexisted, their effects may compete with each other. The polarization curve of the Cl−/SO42− system had a similar overall shape to that of the Cl− system, and both curves exhibited a passivation region.27 In contrast, the polarization curve of the SO42− system differed significantly, and Icorr was a only 0.221 μA cm−2 lower than that of the Cl− system, indicating that Cl− took the lead in corrosion when they coexisted. Cl− was the dominant factor determining the corrosion layer morphology and corrosion rate, while SO42− was not a necessary condition for corrosion.25
The impact of Cl−/SO42−/Ca2+ conditions on the corrosion of copper was found to be similar to that of Cl−/Ca2+ conditions, as evidenced by a negligible difference in Icorr (only 0.0014 μA cm−2). This suggested that both SO42− and Ca2+ have a certain inhibitory effect on the corrosion caused by Cl−. However, when comparing SO42−/Ca2+ conditions with Cl−/SO42− conditions, it was found that the Ecorr of the former shifted negatively by 0.079 V, while Icorr increased by 0.622 μA cm−2 (50%). This implied that the coexistence of SO42− and Ca2+ actually increased the corrosion tendency of copper, contrary to the previously anticipated decrease. The reason for this may be due to a reaction between SO42− and Ca2+, which canceled out their inhibitory effects on each other's corrosion.
 | ||
| Fig. 3 By-product release per unit area under different operating conditions. | ||
Upon addition of Ca2+ in the Cl− system, as indicated in the previous context, the corrosion was inhibited and Icorr decreased. However, the by-product release per unit area increased from 3.64 μg cm−2 to 4.13 μg cm−2, indicating that even the introduction of Ca2+ in Cl− solution restrained the corrosion of copper, it elevated the by-product release of copper. It can be inferred that Ca2+ exerted various influences on the corrosion and by-product release of copper. Similarly, the addition of Ca2+ in SO42− system led to an increase in the by-product release from 1.42 μg cm−2 to 3.13 μg cm−2, a growth of 120.42%, suggesting that Ca2+ not only promoted the corrosion of copper by SO42−, but also facilitated the by-product release of copper. When Cl− and SO42− coexisted, the by-product release increased by 1.01 μg cm−2, or 63.13%, upon the addition of Ca2+. This implied that Ca2+ not only accelerated the corrosion of copper by Cl−/SO42−, but also promoted the by-product release of copper.
Furthermore, it was observed that without considering the effect of Ca2+, the relationship between by-product release and the three different conditions was as follows: Cl− > Cl−/SO42− > SO42−. The by-product release rate in the Cl− condition was 3.64 μg cm−2, which was 60.99% higher than the rate of 1.42 μg cm−2 in the SO42− condition. In the Cl−/SO42− condition, the by-product release rate was 1.60 μg cm−2, which was 56.04% lower than that in the Cl− condition. This suggested that SO42− played a dominant role in by-product release under the Cl−/SO42− condition, and effectively suppressed the by-product release caused by Cl−. The reason for this was that the presence of SO42− immediately changed the solid phase type present in the system containing Cu2+ or Cu(OH)2 solids, resulting in a rapid decrease in by-product release rate in the short term.24 However, the addition of a large amount of Ca2+ could combine with anions in water, significantly inhibiting the Cu(OH)2 phase transformation to a lower solubility and more stable phase caused by chloride or sulfate. Therefore, Ca2+ could increase the solubility of copper ions in water. The study conducted by Yan et al.28 indicates that in the absence of an inhibitor, the corrosion current density of hard tap water exhibited an increase, while the electrochemical resistances were notably decreased compared to those in soft water. This phenomenon led to the disruption of the passive layer and accelerated corrosion.
3.2 EIS analysis
Electrochemical impedance spectroscopy tests were conducted under various conditions, and ZSimpWin software was employed for circuit fitting. The fitted circuit conformed to the Rs(Qb(Rb(QfRp))) model, and the equivalent circuit were illustrated in Fig. 4. The obtained parameters included the solution resistance (Rs), the inner layer membrane resistance (Rp), the outer layer membrane resistance (Rb), as well as the surface diffusivity coefficient (nf) of copper, which is a key parameter for characterizing the surface state.29,30 Typically, when n = 1, the electrode surface is perfectly smooth, and the closer the half-circle in the impedance diagram approaches 1, the more perfect the system is, indicating a closer approximation to an ideal capacitor. Deviation from n = 1 suggests stronger diffusion effects, and the smaller the value of n, the more irregular the half-circle appears. A value of 0.5 < n < 1 represents a rough surface, while 0 < n < 0.5 indicates a porous surface of the electrode.31 The Nyquist plots of the copper electrode impedance under six different conditions were shown in Fig. 5, and the fitting data of the impedance under each condition were listed in Table 4.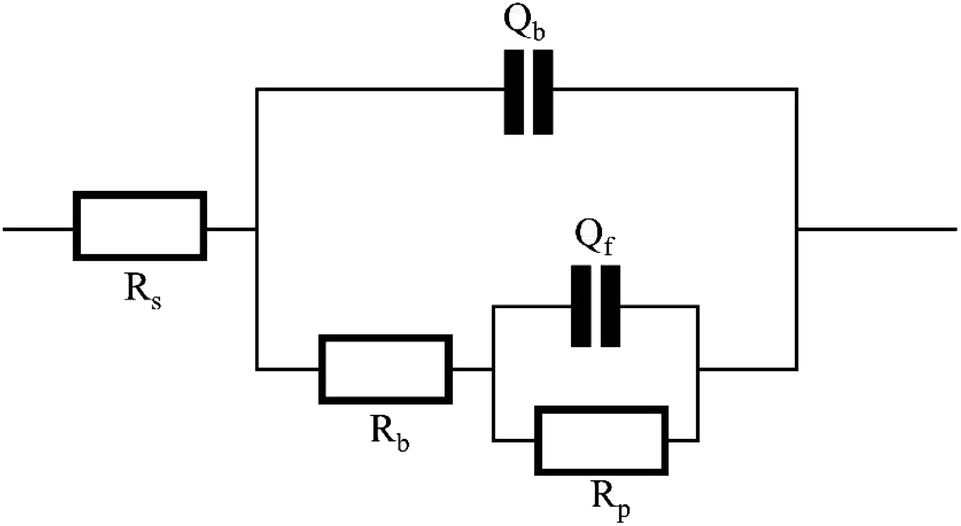 | ||
| Fig. 4 Fits the equivalent circuit diagram. | ||
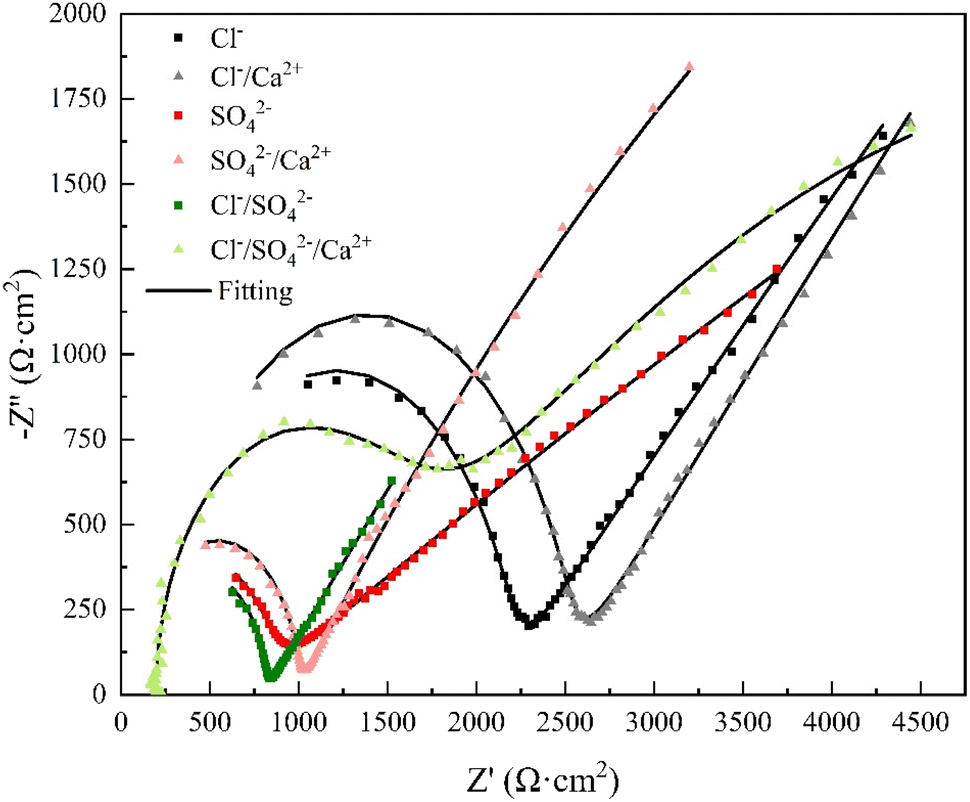 | ||
| Fig. 5 Nyquist diagram of copper electrode. | ||
| Work condition | RS/Ω cm2 | Qb/F cm−2 | Rb/Ω cm2 | Qf/F cm−2 | nf | Rp/Ω cm2 |
|---|---|---|---|---|---|---|
| Cl− | 260.1 | 1.012 × 10−9 | 1864 | 6.464 × 10−5 | 0.4340 | 56![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 900 900 |
| Cl−/Ca2+ | 261.2 | 1.358 × 10−9 | 2199 | 5.967 × 10−5 | 0.5669 | 82200 |
| SO42− | 100.0 | 1.355 × 10−9 | 705.9 | 2.062 × 10−4 | 0.8591 | 89240 |
| SO42−/Ca2+ | 91.69 | 2.099 × 10−9 | 1399.5 | 1.068 × 10−4 | 0.4223 | 16510 |
| Cl−/SO42− | 38.7 | 9.510 × 10 | 713 | 2.091 × 10−4 | 0.4368 | 37550 |
| Cl−/SO42−/Ca2+ | 154.8 | 5.501 × 10 | 1197 | 1.394 × 10−4 | 0.3580 | 15070 |
Based on Table 4, the Rp values under various working conditions were significantly larger than those of Rb and Rs, indicating that Rp was the main resistance for charge transfer. In the Nyquist plot, the depressed semicircle on the left side of each fitting curve represents the high frequency region, while the straight line on the right side corresponds to the low frequency region.28 The capacitive arc in the high frequency region of the Nyquist plot represented the value of the outer layer membrane resistance Rb. As shown in Fig. 5, both the Cl− and Cl−/Ca2+ conditions exhibited a capacitive arc in the high frequency region, but the radius of the arc increased with the addition of Ca2+. Combining this with the data in Table 4, the Rb value for the Cl− condition was 1864 Ω cm2, while the addition of Ca2+ increased the Rb value to 2199 Ω cm2, indicating an increase in the resistance of the outer layer membrane. The Rp value increased by 25300 Ω cm2, which was a 44% increase, indicating a significant increase in the charge transfer resistance of the inner layer membrane and an increase in the difficulty of corrosion. The shift of the corrosion potential to a more positive value and the decrease in the corrosion current density in the previous text also supported this finding. Furthermore, the Cl− condition had a dispersion coefficient nf < 0.5, showing that the anodic by-product release reaction on the surface of the copper electrode was in a porous state, resulting in more pitting corrosion. The Cl−/Ca2+ condition had a dispersion coefficient nf > 0.5, suggesting that the surface of the copper electrode became a rough surface, which suppressed pitting corrosion.
In the SO42−/Ca2+ system, the capacitive arc radius in the high frequency region was greater than that of the SO42− system. Table 4 displayed that the addition of Ca2+ increased the value of Rb from 705.9 Ω cm2 to 1399.5 Ω cm2, an increase of 98.26%, indicating a great rise in the charge transfer resistance of the outer layer film. Furthermore, the value of Rp after Ca2+ addition was 16510 Ω cm2, lower than the value of 89240 Ω cm2 in the SO42− system. This suggested that Ca2+ reduced the charge transfer resistance between the copper substrate and the inner layer product film when SO42− was present, leading to an unstable product film and strengthening the mass transfer of copper ions at the interface and resulting in an increase in by-product release in the solution. Moreover, the addition of Ca2+ caused a decrease in the nf value from 0.8591 to 0.4223, indicating a transformation of the electrode surface from a relatively smooth interface to a porous, rough one. The results of the morphology observation in the subsequent text confirmed this finding. The porous and rough interface implied an exacerbation of copper corrosion and an increase in by-product release.
It can be observed from Fig. 5 that when Cl−/SO42− coexist, the addition of Ca2+ led to an increase of 484 Ω cm2 in Rb, demonstrating that Ca2+ caused corrosion products to adsorb on the copper surface and resulted in a thicker outer layer of the product film. The decrease of 22480 Ω cm2 in Rp value implied a decrease in the charge transfer resistance of the inner layer. The decrease in nf with the addition of Ca2+ suggested that the interface of the corrosion product film became rougher and more porous, and as a result, the degree of corrosion increased, leading to an increase in the amount of by-product release.
Comparing the curves of the Cl− condition and the Cl−/SO42− condition in Fig. 5, the high-frequency arc radius of the latter was generally smaller than that of the former. From the data in Table 4, the Rb value of the Cl− condition was 1864 Ω cm2, while that of the Cl−/SO42− condition was 713 Ω cm2, a decrease of 61.75%. The Rp value of the Cl− condition was 56900 Ω cm2, while that of the Cl−/SO42− condition was 37500 Ω cm2, a decrease of 34.09%. The shift of Ecorr to a more positive value and the decrease of Icorr in the Cl−/SO42− condition both showed that the addition of SO42− reduced the outer product film resistance and the charge transfer resistance of Cl− on the copper electrode, with Cl− playing a dominant role in corrosion. The research results of Shalaby et al.25,32 also confirmed this point.
3.3 The effect of Ca2+ on the morphology of copper samples and composition analysis of corrosion products
 | ||
| Fig. 6 SEM images of copper surface: (a) Cl−, (b) SO42−, (c) Cl−/Ca2+, (d) SO42−/Ca2+, (e) Cl−/SO42−, (f) Cl−/SO42−/Ca2+. (In (a), (1) is the cubic-shaped Cu2O and (2) is the pitting corrosion caused by Cl− on the copper surface). | ||
The smaller ionic radius of Cl− allows it to enter the Cu2O film as an interstitial ion, thereby influencing the formation of copper surface products. When the concentration of Cl− is less than 0.05 mol L−1, the copper surface undergoes uniform corrosion, whereas concentrations above this threshold lead to localized corrosion, commonly known as pitting corrosion. MERKEL's research illustrates that in the range of drinking water quality, the concentration of Cl− is generally below 250 mg L−1, which is less than 0.05 mol L−1, indicating that the predominant form of corrosion is uniform corrosion.35 This is consistent with the corrosion phenomenon in Fig. 6(a). According to Fig. 6(c), the addition of Ca2+ to the Cl− solution caused a change in the morphology of the corrosion products. Although the Cu2O surface on the copper surface was broken, it still exhibited a hexagonal crystal structure with a diameter of approximately 5 μm with small crystals growing unevenly around it. This was in sharp contrast to the stable cuboid products observed in the Cl− solution. Additionally, it could be clearly seen that the copper substrate did not exhibit the small pits observed in the Cl− solution, and was covered with corrosion products. The surface changed from a severely porous state to a rough state, indicating that the addition of Ca2+ weakened the corrosive effect of Cl−. Fateh et al.'s research shows that some of the corrosion inhibitors protect the copper surface through the formation of a protective oxide film and some of them reduce corrosion attack through adsorption and formation of a complex layer.7 The experimental results show that Ca2+ slow down the corrosion caused by Cl− by promoting the formation of oxide film on copper.
According to the EDS results, the ratio of Cu and O elements in the Cl−/Ca2+ solution was similar to that in the Cl− solution, and the macroscopic morphology of the corrosion product was also a dark red solid. Energy spectrum analysis also indicated that the corrosion product was Cu2O.
Within the drinking water quality range, SO42− primarily functions as a corrosion inhibitor.23 Based on Fig. 6(b), it was evident that there was no apparent corrosion on the surface of copper in the SO42− condition, as only a smooth copper substrate was observed in the SEM images. SO42− accelerates the formation of stable corrosion products, leading to the rapid formation of a protective film that inhibits corrosion.19
However, in the SO42−/Ca2+ condition, the majority of the copper surface area was covered with a gravel-like corrosion product with a diameter of approximately 1–4 μm, based on the SEM image in Fig. 6(d). The product in this condition appeared to be finer and consisted of clusters of hexagonal crystals pieces. The reason was that although SO42− in the solution could inhibit corrosion, the addition of Ca2+ could lead to the formation of sparingly soluble CaSO4. When CaSO4 was in contact with the copper surface product, it could cause the corrosion product to break and also create scratches on the copper mental surface, which therefore weakened the inhibitory effect of SO42− on cooper corrosion and increased the corrosion tendency. During this process, some of the products might dissolve and enter the water environment, which was consistent with the previous increase in the amount of dissolved copper. According to the EDS results and the percentage of Cu and O elements, the corrosion product was preliminarily identified as Cu2O. The increase in the Rb value mentioned earlier also indicated an increase in the amount of corrosion product. The decrease in the Rp value demonstrated that Ca2+ can destroy the thin passivation film formed by SO42− on the copper surface, leading to the formation of a loose product layer, which provided a condition for the by-product release of copper.
Based on Fig. 6(e), it could be observed that when Cl−/SO42− coexisted, the surface products were evenly distributed and mainly composed of Cu2O films. Among the different forms of Cu2O, the product composed of cubic structuring Cu2O was smoother, more compact, and had tighter connections between crystals, leading to its strong adhesion to the copper surface and certain inhibitory effect on corrosion.36 When SO42− and Cl− coexisted, they competed in corroding copper. While Cl− reacted with copper to form CuCl2−, SO42− reacted with copper to form Cu4(SO4)(OH)6 that deposited on the copper surface, hindering further reaction of copper and to some extent suppressing the corrosion caused by Cl−. Therefore, although copper underwent corrosion in the Cl−/SO42− condition, the amount of copper leaching into the water was smaller than that in the Cl− condition but greater than that in the SO42− condition.
From Fig. 6(f), it could be observed that the addition of Ca2+ under Cl−/SO42− working conditions led to a roughening of the product surface. The product appeared in cubic form with a diameter of less than 5 μm. The majority of the copper surface was covered by the corrosion product, while the uncovered areas clearly showed the corroded copper substrate with an uneven surface and small cubic products scattered on it. The product film was porous and loose, which failed to effectively isolate corrosive ions and instead allowed local accumulation of corrosive ions, thereby compromising the integrity and compactness of the copper passivation film.37 While part of the metal was protected by the coverage, the other part remained exposed. As a result, the area of the corroding anode was reduced, leading to a situation of large cathode and small anode, which in turn intensified local corrosion of the copper plate.7 The unevenness of the product layer promoted and triggered pitting corrosion of Cl− due to the acceleration of anodic by-product release in the affected area, forming pits that merge and connect into larger corrosion pits. The large difference in crystal grain size and poor crystal stability of the product layer led to the cracking and detachment of the product layer, exposing the copper substrate to water and resulting in intensified corrosion reactions and increased by-product release.
Based on the results from EDS and XPS, the corrosion products in the Cl−/SO42− and Cl− conditions were found to be consistent in which the products were composed of Cu and O elements, and exhibited obvious binding energy peaks of Cu2O. The corrosion products in both conditions were identified as Cu2O. This is consistent with the findings of Wu et al.38 regarding Cu2O. However, in the Cl−/SO42− condition, the crystals were smaller and exhibited a more perfect cubic structure, with uniform particle size between 2 to 3 μm. The crystals were tightly interconnected, resulting in a uniform and stable layer of corrosion products that were less susceptible to detachment. As a result, although copper substrate still corroded in the Cl−/SO42− condition, less copper was dissolved into the water compared to the Cl− condition. This was due to the fact that sulfate ions could lead to intergranular corrosion, which could form another dense film of corrosion particles.24
 | ||
| Fig. 7 XPS spectrum of Cu 2p orbital for corrosion product. | ||
| Condition | Cu | C | O | Cl |
|---|---|---|---|---|
| Cl− | 10.71 | 64.01 | 22.55 | 2.73 |
| Cl−/Ca2+ | 11.26 | 65.46 | 23.28 | — |
| SO42− | 5.49 | 70.33 | 24.18 | — |
| SO42−/Ca2+ | 12.37 | 64.99 | 22.64 | — |
| Cl−/SO42− | 14.17 | 61.17 | 24.66 | — |
| Cl−/SO42−/Ca2+ | 11.19 | 65.20 | 23.6 | — |
Based on Fig. 7, it could be observed that the Cu 2p spectra of the Cl−, SO42−, and Cl−/SO42− working conditions shared similar patterns, with characteristic peaks located around 932.7 eV and 952.3 eV. However, weak satellite peaks at 945 eV could be observed in the Cl− working condition and in the corroded region of the Cl−/SO42− working condition. Combined with the SEM observations and element abundance data in Table 5, it could be inferred that the corrosion product in these cases was Cu2O.39 In the Cl−/SO42− working condition, a small area of the surface remained uncorroded, and thus full spectrum scans were conducted separately for the corroded and uncorroded regions. The results showed that the spectra of the uncorroded region were similar to those of the SO42− working condition, whereas the elemental ratios in the corroded region were similar to those in the Cl− working condition. This suggested that when Cl− and SO42− coexisted, the corrosion product composition was primarily influenced by Cl−.
When Cl− and SO42− coexist in water, Cl− is the dominant factor determining the morphology and corrosion rate of the corrosion layer. SO42− can promote the formation of relatively stable corrosion products, thus inhibiting corrosion. Moreover, SO42− can compete for adsorption sites with Cl−, thereby partially inhibiting Cl− corrosion of copper. Therefore, SO42− is not a necessary condition for corrosion.25 Research results have shown that the morphology of the oxide film on copper surfaces is similar at different concentrations of SO42−, and it only affects the composition of corrosion products in a small number of samples.40 The XPS spectra of the corrosion products in Cl−/Ca2+ and SO42−/Ca2+ exhibited similar shapes, with characteristic peaks that were observed near 932.7 eV and 952.3 eV, and a weak satellite peak that was observed at 945 eV. Combined with the SEM images and results presented in Table 5 and it can be concluded that the corrosion product in these cases was Cu2O.39 Similarly, XPS analysis of the Cl−/SO42−/Ca2+ condition also revealed a distinct Cu2O pattern. Additionally, as shown in Table 5, except for the Cl− condition, the other five samples contained only Cu, O, and C elements. In the Cl− condition, there was a characteristic peak of Cl element, indicating the presence of CuCl in the corrosion product on the copper surface. The reason for the high proportion of C elements in the XPS elemental composition of the SO42− condition might be due to the organic carbon contamination on the sample surface in the air. Moreover, during the XPS measurement, the electron penetration depth was shallow, so the surface material measured had a higher proportion of C element and a lower Cu content.
In various scenarios within the range of drinking water quality, the corrosion products on the surface of copper predominantly consist of Cu2O. However, in chloride-containing conditions, some copper chloride (CuCl) may appear. In environments containing SO42−, the corrosion products may include CuSO4 or Cu4(SO4)(OH)6. In the presence of HCO3−, the corrosion products mainly comprise CuCO3Cu(OH)2. When phosphate ions are present, Cu3(PO4)2 can be formed. Typically, Cu2O is formed through the electrochemical reaction between copper substrate and water, while the formation of other compounds is influenced by the equilibrium of copper with other ions.41
4 Discussion
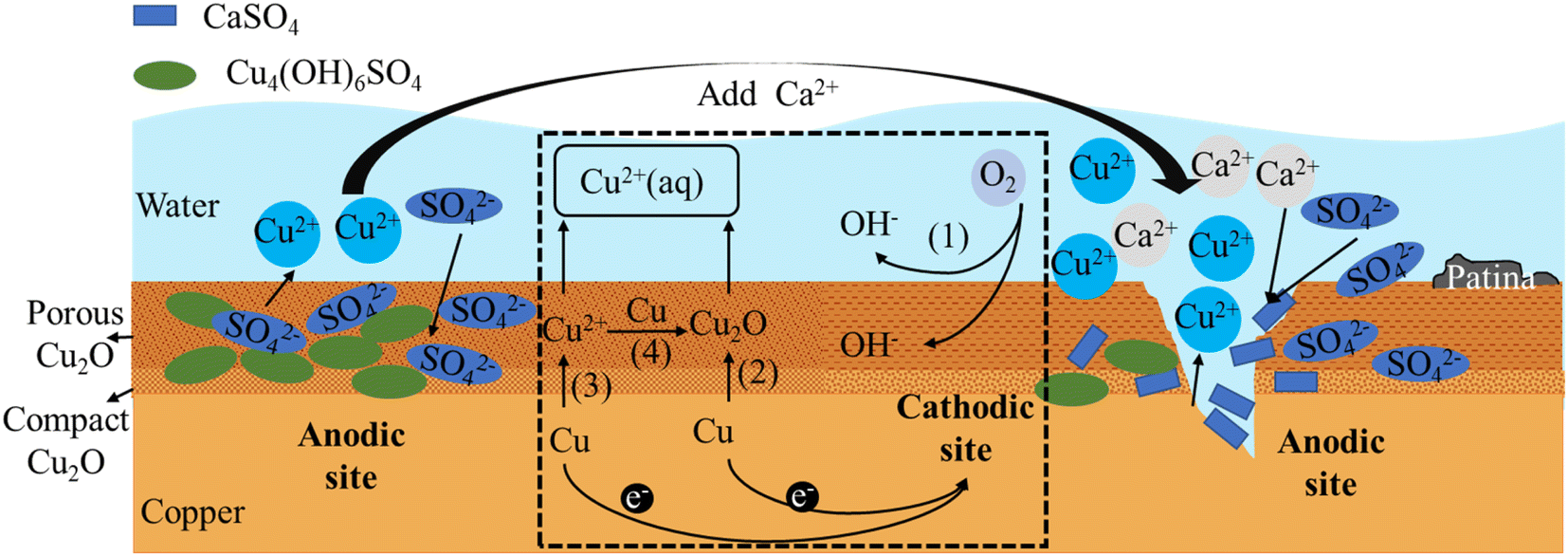
In the field of copper corrosion theory, early scholars Ives and Rawson proposed the Cu2O double layer film theory,42 which suggested that the passive film formed on the copper surface consisted of a dense inner layer and a porous outer layer. The inner layer was made up of dense Cu2O crystals and was very thin, only a few nanometers thick, and the measured value was Rp; the outer layer was made up of irregularly shaped Cu2O crystals of varying sizes, and was much thicker than the inner layer, ranging from tens to hundreds of nanometers thick, and the measured value was Rb. Recent research suggested that the inner layer may also contain a certain number of oxygen vacancies and defects, while the outer layer may also contain other compounds, such as CuO, Cu(OH)2, Cu4(OH)6SO4, etc.According to the research findings from Sections 3.1, 3.2, and 3.3, combined with the dual layer film theory, the principle diagrams of copper corrosion and by-product release were illustrated in Fig. 8 and 9. The dashed box in the figure showed the process of electron transfer and material transformation. On the left side of the dashed box were the anodic reaction diagrams under Cl− and SO42− conditions before the addition of Ca2+. On the right side were the anodic reaction diagrams after the addition of Ca2+.
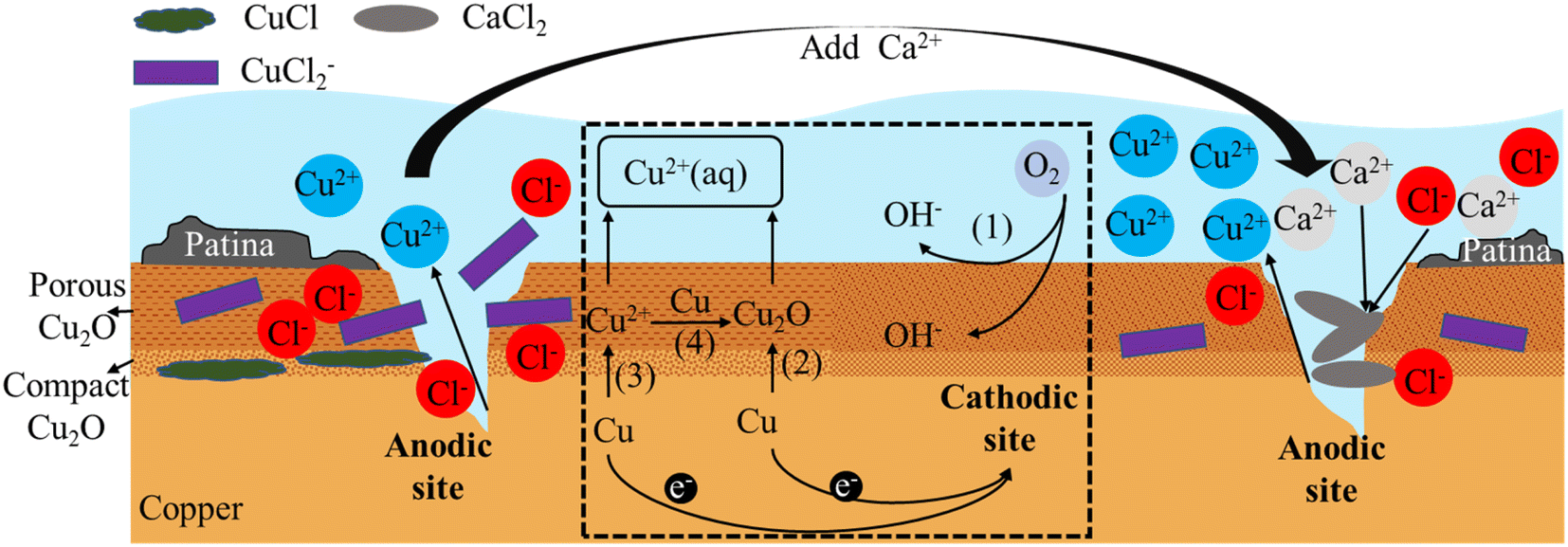 | ||
| Fig. 8 Schematic diagram of copper corrosion and by-product release under Cl− condition. | ||
 | ||
| Fig. 9 Schematic diagram of copper corrosion and by-product release under SO42− condition. | ||
In drinking water, copper was oxidized to Cu(II) or Cu(I), with the latter being slightly soluble in water. Thus, Cu2O film was the primary insoluble product in the copper corrosion process, and Cu2+ was the main soluble substance.43–45 A tremendous electronic barrier was present at the interface between the inner and outer layers of the film, causing an increase in electronic transfer resistance between the porous film and the metal. This resulted in an increase in charge transfer between the outer layer film and the copper substrate, making it more susceptible to further oxidation by oxidants at the electrolyte interface.
Fig. 8 illustrates the mechanisms underlying both one-electron and two-electron processes associated with the by-product release of copper and the formation of the copper film. The cathodic reaction is an oxygen reduction process (represented by (1) in Fig. 8 and reaction (1)). The anodic reaction mainly involves two processes.
The first step is one-electron reaction mechanism (represented by (2) in Fig. 8 and reaction (2)) involves the stepwise formation of slightly soluble cuprite (Cu2O), followed by the subsequent oxidation of cuprite into a soluble Cu(II) species.46,47 The second process is two-electron mechanism (represented by (3) in Fig. 8 and reaction (3)), also known as the by-product release-redeposition mechanism, involves the direct formation of Cu2+ followed by a synproportionation between Cu(0) and Cu(II) to form the sparingly soluble Cu2O48–52(represented by (4) in Fig. 8 and reaction (4)).
| O2 + 2H2O + 4e− → 4OH− | (1) |
| 2Cu + H2O − 2e− → Cu2O + 2H+ | (2) |
| Cu − 2e− → Cu2+ | (3) |
| Cu + Cu2+ + 2OH− → Cu2O + 2H2O | (4) |
The compact inner layer of Cu2O can restrict the further oxidation and corrosion of copper ions, thereby protecting the copper substrate. However, since the outer layer is porous, it cannot prevent oxygen and water from entering the interior, making the copper substrate still susceptible to corrosion. Moreover, the pores and cracks in the outer layer can provide pathways for harmful substances such as chloride ions and sulfate ions to enter the interior, accelerating the corrosion and by-product release of the copper substrate.
In the presence of Cl− or other complexing agents, Cl− ions with a smaller ionic radius can enter the Cu2O film as interstitial ions. When Cl− enters the Cu2O film, new Cu+ ions are generated to maintain electrical neutrality. These ions then react with Cl− to form CuCl deposits between the copper substrate and Cu2O.35 Once a sufficiently thick layer of CuCl is formed, it displays metastable behavior and can release CuCl2− ions into the water, promoting intergranular corrosion. Additionally, the electrical resistance values (Rp and Rb) of the dual-layer film, a mixture of CuCl and Cu2O, decrease,53 leading to faster ion transport and further diffusion of Cu2+ ions into the water.54 This results in more severe corrosion and explains the higher release of soluble copper ions observed in the Cl− condition mentioned above.
Upon introduction of Ca2+ into a Cl− solution, precipitation occurred, resulting in a decrease in the concentration of Cl− and hindering its penetration, thereby slowing down corrosion. The lower concentration of Cl− further reduced its permeation into the system, allowing the Cu2O film to maintain its integrity. Under Ca2+ condition, the charge transfer resistance (Rp) of the film observed in experiments increased by 25300 Ω cm2 compared to the Cl− condition. This increase in Rp resulted in an increase in the electron transfer resistance for reactions (2) and (3), which in turn slowed down copper corrosion and suppressed its occurrence. In this regard, Ca2+ inhibited the corrosive effect of Cl−.
Similarly, as the destruction of the Cu2O film was inhibited and other corrosion products were deposited on the outer layer of the film, an increase in the film resistance Rb by 2199 Ω cm2 was observed in the experiment. This increase resulted in a higher re-deposition resistance of the porous Cu2O film layer, which slowed down the process (4). Consequently, the concentration of copper ions in the solution increased, and the amount of by-product release per unit area increased by 14%. As mentioned earlier, under Cl− conditions, βa < βc, indicates that the cathodic reaction is the controlling factor and the corrosion process is mainly controlled by the dissolved oxygen transfer process. When Ca2+ is added, the electronic transfer resistance of both layers of the film increases, resulting in βa > βc. In this case, the anodic reaction becomes the controlling factor, reflecting the inhibitory effect of Ca2+ on the copper anodic reaction.
SO42− ions can inhibit copper corrosion within the concentration range of drinking water, but extremely high or low concentrations can result in the promotion of copper corrosion. This was related to the concentrations of SO42− and copper ions. When the concentration of SO42− was less than 0.01 mol L−1, it accelerated the formation of stable complexes and quickly formed a stable product film to inhibit corrosion.24 It was found in experiments that the addition of Ca2+ to the SO42− working condition resulted in an increase in Rb value and a decrease in Rp value. The decrease in Rp value was due to Ca2+ binding with SO42−, which reduced the amount of SO42− entering the Cu2O film, and consequently, the stable Cu4(OH)6SO4 generated on the film decreases,24 leading to a decrease in Rp value. As a result, the electron transferred resistance of reactions (2) and (3) decreased, promoting copper corrosion. The increase in Rb value was because the generated CaSO4 is insoluble and adhered to the outer layer of Cu2O film, leading to an increase in Rb value and a slowing down of the redeposition reaction, which in turn increased the copper ion concentration and promoted by-product release. Additionally, Ca2+ competed with Cu2+ for binding with SO42− and OH−, which also increased the copper ion concentration and promoted by-product release. In the presence of SO42−, the anodic reaction was the controlling factor, while the addition of Ca2+ had an effect on both βa and βc, mainly by increasing the anodic reaction rate and slowing down the cathodic reaction rate.
When Cl− and SO42− coexisted, their effects may compete with each other. While Cl− could promote copper corrosion, SO42− could inhibit corrosion.55 In this case, the effect of Cl− may be suppressed by SO42−, resulting in a reduced copper corrosion rate. The corrosion current density was lower than that of Cl− and higher than that of SO42−. The trend in by-product release was consistent with this.
Experimental observations revealed that the introduction of Ca2+ into a Cl− and SO42− coexisting system resulted in a decrease in Rp value. This suggested that Ca2+ initially binded with a large amount of SO42− in the solution, and then binded with the remaining Cl−. As discussed earlier, Cl− now had the opportunity to penetrate the Cu2O film as an interstitial ion and corroded it. At the same time, the stable Cu4(OH)6SO4 complex also reduced, thereby lowering the Rp value, accelerating electron transfer and promoting the corrosion reaction process (2) and (3).
Due to the preferential binding of Ca2+ with SO42− in the solution, CaSO4 was formed and attached to the outer layer of the Cu2O film. Additionally, Ca2+ can inhibit the destruction of the Cu2O film by Cl−. These factors led to an increase in the membrane resistance Rb value, resulting in a reduction in the deposition reaction (4) and an increase in the amount of copper leaching compared to the Cl− and SO42− conditions.
However, in the presence of all three ions (Cl−, SO42−, and Ca2+), their combined effects were complex and depended on various factors such as ion concentrations, pH, and solubility. Cl− and SO42− ions would react with copper separately, while Ca2+ combined with these ions to form insoluble precipitates, thereby affecting copper corrosion and by-product release. Generally, Cl− would promote copper corrosion, while SO42− ions within the range of drinking water quality concentrations would inhibit copper corrosion and by-product release.
In addition, temperature is one of the crucial factors influencing the corrosion process of copper. Variations in temperature can significantly affect the corrosion rate and the mode of corrosion of copper. Generally, as temperature increases, the corrosion rate of copper tends to increase as well.56
In the context of drinking water quality, elevated temperatures accelerate the rates of electrochemical reactions, leading to faster anodic dissolution. This results in a higher transfer rate of copper ions into the solution, meaning that more copper metal dissolves into the solution, thereby increasing the corrosion rate. Increasing temperature also enhances the diffusion rate of oxygen in the solution, leading to an increased oxygen depolarization rate and an elevated cathodic reduction rate.57
Moreover, rising temperatures also influence the physical and chemical properties of the solution, such as solubility, viscosity, and ion diffusion rate.58,59 These changes can impact the concentration distribution and transport rates of corrosive substances within the solution, further influencing the corrosion process of copper.
Lastly, water flow plays a significant role as it can alter the environmental conditions at the interface between the solid copper and the surrounding liquid, facilitating the release of copper from reactive layers.17 Higher water flow velocity can result in easier contact of the corrosive medium with the surface of copper, leading to the removal of the oxide film on the metal surface.60 This exposure of new metal surface further promotes the corrosion process of copper. The increased water flow velocity intensifies the erosive corrosion of copper metal and facilitates the dissolution of cuprous oxide, thereby increasing the release of copper corrosion by-products.38
It can be reasonably inferred that if copper pipes are used in a hot water system with high hardness, the corrosion rate of the pipes may accelerate, leading to the release of more corrosion by-products. Furthermore, a faster water flow velocity will further exacerbate this corrosion phenomenon.
5 Conclusions
The main focus of this study was to investigate the effect of Ca2+ on copper corrosion and by-product release in drinking water, including its impact on the corrosion and by-product release caused by Cl− and SO42− ions on the copper surface, as well as the influence of Ca2+ on copper corrosion and by-product release when Cl− and SO42− ions coexisted. The key findings were as follows:(1) The addition of Ca2+ in the water system had a mitigating effect on the corrosion reaction of copper surfaces compared to Cl− conditions, as evidenced by a positive shift of 0.022 V in Ecorr and a decrease of 0.235 μA cm−2 in Icorr. However, the amount of by-product release increased by 0.5 μg cm−2. The introduction of Ca2+ into the solution resulted in a significant increase in the charge transfer resistance of both the inner and outer layers of the corrosion product film. This could be attributed to the Ca2+ ions binding with Cl− ions, which in turn inhibited the penetration of Cl− ions into the film, thereby maintaining the integrity of the Cu2O film.
(2) The presence of Ca2+ altered the inhibitory effect of SO42− on copper corrosion, promoting corrosion with Ecorr at 0.093 V and an increase in Icorr of 1.021 μA cm−2, resulting in an increase in by-product release by 1.71μg cm−2. This effect was mainly attributed to the reaction between Ca2+ and SO42−, Cu2+ ions, leading to a decrease in the inner layer membrane resistance and an increase in the outer layer membrane resistance. This respectively represented a reduction in the resistance to copper corrosion reaction and an increase in the resistance to deposition reaction.
(3) In the Cl−/SO42− and Cl− conditions, corrosion of copper surfaces was observed, while in the SO42− condition, no significant corrosion was observed. The fitting parameter values for the Cl−/SO42− condition were located between those for the Cl− and SO42− conditions, indicating that Cl− played a dominant role in copper corrosion in the presence of both Cl− and SO42−. In terms of by-product release, SO42− could effectively inhibit the by-product release caused by Cl−, and played a dominant role in this regard. However, in the presence of both Cl− and SO42−, the addition of Ca2+ significantly promoted corrosion on the copper surface, leading to a decrease in Ecorr, an increase in Icorr, and an increase in by-product release.
The study conducted research on the corrosion and by-product release of copper surfaces within a limited time frame of 15 days. However, the structure and material composition of the corrosion product layer on copper surfaces changed with time, indicating the need for further research on the long-term changes in the corrosion product layer of copper surfaces. Conducting such research would provide valuable insights into the corrosion patterns of copper piping systems over time, allowing for a better understanding of the mechanisms and factors that contribute to by-product release. This information could ultimately inform the development of effective corrosion control strategies for copper plumbing systems.
Author contributions
Ping Xu: conceptualization, supervision, project administration, review and editing, validation, funding acquisition. Qiang Fu: data curation, methodology, investigation, data analysis, visualization, original draft preparation, drafts modification. Meihui Zhao: data curation, visualization, methodology, investigation, data analysis. All authors have read and agreed to the published version of the manuscript.Conflicts of interest
The authors declare there is no conflict.Acknowledgements
This study was supported by the National Natural Science Foundation of China (No. 51578035) and National Major Water Pollution and Treatment Project of China (No. 2018ZX07110-008-006). Thanks to Peihan Wu from Emory University for her valuable assistance with English translations and expressions of the manuscript.Notes and references
- U. R. Lenel and P. R. Mudd, Sol. Energy, 1984, 32, 109–120 CrossRef CAS.
- E. G. Stets, C. J. Lee, D. A. Lytle and M. R. Schock, Sci. Total Environ., 2018, 613–614, 1498–1509 CrossRef CAS PubMed.
- G. E. Pizarro, I. T. Vargas, P. A. Pastén and G. R. Calle, Bioelectrochemistry, 2014, 97, 23–33 CrossRef CAS PubMed.
- J. Han, L. Zhang, B. Ye, S. Gao, X. Yao, X. Shi, J. Han, L. Zhang, B. Ye, S. Gao, X. Yao and X. Shi, CCDC Weekly, 2023, 5(13), 297–300 Search PubMed.
- M. Edwards, M. R. Schock and T. E. Meyer, J.–Am. Water Works Assoc., 1996, 88, 81–94 CrossRef CAS.
- N. Boulay and M. Edwards, Water Res., 2001, 35, 683–690 CrossRef CAS PubMed.
- A. Fateh, M. Aliofkhazraei and A. R. Rezvanian, Arabian J. Chem., 2020, 13, 481–544 CrossRef CAS.
- Y. Zhao and R. Mirzaeifar, Appl. Surf. Sci., 2021, 538, 147925 CrossRef CAS.
- G. S. Sajadi, V. Saheb, M. Shahidi-Zandi and S. M. A. Hosseini, Sci. Rep., 2022, 12, 1–13 CrossRef PubMed.
- S. B. Adeloju and H. C. Hughes, Corros. Sci., 1986, 26, 851–870 CrossRef CAS.
- S. B. Adeloju and Y. Y. Duan, Br. Corros. J., 2013, 29, 315–320 CrossRef.
- S. B. Adeloju and Y. Y. Duan, Br. Corros. J., 1994, 29, 309–314 CrossRef CAS.
- S. Grace, D. A. Lytle and M. N. Goltz, J.–Am. Water Works Assoc., 2012, 104, E15–E25 CrossRef.
- D. A. Lytle and J. Liggett, Water Res., 2016, 92, 11–21 CrossRef CAS PubMed.
- J. Dartmann, B. Sadlowsky, T. Dorsch and K. Johannsen, Mater. Corros., 2010, 61, 189–198 CrossRef CAS.
- L. Yohai, W. H. Schreiner, M. Vázquez and M. B. Valcarce, Mater. Chem. Phys., 2013, 139, 817–824 CrossRef CAS.
- I. T. Vargas, D. A. Fischer, M. A. Alsina, J. P. Pavissich, P. Pablo and G. E. Pizarro, Materials, 2017, 10, 1–30 CrossRef PubMed.
- T. H. Merkel and S. O. Pehkonen, J. Am. Chem. Soc., 2013, 41, 21–27 Search PubMed.
- M. Edwards, K. Powers, L. Hidmi and M. R. Schock, Water Supply, 2001, 1, 25–32 CrossRef CAS.
- S. O. Pehkonen, A. Palit and X. Zhang, Corrosion, 2002, 58, 156–165 CrossRef CAS.
- A. E. Broo, B. Berghult and T. Hedberg, Corros. Sci., 1997, 39, 1119–1132 CrossRef CAS.
- T. J. Sorg, M. R. Schock and D. A. Lytle, J.–Am. Water Works Assoc., 1999, 91, 85–97 CrossRef CAS.
- C. Galarce, D. Fischer, B. Díez, I. T. Vargas and G. E. Pizarro, Water, 2020, 12, 1036 CrossRef CAS.
- M. Edwards, K. Powers, L. Hidmi and M. R. Schock, Water Sci. Technol.: Water Supply, 2001, 1, 25–32 CAS.
- H. M. Shalaby, F. M. Al-Kharafi and A. J. Said, Br. Corros. J., 1990, 25, 292–298 CrossRef CAS.
- M. H. Ghandehari, T. N. Andersen and H. Eyring, Corros. Sci., 1976, 16, 123–135 CrossRef CAS.
- D. Lee, Y. Lee and S. Nam, Asian J. Chem., 2008, 20, 5745–5759 CAS.
- X. Yan and J. Sun, Int. J. Electrochem. Sci., 2017, 12, 11580–11593 CrossRef CAS.
- M. A. Amin, J. Appl. Electrochem., 2006, 36, 215–226 CrossRef CAS.
- B. A. Boukamp and H. J. M. Bouwmeester, Solid State Ionics, 2003, 157, 29–33 CrossRef CAS.
- H. Liu and Y. F. Cheng, Electrochim. Acta, 2018, 266, 312–325 CrossRef CAS.
- A. El Warraky, H. A. El Shayeb and E. M. Sherif, Anti-Corros. Methods Mater., 2004, 51, 52–61 CrossRef CAS.
- P. B. Ahirrao, B. R. Sankapal and R. S. Patil, J. Alloys Compd., 2011, 509, 5551–5554 CrossRef CAS.
- D. M. Bastidas, U. Martin, J. M. Bastidas and J. Ress, Mater., 2021, 14, 6168 CrossRef CAS PubMed.
- T. H. Merkel and S. O. Pehkonen, J. Am. Chem. Soc., 2013, 41, 21–27 Search PubMed.
- A. M. Mohammed, S. S. Mohtar, F. Aziz, S. A. Mhamad and M. Aziz, J. Environ. Chem. Eng., 2021, 9, 105138 CrossRef CAS.
- S. B. Adeloju and Y. Y. Duan, Br. Corros. J., 2013, 29, 315–320 CrossRef.
- L. Wu, A. Ma, L. Zhang and Y. Zheng, Corros. Sci., 2022, 201, 110304 CrossRef CAS.
- R. M. Facey and D. W. Smith, J. Cold Reg. Eng., 1995, 9, 23–40 CrossRef.
- D. A. Lytle and M. R. Schock, J.–Am. Water Works Assoc., 2008, 100, 115–129 CrossRef CAS.
- C. Taxén, M. V. Letelier and G. Lagos, Corros. Sci., 2012, 58, 267–277 CrossRef.
- D. J. G. Ives and A. E. Rawson, J. Electrochem. Soc., 1962, 109, 447 CrossRef CAS.
- P. Zhou, M. J. Hutchison, J. R. Scully and K. Ogle, Electrochim. Acta, 2016, 191, 548–557 CrossRef CAS.
- G. Hultquist, P. Szakálos, M. J. Graham, A. B. Belonoshko, G. I. Sproule, L. Gråsjö, P. Dorogokupets, B. Danilov, T. Aastrup, G. Wikmark, G. K. Chuah, J. C. Eriksson and A. Rosengren, Catal. Lett., 2009, 132, 311–316 CrossRef CAS.
- T. Aastrup, M. Wadsak, M. Schreiner and C. Leygraf, Corros. Sci., 2000, 42, 957–967 CrossRef CAS.
- A. Palit and S. O. Pehkonen, Corros. Sci., 2000, 42, 1801–1822 CrossRef CAS.
- Y. Lu, H. Xu, J. Wang and X. Kong, Electrochim. Acta, 2009, 54, 3972–3978 CrossRef CAS.
- N. Boulay and M. Edwards, Water Res., 2001, 35, 683–690 CrossRef CAS PubMed.
- P. Zhou and K. Ogle, Encycl. Interfacial Chem. Surf. Sci. Electrochem., 2018, 11, 478–489 Search PubMed.
- T. Hurlen, M. Yhland, C. Krohn, K. Motzfeldt, O. Theander and H. Flood, Acta Chem. Scand., 1962, 16, 279–282 CrossRef CAS.
- T. Hurlen, E. Nilsson, R. Nilsson, G. E. Olsen, C. Pedersen and J. Toft, Acta Chem. Scand., 1961, 15, 1246–1254 CrossRef CAS.
- H. Lal and H. R. Thirsk, J. Chem. Soc., 1953, 2638–2644 RSC.
- D. B. Harrison, D. M. Nicholas and G. M. Evans, J.–Am. Water Works Assoc., 2004, 96, 67–76 CrossRef CAS.
- Y. Qiu, Q. Chen, F. Ye, X. Cai, D. Zhang and P. Fan, J. Shenzhen Univ., Sci. Eng., 2019, 36, 525–530 CrossRef.
- M. M. El-Naggar, Appl. Surf. Sci., 2006, 252, 6179–6194 CrossRef CAS.
- D. Cheng Kong, C. Fang Dong, K. Xiao and X. Gang Li, Trans. Nonferrous Met. Soc. China, 2017, 27, 1431–1438 CrossRef.
- G. Gunkel, U. Michels and M. Scheideler, Water, 2022, 14, 1246 CrossRef CAS.
- S. S. U. H. Kazmi, Y. Y. L. Wang, Y. E. Cai and Z. Wang, Ecol. Indic., 2022, 143, 109354 CrossRef CAS.
- B. Ma, C. Hu, J. Zhang, M. Ulbricht and S. Panglisch, ACS ES&T Water, 2022, 2, 259–261 Search PubMed.
- J. S. Fries, J. Civ. Hydraul. Eng., 2007, 133, 267–272 CrossRef.
| This journal is © The Royal Society of Chemistry 2023 |