
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceAnion-exchange facilitated selective extraction of sulfate and phosphate by overcoming the Hofmeister bias†
Anamika Gogoia,
Dipjyoti Duttaa,
Beatriz Gil-Hernández c and
Sandeep Kumar Dey*ab
c and
Sandeep Kumar Dey*ab
aMaterial Science and Technology Division, CSIR-North East Institute of Science and Technology, Jorhat 785006, Assam, India. E-mail: sandeep@neist.res.in; Tel: +91-7387633550
bAcademy of Scientific and Innovative Research (AcSIR), Ghaziabad 201002, Uttar Pradesh, India
cDepartmento de Química, Facultad de Ciencias, Sección Química, Universidad de La Laguna, La Laguna, 38206, Tenerife, Spain. E-mail: beagher@ull.es
First published on 31st May 2023
Abstract
Selective recognition and removal of sulfate and phosphates from aqueous media in the presence of highly competing anions is very demanding because of their biological and environmental implications. In this paper, we present the anion recognition approach for the selective and efficient extraction of sulfate by nitrophenyl-functionalized tris-urea receptors (L1–L2) from highly competitive aqueous media with an equivalent concentration of nitrate and other anions. Tetrabutylammonium hydroxide has been used for the first time as a phase transfer anionic extractant for sulfate-exchange from the aqueous phase to the organic phase (dichloromethane) containing a tris-urea receptor (L1–L3). The sulfate extraction efficacy of L2 (≈84–90%) was observed to be higher than those of L1 (≈76–82%) and L3 (≈68–75%) in competitive extraction experiments. In contrast, an analogous nitrophenyl-functionalized tris-thiourea receptor (L4) has been recognized for the selective and efficient extraction of phosphates from aqueous media in the presence of several competing anions including sulfate and nitrate, with ≈85–92% extraction efficiency. In this case, tetrabutylammonium acetate has been used as a phase transfer anionic extractant for phosphate exchange between the two immiscible phases. Due to the higher acidity of tris-thiourea –NH groups in comparison to the analogous tris-urea, tetrabutylammonium hydroxide could deprotonate a hydrogen bond donating –NH group of the thiourea receptor and phosphate extraction was observed to be inefficient in such a case. Several liquid–liquid extraction (LLE) experiments have been carried out to establish the selective removal of sulfate and phosphates by the tripodal receptors from competitive aqueous media having different combinations of two or more anions. The LLE products obtained from organic phases were characterized by NMR (1H, 13C, 31P, and 19F) spectroscopy to affirm the oxoanion selectivity of the receptors and purity of the complexes. The tripodal receptors can easily be recycled for successive extraction cycles by simply washing the LLE products (oxoanion complexes) with a methanol–water (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1, v/v) solvent system followed by filtration.
:1, v/v) solvent system followed by filtration.
Introduction
The design and synthesis of hydrogen bond donor (HBD) anion receptors for the selective recognition and separation of oxoanions, mainly sulfate and phosphates, are of significant relevance because of their importance in biology and ecology.1 The uncontrolled release of anthropogenic sulfate and phosphates is recognized to have an unfavourable impact on the environment.2 The extraction of sulfate from nitrate-rich alkaline aqueous media is a major goal in the remediation of nuclear wastes as sulfate interferes with the vitrification process.3 An excessive presence of phosphates and sulfate in fresh water ecosystems can lead to eutrophication to a significant extent, and an excess of sulfate is also held responsible for the permanent hardness of ground water.4 Liquid–liquid extraction (LLE) and liquid–solid extraction (LSE) of ionic species from water is a rapidly progressing industrial technology for selective anion separation motivated by the advances in understanding of anion recognition chemistry.5,6 The anion recognition approach for the development of synthetic separating agents for sulfate and phosphate extraction can relatively be affordable and highly efficient than selective crystallization technique, which is a comparatively slow process for anion separation in water.3,6Over the past two decades, several HBD receptors have been developed for the selective recognition of sulfate and phosphates in organic and (semi)aqueous media.7,8 However, only a limited number of receptors have been demonstrated to selectively separate sulfate and phosphate from aqueous media in the presence of competing anions.9 Both sulfate and phosphates have higher hydration energies in comparison to chloride, nitrate and other anions (ΔGHE −1080, −465, −350 and −305 kJ mol−1 for SO42−, H2PO4−, Cl− and NO3−, respectively), which makes it challenging for majority of the HBD anion receptors to overcome the Hofmeister bias for sulfate and phosphate separation from water. Nonetheless, Sessler and Moyer et al. has reported an alkylated cyclo[8]pyrrole, fluorinated calix[n]pyrroles (n = 4, 5) and bipyrrole-strapped calix[4]pyrroles which are capable of extracting sulfate from aqueous to organic phase by anion-exchange (1–5, Scheme 2).10 Bowman-James and Sessler et al. has also suggested that the use of HBD macrocycles might allow to overcome the Hofmeister bias for selective sulfate extraction from water in the presence of competing anions (6, Scheme 2).10b The term Hofmeister bias refers to the generally observed order of anion extraction/separation and follows the sequence, ClO4− > I− > NO3− > Br− > Cl− > F− > OH− > CH3CO2− > HPO42− > SO42− > CO32− > PO43−.11 Several ion-pair receptors consisting of urea or squaramide HBD groups have also been reported for the selective extraction of alkali sulfate salts from water.12
The choice of tripodal anion receptors (Scheme 1) for the present studies is based on their higher binding affinity for sulfate or phosphate among various anions.13 The anion binding affinity of the receptors (L1–L4) in solid and solution-states (DMSO-d6) have previously been reported by our group and Gale et al.13 The receptors have been shown to form self-assembled molecular capsules with oxoanions by hydrogen bond (HB) interactions, where the urea-based receptors (L1, L2 and L3) showed higher binding affinity for sulfate and the thiourea-based receptor (L4) showed higher binding affinity for hydrogenphosphate in 1H-NMR titration experiments. To find a possible application of these receptors in anion separation, we envisioned that the tris-urea/thiourea receptors (L1–L4) would facilitate the efficient extraction of sulfate and phosphates by anion-exchange between the two immiscible phases in the presence of a suitable quaternaryammonium salt.
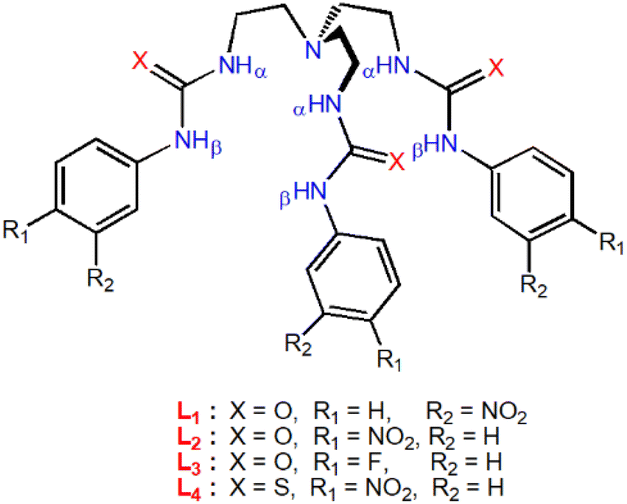 | ||
| Scheme 1 Molecular structures of urea and thiourea-functionalized tripodal anion receptors for selective sulfate and phosphate extraction (synthesis details in the ESI†). | ||
Wu et al. has recently reported a series of second generation tripodal hexa-urea receptors for selective sulfate extraction from an aqueous solution in the presence of competing anions (7, Scheme 2).14 Recently, we have reported that the positional isomers of a novel second-generation tripodal tris-urea receptors can selectively extract phosphate or arsenate from competitive aqueous media depending upon the position of the nitro group on the peripheral phenyl moiety (meta- vs. para-nitrophenyl).15 However, a second generation tripodal anion receptor is generally synthesized in three reaction steps, which would significantly raise the overall cost of LLE process using a quaternaryammonium salt as phase-transfer anionic extractant. We believe that the tren-based urea and thiourea-functionalized first generation tripodal receptors (Scheme 1) could possibly find applications in LLE of tetrahedral oxoanions from water in the presence of competing anions due to their well-established higher binding affinity for sulfate or phosphates. Along this line, Ghosh et al. has demonstrated the efficient extraction of sulfate from water with carbonate complexes of tris-urea receptors in the presence of competing anions (8, Scheme 2).16 The hydrogen bonded carbonate capsules of tris-urea receptors were obtained as crystals from dimethyl sulfoxide solution of the receptor mixed with tetrabutylammonium hydroxide. As crystallization is a slow process for supramolecular self-assembly and often show moderate yield of crystals, it is desirable to come up with a quick and efficient method of sulfate extraction using economically viable HBD receptors and quaternaryammonium salt. Further, tren-based tris-urea receptors can easily be synthesized in a single reaction step with high yields (>80%) under non-inert conditions, in contrast to calix[n]pyrroles, tetra-amide macrocycles, tripodal hexa-urea (Scheme 2) and squaramide-based ion-pair receptors which are often tedious to synthesize due to multi-step reactions.
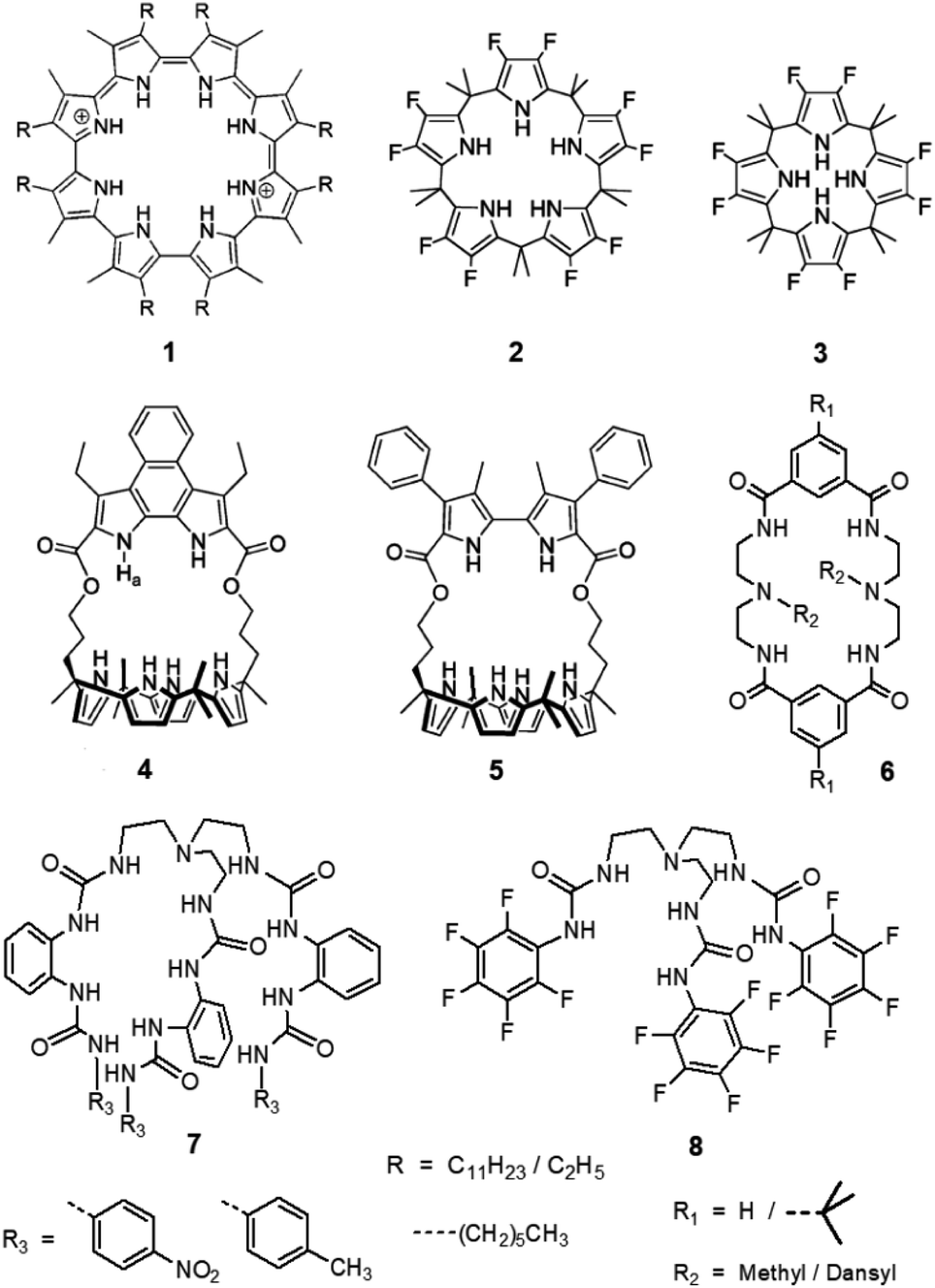 | ||
| Scheme 2 Notable HBD-based sulfate extractors known in the literature.10,14,16 | ||
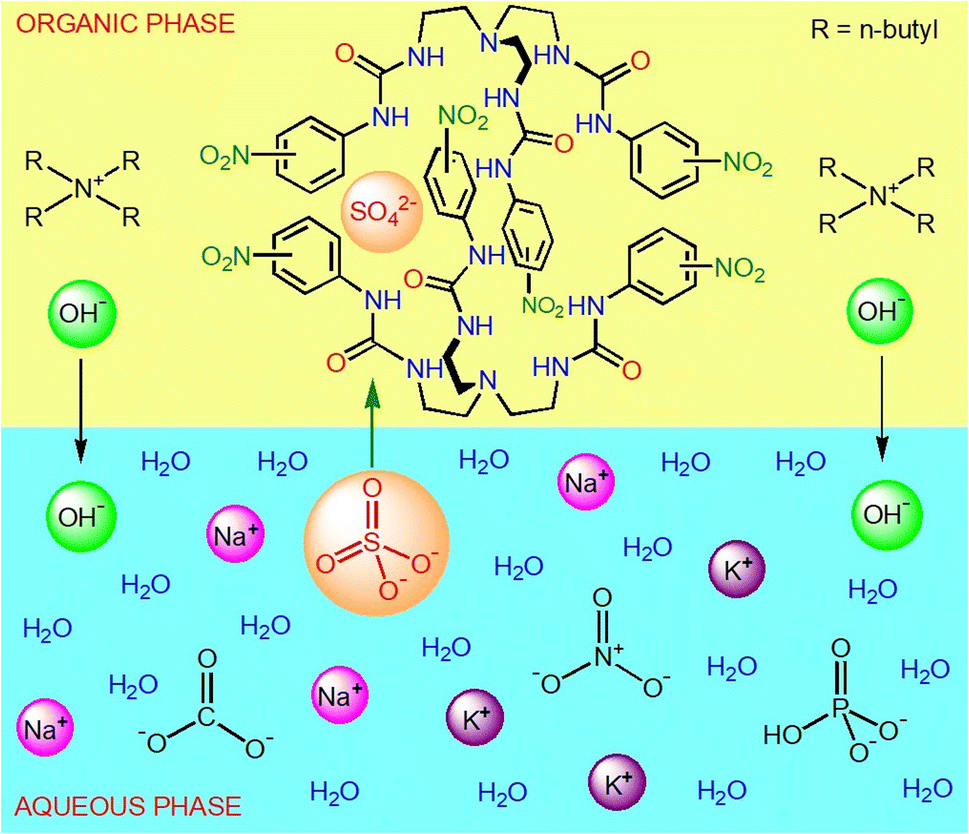
Motivated by the current research interest on the selective separation of oxoanions from highly competitive aqueous media,14–16 herein we report the LLE of sulfate by tris-urea receptors (L1, L2 and L3) from alkaline aqueous media in the presence of several competing anions (SeO42−, HPO42−, HAsO42−, CO32−, CH3CO2−, NO3−, Cl− and F−). Unlike the previous reports on sulfate extraction by HBD receptors using quaternaryammonium salts of chloride or nitrate, here we have used a sulfate selective tris-urea receptor mixed with tetrabutylammonium hydroxide, (n-Bu4N)+OH− for anion-exchange between the immiscible aqueous–organic phases. In contrast to the sulfate selective tris-urea receptors (L1, L2 and L3), tris-thiourea receptor (L4) can selectively and efficiently extract phosphate from an aqueous solution in the presence of competing oxoanions and halides. The differences in oxoanion selectivity of the analogous tris-urea and thiourea receptors (L2 and L4) arises due to the different extent of HBD acidity of –NH groups, where the more acidic tris-thiourea receptor prefers to selectively recognize the more basic phosphate due to receptor–anion complementarity. We have carried out a large set of competitive LLE experiments and NMR (1H, 13C, 31P and 19F) analysis of the LLE products to validate the anion selectivity of each receptor. We have also demonstrated that LLE is a quick and efficient method of obtaining the hydrogen bonded oxoanion complexes of urea and thiourea-based receptors with good yield and purity.
Results and discussions
Both urea and thiourea are strong hydrogen bond donor groups and the receptor–anion interactions can simply be observed in 1H-NMR spectroscopy, where a downfield chemical shift of the urea/thiourea –NH signals suggest hydrogen bonding between a receptor and an anion in the solution-state.14–17 In general, a large downfield shift of the –NH signals correspond to strong receptor–anion binding where the receptor–anion complementarity plays an important role in determining the anion selectivity i.e., the acidity of the urea/thiourea groups of a receptor should complement the basicity of the anion for selective anion recognition. Thus, it is anticipated that each of the anion complexes of tripodal receptors (L1–L4) would generate a distinct 1H-NMR spectrum showing different extent of downfield chemical shift of the –NH (–NHα or –NHβ) signals. The oxoanion (SO42−, SeO42−, HPO42−, HAsO42−, CO32−) complexes of tris-urea receptors (L1–L3) were prepared by LLE (water–dichloromethane) of an oxoanion by a receptor in the presence of two equivalents of (n-Bu4N)+OH− acting as anion-exchanger between the aqueous–organic phases, and the hydrogen bonded complexes were obtained from the organic phase (see ESI†). Due to the higher acidity of thiourea –NH groups, the oxoanion complexes of tris-thiourea receptor (L3) were obtained by LLE (water-dichloromethane) of an oxoanion by the receptor in the presence of two equivalents (n-Bu4N)+CH3CO2− (see ESI†). On the other hand, chloride, acetate, nitrate and hydroxide complexes were obtained from the organic phase after thoroughly washing the dichloromethane solution of a tripodal receptor (L1–L4) and a tetrabutylammonium salt (Cl−, CH3CO2−, NO3− and OH−) with an aqueous solution of alkali metal salt of the respective anions (see ESI†). After complete characterization of the isolated anion complexes by NMR spectroscopy (DMSO-d6), LLE of sulfate and phosphate has been carried out from aqueous media in the presence of one or more competing anions by the tripodal receptors (see ESI†).Anion complexes of tris-ureas
The 1H-NMR spectrum of meta-nitrophenyl functionalized tris-urea receptor (L1) showed the appearance of urea –NH signals at 6.3 and 9.0 ppm for –NHα and –NHβ, respectively (Fig. 1). The aromatic region of the 1H-NMR spectrum of sulfate complex [(n-Bu4N)2(2L1·SO4)] has clearly been observed to be distinctively different when compared to the spectra of other anion complexes (Fig. 1), except having a close similarity with the selenate complex [(n-Bu4N)2(2L1·SeO4)] (Fig. S5 and S6, ESI†). In particular, one of the urea –NH signals (–NHα or –NHβ) have appeared at different chemical shift (δ) value in different anion complexes of L1 (Fig. 1 and Table 1). Notably, the –NHβ signals of [(n-Bu4N)(L1·CH3CO2)] (acetate complex), [(n-Bu4N)(L1·OH)] (hydroxide complex), and [(n-Bu4N)2(2L1·CO3)] (carbonate complex) showed large downfield shifts (Δδ) of 1.0, 1.3 and 1.5 ppm, respectively with respect to the free receptor L1 (Fig. 1 and Table 1). Although the –NHβ signals of sulfate complex and chloride complex [(n-Bu4N)(L1·Cl)] were observed to appear at similar positions (Δδ 0.6 ppm), but the downfield shift of –NHα signals were distinctly different exhibiting larger shift in the sulfate complex (Fig. 1). The downfield shift of –NHα,β signals were observed to be larger in the arsenate complex [(n-Bu4N)2(2L1·HAsO4)] than in the phosphate complex (Table 1, Fig. S12 and S13, ESI†). 31P-NMR spectrum of the phosphate complex showed two signals at 4.5 and 8.8 ppm suggesting the presence of two different phosphate species in the isolated complex from LLE experiment (Fig. S14, ESI†). Negligible downfield shifts of –NHα,β signals were observed in the spectrum of nitrate complex [(n-Bu4N)(L1·NO3)] suggesting weak receptor–anion interactions (Fig. 1).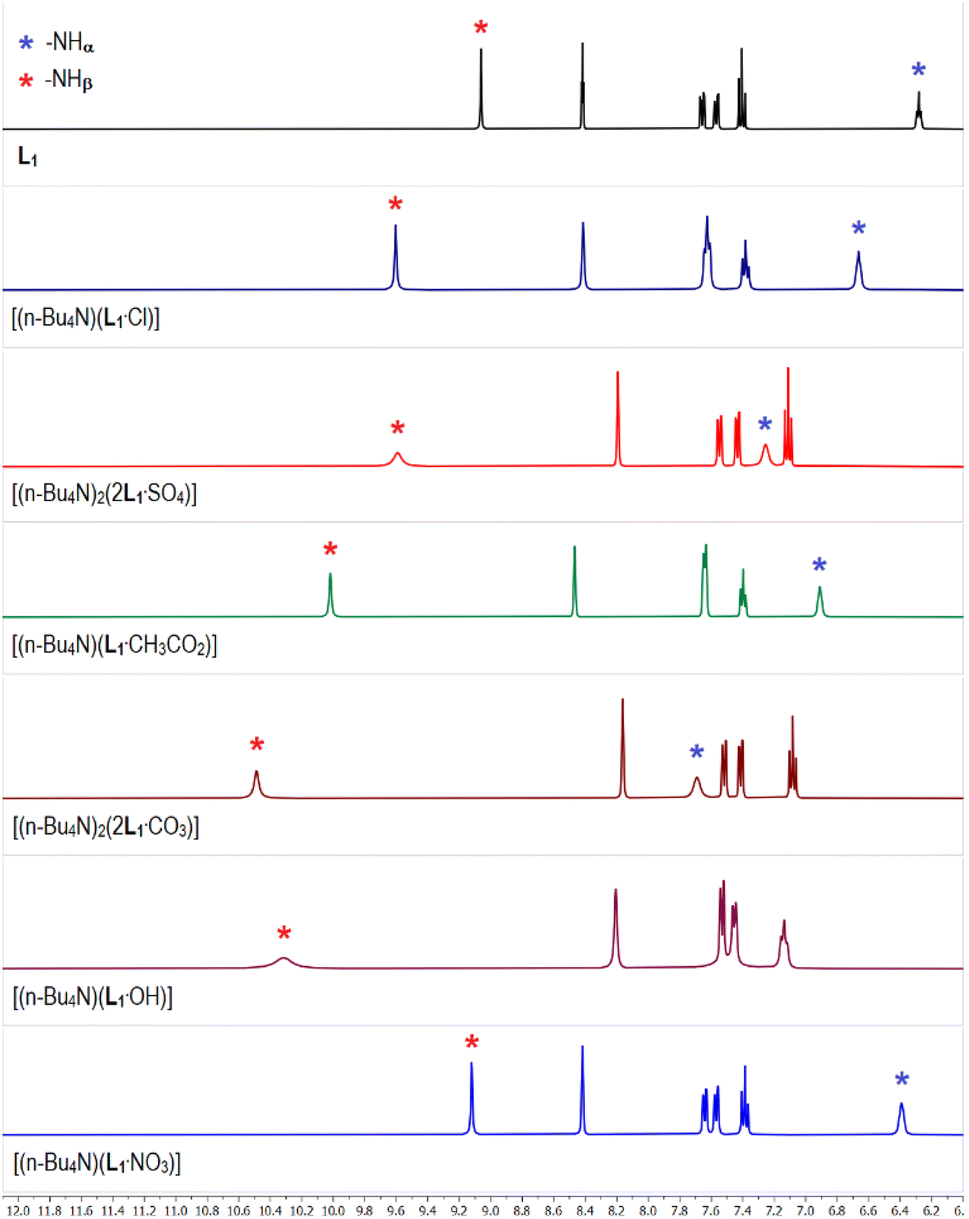 | ||
| Fig. 1 Aromatic region of the 1H-NMR (DMSO-d6) spectra of L1 and the receptor–anion complexes showing variable downfield shift of the urea –NH signals in different complexes relative to L1. (Full spectra are provided in the ESI, Fig. S5–S13†). The 1H-NMR spectra of selenate, phosphate and arsenate complexes of L1 are provided in the ESI.† | ||
| Anion complexes of L1-4 | L1-NHα | L1-NHβ | L2-NHα | L2-NHβ | L3-NHα | L3-NHβ | L4-NHα | L4-NHβ |
|---|---|---|---|---|---|---|---|---|
| Ln (n = 1, 2, 3 or 4) | 6.3 | 9.0 | 6.4 | 9.3 | 6.1 | 8.5 | 8.1 | 10.2 |
| [(n-Bu4N)2(2Ln·SO4)] | 7.3 | 9.6 | 7.4 | 10.0 | 7.0 | 9.1 | 9.1 | 11.0 |
| [(n-Bu4N)2(2Ln·SeO4)] | 7.3 | 9.7 | 7.5 | 10.1 | 6.8 | 9.1 | 9.0 | 11.0 |
| [(n-Bu4N)2(2Ln·HPO4)] | 7.2 | 9.7 | 7.3, 10 | 10.1, 13.0 | 6.8 | 9.1 | 8.8, 11.8 | 10.8, 12.9 |
| [(n-Bu4N)2(2Ln·HAsO4)] | 7.5 | 10.2 | 7.4, 9.7 | 10.3, 13.1 | 6.7 | 9.1 | 9.0, 11.8 | 11.4, 13.0 |
| [(n-Bu4N)2(2Ln·CO3)] | 7.7 | 10.5 | 7.9 | 10.8 | 7.4 | 9.6 | NO | NO |
| [(n-Bu4N)(Ln·CH3CO2)] | 6.9 | 10.0 | 7.4 | 10.5 | 6.8 | 9.4 | 8.9 | 11.3 |
| [(n-Bu4N)(Ln·Cl)] | 6.6 | 9.6 | 6.8 | 9.8 | 6.5 | 9.1 | 8.5 | 10.8 |
| [(n-Bu4N)(Ln·OH)] | 7.2 | 10.3 | 7.7 | 10.6 | NO | 9.4 | NO | NO |
| [(n-Bu4N)(Ln·NO3)] | 6.4 | 9.1 | 6.5 | 9.4 | 6.1 | 8.5 | 8.3 | 10.3 |
The 1H-NMR spectrum of para-nitrophenyl functionalized tris-urea receptor (L2) showed the urea –NH signals at 6.4 and 9.3 ppm for –NHα and –NHβ, respectively (Fig. 2). An appreciable downfield shift (Δδ > 1 ppm) of –NHβ signals were observed in the spectra of acetate, carbonate and hydroxide complexes of L2 (Fig. 2 and S15–S17, ESI†), similar to the analogous anion complexes of L1 (Fig. 1). Notably, the 1H-NMR spectra of sulfate and selenate complexes of L2 were observed to be comparable, and two sets of –NHα,β signals were observed in the spectrum of phosphate complex [(n-Bu4N)2(2L2·HPO4)] suggesting the presence of two different phosphate species (HPO42− and PO43−) in the isolated complex from LLE experiment (Fig. 2 and Table 1). A similar 1H-NMR spectrum has also been observed for the arsenate complex [(n-Bu4N)2(2L2·HAsO4)], as phosphate and arsenate are oxoanions of the same group (Group-15) similar to sulfate and selenate (Group-16) and thus, have similar chemical properties. The percentage of [2L2·PO4]3− and [2L2·HPO4]2− present in the isolated phosphate complex can be quantified by integrating the aromatic –CH signals, where the signals at 7.7 and 7.9 ppm (2 –CH each) have appeared exclusively for the [2L2·PO4]3− and [2L2·HPO4]2−, respectively and the signal at 7.5 ppm is a combination of both the hydrogen bonded adducts (Fig. S20, ESI†). The –CH integral values suggested that the ratio of [2L2·PO4]3− and [2L2·HPO4]2− present in the isolated phosphate complex is ≈40:60. The integral values of urea –NH signals can also be used to calculate the PO43−/HPO42− ratio, but due to the broad nature of –NH signals, the ratio may significantly vary (≈35–40:65–60). Similarly, the ratio of [2L2·AsO4]3− and [2L2·HAsO4]2− present in the isolated arsenate complex was observed to be ≈20:80, as calculated from the –CH (7.7 and 7.9 ppm) and –NH (10.3 and 13.1 ppm) integral values.
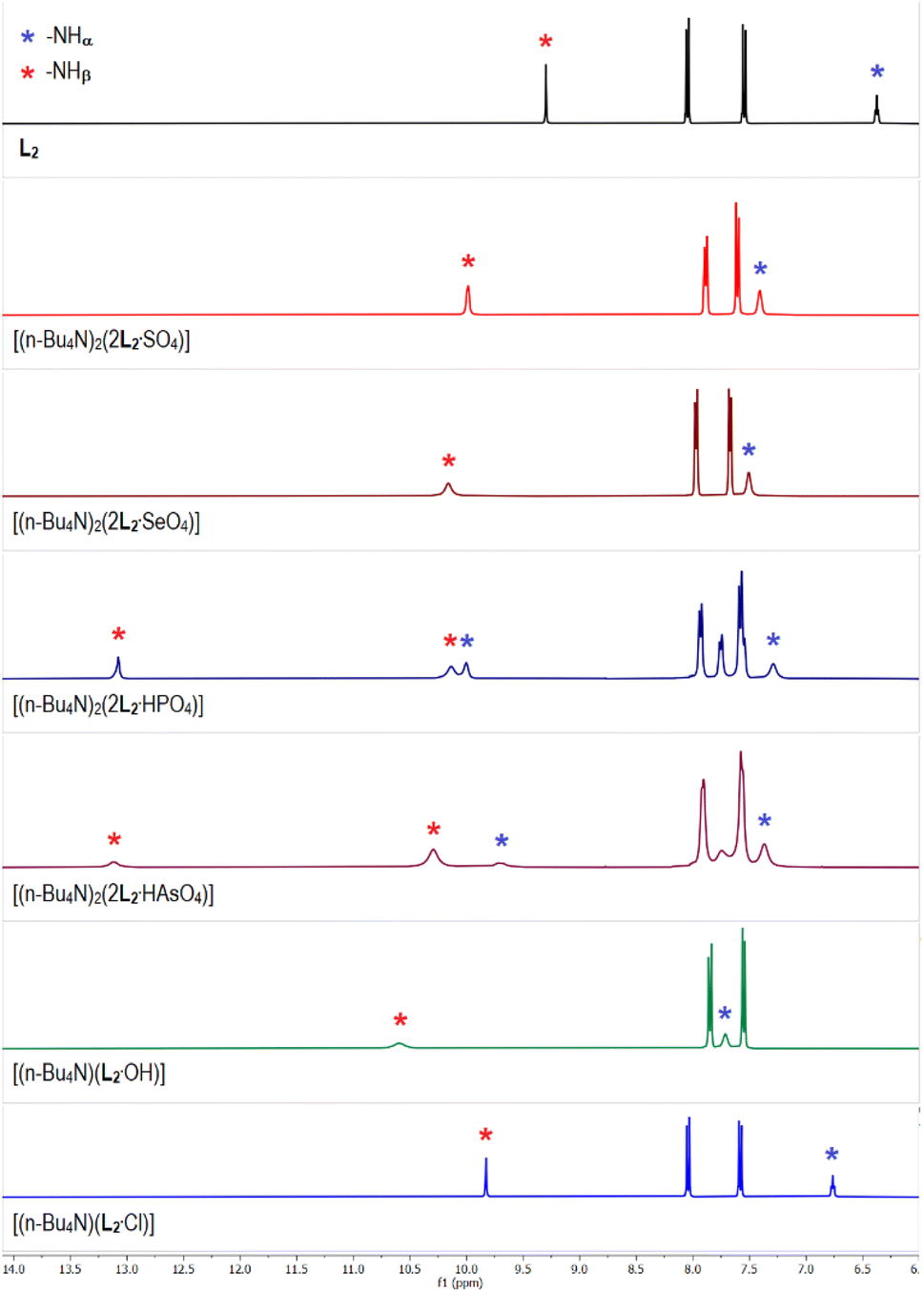 | ||
| Fig. 2 Aromatic region of the 1H-NMR (DMSO-d6) spectra of L2 and the receptor–anion complexes showing variable downfield shift of the urea –NH signals in different complexes relative to L2 (full spectra are provided in the ESI, Fig. S15–S24†). The 1H-NMR spectra of carbonate, acetate and nitrate complexes of L2 are provided in the ESI.† | ||
The 1H-NMR spectrum of fluorophenyl functionalized tris-urea receptor (L3) showed the urea –NH signals at 6.1 and 8.5 ppm for –NHα and –NHβ, respectively suggesting that the –NH groups are less acidic (polarized) in L3 than its nitrophenyl analogue L2 (Fig. S3† and Table 1). The urea –NHα,β signals have experienced different extent of downfield chemical shift in the anion complexes of L3. The carbonate complex [(n-Bu4N)2(2L3·CO3)] has shown the highest shift of –NHα,β signals (Δδ 1.1–1.3 ppm) and negligible shift has been observed in the nitrate complex [(n-Bu4N)(L3·NO3)]. Although the –NHβ signals of sulfate, phosphate and chloride complexes have appeared at similar positions (Δδ 0.6 ppm), but the shift of –NHα signals were observed to be distinctly different (Table 1 and Fig. S26–S35, ESI†).
Single crystals of sulfate complex [(n-Bu4N)2(2L3·SO4)] was obtained from a dimethyl sulfoxide solution of the LLE product (sulfate extraction by L3) at room temperature. The hydrogen bonded complex has crystallized in monoclinic I2/a space group. X-ray structure elucidation revealed the formation of a sulfate encapsulated dimeric capsular assembly of the receptor (Fig. 3). The encapsulated sulfate ion forms sixteen N–H⋯O hydrogen bonds (mean N⋯O distance = 3.12 Å) with the six urea groups of two oppositely oriented receptor molecules (Table S1, ESI†). The dimeric assembly is further stabilized by two aromatic C–H⋯π interactions (C⋯π distance = 3.54 Å) between the fluorophenyl groups of the receptor molecules.
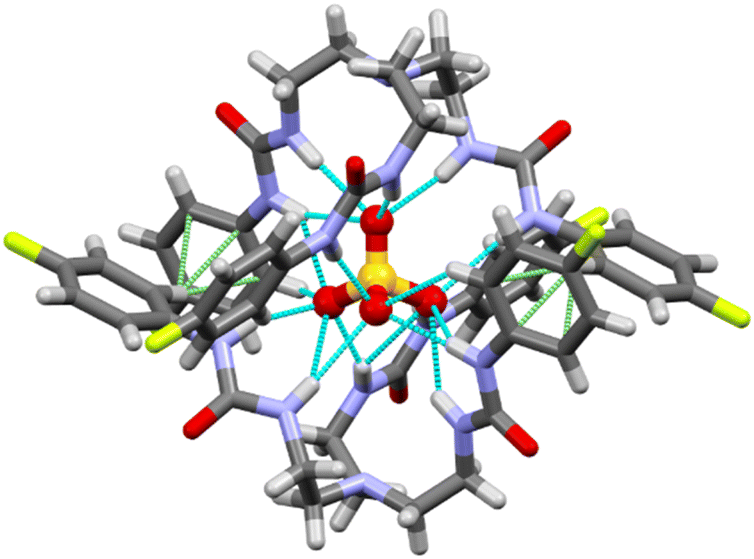 | ||
| Fig. 3 X-ray structure of the sulfate-encapsulated complex [(n-Bu4N)2(2L3·SO4)],‡ tetrabutylammonium cations are not shown for clarity. N–H⋯O hydrogen bonds are shown in blue dotted lines and C–H⋯π interactions are shown in green dotted lines. | ||
Selective extraction of sulfate
Based on the observed anion affinity (association constants) of tris-urea receptors (L1–L3) as determined from quantitative 1H-NMR experiments,13 we have carried out liquid–liquid extraction (LLE) of sulfate by a tris-urea receptor in the presence of several competitive anions (selenate, hydrogenphosphate, nitrate, carbonate, acetate and chloride/fluoride). In the first set of LLE experiments, L1 (100 mg) was dissolved in dichloromethane (20 mL) in the presence of two equivalents of (n-Bu4N+)OH− and an aqueous solution of sulfate and a competing anion (one equiv. each of Na2SO4 and an alkali metal salt of competing anion dissolved in 20 mL of deionized water) was added to the organic media and stirred for an hour. In the second set of LLE experiments, an aqueous solution mixture of sulfate and two competing anions (one equiv. alkali metal salts of (i) SO42−, HPO42− and NO3−, (ii) SO42−, CH3CO2− and CO32−, (iii) SO42−, Cl− and F− dissolved in 20 mL of deionized water) was added to the dichloromethane solution and stirred for an hour. The pH of the aqueous media containing different combinations of alkali metal salts was adjusted to about 10.0 (±0.2) in each case by adding a few drops of dilute NaOH solution, considering the importance of sulfate separation from alkaline nuclear waste media. The receptor–anion complexes were obtained from the dichloromethane phase as yellow powders with ≈76–82% yield (relative to L1) depending on the competing anions. However, in the absence of competing anions, about 85% yield was obtained in sulfate extraction experiment. The 1H-NMR spectra of all the LLE products closely resemble the spectrum of [(n-Bu4N)2(2L1·SO4)] (Δδ –NHα 1.0 ± 0.1 and Δδ –NHβ 0.6 ± 0.1), suggesting the selective extraction of sulfate from competitive aqueous solution (Fig. 4 and S36–S46, ESI†). In a control experiment, the sulfate complex obtained after thoroughly washing a dichloromethane solution of L1 and (n-Bu4N)+HSO4− (two equiv.) with an aqueous solution of Na2SO4 (one equiv.) showed similar 1H-NMR spectrum with four aromatic –CH and two –NH signals (Fig S47, ESI†). 31P, 13C and 19F NMR spectra of the compounds obtained from the above specified set of LLE experiments showed the absence of signals for HPO42−, CO32−, CH3CO2− and F−, and thereby affirming the clean extraction of sulfate ions in the presence of competing anions. The selective extraction of sulfate by L1 has also been achieved in the presence of three competing anions such as, HPO42−, NO3−, CO32− and SeO42−, CH3CO2−, F− (Scheme 3 and Fig. S45 and S46, ESI†). EDX analysis of the compounds were also performed to validate the absence of competing anions like, selenate and arsenate.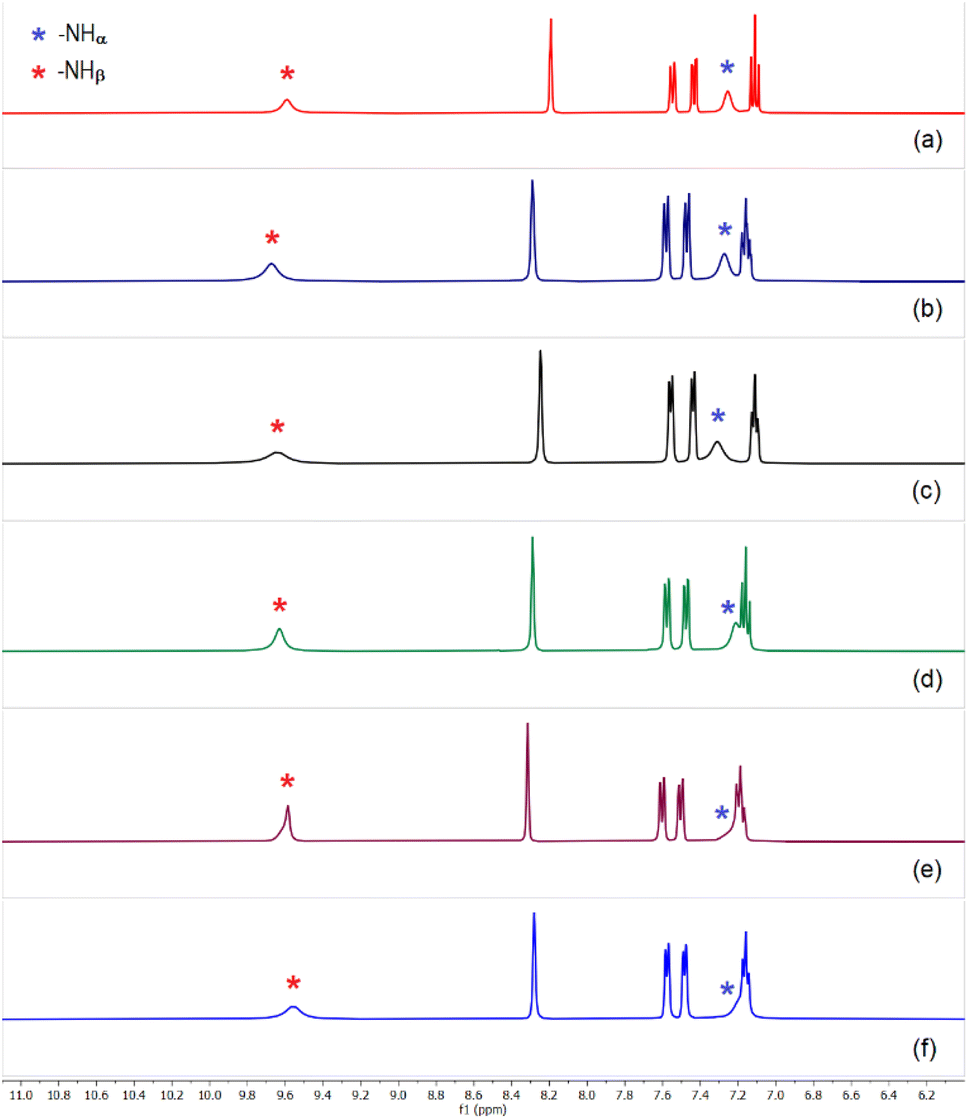 | ||
| Fig. 4 Aromatic region of the 1H-NMR spectra of sulfate complex [(n-Bu4N)2(2L1·SO4)] obtained in the presence of (n-Bu4N)+ hydroxide and (a) selenate, (b) hydrogenphosphate, (c) carbonate and acetate, (d) chloride and fluoride, (e) hydrogenphosphate and nitrate, (f) hydrogenphosphate, carbonate and nitrate, in LLE experiments. (Full spectra are provided in the ESI, Fig. S36–S46†). The spectrum in each case closely resembles the spectrum of sulfate complex, [(n-Bu4N)2(2L1·SO4)] affirming clean sulfate extraction by L1. | ||
 | ||
| Scheme 3 Recognition-guided selective extraction of sulfate by nitrophenyl-functionalized tris-urea receptors from competitive aqueous media at pH = 10. | ||
Similar set of competitive LLE experiments have also been performed with L2 (Scheme 1). Sulfate could easily be extracted from an aqueous to an organic phase within an hour in the presence of a competing oxoanion such as, selenate, hydrogenphosphate, carbonate and acetate at pH ≈ 10 (Fig. 5 and S48–S51, ESI†). Efficient sulfate extraction has also been achieved in the presence of two and three competing anions (CO32−, CH3CO2−/NO3−/Cl− and HPO42−, CO32−, NO3−/Cl−) with ≈84–90% yield of sulfate complex relative to L2 (Fig. 5 and S54–S58, ESI†). It is important to mention that the presence of an equivalent amount of Cl− or NO3− in an aqueous solution of SO42− has shown some degree of partial interference towards sulfate extraction by L1 and L2, within an hour of LLE time (Fig. S40, S52 and S53, ESI†). However, the interference from Cl− and NO3− have been overcome by the addition of an equivalent amount of F−, CO32− or HPO42− to the aqueous solution mixture (Fig. 4, 5, S41–43 and S54–56, ESI†). An additional LLE time of 15–20 minutes has also resulted in clean sulfate extraction by L1 and L2 in the presence of Cl− or NO3− as a competing anion.
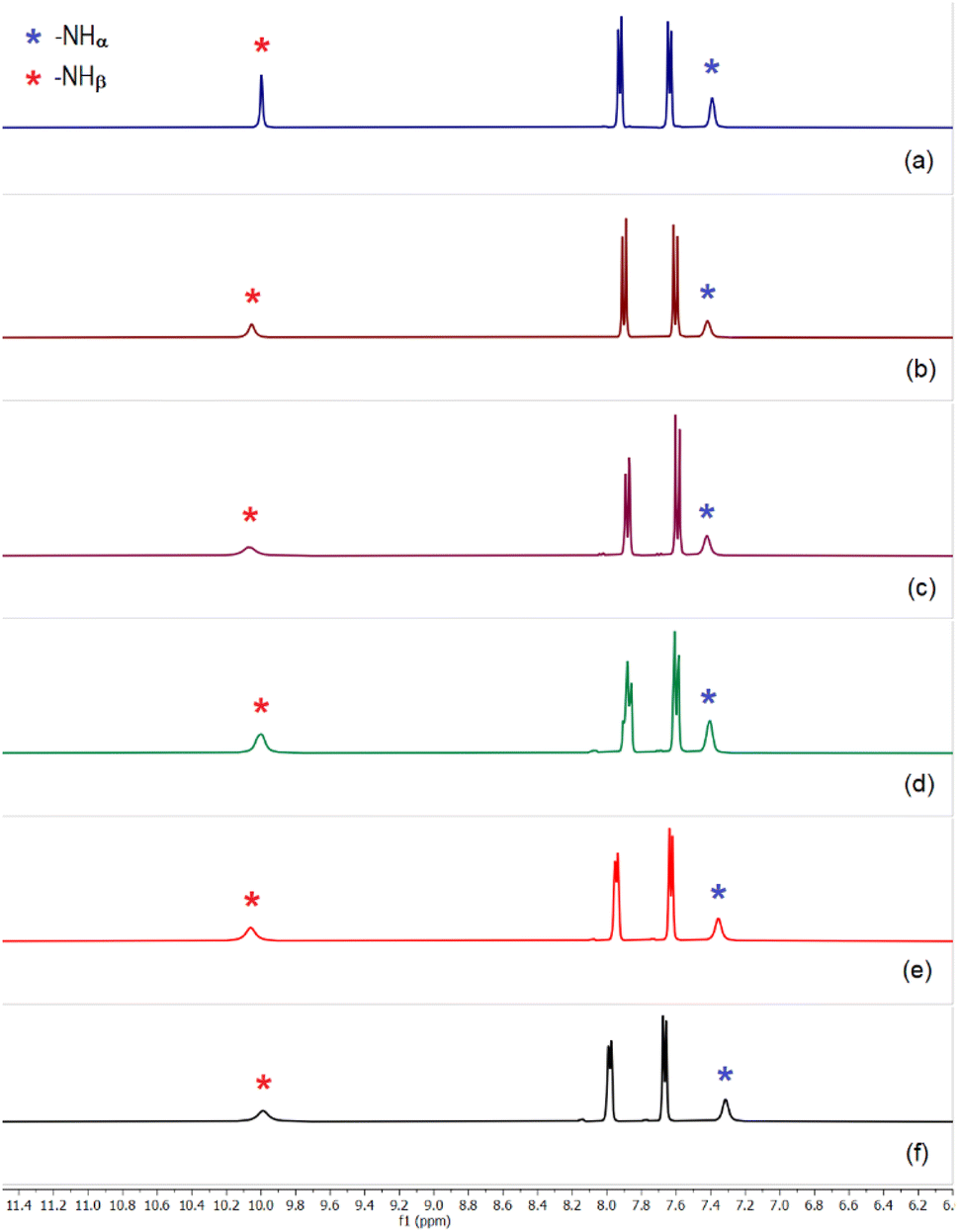 | ||
| Fig. 5 Aromatic region of the 1H-NMR spectra of sulfate complex [(n-Bu4N)2(2L2·SO4)] obtained in the presence of (n-Bu4N)+ hydroxide and (a) hydrogenphosphate, (b) selenate, (c) carbonate, (d) acetate, (e) carbonate and chloride (f) hydrogenphosphate, carbonate and nitrate, in LLE experiments (Full spectra are provided in the ESI, Fig S48–S58†). All the spectra closely resemble the spectrum of [(n-Bu4N)2(2L2·SO4)] affirming clean sulfate extraction by L2. | ||
Unlike L2, sulfate extraction efficiency of fluorophenyl-functionalized tris-urea receptor was comparatively lower (≈68–75% relative to L3) in competitive media and clean sulfate extraction could not be achieved in the presence of an equivalent amount of Cl− or NO3− (Fig. 6 and S67–69, ESI†), even in the presence of CO32− and HPO42− or a longer LLE time (1.5 hours). However, sulfate could efficiently be extracted by L3 in the presence of other competing anions such as, fluoride, hydrogenphosphate, carbonate and acetate within an hour of LLE time at pH ≈ 10 (Fig. 6). Powder XRD patterns of [(n-Bu4N)2(2L3·SO4)] obtained in the presence of competing anions were observed to be in close similarity with the simulated XRD pattern of the crystal structure (see ESI†).
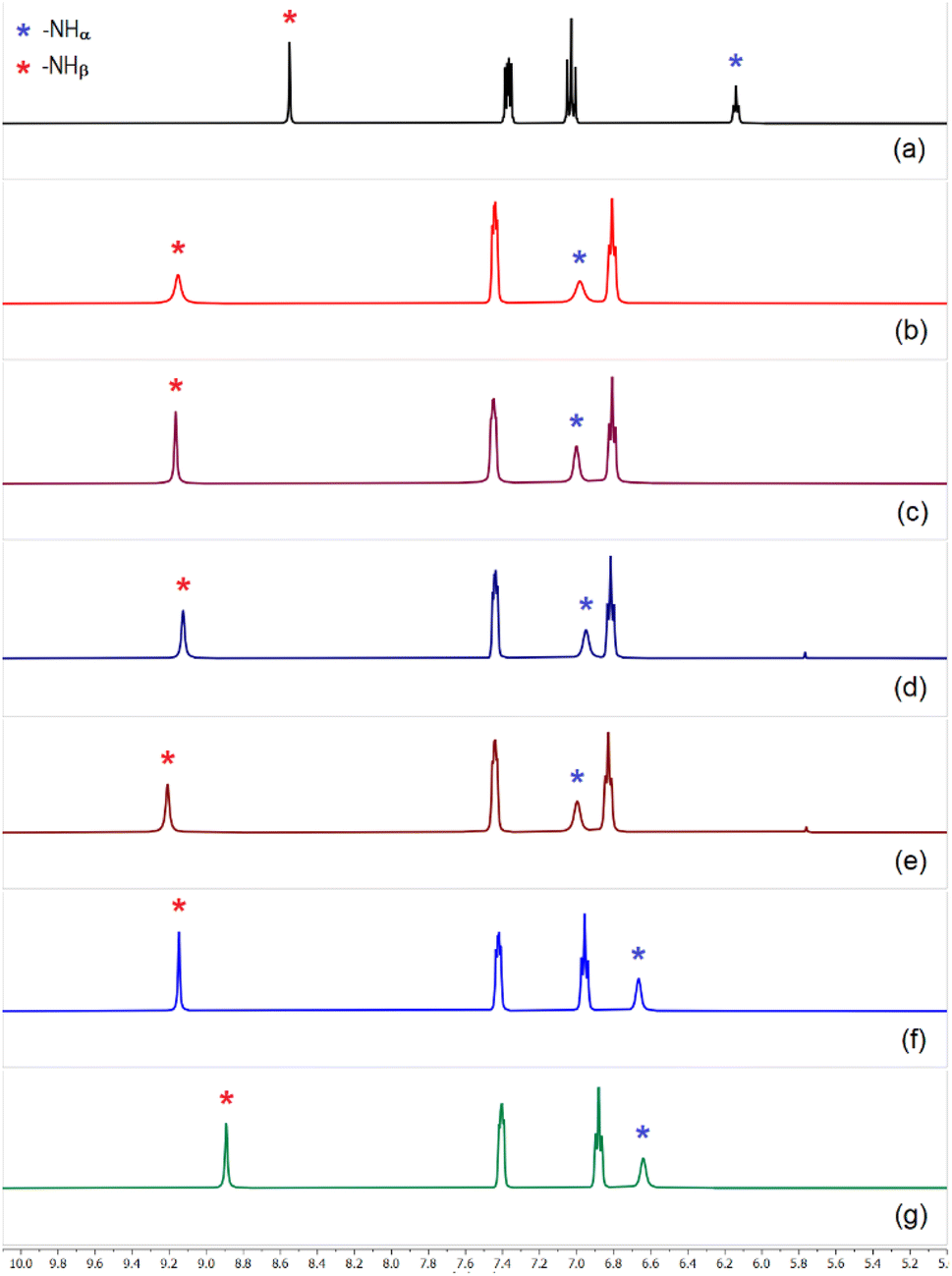 | ||
| Fig. 6 Aromatic region of the 1H-NMR spectra of (a) L3 and spectra of sulfate complex [(n-Bu4N)2(2L3·SO4)] obtained in the presence of (n-Bu4N)+ hydroxide and (b) hydrogenphosphate (c) hydrogenphosphate and fluoride, (d) carbonate and acetate, (e) hydrogenphosphate, carbonate and fluoride, (f) spectrum showing inefficient extraction of sulfate by L3 in the presence of chloride and carbonate, (g) spectrum showing no extraction of sulfate in the presence of nitrate in LLE experiments (full spectra are provided in the ESI, Fig. S59–S69†). | ||
It is important to highlight that the association constants (Ka calculated by 1H-NMR in DMSO-d6) for sulfate binding by the tris-urea receptors (L1–L3) were reported in the order of > 104 M−1 (logKa 4–4.5, error limit 10–15%).13 Competitive LLE experiments have revealed that the nitrophenyl-functionalized tris-urea receptors are better sulfate extractors than their fluorophenyl analogue considering the extraction efficacy and sulfate selectivity. Nitrophenyl bonded to a urea –NH group can induce greater polarization of urea groups (higher –NH acidity) in comparison to halophenyl aromatic and thus, L1 and L2 form stronger hydrogen bonds with anions in the solution-state which resulted in superior sulfate selectivity.
Anion complexes of tris-thiourea
The 1H-NMR spectrum of tris-thiourea receptor L4 showed the –NH signals at 8.1 and 10.2 ppm for –NHα and –NHβ, respectively suggesting that the thiourea –NH groups are more acidic (polarized) than its urea analogues L1–L2 (Fig. 7 and Table 1). The aromatic region of the 1H-NMR spectrum of phosphate complex [(n-Bu4N)2(2L4·HPO4)] has clearly been observed to be distinctively different in comparison to rest of the anion complexes of L4 (Fig. 7), except having a close similarity with the arsenate complex [(n-Bu4N)2(2L4·HAsO4)] (Fig. S70 and 71, ESI†). Similar to the phosphate/arsenate complexes of L2, two sets of –NHα,β signals were also observed in the 1H-NMR spectra of phosphate and arsenate complexes of L4 suggesting the presence of two different phosphate/arsenate species in the isolated complexes from LLE. In a control experiment, the phosphate complex obtained after thoroughly washing a dichloromethane solution of L4 and (n-Bu4N)+H2PO4− (two equiv.) with an aqueous solution of K2HPO4 (one equiv.) showed a similar 1H-NMR spectrum with two sets of –NHα,β signals and three aromatic –CH signals. The aromatic –CH signals at 7.9 and 8.0 ppm (2 –CH each) have appeared for [2L4·HPO4]2−, and the –CH signal at 7.7 ppm (4 –CH) suggested the presence of [2L4·PO4]3− (Fig. 7). The –NHα,β signals for [2L4·PO4]3− were observed to appear at higher δ values (–NHα 11.8 ppm, and –NHβ 12.9 ppm) in comparison to the downfield shift of –NHα,β signals for [L4·HPO4]2− (–NHα 8.8 ppm, and –NHβ 10.8 ppm). The percentage of [2L4·PO4]3− and [2L4·HPO4]2− present in the isolated phosphate complexes has been quantified by integrating the aromatic –CH signals. For example, [2L4·PO4]3− to [2L4·HPO4]2−ratio in complexes obtained in the presence of tetrabutylammonium CH3CO2−, Cl−, F− and NO3− were calculated to be ≈10:90, 20:80, 60:40 and 85:15, respectively (control experiments, ESI†). The 1H-NMR spectrum of carbonate complex [(n-Bu4N)2(2L4·CO3)], showed disappearance of the thiourea –NHα,β signals, indicative of anion-induced broadening of highly polarized –NH peaks (Fig. 7). The 1H-NMR spectra of sulfate complex [(n-Bu4N)2(2L4·SO4)], and selenate complex [(n-Bu4N)2(2L4·SeO4)] were observed to be similar, and an appreciable downfield shift of the –NHα,β signals (Δδ 0.8–1.1 ppm) have been observed in the sulfate/selenate and acetate complexes relative to the free receptor (Fig. 7 and S73–S75 ESI†). The downfield shift of –NHα,β signals in chloride complex [(n-Bu4N)(L4·Cl)] was observed to be comparatively less (Δδ 0.4–0.6 ppm) suggesting weaker receptor–chloride interactions than receptor–oxoanion interactions (Fig. 7). However, negligible downfield shift of the –NHα,β signals in nitrate complex [(n-Bu4N)(L4·NO3)] suggest very feeble interactions of the receptor with a nitrate anion (Fig. 7).
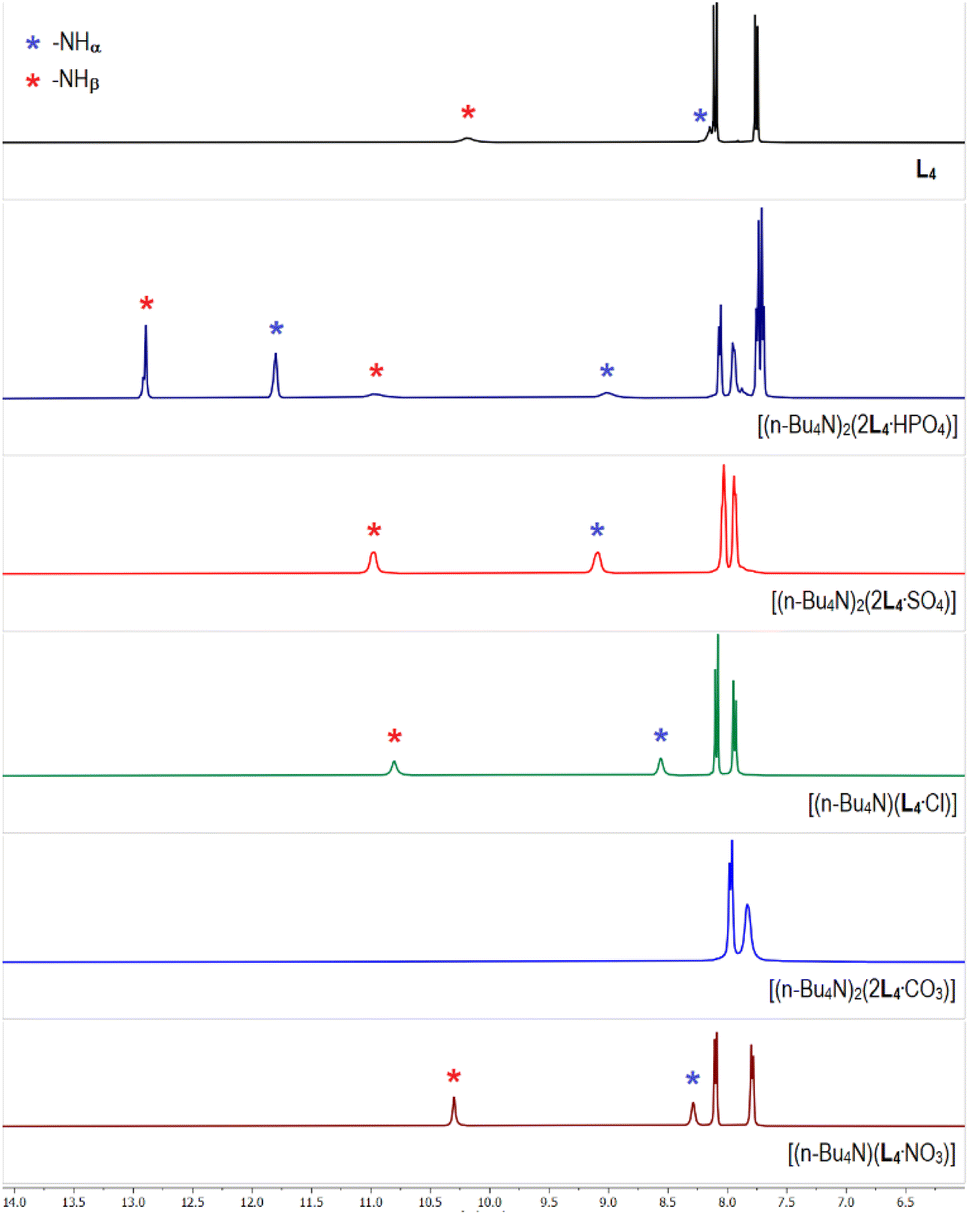 | ||
| Fig. 7 Aromatic region of the 1H-NMR (DMSO-d6) spectra of L4 and the receptor–anion complexes showing variable downfield shift of the thiourea –NH signals in different anion complexes relative to L4. (Full spectra are provided in the ESI, Fig S70–S77†). | ||
Selective extraction of phosphate
Similar to the sulfate extraction experiments, we have carried out liquid–liquid extraction (LLE) of hydrogenphosphate by the tris-thiourea receptor in the presence of a competitive anion (hydrogenarsenate, sulfate, selenate, carbonate, nitrate and chloride). In the first set of LLE experiments, L4 (100 mg) was dissolved in dichloromethane (20 mL) in the presence of three equivalents of (n-Bu4N+)CH3CO2− and an aqueous solution of hydrogenphosphate and a competing anion (one equiv. each of K2HPO4 and an alkali metal salt of competing anion dissolved in 20 mL of deionized water) was added to the organic media and stirred for an hour. In the second set of LLE experiments, an aqueous solution mixture of hydrogenphosphate and two competing anions (one equiv. alkali metal salts of (i) HPO42−, HAsO42− and NO3− (ii) HPO42−, SO42− and Cl− (iii) HPO42−, SO42− and NO3− dissolved in 20 mL of deionized water) was added to the dichloromethane phase and stirred for an hour. The 1H-NMR spectra of the isolated LLE products closely resemble the spectrum of [(n-Bu4N)2(2L4·HPO4)] in each case (Fig. 8), suggesting the selective extraction of phosphate from competitive aqueous media with 85–92% extraction efficiency (relative to L4). The exclusive formation of phosphate complexes in the competitive LLE experiments has further been confirmed by 31P-NMR spectroscopy (Fig. S78–S86, ESI†). The selective extraction of phosphate has also been achieved in the presence of three competing anions (SeO42−, CO32−, Cl− and SO42−, HAsO42−, NO3−) establishing L4 as a highly selective receptor for phosphate extraction (Fig. 8, S88 and S89, ESI†). EDX analysis have also been performed to validate the absence of competing anions such as arsenate and selenate in the LLE products.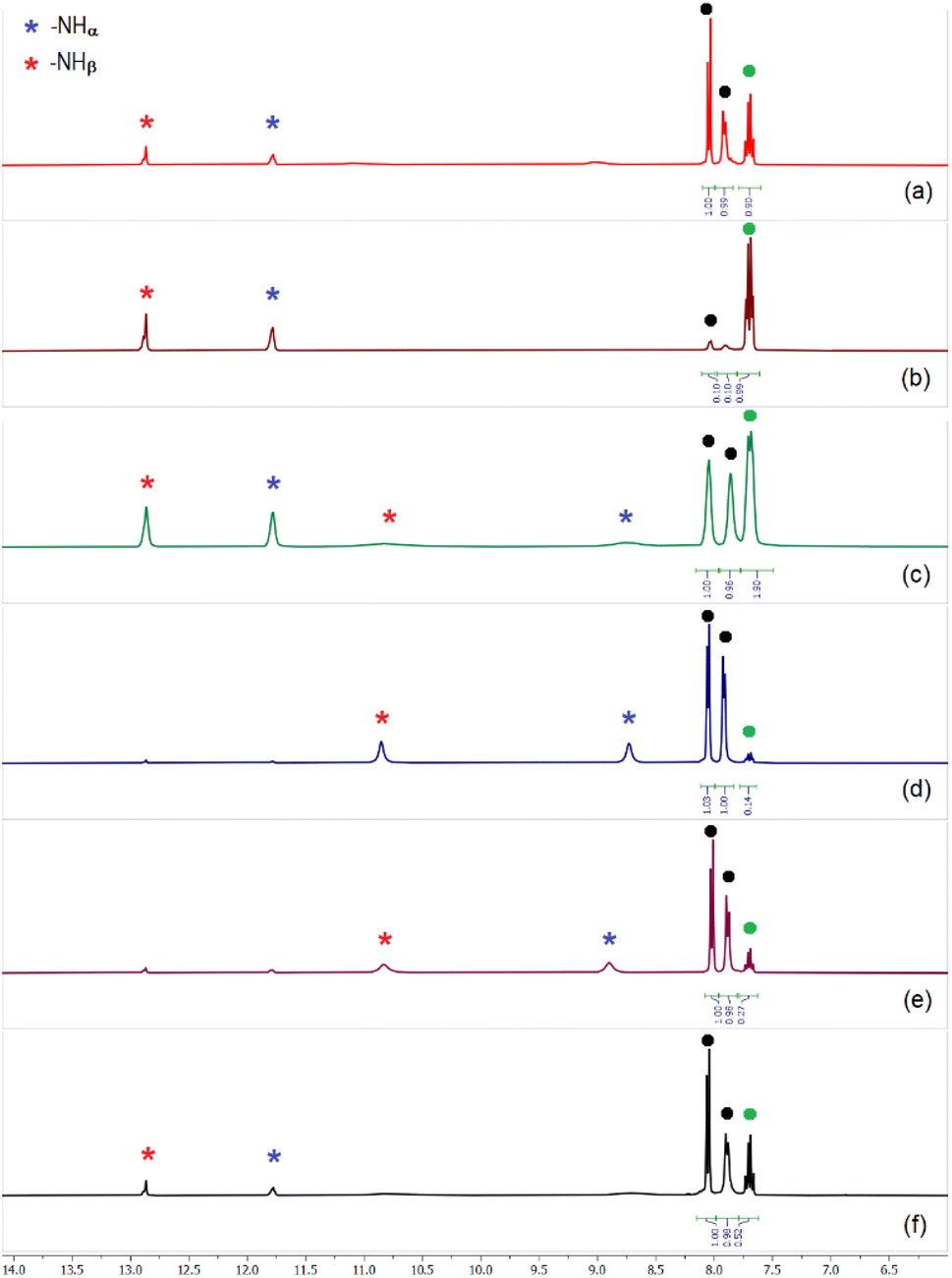 | ||
| Fig. 8 Aromatic region of the 1H-NMR spectra of phosphate complexes obtained in the presence of (n-Bu4N)+ acetate and (a) selenate, (b) carbonate, (c) hydrogenarsenate and nitrate, (d) sulfate and chloride, (e) sulfate and nitrate, (f) selenate, carbonate and chloride, showing the presence of both [2L4-HPO4]2− and [2L4-PO4]3− complexes in different ratios in the isolated LLE products. Black dots represent the –CH signals corresponding to [2L4-HPO4]2− and green dot represent the –CH signal for [2L4-PO4]3−. The [2L4-HPO4]2− to [2L4-PO4]3− ratio can be calculated from the given –CH integrals (full spectra are provided in the ESI, Fig. S78–S89† including 31P-NMR spectra). | ||
It is interesting to note that, depending on the competing anion(s), the ratio of [2L4·PO4]3− to [2L4·HPO4]2− complexes in the LLE products has varied considerably most likely due to the effect of different competing anion(s) on the HPO42− ↔ PO43− equilibrium in aqueous solution. For instance, in the presence of CO32− as a competing anion for phosphate extraction, the [2L4·PO4]3− to [2L4·HPO4]2− ratio was calculated to be 90:10, and in the presence of SeO42− as a competing anion, a ratio of 30:70 was calculated from the aromatic –CH integral values (Fig. 6, S78–S81, ESI†). While ≈50:50 mixture of [2L4·PO4]3− and [2L4·HPO4]2− was obtained in the presence of HAsO42− and NO3− as competing anions, ≈10:90 ratio was observed in the presence of SO42− and Cl−/NO3− as competing anions (Fig. 8 and S82–S87, ESI†).
Sulfate extraction under environmentally relevant conditions
The average distribution of four major anions along the upper Ganga River basin (India) varied approximately as bicarbonate (70%), sulfate (25%), nitrate (3%) and chloride (1%), while the presence of phosphate and fluoride are considerably low.18 Thus, based on the distribution of major anions in Ganga basin, an aqueous solution (20 mL) of four different sodium salts has been prepared containing 7 equiv. of bicarbonate, 2.5 equiv. of sulfate, 0.3 equiv. of nitrate and 0.1 equiv. of chloride with respect to 100 mg of L2 or L4. Tetrabutylammonium acetate (two equiv.) was used as an anion-exchanger in the organic phase for sulfate extraction experiments. As the concentrations of nitrate and chloride were significantly less as compared to sulfate and the presence of carbonate have proven to reduce the interference from nitrate and chloride in the LLE of sulfate, we have observed an efficient extraction of sulfate by L2 and L4 in the presence of excess bicarbonate to obtain the hydrogen bonded sulfate complex (Fig. S90 and S91, ESI†).Another study showed that the average distribution of four major anions in the Brahmaputra River basin groundwater (India) varied approximately as bicarbonate (91%), sulfate (3.5%), chloride (3.5%) and nitrate (1%).19 This implies that the concentration of bicarbonate is 26 times more than the concentration of sulfate and chloride in the groundwater samples tested. Sulfate extraction from an aqueous solution containing equivalent amount of chloride and large excess of bicarbonate (26 equiv.) was not successful with L2 and L4.
For a comparative study, we have also carried out LLE of sulfate and phosphate by L2 and L4 in the absence of competing anions. The extraction efficiency of thiourea receptor L4 was observed to be better than the urea receptor L2 both for sulfate and phosphate extraction, when tetrabutylammonium acetate was used as an anion-exchanger. The sulfate extraction efficiencies of L2 and L4 were observed to be 85% and 90% respectively, considering the formation of a 2:1 receptor–sulfate complex, which were evident from the integral values of 1H-NMR spectra of sulfate complexes (see ESI†). However, sulfate extraction efficiency of L2 exceed 90% when tetrabutylammonium hydroxide was used as an anion-exchanger. The phosphate extraction efficiencies of L2 and L4 were observed to be 82% and 92% respectively, considering the formation of a 2:1 receptor–phosphate complex. The higher extraction efficiency of L4 than L2 is possibly due to the higher acidity of –NH protons (–NH polarization), which implies that the thiourea receptor can form stronger hydrogen bonds with sulfate/phosphate in the solution-state resulting in better extraction efficiency than L2.
Control LLE experiments
In order to establish the best choice of anion-exchanger for the clean and efficient extraction of sulfate by the tris-urea receptors (L1 and L2), we have initially carried out several control LLE experiments using tetrabutylammonium salts of hydroxide, nitrate, acetate, chloride and fluoride. The order of basicity of the anions with respect to hydroxide follows the sequence OH− > F− > CH3CO2− > Cl− > NO3−. Tetrabutylammonium fluoride and acetate have proven to be effective anion-exchangers for sulfate extraction by tris-ureas, similar to (n-Bu4N)+OH−. Whereas, tetrabutylammonium chloride and nitrate have been found to be inefficient anion-exchangers for sulfate extraction from aqueous media (Table 2). Considering the importance of sulfate separation from alkaline nuclear waste media, the use of (n-Bu4N)+OH− for sulfate-exchange from an aqueous phase to the organic phase is well justified, and has proven to be highly efficient for selective sulfate extraction in competitive environment.| n-Bu4N+ salts (anion-exchanger) | L1 (LLE of SO42−) | L2 (LLE of SO42−) | L4 (LLE of HPO42−) |
|---|---|---|---|
| (n-Bu4N)+OH− | ✓ | ✓ | ✗ |
| (n-Bu4N)+CH3CO2− | ✓ | ✓ | ✓ |
| (n-Bu4N)+F− | ✓ | ✓ | ✓ |
| (n-Bu4N)+Cl− | ✗ | ✗ | ✓ |
| (n-Bu4N)+NO3− | ✗ | ✗ | ✓ |
Similarly, control LLE experiments have also been performed to validate a suitable choice of tetrabutylammonium salt for the clean and efficient extraction of phosphate by the tris-thiourea receptor L4. It has been found that the use of tetrabutylammonium hydroxide can deprotonate a more acidic thiourea –NH proton and thus, phosphate extraction by L4 was unachievable in the presence of (n-Bu4N)+OH−. However, tetrabutylammonium salts of acetate, chloride, fluoride and nitrate have proven to be effective anion-exchangers for phosphate extraction by the thiourea receptor (Fig. S92–S96, ESI†) unlike sulfate extraction by tris-urea receptors (Table 2).
So as to validate the anion selectivity order of tripodal receptors in LLE experiments, control experiments were carried out using different combinations of anions (equivalent amounts of two alkali metal salts dissolved in deionized water) in a sequential method. The phosphate selective thiourea receptor can selectively extract chloride in the presence of sulfate/selenate, and sulfate in the presence of selenate/arsenate (Fig. S97–S99, ESI†). The receptor can selectively extract selenate in the presence of arsenate and all of these anions can individually be extracted in the presence of tetrabutylammonium acetate. Thus, the selectivity order for LLE of anions by L4 was established as, HPO42− > Cl− > SO42− > SeO42− > HAsO42− > CH3CO2−. Similarly, the selectivity order for LLE of anions by L1–2 was established as, SO42− > Cl− > HPO42− > SeO42− > HAsO42− > CH3CO2−. The selective extraction of phosphate over selenate and arsenate was further confirmed by 31P-NMR analysis.
Conclusion
In conclusion, we have demonstrated the efficient extraction of sulfate from highly competitive alkaline aqueous media (pH ≈ 10) by nitrophenyl-functionalized tris-urea receptors (L1–L2) in the presence tetrabutylammonium hydroxide (two equiv.) acting as an anion-exchanger between the immiscible aqueous–organic phases. Sulfate can selectively be extracted from the alkaline aqueous media in the presence of nitrate, phosphate and other competing anions establishing L1 and L2 as effective HBD receptors for sulfate removal from aqueous media. Sulfate extraction efficacy of L2 (≈84–90%) were observed to be higher than L1 (≈76–82%) and L3 (≈68–75%) in the presence of two or three competing anions in LLE experiments. Thus, L2 can certainly be considered as the best sulfate extractor as it could selectively extract sulfate from an alkaline aqueous solution containing equivalent amount of sulfate, carbonate/hydrogenphosphate and nitrate/chloride, unlike L3 (Fig. 3–5 and ESI†) which exhibited significant interference from nitrate or chloride towards efficient sulfate extraction.Furthermore, efficient extraction of phosphate from water have also been demonstrated in the presence of several competing anions by using a nitrophenyl-functionalized tris-thiourea receptor, L4 and tetrabutylammonium acetate as an anion-exchanger. Because of the higher acidity of tris-thiourea –NHβ protons in comparison to its analogous urea receptor (L2), tetrabutylammonium hydroxide has been found to deprotonate a thiourea –NH group in LLE experiments and thus, LLE of phosphate was carried out in the presence of a less basic tetrabutylammonium acetate. Tetrabutylammonium acetate and fluoride can also be used for the selective extraction of sulfate by L1–L2, similar to phosphate extraction by L4 (Table 2). Notably, no interference from nitrate and chloride has been observed in the LLE of phosphate by the tris-thiourea receptor.
Overall, we have shown that structurally simple and easy to synthesize first-generation HBD tripodal receptors L2 and L4 can efficiently be employed for the selective extraction of sulfate and phosphate, respectively with up to 90% extraction efficiency depending on the competing anion or anionic mixture. Most importantly, all the receptors can easily be recycled for the successive LLE process by simply dispersing the oxoanion (sulfate/phosphate) complex in methanol–water (1:1, v/v) protic solvent media under stirring (for 15 min), followed by filtration to obtain the recycled product. The easy-to-synthesize tren-based tris-urea/thiourea receptors (mainly L2 and L4) can thus be considered as promising oxoanion receptors for the selective removal of sulfate and phosphates from aqueous waste media.
Finally, from a future perspective, it is of utmost necessity to find a low cost HBD receptor which can selectively and quickly extract sulfate from highly alkaline nitrate-rich aqueous waste media containing varying amount of other competitive ions.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
SKD acknowledges the Department of Science and Technology (DST) New Delhi, India for providing financial support through INSPIRE Faculty Award (DST/INSPIRE/04/2016/001867). We thank the Director of CSIR-NEIST Jorhat for providing access to laboratory and sophisticated analytical instrumentation facilities.Notes and references
- (a) A. Bianchi, K. Bowman-James and E. Garcia Espana, Supramolecular Chemistry of Anions, Wiley-VCH, New York, 1997 Search PubMed; (b) J. L. Sessler, P. A. Gale and W. S. Cho, Anion Receptor Chemistry, Royal Society of Chemistry, Cambridge, UK, 2006 RSC; (c) P. D. Beer and P. A. Gale, Angew. Chem., Int. Ed., 2001, 40, 486 CrossRef CAS.
- (a) L. P. M. Lamers, H. B. M. Tomassen and J. G. M. Roelofs, Environ. Sci. Technol., 1998, 32, 199–205 CrossRef CAS; (b) D. Zak, M. Hupfer, A. Cabezas, G. Jurasinski, J. Audet, A. Kleeberg, R. McInnes, S. M. Kristiansen, R. J. Petersen, H. Liu and T. Goldhammer, Earth-Sci. Rev., 2021, 212, 103446 CrossRef CAS; (c) J. Chen, H. Zhang, L. Liu, J. Zhang, M. Cooper, R. J. G. Mortimer and G. Pan, Sci. Total Environ., 2021, 790, 148010 CrossRef CAS PubMed.
- (a) E. A. Katayev, Y. A. Ustynyuk and J. L. Sessler, Coord. Chem. Rev., 2006, 250, 3004 CrossRef CAS; (b) B. A. Moyer, R. Custelcean, B. P. Hay, J. L. Sessler, K. Bowman-James, V. W. Day and S.-O. Kang, Inorg. Chem., 2013, 52, 3473–3490 CrossRef CAS PubMed.
- (a) H. Weingartner, E. U. Franck, G. Wiegand, N. Dahmen, G. Schwedt, F. H. Frimmel, B. C. Gordalla, K. Johannsen, R. S. Summers, W. Holl, M. Jekel, R. Gimbel, R. Rautenbach and W. H. Glaze, Ullman's Encyclopedia of Industrial Chemistry, Wiley-VCH Verlag GmbH & Co., 2000 Search PubMed; (b) D. W. Schindler, Limnol. Oceanogr., 2006, 51, 356–363 CrossRef; (c) W. K. Dodds, Environ. Sci. Technol., 2009, 43, 12–19 CrossRef CAS PubMed; (d) V. H. Smith and D. W. Schindler, Trends Ecol. Evol., 2009, 24, 201–207 CrossRef PubMed.
- (a) B. A. Moyer and R. P. Singh, Fundamentals and Applications of Anion Separation, Kluwer Academic Plenum, New York, 2004 CrossRef; (b) C. Schalley, Analytical Methods in Supramolecular Chemistry, Wiley-VCH Verlag GmbH & Co., Weinheim, Germany, 2007 Search PubMed.
- I. Ravikumar and P. Ghosh, Chem. Soc. Rev., 2012, 41, 3077–3098 RSC.
- (a) S. Kubik, R. Kirchner, D. Nolting and J. Seidel, J. Am. Chem. Soc., 2002, 124, 12752–12760 CrossRef CAS PubMed; (b) P. A. Gale, J. R. Hiscock, C. Z. Jie, M. B. Hursthouse and M. E. Light, Chem. Sci., 2010, 1, 215–220 RSC; (c) P. G. Young and K. A. Jolliffe, Org. Biomol. Chem., 2012, 10, 2664–2672 RSC; (d) V. J. Dungan, H. T. Ngo, P. G. Young and K. A. Jolliffe, Chem. Commun., 2013, 49, 264–266 RSC; (e) F. Sommer and S. Kubik, Org. Biomol. Chem., 2014, 12, 8851–8860 RSC; (f) L. Qin, A. Hartley, P. Turner, R. B. P. Elmes and K. A. Jolliffe, Chem. Sci., 2016, 7, 4563–4572 RSC; (g) L. Qin, J. R. Wright, J. D. E. Lane, S. N. Berry, R. B. P. Elmes and K. A. Jolliffe, Chem. Commun., 2019, 55, 12312–12315 RSC; (h) J. D. E. Lane, W. J. H. Greenwood, V. W. Day, K. A. Jolliffe, K. Bowman-James and L. Adriaenssens, New J. Chem., 2022, 46, 18119–18123 RSC.
- (a) G. Tumcharern, T. Tuntulani, S. J. Coles, M. B. Hursthouse and J. D. Kilburn, Org. Lett., 2003, 5, 4971–4974 CrossRef CAS PubMed; (b) C. Caltagirone, P. A. Gale, J. R. Hiscock, S. J. Brooks, M. B. Hursthouse and M. E. Light, Chem. Commun., 2008, 3007–3009 RSC; (c) I. Ravikumar, P. S. Lakshminarayanan, M. Arunachalam, E. Suresh and P. Ghosh, Dalton Trans., 2009, 4160–4168 RSC; (d) C. Bazzicalupi, A. Bencini and V. Lippolis, Chem. Soc. Rev., 2010, 39, 3709–3728 RSC; (e) D. Mungalpara, A. Valkonen, K. Rissanen and S. Kubik, Chem. Sci., 2017, 8, 6005–6013 RSC; (f) Z. Huang, C. Jia, B. Wu, S. Jansone-Popova, C. A. Seipp and R. Custelcean, Chem. Commun., 2019, 55, 1714–1717 RSC; (g) W. Zuo, C. Jia, H. Zhang, Y. Zhao, X.-J. Yang and B. Wu, Chem. Sci., 2019, 10, 2483–2488 RSC.
- (a) I. R. Fernando, S. A. Surmann, A. A. Urech, A. M. Poulsen and G. Mezei, Chem. Commun., 2012, 48, 6860–6862 RSC; (b) R. Custelcean, Chem. Commun., 2013, 49, 2173–2182 RSC; (c) B. Akhuli, T. K. Ghosh and P. Ghosh, CrystEngComm, 2013, 15, 9472–9482 RSC; (d) Y.-T. Ke, W.-T. Chou, Y.-F. Chiang, C.-C. Hsieh and Y.-C. Horng, New J. Chem., 2017, 41, 2249–2254 RSC; (e) Y.-T. Ke, W.-T. Chou, Y.-F. Chiang, C.-C. Hsieh and Y.-C. Horng, New J. Chem., 2017, 41, 2249–2254 RSC; (f) N. J. Williams, C. A. Seipp, K. A. Garrabrant, R. Custelcean, E. Holguin, J. K. Keum, R. J. Ellis and B. A. Moyer, Chem. Commun., 2018, 54, 10048–10051 RSC; (g) T. K. Ghosh, R. Dutta, S. Maji, S. Pal and P. Ghosh, Polyhedron, 2019, 172, 74–79 CrossRef CAS; (h) Y.-C. He, Y.-M. Yan, H.-B. Tong, Z.-X. Ren, J.-H. Wang, Y.-B. Zhang, J.-B. Chao and M.-L. Wang, Chem. Commun., 2020, 56, 9364–9367 Search PubMed; (i) L. Qin, S. J. N. Vervuurt, R. B. P. Elmes, S. N. Berry, N. Proschogo and K. A. Jolliffe, Chem. Sci., 2020, 11, 201–207 RSC.
- (a) L. R. Eller, M. Stepien, C. J. Fowler, J. T. Lee, J. L. Sessler and B. A. Moyer, J. Am. Chem. Soc., 2007, 129, 11020–11021 CrossRef CAS PubMed; (b) C. J. Fowler, T. J. Haverlock, B. A. Moyer, J. A. Shriver, D. E. Gross, M. Marquez, J. L. Sessler, M. A. Hossain and K. Bowman-James, J. Am. Chem. Soc., 2008, 130, 14386–14387 CrossRef CAS PubMed; (c) C. J. Borman, R. Custelcean, B. P. Hay, N. L. Bill, J. L. Sessler and B. A. Moyer, Chem. Commun., 2011, 47, 7611–7613 RSC; (d) S. K. Kim, J. Lee, N. J. Williams, V. M. Lynch, B. P. Hay, B. A. Moyer and J. L. Sessler, J. Am. Chem. Soc., 2014, 136, 15079–15085 CrossRef CAS PubMed.
- R. Custelcean and B. A. Moyer, Eur. J. Inorg. Chem., 2007, 1321–1327 CrossRef CAS.
- (a) I. Carreira-Barral, T. Rodríguez-Blas, C. Platas-Iglesias, A. de Blas and D. Esteban-Gómez, Inorg. Chem., 2014, 53, 2554–2568 CrossRef CAS PubMed; (b) T. K. Ghosh, R. Dutta and P. Ghosh, Inorg. Chem., 2016, 55, 3640–3652 CrossRef CAS PubMed; (c) D. Jagleniec, L. Dobrzycki, M. Karbarz and J. Romański, Chem. Sci., 2019, 10, 9542–9547 RSC; (d) M. Zaleskaya, M. Karbarz, M. Wilczek, L. Dobrzycki and J. Romański, Inorg. Chem., 2020, 59, 13749–13759 CrossRef CAS PubMed; (e) M. Zaleskaya, D. Jagleniec and J. Romański, Dalton Trans., 2021, 50, 3904–3915 RSC.
- (a) N. Busschaert, M. Wenzel, M. E. Light, P. Iglesias-Hernández, R. Pérez-Tomás and P. A. Gale, J. Am. Chem. Soc., 2011, 133, 14136–14148 CrossRef CAS PubMed; (b) S. K. Dey, R. Chutia and G. Das, Inorg. Chem., 2012, 51, 1727–1738 CrossRef CAS PubMed; (c) S. K. Dey and G. Das, Dalton Trans., 2012, 41, 8960–8972 RSC; (d) R. Chutia, S. K. Dey and G. Das, Cryst. Growth Des., 2013, 13, 883–892 CrossRef CAS.
- S.-Q. Chen, S.-N. Yu, W. Zhao, L. Liang, Y. Gong, L. Yuan, J. Tang, X.-J. Yang and B. Wu, Inorg. Chem. Front., 2022, 9, 6091–6101 RSC.
- (a) S. K. Dey, Archana, S. Pereira, S. S. Harmalkar, S. N. Mhaldar, V. V. Gobre and C. Janiak, CrystEngComm, 2020, 22, 6152–6160 RSC; (b) S. K. Dey, B. Gil-Hernández, V. V. Gobre, D. Woschko, S. S. Harmalkar, F. R. Gayen, B. Saha, R. L. Goswamee and C. Janiak, Dalton Trans., 2022, 51, 15239–15245 RSC.
- (a) B. Akhuli, I. Ravikumar and P. Ghosh, Chem. Sci., 2012, 3, 1522–1530 RSC; (b) R. Dutta, S. Chakraborty, P. Bose and P. Ghosh, Eur. J. Inorg. Chem., 2014, 4134–4143 CrossRef CAS; (c) U. Manna and G. Das, CrystEngComm, 2021, 23, 512–527 RSC.
- (a) S. K. Dey, A. Basu, R. Chutia and G. Das, RSC Adv., 2016, 6, 26568–26589 RSC; (b) U. Manna and G. Das, Coord. Chem. Rev., 2021, 440, 213931 CrossRef CAS.
- M. K. Sharma, P. Kumar, P. Prajapati, K. Bhanot, U. Wadhwa, G. Tomar, R. Goyal, B. Prasad and B. Sharma, J. Asian Earth Sci., 2022, 8, 100108 Search PubMed.
- S. Verma, A. Mukherjee, R. Choudhury and C. Mahanta, J. Hydrol. Reg. Stud., 2015, 4, 131–153 CrossRef.
Footnotes |
| † Electronic supplementary information (ESI) available: Synthesis of receptors and anion complexes, details of liquid–liquid extraction experiments, 1H-NMR and 31P-NMR spectra. CCDC 2247494. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d3ra01771k |
| ‡ Single crystal X-ray crystallography data of [(n-Bu4N)2(2L3·SO4)] CCDC no. 2247494, F = C86H132F6N16O10S, M = 1696.13, T = 293 K, Space group = I2/a, a = 27.2148(9) Å, b = 13.1020(4) Å, c = 28.9686(11) Å, α = 90°, β = 114.483(4)°, γ = 90°, V = 9400.5(6) Å3, Z = 4, μ = 0.918 mm−1, D = 1.198 g cm−3, F(000) = 3640, θ (max) = 72.503, data completeness = 0.977, measured reflections = 20122, independent reflections = 9108, observed reflections (I > 2 s(I)) = 6797, parameters = 569, R1(F) = 0.0816, wR2(F2) = 0.2943, S = 1.069. |
| This journal is © The Royal Society of Chemistry 2023 |