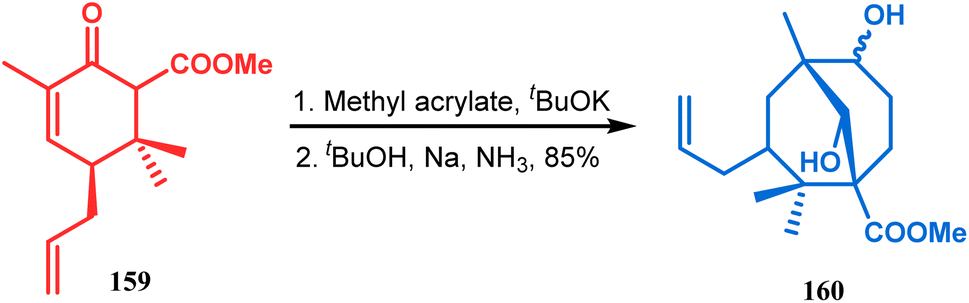DOI:
10.1039/D3RA02003G
(Review Article)
RSC Adv., 2023,
13, 22389-22480
Different routes for the construction of biologically active diversely functionalized bicyclo[3.3.1]nonanes: an exploration of new perspectives for anticancer chemotherapeutics
Received
27th March 2023
, Accepted 9th May 2023
First published on 25th July 2023
Abstract
Cancer is the second most high-morbidity disease throughout the world. From ancient days, natural products have been known to possess several biological activities, and research on natural products is one of the most enticing areas where scientists are engrossed in the extraction of valuable compounds from various plants to isolate many life-saving medicines, along with their other applications. It has been noticed that the bicyclo[3.3.1]nonane moiety is predominant in most biologically active natural products owing to its exceptional characteristics compared to others. Many derivatives of bicyclo[3.3.1]nonane are attractive to researchers for use in asymmetric catalysis or as potent anticancer entities along with their successful applications as ion receptors, metallocycles, and molecular tweezers. Therefore, this review article discusses several miscellaneous synthetic routes for the construction of bicyclo[3.3.1]nonanes and their heteroanalogues in association with the delineation of their anticancer activities with few selective compounds.
 Nilmadhab Roy | Roy received his Master of Science degree from Vidyasagar University (2014), with specialization in Physical Chemistry. He is currently pursuing his PhD from VIT, Vellore, India, under the supervision of Dr Priyankar Paira. His research interest involves “Organometallics and their Nanoconjugates for catalytic and biological applications”. He has 20 publications in ACS, RSC, and Elsevier journals. Currently, he is the member of Society of Biological Inorganic Chemistry. He has received SBIC travel award. |
 Rupankar Paira | Dr. Rupankar Paira completed his Master of Science degree from Indian Institute of Technology (IIT), Kharagpur with specialization in Organic Chemistry. Then, he received his PhD from Indian Institute of Chemical Biology (IICB), Jadavpur University, Kolkata and Post Doctorate degree from Nanyang Technological University (NTU), Singapore. Presently, he is working as Assistant Professor at Maharaja Manindra Chandra College, Kolkata, India. He obtained DST-SERB Young Scientist start-up grant (YSS/2014/000877) in 2014 as well as UGC-MRP grant (ROMRP-ERO-CHEM-2015-16-68950) in 2015. He has 42 publications in various ACS, RSC, Wiley, and Elsevier journals. |
 Priyankar Paira | Dr. Priyankar Paira, MRSC, Associate Professor, Vellore Institute of Technology (VIT), Vellore, Tamil Nadu, India, received his PhD from the Indian Institute of Chemical Biology (IICB), Jadavpur University, Kolkata, and was a post-doctoral candidate from National University of Singapore (NUS). He is a bioinorganic and medicinal chemist with 11 years of experience. He has 100 publications in reputed international journals having 1715 citations and h-index 22 because of his significant contribution to research. He also has 3 patents in medicinal chemistry. He has successfully completed a DST-SERB-YS (YSS/2014/000842) project, DST-SERB project (EMR/2017/000816), and two ICMR projects. At present, he is working on another DST-SERB-CRG project (CRG/2021/002267). In particular, he has always been fascinated by research involving a combination of engineering, medicine, and science, as demonstrated by his publications and collaborations with a number of people from different institutes having diverse research expertise. He is associated as a reviewer with several RSC, ACS, Wiley, and Elsevier journals. Currently, he is serving as a guest editor in International Journal of Medicinal Chemistry and executive guest editor in Current Organic Chemistry. He is also the editorial board member of Current Electrocatalysis, Bentham Science, and Frontiers in Chemistry journals. |
1. Introduction
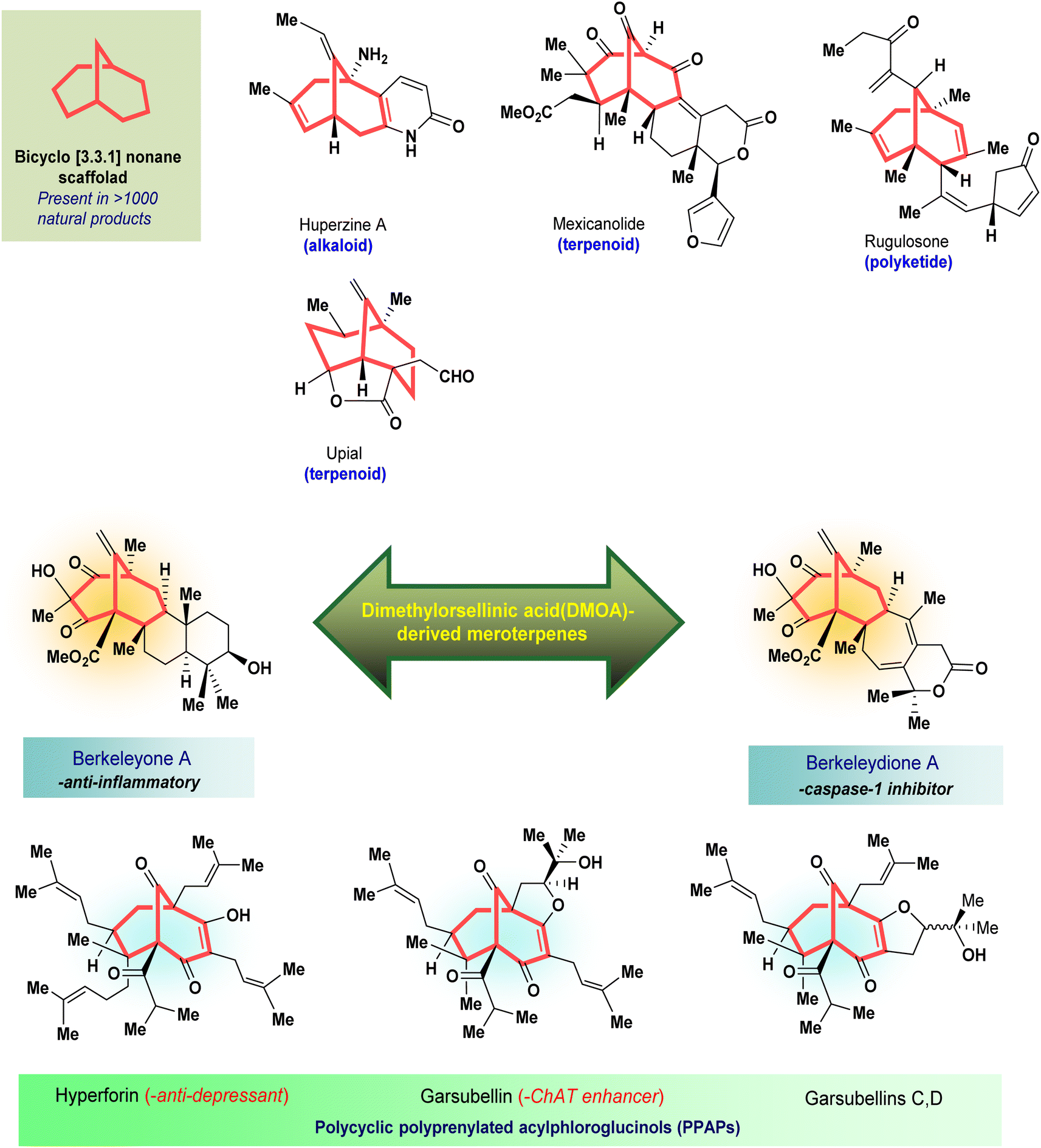
Cleft-shaped entities have long been used as essential chemical tools in molecular recognition studies.1 The real development in this area started during the late 1970s with the discovery of Kagan's ether (2)2 and some other structurally diverse bicyclo[3.3.1]nonane derivatives (3–5),3 along with their successful application as ion receptors, metallocycles, and molecular tweezers.4 The first such molecule, Troger's base (1),5 was developed as early as the late 1880s, yet it continues to fascinate scientists by its uniqueness as an asymmetric catalyst6 as well as a DNA intercalator7 or enzyme inhibitor.8 Even 125 years after its discovery, the science of Troger's base is progressing at a fast pace.9
Apart from these synthetic bicyclo[3.3.1]nonanes, this important core moiety is quite plentiful among several bioactive natural products. For example, alkaloids such as isariotin A, nankakurine A, adaline, huperzine A, and lycodine10 and terpenoids such as upial, spirovibsanin, trifarienols A and B,11 and swietenine12 incorporate this one-carbon-bridged cyclooctane moiety in their structural framework. Some naturally occurring bioactive benzophenones13 and cytostatic metabolites such as gymnastatins F, G, and Q along with their diacetates and triacetates14 also contain this core unit, but bicyclo[3.3.1]nonanes are most abundant among the polyprenylated acylphloroglucinols (PPAPs), for example, clusianone, garsubellin A, aristophenone A, plukenetione, hyperforin, nemorosone, guttiferone A, hypersampsone F, and papuaforin A.15 Recent developments in synthetic chemistry have witnessed the successful application of appropriately modified bicyclo[3.3.1]nonane units as useful precursors for accessing more complex targets (both synthetic and natural products).15 It has also been observed that indole alkaloids containing azabicyclo[3.3.1]nonane architechture play a crucial role as anticancer, antimalarial, antiinflammatory, antiamebic, antileishmanial, antituberculosis, and antiarrhythmic drug candidates. Therefore, this skeleton always acquires a noticeable position in the history of natural products as they possess structural resemblance with the essential amino acid tryptophan as well as its related metabolite, the neurotransmitter serotonin. In particular, these indole alkaloids have gained special attention as they contain azacyclic and tryptophan-derived substructures that are widely regarded as “privileged structures” or efficient substructures suitable for binding to various types of protein receptors with higher responsiveness. Some important indole alkaloids are sarpagine, ajmaline, and macroline, which consist one of the major groups of structurally related indole natural products. These types of new alkaloids are now being isolated with a greater rate from various plant sources throughout the world because of their remarkable biological activity. A book chapter based on “Sarpagine and Related Alkaloids” written by O. A. Namjoshi and J. M. Cook has revealed the synthetic routes as well as their biological evaluation in a precise manner.16
Thus, the synthesis of diversely-functionalized bicyclo[3.3.1]nonanes has gained immense importance in recent times, resulting in the discovery of a large number of synthetic approaches toward this important core moiety. It is worth mentioning that a book chapter by Buchanan17 and a review by Peters18a,c on this important core moiety have appeared during the 1970s. A short review, particularly on the asymmetric synthesis of bicyclo[3.3.1]nonanes, appeared during the beginning of this century.18b,d,e However, this review also reveals the structurally-related bicyclo[3.2.1] octanes19 and bicyclo[4.2.1]nonanes,20 which cover further expansions and developments in the area of both asymmetric (chiral) or nonasymmetric (achiral) bicyclo[3.3.1]nonanes and their heteroanalogues. The chemistry of Troger's base and its analogues is, however, not included in this article as it is already very well-reviewed by several authors.18f–j Hence, in this review article, we have aspired to assemble the recent chemistry of bicyclo[3.3.1]nonanes and their heteroanalogues, both as a synthetic target as well as a synthetic intermediate, discussing its importance in anticancer therapy to unveil a fruitful pathway for the future design of anticancer chemotherapeutics.
2. Conformational features
The chemistry of bicyclo[3.3.1]nonanes is very much dependent on their conformational properties. Thus, great efforts have been dedicated during the last four decades on the broad investigations of their conformational features. Unlike the structurally related bicyclo[3.2.1]octanes, the above hydrocarbons can exist in three possible conformations, namely, a C2v-symmetric twin chair (CC, 30a), Cs-symmetric boat chair (BC, 30b), and C2-symmetric twisted twin boat (BB, 30c).21–23 The destabilizing steric factors present in the BB conformer ruled out the possibility of its existence in a detectable amount. Besides, the high energy difference (ΔG° = 2.3 kcal mol−1) between the most stable CC and the comparatively unstable BC meant that the latter went virtually undetected in NMR even at a low temperature range from −100° to −170 °C without any significant broadening of the NMR signal of the major conformer, as corroborated by statistical calculations.24–27 However, at very high temperature (400 °C) 25% population of BC was established by electron diffraction investigations.24 The corresponding 9-keto analogue, however, presents a different scenario. Although the twisted twin boat form 31c is as unpopulated as 30c,28 the equilibrium between BC (31b) and CC (31a) is less inclined toward CC, with much lower energy barrier (0.95 kcal mol−1) than in 30. Theoretical calculations by several research groups and the lanthanide-induced NMR shift (LIS) investigation determined a 0.9–2.4% population of the BC conformer at −165 °C, as indicated by the significant broadening of the NMR line width of the major isomer.25a,28–32
The CC conformer is, however, not always the predominant entity in all bicyclo[3.3.1]nonanes. Appropriate substitutions in the carbocyclic rings give rise to rather interesting features. For example, 2,4,6,8-tetraaryl-3,7-diazabicyclo[3.3.1]nonanes 32a,b–35a,b always adopt the BC conformation to avoid the 1,3-diaxial steric repulsion between the aryl groups and lone pair–lone pair (lp–lp) repulsion between the nitrogens. However, N-nitrosation imposes more sp2 character on the nitrogens, which lowers the lp–lp repulsion in the CC conformation. Also, this introduces a rather more dominating repulsive factor, the allylic A (1, 3) strain, between the nitroso groups and the neighboring α-aryl groups in the BC conformation, making the CC conformer the chief one.33–38 The conformational behavior of 3-borabicyclo[3.3.1]nonane 36 is rather more fascinating. The pπ–pπ backbonding between the filled p-orbital of oxygen and the vacant p-orbital of boron raises the bond order and restricts rotation around the B–O bond. Thus, repulsive steric factors favor the CC conformer. However, as the temperature increases, the rotation around both B–O and C–phenyl bonds is facilitated, thereby increasing the steric requirements of both methoxy and phenyl groups, resulting in a rise in the BC population, as corroborated by NMR studies. Moreover, when 36 was allowed to form a chelate with pyridine-d5 or dibenzoylmethane (complex 37, 38), boron gets tetracoordinated and its steric requirement rises further, making BC the major conformer.39–41 Simple 9-BBN (39a,b), however, always prefers the CC conformer and even takes part in the palladium-mediated arylation reactions through intermediate 40, also in the CC conformation.41
This kind of preference for the BC conformer is also very common in heavy atom-substituted bicyclo[3.3.1]nonanes. For example, 9-oxa-3,7-dithiabicyclo[3.3.1]nonane (41) as well as 9-oxa-3-selena-7-thiabicyclo[3.3.1]nonane (42) are rich in their BC conformers, mainly due to the lp–lp repulsion of heavy atoms (such as Se and S etc.) present at 3 and 7 positions in the CC conformer. Such phenomena are commonly known as the “Hockey Sticks” effect.42–48
However, 3,7-dithia-1,5-diazabicyclo[3.3.1]nonane (43) shows no such preference for the BC conformer. The presence of two additional stabilizing LP-N–C–S stereoelectronic interactions in this case favors the CC conformer over BC, as justified by QTAIM analysis.49–52 In some cases, the CC conformer is still preferred (e.g., in 44), even in the absence of such stabilizing factors. The stabilization of the CC conformer through improper C–H⋯S hydrogen bonding between S and C7–Hax is supported by the theoretically calculated 1.61 kcal mol−1 energy lowering due to n(S) → σ × (C–H) overlap interaction. Such an interaction is the strongest in 44 compared to the corresponding unsubstituted bicyclo[3.3.1]nonane (30), aza-anagues (45, 46), and oxa-analogues (47, 48).53,54 Protonated 3-aza-bicyclo[3.3.1]nonane (49), however, reveals a different scenario. It forms a dihydrogen bond with significant covalent character due to the close proximity (1.78 Å) between –CH and –HN+ hydrogens, resulting in an energy lowering of 4.24 kcal mol−1.54 Apart from these improper H-bonds, proper hydrogen bonding is also common in appropriately substituted bicyclo[3.3.1]nonanes such as 3-azabicyclo[3.3.1]nonane-2,4-dione and is responsible for its polymorphic property.55
Another interesting conformational feature of bicyclo[3.3.1]nonane arises during the substitution at the bridgehead-methylene group. Unlike the 7,7-diaryl norboranes, which exist as an inseparable mixture of atropisomers at ambient temperature due to the small rotational energy barrier around the C-aryl bonds,56–58 9,9-diarylbicyclo[3.3.1]nonanes could exist in two separate atropisomeric forms, when properly substituted. For example, although the rotational energy barrier is small in 50 and it could not be isolated in its two isomeric forms, 51 and 52 have much higher energy barriers due to an orthogonal propeller-like orientation of the two aryl groups, which enables the separation of their atropisomers.59
3. Synthetic approaches
Bicyclo[3.3.1]nonane first appeared in literature in the form of its aza-analogue, with the discovery of Troger's base (54) (Scheme 1) in 1887 by Julius Troger. He was able to synthesize it simply by reacting p-toluidine with formaldehyde in aqueous HCl. However, it took almost half a century to confirm its actual structure, when Spielman unveiled his work in 1935.60a,b Within a decade, another fascinating feature was established when Prelog successfully separated both the enantiomers of 54 in 1944, showing that chirality can exist in atoms other than carbon. Rapid inversion in a chiral heteroatom center in other unstrained molecules had restricted their separation in isomeric forms. However, it can be stopped and the enantiomers could be separated by introducing conformational strain, as in Troger's base.60c However, more practical syntheses of Troger's base and its analogues came into the literature during the end of the 20th century, apart from a few which were published during 1960s.60a
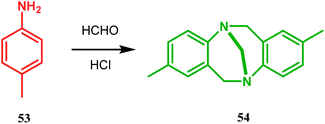 |
| | Scheme 1 Synthesis of Troger's base. | |
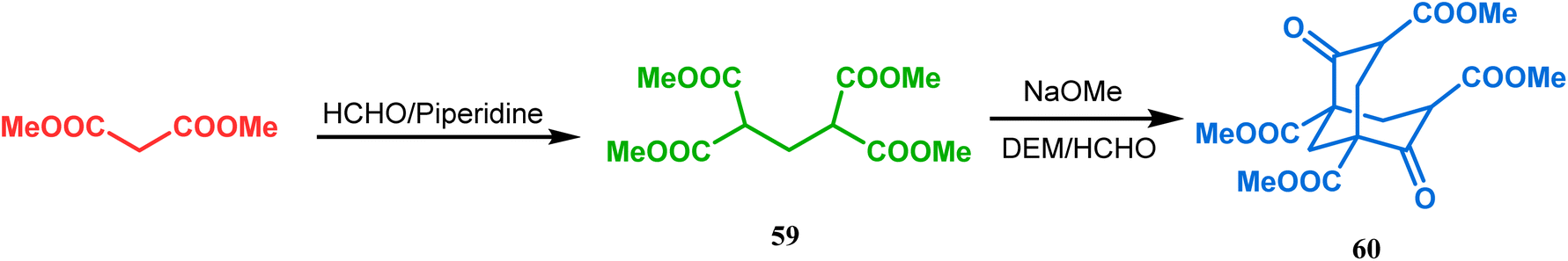
Although the structural uncertainties of Troger's base delayed the development of its chemistry, the progress in the chemistry of other bicyclo[3.3.1]nonanes proceeded smoothly from the very beginning of the 20th century. Condensation between aliphatic or aromatic aldehydes and acetylacetones, followed by acidic dehydration to produce 56 from 55 or 58 from carvone (57, a terpene) and ethyl acetoacetate, are some of those initiative routes to bicyclo[3.3.1]nonanes (Schemes 2 and 3).61–67 The synthesis of Meerwein's ester (60), the formerly used precursor for adamantane synthesis, was also achieved in this period, from dimethyl malonate and formaldehyde via intermediate 59 (Scheme 4).68–70 After these pioneering works on bicyclo[3.3.1]nonanes, the development of numerous synthetic methodologies to access this important moiety has been reported. These could be classified into three major groups: (a) intra- and intermolecular C–C bond formation, (b) intra- and intermolecular C–X bond formation, and (c) ring opening and ring expansion. The following sections of this review deal with all these.
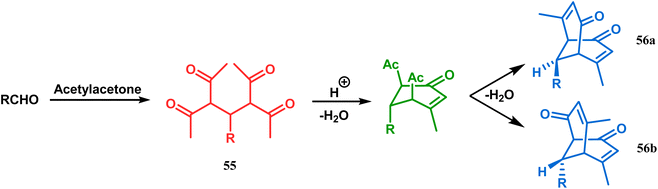 |
| | Scheme 2 Condensation reaction between an aliphatic or aromatic aldehyde and acetylacetone to obtain bicyclo[3.3.1]nonane moieties. | |
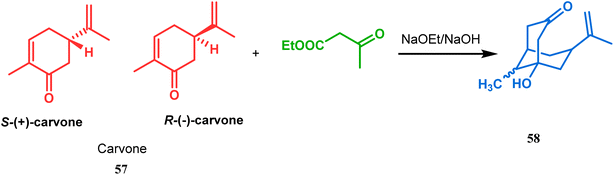 |
| | Scheme 3 Formation of bicyclo[3.3.1]nonane moieties by the reaction between carvone and ethyl acetoacetate (EAA). | |
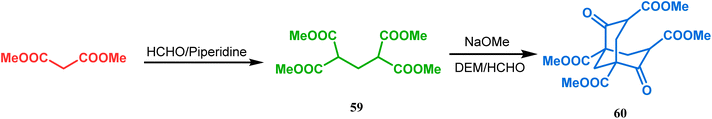 |
| | Scheme 4 Synthesis of Meerwein's ester from dimethyl malonate and formaldehyde. | |
3.1. Intra- and intermolecular C–C bond formation
3.1.1. Aldol condensation. Base-promoted tandem Michael addition-intramolecular aldolizations are well documented in this category. For example, the condensation reaction between dimethyl-1,3-acetonedicarboxylate 61 and enals 62, promoted by piperidine or TBAF, gives high yields of bicyclo[3.3.1]nonenols 63 (Scheme 5).71 Another stereocontrolled route to such nonenols has been unveiled recently by Crowe, where 2-substituted cyclohex-2-enones were successfully condensed with some active methylene group containing esters and amides.
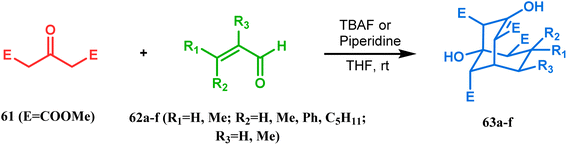 |
| | Scheme 5 Formation of bicyclo[3.3.1] nonenols from the condensation reaction between dimethyl-1,3-acetonedicarboxylate and enals. | |
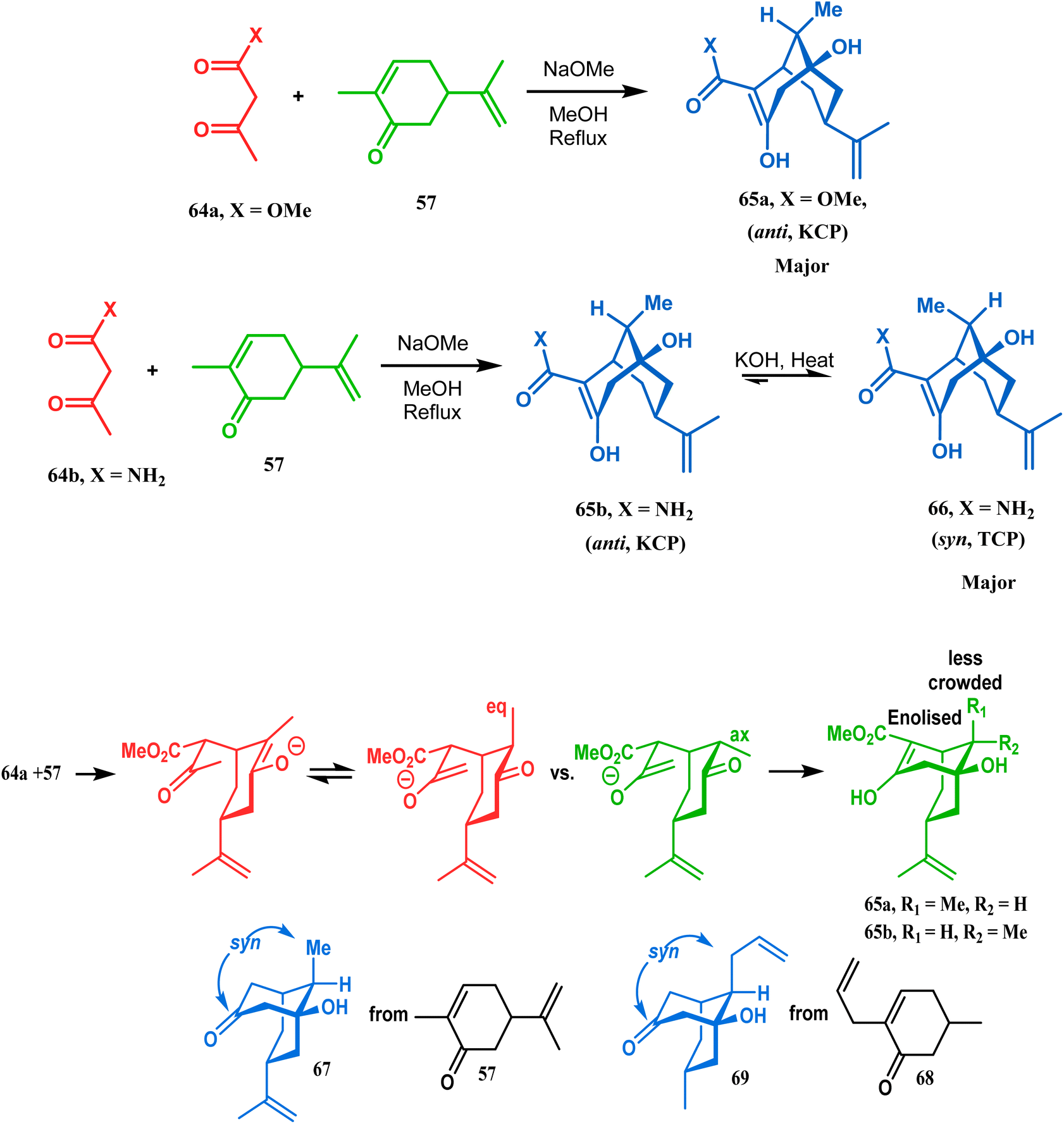
It was observed that the anti-product is always the kinetically-controlled product and the major one, irrespective of the nature of the starting materials. But the syn isomer 66, the thermodynamically-controlled one, becomes the major product (yield 80%) when carvone (57) is refluxed in methanolic KOH with amide 64b (Scheme 6).72
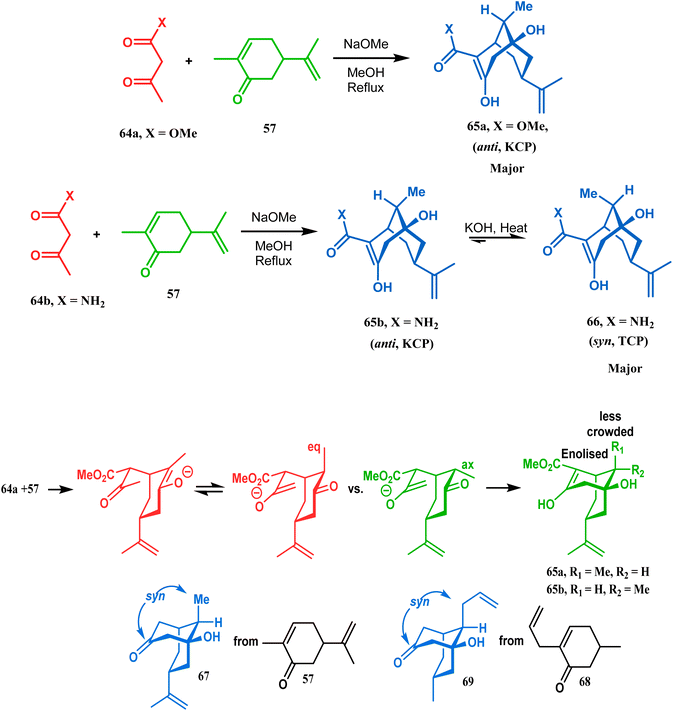 |
| | Scheme 6 A synthetic route for the formation of stereocontrolled bicyclo[3.3.1] nonenols from carvone. | |
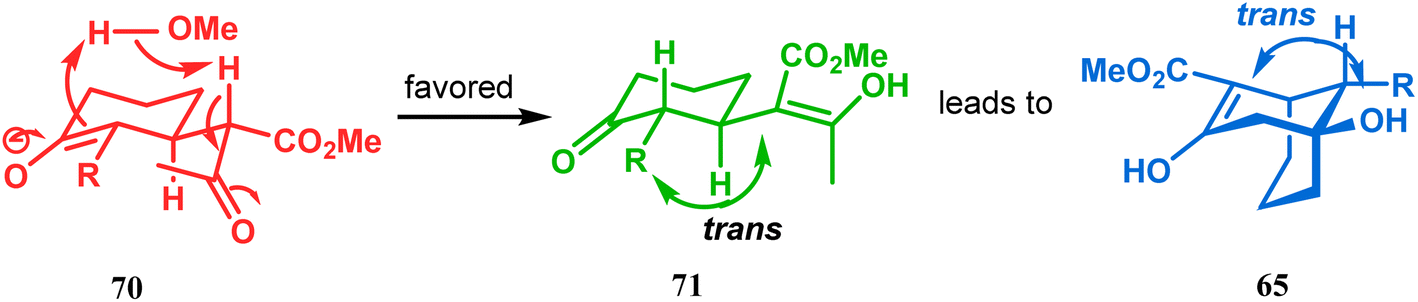
However, these results were in direct conflict with those reported by Kraus and Theobold.73–75 Both the groups obtained the syn isomers (67 and 69) as the major one, supported by the fact that both these syn isomers should be formed from the lower energy conformer, with the methyl/allyl group in the equatorial position. Although a similar result should also be expected in the reaction between 64 and 57, there is also the possibility of an intramolecular proton transfer from 70 to give 71 (Scheme 7), similar to that proposed by Grossman,76 which could account for the unusual stereoselectivity obtained by Crowe.
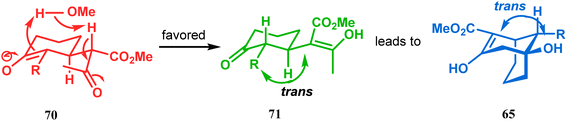 |
| | Scheme 7 Formation of bicyclo[3.3.1] nonenols through intramolecular proton transfer. | |
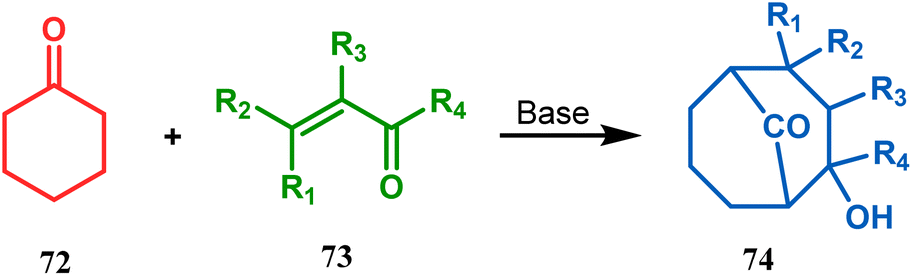
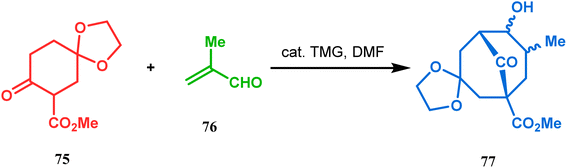
Cyclohexanones (72), when reacted in this manner with α,β-unsaturated aldehydes or ketones (73), yield a bicyclo[3.3.1]nonane (74) with a ketone functionality at the bridgehead position (Scheme 8).71 Similarly, Tückmantel and coworkers showed that β-keto ester (75) could also be annulated with acrolein (76) in the presence of a catalytic amount of TMG (1,1,3,3-tetramethylguanidine) to obtain the bicycle 77 (Scheme 9).10d
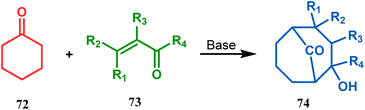 |
| | Scheme 8 Synthesis of bicyclo[3.3.1]nonane from cyclohexanone and α,β-unsaturated aldehydes or ketones. | |
 |
| | Scheme 9 Annulation of β-keto ester with acrolein to from the bicyclo[3.3.1]nonane. | |
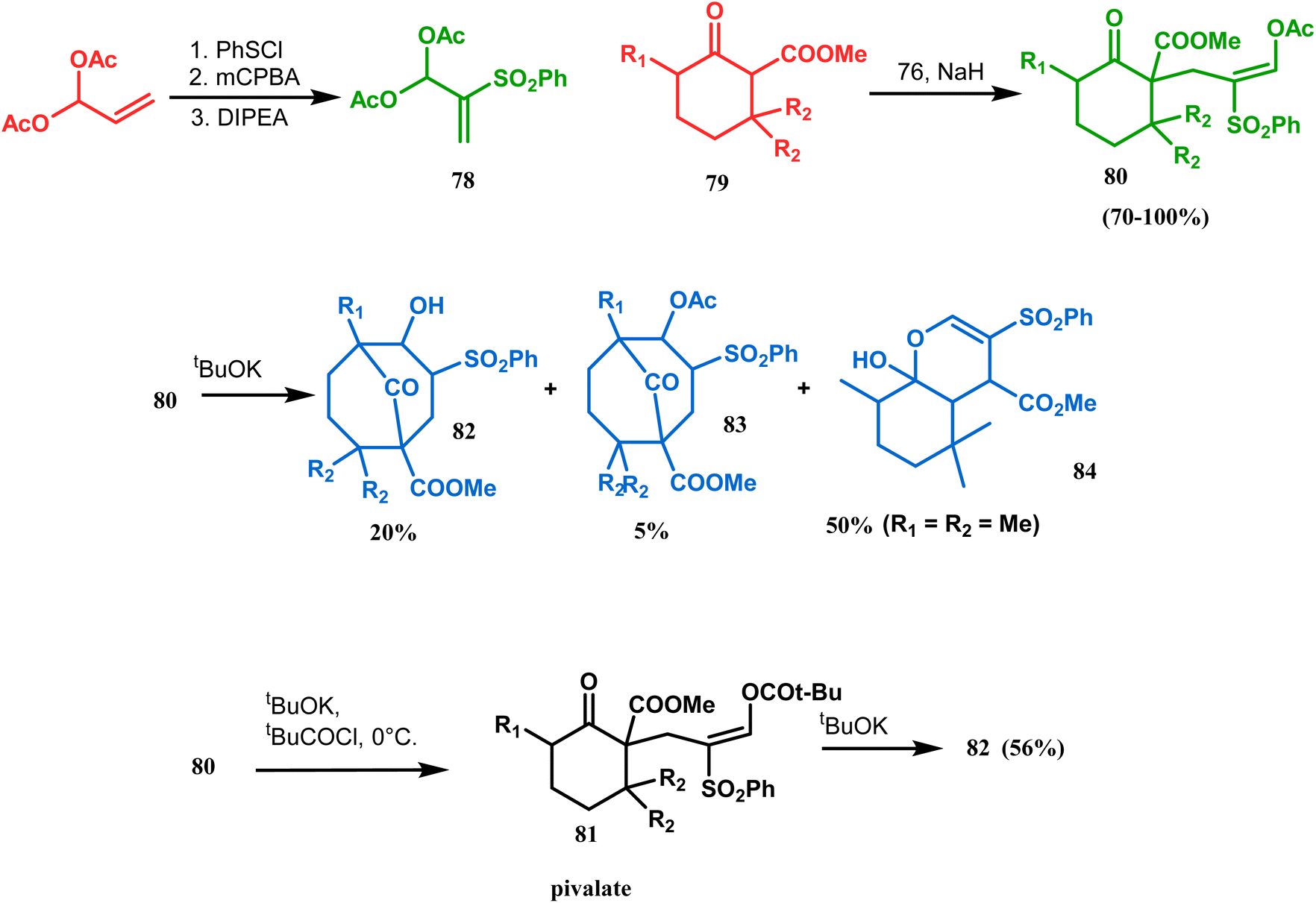
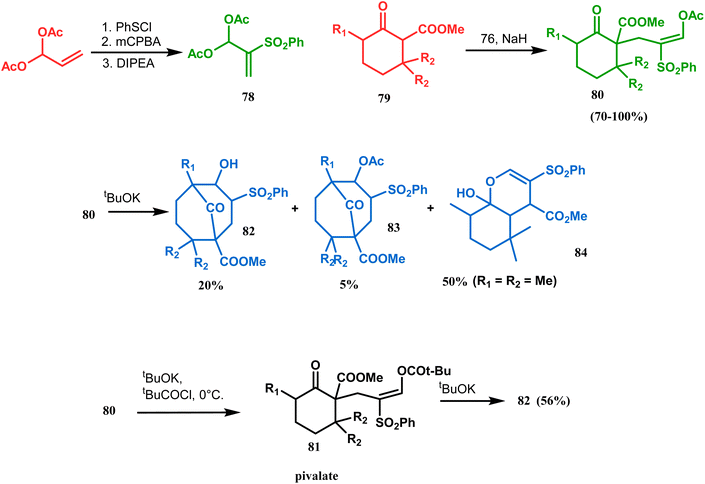
Kraus carried out a detailed investigation of the reaction between diacetoxy sulfone (78) and β-keto esters (79) (Scheme 10).77 The problem of easy deacetylation of 78, leading to unwanted deacetylated product 84 as the major product, was solved by converting 80 into the corresponding pivalate 81. When reacted with potassium tert-butoxide in THF, 79 gave the desired product 82 in 56% yield (Scheme 10). Similar results were also obtained with different R1 and R2.
 |
| | Scheme 10 Reaction of diacetoxy sulfone and β-keto esters. | |
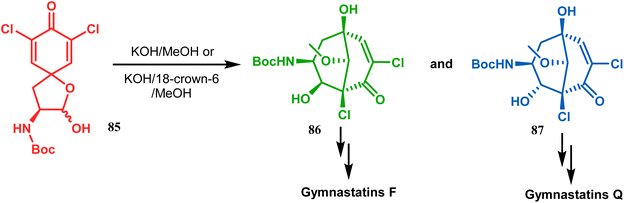
Base-induced tandem Michael addition-intramolecular aldolization is also well explored in natural product synthesis. A classic example in this category is the synthesis of gymnastatins F and Q.78 The synthesis of the key bicyclic core was accomplished by exposing a hemiacetal containing spirodienone (85) to KOH/MeOH or KOH/18-crown-6/MeOH to give 86 and 87, which were then converted to gymnastatins F (yield 36%) and Q (yield 64%), respectively, in a few synthetic steps (Scheme 11).
 |
| | Scheme 11 Synthetic route for the formation of the bicyclic core from a hemiacetal containing spirodienone through tandem Michael addition-intramolecular aldolization. | |
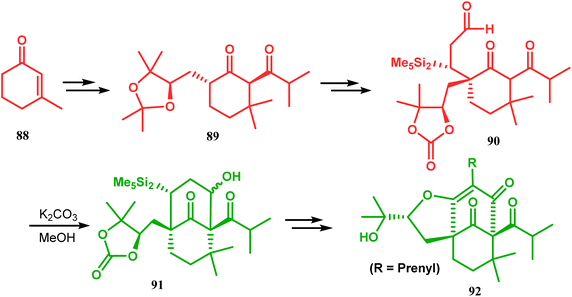
Another application of base-promoted aldolization was unveiled by Usuda during the synthesis of 18-epimer (92) of 8-deprenyl-garsubellin A. The synthetic precursor (90) for this purpose was accessed in a stereocontrolled manner from enone 88 through intermediate 89. Precursor 90, upon-base promoted intramolecular aldol condensation, furnished the bicyclic core 91 (yield 98%) and then led to the formation of tricyclic compound 92 in 69% yield.79 In a similar report, the same authors (Usuda and coworkers) also accessed another garsubellin A analogue (95).80 However, in this case, Al2O3 was used as the base instead of K2CO3 because potassium carbonate leads to the β-elimination of TESOH, forming a mixture of products (Schemes 12 and 13).
 |
| | Scheme 12 Base-promoted alodol condensation for the synthesis of 18-epimer of 8-deprenyl-garsubellin A. | |
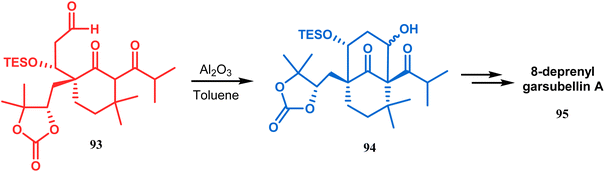 |
| | Scheme 13 Synthetic route for the formation of garsubellin A in the presence of Al2O3. | |
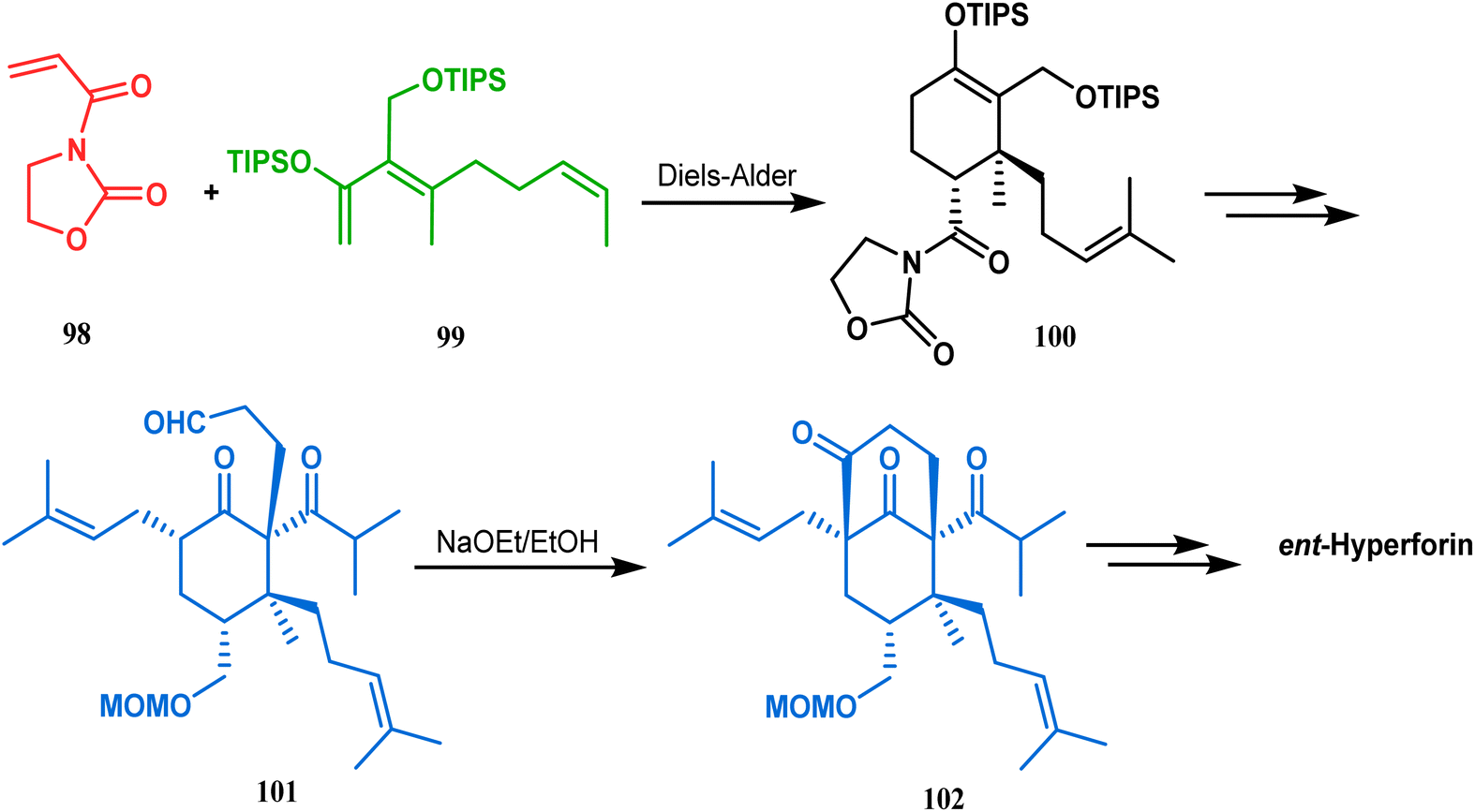
Similarly, the bicyclic core of PPAPs was constructed by Shibasaki's group through a base-promoted intramolecular aldolization technique. It proceeded through the synthesis of the annulation precursor 96, which was then intramolecularly aldolized to the bicycle 97 using NaOEt in EtOH (Scheme 14).81 Very recently, the same methodology was successfully utilized by them to synthesize enantiomerically pure (−)-hyperforin.82 Their targeted aldolization precursor (101) was prepared from the Diels–Alder adduct (100) of 98 and 99 in a stereocontrolled manner. NaOEt-promoted base-catalyzed intramolecular aldolization of 101 furnished the desired bicycle 102 (yield 86%), which was then sequentially converted to enantiomerically pure (−)-hyperforin (Scheme 15).82
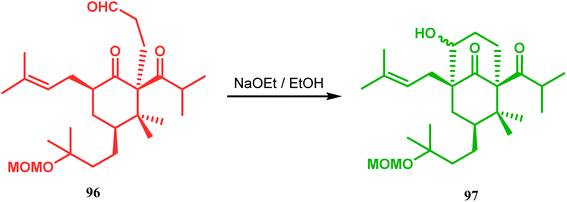 |
| | Scheme 14 Construction of the bicyclic core of polycyclic polyprenylated acylphoroglucinol (PPAs) through base-promoted intramolecular aldolization. | |
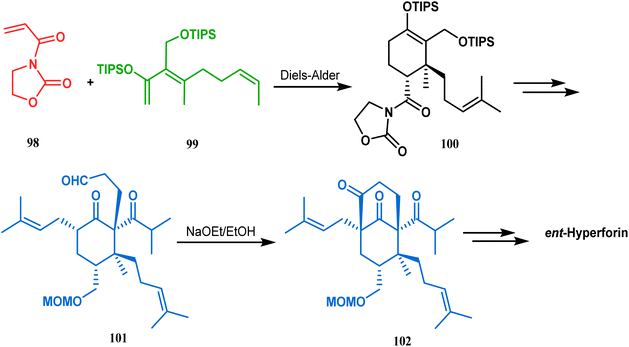 |
| | Scheme 15 Formation of the bicyclic core of (−)-hyperforin through base-promoted intramolecular aldolization. | |
The synthesis of such prenylated bicyclononane core of phloroglucin natural products through base-promoted intramolecular aldolization was extensively investigated by Mehta's group. DIBAL-H-mediated tandem lactone ring opening and intramolecular aldol condensation were the key steps for synthesizing the bicyclo[3.3.1]nonan-9-one cores (105, 106) of garsubellin A, hyperforin, guttiferone A, and hypersampsone F. This is a classic example where the dual characteristic of DIBAL-H, both as a reducing agent (to reduce lactone 103 to a lactol anion) and as a base (to encourage aldol condensation), was successfully applied (Scheme 16).83
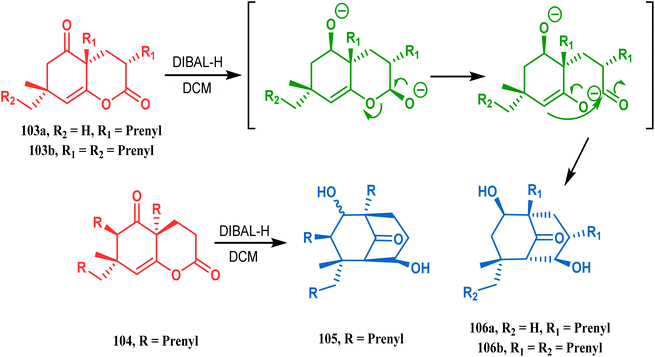 |
| | Scheme 16 Synthetic strategy for obtaining the bicyclo[3.3.1]nonan-9-one cores of garsubellin A, hyperforin, guttiferone A, and hypersampsone F through DIBAL-H-mediated tandem lactone ring opening and intramolecular aldol condensation. | |
A similar attempt was also made by Marazano and coworkers to access models for polyprenylated acylphloroglucinols with a different reducing agent, LiAlH(OtBu)3, instead of DIBAL-H. The desired bicyclo[3.3.1]nonan-9-one (110) was obtained in high yields (82%) by this procedure (Scheme 17).84
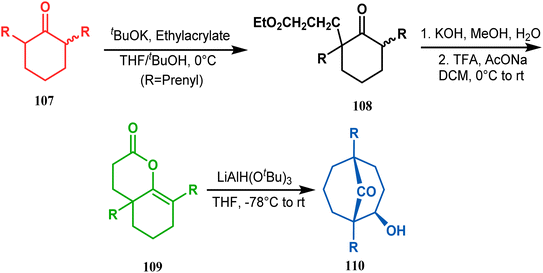 |
| | Scheme 17 Synthesis of polyprenylated acylphloroglucinols via LiAlH(OtBu)3-promoted intramolecular aldol condensation. | |
Apart from base-promoted aldolization techniques, the use of organocatalyzed aldol condensation to achieve the bicyclo[3.3.1]nonane core is well documented. An elaborate study was repored in this regard by Iwabuchi and coworkers. Although their initial attempt to synthesize 114 using L-proline as the catalyst produced the product in low yield and low stereoselectivity, modified proline-analogues 112 and 113 produced 114a and 112b, respectively, with high de and ee (Scheme 18). The result is attributed to the higher availability of catalytically active secondary amine of 112 and 113 than L-proline due to the lesser amount of zwitterion formation in the former two than in the last one. Besides, the hydrophobic environment produced by the tertbutyldiphenylsilyl group and the tetrabutylammonium ion around 112/113 lowered the pKa, thereby raising the nucleophilicity of amine nitrogen, which is required to catalyze the condensation. The reason behind the preference of 112 for 114a and that of 113 for 114b was rationalized from the favored H-bonding and dipole interaction between the developing ions in the transition state.85
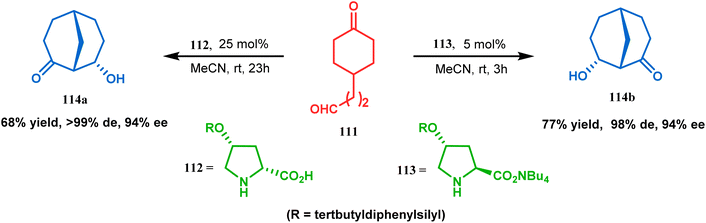 |
| | Scheme 18 Formation of the bicyclo[3.3.1]nonane core via organo-catalyzed aldol condensation. | |
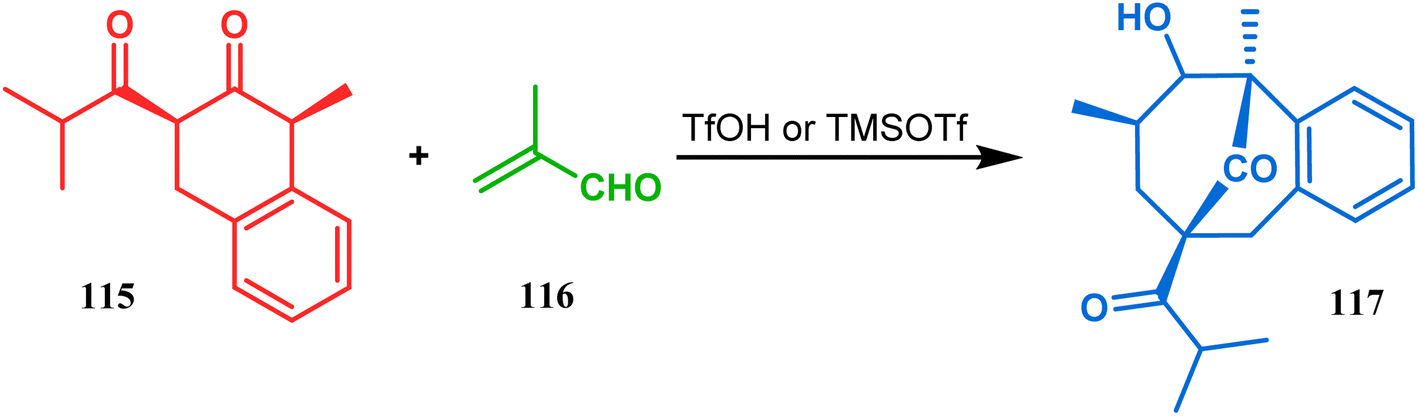
An acid-catalyzed tandem Michael addition-intramolecular aldol-type condensation of diketones to achieve bicyclo[3.3.1]nonenone has been reported by Nicolaou's group. Their studies toward the synthesis of a hyperforin model system involved the treatment of diketone 115 with methyl acrolein (116) in the presence of TfOH or TMSOTf, which promoted Michael addition on the doubly activated carbon of 115, followed by an intramolecular aldolization, leading to bicycle 117 in 63% yield (Scheme 19).86
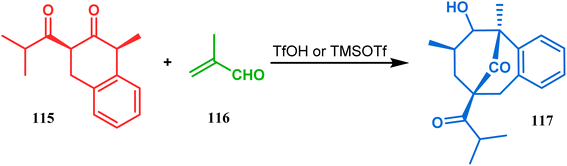 |
| | Scheme 19 Synthesis of hyperforin model via acid-catalyzed tandem Michael addition-intramolecular aldol-type condensation of diketone with methyl acrolein. | |
Acid-promoted routes toward the synthesis of bicyclic core of PPAPs are, however, also followed in Grossman's strategy, which involves an acid-catalyzed intramolecular aldolization of α-silyl enal 118 to furnish bicycle 119 in 72% yield (Scheme 20).87 Another acid-catalyzed methodology, developed by Dixon's group, involves the reaction with ester-carbonyl group, although it does not involve aldolization. Thus, when cyclooctanone derivative 122, obtained from 120 and 121, was treated with TsOH, an intramolecular C-alkylation at the α-position of the carbonyl group occurred, producing the bicyclo[3.3.1]noneone 123 in moderate to high yields (46–80%) (Scheme 21).88 Recently, Y. Kuninobu and coworkers utilized similar reaction partners 124 and 125 and treated them with [ReBr(CO)3(thf)]2 in the presence of TBAF. The reaction proceeded through the formation of cyclooctanone (126), which underwent an intramolecular condensation between the α-methylene of ketone and the ester functionality and produced another bicyclo[3.3.1]nonane-dione (128) (yield 79–93%) (Scheme 22).89
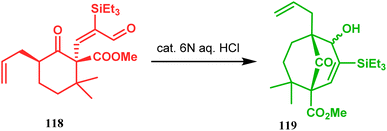 |
| | Scheme 20 Synthesis of the bicyclic core of PPAPs via the acid-catalyzed intramolecular aldolization of α-silylenal. | |
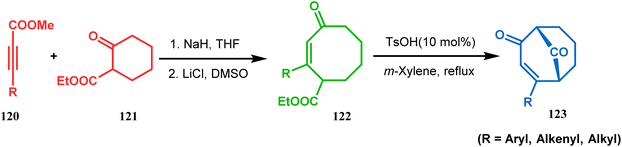 |
| | Scheme 21 Synthesis of bicyclo[3.3.1] noneone via acid-catalyzed intramolecular C-alkylation. | |
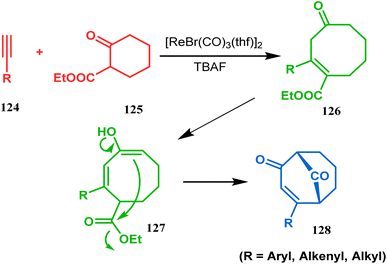 |
| | Scheme 22 Synthesis of bicyclo[3.3.1]nonane-dione via [ReBr(CO)3(thf)]-catalyzed intramolecular condensation. | |
3.1.2. Michael-type addition. The syntheses of such bicyclic ketones were also achieved by Michael addition reaction on acetylenic ω-ketoesters. Miesch and coworkers, while attempting the synthesis of novel oxetane derivatives from acetylenic ω-ketoesters (130) (yield 51%), accidentally found that though use of TBAF produces the desired product, the result was surprisingly modified when TBAF was replaced with tBuOK. The reaction now yielded the bicyclo[3.3.1]nonane core containing tricyclic derivative 131 in 42% yield through a tandem Michael addition–Claisen condensation cascade (Scheme 23).90,91
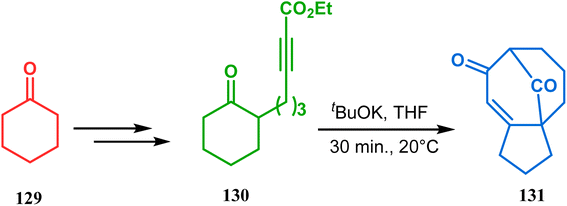 |
| | Scheme 23 Synthesis of the bicyclo[3.3.1]nonane core containing tricyclic derivative through a tandem Michael addition–Claisen condensation cascade in the presence of tBuOK. | |
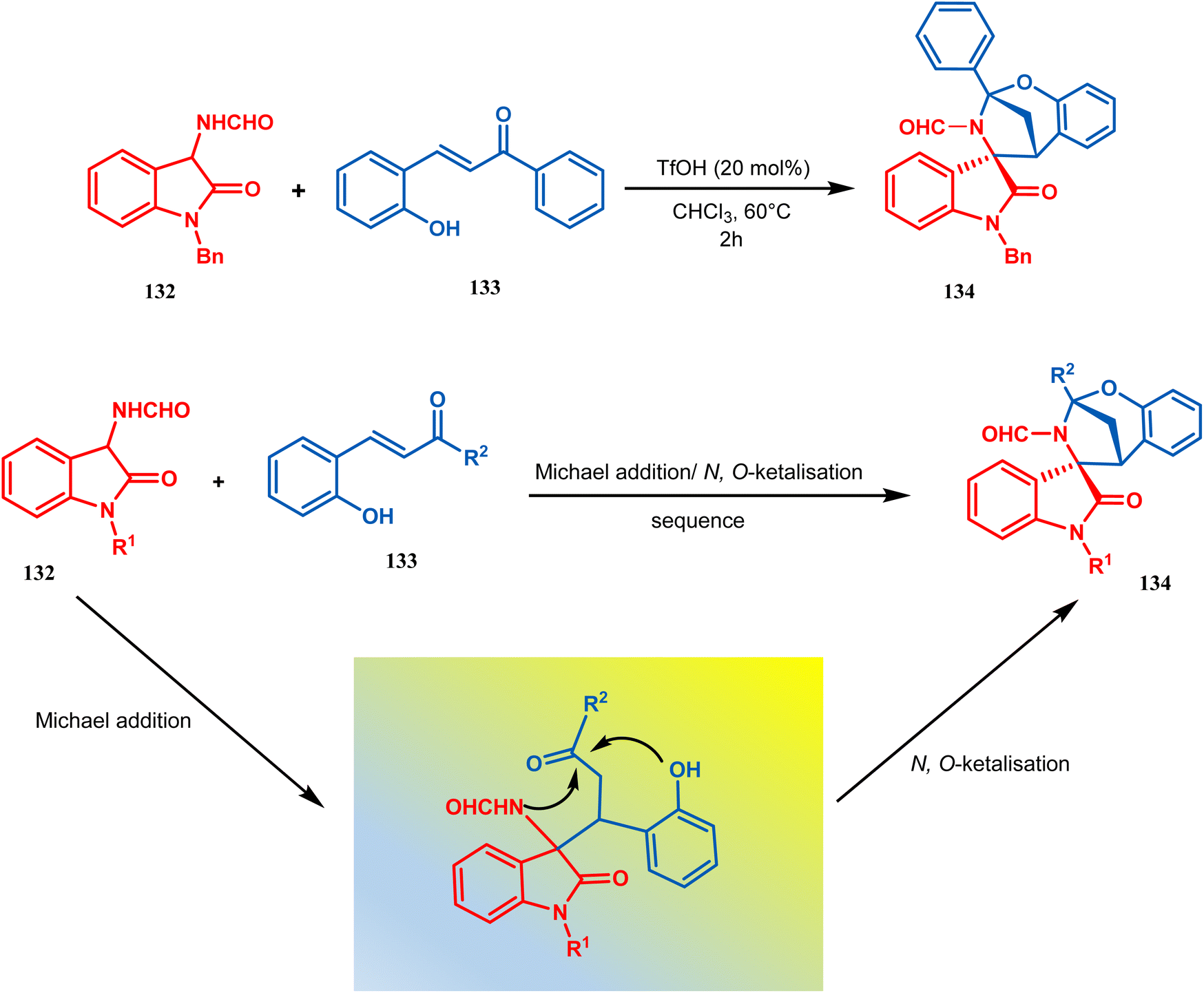
Zhanwei Bu's group developed a new synthetic strategy for the synthesis of bridged cyclic N,O-ketal spirooxindoles via the Michael addition-driven cyclization reaction of 3-hydroxyoxindoles with ortho-hydroxy-chalcones (133). Also, the Michael addition/N,O-ketalization sequence of 3-aminooxindoles (132) with ortho-hydroxychalcones (133) may take place under particular conditions, but 3-amino-oxindole (132) showed relatively lower reactivity compared with 3-hydroxyoxindole. Therefore, they accomplished the Michael addition/N,O-ketalization sequence of 3-amino-oxindoles (132) with ortho-hydroxychalcones under the catalytic influence of TfOH. Thus, they were able to construct a series of diastereoselective bridged cyclic N,O-ketal spirooxindoles (134) with rigid and new skeletons (yield 73%) (Scheme 24).92
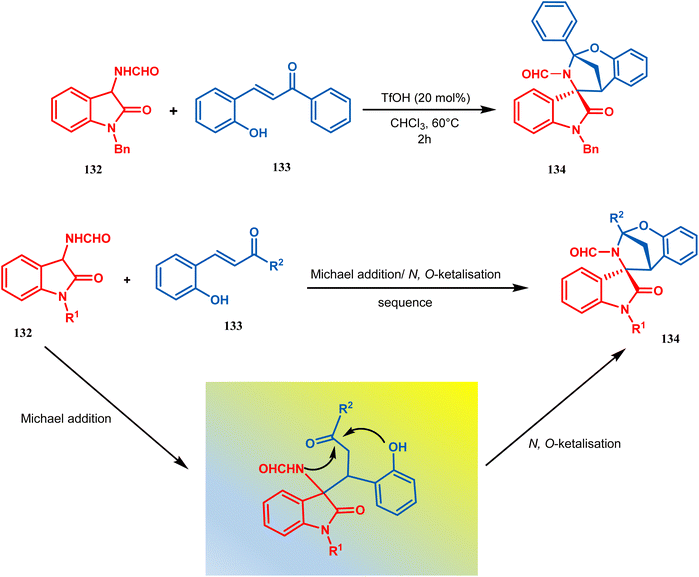 |
| | Scheme 24 Synthesis of diastereoselective bridged cyclic N,O-ketal spirooxindole having a bicyclic core via the Michael addition-driven cyclization reaction. | |
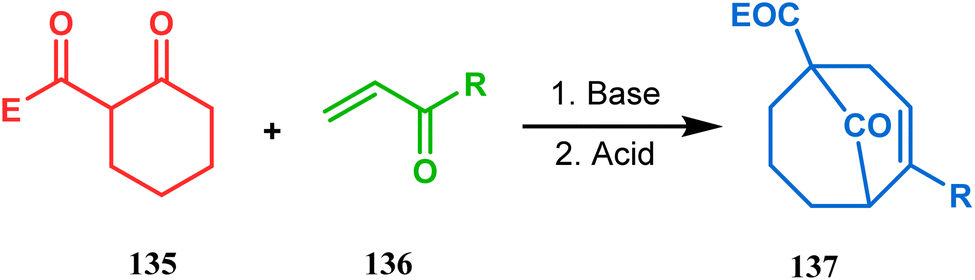
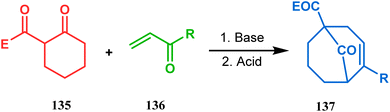
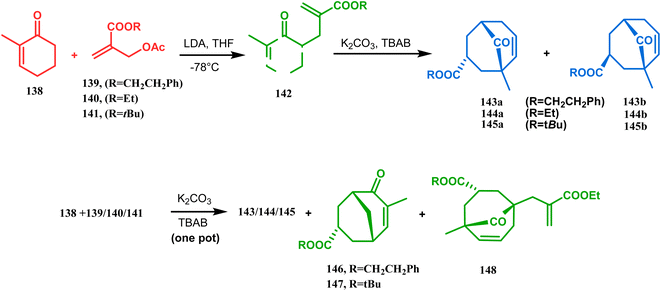
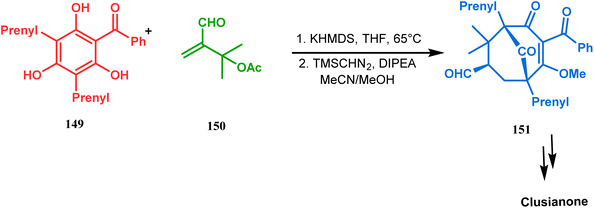
Apart from this, the annulation of β-keto thiolesters or β-keto sulfones93 is also well explored for the synthesis of bicyclo[3.3.1] nonenones. The process involves a base-catalyzed Michael addition between 135 and 136, followed by an acid-catalyzed aldol condensation reaction, forming a β,γ-unsaturated bicyclo[3.3.1]nonenone (137) (Scheme 25). Such nonenones could also be accessed through Michael addition reaction on acrylate derivatives. For example, the β,γ-unsaturated bicyclo[3.3.1]nonenone (143) could be synthesized from 2-cyclohexenone (138) using two successive Michael addition reactions on 139. Although isomerization studies proved that 143b is thermodynamically more stable, the major product becomes 143a (yield 35–36%) via intermediate 142 (yield 50–74%). More interestingly, when a one-pot operation is performed instead of two consecutive operations, a minor amount of α,γ-annulation product (146) (yield 2–7%) is formed along with the α,α′-annulation products (143–145) (Scheme 26). In some cases, for example, with ethyl acrylate derivatives (145), three consecutive Michael addition-derived product 148 with 15% yield is also formed in addition (Scheme 26).94 Applying the same strategy, Porco and coworkers developed the alkylative dearomatization-annulation methodology, which was successfully utilized to achieve the bicyclo[3.3.1]nonane core of clusianone during its total synthesis. The methodology employed LiHMDS or KHMDS to realize the target 151 in 54% yield (Scheme 27) and was also successfully applied to construct multisubstituted bicyclic cores such as clusianone (yield 74%).94
 |
| | Scheme 25 Synthesis of bicyclo[3.3.1]nonenones through the annulation of β-keto thiolesters or β-keto sulfones via base-catalyzed Michael addition and then acid-catalyzed aldol condensation reaction. | |
 |
| | Scheme 26 Synthesis of the bicyclo[3.3.1]nonane core via successive Michael addition reactions. | |
 |
| | Scheme 27 Synthesis of the bicyclo[3.3.1]nonane core of clusianone through alkylative dearomatization-annulation methodology. | |
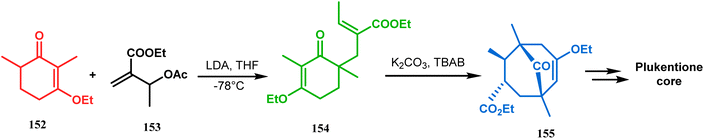
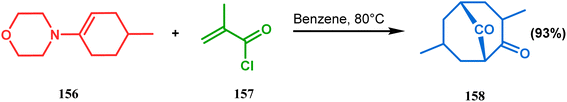
Shortly after the communication by Porco's group, the same strategy was applied by Takagi and coworkers to successfully synthesize the adamantane core of plukentione-type PPAP.95 The cyclohexenone derivative 152 was reacted with acrylate 153 and the annulation precursor 154 (E/Z 25![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1) was obtained in 92% yield. To achieve 155, best results were obtained when 154 was treated with K2CO3 and TBAB. Although the use of cesium carbonate increases the yield of the targeted bicyclic core (155) (from 41–55% yield), the unwanted α,γ-annulation product was also produced as a substantial impurity. However, in both cases, product 155 was obtained as a diastereoisomeric mixture (Scheme 28). Once the bicyclic core was synthesized, a few more synthetic steps led to the desired skeleton.97 In another report by Gambacorta's group, the morpholine derivative 156 was utilized as the Michael donor and methacryloyl chloride (157) as the Michael acceptor. Refluxing in benzene facilitated Michael addition and concomitant cyclization to yield the bicyclo[3.3.1]nonane 158 in 93% yield (Scheme 29).96
:1) was obtained in 92% yield. To achieve 155, best results were obtained when 154 was treated with K2CO3 and TBAB. Although the use of cesium carbonate increases the yield of the targeted bicyclic core (155) (from 41–55% yield), the unwanted α,γ-annulation product was also produced as a substantial impurity. However, in both cases, product 155 was obtained as a diastereoisomeric mixture (Scheme 28). Once the bicyclic core was synthesized, a few more synthetic steps led to the desired skeleton.97 In another report by Gambacorta's group, the morpholine derivative 156 was utilized as the Michael donor and methacryloyl chloride (157) as the Michael acceptor. Refluxing in benzene facilitated Michael addition and concomitant cyclization to yield the bicyclo[3.3.1]nonane 158 in 93% yield (Scheme 29).96
 |
| | Scheme 28 Synthesis of the adamantane core of plukentione-type PPAP. | |
 |
| | Scheme 29 Synthesis of the bicyclo[3.3.1]nonane core via Michael addition and concomitant cyclization reaction. | |
The application of such a reactive starting material pair (cyclohexenone and acrylate) was also extensively studied by Kraus and coworkers. Their route for the total synthesis of papuaforin A unveils one such attempt. In this case, the cyclohexenone derivative 159 was treated with methyl acrylate in the presence of tBuOK, and the intermediate Michael addition product was subjected to Birch reduction/cyclization97,98 to give 160 (Scheme 30), which was then used as the precursor for the synthesis of papuaforin A.15a
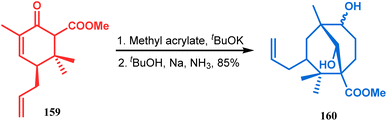 |
| | Scheme 30 Synthesis of papuaforin A via Michael addition, followed by Birch reduction and cyclization. | |
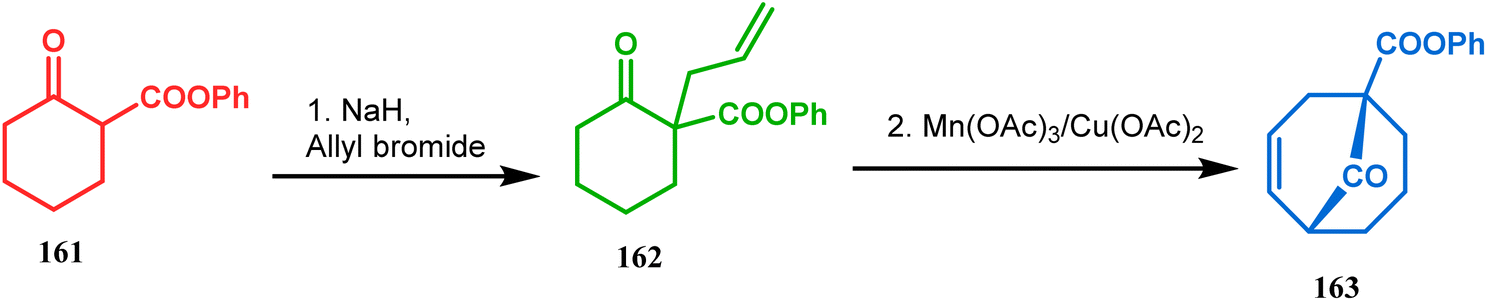
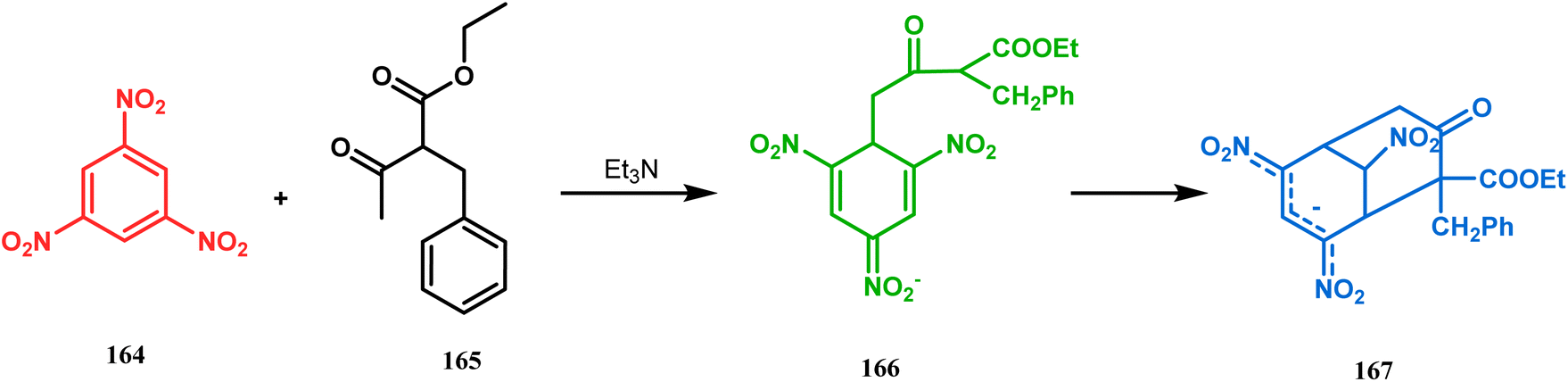
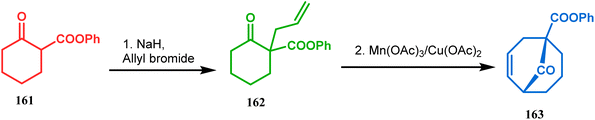
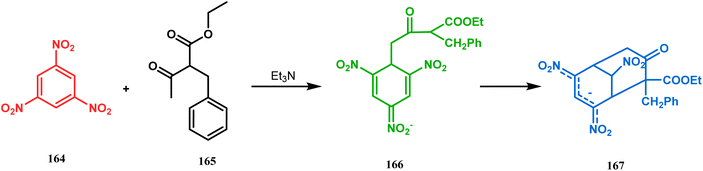
Another application of cyclohexanone by the same group utilizes allyl bromide instead of acrylate derivatives to achieve a similar bicyclo[3.3.1]nonane core. In this case, the synthesis of the bicyclic core of hyperforin and nemorosone was attempted by treating 161 with allyl bromide in the presence of sodium hydride. The intermediate 162 thus formed was then intramolecularly cyclized to 163 by manganic triacetate and cupric acetate having yield of 60% (Scheme 31).15b,99 The use of such 1,3-dicarbonyls as the Michael donor is also exemplified by Kalaivani's group. Thus, 1-benzyl-1-(ethoxycarbonyl)-2-propanone (BEP, 165) was treated with trinitrobenzene (TNB, 164) in the presence of triethyl amine, where TNB acted as the Michael acceptor and the anionic sigma complex 167 was formed (Scheme 32).99
 |
| | Scheme 31 Synthesis of the bicyclo[3.3.1]nonane core by the reaction of allyl bromide and cyclohexanone derivative. | |
 |
| | Scheme 32 Formation of the bicyclo[3.3.1]nonane core by the Michael addition of 1,3-dicarbonyl compound and trinitrobenzene. | |
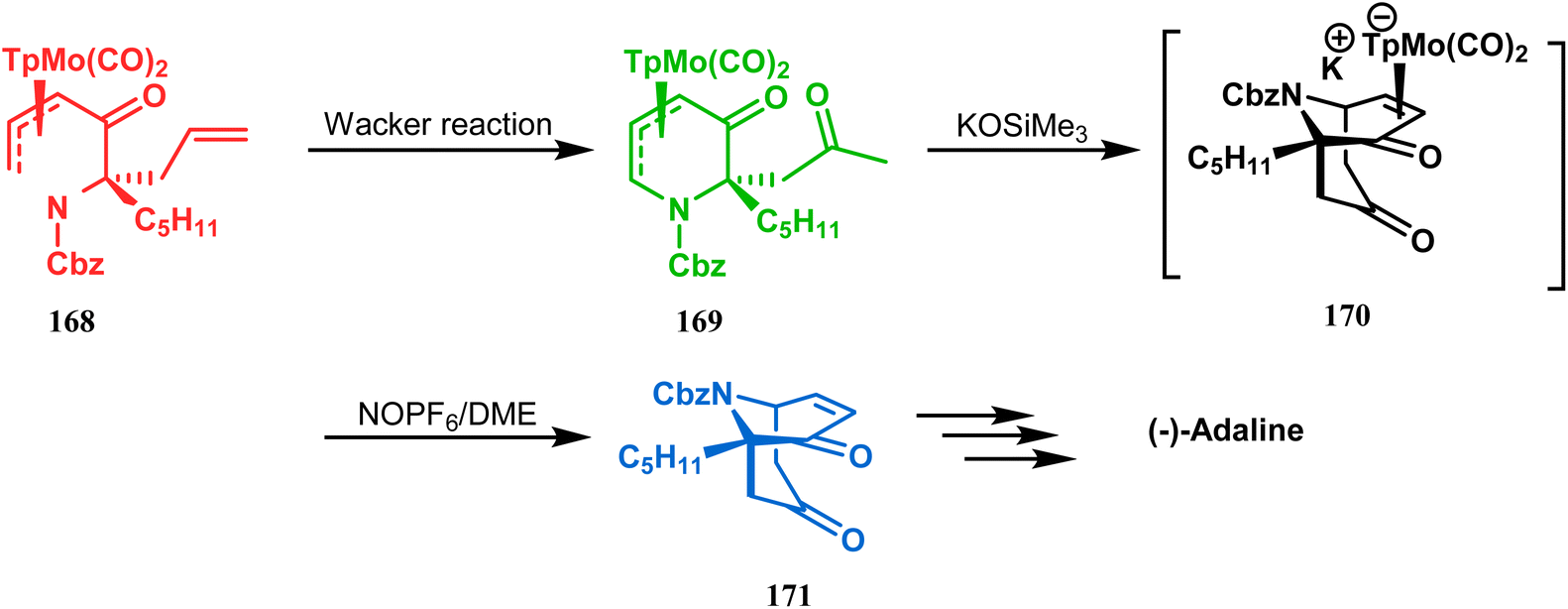
However, Liebeskind and coworkers have described a unique application of a Michael-like addition reaction in the synthesis of natural products using organometallic enantiomeric scaffolding. The authors utilized a TpMo(CO)2(5-oxo-η3-pyridinyl) complex (168) to create the methylketone (169) by Wacker reaction. The Michael-like addition precursor (169) was then reacted with KOSiMe3, which promoted the 1,5-Michael-like bond forming reaction through attack at the neutral η3-allylmolybdenum by the enolized ketomethyl group. The anionic intermediate 170 furnished the bicycle 171 when treated with NOPF6 in DME. A sequential ketalization, reduction, and carbobenzyloxy-deprotection then led to the formation of enantiomerically pure (−)-adaline (Scheme 33).10
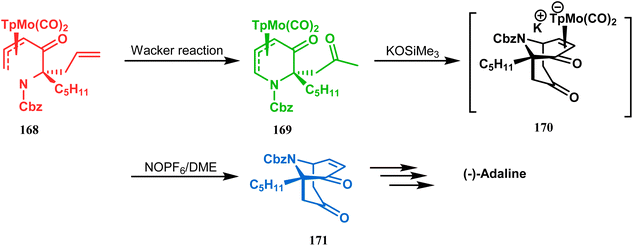 |
| | Scheme 33 Synthetic route for the formation of enantiomerically pure (−)-adaline. | |
3.1.3. Intramolecular cation capture. Apart from these two major classes of intramolecular C–C bond forming approaches, there are several other unique strategies that have applied for the synthesis of the bicyclo[3.3.1]nonane core. One such example is intramolecular carbocation capture. The application of this strategy to obtain the bicyclic core was first demonstrated by Williams's group during the total synthesis of (±)-5,14-bis-epi-spirovibsanin A. Following Bernhardt's method,100–103 initially, they synthesized the racemic enone 173 (yield 70%, 91% ee) from the cyclohexenone derivative 172. The enone (173) was then treated with HCl in methanol, thereby generating a carbocation, which was intramolecularly captured to give the bicyclic ketone 174. Subsequently, through a number of synthetic steps, this bicyclic precursor then produced (±)-5,14-bis-epi-spirovibsanin A (175) (Scheme 34).11 A similar strategy was again used by the same group very recently during the total synthesis of (−)-neovibsanin G and 14-epi-neovibsanin G (181 & 182). However, instead of HCl or H2SO4, the Lewis acid EtAlCl2 was employed this time to carry out the reaction on 176. The desired bicycle (180) was formed due to the epimerization of 178 to 179 to relieve the steric strain between the adjacent carbonyl functionalities. Compound 180 was then sequentially converted into the targeted natural products (Scheme 35).
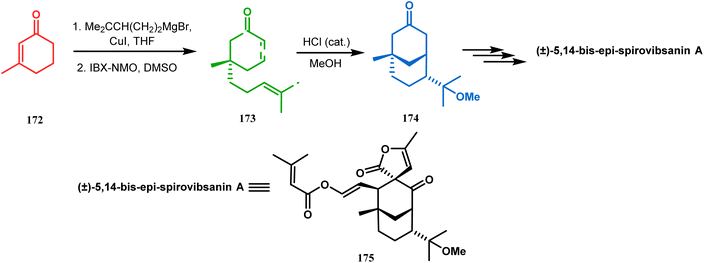 |
| | Scheme 34 Synthesis of (±)-5,14-bis-epi-spirovibsanin A containing the bicyclo[3.3.1]nonane core through intramolecular cation capture. | |
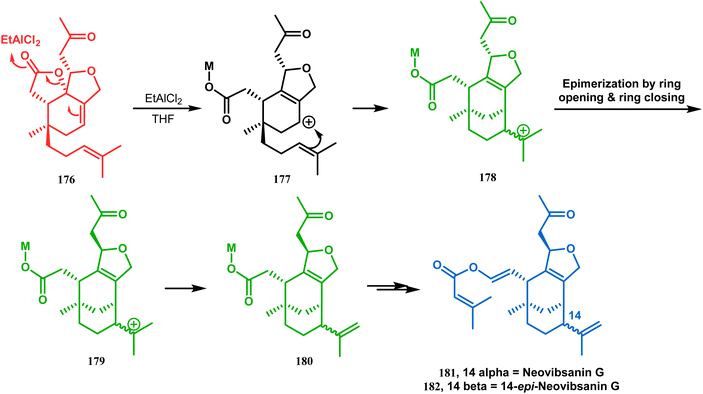 |
| | Scheme 35 Synthesis of (−)-neovibsanin G and 14-epi-neovibsanin G through intramolecular cation capture. | |
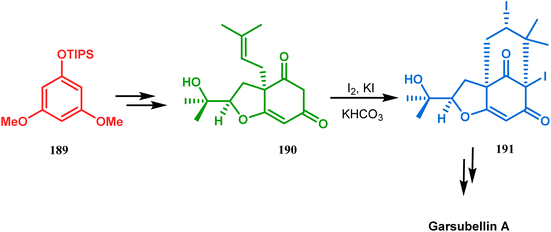
Intramolecular iodonium capture is another such pioneering approach applied by Danishefsky's group for preparing the bicyclo[3.3.1]nonane core during the total synthesis of nemorosone and clusianone. Properly substituted precursor 185, required for their envisioned iodonium formation and carbocyclization, was synthesized from phloroglucinol derivative 183 and reacted with iodine in the presence of KI-KHCO3 to form the desired bicyclic core (188) through iodonium intermediates. During the carbocyclization of 185, two other undesired cyclization products (186, 187) were also formed with yield 78% and 87%, respectively, along with 188. This problem was solved through a high yielding conversion of 186 and 187 into 188 (Scheme 36). However, an identical route toward garsubellin A did not encounter such problems. It is believed that the tetrahydrofuran group present in 190 tilted the conformation of the intermediate iodonium such that the iodinative cyclization proceeded strictly through C–C bond formation, resulting in the exclusive formation of the desired bicycle 191 (Scheme 37). The bicycles (188 and 191) thus formed were then converted to the targeted polyprenylated acylphloroglucinols (PPAPs) through a few more synthetic steps.104,105
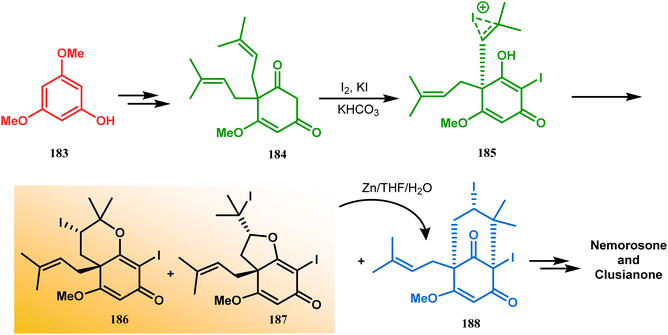 |
| | Scheme 36 Synthesis of nemorosone and clusianone through intramolecular iodonium capture. | |
 |
| | Scheme 37 Synthesis of garsubellin a through intramolecular iodonium capture. | |
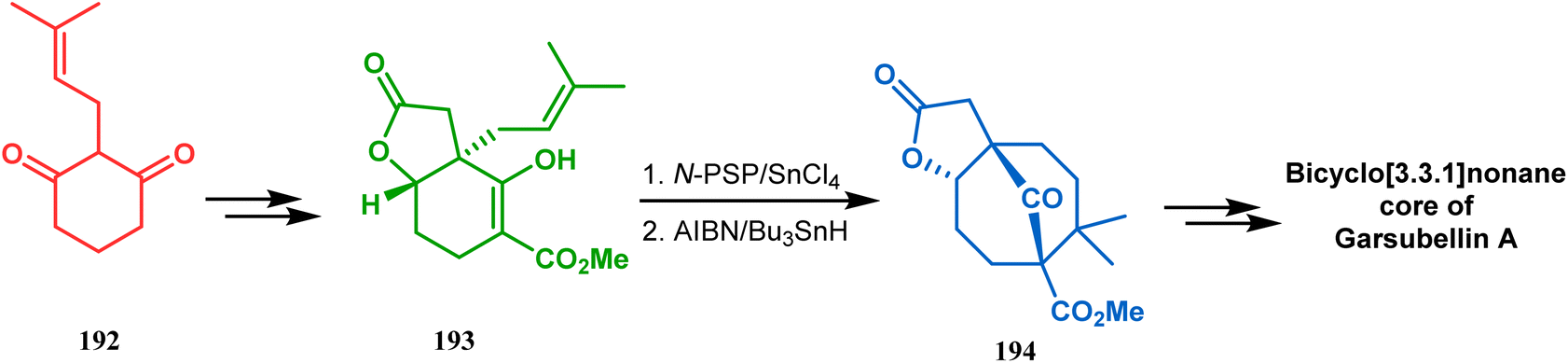
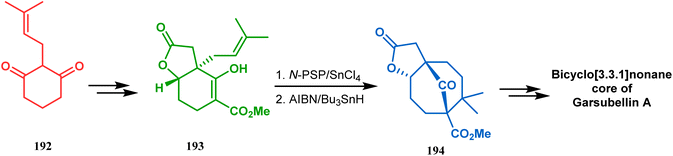
A strategy developed by Nicolaou's group, similar to this iodonium-induced carbocyclization, uses the oxyselenation of olefins.106–108 During the synthesis of the fully functionalized bicyclic core of garsubellin A, this group disclosed a unique application of the N-(phenylseleno)phthalimide/SnCl4 pair for bicyclo[3.3.1]nonane synthesis. The precursor 193, prepared from 1,3-cyclohexadione (192), produced the desired bicycle 194 in 95% yield when treated with N-PSP/SnCl4 in DCM at −23 °C (Scheme 38). The same group utilized this important methodology even in the solid phase synthesis of resin-bound polyfunctionalized bicyclo[3.3.1]nonanes using a one-step loading/cyclization method.
 |
| | Scheme 38 Synthesis of the bicyclo[3.3.1]nonane core of garsubellin A. | |
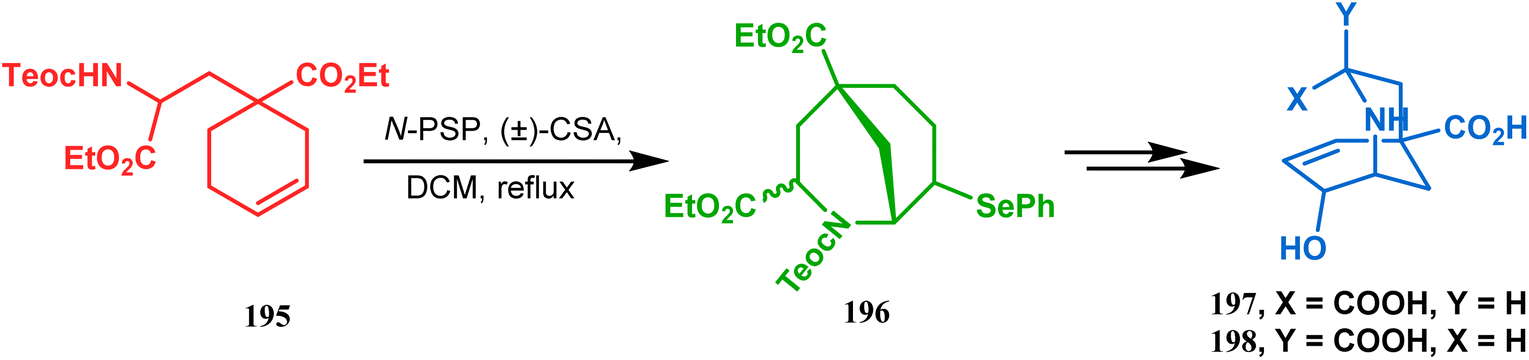
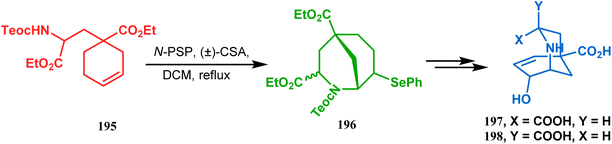
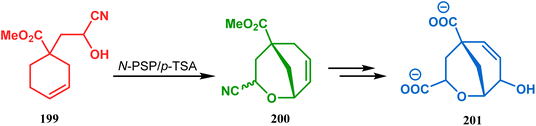
This versatile strategy was also used by Hediger to synthesize a novel class of chorismate mutase inhibitors based on azabicyclo[3.3.1]nonane systems. The key bicyclic core 196 was synthesized from 195 by treating with N-PSP and camphorsulfonic acid (Scheme 39).109 The synthesis of this class of chorismate mutase inhibitors was, however, well investigated by Bartlett's group.110,111 Starting from a cyclohexene derivative 199, they utilized the same N-PSP-mediated strategy to access the ether bicycle 200, which after a few synthetic steps led to 201, a potent inhibitor of chorismate mutase (Scheme 40).
 |
| | Scheme 39 Synthesis of azabicyclo[3.3.1]nonane systems. | |
 |
| | Scheme 40 Synthesis of another potent inhibitor of chorismate mutase. | |
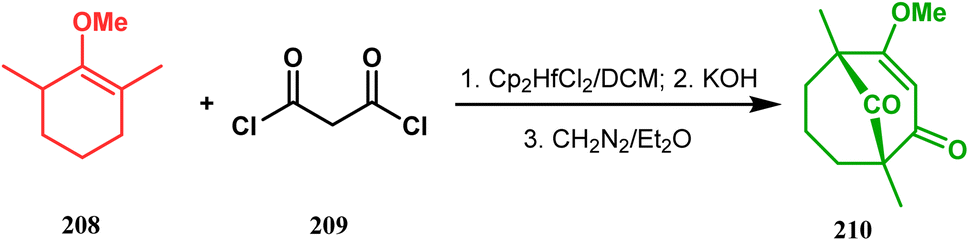
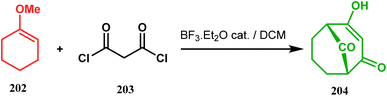
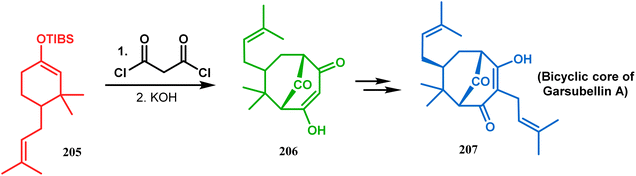
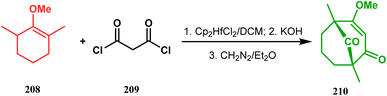
3.1.4. Effenburger-type cyclization. In 1984, Effenberger discovered an efficient route toward the synthesis of bicyclo[3.3.1]nonane ring systems through the reaction of 1-methoxy-1-cyclohexene (202) and malonyl dichloride (203). Since then, this versatile methodology was used by several research groups to achieve synthetic targets containing the bicyclic core 204 (Scheme 41). For example, Stoltz and coworkers applied this strategy on properly functionalized cyclohexanone enol ether 205 and obtained their desired bicycle 206 in 36–55% yield, required for the synthesis of the bicyclic core (207) of garsubellin A in high yield (Scheme 42).112 To achieve this target, the authors reversed the ratio of the starting materials compared to the Effenberger study and used a TBS enol ether instead of methyl enol ether. When a more hindered enol ether (α,α′-disubstituted) was subjected to Effenberger-type cyclization under the similar reaction protocol using TBS enol ether, the reaction failed. However, the methyl enol ether 208 could be cyclized to 210 in 25% yield in the presence of bis(cyclopentadienyl)hafnium dichloride, a Lewis acid mediator (Scheme 43).113 Simpkins and coworkers also proved that the methyl enol ether is the best choice for hindered substrates.
 |
| | Scheme 41 Synthesis of the bicyclo[3.3.1]nonane core following Effenburger-type cyclization. | |
 |
| | Scheme 42 Formation of the bicyclic core of garsubellin A following Effenburger-type cyclization. | |
 |
| | Scheme 43 Synthesis of the bicyclic core with the use of methyl enol ether. | |
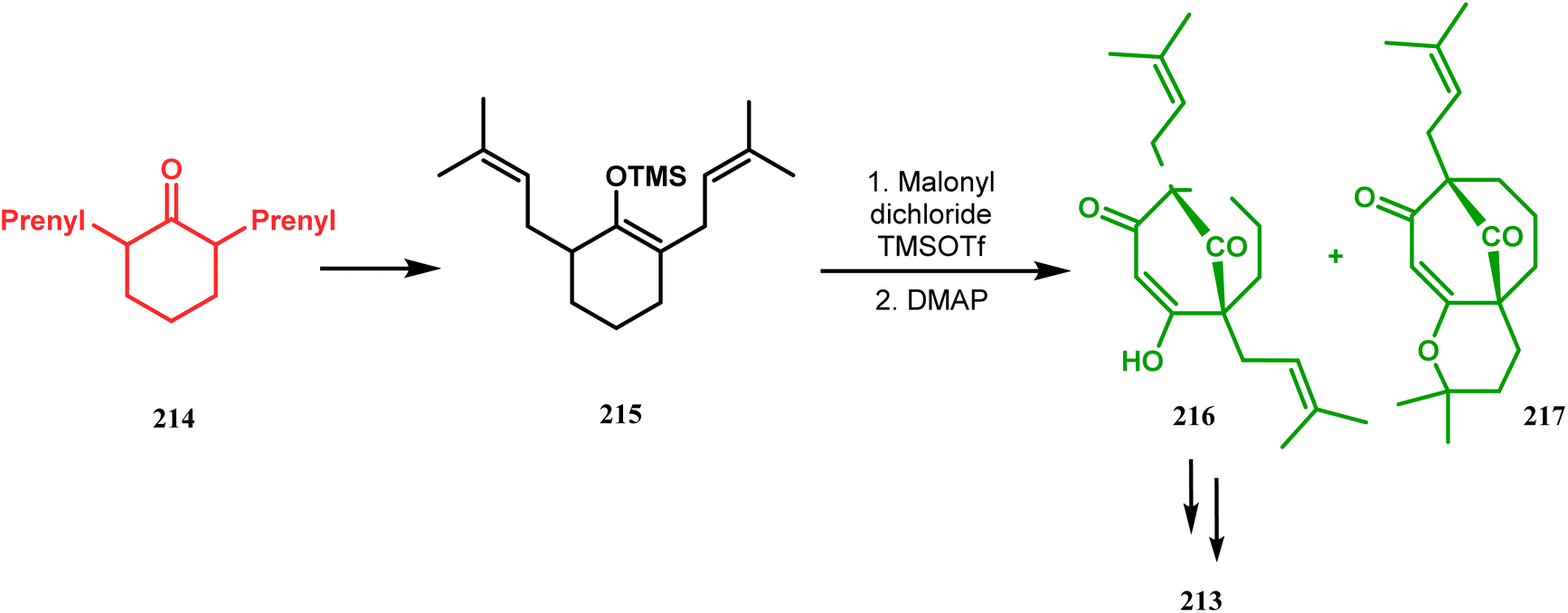
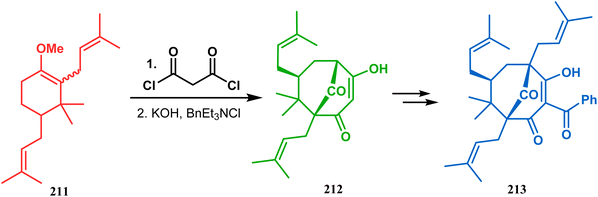
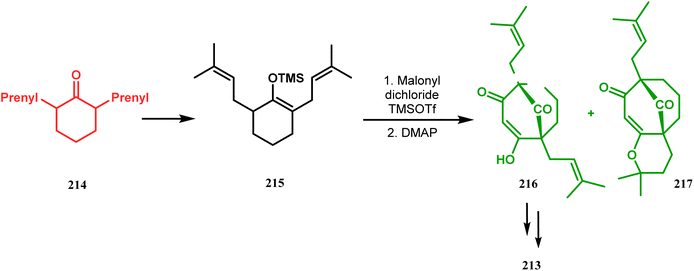
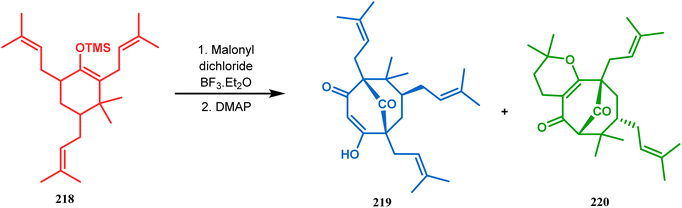
Thus, while attempting the total synthesis of clusianone, the authors subjected an appropriately substituted methyl enol ether 211 to Effenberger cyclization condition and obtained the bicycle 212, which then led to racemic clusianone 213 (yield 90%) in a few synthetic steps (Scheme 44).114 The separation of these racemates was also achieved by them through a bridgehead lithiation strategy using chiral bislithum amides.115 Marazano and coworkers applied this important strategy to achieve the same target (clusianone) starting from a different substrate, a hindered TMS enol ether (215) of 2,6-diprenyl cyclohexanone (214). This α,α′-disubstituted TMS enol ether (215) reacted smoothly with malonyl dichloride and produced the desired bicycle 216 along with another structurally complex unexpected bicyclo[3.3.1]nonane product 217.116 Subsequently, 216 was transformed into clusianone in a few synthetic steps (Scheme 45).117 The results were also similar with a more hindered TMS enol ether 218. When 218 was annulated with malonyl dichloride through Effenberger-type cyclization using BF3·Et2O, the desired bicycle 219 was produced in moderate yields along with another tricyclic derivative 220 (Scheme 46).118
 |
| | Scheme 44 Synthesis of clusianone from substituted methyl enol ether under Effenberger cyclization condition. | |
 |
| | Scheme 45 Synthesis of the bicyclo[3.3.1]nonane core of clusianone using a hindered TMS enol ether of 2,6-diprenyl cyclohexanone. | |
 |
| | Scheme 46 Synthesis of the bicyclic core along with a tricyclic derivative by the reaction of hindered TMS enol ether and malonyl dichloride. | |
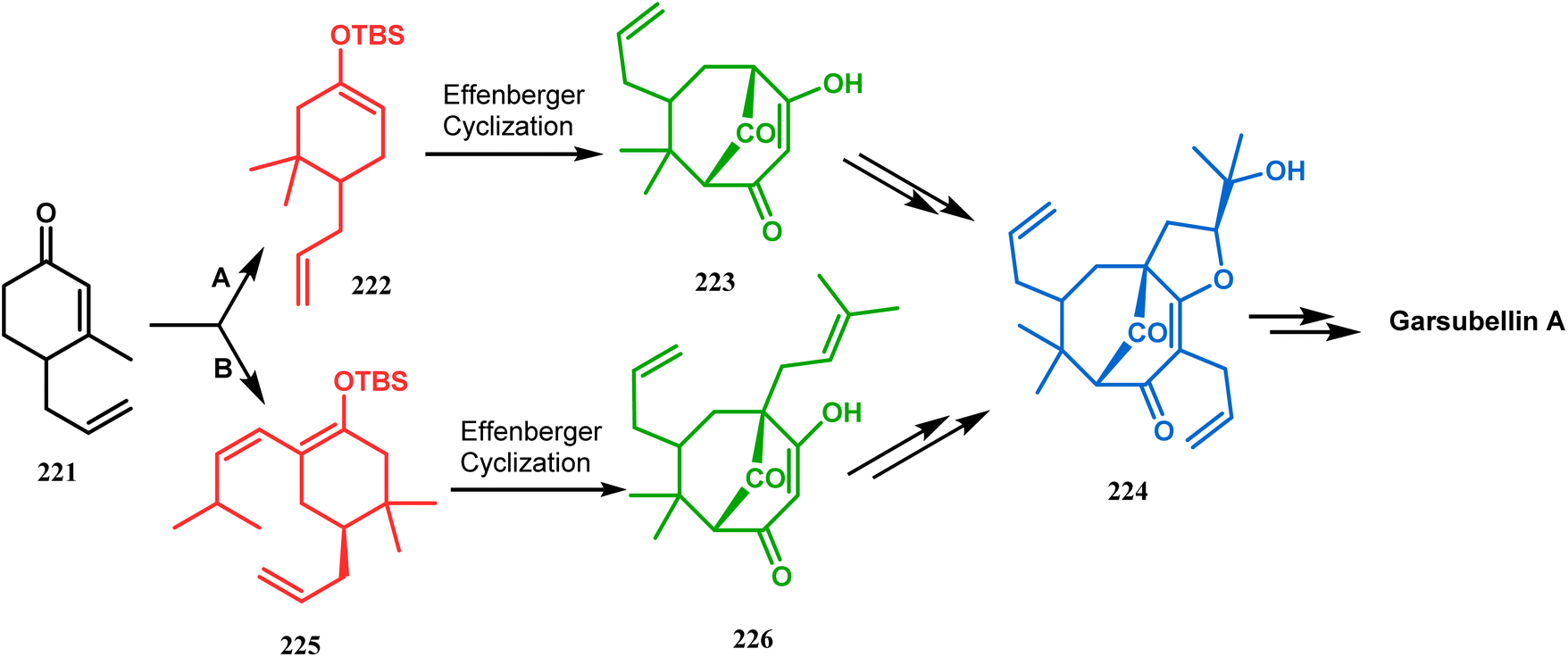
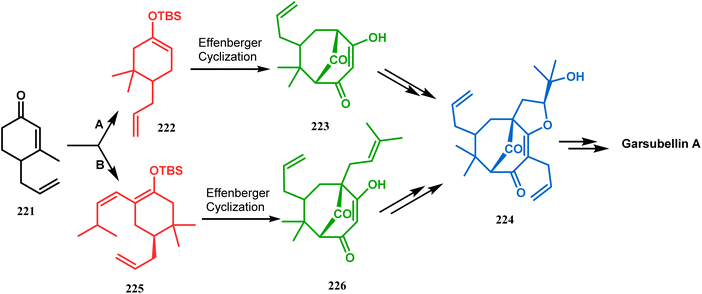
Simpkin's group reported another detailed study on the efficacy of Effenberger-type cyclization in the total synthesis of PPAPs such as garsubellin A, clusianone, and nemorosone. Their target was to synthesize the Danishefsky's bicyclic intermediate 224 in fewer but effective synthetic steps from the TBS enol ether 222 (route A).104,105 Employing the Effenberger cyclization, they then achieved 224 in the shortest route but with a modest yield (Scheme 47).
 |
| | Scheme 47 Synthesis of various PPAPs following Effenberger-type cyclization via the formation of Danishefsky's bicyclic intermediate. | |
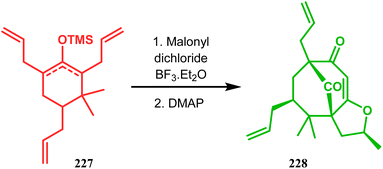
In a modified approach, they minimized the problems with yield-reducing nonstereoselective side-chain reactions, introduced the C-5 prenyl group prior to Effenberger cyclization (route B), and accessed 224 from a different TBS enol ether 225 through the cyclization product 226 (Scheme 47).15c Apart from natural product synthesis, the Effenberger cyclization technique is well explored in the synthesis of other bicyclo[3.3.1]nonane cores. For example, T. J. Blacklock and coworkers described another application of this methodology to synthesize the bicyclo[3.3.1]nonane-2,4,9-trione system 228 from TMS enol ether 227 (Scheme 48).119
 |
| | Scheme 48 Synthesis of the bicyclo[3.3.1]nonane-2,4,9-trione system following the Effenberger cyclization technique. | |
3.1.5. Visible light-driven cyclization. The use of visible light for the synthesis of chemical compounds in the presence of photoredox catalysts has gained incredible success owing to the growing demand for green and sustainable chemistry. A photoredox catalyst is very proficient in transferring light energy to the reacting molecules via a redox neutral pathway. In this synthetic method, single electron transfer (SET) takes place and, thereby, the molecules can be activated depending on the distinctive mode of activation by the photoredox catalyst. Weiqing Xie and coworkers performed the tandem cycloisomerization of 2-aminochalcone (229) with bifunctional nucleophiles in the presence of visible light. This cascade process was accomplished by the irradiation of blue LED at room temperature, which helped to synthesize a structurally-diverse benzo[d][1,3]oxazocine scaffold (231). Benzoxazocine belongs to a family of molecules where both oxygen and nitrogen atoms are embedded in an eight-membered ring, which displays some crucial pharmaceutical properties such as antithrombotic, analgesic, antioxidant, and anticancer activities. Upon irradiation of visible light, 2-amiochalcone took part in tandem E–Z isomerization to yield an intermediate quinolinium molecule, which was subsequently involved in cascade nucleophilic addition as well as cyclization and thus converted to polycyclic benzo[d][1,3]oxazocine (231) in 99% yield (Scheme 49).120
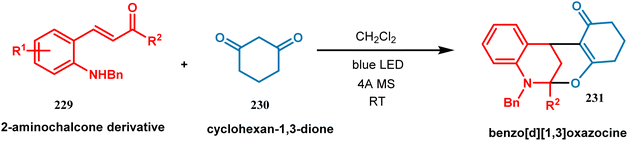 |
| | Scheme 49 Synthesis of polycyclic benzo[d][1,3]oxazocine from 2-aminochalcone derivative upon irradiation of blue LED at room temperature. | |
Xie's group also prepared bioinspired hybrid flavonoids from 2-hydroxychalcone upon irradiation of 24 W CFL with a great yield in the presence of a Brønsted acid. The reaction was proposed to occur through tandem double-bond isomerization and then the dehydration cyclization process of 2-hydroxychalcone (232), giving rise to the flavylium cation, which is transferred to hybrid flavonoids (234) by the attack of nucleophiles in situ with a yield of 33–99% (Scheme 50).121
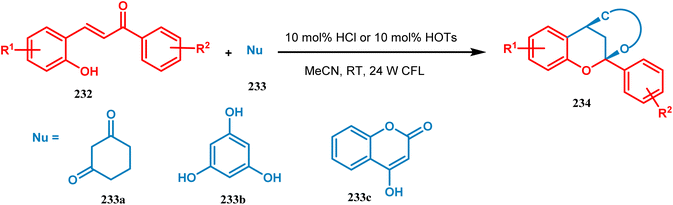 |
| | Scheme 50 Synthesis of bioinspired hybrid flavonoids from 2-hydroxychalcone upon irradiation of 24 W CFL. | |
3.1.6. Catalytic asymmetric synthesis. F. D. Toste and coworkers performed a reaction betweem benzopyrylium salt (235) and 3,5-dimethoxyphenol (236) in the presence of chiral anionic catalyst, where the product was subjected to acid-catalyzed cyclization to construct 2,8-dioxabicyclo[3.3.1]nonane (238) skeleton in 56% yield and 94% ee (Scheme 51). It is noteworthy that this moiety is found in various biologically active natural products.122
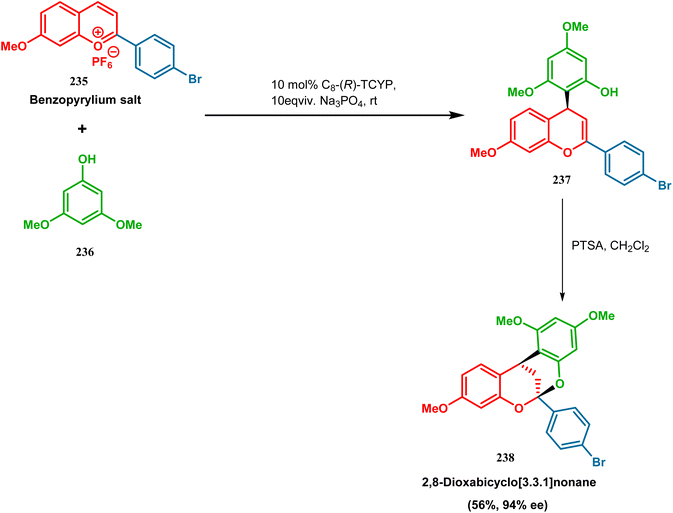 |
| | Scheme 51 Synthesis of 2,8-dioxabicyclo[3.3.1]nonane by the reaction between benzopyrylium salt and 3,5-dimethoxyphenol in the presence of a chiral catalyst. | |
[3.3.1] Bicyclic ketals are an important framework in many biologically important natural products. Few [3.3.1] bicyclic ketals containing compounds such as diinsininol and diinsinin are regarded as effective inhibitors of platelet-activating factor (PAF) that prompt exocytosis compared to the famous PAF antagonist ginkgolide BN 52021. Proanthocyanidin A2 exhibit in vitro selective antiviral efficiency toward the canine distemper virus (CDV) in comparison to ribavirin, thereby making it applicable as a potential anti-CDV compound restricting the replication of that particular virus. Ephedrannin B is also exhibits antiinflammatory effects and subdues the transcription of necrosis factor-a (TNF-a) of the tumor. Therefore, Shi's group was inspired to synthesize chiral heteroannular ketals, which is a great challenge to the organic chemists. They overcame this challenge by accomplishing the reaction in the presence of Pd(II) catalyst and were able to construct highly enantioselective [3.3.1] bicyclic ketals from 2-hydroxyphenylboronic acid (240) and enone 239 in one pot through the asymmetric cascade reaction (Scheme 52).123
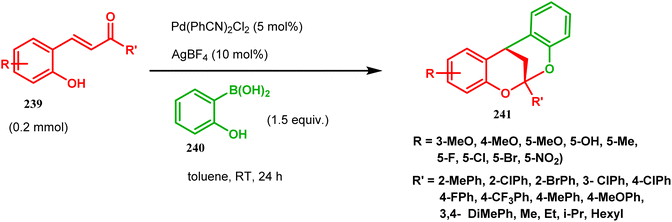 |
| | Scheme 52 One pot asymmetric synthesis of enantioselective [3.3.1] bicyclic ketals from 2-hydroxyphenylboronic acid. | |
3.1.7. Organometallic approaches. Organometallic chemistry has always been used as a powerful tool to achieve synthetic targets with selective stereocontrolled outcomes. Thus, its use to realize complex bicyclo[3.3.1]nonane cores of both natural and nonnatural entities has gained much importance in recent years. The palladium-catalyzed cycloalkenylation technique, developed by Kende118,119 and Saegusa,124 is well explored in this regard. One such example is the palladium acetate-mediated annulation of α-(3-alkenyl)-tethered cyclohexanone TMS-enol ether 242 to a regioisomeric mixture of bicycle 243 (Scheme 53). The issue of stereo- and regioselectivity was addressed by Drouin's group in their intramolecular carbomercuration strategy.125 When this tool was applied on the related TMS-enol ether 244, the bridged bicycle 245 was obtained in regio- and stereochemically pure form (Scheme 53). This methodology, especially the exocyclic vinylmercurial cyclization, witnessed wide application to synthesize diversely-functionalized alkenes.126–130 For example, the strategy was utilized to form the key bicycle 245,11c which in a few synthetic steps was converted into the targeted sesquiterpenes trifarienols A and B by Forsyth's group.
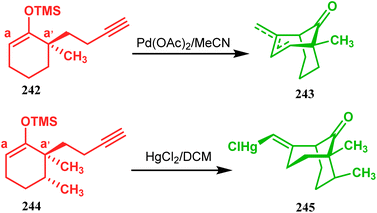 |
| | Scheme 53 Synthesis of bicyclic core by palladium acetate-mediated annulation of α-(3-alkenyl)-tethered cyclohexanone TMS-enol ether. | |
A similar intramolecular alkenylation technique to synthesize such exocyclic double bonds containing bicyclic cores was also unveiled by Honda's team very recently. They synthesized the annulation precursor 247 (yield 89–93%) from commercially available monoprotected 1,4-cyclohexanedione (246) using literature procedures and subjected the TES enol ether to Pd2dba3-catalyzed intramolecular annulation to obtain the desired bicycle 248 along with the intermediate TES enol ether 249, which was converted to 248 in good yields (90–99%) using TBAF (Scheme 54). However, the yield decreases substantially when R![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) H, probably due to facile H–Pd elimination of active hydrogens.131
H, probably due to facile H–Pd elimination of active hydrogens.131
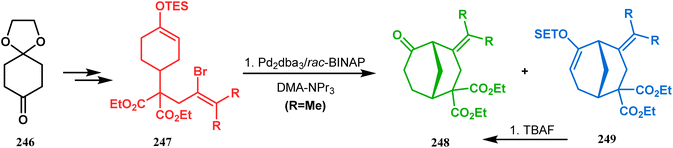 |
| | Scheme 54 Synthesis of exocyclic double bonds containing bicyclic cores through the intramolecular alkenylation technique. | |
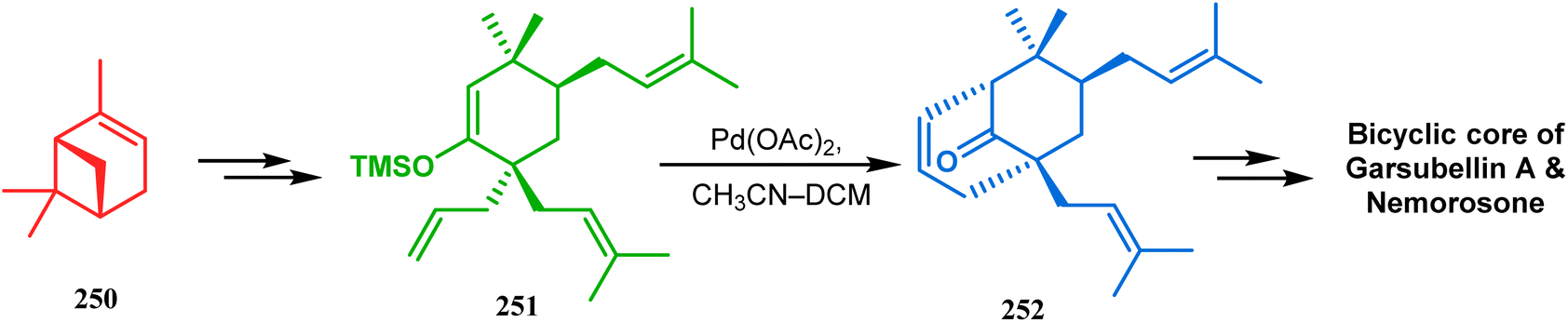
Another intramolecular alkenylation approach to construct the bicyclo[3.3.1]nonane framework was unveiled by Mehta's group. Their idea was to employ α-pinene (250) as the chiron to ensure the desired stereochemistry of the resulting bicyclic core. At the outset, the TMS enol ether 251 was synthesized from 250 in a sequential manner and subjected to Kende's intramolecular alkenylation condition118,119 to form the bicycle 252 in moderate yield (Scheme 55), thereby ensuring a successful enantiospecific route toward the appropriately functionalized bicyclic core of garsubellin A and nemorosone.
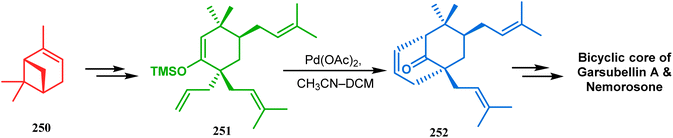 |
| | Scheme 55 Synthesis of bicyclo[3.3.1]nonane framework of garsubellin A and nemorosone through the intramolecular alkenylation technique. | |
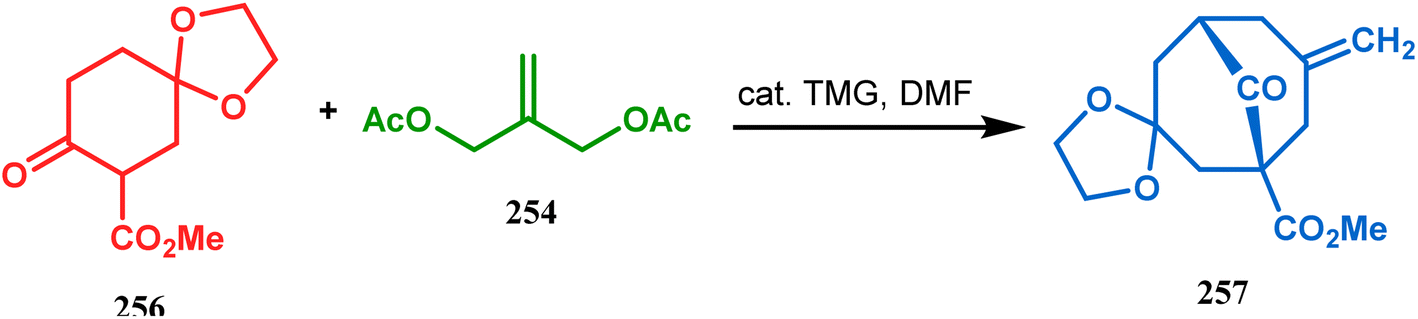
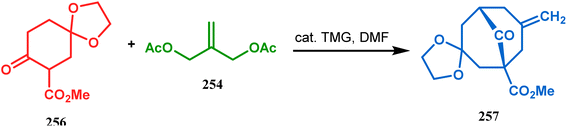
However, the application of palladium chemistry is not limited to intramolecular alkenylation procedures. An intermolecular strategy was also developed by Sivaramakrishnan and coworkers. Their approach toward targeted bicyclo[3.3.1] nonenone 255 employs the Pd-mediated fusion of cyclohexanone derivative 253 with 2-methylene-1,3-propanediol diacetate (254), forming 255 in high yields (Scheme 56).132 Besides, an identical Pd-catalyzed cycloalkylation strategy was also demonstrated by Tuckmental's group, which utilizes the Pd(OAc)2-catalyzed annulation of 256 with 254 to produce bicyclo[3.3.1]nonane 257 (Scheme 57).10d
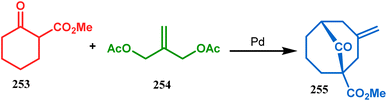 |
| | Scheme 56 Synthesis of bicyclo[3.3.1]nonenone through the Pd-mediated fusion of cyclohexanone derivative with 2-methylene-1,3-propanediol diacetate. | |
 |
| | Scheme 57 Synthesis of bicyclo[3.3.1]nonane through Pd(OAc)2-catalyzed cycloalkylation. | |
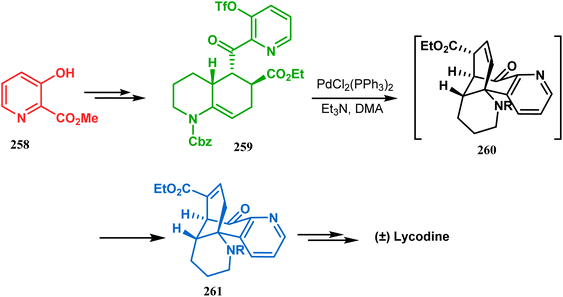
Such a simple palladium-catalyzed technique has been very recently applied by Hirama and Tsukano's group during the total synthesis of lycodine. Starting from 3-hydroxypicolinate (258), they achieved the annulation precursor 259 in a few synthetic steps. With the required precursor in hand, the palladium-catalyzed Mizoroki–Heck cyclization was attempted, resulting in the formation of the desired bicycle 261 through a 6-exo-trig product 260 with only 18% yield. The deactivation of the palladium center by chelation through the pyridyl ketone moiety was attributed to such a low yield of 261, which was cleverly bypassed utilizing a high dilution condition (Scheme 58).10e
 |
| | Scheme 58 Palladium-catalyzed total synthesis of lycodine from 3-hydroxypicolinate having a bicyclic core. | |
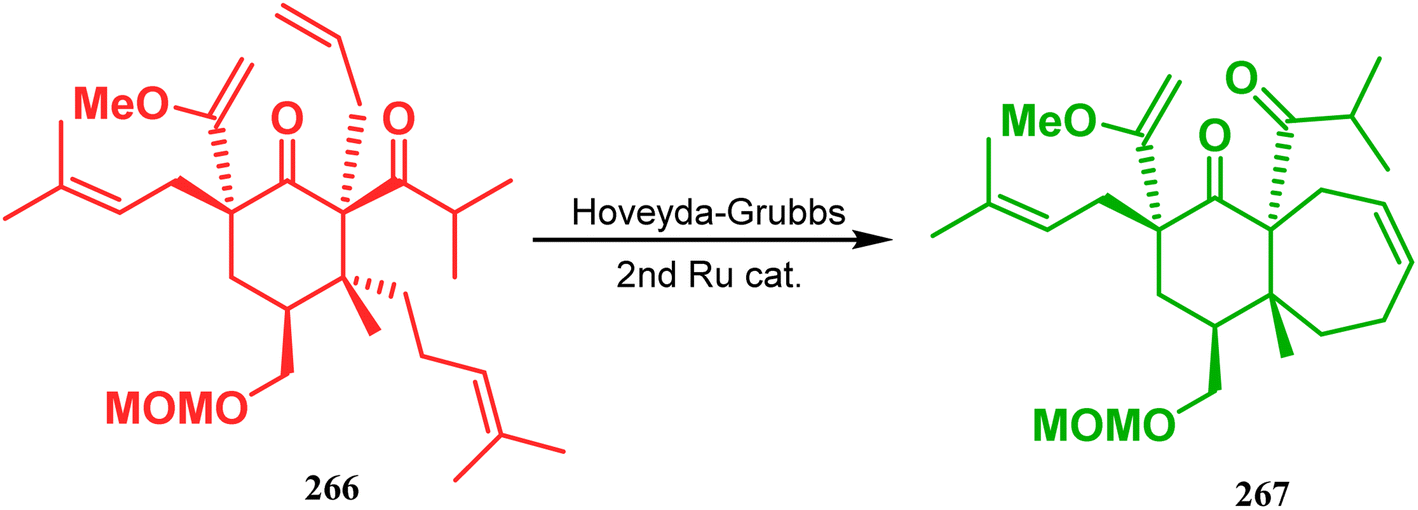
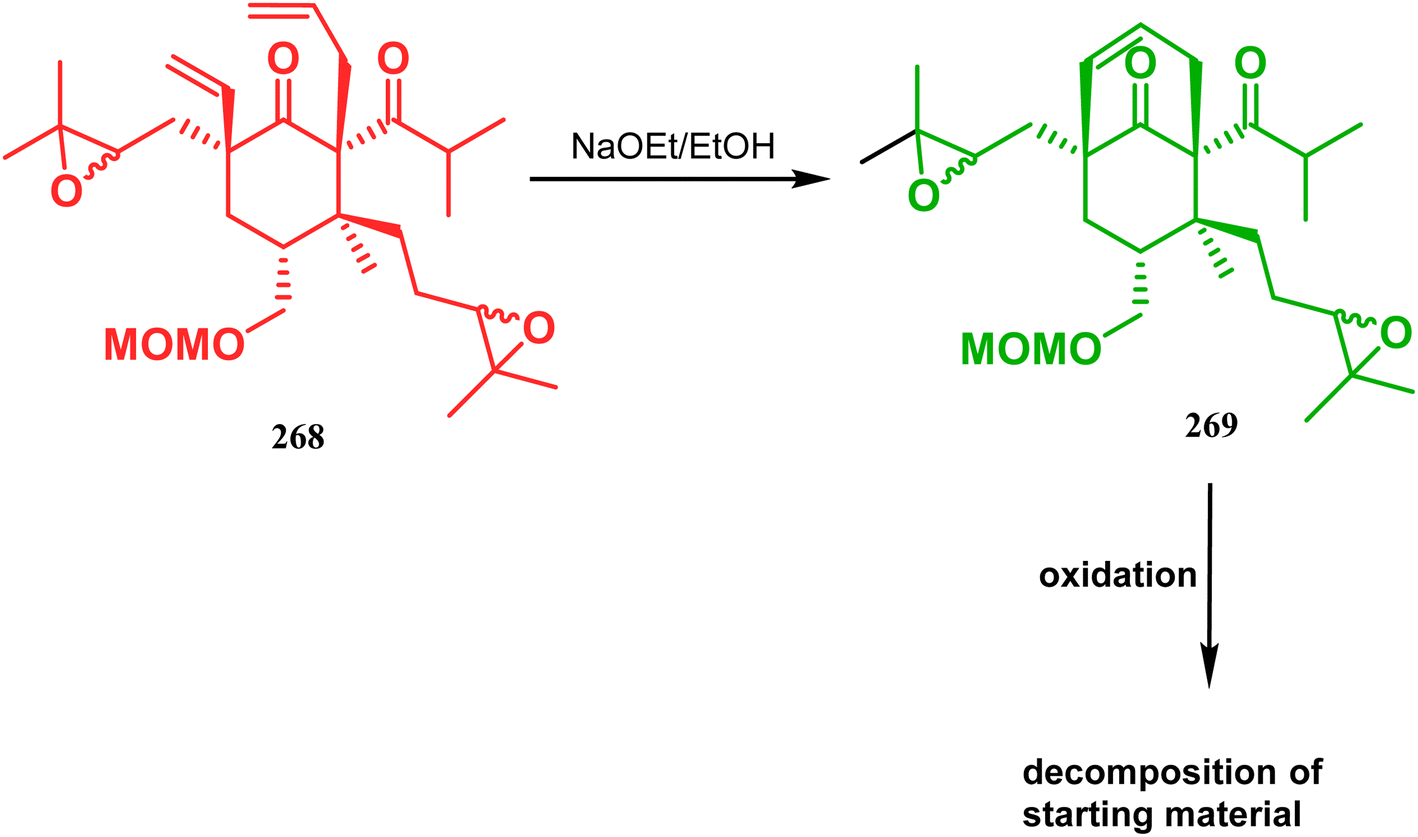
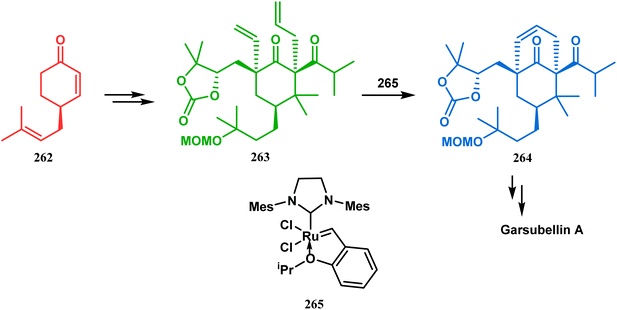
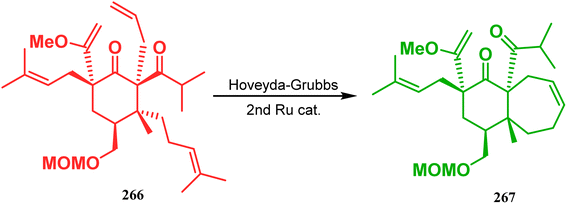
Inspite of such wide variety of palladium-catalyzed strategies, ruthenium and platinum chemistries have also proved their own efficacy to construct bicyclo[3.3.1]nonane cores. One such successful application of Ru catalysis was demonstrated by Shibasaki's group for the total synthesis of racemic garsubellin A133 using ring closing metathesis (RCM) through the Hoveyda–Grubbs catalyst. The necessary cyclohexanone derivative 263, with cis-alkene groups at the α and α′ positions of the ketone, was synthesized from enone 262, and they envisioned ring-closing metathesis using catalyst 265, which was then applied to achieve the bicycle 264 in 92% yield. Sequential allylic oxidation, oxidative cyclization, prenyl regeneration, and Stille coupling then led to the formation of racemic garsubellin A (Scheme 59). However, this strategy did not work for the synthesis of similar bicyclic core of hyperforin due to the presence of excess prenyl groups, leading to a ring-closing metathesis reaction between the terminal vinyl and prenyl groups of 266 to form 267 (Scheme 60),134 which prompted the author to change their route, leading to the synthesis of the Hoveyda–Grubbs precursor 268. Although Hoveyda–Grubbs catalysis was successful in constructing bicycle 269 in 74% yield (Scheme 61),135 the next steps led to the decomposition of the starting materials and again forced the authors to change their strategy. Finally, the authors achieved enantiopure hyperforin through a base-promoted intramolecular aldolization technique.
 |
| | Scheme 59 Construction of the bicyclo[3.3.1]nonane core of garsubellin A using ring-closing metathesis (RCM) in the presence of the Hoveyda–Grubbs catalyst. | |
 |
| | Scheme 60 Ring closing metathesis reaction between the terminal vinyl and prenyl groups in the presence of the Hoveyda-Grubbs catalyst. | |
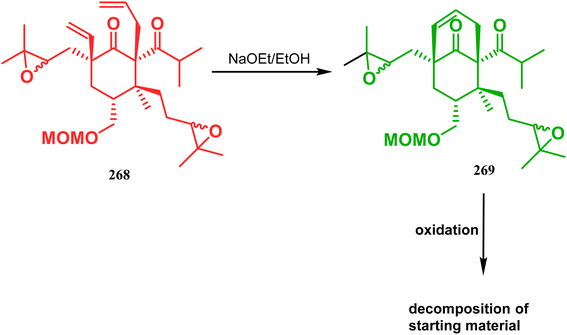 |
| | Scheme 61 Synthesis of the bicyclo[3.3.1]nonane core of hyperforin through base-promoted intramolecular aldolization. | |
Another such nonpalladium catalysis was reported by Gusevskaya's group during the one-pot access toward 4,8-dimethyl-bicyclo[3.3.1]non-7-en-2-ol (271). Their strategy to synthesize the targeted bicycles (due to their potentiality to be used as perfumes) utilized a simple hydroformylation/cyclization reaction of limonene (270) through a platinum-tin combined catalysis (Scheme 62).136
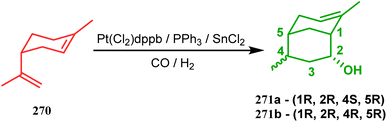 |
| | Scheme 62 Synthesis of 4,8-dimethyl-bicyclo[3.3.1]non-7-en-2-ol through the hydroformylation/cyclization reaction of limonene in the presence of platinum-tin combined catalyst. | |
3.1.8. Radical cyclization. The fascinating chemistry of radicals, unlike carbocations or carboanions, has always attracted the attention of chemists and prompted to explore it in the synthesis of structurally unique cyclic cores. Bicyclo[3.3.1]nonanes are also not an exception. The pioneering and probably the first report in this category appeared in 1987 due to Finlay and Walton.137 They described a transannular radical cyclization of cyclooctenylmethyl bromide (272) to achieve bicyclo[3.3.1]nonane (273) by irradiating a solution of 272 and Bu3SnH in Me(CH2)14Me (Scheme 63). However, a more practical approach and detailed advancement in this area was made by Bonjoch's group and, after a decade of Finlay and Walton's report, they unveiled a new route toward 2-azabicyclo[3.3.1]nonane through a radical ring closure method. Their initial report was a (SiMe3)3SiH/AIBN-mediated radical cyclization of amidocyclohexene 274 to give the desired bicyclic nitrile (275) in good yields (57–70%) (Scheme 64). The corresponding bicyclic carboxylate (277) was obtained with even a better yield when amidocyclohexane 276 was exposed to an identical reaction condition. The amount of radical mediator has shown to have a high influence on the product yield, corroborated by the fact that while 2 equivalents of TTMSS produces a mirror amount of product along with chloro and dichloro analogues, the use of 3.5 equivalents of the same furnishes the desired products in moderate yields.138
 |
| | Scheme 63 Synthesis of bicyclo[3.3.1]nonane through the transannular radical cyclization of cyclooctenylmethyl bromide with the help of Bu3SnH in Me(CH2)14Me. | |
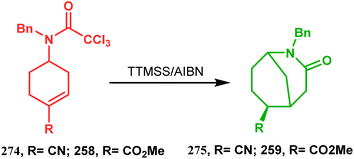 |
| | Scheme 64 Synthesis of 2-azabicyclo[3.3.1]nonane through a radical ring closing reaction in the presence of the (SiMe3)3SiH/AIBN catalyst. | |
In a following report by the same group, the TMS enol ether 278 was treated using a similar protocol to form diastereomeric bicycles 279 (yield 10%) and 280 (yield 21%) along with a β-lactam and another undesired bicycle (Scheme 65).139
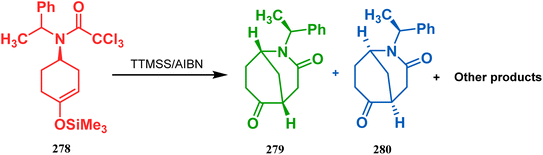 |
| | Scheme 65 Synthesis of diastereomeric bicycles along with a β-lactam from TMS enol ether in the presence of (SiMe3)3SiH/AIBN. | |
The application of radical cyclization is also found in natural product core synthesis. One such report is due to Ward and Caprio. They demonstrated a radical-mediated approach toward the core structure of huperzine A and synthesized the required precursor 282 from a trisubstituted pyridine derivative 281. 282 was then treated with Bu3SnH/AIBN, leading to the bicyclic core structure (283) of Huperzine A through a 6-exo-trig radical cyclization in 97% yield (Scheme 66).140,141
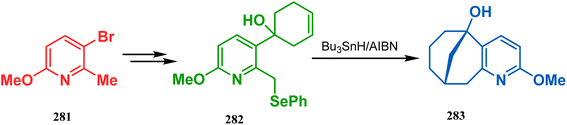 |
| | Scheme 66 Synthesis of the bicyclic core of Huperzine A through a 6-exo-trig radical cyclization in the presence of Bu3SnH/AIBN. | |
3.2. Intra- and intermolecular C–X bond formation
3.2.1. N-Cyclization. Among the several available methods to construct C–X linkage, intramolecular N-alkylation in the presence of a base is very well known for synthesizing heterobicyclo[3.3.1]nonane derivatives. Interest in this area was originated from the potentiality of these bicyclononane derivatives to be used as selective α7 nicotinic ligands, inhibitors, etc.142 Although the synthetic route toward one such entity was reported in as early as the 1950s by Walker's group,143 a much elaborate study was done by Slowinski's group very recently.144 Their journey commenced from 6-methoxynicotinic acid methyl ester (284), which was converted into 288 in a sequential manner. The alkylation precursor was then treated with potassium carbonate in chloroform, leading to the formation of the desired bicycle 289 in 74% yield through an intramolecular alkylation (Scheme 67).
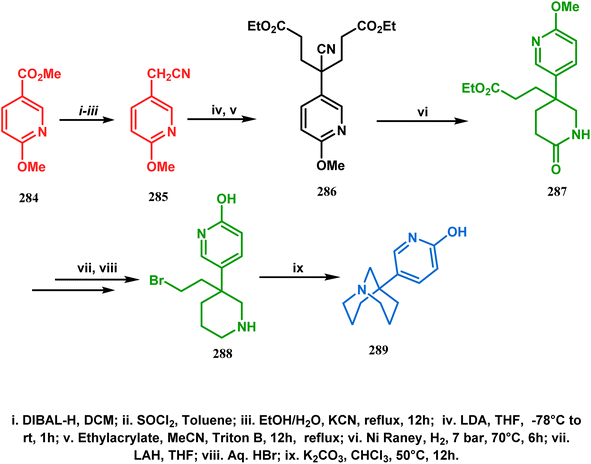 |
| | Scheme 67 Sequential synthesis of the bicyclo[3.3.1]nonane core through intramolecular alkylation reaction. | |
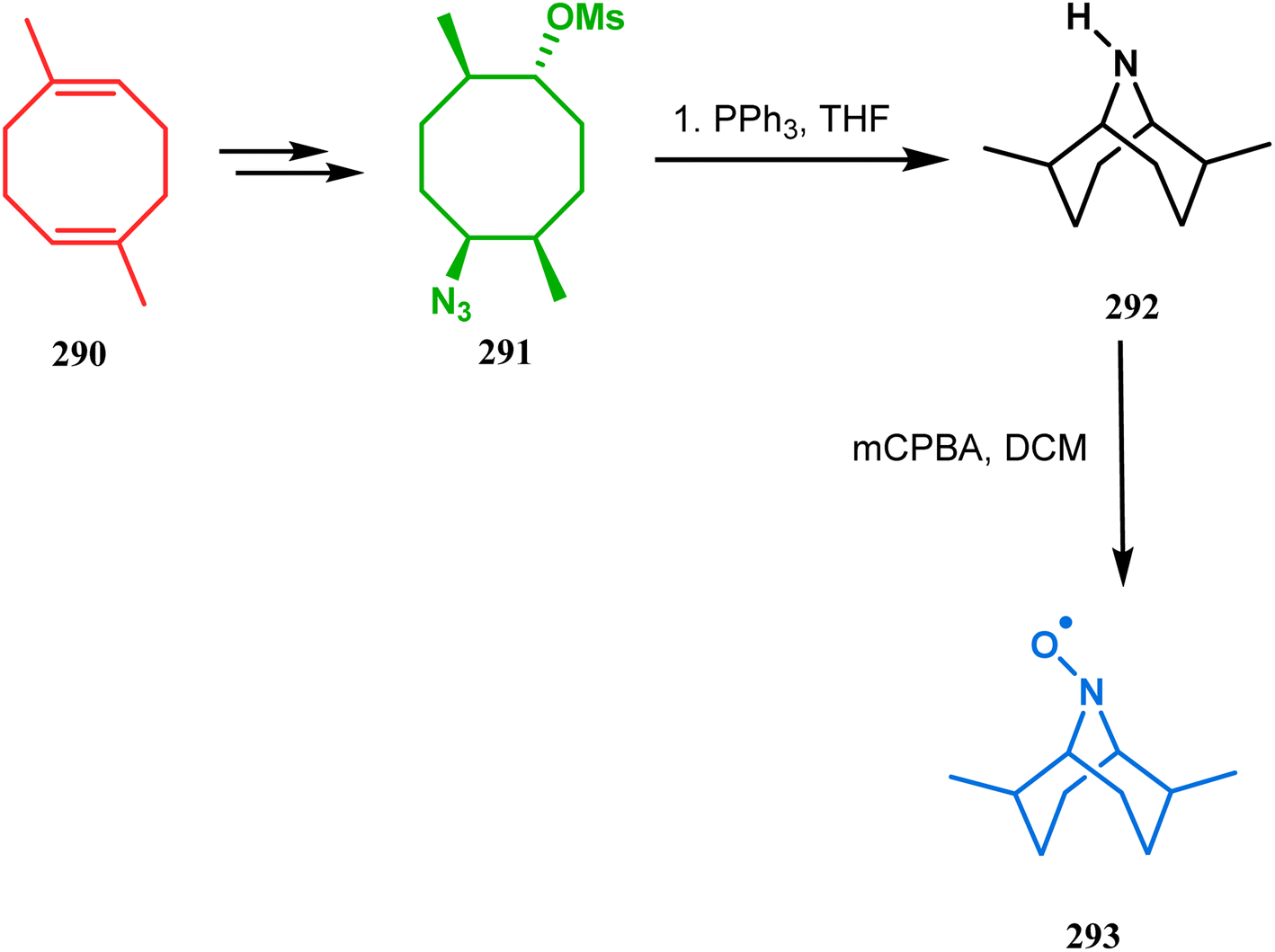
The method was then employed to achieve a series of new chemical entities and was proved to be sufficiently potent as α7 nicotinic ligands, having fascinating selectivity compared to the α4β2 nicotinic receptor. A similar intramolecular substitution, resulting in the formation of another class of azabicyclo[3.3.1]nonane, was also reported in the same decade by Rychnovsky and coworkers.145 The idea was to access some novel C2-symmetric nitroxides to be used as enantioselective oxidants. The synthetic route began with the hydroboration-oxidation of 1,5-dimethyl-1,5-cyclooctadiene (290) to produce the corresponding diol, which in a sequential manner was converted to methanesulfonate 291 in 74% yield. Triphenylphosphine-promoted azide reduction and concomitant cyclization of 291 then produced amine 292 (yield 41%), which on mCPBA oxidation led to the desired nitroxide 293 having a yield of 67% (Scheme 68). The synthesis of the corresponding azabicyclo[2.2.1] heptane nitroxide was reported to be far more difficult.145 To access a more complex bicyclo[3.3.1]nonane nitroxide, the same author utilized another methodology originally developed by Michel and Rassat.146 This study started with the rhenium-catalyzed hydrogen peroxide oxidation of 1,5-cyclooctadiene 294 to furnish diepoxide 295 (yield 50%), which upon reacting with benzyl amine in water produced diol 296 in 91% yield. The attempted oxidation of 297 (yield 91%) remained unsuccessful, and the targeted nitroxide 298 was not isolated due to its instability (Scheme 69).146
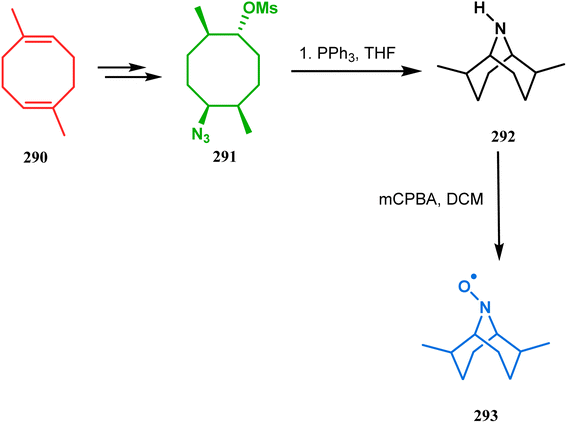 |
| | Scheme 68 Synthesis of nitroxide through triphenylphosphine-promoted azide reduction and then cyclization, followed by mCPBA oxidation. | |
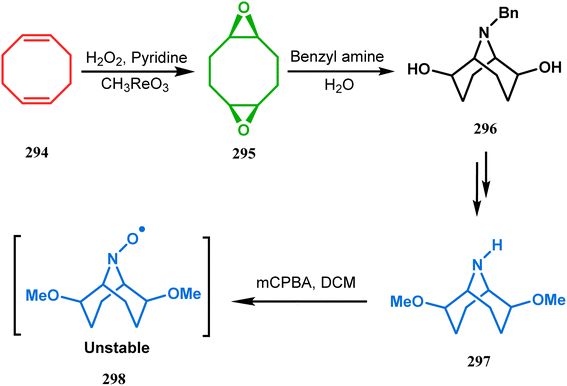 |
| | Scheme 69 Synthesis of nitroxide through the rhenium-catalyzed hydrogen peroxide oxidation of 1,5-cyclooctadiene, followed by mCPBA oxidation. | |
An identical application of benzyl amine through a different route to achieve diazabicyclo[3.3.1]nonanes was also developed by Cingarella's group. Their target was to study the μ-opioid receptor affinity of properly substituted diazabicyclo[3.3.1]nonanes, and it commenced with the α,α′-dibromination of pimelic acid (299) to give 300, followed by the double condensation of benzylamine to furnish N-benzyl-2,6-dicarbomethoxypiperidine (301). This piperidine derivative was then again condensed with the same amine, producing the diazabicycle 304 (Scheme 70).147
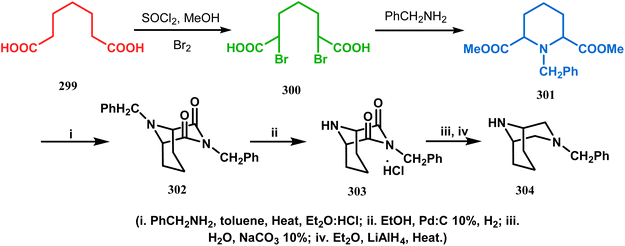 |
| | Scheme 70 Synthesis of diazabicyclo[3.3.1]nonanes through the α,α′-dibromination of pimelic acid, followed by several condensations of benzylamine. | |
A similar condensation strtategy of piperidine-4-ones (305) with chiral amines were also utilized to synthesize bicyclo[3.3.1]nonane skeletons. Kuhl's and Sacchetti's teams described one such useful application of chiral amines in the synthesis of new diamino chiral ligands, and their approach involved the condensation of 305 with formaldehyde and amines in methanol under refluxing condition, thereby forming chiral ligands 306a–d (yield 73–92%) (Scheme 71).
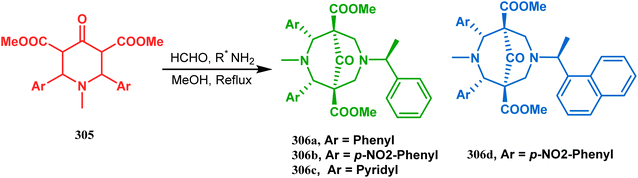 |
| | Scheme 71 Synthesis of the bicyclo[3.3.1]nonane skeletons through the condensation of piperidine-4-ones with formaldehyde and amines in methanol under refluxing condition. | |
These ligands were then successfully employed in the kinetic oxidative resolution of alcohols.148,149 Another application of such double condensation of amines with esters was found in Mattay's report. The employed amine was a macrocyclic amine (307) this time. Thus, when compound 307 was reacted with bicyclic anhydride 308, the double condensation produced the azabicycle 309 (Scheme 72).150
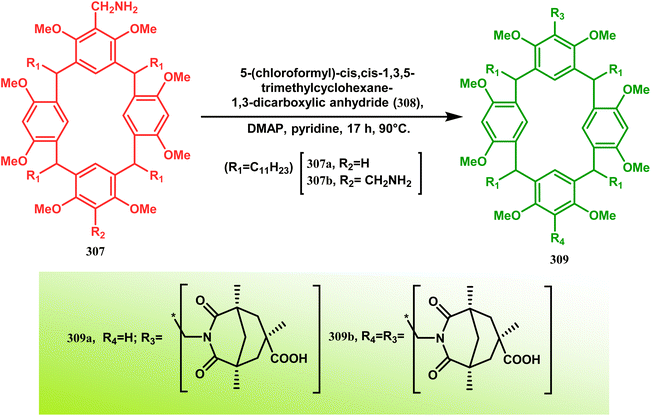 |
| | Scheme 72 Synthesis of azabicycle through the double condensation of a macrocyclic amine with bicyclic anhydride. | |
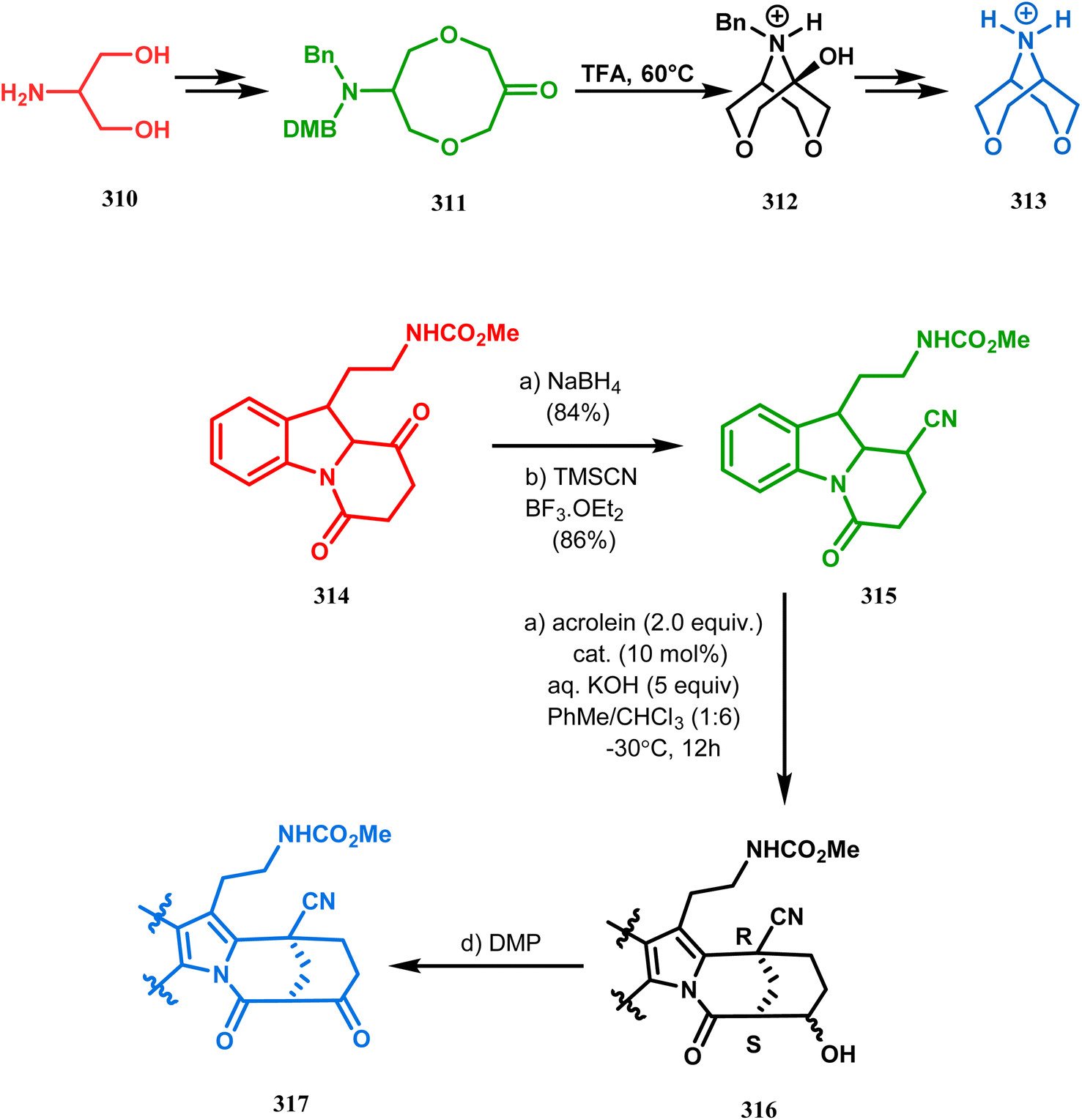
Intramolecular N-cyclization to construct azabicyclo[3.3.1]nonane was also reported by C. M. Park. Beginning with an amine diol 310, the author successfully reached the cyclization precursor 311, which on treatment with trifluoroacetic acid underwent rapid intramolecular cyclization to produce the hemiaminal 312 (yield 80%). The targeted 3,7-dioxa-9-aza-bicyclo[3.3.1]nonane (313) was then synthesized from this hemiaminal in a few synthetic steps having a yield of 99% (Scheme 73a).151a
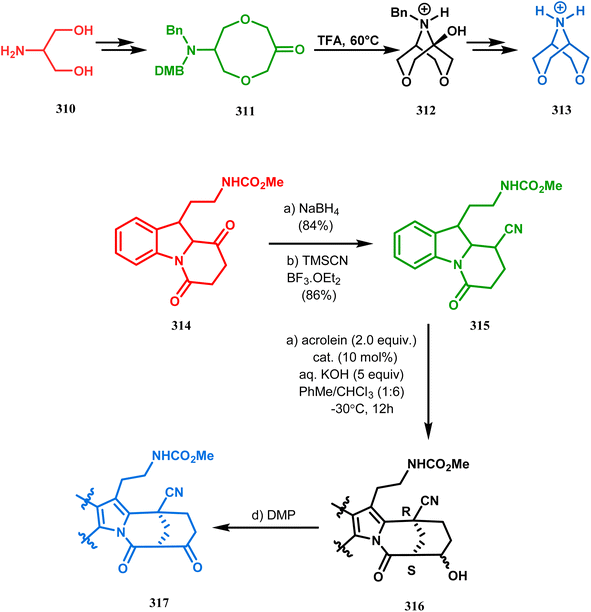 |
| | Scheme 73 (a)Sequential synthesis of 3,7-dioxa-9-aza-bicyclo[3.3.1]nonane through the intramolecular N-cyclization of amine diol in the presence of trifluoroacetic acid (b) synthesis of azabicyclo[3.3.1]nonane moiety using the Michael/aldol cascade reaction. | |
Also, F.-S. Han's research group followed the asymmetric Michael/aldol cascade reaction to synthesize the desired compound (317) (Scheme 73b) in good yield with excellent enantiomeric excess by accomplishing the reaction between 314 and 315 in the presence of NaBH4 and TMSCN in BF3 ether, followed by the reaction with acroleinthe. The yield of the 317 was 56%. It is important to mention that the reaction can be performed at the multigram scale with a considerable yield.151b
P.-Q. Huang and coworkers explored the construction of the tricyclic core from compound 318 by subjecting the diastereomer 318 to catalytic hydrogenolysis in the presence of Boc2O and they isolated alcohol 319 in 62% yield (Scheme 74a). Then, the alcohol 319 was subjected to a Swern oxidation, followed by treatment with TFA to remove the Boc group. On workup using 2 N KOH, the diazatricyclic core 320 was obtained in 51% yield.152a
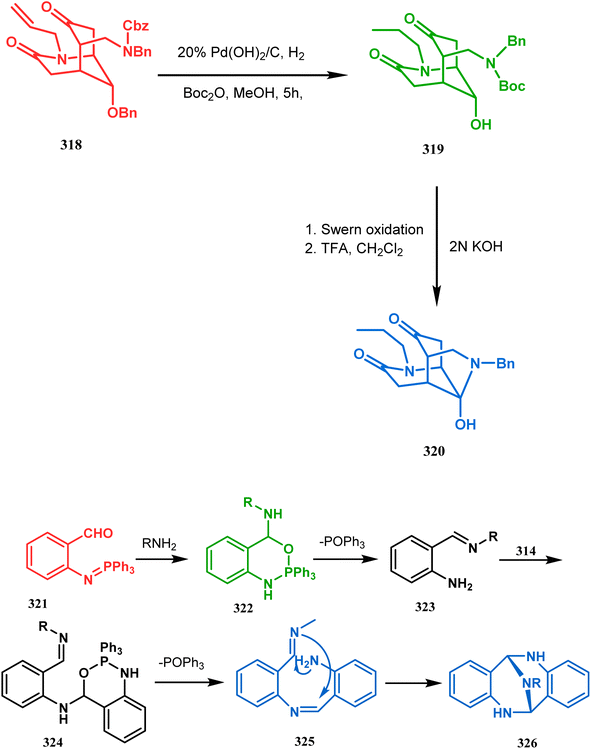 |
| | Scheme 74 (a) The diastereoconvergent synthesis of the diazatricyclic core moiety (b) synthesis of triazabicyclo[3.3.1]nonane through the N-cyclization mechanism. | |
Apart from these aza and diaza analogues of bicyclo[3.3.1]nonane, the triaza analogues are also accessible and, according to Molina's strategy, even the highly unreactive iminophosphorane (321) could be activated toward N-cyclization to achieve the triazabicyclo[3.3.1]nonane (326) (Scheme 74b).
Iminophosphorane (321), originally synthesized by Staudinger reaction, constitutes resonance-stabilized chelates due to its reluctance toward both intra- and intermolecular aza Wittig condensation, even though it has the requisite reactive groups for this reaction. However, when 321 was reacted with primary amines in the presence of catalytic amount of acetic acid, amine 323 was formed through intermediate 322. The aniline derivative 323 thus formed then again reacts with iminophosphorane 321 and proceeds toward the cyclization precursor 325 through intermediate 324. Once compound 325 was formed, two consecutive C–N cyclizations furnished the triazabicyclo[3.3.1]nonane derivative 326 in good yields (40–80%).152b The synthesis of a similar triaza-analogue (329) is reported by Abonia's group. According to their report, it could be achieved via the ammonolysis of alkyl acetoacetates in water.153 Thus, when MeCOCH2CO2Me (327) was treated with aqueous ammonia at room temperature, white crystalline methyl-β-aminocrotonate (328) was found to form rapidly. It was then kept undisturbed for 4 weeks, resulting in the transformation of 328 into another white solid, 1,5-dimethyl-2,6,9-triaza-bicyclo[3.3.1]nonane-3,7-dione (329) (Scheme 75).
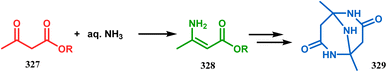 |
| | Scheme 75 Synthesis of 1,5-dimethyl-2,6,9-triaza-bicyclo[3.3.1]nonane-3,7-dione via the ammonolysis of alkyl acetoacetates in water. | |
3.2.2. O-Cyclization. Cyclization through oxygen center is also a useful technique to synthesize heteroanalogues of bicyclo[3.3.1]nonane. Among the few reports available in literature, the acid-catalyzed intramolecular lactonization of dihydropyridines (330) is well investigated. Rudler and coworkers showed that dihydropyridines, which can be obtained from pyridine derivatives, when treated with acids, the appended carboxylic acid group undergo a regioselective intramolecular lactonization reaction and produce oxaza analogues of bicyclo[3.3.1]nonane (332). More interestingly, when these cyclizations were attempted in the presence of halogenated compounds (as Lewis acid), halogens almost unexceptionally get incorporated in the product bicycles (331, 332). Even, when mCPBA was used as the acid catalyst, a hydroxyl group is inserted in the product (333). This cyclization is so facile that even silica gel can catalyze the lactonization procedure (Scheme 76).154
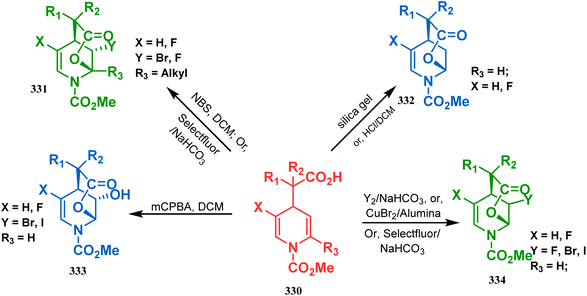 |
| | Scheme 76 Synthesis of the bicyclo[3.3.1]nonane moiety through the acid-catalyzed intramolecular lactonization of dihydropyridines. | |
Petrov and Marshall exemplified another type of acid-catalyzed route toward bicyclo[3.3.1]nonane analogues. Their report on an acid-catalyzed dimerization of 2,2-bis-(trifluoromethyl)-4-alkoxy-thietane (335) demonstrated a trouble-free access toward oxa-bridged dithiabicyclo[3.3.1]nonane (339). It is believed that the reaction proceeds through the ring opening of thietane (335) to form a stable carbocation (336), which upon cyclodimerization gives 337. Acid-promoted MeOH elimination then produced carbocation 338, which was intramolecularly captured by the remaining alkoxy group to form the oxa-bridged dithiabicyclo[3.3.1]nonane (339) (yield 35–50%) (Scheme 77).155
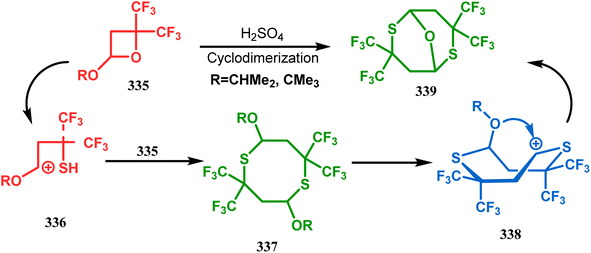 |
| | Scheme 77 Synthesis of oxa-bridged dithiabicyclo[3.3.1]nonane through the acid-catalyzed cyclodimerization of 2,2-bis-(trifluoromethyl)-4-alkoxy-thietane. | |
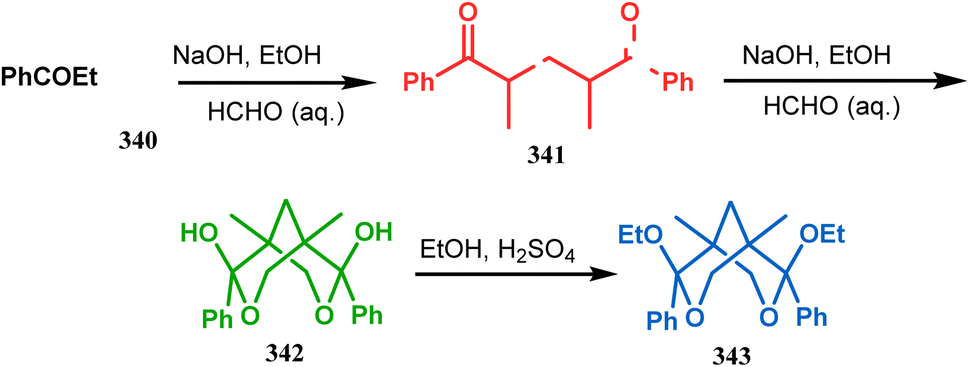
Apart from these reports, E. J. Corey's route toward chelating bis-ethers and bis-amines is also remarkable. According to his findings, when propiophenone (340) was treated with aqueous formalin solution in the presence of a base, the diketone 341 becomes the major product (yield 95%), which on further reaction with formalin solution transforms into bicyclic bis-hemiketal 342 in 55% yield. This bis-ether on treatment with EtOH/H2SO4 transforms into the corresponding bis-ethoxy analogues (343) in 90% yield (Scheme 78).156
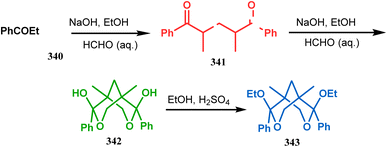 |
| | Scheme 78 Synthesis of bis-ethoxy analogues from propiophenone. | |
Harmata and coworkers described an interesting synthesis of molecular tweezers based on the oxabicyclo[3.3.1]nonane framework. Starting from the dibromo derivative 344, the annulation precursors (345, 346) were synthesized by the stepwise reaction of butyl lithium and benzyl aldehyde, which on SnCl4-mediated cyclization reaction in DCM yielded the bis-kagan's ethers 347 and 348 in good yields (57% and 75%, respectively) (Scheme 79).157–159
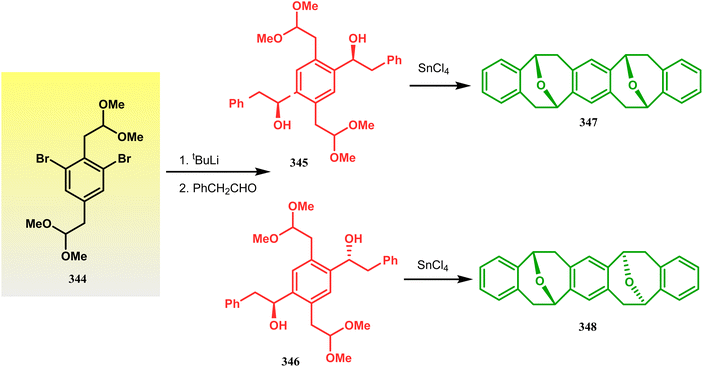 |
| | Scheme 79 Stepwise synthesis of oxabicyclo[3.3.1]nonane-based molecular tweezers in the presence of butyl lithium and benzyl aldehyde, followed by SnCl4-mediated cyclization reaction in DCM. | |
3.2.3. Heavy atom-cyclization. Apart from nitrogen and oxygen cyclizations, the ring closure mechanism is also common with heavy atoms such as sulphur and selenium. Weil,160 Corey,161 and Lautenschlaeger162 were the very first authors to discover one such cyclization. They demonstrated that when 1,5-cyclooctadiene (349) was treated with sulphur or selenium dichloride/dibromide, the bicyclic adducts (351) were produced in high yields through an intermediate (350) (Scheme 80).
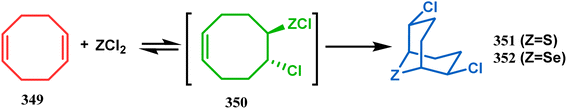 |
| | Scheme 80 Synthesis of sulphur/selenium-containing bicyclo[3.3.1]nonane through the heavy-atom cyclization method. | |
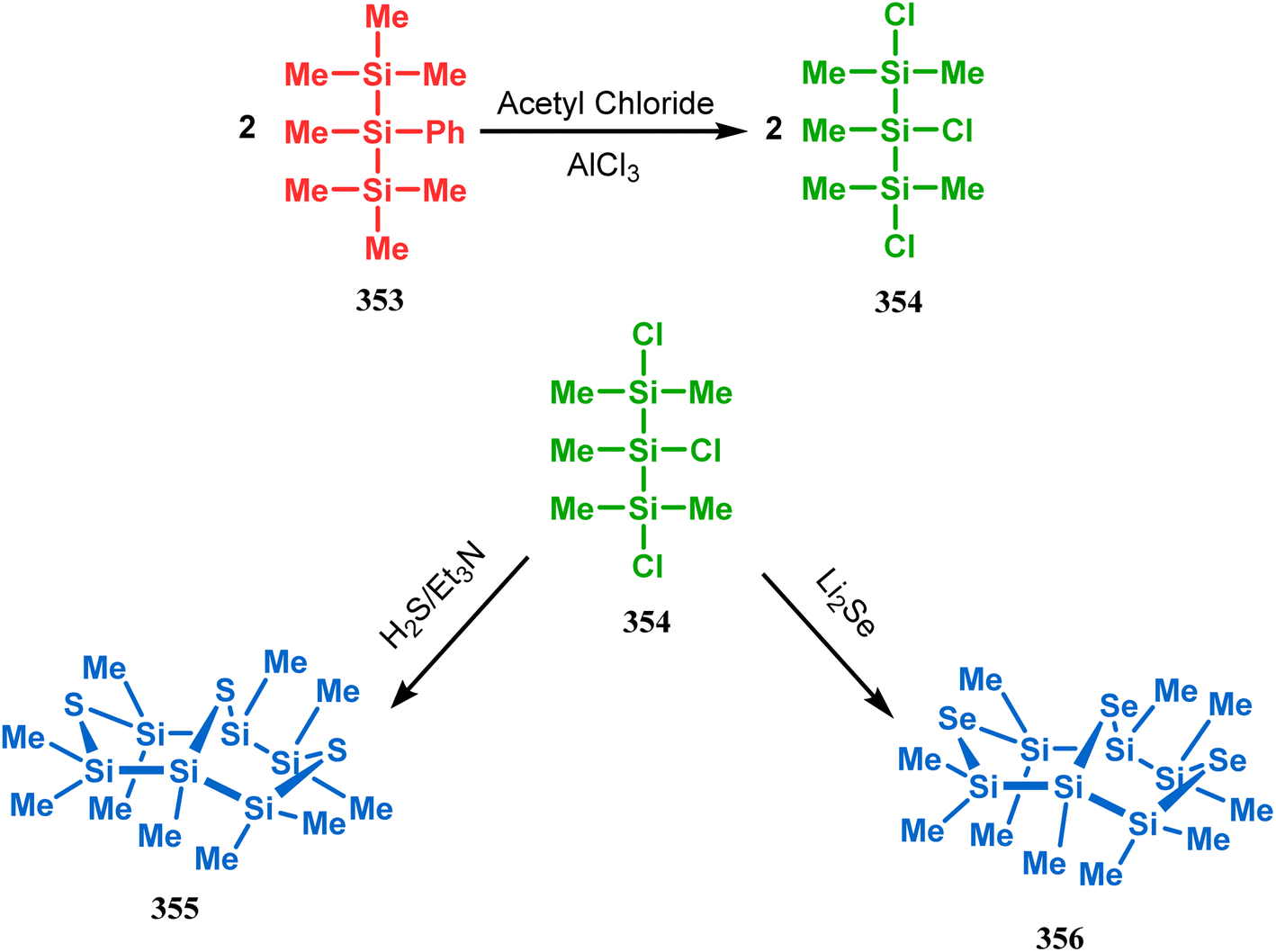
Such entities, having the donor at the 9th position and the leaving group at the β position to the donor, are known as “WCL” electrophiles, after the names of their discoverers. Finn and coworkers very recently developed another application of this methodology to access selenium-containing bicyclo[3.3.1]nonanes and demonstrated that the reaction of 349 with selenium dichloride/dibromide proceeds at a much faster rate to form selenabicycle 352 (Scheme 80).163 Such heavy atom-containing bicyclo[3.3.1]nonanes are found in organosilicon chalcogenides. Herzog and Borrmann showed that organochlorosilanes (354), derived from 2-phenylheptamethyltrisilane (353) on reacting with H2S in the presence of triethylamine led to formation of a bicyclo[3.3.1]nonane skeleton with two trisilane units (355) as the major product along with a bicyclo[3.2.2]nonane skeleton as the minor isomer. However, when Li2Se was reacted with 354 in THF, the corresponding bicyclo[3.3.1]nonane (356) was formed as the sole product (yield 99%). Both these chalcogenides (355, 356) are found to prefer the twin-boat conformer (Scheme 81).164
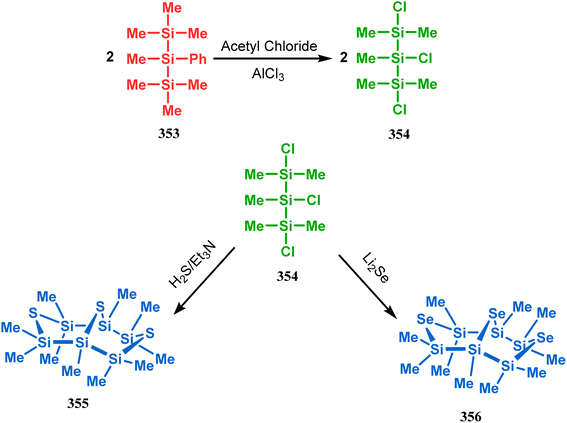 |
| | Scheme 81 Synthesis of the bicyclo[3.3.1]nonane skeleton in the presence of trimethylamine/H2S and in the presence of Li2Se. | |
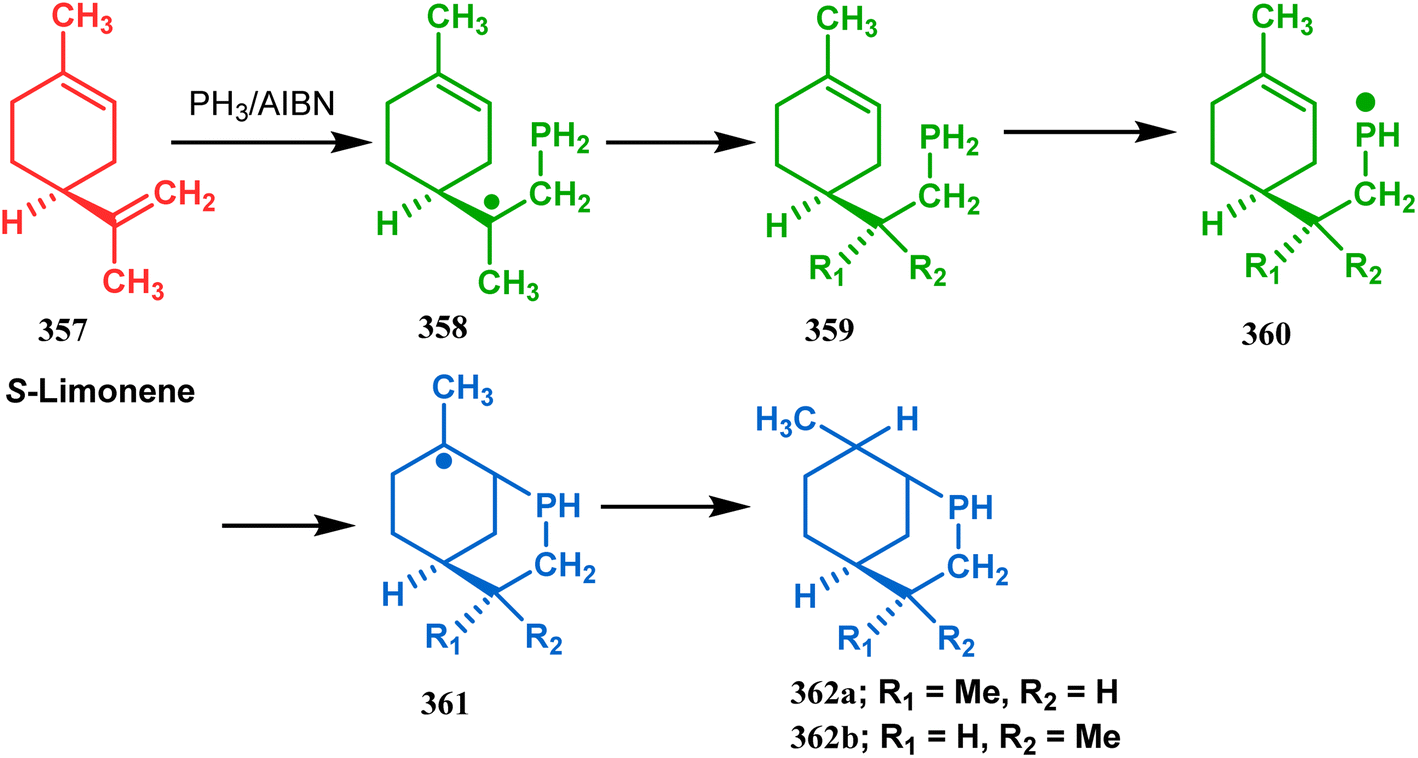
Apart from these sulphur/selenium-containing bicyclo[3.3.1]nonanes, the synthesis of their phosphorus analogues are also well-documented. A unique application of phosphorous-cyclization in this regard was demonstrated by Capretta and coworkers. Their journey began from enantiomerically pure limonene (357), which on treatment with PH3 and AIBN transforms into radical 358 and gets trapped as a mixture of two isomeric phosphines (359) through H-abstraction from PH3. Phosphinyl radical 360 was then generated from 359 and underwent subsequent intramolecular cyclization to give the desired phosphine ligands 362 in >85% yield (Scheme 82).165 These ligands were successfully employed in the cobalt-catalyzed hydroformylation of alkenes.166
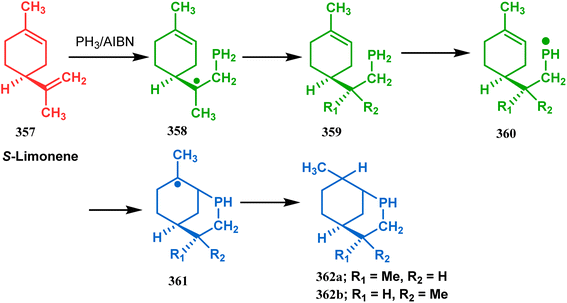 |
| | Scheme 82 Synthesis of phosphorus analogues of sulphur/selenium-containing bicyclo[3.3.1]nonanes on treatment with PH3 and AIBN. | |
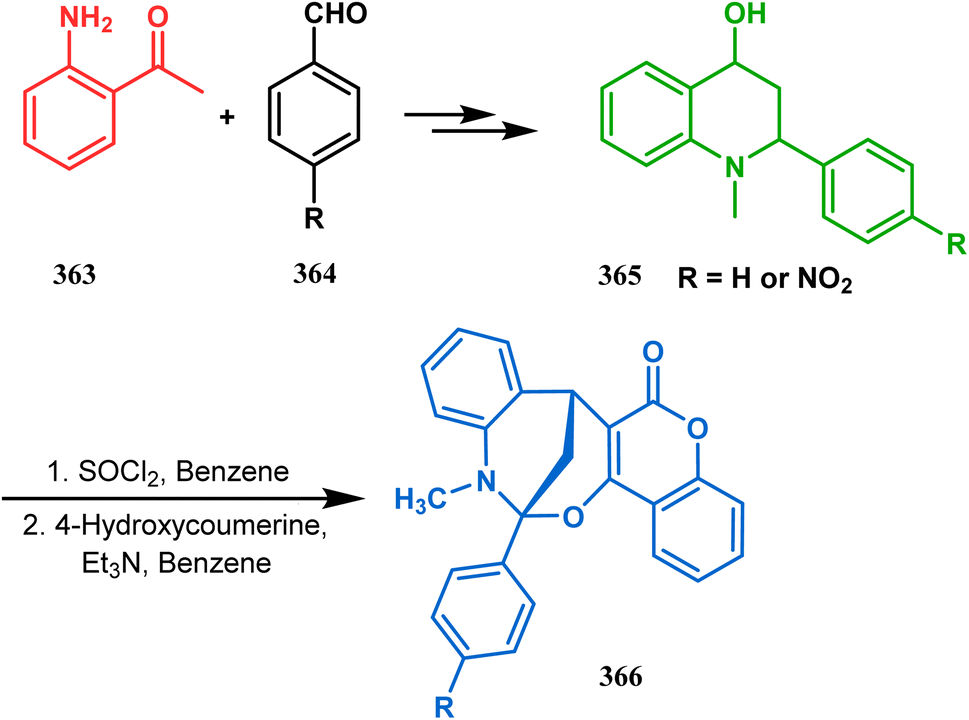
3.2.4. Tandem C-alkylation and heteroatom-cyclization. Tandem C-alkylation & heteroatom-cyclization also constitute a well-explored technique for the synthesis of such heteroanalogues of bicyclo[3.3.1]nonane. The initial advancement of this strategy was made by Yang's team in the year 2006. Starting from 2′-amino acetophenone (363) and benzaldehyde derivatives (364), they synthesized the cyclization precursor 365 (yield 90–92%) effortlessly. C-alkylation and the concomitant O-cyclization of 4-hydroxycoumarin then easily led to the oxazabicyclo[3.3.1]nonane (366) in 43–45% yield (Scheme 83).167
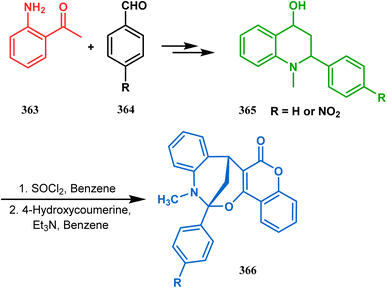 |
| | Scheme 83 Synthesis of oxazabicyclo[3.3.1]nonane through C-alkylation and concomitant O-cyclization of 4-hydroxycoumarin. | |
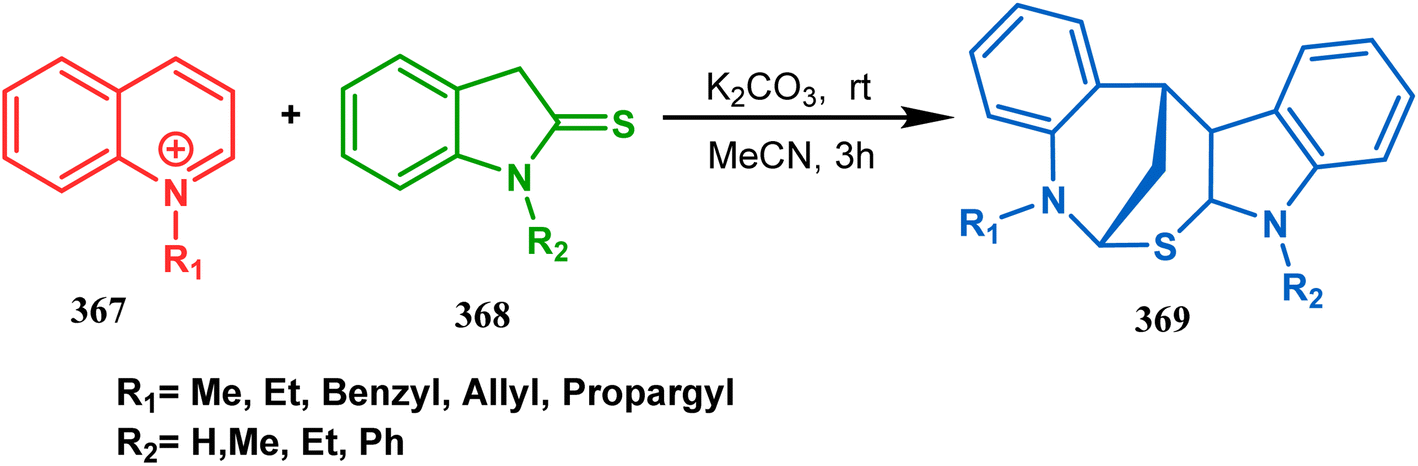
The exploration of this strategy is, however, most extensively done by F. M. Moghaddam's group. Their initial report was a potassium carbonate-mediated single step route toward a thiaazabicyclo[3.3.1]nonane (369). Thus, when indolin-2-thione (367) was treated with N-alkylquinoliniums (368), tandem C-alkylation and intramolecular S-alkylation yielded the bicycle 369 in high yields of 89% (Scheme 84).168
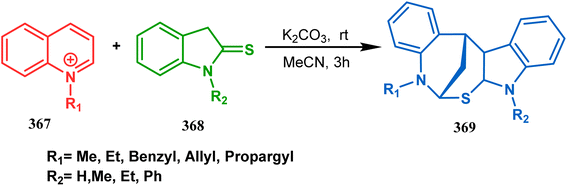 |
| | Scheme 84 Synthesis of thiaazabicyclo[3.3.1]nonane by the reaction of indolin-2-thione and N-alkylquinoliniums through tandem C-alkylation and intramolecular S-alkylation. | |
Within a few months after this communication, the same group disclosed another report where the same N-alkylquinoliniums (367) were exposed to a different binucleophile, 4-hydroxycoumarin (370) this time. This led to a tandem C-alkylation and intramolecular O-cyclization and produced the oxazabicyclo[3.3.1]nonane 371 in excellent yields (72–89%) (Scheme 85a). The strategy also worked well when the coumarin derivative was replaced by a 4-hydroxypyrone derivative (372), giving high yields of bicycle 373 (71–88%) (Scheme 85b).169 This communication was immediately followed by another letter, where a series of novel benzoxazocine derivatives were achieved by applying the same C-alkylation and concomitant O-cyclization strategy. Active methylene containing 1,3-diketones (374) were utilized this time to get condensed with N-alkylquinoliniums (367), resulting in the formation of methylene-bridged benzoxazocines (375) (yield 61–83%) in an identical manner (Scheme 85c).170
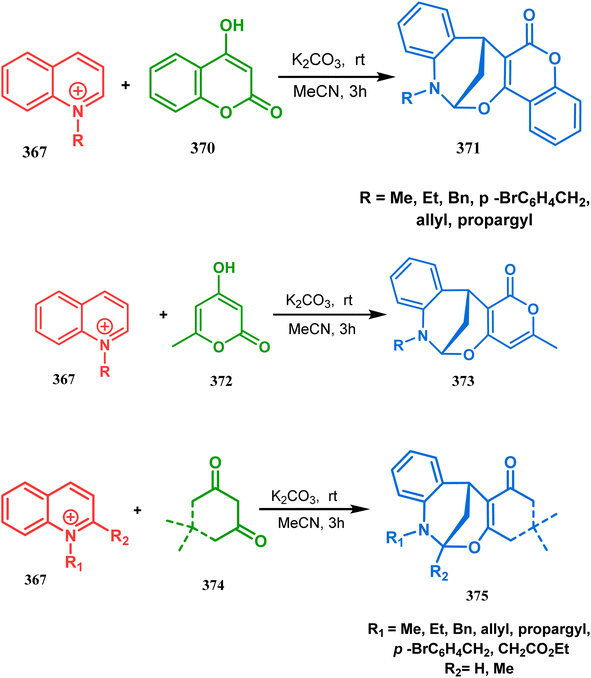 |
| | Scheme 85 (a) Synthesis of oxazabicyclo[3.3.1]nonane by the reaction of 4-hydroxycoumarin and N-alkylquinoliniums through tandem C-alkylation and intramolecular O-cyclization (b): synthesis of oxazabicyclo[3.3.1]nonane by the reaction of 4-hydroxy-pyronederivative and N-alkylquinoliniums through tandem C-alkylation and intramolecular O-cyclization (c) synthesis of methylene-bridged benzoxazocines by the reaction of active methylene-containing 1,3-diketones and N-alkylquinoliniums through tandem C-alkylation and intramolecular O-cyclization. | |
It has also been demonstrated by the same group that 1 3-dihydroxy aromatics (376) could also be successfully annulated with N-alkylquinoliniums (377) using the same methodology to achieve a series of dibenzoxazocines (378) in 15–73% yield, depending on the reaction conditions (Scheme 86a). However, a higher reaction temperature is required to realize these annulations, probably due to the unfavorable dearomatization in the C-alkylation step.171 They has also revealed that even a much less reactive binucleophile such as 2-hydroxynaphthalenes (379) could also be annulated in a similar way to achieve the corresponding methylene-bridged naphthoxazocines (381) in 12–80% yield (Scheme 86b).172
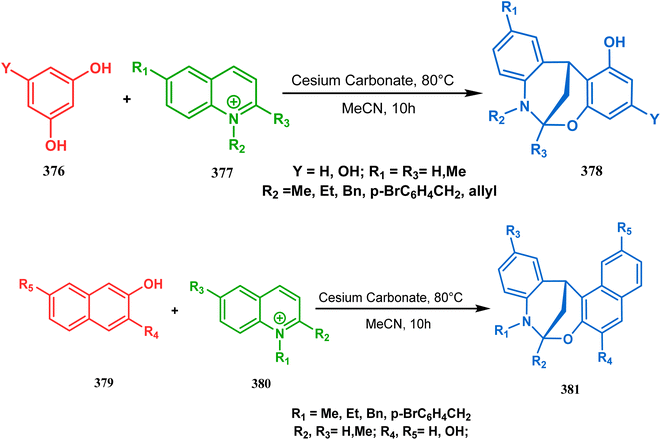 |
| | Scheme 86 (a)Synthesis of dibenzoxazocines by the reaction of 1,3-dihydroxy aromatics with N-alkylquinoliniums through tandem C-alkylation and intramolecular O-cyclization (b) synthesis of methylene-bridged naphthoxazocines by the reaction of 2-hydroxy-naphthalenes with N-alkylquinoliniums through tandem C-alkylation and intramolecular O-cyclization. | |
Apart from the wide ranging investigations of Moghaddam's team, our group also made some contributions toward the synthesis of oxazabicyclo[3.3.1]nonanes. Our initial efforts were dedicated to the synthesis of quinolino/isoquinolino-oxazocines, and through the development of a solvent-free solid-supported methodology, we achieved the target very recently.173 Thus, when hydroxyquinolines (383, 385) and N-alkylquinoliniums (382) were exposed under microwave irradiation using basic alumina as the solid support, the oxazabicylo[3.3.1]nonanes (384, 386) were obtained in excellent yields (85–89% for 384 and 50–92% for 386) (Scheme 87a and b).
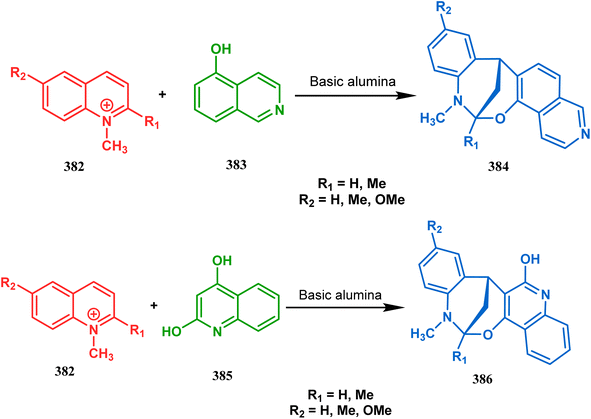 |
| | Scheme 87 (a) Synthesis of oxazabicylo[3.3.1]nonanes by the reaction of hydroxyisoquinoline and N-alkylquinoliniums under microwave irradiation in the presence of basic alumina (b) synthesis of oxazabicylo[3.3.1]nonanes by the reaction of hydroxyquinoline and N-alkylquinoliniums under microwave irradiation in the presence of basic alumina. | |
In a following letter, we also used this environmentally-benign reaction protocol to synthesize diversely-fused dioxa-2-aza-tricyclo[n.3.1.02,n]tetra/pentadecanes. The fused tricyclic oxazaquinolinium salts (388), synthesized by our group very recently in high yields from 8-hydroxyquinoline derivatives (387), was treated with binucleophilic phenol/quinoline/isoquinoline derivatives under the described solvent-free protocol and a series of oxazabicyclo[3.3.1]nonane-containing tricycles (389–391) (yield for 389 73–91%, for 390 87–89%, and for 391 77–83%) were accessed effortlessly (Scheme 88).174 It is believed that the reaction proceeds through an initial C-alkylation on the quinolinium salt to produce an intermediate, followed by an intramolecular O-alkylation, thereby producing 391. It is important to note that all these reactions proceeded with initial C-alkylation, followed by heteroatom-cyclization and not the other way around, thereby demonstrating a particular regioselective nature of this reaction.
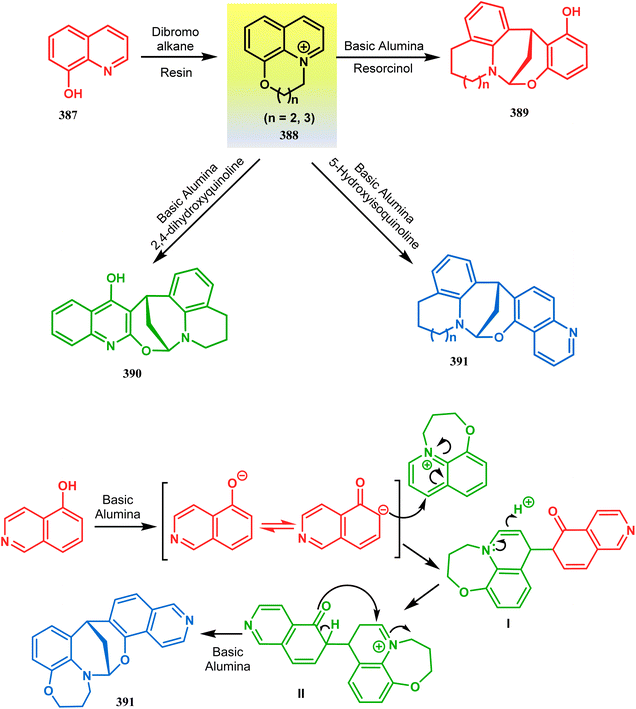 |
| | Scheme 88 Synthesis of oxazabicyclo[3.3.1]nonane-containing tricycles through initial C-alkylation and then intramolecular O-alkylation. | |
Another application of tandem C-alkylation and N-alkylation was unveiled by Sokolov and Meijere's group. Their route toward the cyclization precursor 394 started from methyl-2-(chlorosulfonyl) acetate (392) and p-anisidine (393).175,176 The cyclic sultam (394) thus synthesized was then treated with 1-bromo-3-chloropropane (395) under basic conditions to achieve the desired 1-aza-9-thiabicyclo[3.3.1]nonane derivative (396) in 52–68% yield (Scheme 89). The conformation of this bicyclic sultam was confirmed from X-ray crystallographic analysis, which showed that 396 prefers to stay in its chair–chair conformation, as expected from the fact that an alternate chair-boat conformation should be destabilized by the van der Waals repulsion between the SO2 and the C3 and C7 methylene groups.177
 |
| | Scheme 89 Synthesis of 1-aza-9-thiabicyclo[3.3.1]nonane derivative by the reaction of methyl-2-(chlorosulfonyl) acetate and p-anisidine through tandem C-alkylation and N-alkylation. | |
3.3. Ring opening of other polycycles
3.3.1. Cyclopropane-type ring opening. Studies on the ring-opening reactions of cyclopropane-type systems have been well explored in recent years. In particular, the ring opening of these systems to produce bicyclo[3.3.1]nonane analogues constitutes a useful way toward the synthesis of this important core moiety. During the end of the 20th century, one such important report appeared in the literature due to Srikrishna and coworkers.Their studies on the regioselective cyclopropane cleavage of l-methyltricyclo[4.3.0.02,9]nonan-8-ols unveiled an important route toward chiral bicyclo[3.3.1]nonanes. The ring opening precursor is a tricyclic endo-alcohol (398) (yield 87%) in this case, which was derived from R-carvone (397) in a stereocontrolled manner. It is believed that the mesylate (399) formed from 398 undergoes an E1-type homo-1,4-elimination reaction to give 402 (yield 76%) through a carbocation intermediate 400. Although a Grob-type fragmentation (concerted E2-type elimination) could also explain the outcome, this possibility was ruled out by the fact that the corresponding exo-alcohol (401) (yield 63%) also leads to the same product, thus supporting the E1-type mechanism (Scheme 90).178
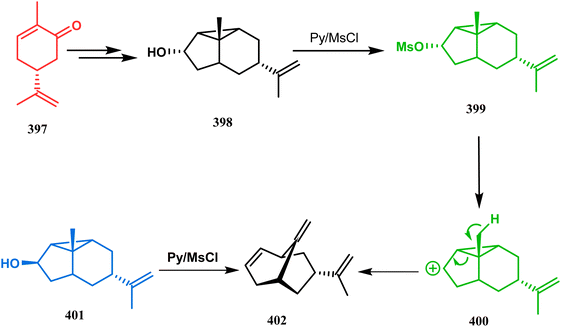 |
| | Scheme 90 Synthesis of chiral bicyclo[3.3.1]nonanes through cyclopropane-type regioselective ring opening of l-methyltricyclo[4.3.0.0]nonan-8-ols. | |
A closely related approach from a similar type of ring opening precursor is also developed by Nakada and coworkers.179,180 They demonstrated a Lewis acid-promoted regioselective ring-opening reaction of the tricycle [4.4.0.05,7]dec-2-ene derivative to achieve a bicyclo[3.3.1]nonane moiety, which has the potential to be used as an intermediate for the synthesis of phloroglucines. Starting from alcohol 403, the required precursor 404 was thus synthesized in a stepwise manner in 93% yield, which upon BF3–OEt2-promoted ring opening reaction in DCM yielded the desired bicycle 405 in excellent yields (33–93%) (Scheme 91). The reaction is believed to proceed via an intramolecular benzyl carbonate attack, leading to cyclopropane ring opening.180
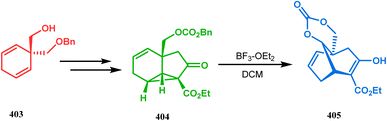 |
| | Scheme 91 Construction of the bicyclo[3.3.1]nonane moiety through the regioselective ring-opening reaction of the tricycle [4.4.0.0]dec-2-ene. | |
In a following letter, within a year, the same group disclosed another application of Lewis acid-mediated cyclopropane ring-opening reaction to achieve a similar goal. This time, the cyclopropanation and ring opening reaction to yield the desired bicycle (408) were done in one-pot sequence. The idea was to facilitate the formation of a more stabilized carbocationic C9 center (by the adjacent methoxy group) to be generated from the methoxy group-triggered ring-opening reaction. Thus, the chronological transformation of 404 into 407 (yield 64%), followed by Cu(OTf)-mediated cyclopropanation and concommitant ZnCl2-promoted ring opening reaction condition, yielded the desired bicycle 408 in 47% yield (Scheme 92).179
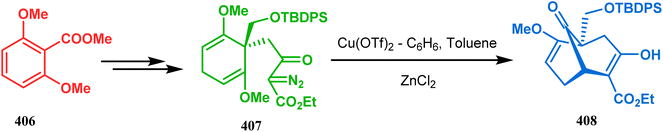 |
| | Scheme 92 Synthesis of the bicycle core through Lewis acid-mediated cyclopropane ring-opening reaction. | |
Wipf and coworkers described a unique example of such a 3-membered ring-opening reaction during aranorosin synthesis. They discovered the fascinating transformation of a spirodiepoxyketone (409) into a bicyclo[3.3.1]non3-en-2-one system (410) in 71% yield, when treated with thiophenols under basic conditions (Scheme 93).181
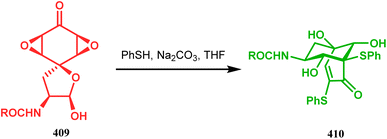 |
| | Scheme 93 Synthesis of the bicyclo[3.3.1] non3-en-2-one system from spirodiepoxyketone through the three-membered ring-opening and rearangement reaction. | |
Stunning structural similarity between 410 and the bicyclic core of natural products gymnastatins F and Q prompted the authors to explore this methodology further. Very recently, the synthesis of densely-functionalized derivatives of this bicyclic system was reported by the same group. For example, spirodiepoxyketones and monoepoxyketones (414–416) obtained in 95% yield from 411–413, respectively, satisfactorily underwent the described ring-opening/rearrangement cascade, yielding the bicyclo[3.3.1]non3-en-2-ones (417–419) in good yields (53–84%) (Schemes 94–96).182 It was also found that although neither electron-donating nor electron-withdrawing substituents in aromatic thiols could affect any parameter of this reaction, the methyl substitution in epoxyketones decelerates product formation.
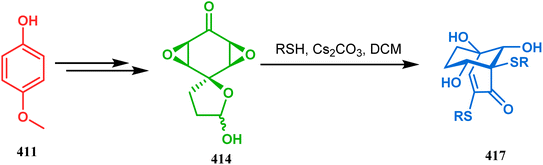 |
| | Scheme 94 Synthesis of bicyclo[3.3.1]non3-en-2-one system from spirodiepoxyketone obtained from 411 through three-membered ring-opening and rearrangement reaction. | |
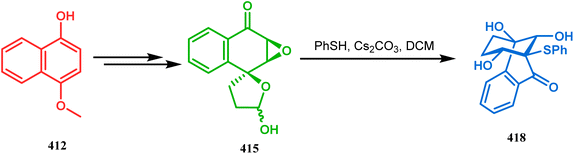 |
| | Scheme 95 Synthesis of bicyclo[3.3.1]non3-en-2-one system from spiromonoepoxyketone obtained from 412 through three-membered ring-opening and rearrangement reaction. | |
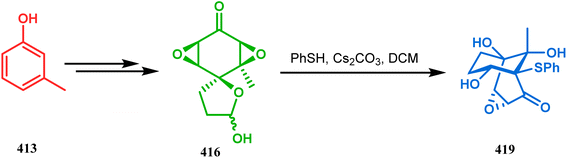 |
| | Scheme 96 Synthesis of the bicyclo[3.3.1]non3-en-2-one system from spiromonoepoxyketone obtained from 413 through three-membered ring-opening and rearrangement reaction. | |
The ring opening of bicycles is also a widely utilized tool to produce azabicyclo[3.3.1]nonanes. A classic example in this catagory should be azabicyclo[3.3.1]nonane analogue synthesis through the ring opening of bicyclic cyclopropanol derivatives. One such report from Chiba and Wang appeared very recently. They designed a useful route toward the 2-azabicyclo[3.3.1]non2-en-1-ol derivatives (429, 433) through Mn(acac)3-catalyzed reaction between bicyclic cyclopropanols (428) with vinyl azides (427, 432). The loss of chirality during the reaction proves that a radical pathway was followed during the course of the reaction. It is believed that the Mn(III) complex promotes the cyclopropane ring opening and forms the reactive radical intermediate 422, which reacts with the vinyl azides and transforms into 424 through the intramolecular radical cyclization of 423 (Scheme 97a).183 A β-methyl group in the vinyl azide retards the reaction, corroborated by the slow reaction rate between 421 and the hindered vinyl azide 425 to give bicycle 426 in 87–89% yield (Scheme 97b).
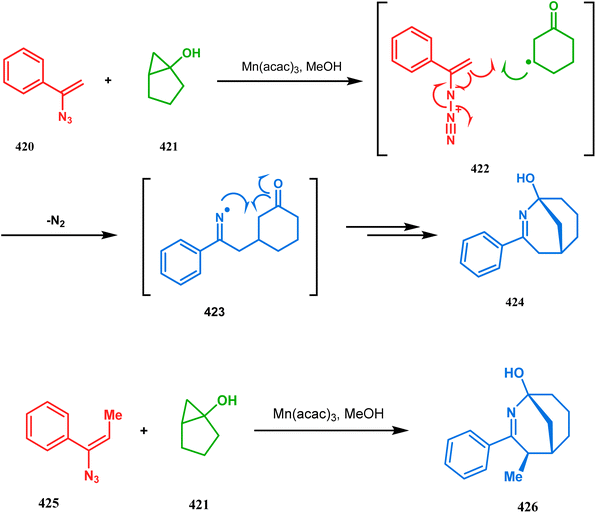 |
| | Scheme 97 (a) Synthesis of 2-azabicyclo[3.3.1]non2-en-1-ol derivative through Mn(acac)3-catalyzed reaction between bicyclic cyclopropanol with vinyl azide (420), followed by intramolecular radical cyclization (b) synthesis of 2-azabicyclo[3.3.1]non2-en-1-ol derivative through Mn(acac)3-catalyzed reaction between bicyclic cyclopropanol with vinyl azide derivative (418), followed by intramolecular radical cyclization. | |
3.3.2. Adamantane-type ring-opening. The ring opening of adamantane-type systems is also a well-explored technique to achieve bicyclo[3.3.1]nonane units. One such example is the synthesis of bicyclo[3.3.1]non6-en-3-ones. Inspite of several available protocols toward this system,184,185 the complexity and lack of generality of those routes prompted Camps and coworkers to unmask a more convenient route to synthesize this important core moiety, leading to the discovery of a unique application of silica gel to achieve 7-alkylbicyclo[3.3.1]non6-en-3-ones. Thus, when 3-alkyl-2-oxaadamant-l-yl-mesylates (427) were treated with silica gel in DCM, the starting material was fragmented and bicycles 430 were obtained through cabocation intermediates 428 and 429 in high yields (71%) (Scheme 98).186
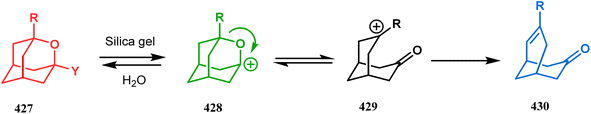 |
| | Scheme 98 Synthesis of 7-alkylbicyclo[3.3.1]non6-en-3-one from 3-alkyl-2-oxaadamant-l-yl-mesylate through the ring-opening reaction. | |
Rossi's group in 1997 demonstrated another fragmentation reaction of adamantane ring systems, yielding 7-methylidenebicyclo[3.3.1]nonane. Although closely related fragmentation reactions of 1-hydroxy-/1-thio-3-bromoadamantanes always require a basic condition,187 Rossi's approach was photochemically based, which promotes a very fast ring-opening reaction of 1,3-dihaloadamantanes (431) with acetophenone enolates (432), forming α-(7-methylidenebicyclo[3.3.1]non2-en-1-yl)acetophenone (435) with high yields. The reaction proceeds with an initial substitution reaction on 431 by 432 to form the monohalo adamantane 433, followed by the ring-opening reaction to produce 434 (Scheme 99) which immediately isomerized to 435. Similar reactions with pinacolone enolate (436) to produce 437 (yield 26–81%) are also fast, whereas the nitromethane anion (438) reacted with 431 in a rather decelerated rate, forming bicycle 439 in 11–67% yield (Scheme 99).188 The irradiation of unsubstituted adamantane, however, shows a different chemistry. Thus, when Albini and coworkers attempted TiO2-mediated photocatalysis of 440, a minor amount of bicyclo[3.3.1]nonanedione (441) was isolated along with two isomeric adamantanols and 2-adamantanone (Scheme 100).189
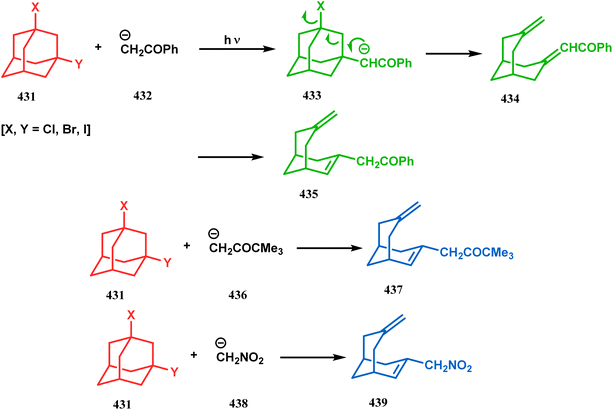 |
| | Scheme 99 Synthesis of 7-methylidenebicyclo[3.3.1]nonanes through the fragmentation reaction of adamantane ring systems. | |
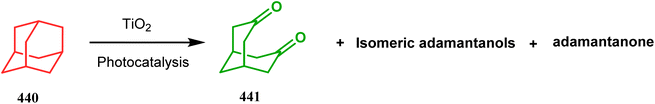 |
| | Scheme 100 Formation of bicyclo[3.3.1]nonanedione through TiO2-mediated photocatalysis of unsubstituted adamantine. | |
The fragmentation reaction of 1,3-disubstituted-adamantanes to produce bicyclo[3.3.1]nonane derivatives has been well investigated in the last 50 years.190–196 Toward this goal, very recently, Skomorokhov and coworkers described the reaction of 1,3-dibromoadamantane (442) with glycols in the presence of sodium glycolate to synthesize bicyclo[3.3.1]nonane derivatives (445, 446) (Scheme 101). It is believed that an intermediate adamantyl carbocation (444), formed by the bromide elimination from 443, yielded the differently substituted bicycles in a stepwise manner.197,198
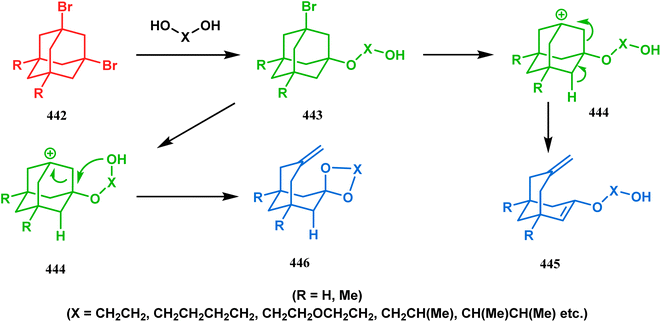 |
| | Scheme 101 Synthesis of bicyclo[3.3.1]nonane derivatives by the reaction of 1,3-dibromoadamantane with glycols in the presence of sodium glycolate. | |
Another simple and convenient synthesis of 1,3,7-trisubstituted bicyclo[3.3.1]nonanes from adamantane derivatives was described by Majerski's group. Their journey commenced from 1-hydroxyadamantane-4-one (447), which, upon sequential ring expansion and oxidation, yielded the ring opening precursor 448, an adamantane-dione derivative, which on treatment with HIO4 yielded the bicyclic 1,3-dicarboxylic acid 449 in 36% yield (Scheme 102).199
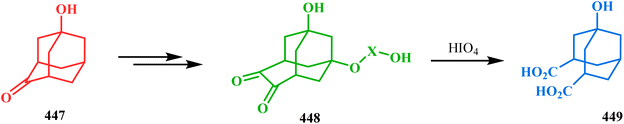 |
| | Scheme 102 Synthesis of 1,3,7-trisubstituted bicyclo[3.3.1]nonane from 1-hydroxyadamantane-4-one and then the synthesis of bicyclic 1,3-dicarboxylic acid on treatment with HIO4. | |
Nurieva and coworkers also employed such adamantyl precursors to achieve their desired colchicine analogues. Thus, adamantane-2-one (450) was first subjected to selenium dioxide-promoted ring-opening reaction, followed by concomitant lactonization to give a tricyclic lactone 451. The base-catalyzed hydrolysis of this lactone then yielded an endo-hydroxy carboxylic acid 452 (Scheme 103).200 The fragmentation of 1,3-adamantanediol (453) is also another useful procedure to achieve functionalized bicyclo[3.3.1]nonanes. Few years ago, Iwabuchi and coworkers demonstrated one such useful application. Following their report, 453 could easily be exposed toward Grob fragmentation to give an exocyclic double bond-containing bicyclo[3.3.1]nonane-3-one (454) derivative,201,202 which was then used as the precursors for the synthesis of differently functionalized bicyclo[3.3.1]nonanes (455) in 73% yield (Scheme 104).
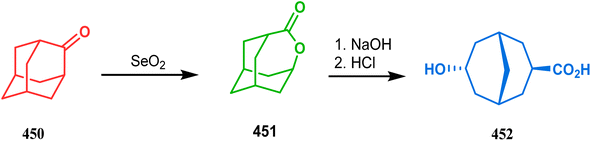 |
| | Scheme 103 Synthesis of endo-hydroxycarboxylic acid from adamantane-2-one. | |
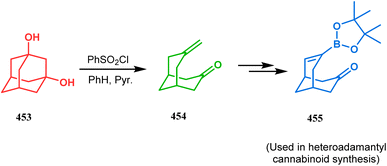 |
| | Scheme 104 Synthesis of differently-functionalized bicyclo[3.3.1]nonane by the fragmentation of 1,3-adamantanediol. | |
3.4. Other routes
3.4.1. Hosomi–Sakurai & carbonyl-ene reaction. The Hosomi–Sakurai reaction and the carbonyl–ene reaction are the two competitive candidates for preparing bicyclo[3.3.1]nonane derivatives from appropriately positioned allyl silane derivatives. Although both of them share an almost similar reaction condition, they differ in their area of application. Honda and coworkers in their two successive articles exemplified this fact in detail. Their initial report was on the total synthesis of upial, a bicyclo[3.3.1]nonane-containing tricyclic terpenoid. The required aldehyde 457 for this purpose was synthesized in a stereocontrolled manner from the optically active ketone 456 stereoselectively. However, the key step of this transformation is the p-toluenesulfonic acid-catalyzed cyclization of aldehyde 457 to give the desired bicycle 458 (yield 74–96%) (Scheme 105). This bicycle was then sequentially converted into upial in a stepwise manner.203 However, the pTsOH-mediated strategy did not work satisfactorily for the synthesis of diastereomeric sesquiterpenes Triferienols A & B. Although the cyclization precursor 460 obtained from ketone 459 produced the desired bicycle 461 through the intramolecular cyclization in 93% yield, an acetal side product also accompanied it.
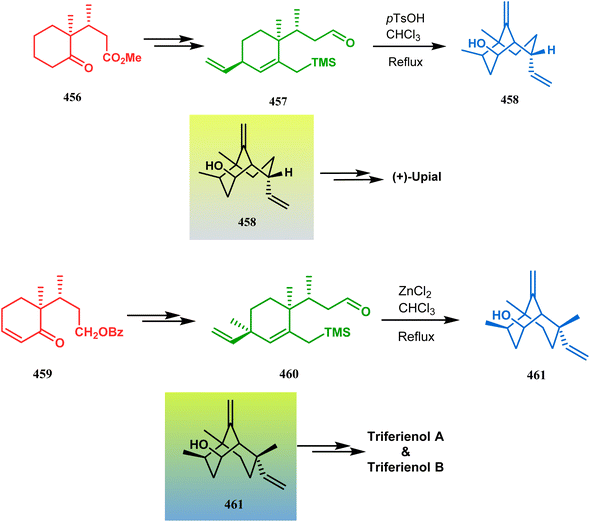 |
| | Scheme 105 Synthesis of bicyclo[3.3.1]nonane derivatives following Hosomi–Sakurai reaction and the carbonyl–ene reaction from appropriately positioned allyl silane derivatives. | |
It is believed that the extra methyl substituent on the allyl group-containing quaternary carbon center leads to a substantial increase in steric crowding during cyclization, therefore raising the possibility of acetal or half-acetal formation. Therefore, an alternate Lewis acid ZnCl2 was introduced, which acted as the initiator of the Hosomi–Sakurai reaction, thereby favoring the cyclization prior to the acetal formation, yielding the desired bicyclic core 461 in excellent yield (93%), which was then transformed into Triferienols A & B in a few steps (Scheme 105).204
3.4.2. Mannich reaction. Brimble and coworkers demonstrated an elaborated investigation on the application of Mannich reaction to synthesize azabicyclo[3.3.1]nonanes.202 Their initial target was to synthesize diversely-substituted dioxazepanes (462) (yield 54–97%) as the Mannich reaction partner of β-keto esters (463). These dioxazepanes, constructed from paraformaldehyde, ethylene glycol, and a primary amine, were then used as the electrophiles for the double Mannich reaction to achieve the desired azabicycles (464). To find the best activator for the desired transformation, a variety of Lewis acids were screened, and methyltrichlorosilane appeared as the best candidate (Scheme 106).205
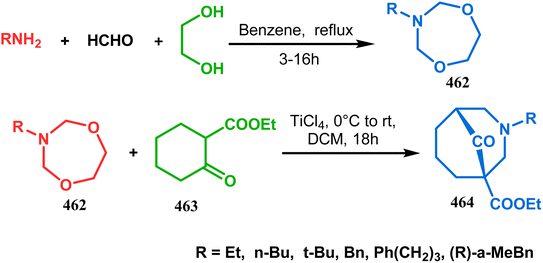 |
| | Scheme 106 Synthesis of azabicyclo[3.3.1]nonane following Mannich reaction. | |
3.4.3. Cycloaddition reaction. Cycloaddition reactions are widely used in the synthesis of natural products. Bicyclo[3.3.1]nonane-containing natural products are also not an exception. In this regard, a unique application of nitrile oxide-allene cycloaddition206 in Hypervolutin A core synthesis was demonstrated by Young and Zeng. Their journey began from the benzylidene ketone 465, which was sequentially transformed into 466 for the synthesis of the cyclization precursor 468. This attempt, however, remained unsuccessful, and an alternate allene derivative 467 was employed in a Lewis acid-promoted Mukaiyama condensation reaction to synthesize 468, a diastereomeric mixture of an allene. This cyclization precursor was then exposed toward phenyl isocyanate to promote the envisioned nitrile oxide-allene cycloaddition reaction, which yielded the desired bicyclo[3.3.1]nonane-containing tricycle 469 in 40% yield (Scheme 107).207 Although, both the diastereomers of 468 were subjected to cycloaddition condition, only the ‘anti’ cycloadduct (carbonyl group and methoxy group opposite to each other) was produced, indicating the reluctant nature of the ‘syn’ diastereoisomer of 468 toward the cycloaddition reaction.
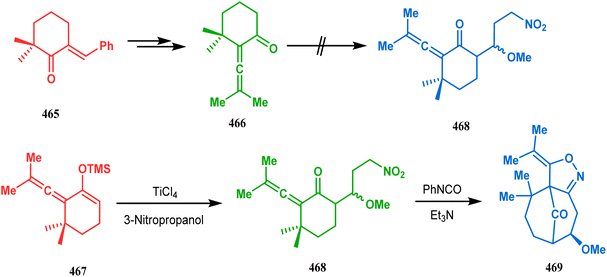 |
| | Scheme 107 Synthesis of the bicyclo[3.3.1]nonane-containing tricycle through nitrile oxide-allene cycloaddition reaction. | |
3.4.4. Double condensation reactions. Multicomponent reactions, especially the 1-step double condensation of two units of 1,3-acetonedicarboxylates with 1,2-dicarbonyl/1,4-dicarbonyl compounds, are a well-explored protocol to achieve bicyclo[3.3.1]nonane-3,7-diones.208–214 Following these reports, Camps and coworkers reported the synthesis of 9-methoxy-9-methylbicyclo[3.3.1]nonane-3,7-dione.215 A similar report on the synthesis of polysubstituted bicyclo[3.3.1]nonane 3,7-diones from cyclohexa-2,5 dienones by the same group appeared during the beginning of this century. This time, a sequential phenyliodonium diacetate (PIDA)-mediated phenol oxidation and double Michael condensation was employed to achieve the desired bicycles. Thus, starting from phenols 470, polysubstituted bicyclo[3.3.1]nonane-dicarboxylates (472) were obtained through quinones 471. These dicarboxylates upon hydrolysis-decarboxylation produced the targeted bicyclic diones (473) in high yields (15–90%) (Scheme 108).216
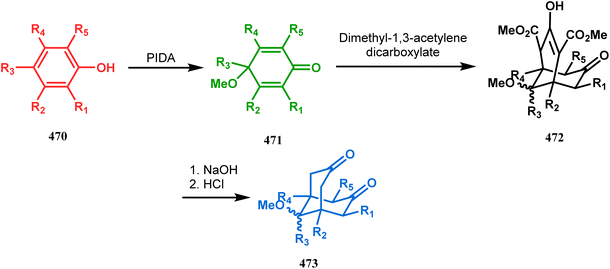 |
| | Scheme 108 Synthesis of polysubstituted bicyclo[3.3.1]nonane-3,7-dione through phenyliodonium diacetate (PIDA)-mediated phenol oxidation and double Michael condensation reaction. | |
In a closely related study by Warnmark and coworkers, condensations between dimethyl malonates and paraformaldehyde have been reported. When 474 and formaldehyde were subjected toward the cyclization protocol, two formaldehyde units get condensed with two dimethyl malonate units, resulting in the synthesis of bicyclo[3.3.1]nonane 475, which upon treatment with HOAc/HCl yielded the racemic mixture of bicyclo[3.3.1]nonane-2,6-dione (476) (Scheme 109).217 To obtain the pure enantiomers, biotransformation technique was then employed. The fact that the use of enzymes and microorganisms to achieve this important system in their enantiomerically pure form are the most popular methodologies218,219 prompted Warnmark's group to apply Baker's yeast to obtain enantiomerically-pure bicyclo[3.3.1]nonane-2,6-dione in a large scale.217 In a similar report by Grauslund and coworkers, genetically engineered Saccharomyces cerevisiae cells were used as the biocatalyst for the kinetic resolution of racemic bicyclo[3.3.1]nonane-2,6-dione.220
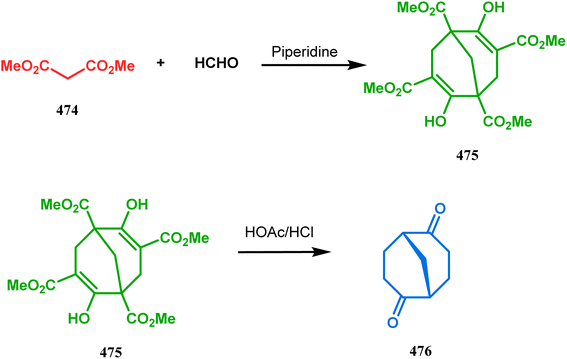 |
| | Scheme 109 Synthesis of bicyclo[3.3.1]nonane-2,6-dione through condensation between dimethyl malonates and paraformaldehyde. | |
3.4.5. Through the solvolysis of other polycycles. During the solvolysis study of 2-bicyclo[3.2.2]nonyl p-toluenesulfonates, Okazaki's group and Schafaer's team found that the solvolysis products of such bicycles are usually enriched with bicyclo[3.3.1]nonanes.221–224 It was found that when 477 was solvolyzed in methanol and 2,2,2-trifluoroethanol (TFE) buffered with 2,6-lutidine, 2-bicyclo[3.3.1]nonene (479) (yield 15–47%) and exo-2-R-bicyclo[3.3.1]nonane (R = methoxy, 480; R = trifluoroethoxy, 481) were formed along with other bicycles in 12–23% yield. The solvolytic data indicated that these products were formed from the 2-bicyclo[3.2.2]nonyl cation (478), thereby unveiling the evidences for the formation of a classical carbocation intermediate (Scheme 110).221
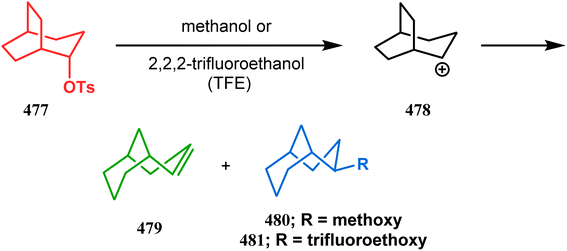 |
| | Scheme 110 Formation of 2-bicyclo[3.3.1]nonene and exo-2-R-bicyclo[3.3.1]nonane through solvolysis. | |
4. Reactivity
4.1. Ring opening & rearrangement
4.1.1. Baeyer–Villiger oxidation. Since the bridged bicyclic lactones are considered as important synthons for the synthesis of related natural product cores, the Bayer–Villiger oxidation of bridged polycyclic ketones have gained substantial importance. Such oxidation protocols on bicyclo[3.3.1]nonane diones are most extensively studied by Stoncius and coworkers. Their initial report was based on the regioselective transformation of differently positioned carbonyl group-containing bicycles to their corresponding lactones. The treatment of diketones 482 and 483 with mCPBA in DCM yielded the oxabicyclo[3.3.2]decanediones 487a,b and 488a,b in 92:8 ratio. The preferential oxidation of the C9 carbonyl group over the C2 carbonyl is determined by kinetic parameters. Thus, although the c isomer is thermodynamically more stable than other isomeric ketolactones (according to AM1 calculations), larger torsional strain release during the formation of tetrahedral intermediate through C9 oxidation than that through C2 oxidation (as the former carbonyl is more distorted from sp2 hybridization than the latter) favored the a and b isomers over c and d. The difference in the migratory aptitudes between C1–C9 and C5–C9 bonds then determines the a:b ratio. Clearly, the electron-withdrawing effect of both carbonyl groups on the former bond made the latter more nucleophilic, resulting in the preferential migration of the C5–C9 bond and thereby favoring the a isomer. This preference was, however, slightly diminished when an ester group was introduced at the C5 position and, as expected, the oxidation of 484 took a much longer completion time and yielded 489a,b in 85:1 ratio.Diketones 486 also demonstrated some interesting features and when exposed to a similar oxidation protocol, the C3 carbonyl group was found to be surprisingly inactive, which could be explained by the steric hindrance caused by the C7 endo-proton.225 The preferential oxidation of the C6 carbonyl yielded oxabicyclo[4.3.1]decanedione 490a regioselectively, as expected from the lower migratory aptitude of the bridging carbon due to the electron withdrawing effect of the β-keto group. The carbonyl groups of 486 are, however, equivalent in nature and thus remove the question of preferential oxidation, but migratory competition remains and the higher migratory aptitude of the bridging carbon (due to higher strain release) produced 491b regioselectively (Scheme 111).226,227 The Bayer–Villiger oxidation of symmetrical dione 492 shows the simplest chemistry and produced the only possible lactone 493.228 The absolute configuration determination of the product lactones was also done by Stoncius's group and IR-VCD spectroscopies and chemical correlations, and the chiroptical properties of these lactones were extensively investigated to assign the configurations.229 Gambacorta's team also successfully synthesized a similar bicyclic lactone 495 from a simpler bicyclic ketone 494 using mCPBA-promoted Bayer–Villiger oxidation. This bicyclic lactone was then employed to synthesize their targeted hydroxyacids 496 and 497 in 95% yield (Scheme 112).230,231
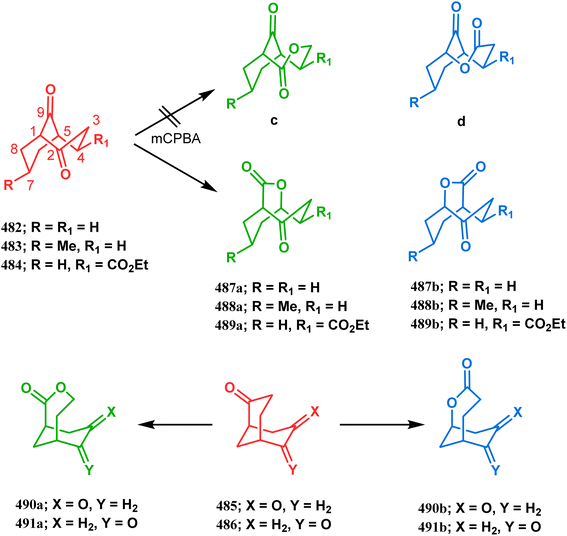 |
| | Scheme 111 Synthesis of bridged bicyclic lactones through Bayer–Villiger oxidation. | |
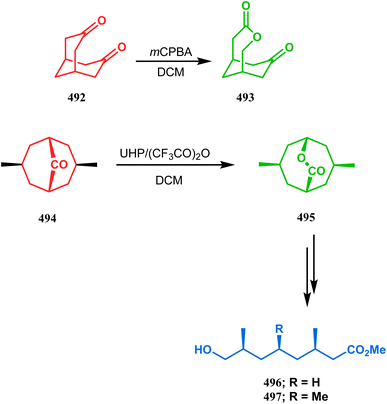 |
| | Scheme 112 Synthesis of hydroxyacids from bicyclic lactone. | |
4.1.2. Other oxidative rearrangements. Studies on the various properties of bicyclo[3.3.1]nonane-diones, especially on its absolute configuration determination, have gained substantial interest in recent times. For example, diketones 498–501 are configured properly using optical rotation and circular dichroism studies.232–244 The reason behind the growing concern in this area is the abundance of this bicycle in natural products and their metabolites245,246 and their use as precursors for the synthesis of other important bicycles. For example, thallium(III) nitrate-promoted oxidative rearrangements are well documented in this regard in the recent literature,247 and its successful application in bicyclo[3.3.1]nonane chemistry was first demonstrated by Butkus and coworkers. During the beginning of this century, his team efficiently synthesized chiral tricyclo[4.3.0.03,8]nonane-4,5-dione from enantiopure bicyclo[3.3.1]nonane-2,6-dione employing this oxidation protocol. Thus, when 498 was treated with Tl(NO3)3 in methanol, the diester 502 was isolated in high yields (85%), which was then sequentially converted into the targeted twistbrendanedione (503) to study its chiroptical properties through ECD and VCD spectroscopies (Scheme 113).248
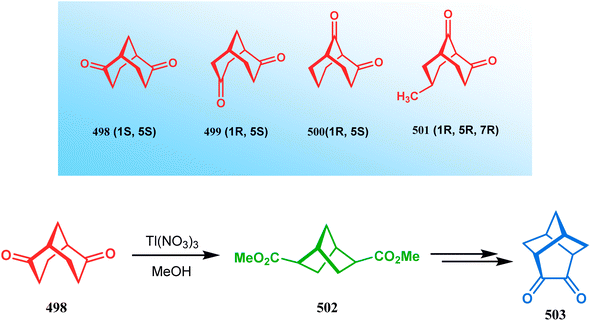 |
| | Scheme 113 Synthesis of tricyclo[4.3.0.0]nonane-4,5-dione from enantiopure bicyclo[3.3.1]nonane-2, 6-dione with the help of Tl(NO3)3 in methanol. | |
However, the Tl(NO3)3 oxidation of other isomeric diketones gives a mixture of products. Thus, the treatment of diketone 500 in a similar oxidation protocol produced the monocarboxylate 504 as a minor product along with other isomeric bicycles. The symmetrical diketone 505 also gives a mixture of compounds, namely, the oxidative rearrangement product 506, its acetal 507, and an oxaadamantane-type tricyclic acetal 508. The formation of 508 was explained by faster transannular cyclization prior to oxidation due to the close proximity of the carbonyl groups. Similar tricyclic acetal (509) was also isolated when another diketone 499 was subjected to an identical oxidation protocol. The oxidation of monoketones, however, describes a simpler chemistry, for example, the thallium nitrate oxidation of monoketones 510a,b mainly gives the diastereomeric mixture of esters 511a,b (Scheme 114).249
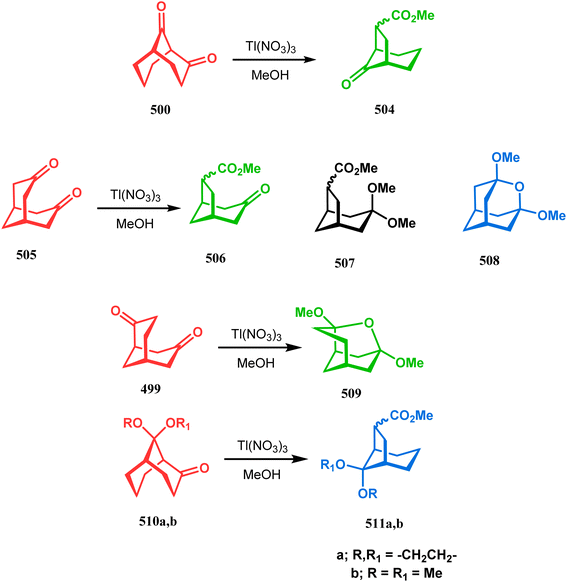 |
| | Scheme 114 Thallium nitrate [Tl(NO3)3] oxidation of isomeric diketones for synthesizing cyclic esters or acetals. | |
Nicolaou and coworkers demonstrated a different type of oxidative fragmentation and concomitant cyclization process on similar bicyclo[3.3.1]nonane diketones. Initially, the OsO4-mediated dihydroxylation on the double bond of 512 gave the dihydroxy-diketone 513. Immediate intramolecular lactonization and concurrent ring opening reaction promoted by 4-DMAP then yielded the envisioned bicyclo[5.2.1] decane (514). The study was further extended on benzo-fused bicyclo[3.3.1]nonane precursor 515, which under similar conditions produced 516, a benzo-fused bicyclo[5.2.1] decane (Scheme 115).86
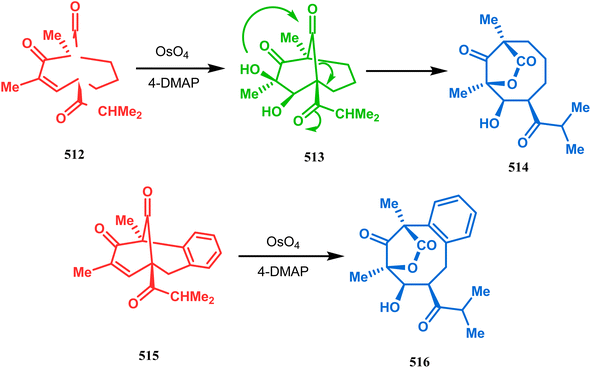 |
| | Scheme 115 Synthesis of bicyclo[5.2.1]decane and benzo-fused bicyclo[5.2.1]decane through the oxidative fragmentation and concomitant cyclization of bicyclo[3.3.1]nonane diketones. | |
4.2. Ring closing cyclizations
4.2.1. Synthesis of adamantane-type ring systems. Ring-closing cyclization technique to achieve adamantyl and heteroadamantyl systems is one of the most popular tools used in bicyclo[3.3.1]nonane chemistry. One of such important example, as exemplified by Makriyannis and coworkers, is the synthesis of heteroadamantyl cannabinoids, known as CB1 and CB2 receptor antagonists. This group employed the preparation of the required vinyl boronate 518 from adamantanol 517 to react with 519, which yielded the cyclization precursor 520. Again, on reduction and intramolecular cyclization, 520 produced the desired oxaadamantyl cannabinoid 522 (Scheme 116).201 Similar oxaadamantane derivatives were also constructed by Vazquez and coworkers to evaluate their potentiality as antivirals, NMDA receptor antagonists, and trypanocidal agents. Starting from diketone 523, secondary oxadamantyl amines 524 were synthesized using an amine condensation-reduction procedure,250–252 which was further derivatized into other secondary and tertiary amines. Furthermore, diketone 523 was also employed to construct oxaadamantanol 525 and was used as the precursor for other oxaadamantyl amines.253,254 Corey and Chau also reported the synthesis of a polysubstituted diazaadamantane derivative. During the synthesis of chelating bis-amines, this heteroadamantane derivative (527) was accidentally synthesized when 526 was stored in a DCM solution (Scheme 117).156
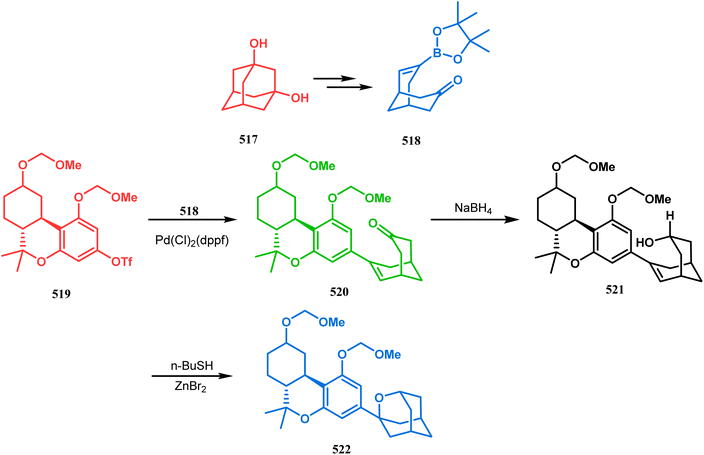 |
| | Scheme 116 Synthesis of the oxaadamantyl cannabinoid system through reduction and intramolecular ring-closing cyclization process. | |
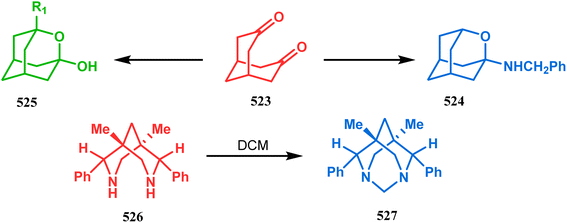 |
| | Scheme 117 Synthesis of oxadamantyl amine, oxaadamantanol, and heteroadamantyl system by ring-closing cyclization. | |
Apart from the syntheses of these heteroadamantyl systems, the transannular cyclization of appropriately substituted bicyclo[3.3.1]nonanes promoted by electrophiles such as bromine and iodine is also well documented.255–258 The contribution of Serguchev and coworkers in this regard is remarkable. During the beginning of this century, his team unveiled their preliminary report on the reaction between 1-chloromethyl-4-fluoro-1,4-diazoniabicyclo[2.2.2]octane bis(tetrafluoroborate) (F-TEDA-BF4) (529) with 3,7-bismethylenebicyclo[3.3.1]nonane (528). Their investigations were carried out in ROH solvents and therefore led to the synthesis of 1-RO-3-fluoromethyladamantanes (530) (R = H, Alk, Ac) along with a slight impurity of 1-fluoro-3-fluoroalkyladamantanes (531) (yield 81–95%) (Scheme 118).259 Interestingly, when the same reaction partners (528 and 529) were reacted in monoglyme, the previously found minor product 531 became major and was isolated in high yields. It is believed that the reaction proceeds through the initial addition of electrolphilic fluorine to the double bond to form an adamantyl carbocation, followed by fluoride capture to give 531 (Scheme 118). The essentiality of monoglyme is attributed to the fact that unlike other solvents (DCM, THF, etc.), it binds to the liberated BF3 during the reaction to form an etherate complex and ensures the release of fluoride ion. The reaction also occurs in nitromethane solvent, but the yield of 531 was low, with 532 as the major product, which could be further derivatized into bromo, chloro, methoxy, or acetoxy adamantanes (533–536) having 50–75% yield (Scheme 119).260
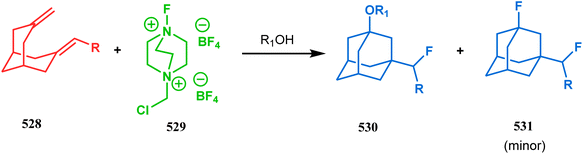 |
| | Scheme 118 Synthesis of 1-RO-3-fluoromethyladamantanes by the reaction between 1-chloromethyl-4-fluoro-1,4-diazoniabicyclo[2.2.2]octane bis(tetrafluoroborate) and 3,7-bismethylenebicyclo[3.3.1]nonane. | |
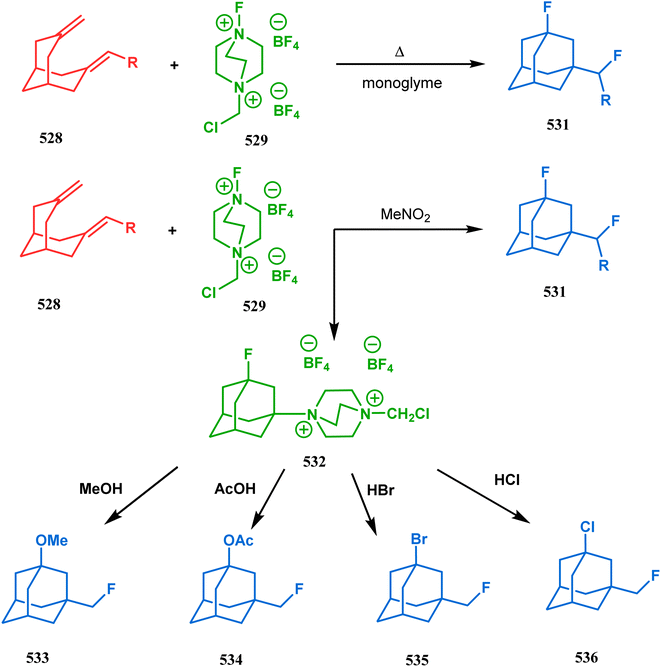 |
| | Scheme 119 Synthesis of bromo, chloro, methoxy, or acetoxy derivatives of adamantanes by the reaction between 1-chloromethyl-4-fluoro-1,4-diazoniabicyclo[2.2.2] octane bis(tetrafluoroborate) and 3,7-bismethylenebicyclo[3.3.1]nonane in monoglymme and nitromethane solvent, followed by treating the product with HBr, HCl, MeOH, and AcOH, respectively. | |
Within a year, the same group published another article, where N-halosuccinimides (537) were employed to synthesize the halo-fluoro-substituted adamantanes from bicyclo[3.3.1]nonane dienes. Thus, when diene 538 was treated with 537 in the presence of Bu4N+H2F3− in DCM, 1-fluoro-3-halomethyladamantanes 539 were produced in a radical pathway. However, unlike NIS, the ability of NBS and NCS to undergo both homolytic and heterolytic fissions lowered the possibility of radical reaction and thereby decreased the product yield from 539a to 539c (yield 25–60%). Similar reactions on substituted dienes (540) demonstrated regioselective fluorination on more substituted double bond and yielded 541 in good yields (50–65%) (Scheme 120). More interestingly, when such reactions were carried out in cyclic ethers, a cascade process, leading to the formation of fluoroalkoxy adamantanes, occurs. Thus, when 538 and 540 was subjected to similar reaction conditions in ethylene oxide (543), oxetane (544), or THF (545), the products formed were fluoroalkoxy-halomethyl adamantanes (546–548). However, a similar reaction in tetrahydropyran (542) produces usual products, possibly due to its higher solubility. Similar haloalkoxyproducts (549) were also obtained when this reaction was performed in polyhalogenated alcohols (Scheme 121).261
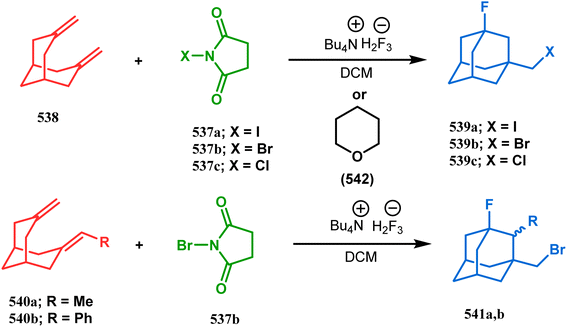 |
| | Scheme 120 Synthesis of halo-fluoro-substituted adamantanes from bicyclo[3.3.1]nonane dienes with the use of N-halosuccinimides in the presence of Bu4N+H2F3− in DCM. | |
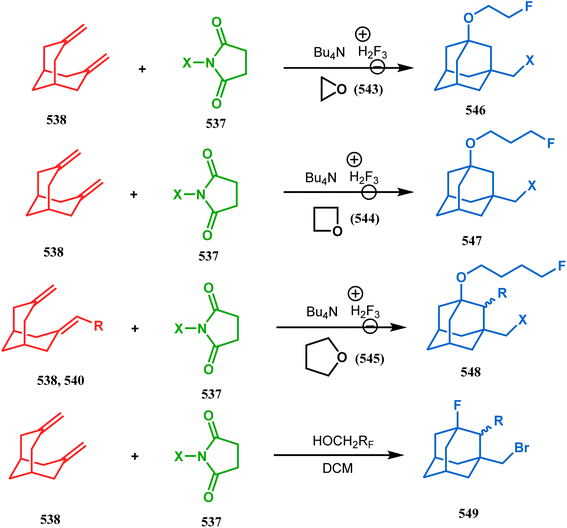 |
| | Scheme 121 Synthesis of fluoroalkoxy adamantanes and halo-fluoro-substituted adamantanes from bicyclo[3.3.1]nonane dienes in ethylene oxide, oxetane, THF, or polyhalogenated alcohols. | |
4.2.2. Synthesis of noradamantane derivatives. The intramolecular cyclization of appropriately functionalized bicyclo[3.3.1]nonanes is also an important tool to produce noradamantane derivatives. Zajac and coworkers made a substantial contribution in this regard. Their initial investigations on 1-sodio-3,5-dichloro-1,3,5-triazine-2,4,6(1H,3H,5H)-trione (NaDCTT) promoted the transformation of diketoxime 550 into the corresponding bis-geminal chloronitro compounds also identified a small amount 3,7-dinitronoradamantane 551 formed during the reaction.262 A more elaborated study in this regard revealed that the employment of mCPBA produces the noradamantane 552 as the sole product with good yields.263 The use of this peracid was further successfully utilized by Zajac's team to synthesize another noradamantane 553 from 9,9-dimethoxybicyclo[3.3.1]nonane-3,7-dione (552) in high yields.264 Reports on the synthesis of a similar noradamantane derivative 555 (yield 85%) from diketone 554 are also found in the literature (Scheme 122).265–269
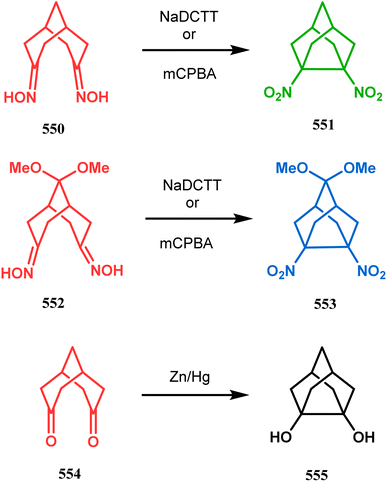 |
| | Scheme 122 Synthesis of different types of noradamantane derivatives. | |
The transformation of 3,7-dimethylenebicyclo[3.3.1]nonane into substituted noradamantanes through the transannular cyclization technique could also be accomplished via a radical-mediated pathway. Serguchev and coworkers described one such unique example employing polyfluoroalkyl radicals. Thus, when 556 was exposed toward UV-radiation or Cu-catalysis with RFI agents {RF = CF3, n-C3F7, CF2COOEt, CF2PO(OEt)2}, corresponding noradamantanes (558) were isolated in quantitative yields. Both the pathways involve the initiation with the formation of the RF radical, which then reacts with 556 to give radical 557. Among the two possible cyclization pathways, namely, the exo-trig and endo-trig cyclization, the former one is more favorable kinetically, and therefore occurs instantly to give noradamantanes 558. Although, endo-trig cyclization could have produced thermodynamically more stable adamantane product (559), the energy barrier between 558 and 559 is too high to be crossed in a high homolyzable solvent such as CBr4; therefore, 558 and 559 are found to form in 9:1 ratio in CBr4 compared to the 3:1 ratio in a less homolyzable solvent CCl4.270 Once this iodomethyl-noradamantane is formed, it was efficiently derivatized into a diverse range of noradamantane derivatives (Scheme 123).
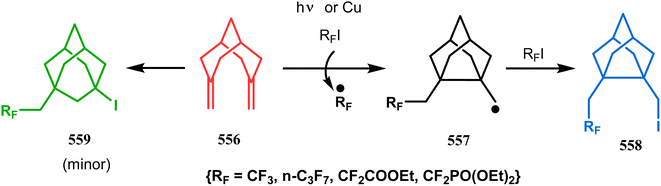 |
| | Scheme 123 Synthesis of substituted noradamantanes by the transformation of 3,7-dimethylenebicyclo[3.3.1]nonane through the transannular cyclization technique via a radical-mediated pathway. | |
The photochemistry of bicyclo[3.3.1]nonanes having parallel exocyclic double bonds in 3 and 7 positions exhibits some unique observations. Several reports on the intramolecular cyclization of such compounds (560) to give cyclobutane-fused noradamantane-type ring systems (561) (Scheme 124) appeared in the literature during the 1970s.271,272 This century also began with one such unique example, exemplified by Averina and coworkers. Their study began from diketodiene 562, which on irradiation transformed into its less stable cis–cis isomeric form (563), and no cycloaddition product was formed. However, when the ether solution of diol 564, obtained by the reduction of 562, was subjected to similar reaction conditions, the product becomes exo-7,endo-10-dihydroxy-2-phenyl-3,4-benzotetracyclo-[4.3.3.18,11.01,6]tridecane (565) (Scheme 125).273
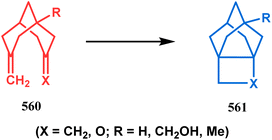 |
| | Scheme 124 Synthesis of cyclobutene-fused noradamantane-type ring systems through the intramolecular cyclization of 560. | |
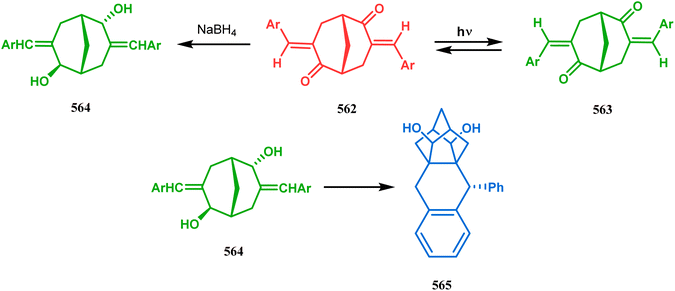 |
| | Scheme 125 Synthesis of exo-7,endo-10-dihydroxy-2-phenyl-3,4-benzotetracyclo [4.3.3.1.0] tridecane from a diketodiene upon irradiation of light. | |
4.2.3. Synthesis of other polycycles. Ring closure reactions of α,α′-dihalobicyclo[3.3.1]nonane-diones under Favorskii reaction condition is a useful protocol to achieve oxatricyclo[4.3.1.03.8]decanes. Butkus and coworkers established this fact efficiently in a pair of subsequent communications. Starting from diketones 566, the Favorskii precursors 567 were synthesized through a simple halogenations reaction. 567, on treatment with NaOMe, formed hemiketal anion 568, which underwent a concomitant cyclization reaction to give the oxatricyclodecanes 569 (Scheme 126).274,275
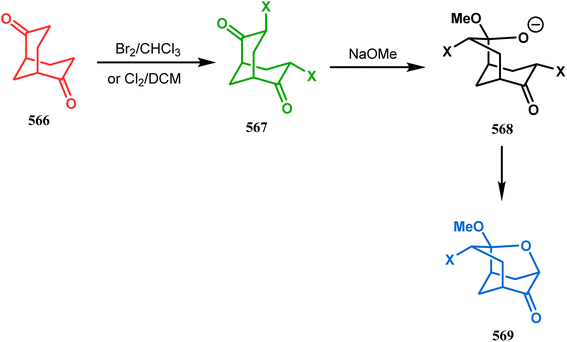 |
| | Scheme 126 Synthesis of oxatricyclo[4.3.1.0]decanes through the ring closure reactions of α,α′-dihalobicyclo[3.3.1]nonane-diones under Favorskii reaction conditions. | |
Another interesting report of intramolecular cyclization of a chelating bis-ether was demonstrated by Corey and Chau. They found that (+) 570 on treatment with catalytic amount of p-TsOH or H2SO4 promotes a fascinating intramolecular dehydration reaction and yielded a crystalline twistane-type tricyclic ether 571, as confirmed by the X-ray crystallographic analysis.156
Synthesis toward a more complicated tetracyclic entity was unveiled by Bechman's team. They demonstrated an efficient route toward C-type bis(diazenes). Their journey began with diketal 572, which was sequentially converted into bicyclo[3.3.1]nonane-diketo-diene 573. The treatment of this precursor with hydrazine then yielded the targeted tetracyclic diazene 574 (Scheme 127).276
 |
| | Scheme 127 Synthesis of twistane-type tricyclic ether and tetracyclic diazene through intramolecular cyclization. | |
Takeuchi and coworkers synthesized propellan-type ring systems from bicyclo[3.3.1]nonane precursors. As described in their communications, the solvolysis of 2-oxobicyclo[3.3.1]non-1-yl triflate (575) could produce propallen 576 as a minor product.277,278 However, other triflate, heptafluorobutyrate, tosylate, and bromo derivatives do not lead to similar propallens (Scheme 128).279
 |
| | Scheme 128 Synthesis of propallen through the solvolysis of 2-oxobicyclo[3.3.1]non-1-yl triflate. | |
4.3. Heterocyclization
Heterocyclic compounds constitute more than half of the entire bioactive molecules. Therefore, the derivatization of newer ring systems into their heterocycle-fused analogues has gained immense importance in recent years; in particular, the fusions of the indole ring system with newer cyclic and acyclic core moieties were investigated extensively by several research groups. Bicyclo[3.3.1]nonanes are also not an exception,280–287 and the remarkable contribution in this regard from Butkus and coworkers is well documented. Their initial reports were on the synthesis of methanocycloocta[b]indoles, which commenced by reacting the racemic dione 577 with phenylhydrazine. The reaction proceeds with an intermediate hydrazone formation to produce the diindolyl product 578.288,289 The synthesis of the corresponding monoindoles is quite difficult and requires a modified approach. Thus, dione 577 was first converted into its monoacetal 579, which on treatment with phenyl hydrazine in the presence of acetic acid yielded the monoindolyl monoketal 580. The desired monoindole 581 was obtained by hydrolyzing 580 (Scheme 129).289
 |
| | Scheme 129 Synthesis of mono and diindolyl products by the reaction of dione and phenylhydrazine. | |
Within a couple of years of this report, the synthesis and absolute configuration determination of a spiro[1,3-benzodioxole-methanocyclooct[b]indole] was demonstrated by the same group. The synthesis of this monoindolyl derivative (585) also required the monoprotection of diketone 582. Thus, the regioselective protection of 582 with catechol (583)290 yielded the protected ketone 585, which on indolization with phenylhydrazine in the presence of HCl (cat. amt.) afforded the chiral 1′H-spiro [1,3-benzodioxole-2,12′-[6′,10′]methanocyclooct [b]indole 585 in high yields (Scheme 130).291
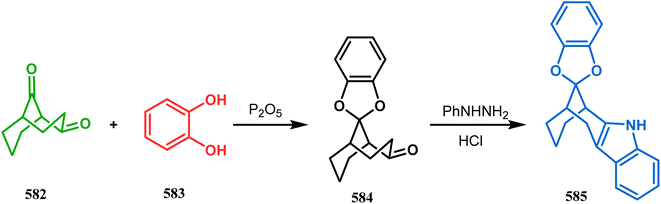 |
| | Scheme 130 Synthesis of chiral 1′H-spiro[1,3-benzodioxole-2,12′-[6′,10′]methanocyclooct [b]indole after the monoprotection of diketone with catechol, followed by indolization with phenylhydrazine in the presence of HCl. | |
A couple of years later, another article from the same team unveiled the synthetic route toward a C2-symmetric methylene-bridged product 588. This time, hydrazine 586 obtained from 4-hydroxy-6-methyl-2(1H)-pyran-2-one292,293 was treated with diketone 577, and the 2,9-diaza-3,10-dimethyl-1,8-dioxo-2,5,6,7,9,12,13,14-octahydro-6,13methanocycloocta[1,2-b:5,6b′] diindole 588 was obtained through the bishydrazone 587 (Scheme 131).
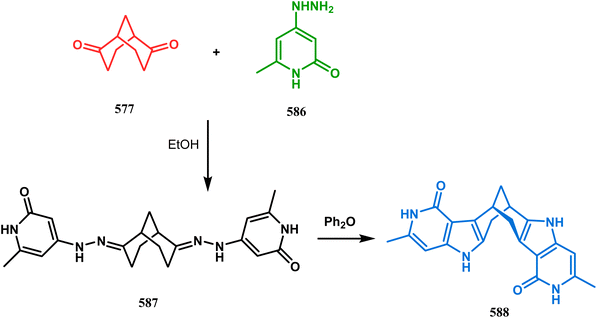 |
| | Scheme 131 Synthesis of 2,9-diaza-3,10-dimethyl-1,8-dioxo2,5,6,7,9,12,13,14-octahydro-6,13-methanocycloocta[1,2-b:5,6b′]diindole. | |
The incorporation of such a self-complementary H-bonding motif 2-pyridone led to the self-assembled aggregation of 588 (Scheme 131),294 similar to the Wallentin's report159 on the self-aggregation of molecular tweezers. A similar approach toward a helical tubular self-aggregated C2-symmetric cleft-shaped molecule was also described by this group recently.295 Furthermore, a closely related report on the synthesis of similar fused heterocyclic ring-containing bicyclo[3.3.1]nonane derivatives was also published recently, where Labanauskas and coworkers efficiently demonstrated the condensation of appropriately substituted bicyclo[3.3.1]nonanones with amino-triazolothione, thiobenzimidazole, thiocarbamide, etc. Thus, when dichloro-dione 589 was condensed with dinucloephiles 590–592, corresponding bicyclo[3.3.1]nonane containing bis-thiazole (593), bis-imidazothiazole (594) and bis-(triazolo-thiadiazine) (595) were obtained in high yields (Scheme 132).
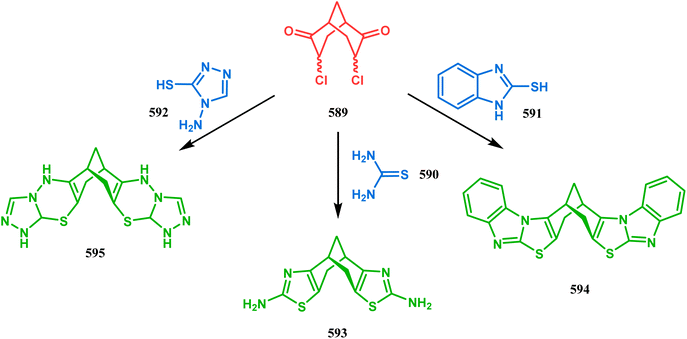 |
| | Scheme 132 Synthesis of bicyclo[3.3.1]nonane containing bis-thiazole, bis-imidazothiazole, and bis-(triazolo-thiadiazine) from dichlorodione upon condensation with dinucloephiles. | |
The corresponding dibromodione however produced the same products (590–592) with 10–20% lower yield, along with some other byproducts. Similar attempts to synthesize single heterocycle-fused bicyclo[3.3.1]nonanes were also successful. Thus, when 3-bromobicyclo[3.3.1]nonane-2-one (596) was refluxed with unchanged dinucleophiles (590–592), the desired singly heterocyclized bicyclo[3.3.1]nonanes (597–599) were obtained in high yields (Scheme 133).296
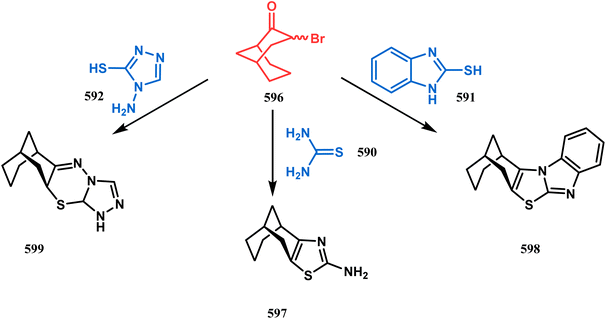 |
| | Scheme 133 Synthesis of heterocycle-fused bicyclo[3.3.1]nonanes by the reaction of 3-bromobicyclo[3.3.1]nonane-2-one with unchanged dinucleophiles under reflux condition. | |
Tacrine–Huperzine A hybrids also constitute an important series of heterocycle-fused bicyclo[3.3.1]nonanes. As described by Camps and coworkers, these entities (605) could easily be synthesized from bicyclo[3.3.1]nonanes (600) by treating them with differently substituted aminobenzonitriles (601).297,298 The reaction proceeds through initial imine formation (602), followed by intramolecular cyclization. Although the regioisomeric enamines (603, 604) could give rise to both the syn and anti aminoquinolines, thermodynamically more stable (due to the anti orientations of endocyclic double bond and heterocyclic ring) enamine 603 undergoes preferential cyclization and yields the anti product 606 (Scheme 134). More interestingly, when enone 607 was treated with aminobenzonitrile 608, instead of forming a new aminoquinoline analogue, a polyfunctionalized adamantane derivative (612) was produced through iminium 610 and adamantyl carbocation 611 (Scheme 135). After their successful syntheses, these Tacrine–Huperzine A hybrids were screened to check their potential for use as acetylcholinesterase inhibitors.297
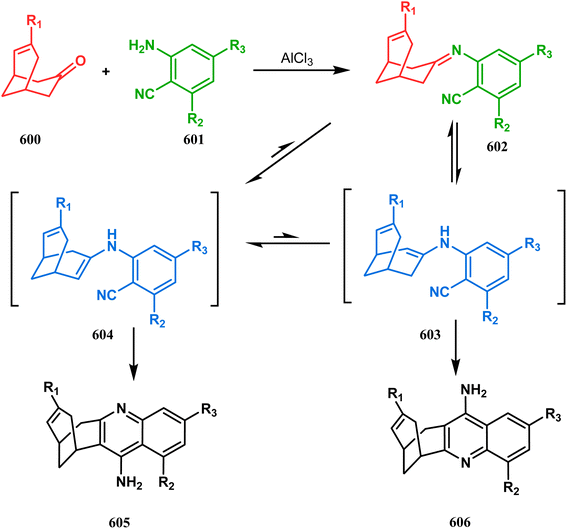 |
| | Scheme 134 Synthesis of heterocycle-fused bicyclo[3.3.1]nonanes from bicyclo[3.3.1]nonanes upon treatment with differently substituted aminobenzonitriles. | |
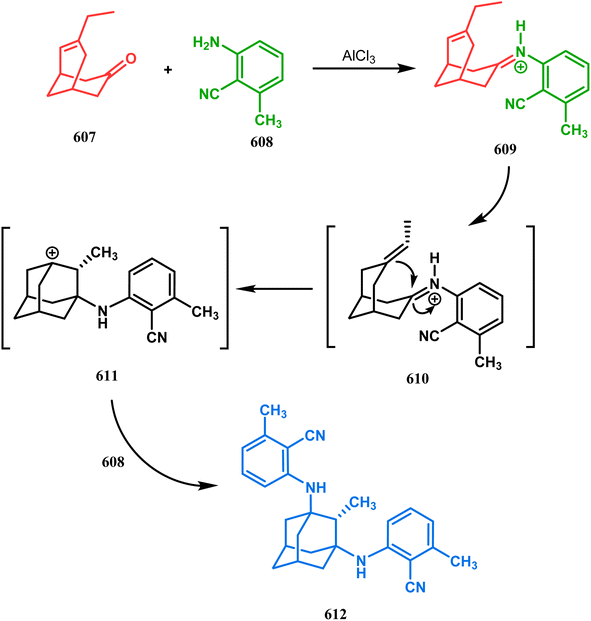 |
| | Scheme 135 Synthesis of polyfunctionalized adamantane derivative from the reaction of bicyclo[3.3.1] nonenone with aminobenzonitrile through iminium and adamantyl carbocation formation. | |
Renard's team also developed some huprine scaffolds to investigate their acetylcholinesterase inhibitory properties. The syntheses of these scaffolds also utilized the same relfluxing procedure as developed by Camps's group. Thus, when different aminobenzonitriles were condensed with similar bicycle[3.3.1]nonenones, a diverse series of heterocycle-fused bicyclo[3.3.1]nonanes were produced, and their AChE inhibitory property was evaluated.299 Within a few months, in another communication, the same group disclosed a modified synthetic approach toward these heterocycle-fused bicyclo[3.3.1]nonanes. This time, oxaadamantane mesylate derivatives (613) were subjected to a one-pot fragmentation–cyclization reaction with aminobenzonitriles (614) under refluxing condition to produce the targeted heterocycle-fused bicyclo[3.3.1]nonanes (615) (Scheme 136).300
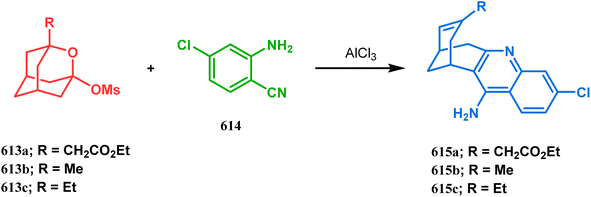 |
| | Scheme 136 Synthesis of heterocycle-fused bicyclo[3.3.1]nonanes from oxaadamantane mesylate derivatives in one-pot fragmentation–cyclization reaction with aminobenzonitriles under refluxing condition. | |
Warnmark and coworkers described an interesting synthetic route toward tweezer 618 to investigate its self-aggregation properties. Initially, diketone 616 was sequentially converted into quinoline-fused amine 617, which upon Pd/C-mediated hydrogenation reaction yielded the desired tweezer 618 (Scheme 137).159
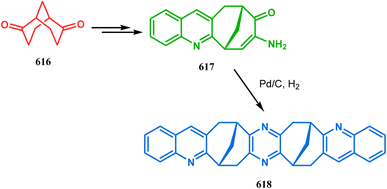 |
| | Scheme 137 Synthesis of tweezer through the sequential conversion of quinoline-fused amine from a diketone, followed by Pd/C-mediated hydrogenation. | |
4.4. Functionalization of bicyclo[3.3.1]nonanes
4.4.1. Reaction with carbonyl groups. The functionalization of bicyclo[3.3.1]nonanone into diverse series of its analogues through the reaction on the ketone functionalities has been thoroughly investigated in the last few decades. Few years ago, during the investigations on the structural features of estrogen receptor, Pike and coworkers found that estrogen receptors contain an unpopulated room inside the binding pocket of the ligand,301 which prompted several research groups to synthesize newer classes of estrogen receptor antagonists. Appropriately substituted bicyclo[3.3.1]nonanes also found their application in this area and are generally synthesized by the McMurry coupling reaction. For example, alkenes 621 were constructed in this manner from bicyclic ketone 619 and another ketone 620 in 54–82% yield (Scheme 138).302–305
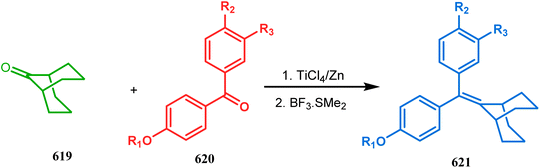 |
| | Scheme 138 Formation of substituted alkenes of bicyclo[3.3.1]nonane from the reaction of bicyclic ketone and any other ketone by McMurry coupling reaction. | |
A similar alkene 623 was synthesized by the Suzuki coupling reaction of 622, which was obtained from ketone 619 with a yield of 61% (Scheme 139). The corresponding saturated analogue (624) could also be obtained from 619 by treating it with phenol in n-BuSH (cat.)/HCl in 67% yield (Scheme 140).304,306 The synthesis of similar entities (626) from ketone 619 was also accomplished by the condensation of Grignard compounds (625) with the carbonyl group, and the yield was 84%. The diketones 627 and 629 could also be exposed to an identical reaction condition to produce corresponding diols 628 (yield 78%) and 630 (yield 83–87%) (Scheme 141).307–309
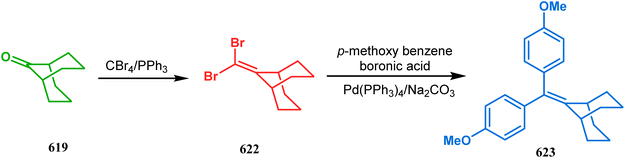 |
| | Scheme 139 Synthesis of substituted alkenes of bicyclo[3.3.1]nonane by the Suzuki coupling reaction. | |
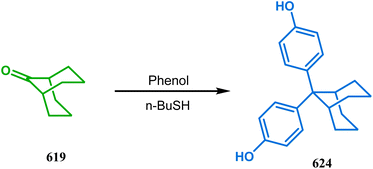 |
| | Scheme 140 Synthesis of saturated analogue of bicyclo[3.3.1]nonane by treating bicyclo[3.3.1]nonanone with phenol in n-BuSH(cat.)/HCl. | |
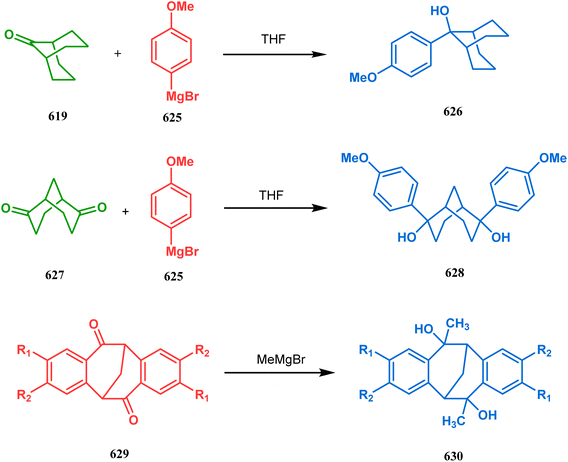 |
| | Scheme 141 Synthesis of various saturated analogues of bicyclo[3.3.1]nonane by the condensation of Grignard compounds with the carbonyl group. | |
Another type of condensation reaction with the carbonyl group involves the olefination of 631. Selective Wittig reaction in one of the carbonyl groups of 631 produced the olefin 632 exclusively at 230 °C under microwave irradiation. However, the same reaction at lower temperature (190 °C) yielded the inseparable mixture (E and Z) of the exocyclic product 633 (Scheme 142).310 Similar syn and anti isomers of bicyclo[3.3.1]nonane-based dioximes were also synthesized and reported in the last century by several groups.311,312
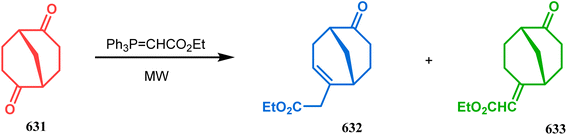 |
| | Scheme 142 Olefination of dione via selective Wittig reaction. | |
Their strategy involved the simple condensation of diketone 634 with hydroxyl amines (635 and 637) to give dioximes 637 and 638 (Scheme 143).311 However, 637 could also be obtained by DMDO-oxidation (dimethyldioxirane) of diamine 639 (Scheme 144).312,313 Grosu and coworkers studied the capability of these entities to form supramolecular aggregates and from their single crystal X-ray crystallographic studies, it was found that the syn isomer of 637 forms a supramolecular wheel (via six hydrogen bondings), and the corresponding anti isomer leads to a cyclic dimer via four hydrogen bonds.314
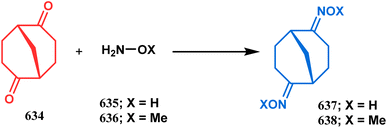 |
| | Scheme 143 Synthesis of bicyclo[3.3.1]nonane-based dioximes through the condensation of diketone with hydroxylamine. | |
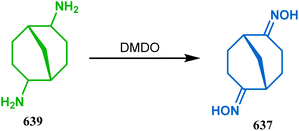 |
| | Scheme 144 Synthesis of bicyclo[3.3.1]nonane-based dioximes from diamine by DMDO oxidation. | |
During the synthesis of novel chiral diene ligands, Hayashi and coworkers described the synthesis of 643, a chiral ligand based on the bicyclo[3.3.1]nonane framework. Starting from dione 640, a couple of phenylation reactions produced the diol 641, which yielded the diene 643 on dehydration in 76–97% yield (Scheme 145a).
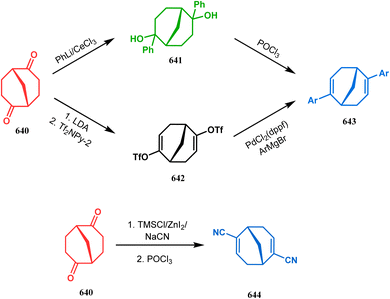 |
| | Scheme 145 (a) Synthesis of bicyclo[3.3.1]nonane-based chiral diene ligands (b) synthesis of dicyanobicyclo[3.3.1]nonane. | |
An alternative route toward this goal proceeded through triflate 642, followed by Pd-catalyzed arylation. This diene was then successfully employed as a chiral ligand in the Suzuki reaction and the addition of aryl/alkenyl boronic acids in α,β-unsaturated ketones.315–317 To synthesize differently functionalized bicyclo[3.3.1]nonane frameworks, the dione 640 is widely employed. For example, Quast and coworkers described a careful cyanation on dione 640 to achieve the dicyanobicyclo[3.3.1]nonane 644 (Scheme 145b).318
Warnmark's group studied the crystal structure of 644 and found the presence of weak hydrogen bonding among the bicyclic units, thereby forming a 2D net-like crystal structure.319 Dione 640 is also used to synthesize its bis-enone (645) and mono-enone analogues (648), as reported by Butkus and coworkers (Scheme 146).320
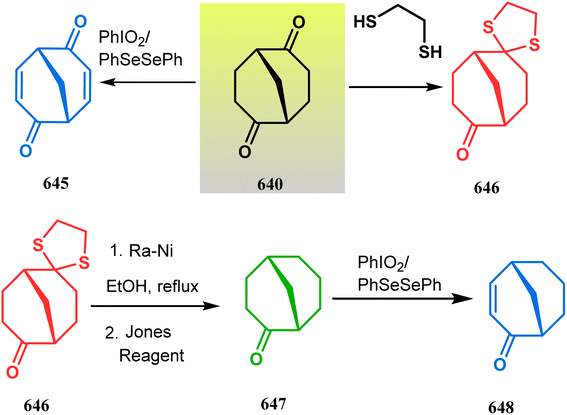 |
| | Scheme 146 Synthesis of mono-enone and bis-enone analogues of bicyclo[3.3.1]nonane. | |
4.4.2. Substitution at the α-position of the carbonyl group. Substitution at the bridgehead position of bridged ketones is always a difficult process as it disobeys Bredt's rule. However, Simpkins and coworkers utilized some efficient lithium amide bases (655a,b) to facilitate metallation at the bridgehead position. During the synthesis of the bicyclic core of garsubellin A, they successfully utilized a rigioselective lithiation procedure to achieve 650 from 649 using LDA as the base (Scheme 147).321
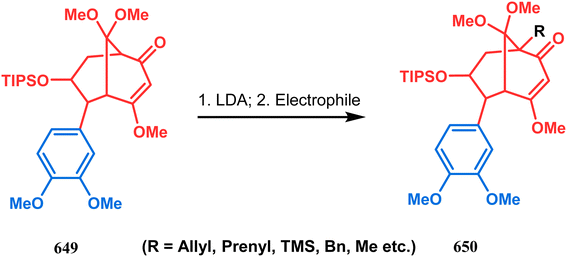 |
| | Scheme 147 Metallation at the bridgehead position during the synthesis of the bicyclic core of garsubellin A. | |
A similar LDA-promoted substitution reaction on analogous bicyclic systems demonstrated different results. It showed that although LDA-mediated TMS-substitution of bicycles 651 produces the corresponding bridgehead substituted products (652) in good yields (Scheme 148a), the same reaction with bicyclo[3.3.1]nonane 653 gives low product yield. Thus, amide bases 655a,b were employed to produce product 654 in high yields (Scheme 148b).322
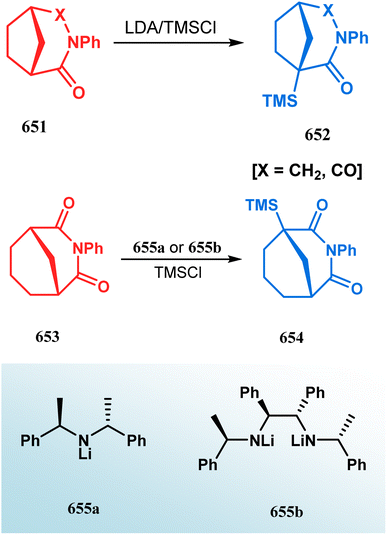 |
| | Scheme 148 (a) Synthesis of bridgehead-substituted product (b) synthesis of bridgehead-substituted product. | |
4.4.3. Amide formation. Amide formation from the amines of bicyclo[3.3.1]nonanes is also well documented in the literature. For example, to study the structural and conformational properties, some amides based on bicyclo[3.3.1]nonane framework were described by Galvez and coworkers. The required amine (657) for this purpose was constructed by reducing the corresponding oxime (656) using lithium aluminium hydride. Once the amine is formed, corresponding amides (658–660) were synthesized using a diverse range of acyl chlorides in DCM (Scheme 149).323
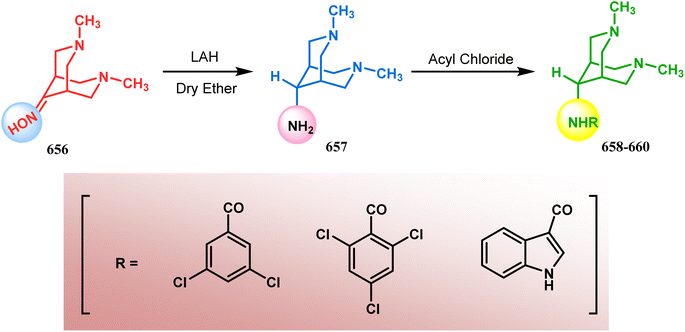 |
| | Scheme 149 Formation of amides from the amines of bicyclo[3.3.1]nonanes. | |
Similar amide formation reactions were also reported by Dutta and coworkers. They first synthesized the optically active amine (661) following their earlier reported procedure,324 which was then separately treated with differently substituted 2,3-epoxypropyl entities (662–664), 3-chloro-4′-fluoro-propiophenone (665), and the corresponding amides (666–669) were isolated in good yields (Scheme 150). These amide linkages were then further modified, and their interaction with dopamine, serotonin, and norepinephrine transporters was investigated.
 |
| | Scheme 150 Synthesis of amides from the reactions of optically-active amine with differently substituted 2,3-epoxypropyl entities and 3-chloro-4′-fluoro-propiophenone. | |
Another report on bicyclo[3.3.1]nonane-based amide formation reaction was published by Gmeiner and coworkers. While searching for selective 5-HT1A superagonists, they synthesized some novel bicyclo[3.3.1]nonane-based pyridylmethylamines (673, 674) from 670 (Scheme 151). The synthesis involves an initial boc-deprotection, followed by amide bond formation with benzoyl chloride to give amide 672, which was then sequentially converted into targeted pyridylmethylamines (673, 674).325 Apart from these amide formation reaction of bicyclo[3.3.1]nonane-amines, the ester formation of the corresponding carboxylic acids is also well documented. Kiryukhin and coworkers described one such example, where carboxylic acid 675 was reacted with 3-hydroxyoxetane 676 and formed the ester 677 (Scheme 152).326
 |
| | Scheme 151 Synthesis of bicyclo[3.3.1]nonane-based pyridylmethylamines. | |
 |
| | Scheme 152 Synthesis of bicyclo[3.3.1]nonane-based ester. | |
4.4.4. Metal complexation. The application of appropriately substituted bicyclo[3.3.1]nonane derivatives for the complexation reaction with rhodium, titanium, and zirconium chlorides or acetates is also reported by several research groups. Brown and coworkers described one such unique example a few years ago. They employed the enantiopure diene 679, obtained from diketone 678, to complex it with the diethylene complex of rhodium acetate (680) and yielded a new rhodium complex (681). Further exposure of 681 with diene 679 for another one hour in TMSOTf produced the sandwiched complex 682. A similar reaction of 679 with the dirhodium complex 683 also gave another complex 684 (Scheme 153).327
 |
| | Scheme 153 Synthesis of rhodium complex. | |
The synthesis of a similar titanium complex was also reported in the literature by Grossman and coworkers. Starting from bicyclo[3.3.1]nonane-2,6-dione (678), this C2-symmetric, chiral ansa–titanocene complex (685) was synthesized in four steps (Scheme 154).328 Halterman and coworkers also attempted the synthesis of similar titanium and zirconium complexes. Their journey also began from dione 678, and the cleft-shaped bis(indene) 686 was constructed for its metallation reaction. Although the crude product obtained from the reaction of 686 with TiCl3 or ZrCl4 showed new signals in the 1HNMR spectrum, the complexes (687, 688) could not be isolated (Scheme 155).
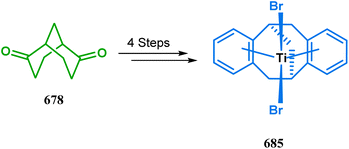 |
| | Scheme 154 Synthesis of titanium complex. | |
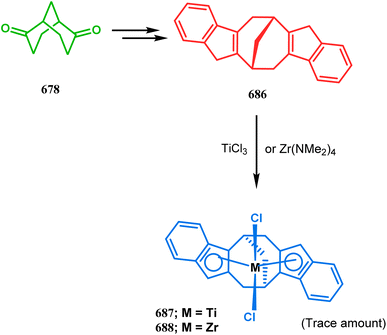 |
| | Scheme 155 Synthesis of titanium and zirconium complex. | |
4.4.5. Other reactions.
4.4.5.1. Aldol condensation. The aldol condensation of bicyclo[3.3.1]nonanones is also reported in the literature. In one such example, Moiseev and coworkers employed KOH to condense dione 689 with aldehydes 690, which yielded the α,β-unsaturated diene-dione 691 in high yields (Scheme 156).329 Interestingly, no monocondensation product was formed during the reaction. Even if the reaction was carried out employing 689 and 690 in 1:1 mole ratio, only the double-condensation product was produced.
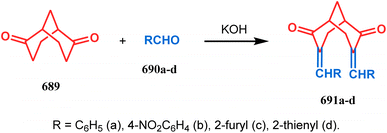 |
| | Scheme 156 Synthesis of bicyclo[3.3.1]nonane-based α,β-unsaturated diene-dione via aldol condensation. | |
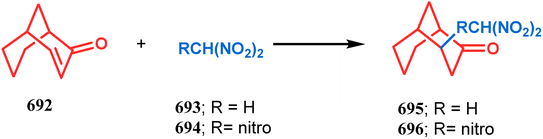 |
| | Scheme 157 Synthesis of bicyclo[3.3.1]nonane-containing nitroalkanes. | |
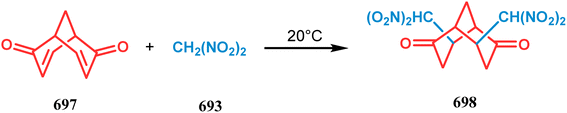 |
| | Scheme 158 Synthesis of bicyclo[3.3.1]nonane-containing nitroalkanes. | |
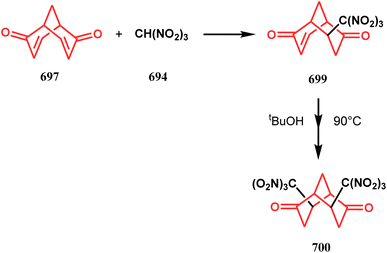 |
| | Scheme 159 Synthesis of bicyclo[3.3.1]nonane-containing nitroalkanes from the reaction of diene-dione with nitroform. | |
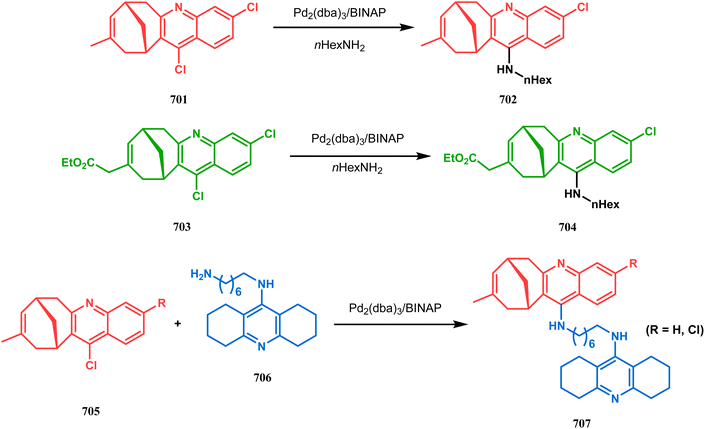 |
| | Scheme 160 Synthesis of aminoquinolines by palladium-catalyzed amination reactions of bicyclo[3.3.1]nonane. | |
Another amination reaction in bicyclo[3.3.1]nonane series was described by Finn and coworkers. They demonstrated the amination of thia-, aza-, and selena-bicyclo[3.3.1]nonane dichlorides (708–711), which proceeds through an internal anchimeric assistance from the ring nitrogen, sulphur, or selenium center. Thus, when dichlorides 708–711 were reacted with benzylamine, bis(amino)-bicyclo[3.3.1]nonanes 716–719 were obtained through a cationic intermediate (712–715) in high yields. A similar reaction with other nuclophiles also proceeds in a similar pathway to produce corresponding substitution products (Scheme 161).163
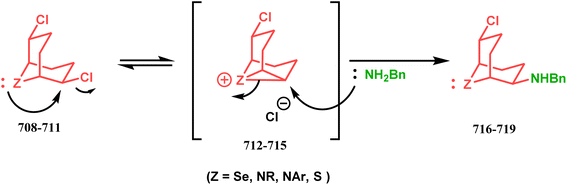 |
| | Scheme 161 Synthesis of bis(amino)-bicyclo[3.3.1]nonanes. | |
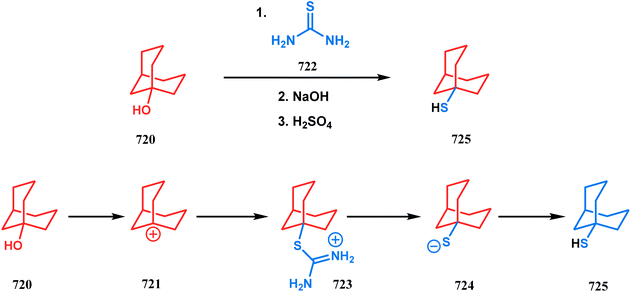 |
| | Scheme 162 Synthesis of bicyclo[3.3.1]nonane-based thiol by the thiourea-mediated thiolation of bicyclo[3.3.1]nonalol. | |
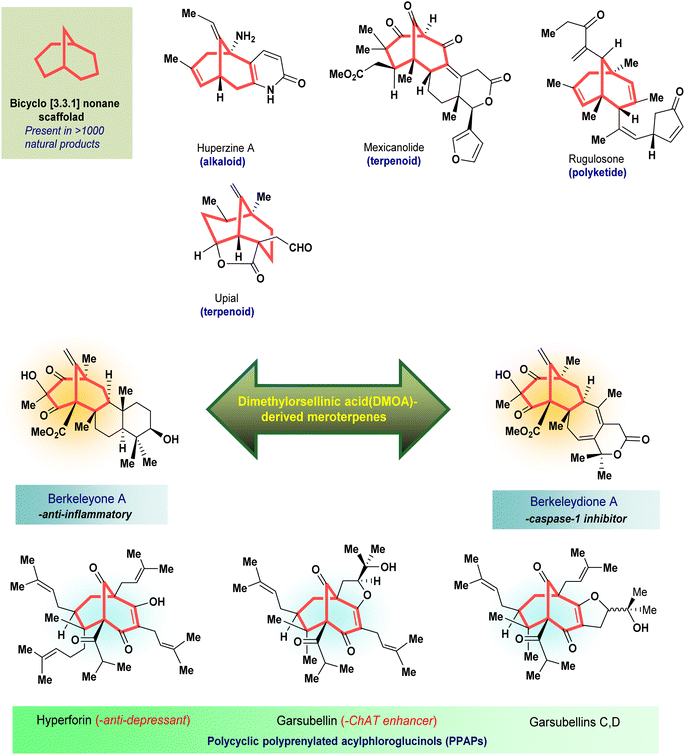 |
| | Fig. 1 A family of meroterpenes as an abundant source of bicyclo[3.3.1]nonane. | |
Hyperforin was the first bicyclic polyprenylated acylphloroglucinol (BPAP) that was isolated in 1971 from a medicinal plant named St. John's wort to treat a plethora of diseases in ancient Greece. As hyperforin was highly admired due to its noticeable antidepressant activity, scientists focused their attention into the discovery and isolation of various natural BPAPs from the plants of Clusiaceae and Hypericaceae family, where all the natural BPAPs are seen to be very efficient to display various biological activities. Keleyone A and berkeleydione are two such crucial derivatives of DMOA that exhibit antiinflammatory and caspase-1 inhibitory potency, respectively. The neuroactive hyperforin and garsubellins are two important polycyclic polyprenylated acylphoroglucinol (PPAs) possessing the bicyclo[3.3.1]nonane scaffold. In practice, PPAs are very competent to bring on different biological activities and are commonly classified as a highly oxygenated acylphloroglucinol with isoprenyl or geranyl side chains. The biosynthesis of BPAPs is accomplished by condensing three malonyl-CoA units and one acyl-CoA unit to construct the polyketide moiety, which is subsequently converted into acylphloroglucinol through Dieckmann cyclization. After that, the prenylation of this moiety produces monocyclic polyprenylated acylphloroglucinols (MPAPs), which in turn undergoes several cyclizations to form the varied frameworks of BPAPs and their analogues. Inspite of having astounding anticancer, antidepressant, antiinflammatory, antioxidant, and antimicrobial activities of all the BPAPs, scientists are facing challenges to find out lead compounds among all the BPAPs. The most important and extensively studied biological function of BPAPs is its incredible anticancer activity. The most promising anticancer activity is observed for hyperfoin, nemorosone, guttiferones, oblongifolin C, clusianone, and garcinol. Most of these BPAPs have been found to reveal their anticancer potential through the induction of cancer cell death or by the inhibition of cancer cell survival, adhesion, proliferation, invasion, angiogenesis, as well as metastasis, triggering some signaling pathways.335
It has been studied that hyperforin conveys its anticancer activities mostly toward leukaemia, colorectal carcinoma, nonsmall cell lung cancer, and muscle-invasive bladder cancer as a result of provoking mitochondrial intrinsic/extrinsic pathways through the release of cytochrome C and the reduction of mitochondrial membrane potential (MMP). Hence, it is very much efficient to endorse the upregulation of proapoptotic proteins (Bak, Bad, procaspase-3/9) and the suppression of antiapoptotic proteins such as Mcl-1, XIAP, and C-FLIP. Hyperforin is best suited for the treatment of acute myeloid leukaemia (AML) and chronic lymphoid leukaemia (CLL), triggering apoptosis through the modulation of the PI3K/Akt signaling pathway, where it destroys the kinase activity of serine/threonine protein kinase B (PKB)/Akt1 and thereby encourages the activation of proapoptotic Bad and procaspases-9/-3. In case of CCL, hyperfoin is seen to upregulate Noxa, which in turn stimulates the emergence of proapototic Bak protein from Mcl-1 and thus helps the apoptogenic factors to be released from the mitochondria, leading to cell death. In line with this, hyperforin downregulates the NF-κβ P65 and thereby restrains the expression of antiapoptotic and tumor growth proteins. Also, it facilitates the cell cycle arrest at the G1 phase by hindering the formation of cyclin D1. Moreover, hyperforin is adept at inducing apoptosis, creating endoplasmic reticulum (ER) stress and damaging DNA through the increase in ROS level and calcium signaling in the cytoplasm of cancer cells. It has been detected that vascular endothelial growth factor (VEGF) and matrix metalloproteinases-9 (MMP-9) protein level are suppressed on the treatment of hypoforin along with the suppression of P-glycoprotein expression, which indicates the high anticancer proficiency of hyperforin. It can be mentioned that hyperforin can also be ascribed to have antiangiogenic, antigenotoxic, and anticlastogenic properties (Fig. 2).336
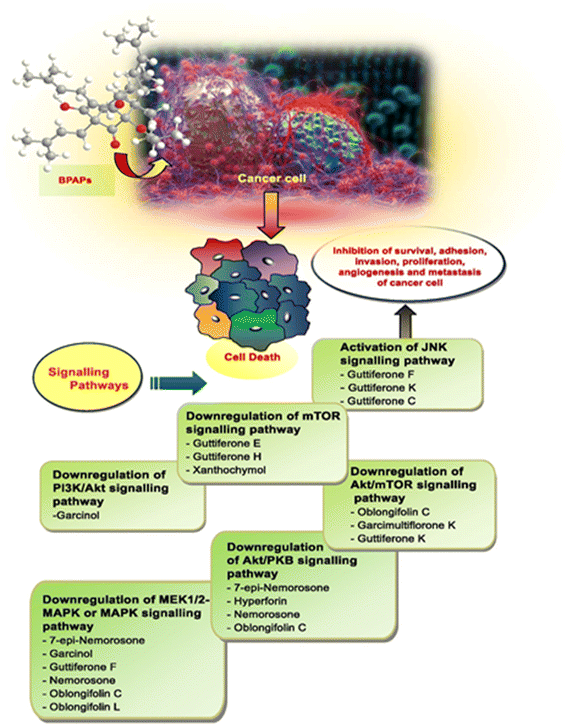 |
| | Fig. 2 Different anticancer signaling pathways induced by various BPAPs. | |
Nemorosone is also a potent anticancer compound containing the bicylo[3.3.1]nonane moiety. It is isolated from the flora resin of Clusia rosea plant (Fig. 3). This naturally obtained compound has been found to induce apoptosis by activating the unfolded protein response (UPR) in case of pancreas cancer. Nemorosone is seen to be very active to resist the progression of cell cycle in leukaemia cells after targeting the Akt/PKB signal transducer in association with diminishing the formation of cyclins A, B1, D1, E, as well as c-Myb levels. It is also very potent against neuroblastoma cells by triggering the activity of caspase-3 and then obstructing the kinase activity of ERK1/2 or regulating the Akt/PKB signaling pathway. To prevent oestrogen receptor alpha positive (ERα+) breast cancer, nemorosone may successfully be used as an adjuvant owing to its inhibitory action toward 17-β-estradiol (E2) without any genotoxicity. The mechanistic study reveals that nemorosone can destroy the ERα+ cells by arresting the progression of cell cycle at the G0/G1 phase by modifying the expression of Akt, ERK1/2, and other genes associated with cell cycle, apoptosis, or hormone receptor. Moreover, nemorosone can act as a chemosensitizer toward the doxorubicin-resistant colon carcinoma cells (LoVo Dox) and can bring on apoptosis through cell cycle arrest with the production of profuse ROS and changing of inner mitochondrial membrane potential (MMP).337 The proliferation and then metastasis of human colorectal carcinoma (CRC) cells were found to be inhibited on the treatment of nemorosone as it is capable of diminishing the epithelial–mesenchymal transition (EMT)-related markers in CRC cells.338 7-epi-nemorosone, an epimer of nemorosone is also efficient to cause the apoptosis of cancer cells. It has been studied that 7-epi-nemorosone can suppress the expression of androgen receptor (AR) and the production of prostate-specific antigen (PSA) (Fig. 3).
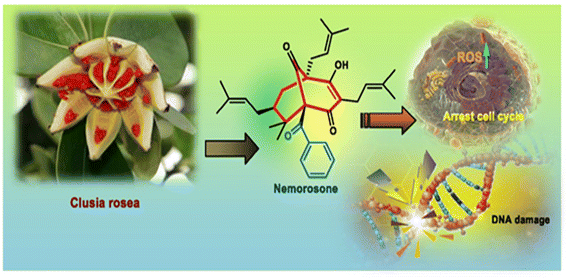 |
| | Fig. 3 Nemorosone isolated from Clusia rosea and its anticancer activity. | |
Therefore, it can alter the MEK1/2 as well as Akt/PKB transducers in androgen-dependent prostate carcinoma cells (LNCaP) along with the downregulation of cyclins D1/D3 and cyclin-dependent kinase (CDK) 4/6. The anticancer potential of nemorosone against two human colorectal cancer cell lines, HT-29 and LoVo, was tested. The corresponding IC50 values have been provided in Table 1.
Table 1 IC50 values for treating nemorosone against HT-29 and LoVo cells
| IC50 (μM) |
| HT-29 |
LoVo |
| 24 h |
57.1 ± 3.7 |
64.3 ± 4.7 |
| 48 h |
33.4 ± 2.8 |
35.9 ± 9.1 |
| 72 h |
25.7 ± 3.3 |
22.8 ± 6.2 |
The potential anticancer compound, guttiferone A, which possesses the bicyclo[3.3.1]nonane core, was first isolated from the root of Symphonia globulifera and recently isolated from Garcinia livingstonei and Garcinia macrophylla. The remarkable anticancer property of this compound was attracted by the researchers; hence, they were eager to study its mechanisms of action in cancer cells. It has been observed that this compound can act as an inhibitor of serine and cysteine proteases and accelerates the production of ROS, which leads to a decrease in mitochondrial membrane potential (MMP) in MCF-7 breast cancer cells.339 Consequently, proapoptotic Bax protein becomes upregulated, whereas antiapoptotic Bcl-2 becomes downregulated on the treatment of guttiferone A. It also conveys cytotoxity toward hepatocellular carcinoma. Its anticancer activity was accounted for ATP depletion, NADPH depletion, decrease in MMP, uncoupling of membrane, Ca2+ efflux, cyclosporine A/EGTA-insensitive membrane permeabilization, and ROS accumulation.340 The IC50 value was found to be 15 μM against the MCF-7 breast cancer cell lines upon treatment of guttiferone A in a dose-dependant manner (Fig. 4).
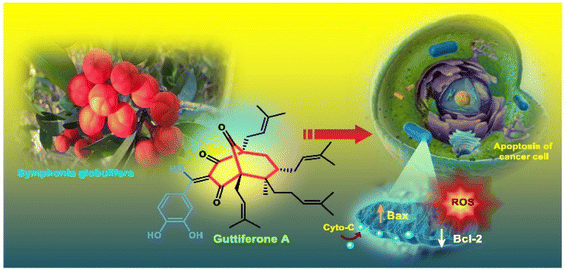 |
| | Fig. 4 Guttiferone A isolated from Symphonia globulifera and its anticancer activity. | |
Garcinol is also an important polyisoprenylated benzophenone containing the bicyclo[3.3.1]nonane moiety; this phytochemical can be extracted from the rind of Garcinia indica fruit, widely renowned as Kokum or Mangosteen as well as from the leaves of this plant, a medicinal plant profusely found in tropical regions. Among all the chemical components obtained from this fruit extract including citric acid, oxalic acid, hydroxycitric acid (HCA), and hydroxycitric acid lactone, the benzophenone derivatives such as garcinol and its isomer isogarcinol are very important in context to their diverse biological activities (Fig. 5). The anticarcinogenic capabilities of garcinol seem to be evolved due to its antiinflammatory, antioxidative, antiangiogenic, and proapoptotic activities, rendering remarkable epigenetic influences through the inhibition of histone acetyltransferases (HATs) enzymes and posttranscriptional deregulation in the expression of miRNA profiles accountable to carcinogenesis.341 In light of the in vitro and in vivo studies, it has been revealed that the antineoplastic efficacies of garcinol is manifested in consequence of inhibition of various cellular incidents with the regulation of transcription factors JAK/STAT3 and NF-κB in tumor cells, thereby seizing the rapid escalation of malignant cells. Therefore, the potential anticancer aptitude of garcinol has recently been reflected in different oncological reformations in breast cancer, colon cancer, hepatocellular carcinoma, leukemia, prostate cancer, pancreatic cancer, head and neck cancer, etc., and this behavior of garcinol has drawn it toward preclinical trials. Although a clean perception of its mechanism of action is still a matter of profound research, it has been notified that garcinol is capable of suppressing tumorigenesis by diminishing the expression of COX2, iNOS, NF-κB, and STAT3 function along with the inhibition of histone deacetylase 11 (HDAC11) among all other HDACs. Also, it can restrain the function of 5-lypoxygenase and microsomal prostaglandin E2 synthase 1(mPGES-1).342
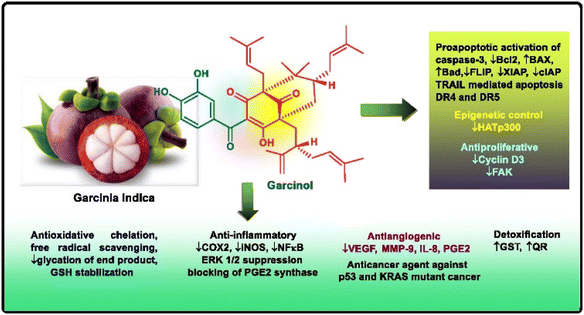 |
| | Fig. 5 Different mechanistic targets for the execution of anticancer activities by garcinol or camboginol. | |
Esophageal cancer is also a very serious type of cancer and common cause of death due to its poor prognosis as well as irregular geographical distribution. The two main histological types of esophageal cancers are (1) esophageal adinocarcinoma and (2) esophageal squamous cell carcinoma (ESCC). The ESCC type of esophageal cancer is seen to pervade mostly in Eastern Asia and Africa. The statistical report from United States, China, and Europe reveals that less than 21% of esophageal cancer-affected people can survive upto 5 years. Cancer cell metastasis is assumed to be the prime cause of death in esophageal cancer. Garcinol has been seen to be very active to hold back the metastasis and is thus potent toward esophageal cancer.343
As garcinol is a histone acetyltransferase (HAT) inhibitor, it is very much efficient to bring on cell cycle arrest and apoptosis in the cancer cell (Fig. 6). Besides, garcinol is also very potent to resist angiogenesis and metastasis, two important hallmarks of cancer by modulating several proinflammatory signaling pathways, which leads to the inhibition of angiogenesis in malignant cells. Upon the intraperetoneal administration of garcinol in xenograft mice model, Li et al. observed that garcinol can deregulate the activity of oncogenic transcription factors such as STAT3/NF-κB in a dose as well as time-dependant manner in case of head and neck carcinoma (HNSCC). On the other hand, the inactivation of STAT3/NF-κB can pin down the overexpression of several kinases such as janus kinase 1/2 (JAK1/2), TGF-β-activated kinase 1 (TAK1), c-Src, and inhibitor of Iκβ kinase (IKK) in HNSCC cells and garcinol was successfully reported to hinder the growth in an athymic nu/nu mice. Therefore, a number of studies have been accomplished with the potency of garcinol against different types of cancers (Table 2).
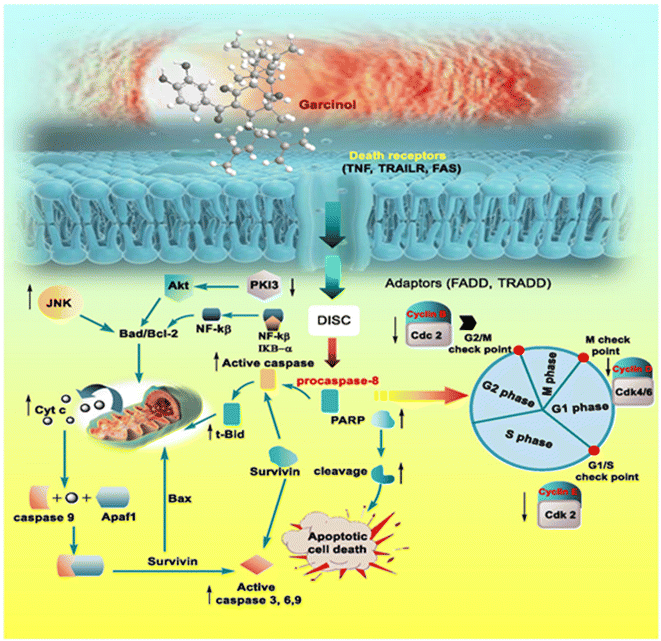 |
| | Fig. 6 Schematic representation of the mechanism of action of garcinol (↑ upregulation and ↓ downregulation) on treatment against cancer cells. | |
Table 2 Anticancer effects of garcinol observed in in vitro studies
| Types of cancer |
Effects |
Garcinol concentration |
| Melanoma, glioblastoma, cervical cancer, breast cancer, leukemia, lung cancer, hepatocellular carcinoma, pancreatic cancer, colon cancer, prostate cancer |
Increased apoptosis |
2.5–50 μM |
| Melanoma, hepatocellular carcinoma, leukemia, colon cancer, pancreatic cancer |
↑Caspase-3, ↑caspase-9 |
0–50 μM |
| Breast cancer, hepatocellular carcinoma, lung cancer |
Cell cycle arrest, ↓cyclinsB, D1, D3, and E |
0–50 μM, 500 ppm |
| Melanoma, glioblastoma, breast cancer, hepatocellular carcinoma, colon cancer |
↑Bax, ↑Bad, Bcl-2, Bcl-xl |
0–50 μM |
| Breast cancer, oral squamous cell carcinoma, prostate cancer, pancreatic cancer |
↓NF-κB signaling pathway |
0–50 μM |
| Breast cancer, gallbladder cancer, pancreatic cancer, prostate cancer, colon cancer |
↓MMP2, ↓MMP9 |
0–30 μM |
| Breast cancer, pancreatic cancer, prostate cancer, hepatocellular carcinoma |
p-STAT3 and STAT3 signaling pathway |
0–50 μM |
| Oral squamous cell carcinoma, breast cancer, hepatocellular carcinoma, colon cancer, prostate cancer, pancreatic cancer |
↓VEGF |
0–25 μM |
| Hepatocellular carcinoma, prostate cancer, pancreatic cancer |
↓IL-6 |
0–25 μM |
| Glioblastoma, lung cancer, breast cancer, pancreatic cancer |
↑mi RNA |
0–40 μM |
| Esophageal cancer, breast cancer |
HAT inhibition |
0–50 μM |
Isogarcinol or cambogin, which is known to be capable of bringing about astounding anticancer activity, also belongs to polycyclic polyprenylated acylphoroglucinol (PPAs) group having bicyclo[3.3.1]nonane as a core moiety (Fig. 7). This valuable compound can also be extracted from Garcinia genus, which was traditionally used for treating cancer throughout Southern Asia. Cambogin was found to have remarkable anticancer activity due to its impressive proapoptotic effects on medulloblastoma as well as breast cancer cells. Among all the types of cancer, breast cancer is now assumed to be the most serious type of cancer in women, which is mainly seen to be come out due to some risk factors such as obesity, hormone replacement therapy during menopause, consumption of alcohol, and ionizing radiation. Recently, it is unfortunate that the multifarious growth of breast tumor and bone metastasis can not be restrained by simple mastectomy and chemotherapy. Therefore, the deterrence and healing of breast cancer necessitates the urgent development of novel therapeutic approaches with the use of effective chemotherapeutic agents. Xu et al. isolated a number of polycyclic polyprenylated acylphoroglucinol (PPAs) from Garcinia genus and assessed their effect toward the proliferation of breast cancer cells.344 However, cambogin is the best among the tested compounds that exhibits notable inhibitory potential toward the rapid growth of breast cancer cells. After the treatment of cambogin against a number of cancer cell lines such as HeLa (human cervical carcinoma), HepG2 (human hepatic carcinoma), A549 (human lung carcinoma), HCT116 (human colon carcinoma), SK-BR-3 (ER− PR− HER2+), MCF-7(ER+PR+HER2−), and triple negative breast cancer (TNBC) cell line, MDA-MB-468 (ER− PR− HER2−), it was identified that cambogin was much more adept in showing the best cytotoxicity toward breast cancer cell lines, whereas cambogin was reluctant toward normal HMEC-1 cells at a similar dosage.345
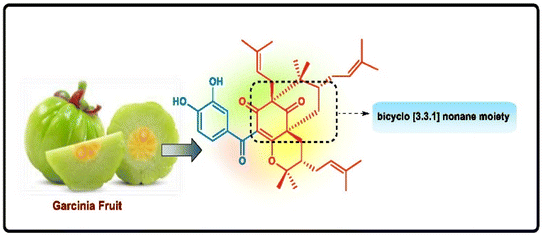 |
| | Fig. 7 The chemical structure of cambogin (isogarcinol) extracted from garcinia fruit having bicyclo[3.3.1]nonane core moiety. | |
It was also apparent that MCF-7 cells were very much susceptible to cambogin treatment in a time- and dose-dependant manner. In addition, MDA-MB-468 and SK-BR-3 cells were also compelled not to be proliferated under the time- and dose-dependant treatment of cambogin.
Oblongifolin C is another naturally-obtained biologically active compound with the bicyclo[3.3.1]nonane core moiety. It is mainly extracted from Garcinia yunnanensis Hu, and this compound has been observed to be very competent to exhibit promising anticancer activity in vitro as well as in vivo (Fig. 8). It is well known to inhibit human silent information regulator 1 and 2 (SIRT1 and SIRT2), where SIRT1 is much more susceptible to oblongifolin C than SIRT2. It is efficient to restrict the proliferation of cancer cells, thereby causing cellular apoptosis through the activation of proapoptotic Bax protein along with mitochondrial dysfunction. Oblongifolin C has been seen to increase the sensitivity of gemcitabine-resistant pancreatic cancer cells by suppressing the Src, ERK/MAPK, Akt pathways and regulating the action of the proteins that are responsible for cell cycle progression.
 |
| | Fig. 8 The chemical structure of cambogin oblongifolin C extracted from Garcinia yunnanensis Hu having bicyclo[3.3.1]nonane core moiety. | |
In human colorectal cells, oblongifolin C, along with guttiferone K, can cause apoptosis by enhancing the profuse production of ROS and increase the phosphorylation of the JNK protein. Moreover, oblongifolin C can create ER stress, which may lead to the destruction of cancer cells by triggering the transcription factor CHOP and activating the JNK kinases. Oblongifolin C is a very powerful anticancer agent as it causes DNA damage by rupturing the DNA double-strand and inhibits the repair mechanism to mend the damage of DNA. It is very efficient to restrain the metastasis of cancer through keratin 18/tubulin, MEK/ERK, and Akt/mTOR signaling pathways. Also, it can inhibit the autophagic flux by obstructing the autophagosome-lysosome fusion and by changing the lysosomal proteolytic activity (Fig. 9).346
 |
| | Fig. 9 Mechanism of anticancer action induced by oblongifolin C. | |
Guttiferone k is an anticancer natural compound that is also obtained from Garcinia yunnanensis Hu having the bicyclo[3.3.1]nonane core moiety. It accelerates c-MYC protein degradation and stabilizes the FBXW7 protein levels. Consequently, it exerts anticancer activity through the obstruction of cell cycle re-entry in quiescent cancer cells. In line with this, it can trigger Akt-mTOR-mediated autophagy. It is seen to be very proficient to hinder the metastasis in human hepatocellular carcinoma in vitro as well as in vivo through the modulation of actin binding protein profiling 1 (PFN 1).347
Cluisianone and its derivative, which is isolated from Garcinia parvifolia, can produce excellent cancer annihilation property that induces mitochondrial dysfunction and apoptotic cell death. It compels the downregulation of the β-tubulin proteins in cancer cells and impairs the activity of CDK1 and cyclin B1 to inhibit the progression of the cell cycle.348 Its active epimer, 7-epi-cluisianone, succeeded in bringing about cell death in 25 cancer cell lines out of 60 human cancer cell types through the modification of the immune system, prevention of angiogenesis, along with cancer cell invasion in the body.
The remarkable pharmacological activities of some alkaloids, terpenoids, dibenzoyl glycosides, and flavonoids-containing bicyclo[3.3.1]nonane moiety isolated from the roots of sophora flavescens, a well known deciduous shrub, attracted the researchers for use in the treatment of cancer. Therefore, Zhang et al.349 synthesized highly potential sophopterocarpan-A (741) having a benzotetrahydrofuran-fused bicyclo[3.3.1]nonane ring, which triggered the autophagic pathway to destroy the cancer cells (Fig. 10, Scheme 163). The in vitro anticancer proficiency of sophopterocarpan A was monitored against breast cancer cell line (MCF-7), human lung carcinoma cell line (A549), and human liver cancer cell line (HepG2). It was found that this compound was highly competent to inhibit the growth of MCF-7 cells rather than other cancer cell lines, revealing an IC50 of 29.36 μM. The tumor growth inhibition potential of this compound was found to take place through the autophagy mechanism, where cellular dilapidation eliminated the cancer endorsing factors and thereby led to the demolition of cancer cells. This autophagic mechanism was analyzed in the autophagy-detecting system with respect to curcumin as the positive control.350
 |
| | Fig. 10 Sophopterocarpan-A isolated from the roots of Sophora flavescenes and its anticancer activity. | |
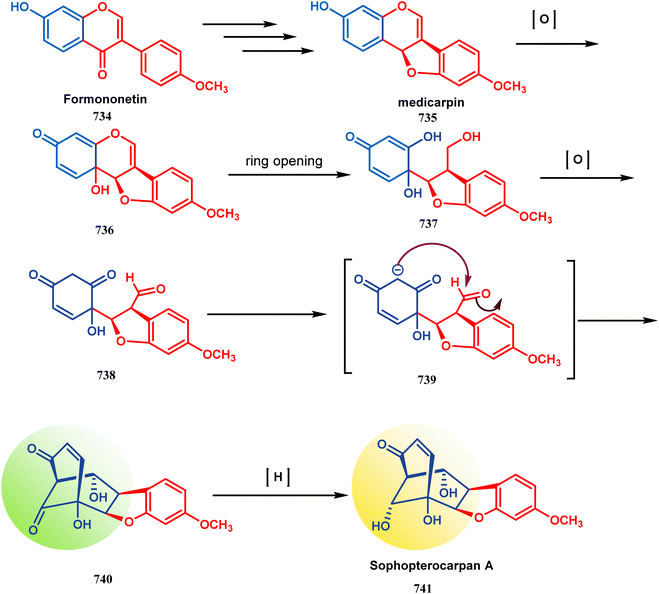 |
| | Scheme 163 Synthesis of sophopterocarpan-A. | |
Human liver cancer is now being considered as a very crucial cause of death throughout the world. Oxidative stress due to the excessive production of reactive oxygen species (ROS) in cells has been diagnosed as one of the various hallmarks of cancer. In this case, researchers have found out that the antioxidant can act as a protective substance to prevent multistage carcinogenesis because a living organism has its own enzymatic as well as nonenzymatic antioxidant functions, which can withstand against oxidative damages induced by ROS. Therefore, much more attention had been paid for seeking out some synthetic antioxidants capable of targeting various signaling pathways in cancer. Researchers observed that naturally obtained diterphenoid or norditerphenoid alkaloids possessing 3-azabicyclo[3.3.1]nonane pharmacophore have many biological activities. It was found that the derivatives of 2,4-diaryl-3-azabicyclo[3.3.1]nonan-9-one exhibited excellent antitumor activity along with antimicrobial activity. To correlate the antioxidant property and cytotoxicity of the natural product, T. Balasankar et al. synthesized electron-withdrawing halogen group-substituted 2,4-diaryl-3-azabicyclo[3.3.1]nonan-9-one, and they observed strong cytotoxicity but poor antioxidant properties of the synthesized compounds, which may be due to the prooxidant effect (Scheme 164).350 But with the substitution of electron-donating groups (–CH3, –OCH3, –CH(CH3)2) in the aryl rings of azabicyclo[3.3.1]nonan-9-ones, the antioxidant property was increased and cytotoxicity was decreased.
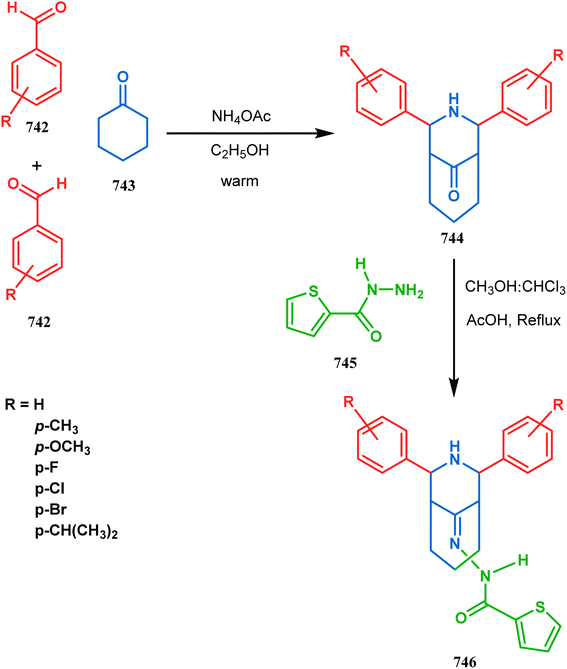 |
| | Scheme 164 Synthesis of halogen group-substituted 2,4-diaryl-3-azabicyclo[3.3.1]nonan-9-one. | |
To know the antagonist drug potency, they recorded the IC50 values of the compounds by treating the compounds against human liver hepatocellular carcinoma (HepG2) cells. The introduction of electron-withdrawing –F, –Cl, and –Br substitution at the para position of diaryl rings in the azabicyclo[3.3.1]nonan-9-ones exhibited strong cytotoxicity compared to the substitution of the electron donating –CH3, –OCH3, and –CH(CH3)2 groups. Among all the compounds, the fluoro (-F)-substituted compound showed the best cytotoxicity (3.76 μg mL−1) for 48 h of incubation. The image of the compounds-treated apoptotic cells using Hoechst stain under a fluorescent microscope revealed considerable cell shrinkage and chromatin condensation (pyknosis) along with the disintegration of the nucleus. Also, they studied the free radical scavenging activity of all these compounds against DPP![[H with combining macron]](https://www.rsc.org/images/entities/char_0048_0304.gif) , O, and O
, O, and O![[2 with combining macron]](https://www.rsc.org/images/entities/char_0032_0304.gif) − radicals. It was recorded that the methoxy (–OCH3)–substituted compound showed the best antioxidant property (Scheme 165, Table 3).
− radicals. It was recorded that the methoxy (–OCH3)–substituted compound showed the best antioxidant property (Scheme 165, Table 3).
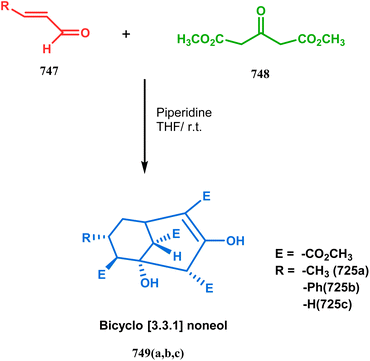 |
| | Scheme 165 Synthesis of bicyclo[3.3.1]nonenols. | |
Table 3 Bicyclo[3.3.2]nonene derivatives (749–752)
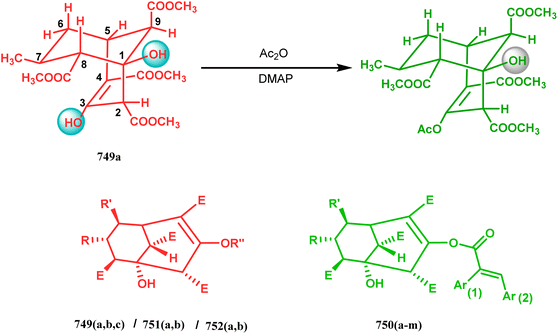
|
| Compd |
R |
R′ |
R′′ |
Compd |
R |
Ar(1) |
Ar(2) |
| 749a |
Me |
H |
H |
750a |
Me |
H |
Ph |
| 749b |
Ph |
H |
H |
750b |
Me |
H |
4-Chlorophenyl |
| 749c |
H |
H |
H |
750c |
Me |
Ph |
Ph |
| 751a |
Me |
H |
–COPh |
750d |
Me |
Ph |
4-Chlorophenyl |
| 751b |
Ph |
H |
–COPh |
750e |
Me |
Ph |
2,6-Difluorophenyl |
| 752a |
Me |
Me |
H |
750f |
Me |
4-Chlorophenyl |
Ph |
| 752b |
Ph |
Me |
H |
750g |
Me |
2,4-Dichlorophenyl |
Ph |
| — |
— |
— |
— |
750h |
Me |
4-Nitrophenyl |
Ph |
| — |
— |
— |
— |
750i |
Me |
4-Methoxyphenyl |
Ph |
| — |
— |
— |
— |
750j |
Me |
4-Chlorophenyl |
4-Chlorophenyl |
| — |
— |
— |
— |
750k |
Me |
2,4-Dichlorophenyl |
2,4-Dichlorophenyl |
| — |
— |
— |
— |
750l |
Ph |
H |
4-Chlorophenyl |
| — |
— |
— |
— |
750m |
Ph |
4-Chlorophenyl |
4-Chlorophenyl |
As bicyclo[3.3.1]nonane is a very important framework in biologically-active natural products, it always deserves the evaluation of its biological importance. Therefore, J. Valgeirsson and coworkers synthesized bicyclo[3.3.1]nonenols, and their anticancer activities were screened against various human cancer cell lines (Scheme 164). The prepared bicyclo[3.3.1]noneols were sent to the National Cancer Institute (NCI) to justify their in vitro cytotoxic behavior against 60 tumor cell lines. The compounds showed anticancer activity in the GI50 range of 1–100 μM (Table 4).351
Table 4 GI50 values (μM) of bicyclo[3.3.1]nonenes against different human cancer cell lines
| Compds. |
MOLT-4 (leukemia) |
H522 (lung) |
KM12 (colon) |
SF-539 (CNS) |
M14 (melanoma) |
SK-OV-3 (ovarian) |
A498 (renal) |
DU-145 (prostate) |
MCF-7 (breast) |
| 749a |
26.3 |
25.2 |
23.5 |
18.4 |
20.5 |
26.0 |
19.4 |
20.2 |
34.8 |
| 749b |
11.6 |
6.71 |
11.7 |
12.6 |
10.8 |
17.4 |
17.2 |
12.9 |
5.93 |
| 749c |
29.4 |
10.6 |
21.6 |
18.6 |
22.6 |
19.2 |
16.5 |
44.7 |
35.6 |
| 750a |
>100 |
5.59 |
27.9 |
19.8 |
25.5 |
21.4 |
13.3 |
18.0 |
19.3 |
| 750b |
15.8 |
6.81 |
34.1 |
17.6 |
23.2 |
24.6 |
— |
39.4 |
— |
| 750c |
— |
45.4 |
>100 |
>100 |
>100 |
>100 |
>100 |
>100 |
>100 |
| 750d |
>100 |
5.99 |
>100 |
>100 |
>100 |
>100 |
5.55 |
>100 |
>100 |
| 750e |
>100 |
6.76 |
>100 |
>100 |
>100 |
>100 |
|
>100 |
— |
| 750f |
79.2 |
>100 |
>100 |
>100 |
>100 |
>100 |
>100 |
>100 |
>100 |
| 750g |
>100 |
>100 |
>100 |
>100 |
>100 |
>100 |
>100 |
>100 |
>100 |
| 750h |
3.53 |
77.2 |
>100 |
51.5 |
>100 |
>100 |
>100 |
>100 |
>100 |
| 750i |
>100 |
35.3 |
>100 |
>100 |
>100 |
80.3 |
>100 |
>100 |
>100 |
| 750j |
3.62 |
10.8 |
26.0 |
>100 |
>100 |
>100 |
>100 |
>100 |
>100 |
| 750k |
>100 |
64.1 |
>100 |
>100 |
>100 |
>100 |
>100 |
>100 |
>100 |
| 750l |
3.45 |
1.96 |
4.33 |
3.41 |
3.46 |
24.6 |
6.04 |
5.80 |
— |
| 750m |
2.22 |
3.84 |
>100 |
>100 |
30.1 |
64.0 |
— |
>100 |
— |
| 750a |
46.1 |
13.2 |
28.4 |
14.5 |
28.3 |
29.5 |
26.4 |
59.2 |
29.0 |
| 750b |
3.64 |
— |
8.6 |
2.20 |
1.95 |
10.6 |
16.3 |
9.40 |
3.02 |
| 752a |
>100 |
26.9 |
34.7 |
35.7 |
67.5 |
49.4 |
>100 |
>100 |
38.1 |
| 752b |
86.0 |
18.9 |
>100 |
>100 |
88.1 |
>100 |
— |
>100 |
— |
5. Conclusion and future perspectives
The synthesis, derivatization, and bioevaluation of bicyclo[3.3.1]nonane and its heteroanalogues have always been the center of growing interest. Although this important moiety was discovered more than a century ago, chemists are still developing so far unknown enities based on this important framework, while biologists continue to unveil newer areas where its phamacophoric potential is being tested. Also, the global necessity of employment of green chemistry in recent times has witnessed the fast-growing efforts of researchers to investigate the possible environment-friendly modifications of these methodologies using several green tools, including solventless techniques, solid-supports, microwave irradiations, and water as the solvent. Besides, several occurrence of bicyclo[3.3.1]nonane in a numerous bioactive natural products and its anticancer activity have attracted the attention of several research groups worldwide to develop novel synthetic routes for the construction of this bicycle with selective functionalization, both from asymmetric and nonasymmetric point-of-view to design anticancer scaffolds. It can be hoped that the chemistry of bicyclo[3.3.1]nonanes will be beneficial for developing selective anticancer drugs in the imminent future.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
The authors are grateful to VIT for ‘VIT SEED GRANT’. We acknowledge DST, New Delhi, India for DST-FIST project. Authors are grateful to Department of Science and Technology, Government of India for supporting the work through the DST-SERB CRG project grant (CRG/2021/002267).
References
- M. Demeunynck and A. Tatibouet, Recent Developments in Tröger’s Base Chemistry, In Progress in Heterocyclic Chemistry, ed. G. W. Gribble and T. L. Gilchrist, Pergamon, Oxford, UK, 1999, vol. 11, p. 1 Search PubMed.
- J. S. Chen, W. H. Watson, J. Kagan, D. A. Agdeppa and S.-A. Chen, The structure of diethyl (1R*,4S*,5R*,8R*)-4,8-dimethyl-2,3;6,7-dibenzo-9-oxabicyclo[3.3.1]nona-2,6-diene-4,8-dicarboxylate, Acta Crystallogr., Sect. B: Struct. Crystallogr. Cryst. Chem., 1978, 34, 3627 CrossRef.
-
(a) P. A. Levkin, Y. A. Strelenko, K. A. Lyssenko, V. Schurig and R. G. Kostyanovsky, Temperature-dependent racemic compound-conglomerate crystallization of 2,3:6,7-dibenzobicyclo[3.3.1]nona-2,6-diene-4,8-dion, Tetrahedron: Asymmetry, 2003, 14, 2059 CrossRef CAS;
(b) P. Molina, A. Arques, A. Tarraga and M. del Rosario Obon, Synthesis and X-ray crystallographic study of 6,12-epiiminodibenzo[b,f][1,5]diazocines, Tetrahedron, 1998, 54, 997 CrossRef CAS;
(c) S. Stoncius, E. Butkus, A. Zyilinskas, K. Larsson, L. Ohrstrom, U. Berg and K. J. Warnmark, Design and Synthesis of a C2-Symmetric Self-Complementary Hydrogen-Bonding Cleft Molecule Based on the Bicyclo[3.3.1]nonane and 4-Oxo-5-azaindole Framework. Formation of Channels and Inclusion Complexes in the Solid, Org. Chem., 2004, 69, 5196 CrossRef CAS.
-
(a) K. Naemura, R. Fukunaga, M. Komatsu, M. Yamanaka and H. Chikamatsu, Synthesis and Enantiomer Recognition of Dipodands and Crown Ethers Containing the 2,3:6,7-Dibenzobicyclo[3.3.1]nona-2,6-diene Residue as the Chiral Subunit, Bull. Chem. Soc. Jpn., 1989, 62, 83 CrossRef CAS;
(b) M. Harmata and M. Kahraman, Synthesis of a chiral molecular square with two organic corners, Tetrahedron Lett., 1999, 40, 4133 CrossRef CAS;
(c) M. Harmata and T. Murray, Molecular clefts. 1. Synthetic methodology for the preparation of analogs of Kagan's ether, J. Org. Chem., 1989, 54, 3761 CrossRef CAS.
- J. Tröger, Ueber einige mittelst nascirenden Formaldehydes entstehende Basen, J. Prakt. Chem., 1887, 36, 225 CrossRef.
-
(a) M. Harmata and M. Kahraman, Congeners of Troeger's base as chiral ligands, Tetrahedron: Asymmetry, 2000, 11, 2875 CrossRef CAS;
(b) H. U. Blaser, H. P. Jalett, W. Lottenbach and M. Studer, Heterogeneous Enantioselective Hydrogenation of Ethyl Pyruvate Catalyzed by Cinchona-Modified Pt Catalysts: Effect of Modifier Structure, J. Am. Chem. Soc., 2000, 122, 12675 CrossRef CAS.
- C. Bailly, W. Laine, M. Demeunynck and J. Lhomme, Enantiospecific Recognition of DNA Sequences by a Proflavine Tröger Base, Biochem. Biophys. Res. Commun., 2000, 273, 681 CrossRef CAS.
- R. A. Johnson, R. R. Gorman, R. J. Wnuk, N. J. Crittenden and J. W. Aiken, Troeger's base. An alternate synthesis and a structural analog with thromboxane A2 synthetase inhibitory activity, J. Med. Chem., 1993, 36, 3202 CrossRef CAS PubMed.
-
(a) A. Sharma, C. Besnard, L. Gǵeńee and J. Lacour, Asymmetric synthesis of ethano-Tröger bases using CuTC-catalyzed diazo decomposition reactions, Org. Biomol. Chem., 2012, 10, 966 RSC;
(b) J. Artacho, E. Ascic, T. Rantanen, J. Karlsson, C. Wallentin, R. Wang, O. F. Wendt, M. Harmata, V. Snieckus and K. Warnmark, Twisted Amide Analogues of Tröger’s Base, Chem. – Eur. J., 2012, 18, 1038–1042 CrossRef CAS PubMed.
-
(a) R. Haritakun, P. Srikitikulchai, P. Khoyaiklang, M. Isaka and A. -D. Isariotins, Alkaloids from the Insect Pathogenic Fungus Isaria tenuipes BCC 7831, J. Nat. Prod., 2007, 70, 1478–1480 CrossRef CAS PubMed;
(b) Y. Hirasawa, H. Morita and J. Kobayashi, Nankakurine A, a Novel C16N2-Type Alkaloid from Lycopodium hamiltonii, Org. Lett., 2004, 6, 3389–3391 CrossRef CAS PubMed;
(c) T. C. Coombs, Y. Zhang, E. C. G. Amblard and L. S. Liebeskind, Organometallic Enantiomeric Scaffolding.
Sequential Semipinacol/1,5-“Michael-like” Reactions as a Strategic Approach to Bridgehead-Quaternary Center Aza[3.3.1]Bicyclics: Application to the Total Synthesis of (−)-Adaline, J. Am. Chem. Soc., 2009, 131, 876–877 CrossRef CAS PubMed;
(d) A. P. Kozikowski, G. Campania and W. Tückmantel, An approach to open chain and modified heterocyclic analogues of the acetylcholinesterase inhibitor, huperzine A, through a bicyclo[3.3.1]nonane intermediate, Heterocycles, 1994, 39, 101–116 CrossRef CAS;
(e) C. Tsukano, L. Zhao, Y. Takemoto and M. Hirama, Concise Total Synthesis of (±)-Lycodine, Eur. J. Org. Chem., 2010, 22, 4198–4200 CrossRef.
-
(a) J. F. Blount, R. W. Dunlop, K. L. Erickson, R. J. Wells and J. Robert, Two diterpenes with new carbocyclic ring systems from an Australian collection of the brown alga Dictyota dichotoma, Aust. J. Chem., 1982, 35, 145–163 CrossRef CAS;
(b) G. Schulte, P. J. Scheuer and O. J. McConnell, Upial, a sesquiterpenoid bicyclo[3.3.1]nonane aldehyde lactone from the marine sponge Dysidea fragilis, J. Org. Chem., 1980, 45, 552–554 CrossRef CAS;
(c) H. Huang and C. J. Forsyth, Synthesis of the Sesquiterpenes Trifarienols A and B via Anti-Selective .alpha.'-Intramolecular Carbomercuration, J. Org. Chem., 1995, 60, 5746–5747 CrossRef CAS;
(d) M. J. Gallen and C. M. Williams, Total Synthesis of (±)-5,14-bis-epi-Spirovibsanin A, Org. Lett., 2008, 10, 713–715 CrossRef CAS PubMed.
- S. Dewanjee, A. Maiti, A. K. Das, S. C. Mandal and S. P. Dey, Swietenine: A potential oral hypoglycemic from Swietenia macrophylla seed, Fitoterapia, 2009, 80, 249–251 CrossRef CAS PubMed.
- S. Baggett, P. Protiva, E. P. Mazzola, H. Yang, E. T. Ressler, M. J. Basile, B. Weinstein and E. J. Kennelly, Bioactive Benzophenones from Garcinia xanthochymus Fruits, J. Nat. Prod., 2005, 68, 354–360 CrossRef CAS PubMed.
- T. Amagata, K. Minoura, A. Numata and F. −H. Gymnastatins, Cytostatic Metabolites from the Sponge-Derived Fungus Gymnascella dankaliensis, J. Nat. Prod., 2006, 69, 1384–1388 CrossRef CAS PubMed.
-
(a) G. A. Kraus and I. Jeon, Progress towards the synthesis of papuaforin A: selective formation of α-bromoenones from silyl enol ethers, Tetrahedron Lett., 2008, 49, 286–288 CrossRef CAS PubMed;
(b) G. A. Kraus, T. H. Nguyen and I. Jeon, Synthesis of the core bicyclic system of hyperforin and nemorosone, Tetrahedron Lett., 2003, 44, 659–661 CrossRef CAS;
(c) N. M. Ahmad, V. Rodeschini, N. S. Simpkins, S. E. Ward and A. J. Blake, Synthesis of Polyprenylated Acylphloroglucinols Using Bridgehead Lithiation: The Total Synthesis of Racemic Clusianone and a Formal Synthesis of Racemic Garsubellin, J. Org. Chem., 2007, 72, 4803–4815 CrossRef CAS;
(d) M. Taher, M. Idris, F. Ahmad and D. Arbain, A polyisoprenylated ketone from Calophyllum enervosum, Phytochemistry, 2005, 66, 723–726 CrossRef CAS PubMed;
(e) R. Takagi, Y. Inoue and K. Ohkata, Construction of the Adamantane Core of Plukenetione-Type Polycyclic Polyprenylated Acylphloroglucinols, J. Org. Chem., 2008, 73, 9320–9325 CrossRef CAS PubMed;
(f) G. Mehta, M. K. Bera and S. Chatterjee, A stereodefined approach towards the bicyclo[3.3.1]nonan-9-one core of the phloroglucin natural products guttiferone A and hypersampsone F, Tetrahedron Lett., 2008, 49, 1121–1124 CrossRef CAS;
(g) N. Biber, K. Mows and B. Plietker, The total synthesis of hyperpapuanone, hyperibone L, epi-clusianone and oblongifolin A, Nat. Chem., 2011, 3, 938–942 CrossRef CAS PubMed.
- O. A. Namjoshi and J. M. Cook, Sarpagine and related alkaloids, The Alkaloids, 2016, vol. 76, pp. 1099–4831.(Book Chapter) Search PubMed.
- G. L. Buchanan and D. Ed. Lloyd, Logos Press: London, The Biosynthesis of Carbocyclic Compounds, Topics in Carbocyclic Chemistry, 1969, vol. 1, p. 199 Search PubMed.
-
(a) J. A. Peters, Synthesis of Bicyclo[3.3.1]nonanes, Synthesis, 1979, 1979(5), 321–336 CrossRef;
(b) E. Butkus, Stereocontrolled Synthesis and Reactions of Bicyclo [3.3.1]nonanes, Synlett., 2001, 1827 CrossRef CAS;
(c) J. Bonjoch, F. Diaba and B. Bradshaw, Synthesis of 2-Azabicyclo[3.3.1]nonanes, Synthesis, 2011, 0993–1018 CrossRef CAS;
(d) R. Jeyaraman and S. Avila, Chemistry of 3-Azabicyclo[3.3.1]nonanes, Chem. Rev., 1981, 81, 149–174 CrossRef CAS;
(e) J. Bosch and J. Bonjoch, Synthesis of 2-Azabicyclo[3.3.1]nonanes, Heterocycles, 1980, 14, 505–529 CrossRef CAS;
(f) J. J. Turner and M. M. Harding, Synthesis and Properties of Chiral Molecular Clefts Related to Tröger's Base, Supramol. Chem., 2005, 17, 369–375 CrossRef CAS;
(g) M. Valk, R. M. Strongin and V. Kral, Tröger's Base Derivatives—New Life for Old Compounds, Supramol. Chem., 2005, 17, 347 CrossRef;
(h) M. Demeunynck and A. Tatibouet, Progress in Heterocyclic Chemistry, ed. G. W. Gribble and T. L. Gilchrist, Elsevier Science Ltd., Oxford, 1999, vol. 11, p. 1 Search PubMed;
(i) B. G. Bag, The chemistry of a non-natural product: Tröger's base, Curr. Sci., 1995, 68, 279 CAS;
(j) B. Dolensky, J. Elguero, V. Kral, C. Pardo and M. Valk, Current Tröger's Base Chemistry, Adv. Heterocycl. Chem., 2007, 93, 1–56 CrossRef CAS.
- M.-H. Filippini and J. Rodriguez, Synthesis of Functionalized Bicyclo[3.2.1]octanes and Their Multiple Uses in Organic Chemistry, Chem. Rev., 1999, 99, 27–76 CrossRef CAS PubMed.
- J. Casanova, G. Koukoua and B. Waegell, BICYCLO[4.2.1]nonane skeleton-conformational-analysis, synthesis, reactivity, Bull. Soc. Chim. Fr., 1990, 127, 528 Search PubMed.
- N. L. Allinger, M. T. Tribble, M. A. Miller and D. H. Wertz, Conformational analysis. LXXI. Calculation of nuclear magnetic resonance spectra by semiempirical methods. I. Proton chemical shifts in hydrocarbons, J. Am. Chem. Soc., 1971, 93, 1637 CrossRef CAS.
- E. M. Engler, J. D. Andose and P. V. R. Schleyer, Critical evaluation of molecular mechanics, J. Am. Chem. Soc., 1973, 95, 8005 CrossRef CAS.
- E. Osawa, K. Aigami and Y. Imamoto, Application of Force Field Calculations to Organic Chemistry. IV: Steric Analysis of Synthesis and Structure of 1,4- Dihydroxytricyclo (6.4.0.0) Dodecane-7,10-Dione. Dynamic Conformational Calculations of its Hydrocarbon Skeleton and related systems (Bicyclo (3.3.1) nonane and Bicyclo (3.3.2) Decane), J. Chem. Soc., Perkin Trans. 2, 1979, 172 Search PubMed.
- V. S. Mastryukov, M. V. Popik, O. V. Dorofeeva, A. V. Golubinskii, L. V. Vilkov, N. A. Belikova and N. L. Allinger, Chair-boat equilibriums in bicyclo[3.3.1]nonane at 65 and 400.degree.C studied by electron diffraction and molecular mechanics, J. Am. Chem. Soc., 1981, 103, 1333 CrossRef CAS.
-
(a) F. A. L. Anet and V. J. Basus, Detection of a crown family conformation in cyclooctane by proton and carbon-13 nuclear magnetic resonance, J. Am. Chem. Soc., 1973, 95, 4424 CrossRef CAS;
(b) F. A. L. Anet and V. J. Basus, Limiting equations for exchange broadening in two-site NMR systems with very unequal populations, J. Magn. Reson., 1978, 32, 339 CAS;
(c) N. Okazawa and T. S. Sorensen, The line-shape analysis of nuclear magnetic resonance peaks broadened by the presence of a 'hidden' exchange partner, Can. J. Chem., 1978, 56, 2737 CrossRef CAS.
- R. M. Paroli, D. F. R Gilson and I. S. Butler, Spectroscopic and Differential Scanning Calorimetric Studies of the Order-Disorder Phase Transition in Bicyclononanone, J. Solid State Chem., 1998, 136, 16–20 CrossRef CAS.
- C. Jaime, E. Osawa, Y. Takeuchi and P. Camps, Application of empirical potential energy calculations to organic chemistry. Part 19. Conformational preference in 2,4-dimethoxybicyclo[3.3.1]nonan-9-one and related molecules. Analysis of vicinal NMR coupling constants in multiple rotor system by combined molecular mechanics and generalized Karplus equation, J. Org. Chem., 1983, 48, 4514 CrossRef CAS.
- K. Gundertofte, T. Liljefors, P.-O. Norrby and I. Pettersson, A comparison of conformational energies calculated by several molecular mechanics methods, J. Comput. Chem., 1996, 17, 429–449 CrossRef CAS.
- D. J. Raber, C. M. Janks, M. D. Johnston Jr. and N. K. Raber, Structure elucidation with lanthanide induced shifts. 9. Bicyclo [3.3.1]nonan-9-one, Tetrahedron Lett., 1980, 21, 677 CrossRef CAS.
- A. J. Mora and A. N. Fitch, The low-temperature structures of bicyclo[3.3.1]nonan-9-one and 3-azabicyclo[3.2.2]nonane, Z. Kristallogr., 1999, 214, 480 CAS.
-
(a) F. A. L. Anet, C. H. Bradley and G. W. Buchanan, Direct detection of the axial conformer of methylcyclohexane by 63.1MHz carbon-13 nuclear magnetic resonance at low temperatures, J. Am. Chem. Soc., 1971, 93, 258 CrossRef CAS;
(b) K. B. Wiberg, J. D. Hammer, H. Castejon, W. F. Bailey, E. L. DeLeon and R. M. Jarret, Conformational Studies in the Cyclohexane Series. 1. Experimental and Computational Investigation of Methyl, Ethyl, Isopropyl, and tert-Butylcyclohexanes, J. Org. Chem., 1999, 64, 2085 CrossRef CAS PubMed;
(c) L. Lunazzi, A. Mazzanti, D. Casarini, O. De Lucchi and F. Fabris, Conformational Studies by Dynamic NMR. 70.1 Stereomutations of Homochiral Dicarvone in Solution and in the Solid State, J. Org. Chem., 2000, 65, 883 CrossRef CAS.
- S. Grilli, L. Lunazzi and A. Mazzanti, Conformational Studies by Dynamic NMR. 76.1 Stereodynamics of Ring Inversion of Bicyclo[3.3.1]nonan-9-one, J. Org. Chem., 2000, 65, 3563–3565 CrossRef CAS PubMed.
-
(a) R. Jeyaraman and S. Avila, Chemistry of 3-azabicyclo[3.3.1]nonanes, Chem. Rev., 1981, 81, 149 CrossRef CAS;
(b) N. S. Zefirov and V. A. Palyulin, Conformational analysis of bicyclo [3.3. 1] nonanes and their hetero analogs, Top. Stereochem., 1991, 20, 171 CAS.
- H. Quast and B. Mü ller, Stereochemie von Tetraaryl-3, 7-diazabicyclo [3.3. 1] nonanen und Tetraaryl-1, 3-diazaadamantanen, Chem. Ber., 1980, 113, 2959 CrossRef CAS.
-
(a) L. M. Jackman, T. S. Dunne, B. Müller and H. Quast, Conformation in solution of tetraaryl-3, 7-diazabicyclo[3.3.1]nonanes and tetra-and pentaaryl-1, 3-diazaadamantanes. A nuclear magnetic resonance study, Chem. Ber., 1982, 115, 2872 CrossRef CAS;
(b) H. Quast, B. Müller, E.-M. Peters, K. Peters and H. G. von Schnering, Kristall- und Molekülstruktur von Tetraaryl-3,7-diazabicyclo- [3.3.1]nonanen und Tetra- und Pentaaryl-1,3-diaza-adamantanen. Sessel/Boot-Konformation der Tetraaryl-3,7-diazabicyclo[3.3.1]nonane, Chem. Ber., 1982, 115, 3631 CrossRef CAS.
-
(a) N. L. Allinger, Conformational analysis. 130. MM2. A hydrocarbon force field utilizing V1 and V2 torsional terms, J. Am. Chem. Soc., 1977, 99, 8127 CrossRef CAS;
(b) N. L. Allinger and Y. H. Yuh, QCPE, 1980, No 395 Search PubMed.
- P. H. McCabe, N. J. Milne and G. A. Sim, Conformations of derivatives of 3,7-diazabicyclo[3.3.1]nonan-9-one. Comparison of 3-ethoxycarbonyl-7-methyl-1,5-diphenyl-3,7-diazabicyclo[3.3.1]nonan-9-one and 3,7-di(ethoxycarbonyl)-1,5-diphenyl-3,7-diazabicyclo[3.3.1]nonan-9-one: effect of a nucleophile ⋯ electrophile interaction on molecular geometry, J. Chem. Soc., Perkin Trans. 2, 1989, 831–834 RSC.
- M. Gdaniec, M. Pham and T. Połoński, Conformation and Stereodynamics of N, N′-Dinitroso-2,4,6,8-tetraaryl-3,7-diazabi-cyclo[3.3.1]nonanes, J. Org. Chem., 1997, 62, 5619–5622 CrossRef CAS.
- B. M. Mikhailov and Y. N. Bubnov, Organoboron Compounds in Organic Synthesis, Harwood Academy Science Publishing, Chur, 1984 Search PubMed.
- H. Gunter, NMR-Spectroscopie, Georg Thieme Verlag, Stuttgart, 1973 Search PubMed.
-
(a) M. E. Gurskii, I. D. Gridnev, Y. N. Bubnov, A. Pelter and P. Rademacher, Conformational behavior of 3-borabicyclo [3.3. 1] nonanes: 1. Study of molecular dynamics in 3-methoxy-7α-phenyl-1, 5-dimethyl-3-borabicyclo [3.3. 1] nonane, J. Organomet. Chem., 1999, 590, 227–233 CrossRef CAS;
(b) A. Fiirstner and G. Seidel, Palladium-catalyzed arylation of polar organometallics mediated by 9-methoxy-9-borabicyclo [3.3. 1] nonane: Suzuki reactions of extended scope, Tetrahedron, 1995, 51, 11165–11176 CrossRef.
- M. E. Gurskii, K. A. Lyssenko, A. L. Karionova, P. A. Belyakov, T. V. Potapova, M. Y. Antipin and Y. N. Bubnov, Unique stereochemistry of 3-borabicyclo [3.3. 1] nonane derivatives, Russ. Chem. Bull., 2004, 53, 1963–1977 CrossRef CAS.
- K. A. Lyssenko, M. Y. Antipin, M. E. Gurskii, Y. N. Bubnov, A. L. Karionova and R. Boese, Characterization of the B⋯π-system interaction via topology of the experimental charge density distribution in the crystal of 3-chloro-7α-phenyl-3-borabicyclo[3.3.1]nonane, Chem. Phys. Lett., 2004, 384, 40–44 CrossRef CAS.
- N. S. Zefirov and V. A. Palyulin, In Topics in Stereochemistry, John Wiley & Sons, Inc., Hoboken, NJ, 1991, vol. 20, pp. 171–230 Search PubMed.
- J. Choo, S. Kim, H. Joo and Y. J. Kwon, The molecular structure and conformation of bicyclo [3.3. 1] nonan-9-one: ab initio and DFT calculations, J. Mol. Struct.: THEOCHEM, 2002, 619, 113–120 CrossRef CAS.
- N. S. Zefirov, The problem of conformational effects, Tetrahedron, 1977, 33, 3192 CrossRef.
- S. A. Pisarev and V. A. Palyulin, Conformational effects of 1, 5, 9-substitution in symmetric bicyclo [3.3.1] nonane analogues, Mendeleev Commun., 2021, 31, 612–614 CrossRef CAS.
- R. Gleiter, M. Kobayashi, N. S. Zefirov and V. A. Palyulin, Photoelectron-spectra and interaction of lone e pairs in 3, 7, 9-triheteroderivatives of bicyclo [3.3.1] nonane, Proc. Natl. Acad. Sci., 1977, 235, 347–350 CAS.
- D. Ferro-Costas, N. Otero, A. M. Grana and R. A. Mosquera, A QTAIM- based energy partitioning for understanding the physical origin of conformational preferences: Application to the Z effect in O=C-X-R and related units, J. Comput. Chem., 2012, 33, 2533–2543 CrossRef CAS.
- Y. Tsubomoto, S. Hayashi, W. Nakanishi, L. K. Mapp and S. J. Coles, High-resolution X-ray diffraction determination of the electron density of 1-(8-PhSC10H6)SS(C10H6SPh-8′)-1′ with the QTAIM approach: evidence for S4 σ(4c-6e) at the naphthalene peri-positions, RSC Adv., 2018, 8, 9651–9660 RSC.
- I. S. Bushmarinov, M. Yu. Antipin, V. R. Akhmetova, G. R. Nadyrgolova and K. A. Lyssenko, Stereoelectronic Effects in N-C-S Systems: Experimental and ab Initio AIM Study, J. Phys. Chem. A, 2008, 112, 5017–5023 CrossRef CAS.
- I. S. Bushmarinov, I. V. Fedyanin, K. A. Lyssenko, V. L. Lapteva, S. A. Pisarev, V. A. Palyulin, N. S. Zefirov and M. Y. Antipin, The “Hockey Sticks” Effect Revisited: The Conformational and Electronic Properties of 3,7-Dithia-1,5-diazabicyclo[3.3.1]nonane from the QTAIM Perspective, J. Phys. Chem. A, 2011, 115, 12738–12745 CrossRef CAS.
-
(a) R. Jeyaraman and S. Avila, Chemistry of 3-azabicyclo [3.3. 1] nonanes, Chem. Rev., 1981, 81, 149 CrossRef CAS;
(b) N. S. Zefirov and V. A. Palyulin, Conformational analysis of bicyclo [3.3. 1] nonanes and their hetero analogs, Top. Stereochem., 1991, 20, 171 CAS.
- A. Kolocouris, CHax⋯Yax Contacts in Cyclohexane Derivatives Revisited−Identification of Improper Hydrogen-Bonded Contacts, J. Org. Chem., 2009, 74, 1842–1849 CrossRef CAS PubMed.
- A. T. Hulme, A. Johnston, A. J. Florence, P. Fernandes, K. Shankland, C. T. Bedford, G. W. A. Welch, G. Sadiq, D. A. Haynes, W. D. S. Motherwell, D. A. Tocher and S. L. Price, Search for a Predicted Hydrogen Bonding Motif− A Multidisciplinary Investigation into the Polymorphism of 3-Azabicyclo [3.3. 1] nonane-2, 4-dione, J. Am. Chem. Soc., 2007, 129, 3649–3657 CrossRef CAS PubMed.
- A. G. Martinez, J. O. Barcina, A. Albert, F. H. Cano and L. R. Subramanian, 7, 7-Diphenylnorbornane: The first cofacial diphenylmethane derivative, Tetrahedron Lett., 1993, 34, 6753 CrossRef CAS.
-
(a) J. C. Barnes, J. D. Paton, J. R. Damewood and K. Mislow, Crystal and molecular structure of diphenylmethane, J. Org. Chem., 1981, 46, 4975 CrossRef CAS;
(b) M. Feigel, Phenyl rotation in diphenylether and diphenylmethane calculated with ab initio methods, J. Mol. Struct.: THEOCHEM, 1996, 36, 83 CrossRef;
(c) S. Grilli, L. Lunazzi and A. Mazzanti, Conformational
Studies by Dynamic NMR. 83.1 Correlated Enantiomerization Pathways for the Stereolabile Propeller Antipodes of Dimesityl Substituted Ethanol and Ethers, J. Org. Chem., 2001, 66, 5853 CrossRef CAS PubMed.
- A. G. Martinez, J. O. Barcina, A. F. Cerezo and R. G. Rivas, Hindered rotation in diphenylmethane derivatives. Electrostatic vs charge-transfer and homoconjugative aryl− aryl interactions, J. Am. Chem. Soc., 1998, 120, 673 CrossRef.
- D. Casarini and C. Rosini, Conformational Studies by Dynamic NMR. 93.1 Stereomutation, Enantioseparation, and Absolute Configuration of the Atropisomers of Diarylbicyclononanes, J. Org. Chem., 2003, 68, 1815–1820 CrossRef CAS.
-
(a) S. Sergeyev, Recent Developments in Synthetic Chemistry, Chiral Separations, and Applications of Tröger's Base Analogues, Helv. Chim. Acta, 2009, 92, 415–444 CrossRef CAS;
(b) M. A. Spielman, The structure of Troeger's base, J. Am. Chem. Soc., 1935, 57, 583 CrossRef CAS;
(c) V. Prelog and P. Wieland, The resolution of Troger's base into its optical antipodes, a note on the stereochemistry of trivalent nitrogen, Helv. Chim. Acta, 1944, 27, 1127 CrossRef CAS.
- J. A. Peters, Synthesis of Bicyclo [3.3. 1] nonanes, Synthesis, 1979, 321–336 CrossRef CAS.
- E. Knoevenagel, 1, 5-Diketone, Justus Liebigs Ann. Chem., 1894, 281, 25 CrossRef.
- E. Knoevenagel, Ueber Condensationsproducte von Acetylaceton mit Aldehyden, Ber. Dtsch. Chem. Ges., 1903, 36, 2136 CrossRef CAS.
- P. A. Knott and J. M. Mellor, Synthesis of bicyclo[3,3,1]nona-3,7-diene-2,6-diones and bicyclo[3, 3,1]-nona-3,6- diene-2,8-diones, J. Chem. Soc. C, 1971, 670–674 RSC.
- D. W. Theobald, On the reaction of acetoacetic ester with (+)-carvone: An oxabicyclo [3.3. 1] nonene and some bicyclo [3.3. 1] nonanolones, Tetrahedron, 1969, 25, 3139 CrossRef CAS.
- P. Rabe, Zur Kenntniss der 1, 5-Diketone. Ueber die Umlargerung von 1, 5-Diketonen in cyklische Ketonalkohole and uber die Synthese bicyklischer Ketonalkohole mit Bruckenbindung, Justus Liebigs Ann. Chem., 1908, 360, 265 CrossRef.
- P. Rabe, Synthesen bicyclischer Systeme mit Brückenbindung. III. Mittheilung: Ueber die Anlagerung von Acetessigester an Methyl-cyclohexenon, Ber. Dtsch. Chem. Ges., 1904, 37, 1671 CrossRef CAS.
- H. Meerwein and W. Schurmann, Uber eine Synthese von Abkommlingen des Bicyclo-[1, 3, 3]-nonans, Justus Liebigs Ann. Chem., 1913, 398, 196 CrossRef CAS.
- H. Meerwein, German Patent 277467, 1913Chem. Zentralbl., 1914, 2, 740 Search PubMed.
- H. Meerwein, F. Kiel, G. Klosgen and E. Schoch, Über bicyclische und polycyclische Verbindungen mit Brückenbindung. Über das Bicyclo-[1, 3, 3]-nonan und seine Abkömmlinge, J. Prakt. Chem., 1922, 104, 161 CrossRef CAS.
- K. Aoyagi, H. Nakamura and Y. Yamamoto, A Concise and Stereospecific One-Shot Synthesis of Bicyclo[3.3.1]nonenols from Dimethyl 1,3-Acetonedicarboxylate and Enals via the Sequential Michael Addition−Intramolecular Aldolization, J. Org. Chem., 1999, 64, 4148–4151 CrossRef CAS.
- D. Wang and W. E. Crowe, One-carbon bridge stereocontrol in Robinson annulations leading to bicyclo [3.3. 1] nonanes, Org. Lett., 2010, 12, 1232–1235 CrossRef CAS PubMed.
- D. W. Theobald, On the reaction of acetoacetic ester with (+)-carvone: An oxabicyclo [3.3. 1] nonene and some bicyclo [3.3.1] nonanolones, Tetrahedron, 1969, 25, 3139 CrossRef CAS.
- G. A. Kraus and Y. S. Hon, Bridgehead intermediates in organic synthesis: two direct syntheses of (.+-.)-lycopodine, J. Am. Chem. Soc., 1985, 107, 4341 CrossRef CAS.
- G. A. Kraus and Y. S. Hon, The Total Synthesis of Lycopodine using Bridgehead Intermediates, Heterocycles, 1987, 25, 377 CrossRef CAS.
- R. B. Grossman and S. V. Ley, Chemistry of insect antifeedants from Azadirachta indica (Part 17): Synthesis of model compounds of azadirachtin. Unusual effect of remote substituents on the course of the oxidative ring contraction reaction, Tetrahedron, 1994, 50, 11553 CrossRef CAS.
- G. A. Kraus and I. Jeon, Preparation of complex bridged bicyclic ring systems from 3,3-diacetoxy-2-phenylsulfonylpropene and β-keto esters, Tetrahedron, 2005, 61, 2111–2116 CrossRef CAS.
- K. Murayama, T. Tanabe, Y. Ishikawa, K. Nakamura and S. Nishiyama, A synthetic study on gymnastatins F and Q: the tandem Michael and aldol reaction approach, Tetrahedron Lett., 2009, 50, 3191–3194 CrossRef CAS.
- H. Usuda, M. Kanai and M. Shibasaki, Studies toward the Total Synthesis of Garsubellin A: A Concise Synthesis of the 18-epi-Tricyclic Core, Org. Lett., 2002, 4, 859–862 CrossRef CAS.
- H. Usuda, M. Kanai and M. Shibasaki, Studies toward the total synthesis of garsubellin A: synthesis of 8-deprenyl-garsubellin A, Tetrahedron Lett., 2002, 43, 3621–3624 CrossRef CAS.
- Y. Shimizu, A. Kuramochi, H. Usuda, M. Kanai and M. Shibasaki, A new approach for the construction of a highly congested bicyclic system in polycyclic polyprenylated acylphloroglucinols (PPAPs), Tetrahedron Lett., 2007, 48, 4173–4177 CrossRef CAS.
- Y. Shimizu, S.-L. Shi, H. Usuda, M. Kanai and M. Shibasaki, The first catalytic asymmetric total synthesis of ent-hyperforin, Tetrahedron, 2010, 66, 6569–6584 CrossRef CAS.
-
(a) G. Mehta and M. K. Bera, A concise approach towards the bicyclo [3.3. 1] nonan-9-one core present in the phloroglucin natural product hyperforin, Tetrahedron Lett., 2008, 49, 1417–1420 CrossRef CAS;
(b) G. Mehta and M. K. Bera, A rapid acquisition of the bicyclo [3.3. 1] nonan-9-one core present in garsubellin A and related phloroglucins, Tetrahedron Lett., 2006, 47, 689–692 CrossRef CAS;
(c) G. Mehta, M. K. Bera and S. Chatterjee, A stereodefined approach towards the bicyclo [3.3. 1] nonan-9-one core of the phloroglucin natural products guttiferone A and hypersampsone F, Tetrahedron Lett., 2008, 49, 1121–1124 CrossRef CAS.
- T. Pouplin, B. Tolon, P. Nuhant, B. Delpech and C. Marazano, Synthetic Studies Towards Bridgehead Diprenyl-Substituted Bicyclo[3.3.1]nonane-2,9-diones as Models for Polyprenylated Acylphloroglucinol Construction, Eur. J. Org. Chem., 2007, 5117–5125 CrossRef CAS.
- N. Itagaki, M. Kimura, T. Sugahara and Y. Iwabuchi, Organocatalytic Entry to Chiral Bicyclo[3.n.1]alkanones via Direct Asymmetric Intramolecular Aldolization, Org. Lett., 2005, 7, 4185–4188 CrossRef CAS PubMed.
- K. C. Nicolaou, G. E. A. Carenzi and V. Jeso, Construction of Highly Functionalized Medium-Sized Rings: Synthesis of Hyperforin and Perforatumone Model Systems, Angew. Chem., Int. Ed., 2005, 44, 3895–3899 CrossRef CAS PubMed.
- R. Ciochina and R. B. Grossman, A New Synthetic Approach to the Polycyclic Polyprenylated Acylphloroglucinols, Org. Lett., 2003, 5, 4619–4621 CrossRef CAS PubMed.
- I. N. Michaelides, B. Darses and D. J. Dixon, Acid-Catalyzed Synthesis of Bicyclo[3.n.1]alkenediones, Org. Lett., 2011, 13, 664–667 CrossRef CAS PubMed.
- Y. Kuninobu, J. Morita, M. Nishi, A. Kawata and K. Takai, Rhenium-catalyzed formation of bicyclo [3.3. 1] nonene frameworks by a reaction of cyclic β-keto esters with terminal alkynes, Org. Lett., 2009, 11, 2535–2537 CrossRef CAS PubMed.
- A. Klein and M. Miesch, New cascade reactions starting from acetylenic ω-ketoesters: an easy access to electrophilic allenes and to 1, 3-bridgehead ketones, Tetrahedron Lett., 2003, 44, 4483–4485 CrossRef CAS.
- H. J. Liu, L. K. Ho and H. K. Lai, Annelation of β-keto thiolesters and synthetic application, Can. J. Chem., 1981, 59, 1685 CrossRef CAS.
- J. Guo, X. Bai, Q. Wang and Z. Bu, Diastereoselective Construction of Indole-Bridged Chroman Spirooxindoles through a TfOH catalyzed Michael Addition-Inspired Cascade Reaction, J. Org. Chem., 2018, 83, 3679–3687 CrossRef CAS PubMed.
- R. Takagi, T. Nerio, Y. Miwa, S. Matsumura and K. Ohkata, Construction of the bicyclo[3.3.1]nonenone core by successive Michael reactions of 2-cyclohexenone derivatives, Tetrahedron Lett., 2004, 45, 7401–7405 CrossRef CAS.
- J. Qi and J. A. Porco Jr., Rapid Access to Polyprenylated Phloroglucinols via Alkylative Dearomatization−Annulation: Total Synthesis of (±)-Clusianone, J. Am. Chem. Soc., 2007, 129, 12682–12683 CrossRef CAS PubMed.
- R. Takagi, Y. Inoue and K. Ohkata, Construction of the adamantane core of Plukenetione-type polycyclic polyprenylated acylphloroglucinols, J. Org. Chem., 2008, 73, 9320–9325 CrossRef CAS PubMed.
- A. Gambacorta, D. Tofani, A. Tafi and M. A. Farah, Chair–boat equilibrium as driving force
in epimerization of 3,7-dimethylbicyclo[3.3.1]nonan-2,9-dione derivatives. Stereocontrolled synthesis of the 3-exo,7-exo- and 3-endo,7-exo-dimethylbicyclo[3.3.1]nonan-9-ones, Tetrahedron, 2001, 57, 5435–5444 CrossRef CAS.
- G. A. Kraus, E. Dneprovskaia, T. H. Nguyen and I. Jeon, Synthesis of a model system for the preparation of phloroglucinol containing natural products, Tetrahedron, 2003, 59, 8975–8978 CrossRef CAS.
- R. K. Boeckman Jr., A. Arvanitis and M. E. Voss, Synthetic studies directed toward naturally occurring cyclooctanoids. 1. A total synthesis of (.+-.)-ceroplastol I, J. Am. Chem. Soc., 1989, 111, 2737–2739 CrossRef.
- D. Kalaivani, M. Vasuki and S. Santhi, Mechanism and linear free energy relationships in the kinetics of formation of bicyclo[3.3.1]nonane derivatives from 1,3,5-trinitrobenzene,phenyl-substituted1-benzyl-1-(ethoxycarbonyl)-2-propanones, and triethylamine, Int. J. Chem. Kinet., 2011, 43, 467–473 CrossRef CAS.
- R. Heim, S. Wiedemann, C. M. Williams and P. V. Bernhardt, Expedient construction of the vibsanin E core without the use of protecting groups, Org. Lett., 2005, 7, 1327 CrossRef CAS PubMed.
- D. P. Tilly, C. M. Williams and P. V. Bernhardt, Construction of the cyclovibsanin core via a biogenetically modeled approach, Org. Lett., 2005, 7, 5155 CrossRef CAS PubMed.
- B. D. Schwartz, D. P. Tilly, R. Heim, S. Wiedemann, C. M. Williams and P. V. Bernhardt, Towards the Total Synthesis of Vibsanin E, 15-O-Methylcyclovibsanin B, 3-Hydroxyvibsanin E, Furanovibsanin A, and 3-O-Methylfuranovibsanin A, Eur. J. Org. Chem., 2006, 3181 CrossRef CAS.
- J. Y. W. Mak and C. M. Williams, Enantioselective total synthesis of (-)-neovibsanin G and (-)-14-epi-neovibsanin G, Chem. Commun., 2012, 48, 287–289 RSC.
- D. R. Siegel and S. J. Danishefsky, Total synthesis of garsubellin A, J. Am. Chem. Soc., 2006, 128, 1048–1049 CrossRef CAS PubMed.
- C. Tsukano, D. R. Siegel and S. J. Danishefsky, Differentiation of nonconventional “carbanions”—the total synthesis of nemorosone and clusianone, Angew. Chem., Int. Ed., 2007, 46, 8840–8844 CrossRef CAS PubMed.
-
(a) W. P. Jackson, S. V. Ley and A. J. Whittle, Selenium-mediated cyclization reactions of alkenyl-substituted β-ketoesters, Chem. Commun., 1980, 1173–1174 RSC;
(b) W. P. Jackson, S. V. Ley and J. A. Morton, Cyclisation reactions of alkenyl β-ketoesters involving a novel phenylseleno group migration, Tetrahedron Lett., 1981, 22, 2601–2604 CrossRef CAS.
- K. C. Nicolaou, J. A. Pfefferkorn, S. Kim and H. X. Wei, Synthesis of the fully functionalized bicyclic core of garsubellin A, J. Am. Chem. Soc., 1999, 121, 4724–4725 CrossRef CAS.
- K. C. Nicolaou, J. A. Pfefferkorn, G.-Q. Cao, S. Kim and J. Kessabi, A facile method for the solution and solid-phase synthesis of substituted [3.3. 1] bicycles, Org. Lett., 1999, 1, 807–810 CrossRef CAS PubMed.
- M. E. Hediger, Design, synthesis, and evaluation of aza inhibitors of chorismate mutase, Bioorg. Med. Chem., 2004, 12, 4995–5010 CrossRef CAS PubMed.
- P. A. Bartlett and C. R. Johnson, An inhibitor of chorismate mutase resembling the transition-state conformation, J. Am. Chem. Soc., 1985, 107, 7792 CrossRef CAS.
-
(a) P. A. Bartlett, Y. Nakagawa, C. R. Johnson, S. H. Reich and A. J. Luis, Chorismate mutase inhibitors: synthesis and evaluation of some potential transition-state analogs, Org. Chem., 1988, 53, 3195 CrossRef CAS;
(b) W. W. Smith and P. A. Bartlett, An improved synthesis of the transition state analog inhibitor of chorismate mutase, J. Org. Chem., 1993, 58, 7308 CrossRef CAS.
- S. J. Spessard and B. M. Stoltz, Progress toward the synthesis of garsubellin A and related phloroglucins: the direct diastereoselective synthesis of the bicyclo [3.3. 1] nonane core, Org. Lett., 2002, 4, 1943–1946 CrossRef CAS PubMed.
- D. Casarini and C. Rosini, Conformational Studies by Dynamic NMR. 93.1 Stereomutation, Enantioseparation, and Absolute Configuration of the Atropisomers of Diarylbicyclononanes, J. Org. Chem., 2003, 68, 1815–1820 CrossRef CAS.
- V. Rodeschini, N. S. Simpkins and C. Wilson, Kinetic resolution in a bridgehead lithiation mediated by a chiral Bis-lithium amide: Assignment of the absolute configuration of clusianone, J. Org. Chem., 2007, 72, 4265–4267 CrossRef CAS PubMed.
- V. Rodeschini, N. M. Ahmad and N. S. Simpkins, Synthesis of (+/−)-Clusianone: High-Yielding Bridgehead and Diketone Substitutions by Regioselective Lithiation of Enol Ether Derivatives of Bicyclo[3.3.1]nonane-2,4,9-triones, Org. Lett., 2006, 8, 5283–5285 CrossRef CAS.
- P. Nuhant, M. David, T. Pouplin, B. Delpech and C. Marazano, α, α′-Annulation of 2, 6-Prenyl-Substituted Cyclohexanone Derivatives with Malonyl Chloride: Application to a Short Synthesis of (±)-Clusianone. Formation and Rearrangement of a Biogenetic-Like Intermediate, Org. Lett., 2007, 9, 287–289 CrossRef CAS PubMed.
- N. M. Ahmad, V. Rodeschini, N. S. Simpkins, S. E. Ward and A. J. Blake, Synthesis of polyprenylated acylphloroglucinols using bridgehead lithiation: the total synthesis of racemic clusianone and a formal synthesis of racemic garsubellin A, J. Org. Chem., 2007, 72, 4803–4815 CrossRef CAS PubMed.
- A. S. Kende, B. Roth and P. J. Sanfilippo, Facile, palladium (II)-mediated synthesis of bridged and spirocyclic bicycloalkenones, J. Am. Chem. Soc., 1982, 104, 1784 CrossRef CAS.
- A. S. Kende, B. Roth, P. J. Sanfilippo and T. J. Blacklock, Mechanism and regioisomeric control in palladium (II)-mediated cycloalkenylations. A novel total synthesis of (.+-.)-quadrone, J. Am. Chem. Soc., 1982, 104, 5808–5810 CrossRef CAS.
- Y.-Q. Gao, Y. Hou, L. Zhu, J. Chen, R. Li, S.-Y. Zhang, Y.-P. He and W. Xie, Visible-Light Driven Synthesis of Polycyclic Benzo[d][1,3]oxazocine From 2-Aminochalcone, Chem. Commun., 2020, 56, 6739–6742 RSC.
- Y.-Q. Gao, Y. Hou, L. Zhu, G. Chen, D. Xu, S.-Y. Zhang, Y.-P. He and W. Xie, A bio-inspired synthesis of hybrid flavonoids from 2-hydroxychalcone driven by visible light, RSC Adv., 2019, 9, 29005 RSC.
- Z. Yang, Y. He and F. D. Toste, Biomimetic Approach to the Catalytic Enantioseiective Synthesis of Flavonoids, J. Am. Chem. Soc., 2016, 138, 9775–9778 CrossRef CAS PubMed.
- F. Wang, F. Chen, M. Qu, T. Li, Y. Liu and M. Shi, A Pd(II)-catalyzed asymmetric approach toward chiral [3.3.1]-bicyclic ketals using 2-hydroxyphenylboronic acid as a pro-bis(nucleophile), Chem. Commun., 2013, 49, 3360 RSC.
- Y. Ito, H. Aoyama, T. Hirao, A. Mochizaki and T. Saegusa, Cyclization reactions via oxo-. pi.-allylpalladium (II) intermediates, J. Am. Chem. Soc., 1979, 101, 494 CrossRef CAS.
- J. Drouin, M. A. Boaventura and J. M. Conia, Cyclization of acetylenic carbonyl compounds via their silyl enol ether derivatives: a new intramolecular C-vinylation induced by mercury(II) salts. Stereochemistry and functionalization of the intermediate vinylmercurial, J. Am. Chem. Soc., 1985, 107, 1726–1729 CrossRef CAS.
- H. Huang and C. J. Forsyth, A stereoselective total synthesis of (±)-erythrodiene, Tetrahedron Lett., 1993, 34, 7889–7890 CrossRef.
- H. Huang and C. J. Forsyth, Anti Selective Spirocarbomercuration: Synthesis and Stereochemistry of the Spirobicyclic Sesquiterpenes Spirojatamol and Erythrodiene, J. Org. Chem., 1995, 60, 2773–2779 CrossRef CAS.
- M. A. Boaventura and J. Drouin, Cyclisation de composés acétyléniques carbonyls via leur ether d’énol silylé. II. Synthése et réactivité de quelques ethers d’énols derives d’aldéhydes ou de cétones acétyléniques terminaux, Bull. Soc. Chim. Fr., 1987, 1015–1026 CAS.
- M. A. Boaventura, J. Drouin, F. Theobald and N. Rodier, La cyclisation des composés carbonyls acétyléniques via leurs ethers d’énol silyliques: une nouvelle C-vinylation intramoléculaire induite par les sels mercuriques. Détermination radiocristallographique de la stéréochimie de l’intermédiaire vinylmercurique, Bull. Soc. Chim. Fr., 1987, 1006–1014 CAS.
- C. J. Forsyth and J. Clardy, Total syntheses of (+)-and (-)-didemnenones A and B. Anti selectivity in the intramolecular carbomercuration reaction, J. Am. Chem. Soc., 1990, 112, 3497–3505 CrossRef CAS.
- H. Shigehisa, T. Jikihara, O. Takizawa, H. Nagase and T. Honda, An exceptional palladium-catalyzed alkenylation of silyl enol ether in the absence of a fluoride additive, Tetrahedron Lett., 2008, 49, 3983–3986 CrossRef CAS.
-
(a) D. Gravel, S. Benoît, H. Kumanovic and H. Sivaramakrishnan, On the palladium catalyzed reaction of methallyl-1, 1-diacetate with cyclic β-ketoesters. Intervention of hidden mechanisms, Tetrahedron Lett., 1992, 33, 1403 CrossRef CAS;
(b) D. Gravel, S. Benoît, H. Kumanovic and H. Sivaramakrishnan, Novel palladium catalyzed bicycloannulation of monoactivated cyclic ketones using a 1, 3-allylic diacetate and an enolzying catalyst, Tetrahedron Lett., 1992, 33, 1407 CrossRef CAS.
- A. Kuramochi, H. Usuda, K. Yamatsugu, M. Kanai and M. Shibasaki, Total synthesis of (±)-garsubellin A, J. Am. Chem. Soc., 2005, 127, 14200–14201 CrossRef CAS PubMed.
- Y. Shimizu, A. Kuramochi, H. Usuda, M. Kanai and M. Shibasaki, A new approach for the construction of a highly congested bicyclic system in polycyclic polyprenylated acylphloroglucinols (PPAPs), Tetrahedron Lett., 2007, 48, 4173–4177 CrossRef CAS.
- Y. Shimizu, S.-L. Shi, H. Usuda, M. Kanai and M. Shibasaki, The first catalytic asymmetric total synthesis of ent-hyperforin, Tetrahedron, 2010, 66, 6569–6584 CrossRef CAS.
- A. de O. Dias, R. Augusti, E. N. dos Santos and E. V. Gusevskaya, Convenient one-pot synthesis of 4, 8-dimethyl-bicyclo [3.3. 1] non-7-en-2-ol via platinum/tin catalyzed hydroformylation/cyclization of limonene, Tetrahedron Lett., 1997, 38, 41–44 CrossRef.
- McC. Finlay and J. C. Walton, Formation of bicyclo [3.2. 1] octane, bicyclo [4.2. 1] nonane, and bicyclo [3.3. 1] nonane by transannular radical cyclisations, Chem. Commun., 1987, 19, 1456–1457 Search PubMed.
- J. Quirante, C. Escolano, M. Massot and J. Bonjoch, Synthesis of 2-azabicyclo[3.3.1]nonanes by means of (carbamoyl)dichloromethyl radical cyclization, Tetrahedron, 1997, 53, 1391–1402 CrossRef CAS.
- J. Quirante, M. Torra, F. Diaba, C. Escolano and J. Bonjoch, Synthesis of enantiopure 2-azabicyclo [3.3.1] nonanes by a radical ring closure, Tetrahedron, 1999, 10, 2399–2410 CrossRef CAS.
- J. Ward and V. Caprio, A radical mediated approach to the core structure of huperzine A, Tetrahedron Lett., 2006, 47, 553–556 CrossRef CAS.
- J. Ward and V. Caprio, Synthesis of the bicyclo [3.3.1] nonane core of huperzine A and novel pyridine-fused tricycles by cyclisation of pyridine-based radicals, Heterocycles, 2009, 79, 791–804 CrossRef CAS PubMed.
- N. Z. Olga, V. N. Evgeniya, I. C. Vladimir, S. S. Irina, I. P. Danil, V. O. Mikhail and V. G. Natalia, Design, synthesis and biotest of a bicyclo [3.3. 1] nonane analogue of 2-amino-5, 6-dihydro-4H-1, 3-thiazine, Mendeleev Commun., 2010, 20, 323–325 CrossRef.
- G. M. Badger, J. W. Cook and T. Walker, The synthesis of piperidine derivatives. Part III. 5-Phenyl-1-azabicyclo[3.3.1] nonane, J. Chem. Soc., 1949, 1141–1144 RSC.
- F. Slowinski, O. B. Ayad, J. Vache, M. Saady, O. Leclerc and A. Lochead, Synthesis of new bridgehead substituted azabicyclo-[2.2. 1] heptane and-[3.3. 1] nonane derivatives as potent and selective α7 nicotinic ligands, Org. Lett., 2010, 12, 5004–5007 CrossRef CAS PubMed.
- B. Graetz, S. Rychnovsky, W.-H. Leu, P. Farmer and R. Lin, C2-Symmetric nitroxides and their potential as enantioselective oxidants, Tetrahedron: Asymmetry, 2005, 16, 3584–3598 CrossRef CAS.
- P. Michel and A. Rassat, An easy access to 2, 6-dihydroxy-9-azabicyclo, J. Org. Chem., 2000, 65, 2572–2573 CrossRef CAS PubMed.
- G. A. Pinna, G. Murineddu, M. M. Curzu, S. Villa, P. Vianello, P. A. Borea, S. Gessi, L. Toma, D. Colombo and G. Cignarella, Synthesis, modelling,
and μ-opioid receptor affinity of N-3 (9)-arylpropenyl-N-9 (3)-propionyl-3, 9-diazabicyclo [3.3. 1] nonanes, ILFarmaco, 2000, 55, 553–562 CrossRef CAS PubMed.
- G. Lesma, T. Pilati, A. Sacchetti and A. Silvani, New chiral diamino ligands as sparteine analogues. Application to the palladium-catalyzed kinetic oxidative resolution of 1-phenyl ethanol, Tetrahedron: Asymmetry, 2008, 19, 1363–1366 CrossRef CAS.
- U. Kuhl, W. Englberger, M. Haurand and U. Holzgrabe, Diazabicyclo [3.3. 1] nonanone-type Ligands for the Opioid Receptors, Arch. Pharm. Pharm. Med. Chem., 2000, 333, 226–230 CrossRef CAS.
- I. Stoll, A. Mix, A. B. Rozhenko, B. Neumann, H.-G. Stammler and J. Mattay, Kemp's triacid attached to octa-O-methyl resorc [4] arenes: conformations in solution and comparative binding studies with various 2-amino pyridines, Tetrahedron, 2008, 64, 3813–3825 CrossRef CAS.
-
(a) C.-M. Park, Concise synthesis of 3, 7-dioxa-9-aza-bicyclo [3, 3, 1]-nonane, J. Org. Chem., 2006, 71, 413–415 CrossRef CAS PubMed;
(b) D.-X. Tan, J. Zhou, C.-Y. Liu and F.-S. Han, Enantioselective Total Synthesis and Absolute Configuration Assignment of (+)-Tronocarpine Enabled by an Asymmetric Michael/Aldol Reaction, Angew. Chem., 2020, 59, 3834–3839 CrossRef CAS PubMed.
-
(a) R.-F. Yang and P.-Q. Huang, Studies towards an Enantioselective Total Synthesis of Sarain A: A Concise Asymmetric Construction of the Diazatricyclic Core, Chem. - Eur. J., 2010, 16, 10319–10322 CrossRef CAS PubMed;
(b) P. Molina, A. Arques, A. Tarraga and M. R. Obon, Synthesis and X-ray crystallographic study of 6, 12-epiiminodibenzo [b, f][1, 5] diazocines, Tetrahedron, 1998, 54, 997–1004 CrossRef CAS.
- R. Paredes, R. Abonia, J. Cadavid, R. Moreno-Fuquen, A. Jaramillo, A. Hormaza, A. Ramirez and A. Kennedy, A mechanistic study of the ammonolysis of alkyl acetoacetates in water. Formation of 1, 5-dimethyl-2, 6, 9-triaza-bicyclo [3.3. 1] nonane-3, 7-dione as the main product, Tetrahedron, 2002, 58, 55–60 CrossRef CAS.
- H. Rudler, A. Parlier, L. Hamon, P. Herson, P. Chaquin and J.-C. Daran, Straightforward access to tetrahydropyridine and piperidine-fused fluorolactones from pyridines and bis (trimethylsilyl) ketene acetals, Tetrahedron, 2009, 65, 5552–5562 CrossRef CAS.
- V. A. Petrov and W. Marshall, Acid catalyzed cyclodimerisation of 2, 2-bis (trifluoromethyl)-4-alkoxy-oxetanes and-thietanes. Synthesis of 2, 2, 6, 6-tetrakis(trifluoromethyl)-4,8-dialkoxy-1,5-dioxocanes and 3, 3, 7, 7- tetrakis(trifluoromethyl)-9-oxa-2, 6-dithia-bicyclo[3.3.1] nonane, Beilstein J. Org. Chem., 2010, 6, 46 Search PubMed.
- F. H. V. Chau and E. J. Corey, Short and simple synthesis of chelating bis-ethers and bis-amines in the bicyclo [3.3. 1] nonane series, Tetrahedron Lett., 2006, 47, 2581–2583 CrossRef CAS.
- J. Fleischhauert, M. Harmatatt, M. Kahramantt, A. Koslowskit and C. J. Welch, The determination of the absolute configuration of a chiral molecular tweezer using CD spectroscopy, Tetrahedron Lett., 1997, 38, 8655–8658 CrossRef.
- M. Harmata and C. L. Barnes, Molecular clefts. 3. The crystal structure of a chiral molecular tweezer and its guest, J. Am. Chem. Soc., 1990, 112, 5655 CrossRef CAS.
- C.-J. Wallentin, T. Wixe, O. F. Wendt, K.-E. Bergquist and K. Warnmark, Synthesis and Self-Aggregation of Enantiopure and Racemic Molecular Tweezers Based on the Bicyclo [3.3. 1] nonane Framework, Chem. – Eur. J., 2010, 16, 3994–4002 CrossRef CAS.
- E. D. Weil, K. J. Smith and R. J. Gruber, Transannular addition of sulfur dichloride to cyclooctadienes, J. Org. Chem., 1966, 31, 1669–1682 CrossRef CAS.
- E. J. Corey and E. Block, New synthetic approaches to symmetrical sulfur-bridged carbocycles, J. Org. Chem., 1966, 31, 1663–1668 CrossRef CAS.
- F. K. Lautenschlaeger, The reaction of sulfur dichloride with cis,cis-1,5-cyclooctadiene, Can. J. Chem., 1966, 44, 2813–2817 CrossRef CAS.
- A. A. Accurso, S.-H. Cho, A. Amin, V. A. Potapov, S. V. Amosova and M. G. Finn, Thia-, aza-, and selena [3.3. 1] bicyclononane dichlorides: Rates vs internal
nucleophile in anchimeric assistance, J. Org. Chem., 2011, 76, 4392–4395 CrossRef CAS PubMed.
- U. Herzog and H. Borrmann, Organosilicon chalcogenides with trisilane units-bicyclo [3.3.1]nonanes, bicyclo[3.2.2.]nonanes and spiro[4.4]nonanes, J. Organomet. Chem., 2004, 689, 564–574 CrossRef CAS.
- A. Robertson, C. Bradaric, C. S. Frampton, J. McNulty and A. Capretta, Novel chiral phosphines derived from limonene: the synthesis and structure of 4, 8-dimethyl-2-phosphabicyclo [3.3. 1] nonane, Tetrahedron Lett., 2001, 42, 2609–2612 CrossRef CAS.
- A. Polas, J. D. E. T. Wilton-Ely, A. M. Z. Slawin, D. F. Foster, P. J. Steynberg, M. J. Green and D. J. Cole-Hamilton, Limonene-derived phosphines in the cobalt-catalysed hydroformylation of alkenes, Dalton Trans., 2003, 4669–4677 RSC.
- J. F. Wang, Y. X. Liao, P. Y. Kuo, Y. H. Gau and D. Y. Yang, Synthesis and characterization of [1] benzopyrano [4, 3-d][1,3] benzooxazocin-13-one and its derivatives, Synlett, 2006, 17, 2791 Search PubMed.
- F. M. Moghaddam, Z. Mirjafary, H. Saeidian, S. Taheri, D. Doulabi and M. Kiamehr, Facile entry to polycyclic indolylhydroquinoline skeletons via tandem C-alkylation and intramolecular S-alkylation, Tetrahedron, 2010, 66, 134–138 CrossRef CAS.
- F. M. Moghaddam, Z. Mirjafary, H. Saeidian, S. Taheri and B. Soltanzadeh, Synthesis of eight-membered hydroquinolines related to alkaloid skeletons via addition of 4-hydroxycoumarin or 4-hydroxypyran-2-one to quinolinium salts, Tetrahedron, 2010, 66, 3678–3681 CrossRef CAS.
- F. M. Moghaddam, Z. Mirjafary, H. Saeidian, S. Taheri and M. R. Khodabakhshi, A new and convenient approach to heterotetracyclic benzoxazocines through addition of 1, 3-dicarbonyl compounds to quinolinium salts, Tetrahedron Lett., 2010, 51, 2704–2707 CrossRef CAS.
- F. M. Moghaddam, H. Saeidian, Z. Mirjafary, S. Taheri and M. Kiamehr, The synthesis of dibenzazocines via tandem dinucleophilic addition of phenols to quinolinium salts, Arkivoc, 2010, 11, 91–100 Search PubMed.
- F. M. Moghaddam, S. Taheri, Z. Mirjafary, H. Saeidian, M. Kiamehr and M. Tafazzoli, A Facile Synthesis of Bridged Polycyclic Naphthooxazocine Skeletons: Eight-Membered-Ring Constructions via Tandem Dinucleophilic Addition of Naphthalenols to Quinolinium Salts, Helv. Chim. Acta, 2011, 94, 142–147 CrossRef CAS.
- S. Mondal, R. Paira, A. Maity, S. Naskar, K. B. Sahu, A. Hazra, P. Saha, S. Banerjee and N. B. Mondal, Basic alumina supported tandem synthesis of bridged polycyclic quinolino/isoquinolinooxazocines under microwave irradiation, Tetrahedron Lett., 2011, 52, 4697–4700 CrossRef CAS.
- R. Paira, S. Mondal, A. Maity, K. B. Sahu, S. Naskar, P. Saha, A. Hazra, S. Kundu, S. Banerjee and N. B. Mondal, An easy access to diversely fused dioxa-2-aza-tricyclo [n. 3.1. 02, n] tetra/pentadecanes under solvent-free condition, Tetrahedron Lett., 2011, 52, 5516–5520 CrossRef CAS.
- V. A. Rassadin, A. A. Tomashevskiy, V. V. Sokolov and A. A. Potekhin, Khim. Geterotsykl. Soed., 2008, 605–617 (Chem. Heterocycl. Compd., 2008, 44, 474–485) Search PubMed.
- V. A. Rassadin, A. A. Tomashevskiy, V. V. Sokolov, A. Ringe, J. Magull and A. Meijere, Facile Access to Bicyclic Sultams with Methyl 1-Sulfonylcyclopropane-1-carboxylate Moieties, Eur. J. Org. Chem., 2009, 2635–2641 CrossRef CAS.
- V. A. Rassadin, D. S. Grosheva, A. A. Tomashevskiy, V. V. Sokolov, D. S. Yufit, S. I. Kozhushkov and A. de Meijere, Bicyclic sultams with a nitrogen at the bridgehead and a sulfur atom in the apex position: Facile preparation and conformational properties, Eur. J. Org. Chem., 2010, 3481–3486 CrossRef CAS.
- A. Srikrishna and D. Vijaykumar, Facile
formation of chiral bicyclo [3.3. 1] nonenes via regioselective cyclopropane cleavage of 1-methyltricyclo [4.3. 0.02, 9] nonan-8-ols, Tetrahedron Lett., 1998, 39, 5833–5834 CrossRef CAS.
- M. Abe and M. Nakada, Synthetic studies on phloroglucins: a new approach to the bicyclo[3.3.1]nonane system via the regioselective ring-opening of the methoxycyclopropane, Tetrahedron Lett., 2007, 48, 4873–4877 CrossRef CAS.
- M. Abe and M. Nakada, New construction of the bicyclo [3.3. 1] nonane system via Lewis acid promoted regioselective ring-opening reaction of the tricyclo [4.4. 0.05, 7] dec-2-ene derivative, Tetrahedron Lett., 2006, 47, 6347–6351 CrossRef CAS.
- P. Wipf, P. Jeger and Y. Kim, Thiophilic ring-opening and rearrangement reactions of epoxyketone natural products, Bioorg. Med. Chem. Lett., 1998, 8, 351 CrossRef CAS PubMed.
- J. T. Hammill, J. Contreras-García, A. M. Virshup, D. N. Beratan, W. Yang and P. Wipf, Synthesis and chemical diversity analysis of bicyclo[3.3.1]non-3-en-2-ones, Tetrahedron, 2010, 66, 5852–5862 CrossRef CAS PubMed.
- Y.-F. Wang and S. Chiba, Mn (III)-mediated reactions of cyclopropanols with vinyl azides: synthesis of pyridine and 2-azabicyclo [3.3. 1] non-2-en-1-ol derivatives, J. Am. Chem. Soc., 2009, 131, 12570–12572 CrossRef CAS.
-
(a) T. Sasaki, S. Eguchi and T. Tom, Synthesis of adamantane derivatives. XII. Schmidt reaction of adamantan-2-one, J. Org. Chem., 1970, 35, 4109–4114 CrossRef CAS;
(b) J. G. Henkel and J. T. Hane, Efficient synthesis of barbaralane, J. Org. Chem., 1983, 48, 3858–3859 CrossRef CAS.
-
(a) K. Kimoto, T. Imagawa and M. Kawanisi, Reduction of 7-Methylenebicyclo [3.3.1] nonan-3-one and Related Compounds: Structural Investigation of the Products, Bull. Chem. Soc. Jpn., 1972, 45, 3698–3702 CrossRef CAS;
(b) J.-H. Liu and P. Kovacic, Chemistry of adamantanes and related compounds. Diazotization of endo-7-aminomethylbicyclo [3.3. 1] nonan-3-one and endo-3-aminomethylbicyclo [3.3. 1] nonane, J. Org. Chem., 1973, 38, 3462–3466 CrossRef CAS.
- P. Camps, R. E. Achab, M. Font-Bardia, D. G∼irbig, J. Morral, D. Mufioz-Torrero, X. Solans and M. Simon, Easy synthesis of 7-alkylbicyclo [3.3. 1] non-6-en-3-ones by silica gel-promoted fragmentation of 3-alkyl-2-oxaadamant-1-yl mesylates, Tetrahedron, 1996, 52, 5867–5880 CrossRef CAS.
- W. Fischer and C. A. Grog, Infrared and Raman spectroscopic studies of benzothiazolinic spiropyrans and merocyanines, Helv. Chim. Acta, 1978, 1588 CrossRef CAS.
- A. E. Lukach, A. N. Santiago and R. A. Rossi, Reactions of 1,3-Dihaloadamantanes with Carbanions in DMSO: Ring-Opening Reactions to Bicyclo[3.3.1]nonane Derivatives by the SRN1 Mechanism, J. Org. Chem., 1997, 62, 4260–4265 CrossRef CAS PubMed.
- L. Cermenati, D. Dondi, M. Fagnoni and A. Albini, Titanium dioxide photocatalysis of adamantine, Tetrahedron, 2003, 59, 6409–6414 CrossRef CAS.
- R. C. Fort Jr., Adamantane. The Chemistry of Diamond Molecules, Marcel Dekker, New York, 1976 Search PubMed.
- E. I. Bagrii, Adamantany (Adamantanes), Nauka, Moscow, 1989 Search PubMed.
- C. A. Grob and W. Baumann, Helv. Chim. Acta, 1955, 38, 594 CrossRef CAS.
- C. A. Grob, Das Prinzip der Äthylogie in der organischen Chemie, Experientia, 1957, 13, 126 CrossRef CAS.
- H. Stetter and P. Tacke, Neue Synthesen von 2.4-disubstituierten s-Triazinen, Angew. Chem., 1962, 74, 354 CrossRef.
- C. A. Grob and W. Schwartz, Die synchrone Fragmentierung von γ-Amino-cycloalkylhalogeniden 1. Teil. Die Solvolyse von 1-Amino-3-brom-adamantanen. Fragmentierungs-Reaktionen. 10. Mitteilung, Helv. Chim. Acta, 1964, 47, 1870 CrossRef CAS.
- A. K. Shiryaev, I. K. Moiseev and A. V. Bartsev, A New Fragmentation of N-Substituted 1-Halo-3-aminoadamantanes (I), Zh. Obshch. Khim., 1990, 60, 2800 CAS.
- M. Y. Skomorokhov and Y. N. Klimochkin, Cyclization of 7-Methylene-3-[2-(4-nitrobenzoyloxy) ethoxy]-bicyclo[3.3.1]non-2-ene in the Presence of Acids, Russ. J. Org. Chem., 2001, 37, 1050 CrossRef CAS.
- M. Y. Skomorokhov and Y. N. Klimochkin, Reaction of Dibromoadamantanes with Glycols in the Presence
of Sodium Glycolate, Russ. J. Org. Chem., 2011, 47, 1811–1816 CrossRef CAS.
- L. Klaić, J. Veljković and K. Mlinarić-Majerski, Convenient synthesis of novel 1, 3, 7-trisubstituted bicyclo [3.3. 1] nonane derivatives, Synth. Commun., 2002, 32, 89–97 CrossRef.
- E. V. Nurieva, I. S. Semenova, V. N. Nuriev, D. V. Shishov, I. I. Baskin, O. N. Zefirova and N. S. Zefirov, Diels-alder reaction as a synthetic approach to bicyclo [3.3. 1] nonane colchicine analogs, Russ. J. Org. Chem., 2010, 46, 1892–1895 CrossRef CAS.
- D. D. Dixon, D. Sethumadhavan, T. Benneche, A. R. Banaag, M. A. Tius, G. A. Thakur, A. Bowman, J. T. Wood and A. Makriyannis, Heteroadamantyl cannabinoids, J. Med. Chem., 2010, 53, 5656–5666 CrossRef CAS PubMed.
- M. Shibuya, M. Tomizawa, I. Suzuki and Y. Iwabuchi, 2-Azaadamantane N-Oxyl (AZADO) and 1-Me-AZADO: Highly Efficient Organocatalysts for Oxidation of Alcohols, J. Am. Chem. Soc., 2006, 128, 8412–8413 CrossRef CAS PubMed.
- K. Takahashi, M. Watanabe and T. Honda, Highly Efficient Stereocontrolled Total Synthesis of (+)-Upial, Angew. Chem., Int. Ed., 2008, 47, 131–133 CrossRef CAS PubMed.
- K. Takahashi, R. Akao and T. Honda, Efficient diastereoselective synthesis of trifarane-type sesquiterpenes, trifarienols A and B, J. Org. Chem., 2009, 74, 3424–3429 CrossRef CAS PubMed.
- K. Sparrow, D. Barker and M. A. Brimble, An efficient synthesis of 3-alkyl-1, 5, 3-dioxazepanes and their use as electrophiles in double-Mannich reactions, Tetrahedron, 2012, 68, 1017–1028 CrossRef CAS.
-
(a) T. Mukaiyama and T. Hoshino, The Reactions of Primary Nitroparaffins with Isocyanates, J. Am. Chem. Soc., 1960, 82, 5339 CrossRef CAS;
(b) S. C. Sharma and K. B. G. Torssell, Silyl nitronates in organic synthesis. Routes to heterocycles, Acta Chem. Scand., 1979, 33, 379 CrossRef.
- D. G. J. Young and D. Zeng, A Preliminary Approach to Nonenolizable β,β-Tricarbonyls: Assembly of a Hyperevolutin Prototype, J. Org. Chem., 2002, 67, 3134–3137 CrossRef CAS PubMed.
- S. H. Bertz and G. Dabbagh, Synthesis of Bicyclo [3.3.1] nonane Derivatives under Physiological Conditions, Angew. Chem., Int. Ed. Engl., 1982, 21, 306–307 CrossRef.
- S. H. Bertz, Series on chemistry under physiological conditions. Part 7. Tetramethyl 3,7-dihydroxybicyclo[3.3.1]nona-2,6-diene-2,4,6,8-tetracarboxylate: a useful companion to Meerwein's ester. Topological analysis of bicyclo[3.3.1]nonane synthesis, J. Org. Chem., 1985, 50, 3585–3592 CrossRef CAS.
- R. D. J. Sands, Reaction of dimethyl 3-oxoglutarate with 1,3-dicarbonyl compounds, J. Org. Chem., 1983, 48, 3362–3363 CrossRef CAS.
- I. A. McDonald and A. S. Dreiding, Triasteranetrione, Helv. Chim. Acta, 1973, 56, 2523–2534 CrossRef CAS.
- H. Stetters and J. Lennartz, Über Verbindungen mit Urotropin-Struktur, LIX. Ringschlüsse auf der Basis des Bicyclo [3.3. 1] nonan-3, 7, 9-trions, Liebigs Ann. Chem., 1977, 1807–1816 CrossRef.
- H. Stetter and J. Mayer, Über Verbindungen mit Urotropin-Struktur, XV. Synthese des 2-Oxa-adamantan-Ringsystems, Chem. Ber., 1959, 92, 2664–2666 CrossRef CAS.
- G. Aranda, J. M. Bernassau, M. Fetizon and I. Hanna, Conformational analysis, synthesis, and carbon-13 spectroscopy of 9, 9-dimethylbicyclo [3.3. 1] nonane derivatives, J. Org. Chem., 1985, 50, 1156–1161 CrossRef CAS.
- P. Camps, R. El Achab, M. Font-Bardia, D. GoÈrbig, J. Morral, D. MunÄoz-Torrero, X. Solans and M. Simon, Easy synthesis of 7-alkylbicyclo [3.3. 1] non-6-en-3-ones by silica gel-promoted fragmentation of 3-alkyl-2-oxaadamant-1-yl mesylates, Tetrahedron, 1996, 52, 5867–5880 CrossRef CAS.
- P. Camps, A. GonzaÂlez, D. MunÄoz-Torrero, M. Simon, A. ZuÂnÄiga, M. A. Martins, M. Font-Bardia and X. Solans, Synthesis of Polysubstituted Bicyclo [3.3. 1] nonane-3, 7-diones from Cyclohexa-2, 5-dienones and Dimethyl 1, 3-Acetonedicarboxylate, Tetrahedron, 2000, 56, 8141–8151 CrossRef CAS.
- C.-J. Wallentin, E. Orentas, E. Butkus and K. Wärnmark, Baker's yeast for sweet dough enables large-scale synthesis of enantiomerically pure bicyclo [3.3. 1] nonane-2, 6-dione, Synthesis, 2009, 5, 0864–0867 Search PubMed.
- K. Naemura, H. Ida and R. Fukuda, Lipase YS-catalyzed enantioselective transesterification of alcohols of bicarbocyclic compounds, Bull. Chem. Soc. Jpn., 1993, 66, 573 CrossRef CAS.
- M. Miyazawa, M. Nobata, S. Okamura, O. Muraoka, G. Tanabe and H. Kameoka, Stereoselective reduction of (±)-bicyclo [3.3. 1] nonane-2, 6-dione by microorganisms, J. Chem. Technol. Biotechnol., 1998, 71, 281–284 CrossRef CAS.
- M. Carlquist, C.-J. Wallentin, K. Wärnmark and M. F. Gorwa-Grauslund, Genetically engineered Saccharomyces cerevisiae for kinetic resolution of racemic bicyclo [3.3. 1] nonane-2, 6-dione, Tetrahedron: Asymmetry, 2008, 19, 2293–2295 CrossRef CAS.
- T. Okazaki, E. Terakawa, T. Kitagawa and K. Takeuchi, Solvolysis of 2-Bicyclo[3.2.2]nonyl p-Toluenesulfonate. Evidence for the Formation of Classical Carbocation Intermediates, J. Org. Chem., 2000, 65, 1680–1684 CrossRef CAS.
- J. P. Schaefer, L. S. Endres and M. D. Moran, Bicyclo [3.3. 1] nonanes. III. Preparation and reactions of bicyclo [3.2. 2] nonanes, J. Org. Chem., 1967, 32, 3963 CrossRef CAS.
- P. Rademacher and R. F. Wiesmann, Transanulare Wechselwirkungen in difunktionellen mittleren Ringen, 4. Spektroskopische und theoretische Untersuchungen an bicyclischen Boraalkenen, Chem. Ber., 1994, 127, 509 CrossRef CAS.
- J. P. Schaefer, J. C. Lark, C. A. Flegal and L. M. Honig, Bicyclo [3.3. 1] nonanes. II. Synthesis and reactions of simple derivatives, J. Org. Chem., 1967, 32, 1372 CrossRef CAS.
-
(a) T. Momose, S. Atarashi and O. Muraoka, Novel steric factors operating on the Baeyer-villiger oxidationof bicyclo [3,3,1] Nonan-3-one system, Tetrahedron Lett., 1974, 15, 3697–3700 CrossRef;
(b) T. Momose, O. Muraoka, S. Atarashi and T. Horita, Bicyclo [3. 3. 1] nonanes as Synthetic Intermediates. IV. Behavior of Bicyclo [3. n. 1] alkan-3-ones toward the Baeyer-Villiger Oxidation, Chem. Pharm. Bull., 1979, 27, 222 CrossRef CAS;
(c) T. Momose and S. Atarashi, Bicyclo [3. 3. 1] nonanes as Synthetic Intermediates. V. The Baeyer-Villiger Oxidation of Bicyclo [3. 3. 1] nonane-3, 7-dione and Its Congeners, Chem. Pharm. Bull., 1979, 27, 824 CrossRef CAS.
- E. Butkus and S. Stoncius, Stereoselective Baeyer–Villiger oxidation of some bridged bicyclic diketones, J. Chem. Soc., Perkin Trans. 1, 2001, 1885–1888 RSC.
- E. Butkus, S. Stoncius and A. Zilinskas, Chirality, 2001, 13, 694–698 CrossRef CAS PubMed.
- S. Stoncius, U. Berg and E. Butkus, Chiral bicyclic keto lactones: Determination of the absolute configuration by the study of chiroptical properties and chemical correlation, Tetrahedron: Asymmetry, 2004, 15, 2405–2413 CrossRef CAS.
- P. J. Stephens, D. M. McCann, F. J. Devlin, T. C. Flood, E. Butkus, S. Stoncius and J. R. Cheeseman, Determination of Molecular Structure Using Vibrational Circular Dichroism Spectroscopy: The Keto-lactone Product of Baeyer−Villiger Oxidation of (+)-(1R,5S)-Bicyclo[3.3.1]nonane-2,7-dione, J. Org. Chem., 2005, 70, 3903–3913 CrossRef CAS PubMed.
- A. Gambacorta, D. Tofani, P. Lupattelli and A. Tafi, Desymmetrisation of meso-methylcyclooctanones. Highly enantioselective synthesis of C8 syn-isoprenoid and syn, syn-deoxypropionate subunits from a bicyclo[3.3.1]nonane precursor, Tetrahedron Lett., 2002, 43, 2195–2198 CrossRef CAS.
- A. Gambacorta, S. Turchetta and M. E. Farah, BICYCLO[3.3.1]nonane approach to polymethyl alternating systems, the syn-and anti-1,5-dimethyl systems, Gazz. Chim. Ital., 1992, 122, 527–529 CAS.
- G. Snatzke and B. Wolfram, Circulardichroismus—L: Chiroptische eigenschaften von thiaadamantanderivaten, Tetrahedron, 1972, 28, 655 CrossRef CAS.
- H. Gerlach, Racematspaltung und Bestimmung der absoluten Konfiguration von 2, 6-disubstituierten Bicyclo [3.3. 1] nonanen, Helv. Chim. Acta, 1978, 61, 2773 CrossRef CAS.
- U. Berg and E. Butkus, Enantiomer separation and circular dichroism spectra of bicyclo[3.3.1] nonanedione, J. Chem. Res., Synop., 1993, 116–117 CAS.
- U. Berg and E. Butkus, An analysis of the circular dichrosm of bicyclo [3.3.1] nonanediones, J. Chem. Res., Synop., 1994, 356–357 CAS.
- E. Butkus, S. Stoncius and A. Zilinskas, Determination of the absolute configuration of bicyclo [3.3. 1] nonane-2, 7-dione by circular dichroism spectroscopy and chemical correlation, Chirality, 2001, 13, 694 CrossRef CAS PubMed.
- G. Hoffmann and R. Wiartalla, Ein rascher Zugang zu optisch aktiven Adamantan-Derivaten, Tetrahedron Lett., 1982, 23, 3887 CrossRef CAS.
- E. Butkus, U. Berg, A. Zilinskas, R. Kubilius and S. Stoncius, Enantiomer separation and absolute configuration of densely functionalized 2-oxatricyclo [4.3. 1.03, 8] decanes by CD spectroscopy and chemical correlation, Tetrahedron: Asymmetry, 2000, 11, 3053 CrossRef CAS.
- E. Butkus, A. Zilinskas, S. Stoncius, R. Rozenbergas, M. Urbanova, V. Setnicka, P. Bour and K. Volka, Synthesis and chiroptical properties of enantiopure tricyclo [4.3. 0.03, 8] nonane-4, 5-dione (twistbrendanedione), Tetrahedron: Asymmetry, 2002, 13, 633 CrossRef CAS.
- J. Malinauskiene, P. Kadziauskas, A. Malinauskas and J. Kulys, Enzymatic Enantioseparation of Bicyclo [3.3. 1] nonane-2, 6-diones, Monatsh. Chem., 1999, 130, 1513 CrossRef CAS.
- E. Butkus, U. Berg, J. Malinauskiene and J. Sandström, Synthesis and Chiroptical Properties of Methanocycloocta[b]indoles, Synthesis and Chiroptical Properties of Methanocycloocta[b]indoles, J. Org. Chem., 2000, 65, 1353 CrossRef CAS PubMed.
-
(a) D. A. Lightner and J. E. Gurst, Organic Conformational Analysis and Stereochemistry, Wiley, New York, 2000, ch. 4, pp. 63–94 Search PubMed;
(b) D. A. Lightner, In Circular Dichroism: Principles and Applications, ed. Berova, N., Nakanishi, K. and Woody, R. W., Wiley, New York, 2000, ch. 10, pp. 261–303 Search PubMed.
- P. J. Stephens, D. M. McCann, E. Butkus, S. Stoncius, J. R. Cheeseman and M. J. Frisch, Determination of absolute configuration using concerted ab initio DFT calculations of electronic circular dichroism and optical rotation: bicyclo [3.3. 1] nonane diones, J. Org. Chem., 2004, 69, 1948–1958 CrossRef CAS.
- V. Rodeschini, N. S. Simpkins and C. Wilson, Kinetic resolution in a bridgehead lithiation mediated by a chiral Bis-lithium amide: Assignment of the absolute configuration of clusianone, J. Org. Chem., 2007, 72, 4265–4267 CrossRef CAS PubMed.
- E. Butkus, Stereocontrolled synthesis and reactions of bicyclo [3.3. 1] nonanes, Synlett, 2001, 1827 CrossRef CAS.
-
(a) B. M. Fraga, Natural sesquiterpenoids, Nat. Prod. Rep., 2002, 19, 650 RSC;
(b) H. Usuda, M. Kanai and M. Shibasaki, Studies toward the total synthesis of garsubellin A: synthesis of 8-deprenyl-garsubellin A, Tetrahedron Lett., 2002, 43, 3621 CrossRef CAS;
(c) S. J. Sepssard and B. M. Stoltz, Progress toward the synthesis of garsubellin A and related phloroglucins: the direct diastereoselective synthesis of the bicyclo [3.3. 1] nonane core, Org. Lett., 2002, 4, 1943 CrossRef PubMed.
- H. M. C. Ferraz, L. F. Silva Jr. and T. O. Vieira, Thallium (III) in organic synthesis, Synthesis, 1999, pp. 2001–2023 Search PubMed.
- E. Butkus, A. Zilinskas, S. Stoncius, R. Rozenbergas, M. Urbanova, V. Setnicka and P. B. K. Volka, Synthesis and chiroptical properties of enantiopure tricyclo [4.3. 0.03, 8] nonane-4, 5-dione (twistbrendanedione), Tetrahedron: Asymmetry, 2002, 13, 633–638 CrossRef CAS.
- R. Kubilius, G. Bagdziunas and E. Butkus, Ring contraction/transannular cyclization of chiral bicyclo [3.3. 1] nonanediones mediated by thallium (III) nitrate, Tetrahedron Lett., 2011, 52, 346–348 CrossRef CAS.
- M. D. Duque, P. Camp, L. Profire, S. Montaner, S. Vázquez, F. X. Sureda, J. Mallol, M. López-Querol, L. Naesens, E. De Clercq, S. R. Prathalingam and J. M. Kelly, Synthesis and pharmacological evaluation of (2-oxaadamant-1-yl) amines, Bioorg. Med. Chem., 2009, 17, 3198–3206 CrossRef CAS PubMed.
- A. G. Geigy, Patent GB 1123609, 1968.
- A. R. Gagneux and R. Meier, 1-Substituted 2-heteroadamantanes, Tetrahedron Lett., 1969, 17, 1365 CrossRef PubMed.
- H. Stetter and P. Tacke, Uber Verbindungen mit Urotropin-Struktur, XXVII Uber eine Fragmentierung in der Adammantan-Reihe, Chem. Ber., 1963, 25, 694 CrossRef.
-
(a) P. Camps, R. El Achab, M. Font-Bardia, D. M. Görbig, J. Morral, D. Muñoz-Torrero, X. Solans and M. Simon, Easy synthesis of 7-alkylbicyclo [3.3. 1] non-6-en-3-ones by silica gel-promoted fragmentation of 3-alkyl-2-oxaadamant-1-yl mesylates, Tetrahedron, 1996, 52, 5867 CrossRef CAS;
(b) P. Camps, R. El Achab, D. M. Görbig, J. Morral, D. Muñoz-Torrero, A. Badia, J. E. Baños, N. M. Vivas, X. Barril, M. Orozco and F. J. Luque, Synthesis, in Vitro Pharmacology, and Molecular Modeling of Very Potent Tacrine−Huperzine A Hybrids as Acetylcholinesterase Inhibitors of Potential Interest for the Treatment of Alzheimer's Disease, J. Med. Chem., 1999, 42, 3227 CrossRef CAS PubMed.
- P. A. Krasutskii, Y. A. Serguchev, A. G. Yurchenko and A. V. Khotkevich, Synthesis, in Vitro Pharmacology, and Molecular Modeling of Very Potent Tacrine–Huperzine A Hybrids as Acetylcholinesterase Inhibitors of Potential Interest for the Treatment of Alzheimer's Disease, Teor. Eksp. Khim., 1983, 19, 229–232 CAS.
- P. A. Krasutskii, A. V. Khotkevich, Y. A. Serguchev and A. G. Yurchenko, Mechanism of transannular bromination reactions of diolefins of the bicyclo [3.3. 1] nonane series, Teor. Eksp. Khim., 1985, 21, 52–56 CAS.
- Y. A. Serguchev, A. V. Khotkevich and P. A. Krasutskii, Dokl. Akad. Nauk. UkrSSR Ser. B., 1985, 51–54 (Chem. Abstr., 1986, 105, 152577t) CAS.
- P. A. Krasutskii, N. B. Ambrosienko, V. N. Rodionov, A. G. Yurchenko, Z. N. Parnes and G. I. Bolestova, Zh. Org. Khim., 1985, 21, 1465–1470 (Chem. Abstr., 1986, 104, 186041h) CAS.
- Y. A. Serguchev, L. F. Lourie and M. V. Ponomarenko, Selective transannular cyclization of 3, 7-bismethylenebicyclo [3.3.1] with F-TEDA-BF4 in protic, Mendeleev Commun., 2000, 10, 121–123 CrossRef.
- Y. A. Serguchev, L. F. Lourie and M. V. Ponomarenko, Difluorination ability of F-TEDA-BF4 in the transannular cyclization of bicyclo[3.3.1]nonane dienes in monoglyme, Mendeleev Commun., 2002, 12, 1–2 CrossRef.
- Y. A. Serguchev, M. V. Ponomarenko, L. F. Lourie and A. N. Chernega, Synthesis of halo-fluoro-substituted adamantanes by electrophilic transannular cyclization of bicyclo [3.3. 1] nonane dienes, J. Fluorine Chem., 2003, 123, 207–215 CrossRef CAS.
- T. R. Walters, W. W. Zajac Jr. and J. M. Woods, New reagents for the synthesis of gem-halonitro compounds from oximes, J. Org. Chem., 1991, 56, 316 CrossRef CAS.
- P. Camps and D. Munoz-Torrero, Alternative syntheses of bridgehead polycyclic 1,2-diamines and 2-aminoalcohols from di- and mono-oximes of some bicyclic diketones: Highly improved synthesis of tricyclo[3.3.1.03,7]nonane-3,7-diamine, Tetrahedron Lett., 1994, 35, 3187 CrossRef CAS.
- W. W. Zajac Jr., U.S. Pat., 5105301, 1992 Search PubMed.
- W. T. Borden and T. Ravindranathan, Transannular ring closure by reduction of cyclooctane-1, 5-diones. Synthesis of a bisnoradamantan-1-o1, J. Org. Chem., 1971, 36, 4125 CrossRef CAS.
-
(a) G. L. Baughman, Dibromination of Adamantane, J. Org. Chem., 1964, 29, 238 CrossRef CAS;
(b) H. Stetter and P. Tacke, Uber Verbindungen mit Urotropin-Struktur, XXVII Uber eine Fragmentierung in der Adammantan-Reihe, Chem. Ber., 1963, 25, 694 CrossRef.
- J. A. Zalikowski, K. E. Gilbert and W. T. Borden, Oxidation of 7-(hydroxymethyl) bicyclo [3.3. 1] nonan-3-ol. Convenient synthesis of bicyclo [3.3. 1] nonane-3, 7-dione, J. Org. Chem., 1980, 45, 346 CrossRef CAS.
- S. H. Bertz, Tetramethyl 3,7-dihydroxybicyclo[3.3.1] nona-2,6-diene-2,4,6,8-tetracarboxylate: a useful companion to meerwein's ester. Topological analysis of bicyclo[3.3.1] nonane synthesis, J. Org. Chem., 1985, 50, 3585–3592 CrossRef CAS.
- S. Ioannou and A. V. Nicolaides, An improved synthesis of diiodonoradamantane, Tetrahedron Lett., 2009, 50, 6938–6940 CrossRef CAS.
- M. V. Ponomarenko, Y. A. Serguchev, B. V. Ponomarenko, G.-V. Roschenthaler and A. A. Fokin, Experimental and theoretical studies on the transannular cyclizations of 3, 7-dimethylenebicyclo [3.3. 1] nonane with polyfluoroalkyl radicals, J. Fluorine Chem., 2006, 127, 842–849 CrossRef CAS.
- T. Mori, K. Kimoto, M. Kawanisi and H. Nozaki, Preparation and reactions of 1,5-polymethylene-bridged 3,7-dimethylenebicyclo[3.3.1]nonan-9-ones, Tetrahedron Lett., 1969, 10, 3653 CrossRef.
- A. G. Yurchenko, A. T. Veroshchenko and F. N. Stepanov, Photoisomerization of 3, 7-dimethylenebicyclo [3.3.1] nonane, Zh. Org. Khim., 1970, 6, 189–194 CAS.
- N. V. Averina, G. S. Borisova, A. A. Borisenko and N. S. Zefirov, Photochemistry of 3, 7-Bis(arylmethylene)bicyclo[3.3.1]nonane Derivatives, Russ. J. Org. Chem., 2001, 37, 957–963 CrossRef CAS.
- E. P. Butkus, A. J. Zilinskas, P. P. Kadziauskas and R. R. Kubilius, Russ. Chem. Bull., 1997, 46(2), 328–330 CrossRef CAS.
- E. Butkus, R. Kubilius, S. Stoncius and A. Z. ilinskas, ntramolecular ring closure via ether bond in reaction of α, α′-halogeno bicyclo [3.3.1] nonanediones under basic conditions, J. Chem. Soc., Perkin Trans. 1, 1999, 1431–1436 RSC.
- E. Beckmann, N. Bahr, O. Cullmann, F. Yang, M. Kegel, M. Vögtle, K. Exner, M. Keller, L. Knothe and H. Prinzbach, Proximate,“Parallel-In-Plane” Preoriented Bis (diazenes)− In-Plane Delocalized Bis (homoconjugated) 4N/5 (6) e Anions, Eur. J. Org. Chem., 2003, 4248–4264 CrossRef CAS.
- K. Takeuchi, Y. Ohga, M. Yoshida, K. Ikai, T. Shibata, M. Kato and A. Tsugeno, Solvolyses of 2-Oxo Bridgehead Compounds: A Critical Examination of π-Conjugative Stabilization of α-Carbonyl Carbocations, J. Org. Chem., 1997, 62, 5696 CrossRef CAS.
- K. Takeuchi, Y. Ohga, K. Tokunaga and A. Tsugeno, Formation of a propellanone in the solvolysis of a 2-oxo bicyclic bridgehead compound, Tetrahedron Lett., 1996, 37, 8185 CrossRef CAS.
- K. Tokunaga, S. Tachibana, T. Okazaki, Y. Ohga, T. Kitagawa and K. Takeuchi, Structural factors for the formation of propellane-type products in the solvolysis of bicyclic bridgehead compounds, J. Org. Chem., 2000, 65, 4964–4972 CrossRef CAS PubMed.
- T. Y. Baranova, O. N. Zefirova, N. V. Averina, V. V. Boyarskikh, G. S. Borisova, N. V. Zyk and N. S. Zefirov, Russ. J. Org. Chem., 2007, 43, 1196–1201 CrossRef CAS.
- M. L. Trudell and J. M. Cook, Total synthesis of (.+-.)-suaveoline, J. Am. Chem. Soc., 1989, 111, 7504 CrossRef CAS.
- X. Fu and J. M. Cook, General approach for the synthesis of ajmaline-related alkaloids. Enantiospecific total synthesis of (-)-suaveoline, (-)-raumacline, and (-)-Nb-methylraumacline, J. Org. Chem., 1993, 58, 661 CrossRef CAS.
- P. D. Bailey, K. M. Morgan, D. I. Smith and J. M. Vernon, New asymmetric routes to ajmaline and suaveoline indole alkaloids, J. Chem. Soc., Perkin Trans. 1, 2000, 3566 RSC.
- P. D. Bailey, K. M. Morgan and D. I. Smith, The total synthesis of (−)-suaveoline, J. Chem. Soc., Perkin Trans. 1, 2000, 3578 RSC.
- T. Yu. Baranova, O. N. Zefirova, N. V. Averina, V. V. Boyarskikh, G. S. Borisova, N. V. Zyk and N. S. Zefirov, Synthetic approach to preparation of indole derivatives fused with a bicyclo [3.3.1] nonane framework, Russ. J. Org. Chem., 2007, 43, 1196–1201 CrossRef CAS.
- E. Butkus, U. Berg, J. Malinausiene and J. Sandstrom, Synthesis and Chiroptical Properties of Methanocycloocta[b]indoles, J. Org. Chem., 2000, 65, 1353 CrossRef CAS.
- H. Klusacek, H. Musso and V. Asterane, Studien in der Tricyclo[3.3.1.02.8]nonan-Reihe, Chem. Ber., 1970, 103, 3066 CrossRef CAS.
- P. Kadziauskas, E. Butkus, J. Vasiulyte, N. V. Averina and N. S. Zefirov, Synthesis of indoles condensed with a bicyclo [3.3.1]nonane skeleton, Chem. Heterocycl. Compd., 1979, 315–318 CAS.
- E. Butkus, U. Berg, J. Malinauskieneù and J. Sandstrom, Synthesis and Chiroptical Properties of Methanocycloocta[b]indoles, J. Org. Chem., 2000, 65, 1353–1358 CrossRef CAS PubMed.
- U. Berg, E. Butkus and A. Stončius, Stereochemistry of the reduction of bicyclo[3.3.1]nonane-2,9-dione by complex hydrides, J. Chem. Soc., Perkin Trans. 2, 1995, 97–102 RSC.
- E. Butkus, J. Malinauskiene and S. Stoncius, Synthesis, chiroptical properties and absolute configuration of spiro [1,3-benzodioxole-methanocyclooct[b]indole], Org. Biomol. Chem., 2003, 1, 391–394 RSC.
- C.-S. Wang, Structure and chemistry of 4-hydroxy-6-methyl-2-pyridone, J. Heterocycl. Chem., 1970, 7, 389–392 CrossRef CAS.
- E. Bisagni, C. Ducrocq and A. Civier, Aza-indoles—IV: Méthode générale de synthèse des aza-5 indoles fonctionnalisés sur leur sommet 4 et de derivés polycycliques apparentés, Tetrahedron, 1976, 32, 1383–1390 CrossRef CAS.
- S. Stoncius, E. Butkus, A. Zÿilinskas, K. Larsson, L. Ohrstrom, U. Berg and K. Warnmark, Design and Synthesis of a C2-Symmetric Self-Complementary Hydrogen-Bonding Cleft Molecule Based on the Bicyclo[3.3.1]nonane and 4-Oxo-5-azaindole Framework. Formation of Channels and Inclusion Complexes in the Solid State, J. Org. Chem., 2004, 69, 5196–5203 CrossRef CAS PubMed.
- S. Stoncius, E. Orentas, E. Butkus, L. Ohrstrom, O. F. Wendt and K. Warnmark, An Approach to Helical Tubular Self-Aggregation Using C2-Symmetric Self-Complementary Hydrogen-Bonding Cavity Molecules, J. Am. Chem. Soc., 2006, 128, 8272–8285 CrossRef CAS.
- L. Labanauskas, A. Zilinskas, S. Visniakova, G. Urbelis, O. Gedrimaite, R. Rozenbergas and A. Podgursky, Synthesis of bicyclo[3.3.1]nonane derivatives containing fused heterocyclic rings, ARKIVOC, 2008, 15, 256–261 Search PubMed.
- P. Camps, R. El Achab, D. M. Gorbig, J. Morral, D. Munoz-Torrero, A. Badia, J. E. Banos, N. M. Vivas, X. Barril, M. Orozco and F. J. Luque, Synthesis, in Vitro Pharmacology, and Molecular Modeling of Very Potent Tacrine−Huperzine A Hybrids as Acetylcholinesterase Inhibitors of Potential Interest for the Treatment of Alzheimer's Disease, J. Med. Chem., 1999, 42, 3227–3242 CrossRef CAS PubMed.
- A. Badia, J. E. Banos, P. Camps, J. Contreras, D. M. Gorbig, D. Munoz-Torrero, M. Simon and N. M. Vivas, Synthesis and evaluation of tacrine–Huperzine a hybrids as acetylcholinesterase inhibitors of potential interest for the treatment of alzheimer's disease, Bioorg. Med. Chem., 1998, 6, 427–440 CrossRef CAS PubMed.
- C. Ronco, G. Sorin, F. Nachon, R. Foucault, L. Jean, A. Romieu and P.-Y. Renard, Synthesis and structure–activity relationship of Huprine derivatives as human acetylcholinesterase inhibitors, Bioorg. Med. Chem., 2009, 17, 4523–4536 CrossRef CAS.
- C. Ronco, L. Jean and P.-Y. Renard, Improved synthetic pathway for the derivatization of huprin, Tetrahedron, 2010, 66, 7399–7404 CrossRef CAS.
- A. C. Pike, A. M. Brzozowski, R. E. Hubbard, T. Bonn, A. G. Thorsell, O. Engstrom, J. Ljunggren, J. Gustafsson and M. Carlquist, Structure of the ligand-binding domain of oestrogen receptor beta in the presence of a partial agonist and a full antagonist, EMBO J., 1999, 18, 4608–4618 CrossRef CAS PubMed.
- P. L. Coe and C. E. Scriven, Crossed coupling of functionalised ketones by low valent titanium (the McMurry reaction): a new stereoselective synthesis of tamoxifen, J. Chem. Soc., Perkin Trans. 1, 1986, 475–477 RSC.
- J. E. McMurry and L. R. Krepski, Synthesis of unsymmetrical olefins by titanium(0) induced mixed carbonyl coupling. Some comments on the mechanism of the pinacol reaction, J. Org. Chem., 1976, 41, 3929–3930 CrossRef CAS.
- R. S. Muthyala, S. Sheng, K. E. Carlson, B. S. Katzenellenbogen and J. A. Katzenellenbogen, Bridged Bicyclic Cores Containing a 1,1-Diarylethylene Motif Are High-Affinity Subtype-Selective Ligands for the Estrogen Receptor, J. Med. Chem., 2003, 46, 1589–1602 CrossRef CAS PubMed.
- K. J. Kieser, D. W. Kim, K. E. Carlson, B. S. Katzenellenbogen and J. A. Katzenellenbogen, Characterization of the Pharmacophore Properties of Novel Selective Estrogen Receptor Downregulators (SERDs), J. Med. Chem., 2010, 53, 3320–3329 CrossRef CAS.
- D.-J. Liaw, B.-Y. Liaw and C.-Y. Chung, Synthesis and characterization of new adamantane-type cardo polyamides, Acta Polym., 1999, 50, 135–140 CrossRef CAS.
- R. S. Muthyala, K. E. Carlson and J. A. Katzenellenbogen, Exploration of the bicyclo[3.3.1]nonane system as a template for the development of new ligands for the estrogen receptor, Bioorg. Med. Chem. Lett., 2003, 13, 4485–4488 CrossRef CAS PubMed.
- M. Uemura, K. Isobe and Y. Hayashi, (η6-arene) tricarbonylchromium complex in organic synthesis: stereoselective alkylation at benzylic position of (η6-Arene) tricarbonylchromium, Tetrahedron Lett., 1985, 26, 767 CrossRef CAS.
- D.-J. Liaw, B.-Y. Liaw and C.-Y. Chung, Synthesis and characterization of new adamantane-type cardo polyamides, Acta Polym., 1999, 50, 135 CrossRef CAS.
- J. Wu, D. Li, H. Wu, L. Sun and W.-M. Dai, Microwave-assisted regioselective olefinations of cyclic mono- and di-ketones with a stabilized phosphorus ylid, Tetrahedron, 2006, 62, 4643–4650 CrossRef CAS.
- A. Strenge and P. Rademacher, Spectroscopic and Theoretical Investigations of Monocyclic Dioximes and Dimethoximes with Six-, Eight-, and Ten-Membered Ring, Eur. J. Org. Chem., 1999, 1601–1609 CrossRef CAS.
- P. Camps, D. Muñoz-Torrero and V. Muñoz-Torrero, Unusual oxidation of bridgehead polycyclic 1, 2-diamines and 2-aminoalcohols with dimethyldioxirane: Formation of dioximes and monooximes by cleavage of the central carbon-carbon bond, Tetrahedron Lett., 1995, 36, 1917–1920 CrossRef CAS.
- P. Camps and D. Muñoz-Torrero, Alternative synthesis of bridgehead polycyclic 1,2-diamines and 2-aminoalcohols from di- and mono-oximes of some bicyclic diketones: Highly improved synthesis of tricycle[3.3.1.0]nonane-3,7-diamine, Tetrahedron Lett., 1994, 35, 3187–3190 CrossRef CAS.
- N. Tosa, A. Bende, R. A. Varga, A. Terec, I. Bratu and I. Grosu, H-Bond-Driven Supramolecular Architectures of the Syn and Anti Isomers of the Dioxime of Bicyclo[3.3.1]nonane-3,7-dione, J. Org. Chem., 2009, 74, 3944–3947 CrossRef CAS.
- Y. Otomaru, A. Kina, R. Shintani and T. Hayashi, C2-Symmetric bicyclo[3.3.1]nona-2,6-diene and bicyclo[3.3.2]deca-2,6-diene: new chiral diene ligands based on the 1,5-cyclooctadiene framework, Tetrahedron: Asymmetry, 2005, 16, 1673–1679 CrossRef CAS.
- R. Shintani, Y. Ichikawa, K. Takatsu, F.-X. Chen and T. Hayashi, Tuning the Chiral Environment of C2-Symmetric Diene Ligands: Development of 3,7-Disubstituted Bicyclo[3.3.1]nona-2,6-dienes, J. Org. Chem., 2009, 74, 869–873 CrossRef CAS PubMed.
- Y. Otomaru, N. Tokunaga, R. Shintani and T. Hayashi, C2-Symmetric Bicyclo[3.3.1]nonadiene as a Chiral Ligand for Rhodium-Catalyzed Asymmetric Arylation of N-(4-Nitrobenzenesulfonyl)arylimines, Org. Lett., 2005, 7, 307–310 CrossRef CAS.
- H. Quast, M. Witzel, E.-M. Peters, K. Peters and H. G. von Schnering, Syntheses and structures of 3,7-substituted barbaralanes, Liebigs Ann., 1995, 725–738 CrossRef CAS.
- C.-J. Wallentin, E. Orentas, M. T. Johnson, N. B. Bathori, E. Butkus, O. F. Wendt, K. Warnmark and L. Ohrstrom, Synthetic and crystallographic studies of bicyclo[3.3.1]nonane derivatives: from strong to weak hydrogen bonds and the stereochemistry of network formation, CrystEngComm, 2012, 14, 178 RSC.
- E. Orentas, G. Bagdziunas, U. Berg, A. Zilinskas and E. Butkus, Enantiospecific Synthesis and Chiroptical Properties of Bicyclic Enones, Eur. J. Org. Chem., 2007, 4251–4256 CrossRef CAS.
- N. M. Ahmad, V. Rodeschini, N. S. Simpkins, S. E. Ward and C. Wilson, Synthetic studies towards garsubellin A: synthesis of model systems and potential mimics by regioselective lithiation of bicyclo[3.3.1]nonane-2,4,9-trione derivatives from catechinic acid, Org. Biomol. Chem., 2007, 5, 1924–1934 RSC.
- G. M. P. Giblin, D. T. Kirk, L. Mitchell and N. S. Simpkins, Bridgehead Enolates: Substitution and Asymmetric Desymmetrization of Small Bridged Carbonyl Compounds by Lithium Amide Bases, Org. Lett., 2003, 5, 1673–1675 CrossRef CAS.
- M. J. Fernfindez, R. M. Huertas, E. Galvez, J. Server-Carrio, M. Martinez-Ripoll and J. Bellanato, Synthesis and structural and conformational study of some amides derived from 3,7-dimethyl-3,7-diazabicyclo[3.3.1]nonan-9-amine, J. Mol. Struct., 1995, 355, 229–238 CrossRef.
-
(a) R. Kolhatkar, C. D. Cook, S. K. Ghorai, J. Deschamps, P. M. Beardsley, M. E. Reith and A. K. Dutta, Further Structurally Constrained Analogues of cis-(6-Benzhydrylpiperidin-3-yl)benzylamine with Elucidation of Bioactive Conformation: Discovery of 1,4-Diazabicyclo[3.3.1]nonane Derivatives and Evaluation of Their Biological Properties for the Monoamine Transporters, J. Med. Chem., 2004, 47, 5101–5113 CrossRef CAS PubMed;
(b) M. Mishra, R. Kolhatkar, J. Zhen, I. Parrington, M. E. A. Reith and A. K. Dutta, Further structural optimization of cis-(6-benzhydryl-piperidin-3-yl)-benzylamine and 1,4-diazabicyclo[3.3.1]nonane derivatives by introducing an exocyclic hydroxyl group: Interaction with dopamine, serotonin, and norepinephrine transporters, Bioorg. Med. Chem., 2008, 16, 2769–2778 CrossRef CAS PubMed.
- S. Bollinger, H. H€ubner, F. W. Heinemann, K. Meyer and P. Gmeiner, Novel Pyridylmethylamines as Highly Selective 5-HT1A Superagonists, J. Med. Chem., 2010, 53, 7167–7179 CrossRef CAS PubMed.
- M. V. Kiryukhin, E. V. Nurieva, D. V. Shishov, V. N. Nuriev, N. V. Zyk, N. S. Zefirov and O. N. Zefirova, Seriya 2: Khimiya, 2007, 48, 342–346 CAS.
- M. Mayr, C. J. R. Bataille, S. Gosiewska, J. A. Raskatov and J. M. Brown, Synthesis and rhodium complexation of enantiomerically enriched bicyclo [3.3. 1] nona-2, 6-diene, Tetrahedron: Asymmetry, 2008, 19, 1328–1332 CrossRef CAS.
-
(a) R. B. Grossman, J. C. Tsai, W. M. Davis, A. Gutierrez and S. L. Buchwald, Synthesis and structure of a C2-symmetric, doubly bridged ansa-titanocene complex, Organometallics, 1994, 13, 3892–3896 CrossRef CAS;
(b) R. L. Halterman, A. Tretyakov, D. Combs, J. Chang and M. A. Khan, Synthesis and Structure of C2-Symmetric, Doubly Bridged Bis(indenyl)titanium and -zirconium Dichlorides, Organometallics, 1997, 16, 3333–3339 CrossRef CAS.
- A. A. Pimenov, N. V. Makarova, M. N. Zemtsova and I. K. Moiseev, Synthesis of α, β- Unsaturated Ketones on the Basis of Bicyclo [3.3.1] nonane-2, 6-dione, Russ. J. Org. Chem., 2001, 37, 450–451 CrossRef CAS.
- G. A. Smirnov, V. V. Sevost'yanova and T. A. Klimova, Izv. Akad. Nauk, Ser. Khim., 1981, 2624 (Bull. Acad. Sci. USSR, Div. Chem. Sci. (Engl. Transl.), 1981, 30) CAS.
- M. M. Krayushkin, V. V. Sevostyanova, V. N. Yarovenko and I. V. Zavarzin, Reactions of α,β-unsaturated ketones of the bicyclo[3.3.1]nonane series with nitro alkanes, Russ. Chem. Bull. Int. Ed., 2009, 58, 2175–2177 CrossRef CAS.
- C. Ronco, L. Jean, H. Outaabout and P. Y. Renard, Palladium-Catalyzed Preparation of N-Alkylated Tacrine and Huprine Compounds, Eur. J. Org. Chem., 2011, 302–310 CrossRef CAS.
- B. A. Tkachenko, N. A. Fokina, L. V. Chernish, J. E. P. Dahl, S. Liu, R. M. K. Carlson, A. A. Fokin and P. R. Schreiner, Functionalized Nanodiamonds Part 3: Thiolation of Tertiary/Bridgehead Alcohols, Org. Lett., 2006, 8, 1767–1770 CrossRef CAS PubMed.
- X. Shen, C. P. Ting, G. Xu and T. J. Maimone, Programmable meroterpene synthesis, Nat. Commun., 2020, 11, 14354–14355 Search PubMed.
- Y. Phang, X. Wang, Y. Lu, W. Fu, C. Zheng and H. Xu, Bicyclic polyprenylated acylphloroglucinols and their derivatives: structural modification, structure-activity relationship, biological activity and mechanism of action, Eur. J. Med. Chem., 2020, 205, 112646 CrossRef CAS PubMed.
- R. J. Wolf, R. A. Hilger, J. D. Hoheisel, J. Warner and F. Holtrup, In Vivo Activity and Pharmacokinetics of Nemorosone on Pancreatic Cancer Xenografts, PLoS One, 2013, 8, 74555 CrossRef.
- Y. Frion-Herrera, D. Gabbia, M. Scaffidi, L. Zagni, O. Cuesta-Rubio, S. De Martin and M. Carrara, The Cuban Propolis Component Nemorosone Inhibits Proliferation and Metastatic Properties of Human Colorectal Cancer Cells, Int. J. Mol. Sci., 2020, 21, 1827 CrossRef CAS PubMed.
- H. M. Wu and Y. M. Li, In vitro antitumor activity of guttiferone-A in human breast cancer cells is mediated via apoptosis, mitochondrial mediated oxidative stress and reactive oxygen species production, J. BUON, 2017, 22, 1500 Search PubMed.
- G. L. Pardo-Andreu, Y. Nunez-Figueredo, V. G. Tudella, O. Cuesta-Rubio, F. P. Rodrigues, C. R. Pestana, S. A. Uyemura, A. M. Leopoldino, L. C. Alberici and C. Curti, The anti-cancer agent guttiferone-A permeabilizes mitochondrial membrane: Ensuing energetic and oxidative stress implications, Toxicol. Appl. Pharmacol., 2011, 253, 282 CrossRef CAS PubMed.
- P. Kopytko, K. Piotrowska, J. Janisiak and M. Tarnowski, Garcinol—A Natural Histone Acetyltransferase Inhibitor and New Anti-Cancer Epigenetic Drug, Int. J. Mol. Sci., 2021, 22, 2828 CrossRef CAS PubMed.
- S. I. Son, D. Su, T. T. Ho and H. Lin, Garcinol Is an HDAC11 Inhibitor, ACS Chem. Biol., 2020, 15, 2866–2871 CrossRef CAS PubMed.
- V. Aggarwal, H. Singh Tuli, J. Kaur, D. Aggarwal, G. Parashar, N. Chaturvedi Parashar, S. Kulkarni, G. Kaur, K. Sak, M. Kumar and K. S. Ahn, Garcinol Exhibits Anti-Neoplastic Effects by Targeting Diverse Oncogenic Factors in Tumor Cells, Biomedicines, 2020, 8, 103 CrossRef CAS PubMed.
- S. H. Lim, H. S. Lee, C. H. Lee and C.-I. Choi, Pharmacological Activity of Garcinia indica (Kokum): An Updated Review, Pharmaceuticals, 2021, 14, 1338 CrossRef CAS PubMed.
- K. Shen, J. Xie, H. Wang, H. Zhang, M. Yu, F. Lu, H. Tan and H. Xu, Mol. Cancer Ther., 2015, 14, 1048 CrossRef PubMed.
- H. Sui, H. Tan, J. Fu, Q. Song, R. Jia, L. Han, Y. Lv, H. Zhang, D. Zheng and L. Dong, The active fraction of Garcinia yunnanensis suppresses the progression of colorectal carcinoma by interfering with tumorassociated macrophage-associated M2 macrophage polarization in vivo and in vitro, FASEB J., 2020, 34, 7387 CrossRef CAS PubMed.
- K. Shen, Z. Xi, J. Xie, H. Wang, C. Xie, C. S. Lee, P. Fahey, Q. Dong and H. Xu, Guttiferone K suppresses cell motility and metastasis of hepatocellular carcinoma by restoring aberrantly reduced profiling, Oncotarget, 2016, 7, 56650 CrossRef PubMed.
- F. H. Reis, G. L. Pardo-Andreu, Y. Nunez-Figueredo, O. Cuesta-Rubio, J. Martin-Prida, S. A. Uyemura, C. Curti and L. C. Alberici, Clusianone, a naturally occurring nemorosone regioisomer, uncouples rat liver mitochondria and induces HepG2 cell death, Chem.-Biol. Interact., 2014, 212, 20 CrossRef CAS PubMed.
- W. F. Taylor, M. Yanez, S. E. Moghadam, M. Moridi Farimani, S. Soroury, S. N. Ebrahimi, M. Tabefam and E. Jabbarzadeh, 7-epi-Clusianone, a Multi-Targeting Natural Product with Potential Chemotherapeutic, Immune-Modulating, and Anti-Angiogenic Properties, Molecules, 2019, 24, 4415 CrossRef CAS PubMed.
- H. Zhu, Y.-N. Yang, K. Xu, J. Xie, Z.-M. Feng, J.-S. Jiang and P.-C. Zhang, Org. Biomol. Chem., 2013, 1–4 Search PubMed.
- M. Manimaran, A. Ganapathi and T. Balasankar, Synthesis, Spectral, Anti-Liver Cancer and Free Radical Scavenging Activity of New Azabicyclic Thienoyl Hydrazone Derivatives, Open J. Med. Chem., 2015, 5, 33–47 CAS.
- J. K. F. Garrison, S. Jonsson and J. Valgeirsson, Synthesis and antitumor activity of bicyclo[3.3.1]nonenol derivatives, Bioorg. Med. Chem., 2004, 12, 5563–5569 CrossRef.
|
| This journal is © The Royal Society of Chemistry 2023 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a  *ab and
Priyankar Paira
*ab and
Priyankar Paira