
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicencePreparation of hydroxyapatite coated porous carbon nanofibres for DEX loading and enhancing differentiation of BMSCs
Liujia Lan†
a,
Qian Zhang†a,
Huiyun Zhang†b,
Xiaochuan Yanga,
Suying Lia,
Guang Li c,
Yi Luoa,
Du Nie*a,
Guangyu Zhang*a and
Jiamu Dai*a
c,
Yi Luoa,
Du Nie*a,
Guangyu Zhang*a and
Jiamu Dai*a
aSchool of Textile and Clothing, Key Laboratory of Neuroregeneration of Jiangsu and Ministry of Education, Co-innovation Center of Neuroregeneration, Nantong University, Nantong, 226001, China. E-mail: flory2114@hotmail.com; zgyu85@ntu.edu.cn; jmdai@ntu.edu.cn
bDongfang Hospital Affiliated to Beijing University of Chinese Medicine, Beijing 100078, China
cState Key Laboratory for Modification of Chemical Fibers and Polymer Materials, College of Materials Science and Engineering, Donghua University, Shanghai 201620, China
First published on 20th October 2023
Abstract
The proliferation and differentiation of bone mesenchymal stem cells (BMSCs) in vitro are the key properties of bone tissue engineering for biomaterials. In this study, hydroxyapatite (HA) coated porous carbon nanofibres (PCNFs) were prepared to load dexamethasone (DEX) and further improve the differentiation ability of the BMSCs. Various characterisations were applied to reveal the DEX loading efficacy and biocompatibility, especially the differentiation strength. The results showed that HA could be successfully coated on the PCNFs by pretreating the surface using PEG conjugation. With an increase of HA, the particle diameter increased and the DEX loading decreased. In vitro experiments proved higher cell viability, alkaline phosphatase (ALP) activity, calcium nodule secretion ability and the RUNX2 protein expression, indicating that the as-prepared was of great biocompatibility and optimised osteoconductivity, which was attributed to the componential imitation to natural bone and the accelerated BMSCs differentiation. Consequently, the novel DEX loaded and HA coated PCNFs can provide potential applications in bone tissue regeneration.
Introduction
In recent research, advanced bioactive materials have been developed to replace current main strategies of autografts and allografts for repairing bone defects.1 Whereas, due to the inherent drawback, the clinical application of these strategies has been strongly limited.2 Thus, novel materials are continuing to be researched as alternatives for bone tissue regeneration. Over the past decades, numerous bioactive nanomaterials have been developed for use in this field, such as bioceramics,3,4 hydroxyapatite (HA),5,6 metals,7,8 silica nanoparticles,9,10 as well as supplementing their use with metal elements,11,12 and bioactive molecules.12–14 However, carbon nanomaterials have been proposed as a potential alternative, because of their strong interfacial bonding strength with the substrate.14–16However, a lot of research is under way to overcome various problems arising from single components of the previously mentioned materials. For example, growth factors are incorporated with a carrier to act as a drug delivery system for improving bone regeneration,17,18 and HA has been applied as a coating or filler to make the matrix more bioactive and osteoconductive which is attributed to the main inorganic composition of natural bone.19,20 Dexamethasone (DEX), a synthetic glucocorticoid, has been applied to induce osteogenic differentiation of bone marrow stem cells (BMSCs).21,22 However, DEX in high concentrations can suppress osteoblast proliferation and even induce toxic side effects.23 To deal with this issue, drug carriers have been proposed which absorb the DEX and release it at an appropriate rate.24,25
In this study, porous carbon nanofibres (PCNFs) were prepared for use as the matrix and drug carrier for the HA coating and DEX loading to improve the osteoconduction activity (see Scheme 1). To enhance the bonding strength between the PCNFs and HA due to the high hydrophobicity of the PCNFs, acid treatment and polyethylene glycol (PEG) conjugation were applied to improve the hydrophilicity, and then the PCNFs could easily capture Ca2+ and further induce the formation of the HA coating.26 In addition, due to its great adsorption ability,27 DEX was loaded onto the PCNFs to construct the drug delivery system. Various characterisations were carried out to study the HA coating, and the DEX loading and release profile. In addition, in vitro experiments were used to reveal the biocompatibility, alkaline phosphatase (ALP) activity and calcium nodule secretion to prove the osteoconductivity under the dual influences of the HA coating and the DEX loading.
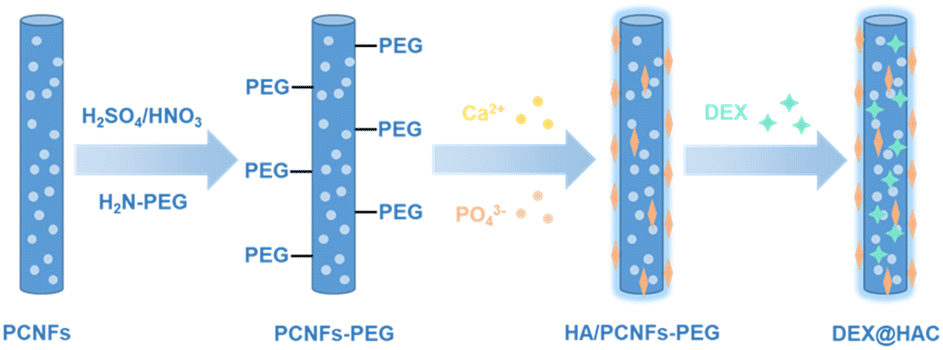 | ||
| Scheme 1 Schematic illustration of the DEX@HA/PCNFs-PEG preparation. | ||
Experimental
Fabrication of porous carbon nanofibres
The PCNFs were prepared using an electrospinning technique reported in our previous work.28 Briefly, polyacrylonitrile (PAN) and polymethylmethacrylate (PMMA), with a mass ratio of 7![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :3, were dissolved in N,N-dimethylformamide (DMF) to obtain a blended solution with concentration of 10 wt%. For electrospinning, the blend solution was loaded into a plastic syringe under conditions including a voltage of 18 kV, extrusion rate of 0.9 ml h−1 and collecting distance of 18 cm between the needle tip and aluminium foil. After vacuum drying overnight at 60 °C, the nanofibres were pre-oxidised at 280 °C for 2 h, and carbonised at 800 °C for 2 h to obtain the PCNFs. For comparison, carbon nanofibres without pores (CNFs) were also prepared by removing PMMA from the electrospinning solution.
:3, were dissolved in N,N-dimethylformamide (DMF) to obtain a blended solution with concentration of 10 wt%. For electrospinning, the blend solution was loaded into a plastic syringe under conditions including a voltage of 18 kV, extrusion rate of 0.9 ml h−1 and collecting distance of 18 cm between the needle tip and aluminium foil. After vacuum drying overnight at 60 °C, the nanofibres were pre-oxidised at 280 °C for 2 h, and carbonised at 800 °C for 2 h to obtain the PCNFs. For comparison, carbon nanofibres without pores (CNFs) were also prepared by removing PMMA from the electrospinning solution.
Surface treatment of the porous carbon nanofibres
A process of acidification was carried out to improve the hydrophilicity of PCNFs before coating with HA. Generally, PCNFs were dispersed in a mixed acid of sulfuric acid and nitric acid (volume ratio of 1:3) for 4 h at 70 °C. Then the mixture was poured into a large amount of deionised water followed by filtration. Finally, the PCNFs were washed three times with deionised water and then vacuum dried at 60 °C.
Next, 100 mg of amino-terminated polyethylene glycol (PEG-NH2) was dissolved in 10 ml of DMF, 20 mg of PCNFs were then added and ultrasonically dispersed for 10 min. After further addition of 10 ml of 4-dimethylaminopyridine (DMAP) and 50 mg of dicyclohexylcarbodiimide (DCC), the blend solution was stirred for 24 h at 60 °C for the PEG conjugation to occur. Finally, the black sample was centrifuged and washed three times with deionised water, and then vacuum dried overnight.
Fabrication of hydroxyapatite coated porous carbon nanofibres
Subsequently, the PCNFs were dispersed in 0.05 M Ca(NO3)2 solution for 4 h by ultrasonication and stirring. Then, an equal volume of 0.03 M NaH2PO4 solution was added dropwise to the solution with the pH adjusted to 10 using ammonium hydroxide. After stirring for another 4 h, the solution was centrifuged at 1 × 104 rpm for 5 min, and then the as-prepared HA/PCNFs was washed three times and then vacuum dried. Using this method, different ratios of PCNF/HA could be prepared by controlling the concentrations of Ca(NO3)2 and NaH2PO4. In this study, samples with different mass ratios of PCNF and HA (1:0.5, 1:1 and 1:2) were prepared, and were denoted as 0.5HAC, 1HAC and 2HAC, respectively.
Characterisations
Morphological observations were obtained using field emission scanning electron microscopy (FESEM, Zeiss GeminiSEM 300, Germany) and transmission electron microscopy (TEM, FEI Talos F200X G2, USA). The FTIR spectra with wavenumbers from 400 cm−1 to 4000 cm−1 were determined using a spectrophotometer (ThermoScientific, Nicolet iS 50, USA). The particle size was studied using a zeta potential and particle size analyser (Brookhaven, NanoBrook 90plus Zeta, USA).DEX loading and in vitro release
For the DEX loading, 2 mg of DEX was dissolved in 1.5 ml of ethanol, which was then added to 13.5 ml of phosphate buffer solution (PBS) containing 20 mg of PCNFs or HA/PCNFs, with ultrasonic dispersion. After stirring for 12 h, the samples were centrifuged and washed with PBS to obtain the DEX@HA/PCNFs. The supernatant and washing solutions were collected and analysed by UV-Vis spectrophotometry at 242 nm to calculate the DEX loading efficacy.To study the DEX release behaviour, 4 ml of the suspension with a concentration of 0.25 mg ml−1 in PBS was placed in a shaker at 37 °C and 100 rpm. At predetermined time points, all the samples were centrifuged at 10000 rpm for 5 min, and 2 ml of the supernatant was withdrawn for drug release analysis, and then an equal volume of fresh PBS was added back to the dispersion. The amount of released DEX was further determined by UV-Vis spectrophotometry at 242 nm.
Cell culture
The BMSCs were cultured with DMEM medium with a low glucose concentration (HyClone, USA) containing 10% foetal bovine serum (Gibco, USA), 100 U ml−1 of penicillin, and 100 μg ml−1 of streptomycin (Gibco, USA) at 37 °C in a humidified CO2 incubator.Cytotoxicity test
The MTT assay was applied to study the cytotoxicity of the samples. Briefly, BMSCs were seeded in 96-well plates with a density of 5 × 103 cells per well and cultured for 24 h. Next, the BMSCs were washed with PBS and treated with samples of various concentrations (6.25, 12.5, 25 and 50 μg ml−1). After incubating for 24 h and 48 h, the cells were washed with PBS again, and then re-incubated with 90 μl of fresh basic medium containing 10 μl of MTT (5 mg ml−1) per well for 4 h. Then, 100 μl of DMSO per well was added to replace the removed medium and the plate was then placed in a 100 rpm shaker for 10 min at 37 °C. Finally, the absorbance of the solution was scanned with all-wavelength microplate reader (Tecan, Infinite M Nano, Switzerland) to determine the viability of cells.Osteoconductive culture
For the osteoconductivity assay, the BMSCs were firstly cultured in a well plate with normal medium until the cells had nearly overspread the well, then the normal medium was replaced by osteoconductive medium (normal medium supplemented with 10 mM of β-glycerol phosphate and 50 μg ml−1 of L-ascorbic acid) with samples of different concentrations (6.25, 12.5 and 25 μg ml−1), and the pure osteoconductive medium was used as control. The medium used in this experiment was replaced every 3 days.Alkaline phosphate activity test
The ALP activity of the BMSCs treated with different samples was evaluated using ALP assay kit (Beyotime Institute of Biotechnology, China). Briefly, the BMSCs were lysed with cell lysis buffer at predetermined time points. Next, 50 μl of the cell lysates and 50 μl of the substrate solutions were mixed in a 96-well plate and incubated at 37 °C for 10 min. Then, the stop solution was added to terminate the reaction and the samples were scanned at 405 nm with a microplate reader. For normalisation, the total protein concentration was measured using a protein assay kit (Beyotime Institute of Biotechnology, China).Alizarin Red S staining
After incubation in conducting medium for 14 days with different samples of 25 μg ml−1, the BMSCs were fixed with 4% glutaraldehyde for 30 min and then washed with PBS. Washing with deionised water was further used to remove residual salts. For the staining of the BMSCs, Alizarin Red S (ARS) solution with a density of 2% (w/v) was firstly adjusted to a pH value of about 4.1–4.3 and this was followed by cell staining at room temperature. After staining for 10 min, the ARS solution was removed and the cells were washed with deionised water until there was no residual ARS. The stained cells were photographed with a digital camera (Canon, EOS 550D). To quantitively analyse the stained matter, 10% acetic acid was added per well and the cells were incubated overnight. The solution was collected and centrifuged at 10000 rpm for 15 min. After neutralisation with ammonium hydroxide, the supernatant was scanned by a microplate reader at 405 nm.
RUNX2 staining
After treating the BMSCs with the same protocol as that for the osteoconductivity tests for 21 days, the cells were fixed with 4% paraformaldehyde solution for 20 min. Then, 0.1% Triton X-100 solution was used to permeabilise the cells for 5 min, which were then blocked by nonspecific binding with 10% normal donkey serum for another 30 min. Afterwards, the primary antibody of RUNX2 (D1L7F) rabbit mAb (Cell Signaling Technology, Beverly, MA, USA) was added to the cells and incubated overnight at 4 °C. After washing three times with PBS, the secondary antibody of donkey anti-rabbit IgG H&L (Alexa Fluor 405, 1:200 dilution, Abcam, Cambridge, UK) was further added to the cells for a 1 h treatment. Finally, after washing three times with PBS, propidium iodide (PI) was used to stain the cells for the confocal laser microscopy observation.
Statistical analysis
All the values are presented as mean ± standard deviation. A one-way analysis of variance (one-way ANOVA) was used in the statistical analysis. The statistical analysis criteria were **P < 0.01 and *P < 0.05.Results and discussion
Fabrication and characterisation of HA coated PCNFs
In Fig. 1, the FESEM and TEM images of the CNFs and PCNFs, respectively are displayed. The fibre diameter was about 200 nm. Obviously, the surface of the CNFs was smooth and the inside was solid according to the FESEM (Fig. 1a) and TEM (Fig. 1c) images. However, mesopores were found on the surface of the PCNFs (Fig. 1b), and long voids were oriented along the inside axis of the nanofibres (Fig. 1d). The position of the pores matched the location of the PMMA in the original fibres before the carbonisation process, which would be degraded under high temperature and also be removed to the outside to construct the porous structure in the nanofibre. In addition, PAN would be converted into the carbon framework and would remain to support the whole nanofibre structure.29 | ||
| Fig. 1 The FESEM images of (a) CNFs and (b) PCNFs, and the TEM images of (c) CNFs and (d) PCNFs. | ||
To prove the success of the PEG conjugation, samples were analysed by FTIR. Compared with the PCNFs, a peak at 3400 cm−1 was attributed to –OH due to acid treatment on the PCNFs,30,31 and the peaks at 1735 cm−1 and 1616 cm−1 were assigned to –NH–CO– from the reaction of the PCNFs and PEG-NH2 (Fig. 2a).32,33 Obviously, the PEG was successfully conjugated onto the PCNFs. In addition, the DEX loaded PCNFs-PEG showed several new peaks corresponding to the DEX spectrum when compared to the PCNFs-PEG (Fig. 2b), indicating that DEX could be absorbed onto the matrix well.
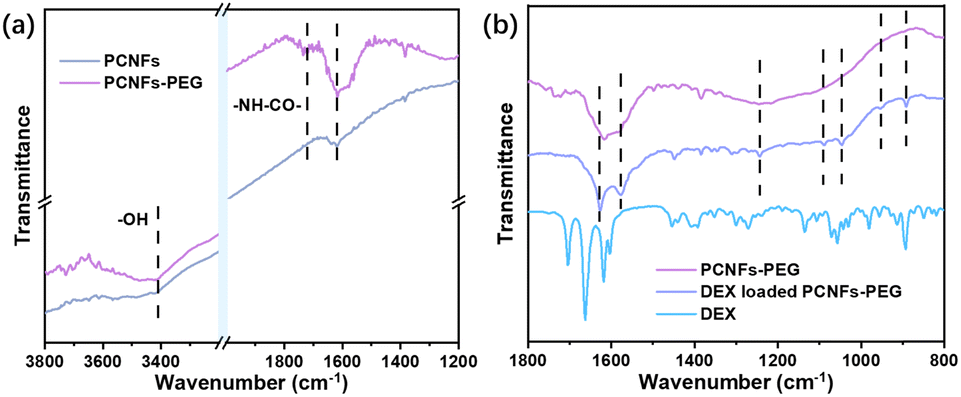 | ||
| Fig. 2 (a) The FTIR spectra of the PCNFs and PCNFs-PEG. (b) The FTIR spectra of the PCNFs-PEG, DEX loaded PCNFs-PEG and pristine DEX. | ||
After incorporation with the HA, the morphology change of PCNF was evaluated by FESEM as well and the images are shown in Fig. 3. On the surface of the 0.5HAC (Fig. 3a and d), a few small HA particles happened to adhere on the PCNF, and other particles were also found in the gap between the fibres. However, the HA particles on 1HAC (Fig. 3b and e) began to form stick shapes to connect the fibres over a large distance, which would gather nanofibres together to obtain larger sized particles. Furthermore, the HA particles on 2HAC (Fig. 3c and f) almost fully covered the surface of the PCNF, and the stick shape was turned into a network to strongly connect the fibres which would further develop much larger particles based on 1HAC.
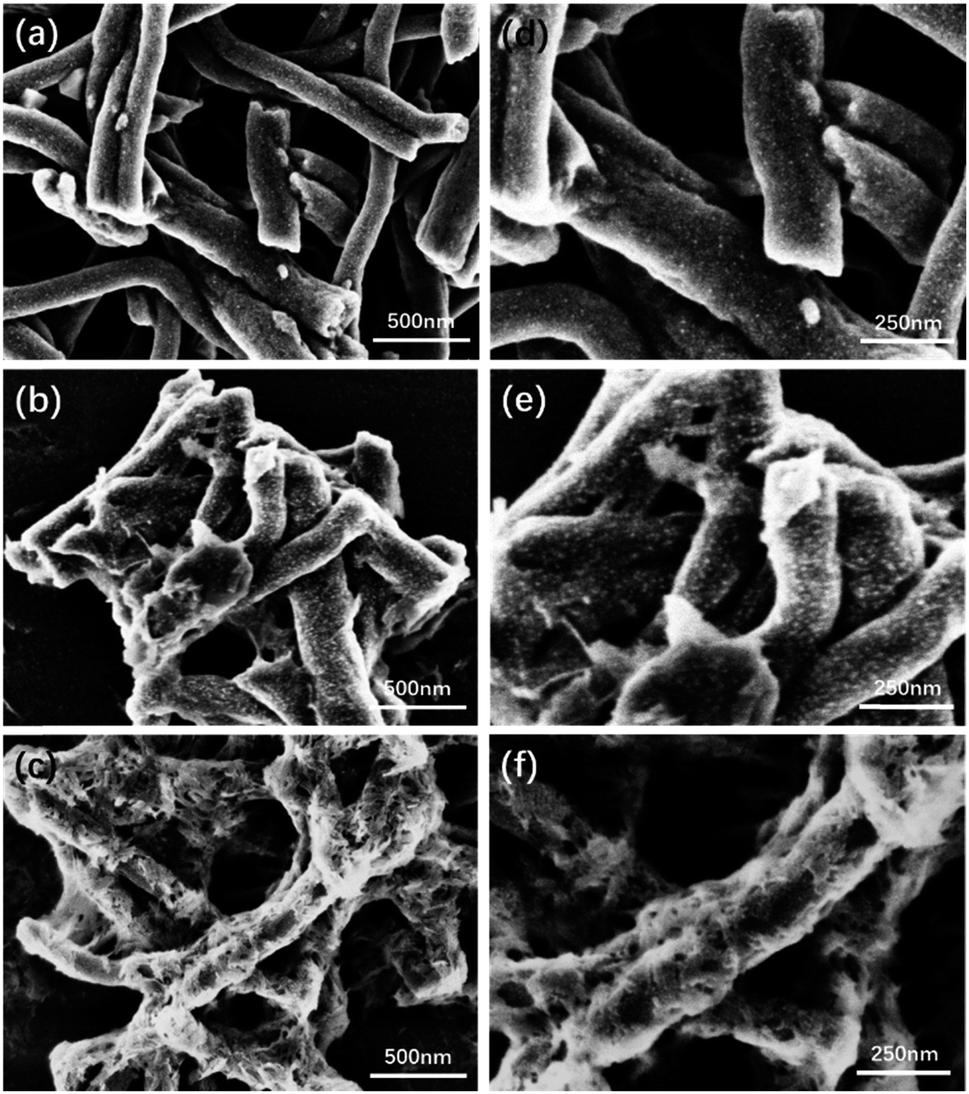 | ||
| Fig. 3 The FESEM images of (a) 0.5HAC, (b) 1HAC and (c) 2HAC. (d), (e) and (f) are the high magnification images of (a), (b) and (c), respectively. | ||
The particle sizes of the PCNFs before and after HA coating were further determined to reveal the influence of the amount of coating. As shown in Fig. 4, the average particle size of the PCNFs was about 350 nm, which became larger as the amount of HA increased. For 2HAC, the particle size was about 500 nm.
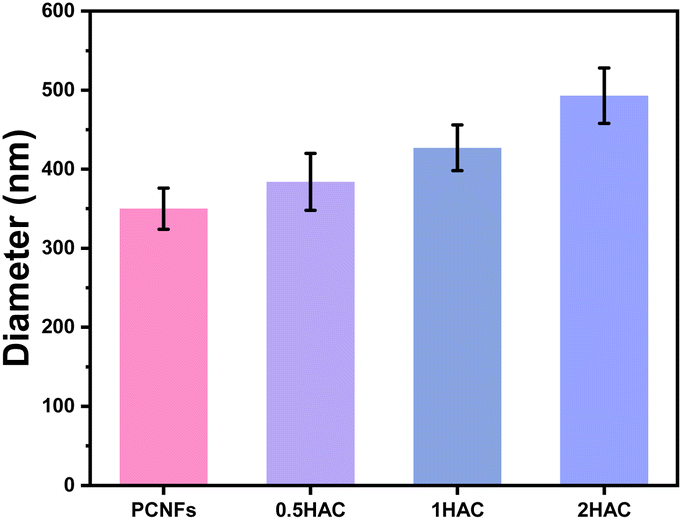 | ||
| Fig. 4 Particle sizes of PCNFs, 0.5HAC, 1HAC and 2HAC. | ||
To investigate the stability in an aqueous environment, original PCNFs, acid treated PCNFs, and 1HAC were ultrasonically dispersed in PBS and left for 10 min. As shown in Fig. 5, after dispersion in PBS for 10 min, the PCNFs almost sank to the bottom, whereas the dispersion of acid treated PCNFs and 1HAC still remained non-transparent black. These results revealed the stability of the PCNFs in an aqueous environment was significantly improved.
 | ||
| Fig. 5 Photograph of (a) PCNFs, (b) acid treated PCNFs and (c) 1HAC after dispersion in PBS for 10 min. | ||
Cytotoxicity of HA coated PCNFs
Examining the cytotoxicity is essential to evaluate the biocompatibility and further osteogenic differentiation activity of the prepared particles. Firstly, the CNFs were applied as the control group in a study of the influence of the pores on cytotoxicity using the MTT assay. As shown in Fig. 6a and b, after incubation with the BMSCs for 24 h and 48 h at various concentrations (3.125, 6.25, 12.5, 25 and 50 μg ml−1), the cell viability of the CNFs and PCNFs showed no significant difference, indicating that the pores would not affect the cytotoxicity of the CNF matrix. Subsequently, the HAC were further measured to evaluate the effect of the HA coating. It was found that the PCNFs and HAC showed no obvious cytotoxicity difference after 24 h incubation (Fig. 6c). However, HAC showed a relatively higher cell viability than the PCNFs at a higher concentration such as 50 μg ml−1 after incubation for 4 h (Fig. 6d). Significantly, the cell viability of 2HAC reached about 111.8% after incubating for 48 h, whereas that of the PCNF was 94.3%. The results further indicated that PCNF would not cause obvious cytotoxicity on the BMSCs in vitro, and the HA coating on PCNF could improve the biocompatibility. Therefore, PCNF and HAC are highly biocompatible and convenient for use in the following experiments.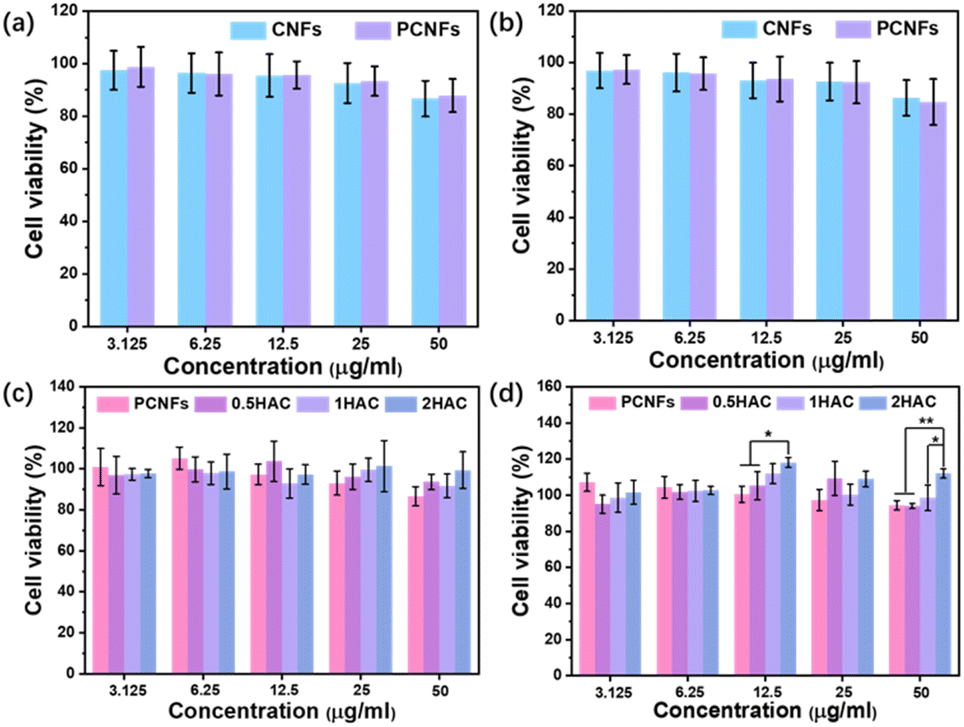 | ||
| Fig. 6 Cell viability of BMSCs cultured with CNFs and PCNFs for (a) 24 h and (b) 48 h. Cell viability of BMSCs cultured with different concentrations of PCNFs and HA coated PCNFs for (c) 24 h and (d) 48 h. | ||
The ALP activity test of HA coated PCNFs
To improve osteoconductivity, HA was incorporated on the surface of PCNF to improve the osteogenic differentiation activity of the BMSCs. After treatment with different samples (with or without HA coating) of various concentrations for different culture periods, the ALP activity of the BMSCs was evaluated as a marker of osteogenic differentiation, which would be upregulated during the early part of osteogenesis. Fig. 7 shows the in vitro ALP activity of BMSCs treated with different samples for various culture periods. It can be seen that the ALP activity of all the test groups increased as the culture time was prolonged.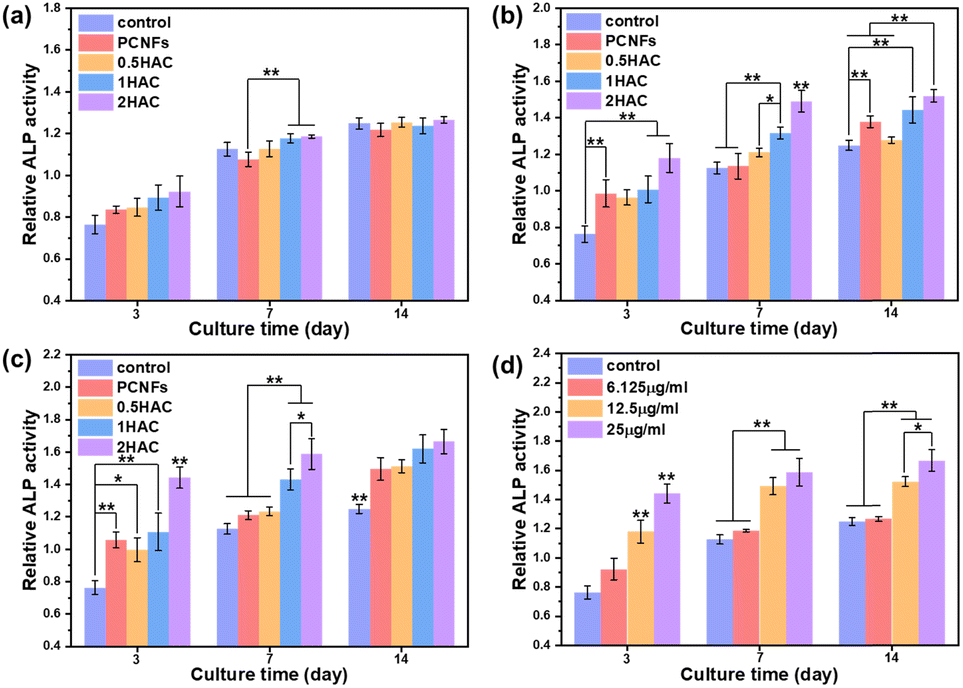 | ||
| Fig. 7 Investigation of the in vitro ALP activity of PCNFs and HA coated PCNFs at concentrations of (a) 6.25 μg ml−1, (b) 12.5 μg ml−1 and (c) 25 μg ml−1. (d) The ALP activity with different concentrations of 2HAC. All data were analysed using a one-way ANOVA and compared against the osteoconductive group. | ||
Osteoconductivity of DEX loaded HA/PCNFs
As a type of porous carbon material, the PCNFs could present a great adsorbing ability for some small molecules. Herein, HA/PCNFs were applied as a drug carrier on which to load DEX, which can limit cell proliferation and induce calcium nodule secretion. Initially, the pH value was first evaluated to study the influence on DEX adsorption in PCNFs. Obviously, at pHs of 3, 7 and 11, the DEX adsorption showed no significant difference (Fig. 8a), revealing that the surface charge of the PCNFs and DEX had no obvious influence on drug adsorption. In addition, the DEX loading efficacy decreased with increase of HA coating (Fig. 8b), and this was attributed to the decreased amount of the PCNFs and low absorption ability of HA, which further induced the quicker DEX release rate (Fig. 8c). Herein, CNFs without pores were used as the control group for determining the influence of the pores on DEX adsorption. Obviously, the PCNFs showed a much higher DEX adsorption capacity than the CNFs. As our previous work described,34 the porosity was significantly improved by adding thermo-degradable PMMA into PAN solution. Therefore, the enlarged specific surface area of the PCNFs could provide more sites for DEX adsorption. Next, the influence of the pH value on DEX release behaviour from 1HAC was also studied. Obviously, the release rate at pH 3 is significantly faster than those of 7 and 11. In an acidic environment, the coated HA would be dissolved to quickly release adsorbed DEX over a short period of time. However, 1HAC in an alkaline environment showed a similar DEX release to that in a neutral solution. In an alkaline solution, HA was more stable and this avoided the release of a burst of DEX as happened under acidic conditions, and the alkaline environment itself showed no significant influence.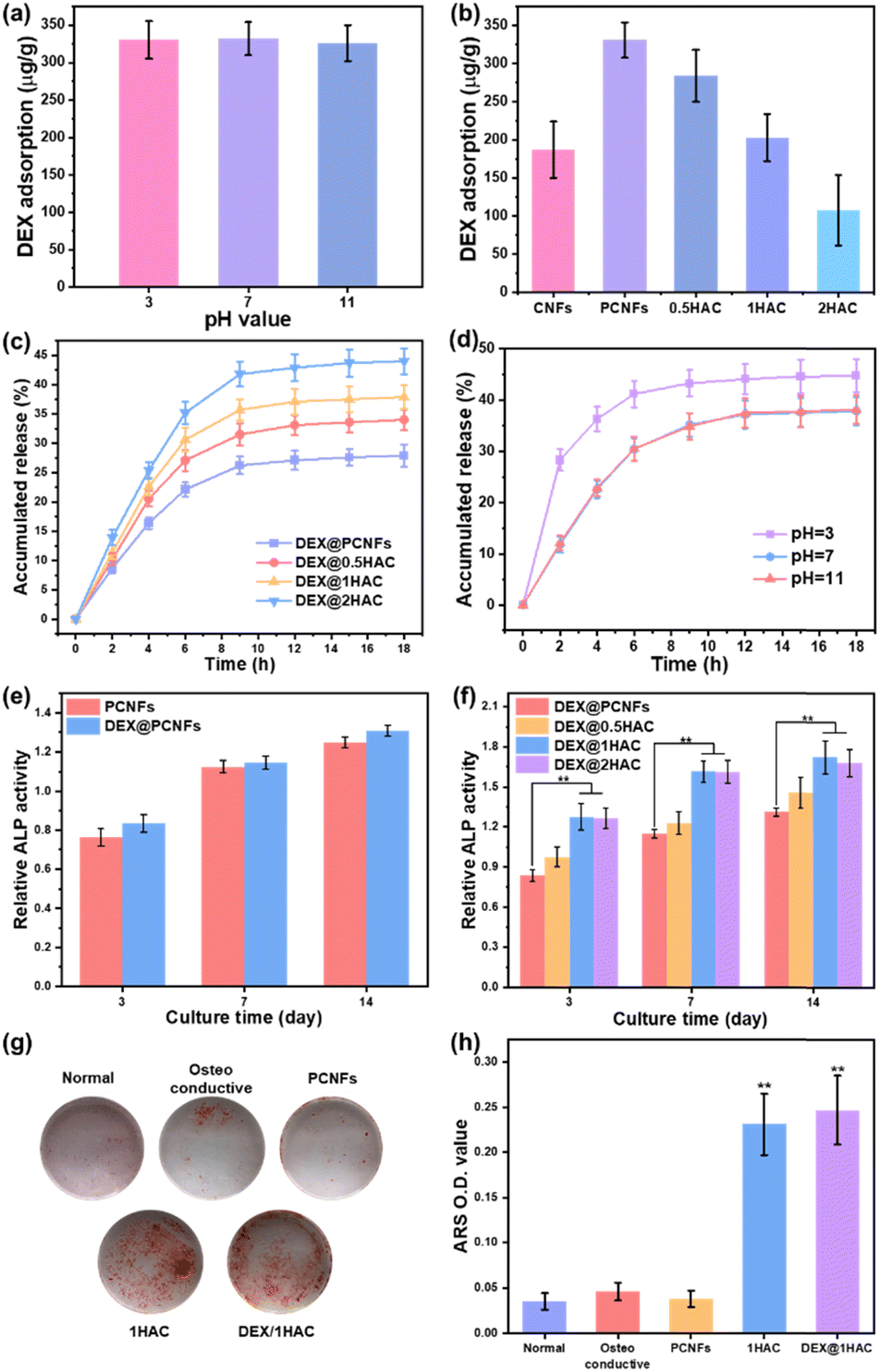 | ||
| Fig. 8 (a) The DEX adsorption of PCNFs at different pH values. DEX (b) adsorption and (c) release profiles of PCNFs, 0.5HAC, 1HAC and 2HAC. (d) The DEX release profile of 1HAC at different pH values (3, 7 and 11). (e) The in vitro ALP activity of PCNFs with or without DEX loading. (f) The in vitro ALP activity of DEX loaded particles. (g) Photographs of ARS stained BMSCs after treatment with different samples for 14 days. (h) The OD value of the mineralised matrix dissolved from (g). All the data were analysed using a one-way ANOVA against the osteoconductive group. | ||
Next, the ALP activity of the DEX@HA/PCNFs was also evaluated (Fig. 8e and f) to study the influence of DEX. Predictably, PCNFs with DEX loading showed a slightly higher ALP activity, which would be much higher when based on the incorporation of the HA coating. However, 1HAC and 2HAC presented similar ALP activity to that of the BMSCs, indicating a balance of DEX loading and HA coating.
To minimise the particle size, 1HAC and DEX@1HAC were chosen to study the calcium nodule secretion behaviour on BMSCs. After incubation for 14 days with different treatments, the cells were immersed in ARS to stain the secreted calcium nodules with a red colour. As shown in Fig. 8g, no obvious calcium nodules could be found for the cells cultured with normal medium. However, after changing to an osteoconductive medium, a small amount of the ARS-stained calcium nodules could be observed. When the PCNFs were added, the amount of red coloured cells was not significantly increased, indicating that PCNFs could not show osteoconductivity to the BMSCs. After HA coating, the HA/PCNFs could obviously increase the quality of the calcium nodules, as well as the particles with DEX loading. To quantitively analyse the secretion ability of the calcium nodules, the stained matter was dissolved by 1% acetic acid and scanned with a microplate reader to obtain the optical density (OD) values at wavelength of 450 nm (Fig. 8h). It was noted that 1HAC and DEX@1HAC presented much higher OD values than other samples, which agreed with the results shown in Fig. 8g. The previous findings revealed that the HA coating and the DEX loading could upregulate the ALP level and promote the mineralised matrix formation.
The expression of the osteo-related proteins was more active during the osteogenic differentiation process. Herein, the RUNX-related transcription factor 2 (RUNX2), one of the earliest signs of osteoblastic differentiation, was chosen and evaluated to reveal its osteogenic activity. After treatment with different samples for 21 days and RUNX2 staining, the cells were observed and are shown in Fig. 9. Obviously, cells cultured in an osteoconductive medium presented positive RUNX2 protein when compared to those cells in normal medium. Significantly, a stronger fluorescence intensity was observed on 1HAC and DEX/1HAC, indicating that HA and adsorbed DEX could stimulate RUNX2 protein expression when comparing to that of PCNFs. Therefore, the immunostaining of RUNX2 further proved the previous results on improving bone tissue regeneration.
 | ||
| Fig. 9 The RUNX2 immunostaining of BMSCs treated with different samples for 21 days (blue for RUNX2, red for cell nuclei, scale bar = 50 μm). | ||
Conclusions
In this study, HA coated and DEX loaded PCNFs were successfully prepared for bone tissue regeneration. With acid treatment and conjugation of PEG, the hydrophobicity of the PCNFs was overcome following the HA coating. Benefiting from the adsorbing ability, DEX could be efficiently loaded on PCNFs, which would decrease as the HA coating increased. In vitro experiments revealed that the HA coating and loaded DEX could upregulate the BMSCs proliferation rate, the ALP level, the calcium nodule secretion and the RUNX2 protein expression on the PCNFs, which reached optimal of HA/PCNFs with a mass ratio of 1:1. Therefore, the HA coated and DEX loaded PCNFs showed a promising ability, because of the biocompatibility and osteoconductivity, for bone regeneration applications.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This study was financially supported by the National Key Research and Development Program of China (Grant No. 2016YFB0303101), the National Natural Science Foundation of China (Grant No. 51803094, 81702723), Higher Education of Jiangsu Province (No. 23KJA540001), Jiangsu University “Qinglan Project”, Science and Technology Project of Nantong City (Grant No. MS12021006, MS2022077). The authors also thank Nantong University for the use of various pieces of characterisation equipment.Notes and references
- B. J. R. F. Bolland, K. Partridge, S. Tilley, A. M. R. New, D. G. Dunlop and R. O. C. Oreffo, Regener. Med., 2006, 1, 457–467 CrossRef CAS PubMed.
- M. A. Woodruff, C. Lange, J. Reichert, A. Berner, F. Chen, P. Fratzl, J.-T. Schantz and D. W. Hutmacher, Mater. Today, 2012, 15, 430–435 CrossRef CAS.
- T. J. Levingstone, S. Herbaj and N. J. Dunne, Nanomaterials, 2019, 9, 1570 CrossRef CAS PubMed.
- W. Ma, W. Wang, F. Liu, Y. Kong, B. Xia, H. Yang, H. Zhao, L. Wang, K. Li, Y. Li, Y. Sang, H. Liu, X. Wang and J. Qiu, Composites, Part B, 2023, 255, 110612 CrossRef CAS.
- Z. Liu, S. Yamada, Y. Otsuka, T. G. Peñaflor Galindo and M. Tagaya, Dalton Trans., 2022, 51, 9572–9583 RSC.
- Y. Qiu, X. Xu, W. Guo, Y. Zhao, J. Su and J. Chen, ACS Biomater. Sci. Eng., 2020, 6, 2323–2335 CrossRef CAS PubMed.
- X. Zhang, G. Cheng, X. Xing, J. Liu, Y. Cheng, T. Ye, Q. Wang, X. Xiao, Z. Li and H. Deng, J. Phys. Chem. Lett., 2019, 10, 4185–4191 CrossRef CAS PubMed.
- H. Liang, C. Jin, L. Ma, X. Feng, X. Deng, S. Wu, X. Liu and C. Yang, ACS Appl. Mater. Interfaces, 2019, 11, 41758–41769 CrossRef CAS PubMed.
- F. Kurtuldu, N. Mutlu, M. Michálek, K. Zheng, M. Masar, L. Liverani, S. Chen, D. Galusek and A. R. Boccaccini, Mater. Sci. Eng. C, 2021, 124, 112050 CrossRef CAS PubMed.
- C. Lei, Y. Cao, S. Hosseinpour, F. Gao, J. Liu, J. Fu, R. Staples, S. Ivanovski and C. Xu, Nano Res., 2021, 14, 770–777 CrossRef CAS.
- J. P. O'Connor, D. Kanjilal, M. Teitelbaum, S. S. Lin and J. A. Cottrell, Materials, 2020, 13, 2211 CrossRef PubMed.
- C. Xu, Y. Cao, C. Lei, Z. Li, T. Kumeria, A. K. Meka, J. Xu, J. Liu, C. Yan, L. Luo, A. Khademhosseini, A. Popat, Y. He and Q. Ye, ACS Appl. Nano Mater., 2020, 3, 1457–1467 CrossRef CAS.
- B. Safari, S. Davaran and A. Aghanejad, Int. J. Biol. Macromol., 2021, 175, 544–557 CrossRef CAS PubMed.
- W. Bu, X. Xu, Z. Wang, N. Jin, L. Liu, J. Liu, S. Zhu, K. Zhang, R. Jelinek, D. Zhou, H. Sun and B. Yang, ACS Appl. Mater. Interfaces, 2020, 12, 50287–50302 CrossRef CAS PubMed.
- M. K. Haider, L. Sun, A. Ullah, S. Ullah, Y. Suzuki, S. Park, Y. Kato, Y. Tamada and I. S. Kim, Mater. Today Commun., 2021, 27, 102259 CrossRef CAS.
- M. Świętek, A. Brož, J. Tarasiuk, S. Wroński, W. Tokarz, A. Kozieł, M. Błażewicz and L. Bačáková, Mater. Sci. Eng. C, 2019, 104, 109913 CrossRef PubMed.
- Z. Lv, T. Hu, Y. Bian, G. Wang, Z. Wu, H. Li, X. Liu, S. Yang, C. Tan, R. Liang and X. Weng, Adv. Mater., 2023, 35, 2206545 CrossRef CAS PubMed.
- S. S. Lee, J. H. Kim, J. Jeong, S. H. L. Kim, R. H. Koh, I. Kim, S. Bae, H. Lee and N. S. Hwang, Biomaterials, 2020, 257, 120223 CrossRef CAS PubMed.
- F. Xing, Z. Chi, R. Yang, D. Xu, J. Cui, Y. Huang, C. Zhou and C. Liu, Int. J. Biol. Macromol., 2021, 184, 170–180 CrossRef CAS PubMed.
- B. Yuan, H. Chen, R. Zhao, X. Deng, G. Chen, X. Yang, Z. Xiao, A. Aurora, B. A. Iulia, K. Zhang, X. Zhu, A. V. Iulian, S. Hai and X. Zhang, Bioact. Mater., 2022, 18, 354–367 CrossRef CAS PubMed.
- J. Cui, X. Yu, B. Yu, X. Yang, Z. Fu, J. Wan, M. Zhu, X. Wang and K. Lin, Adv. Healthcare Mater., 2022, 11, 2200571 CrossRef CAS PubMed.
- N. Chauhan, P. Gupta, L. Arora, D. Pal and Y. Singh, Mater. Sci. Eng. C, 2021, 130, 112463 CrossRef CAS PubMed.
- X. Zhou, P. Liu, W. Nie, C. Peng, T. Li, L. Qiang, C. He and J. Wang, Int. J. Biol. Macromol., 2020, 149, 116–126 CrossRef CAS PubMed.
- F. FotouhiArdakani, M. Mohammadi and S. Mashayekhan, Appl. Phys. A: Mater. Sci. Process., 2022, 128, 654 CrossRef CAS.
- M. Mohseni, P. Shokrollahi, F. Shokrolahi, S. Hosseini, L. Taghiyar and A. Kamali, Int. J. Pharm., 2022, 626, 122196 CrossRef CAS PubMed.
- H. Samadian, H. Mobasheri, M. Azami and R. Faridi-Majidi, Sci. Rep., 2020, 10, 14853 CrossRef CAS PubMed.
- D. Nie, P. Wang, C. Zang, G. Zhang, S. Li, R. Liu, Y. Zhang, G. Li, Y. Luo, W. Zhang and J. Dai, ACS Omega, 2022, 7, 2198–2204 CrossRef CAS PubMed.
- G. Li, T. Xie, S. Yang, J. Jin and J. Jiang, J. Phys. Chem. C, 2012, 116, 9196–9201 CrossRef CAS.
- J. Song, G. Li and J. Qiao, Electrochim. Acta, 2015, 177, 174–180 CrossRef CAS.
- F. Li, L. Liu, Z. Yang and S. Li, J. Hazard. Mater., 2021, 401, 123339 CrossRef CAS PubMed.
- Y. Wang, Y. Mei, Q. Wang, W. Wei, F. Huang, Y. Li, J. Li and Z. Zhou, Composites, Part B, 2019, 162, 54–61 CrossRef CAS.
- R. A. Lusiana, V. D. A. Sangkota, N. A. Sasongko, G. Gunawan, A. R. Wijaya, S. J. Santosa, D. Siswanta, M. Mudasir, M. N. Z. Abidin, S. Mansur and M. H. D. Othman, Int. J. Biol. Macromol., 2020, 152, 633–644 CrossRef CAS PubMed.
- R. Li, Y. Wu, Z. Bai, J. Guo and X. Chen, RSC Adv., 2020, 10, 42120–42127 RSC.
- J. Dai, Y. Luo, D. Nie, J. Jin, S. Yang, G. Li, Y. Yang and W. Zhang, Chem. Eng. J., 2020, 397, 125402 CrossRef CAS.
Footnote |
| † These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2023 |