
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceAssembling phenyl-modified colloidal silica on graphene oxide towards ethanol redispersible graphene oxide powder
Jian Huang *a,
Qian Zhanga,
Zhengcai Yanga,
Hailong Hua,
Mesfin Manukaa,
Yuting Zhaoa,
Xin Wangb,
Wufeng Wangc,
Rong Yangc,
Shouwei Jiand,
Hongbo Tana,
Xiangguo Lia,
Yang Lva,
Pei Tanga and
Baoguo Mad
*a,
Qian Zhanga,
Zhengcai Yanga,
Hailong Hua,
Mesfin Manukaa,
Yuting Zhaoa,
Xin Wangb,
Wufeng Wangc,
Rong Yangc,
Shouwei Jiand,
Hongbo Tana,
Xiangguo Lia,
Yang Lva,
Pei Tanga and
Baoguo Mad
aState Key Laboratory of Silicate Materials for Architectures, Wuhan University of Technology, Wuhan 430070, China. E-mail: JHuang@whut.edu.cn
bCollege of Civil and Transportation Engineering, Shenzhen University, Shenzhen, 518000, China
cWuhan University of Technology Advanced Engineering Technology Research Institute of Zhongshan City, Zhongshan, 528400, China
dSchool of Materials Science and Engineering, Wuhan University of Technology, Wuhan 430070, China
First published on 4th July 2023
Abstract
Recently, ethanol has shown promising potential in the large-scale reduction of graphene oxide (GO) into graphene. However, dispersion of GO powder in ethanol is a challenge due to its poor affinity, which hinders permeation and intercalation of ethanol between GO molecule layers. In this paper, phenyl-modified colloidal silica nanospheres (PSNS) were synthesized by phenyl-tri-ethoxy-silane (PTES) and tetra-ethyl ortho-silicate (TEOS) using a sol–gel method. PSNS was then assembled onto a GO surface to form a PSNS@GO structure by possible non-covalent π–π stacking interactions between the phenyl groups and GO molecules. The surface morphology, chemical composition, and dispersion stability were analyzed by scanning electron microscopy, Fourier transform infrared spectroscopy, thermogravimetry, Raman spectroscopy, X-ray diffractometry, nuclear magnetic resonance, and particle sedimentation test. The results showed that the as-assembled PSNS@GO suspension had excellent dispersion stability with an optimal PSNS concentration of 5 vol% PTES. With the optimized PSNS@GO, ethanol can permeate between the GO layers and intercalate along with PSNS particles via formation of hydrogen bonds between assembled PSNS on GO and ethanol, achieving a stable dispersion of GO in ethanol. The optimized PSNS@GO powder remained redispersible after drying and milling according to this interaction mechanism which is favorable for large scale reduction processes. Higher PTES concentration may result in agglomeration of PSNS and formation of wrapping structures of PSNS@GO after drying and worsen its dispersion capability.
1 Introduction
Due to its excellent electrical conductivity, thermal conductivity, mechanical strength, and extremely high specific surface area, graphene has shown great application prospects in many fields.1–7 The preparation of graphene has also been widely studied,8,9 among which the reduction of graphene oxide (GO) into graphene is the most feasible method for large-scale preparation of graphene. At present, the commonly used reducing agents of GO are hydrazine,10,11 sodium borohydride,12 and hydroiodic acid.13 These reducing agents are toxic, explosive, and highly corrosive. The subsequent waste liquid treatment cost is also very high, and it is difficult to achieve large-scale production and industrial applications. Therefore, it is significant to find a green reducing agent to achieve large-scale reduction of GO into graphene.Recently, some researchers have proposed a simple and green reduction method to use supercritical alcohols (such as ethanol, etc.) as reducing agents to achieve graphene from GO.14–16 There are also studies on the preparation of graphene with a perfect layered structure by thermal reduction of ethanol.17,18 These studies have verified that GO can be reduced into graphene in ethanol, which provides a green reduction pathway for GO to prepare graphene.
Although GO could readily disperse in water to form a stable homogeneous solution,19 its dispersion in ethanol is still a challenge. This is because the hydrogen bond energy between epoxy, hydroxyl and carboxyl functional groups of GO and water is higher than that of GO and ethanol. The enthalpy of GO dissolution in water is smaller than in ethanol,20 which means GO has a better affinity with water than ethanol.21 In the case of GO powder dispersing in water, water molecules could insert into the interstitials of GO powder where water could freely pass through GO molecule layers by formation of hydrogen bonds,22–24 intercalating GO powder into individual molecule layers. However, in the case of GO powder dispersing in ethanol, the intercalation of ethanol in GO molecules is strongly hindered. It was reported that in an ideal state, only one ethanol monolayer could form between neighboring GO molecules.23 It is much harder for ethanol to further insert into the interstitials of GO powder than water due to insufficient hydrogen interactions. Therefore, the separation and dispersion of GO powder in ethanol is much more difficult than in water.25 It is of great significance to seek a method that enables stable dispersion of GO in ethanol for further improving the use of ethanol in large-scale GO reduction into graphene.
Application of supramolecular interactions including π–π interaction, hydrogen bonds, electrostatic interactions et al. to stabilize GO dispersion in various liquid medium is a solution to the incompatibility between GO and the solvent.26,27 Most studies based on this strategy applied small molecules28–30 that possess functional groups that can interact with GO as well as the liquid medium. These small molecules act as coupling agents that can connect with GO by supramolecular interactions as well as the liquid medium, so as to intercalate the GO layers, realizing GO dispersion29 in liquid medium by constructing stable supramolecular compound structures.30 However, these compound structures are weakly bounded and constructed in dilute solutions. There is little study on the stability of these compound structure in concentrated solutions or after drying into powders.
Physical insertion of solid particles is another strategy to stabilize GO dispersion. Compared with dispersion strategy by supramolecular modification, solid particles are much larger and could provide more space between GO sheets. The dispersion stability remained using this strategy in a concentrated suspension and even in a solid state. In Sun's research,31 silica fume of ultra-fine size was inserted between GO sheets, and GO agglomeration was suppressed in solidified Portland cement. Yang et al.32 used Mg2+/Al3+ decomposed from slag to form layered double hydroxides on the GO sheets. The distance of GO was enlarged, and the aggregation of GO was restrained in a solidified alkaline activated slag cement. Except for inorganic particles, organic particles were also utilized to disperse GO in a polymer matrix. Xiao et al.33 grafted polystyrene onto the GO nanosheets via electrostatic adsorption, conducted by an in situ demulsification process. The dispersion stability of GO in polystyrene was improved.
In this study, attempts of combining both supramolecular interactions and insertion of solid particles were carried out. Solid phenyl-modified colloidal silica nanospheres (PSNS) were synthesized. The π–π interaction between sp2 carbon rings of PSNS and GO could lead to the formation of PSNS@GO structures. By optimizing the amount of benzene rings on PSNS, solid PSNS particles achieved redispersion of dried PSNS@GO powders.
2 Experimental
2.1 Materials
Tetra-ethyl ortho-silicate (TEOS), phenyl-tri-ethoxy-silane (PTES), ethanol (EtOH, i.e., C2H5OH) with a purity of 99%, and aqueous ammonia (NH4OH; 25–28% NH3 basis) were purchased from Sinopharm Chemical Reagent Co. Ltd. 1–2 layers of GO were bought from Suzhou Tanfeng Graphene Oxide Technology Co., Ltd. The purity of GO was greater than 95%, the diameter of the sheet layer was 10–50 μm, the thickness was about 1 nm, and the specific surface area was 100–300 m2 g−1. Water was deionized in-house to a resistivity of 16.8 MΩ cm. All chemicals were used without further purification.2.2 Preparation of the PSNS
Using the Stöber method,34,35 PSNS was prepared by a one-pot two-step feeding process. Solution A was a mixture of 10 mL aqueous ammonia, 30 mL deionized water, and 100 mL of ethanol. Solution B was a mixture of 10 mL TEOS and 10 mL ethanol. After adding solution B into solution A at a speed of 2 mL min−1, the reaction mixture was stirred for 3 h at a mechanical stirring speed of 200 rpm and 30 °C. For the preparation of pure colloidal silica nanospheres (SNS), the reaction product was prepared without the addition of PTES and named S-1. The modification of SNS was carried out as follows. After 3 h of reaction, 5 mL of ammonia was immediately added, and the dispersant PTES (5 vol%, 10 vol% or 20 vol% of TEOS) dissolved in 10 mL ethanol was injected into the stirred solution at a speed of 2 mL min−1 by a peristaltic pump to modify the surface of SNS and named S-2, S-3, and S-4, respectively. The reaction was allowed to continue stirring for an additional 2 h after the addition of the dispersant. After the reaction, the particles were separated by centrifugation in every step of the solid–liquid separation and washed with deionized water and ethanol by ultrasonication (120 W, 40 kHz). Finally, PSNS powders were dried at 50 °C for 12 h for various characterizations.2.3 Assembling of PSNS@GO
Firstly, 0.5 g SNS and PSNS (S-1, S-2, S-3, and S-4), respectively, were dispersed in 50 mL ethanol, to which 0.5 g GO powder was added by mechanical stirring at 200 rpm and 60 °C for 3 h. Finally, the dispersion samples were dried at 80 °C for 12 h for various characterizations and named Sg-1, Sg-2, Sg-3, and Sg-4, respectively. A control sample, Sg-0, was prepared by dispersing 0.5 g GO in 50 mL ethanol, without PSNS. The preparation process is shown in Fig. 1.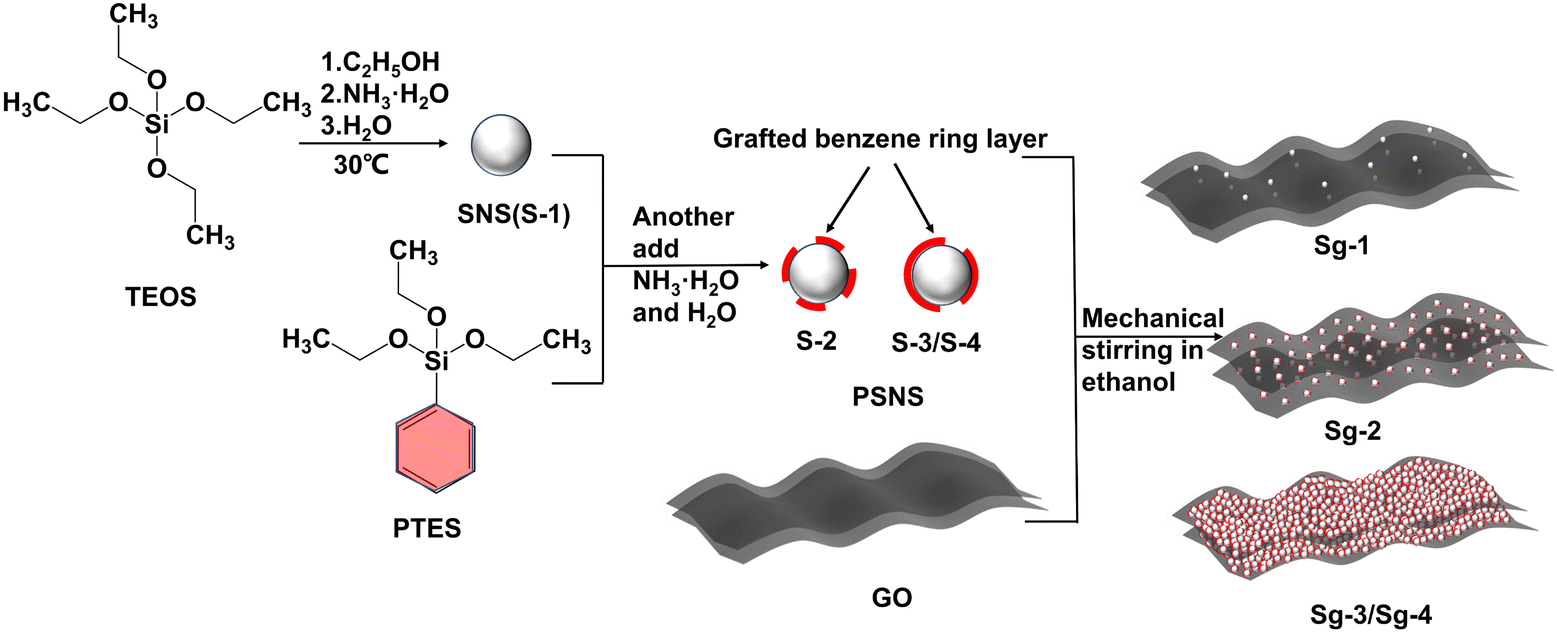 | ||
| Fig. 1 Schematic preparation process of PSNS@GO. | ||
2.4 Dispersion stability evaluation of PSNS@GO in ethanol
The first dispersion and redispersion stability of GO in ethanol was examined by sedimentation experiments. It was carried out in 15 mL glass vials. For the first dispersion stability evaluation, the assembled PSNS@GO suspension was transferred to glass vials and the dispersion stability was observed after mechanical stirring at room temperature for 30 min. Then, the assembled PSNS@GO samples were dried into films. For redispersion, 0.12 g PSNS@GO dry film particles (Sg-0, Sg-1, Sg-2, Sg-3, and Sg-4) were milled and dispersed in 12 mL ethanol by mechanical stirring at room temperature for 30 min. After a period of time, the dispersion stability was estimated based on photographs of dispersed particles after standing with reference. The height of the supernatant (Hn) and the total height of the suspension (H0) are measured to calculate the settlement rate, SR = (Hn/H0) × 100%.352.5 Characterization of PSNS@GO
The surface morphology of PSNS and PSNS@GO was observed by QUANTA FEG 450 Scanning Electron Microscope (SEM). The interlayer spacing of the sample was obtained by using the X-ray diffraction analyzer model Empyrean produced in the Netherlands. The change of organic functional groups was investigated using a Nexus Intelligent Fourier transformed infrared spectrometer from Thermo Nicolet Co., Ltd in a transmittance mode. The topological structures of dried PSNS@GO films were investigated by micro-CT (Xradia 510 Versa), the colormap of the image was in the range of 37![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 to 40000 with 0.3 of opacity in volume rendering. The concentration of carbon atoms in different chemical environments in the sample was studied by 13C solid-state nuclear magnetic resonance (NMR) spectroscopy (Bruker, Advance, III400), it was carried out at 10 MHz and at a 25 kHz spinning speed. The structural changes of GO were detected by Raman spectrometer. The thermal stability was investigated using a thermal gravimetry (TG)-differential scanning calorimetry (STA449F3) instrument at a heating rate of 10 °C min−1 in which air was used as purging gas.
000 to 40000 with 0.3 of opacity in volume rendering. The concentration of carbon atoms in different chemical environments in the sample was studied by 13C solid-state nuclear magnetic resonance (NMR) spectroscopy (Bruker, Advance, III400), it was carried out at 10 MHz and at a 25 kHz spinning speed. The structural changes of GO were detected by Raman spectrometer. The thermal stability was investigated using a thermal gravimetry (TG)-differential scanning calorimetry (STA449F3) instrument at a heating rate of 10 °C min−1 in which air was used as purging gas.
3 Results and discussion
3.1 Morphology and chemical composition of PSNS
The surface morphology of PSNS was observed by SEM. As shown in Fig. 2, the sizes of PSNS are relatively uniform with diameter in the range of 200–300 nm. It can be seen from Fig. 2(a)–(c) that S-1, S-2, and S-3 are perfectly spherical without adhesions. However, for S-4, severe adhesions can be found between the PSNS possibly due to cross-linking of silicate oligomers formed between PSNS by excess PTES. | ||
| Fig. 2 SEM images of (a) S-1, (b) S-2, (c) S-3 and (d) S-4. | ||
FTIR spectra of PSNS are presented in Fig. 3. For all of the samples, there are obvious absorption peaks at 470 cm−1, 800 cm−1 and 1100 cm−1, which correspond to the bending vibration, symmetrical stretching vibration, and anti-stretching vibration of Si–O–Si bond.36 The absorption peak at 957 cm−1 is attributed to the tensile vibration of the Si–O bond in Si–OH.36 The peak at around 1640 cm−1 is assigned to H–O–H bending vibrations of weakly bound H2O adsorbed on the nanoparticle surface of PSNS. The typical broad band shown for samples at 3430 cm−1 associated with hydrogen-bonded silanol groups with absorbed molecular water and ethanol.
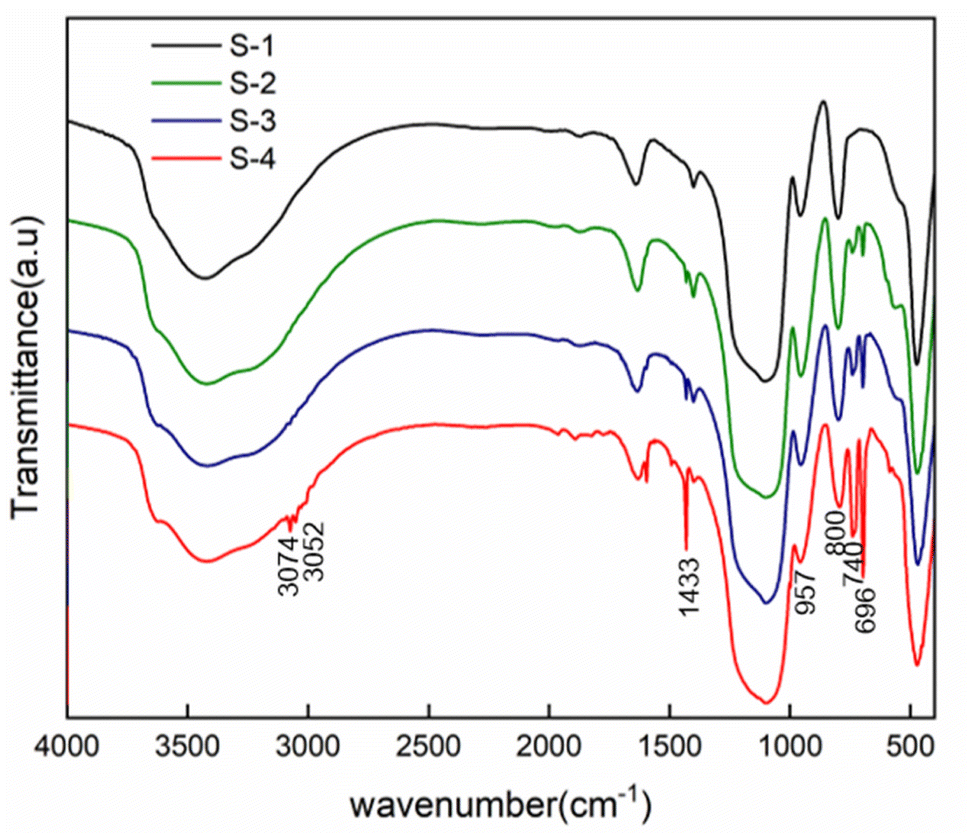 | ||
| Fig. 3 FTIR spectra of PSNS samples. | ||
Compared with sample S-1, obvious new absorption peaks at 696 cm−1, 740 cm−1, 1433 cm−1, 3052 cm−1 and 3074 cm−1 can be found for samples S-2, S-3, and S-4. The absorption peak positions at 740 cm−1 and 696 cm−1 are attributed to the out of plane bending of the C–H bond on the monosubstituted benzene ring;37 1433 cm−1 is own to stretching vibration absorption peak of the benzene ring skeleton;38 3052 cm−1 and 3074 cm−1 are the C–H bond stretching vibration absorption peaks on the benzene ring,38 which is a sign of presence of phenyl functional groups on the surface of SNS after washing and drying.
3.2 Characterization of PSNS@GO
SEM was used to analyze the assembled structure between different PSNS and GO after drying and milling, and the results are shown in Fig. 4. Unmodified silica nanospheres intercalated with GO are shown in Fig. 4(a). It can be seen that only a small amount of SNS was assembled onto GO sheets. In contrast, Fig. 4(b)–(d) show that the PSNS could be assembled onto the GO sheets in large amounts, even after suffering severe mechanical milling. This indicates that the attraction between PSNS and GO increases with the introduction of phenyl functional groups, which play an important role in the non-covalent assembly between PSNS and GO. This also demonstrates possible existence of π–π interactions between PSNS and GO. For sample Sg-2, when the addition of PTES was 5 vol% of TEOS, the prepared PSNS (S-2) was assembled as monolayers on the surface of GO sheets and had good dispersion on GO sheets (Fig. 4(b)). For sample Sg-3, thicker layer of PSNS was formed and assembled as shown in Fig. 4(c). And severe agglomeration appeared between Sg-4 solid particles (Fig. 4(d)). Obviously, PSNS with an appropriate amount of phenyl functional groups can be more effectively assembled on the surface of GO sheets. As the content of phenyl functional groups in PSNS further increases, PSNS@GO solid particles tend to agglomerate. | ||
| Fig. 4 SEM images of PSNS@GO samples of (a) Sg-1, (b) Sg-2, (c) Sg-3 and (d) Sg-4. | ||
In order to compare the effect of non-covalent functionalization on the interlayer spacing of GO, the PSNS@GO powder samples were characterized by XRD. As shown in Fig. 5, raw GO has a high intensity, narrow diffraction peak at 2θ = 9.60°, with a layer spacing of 0.91 nm, as calculated by the Bragg equation, which is larger than the layer spacing of graphene at 0.335 nm.39 This is due to the large number of oxygen-containing groups on the GO sheets, which significantly increase the interlayer spacing compared with graphene.
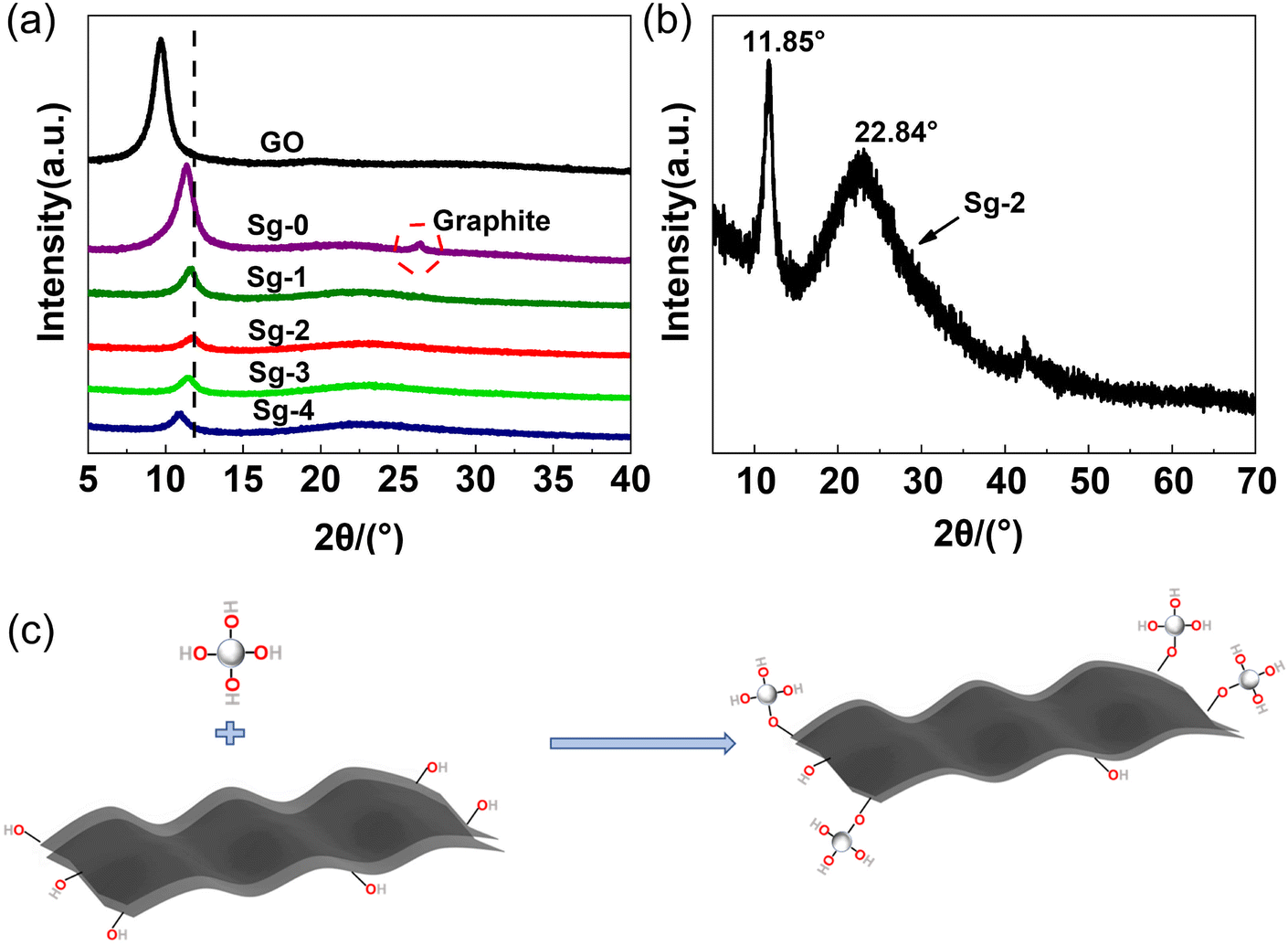 | ||
| Fig. 5 (a) XRD patterns of GO powders and PSNS@GO samples, (b) enlarged XRD patterns of PSNS@GO sample Sg-2, (c) schematic diagram of the reaction between the silica hydroxyl groups on silica nanospheres and the oxygen-containing functional groups on GO sheets. | ||
Sample Sg-0 is unmodified GO dispersed in ethanol and dried directly. As it can be seen, Sg-0 has a narrow diffraction peak at 2θ = 11.34°, with a layer spacing of 0.78 nm. Compared with raw GO powder, the XRD diffraction peak of Sg-0 significantly shifted to higher degree, and a new diffraction peak appears at 26.46°. This is possible due to that some of the oxygen-containing groups of the GO are eliminated during the drying process,40 which reduces the distance between the GO layers. At the same time, the GO sheets are irreversibly agglomerated during the drying process, and they are stacked into multilayer graphene or graphite sheets as indicated by the new diffraction peak appears at 26.46°.
The XRD diffraction peak of PSNS@GO also shifted to higher degree, but there is no diffraction peak around 26°. It confirmed that the silica nanospheres effectively inhibited the tendency of GO to agglomerate during the drying process. There are broad diffraction peaks at 22.84° in samples Sg-1, Sg-2, Sg-3 and Sg-4, which indicates that the PSNS are amorphous.41 These samples have diffraction peaks at 11.62°, 11.85°, 11.47°, and 10.87°, respectively, and the corresponding interlayer spacings gradually increased from 0.76 nm to 0.81 nm. The layer spacing of Sg-2 decreased the most, while the Sg-4 sample decreased the least compared with GO. It is possible that the chemical composition of the silica surface has an effect on the layer spacing of GO. The reduction of the GO interlayer spacing may be influenced by the hydroxyl group content on the surface of silica nanospheres. The silica hydroxyl groups on the surface of the silica nanospheres promote the removal of oxygen-containing groups from the GO surface as Fig. 5(c), leading to the reduction of the GO interlayer spacing. Compared with Sg-1, more silica hydroxyl groups are brought in Sg-2 due to the introduction of PTES, leading to more depletion of oxygen-containing groups on the GO surface and a decrease in the layer spacing. When the PTES concentration is higher than 5 vol%, the PSNS itself will undergo condensation that will consume silicone hydroxyl groups, and the ability to reduce the GO interlayer spacing is reduced as the silanol content on the PSNS surface decreases. The reaction between the silanol groups on the PSNS surface and the oxygen-containing groups on the edges of the GO sheets may explain this phenomenon.
Chemical structure changes during the surface modification of GO can be followed by Raman spectroscopy. Fig. 6 shows the Raman spectra of raw GO powder, Sg-0, Sg-1, Sg-2, Sg-3, and Sg-4. As can be seen, the Raman spectrum of the pure GO displays a relatively strong band at 1346 cm−1 (D band) and 1580 cm−1 (G band), which is attributed to the vibrations of defective and disordered sp3 carbon atoms and the vibrations of sp2 carbon atoms in the two-dimensional hexagonal lattice of graphite, respectively.42 The intensity ratio of D band to G band (ID/IG), was calculated as a measure of the quality of graphitization or defective disorders on GO. The ID/IG value is 1.12 for raw GO, which shows high defect region on GO.
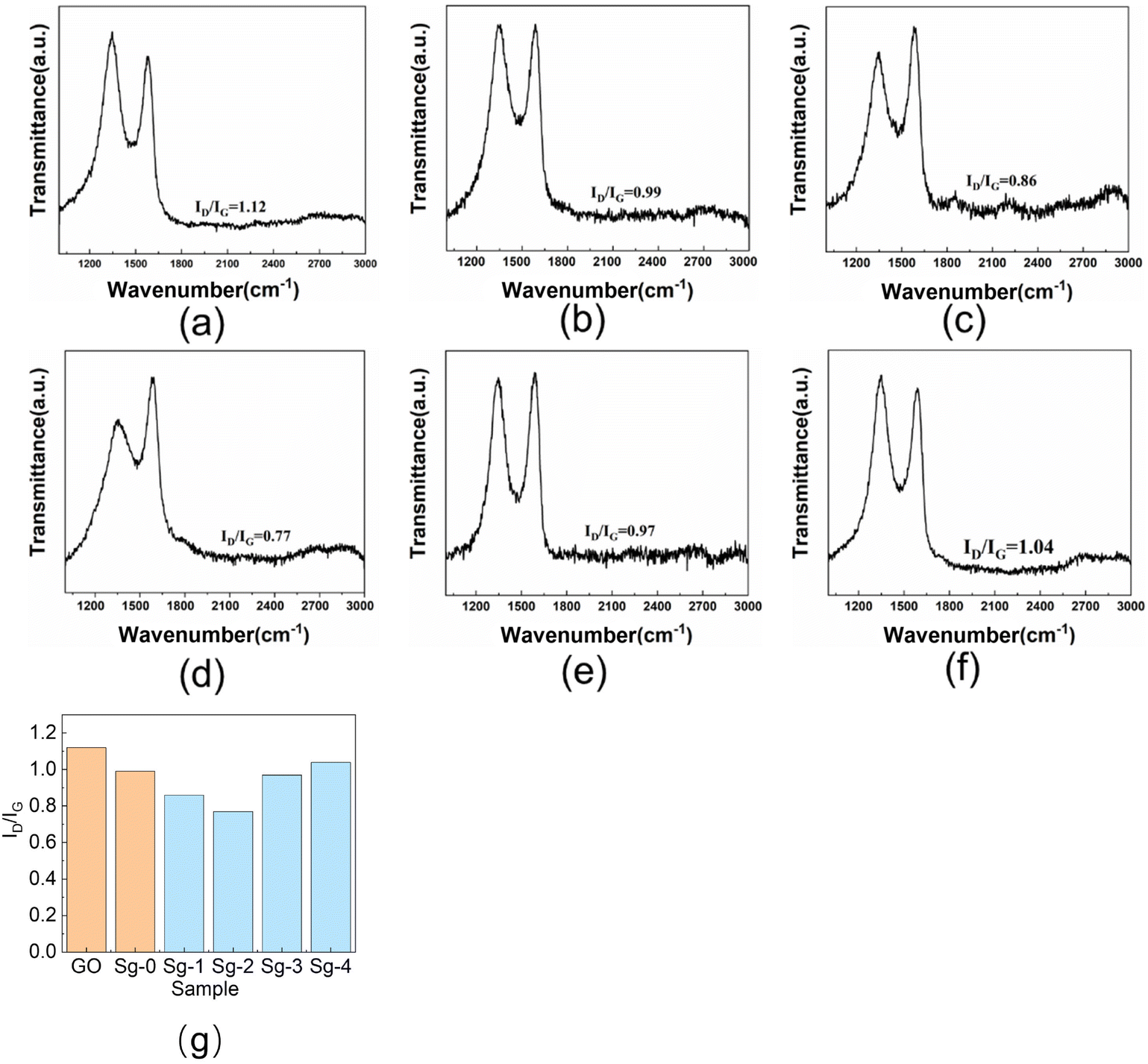 | ||
| Fig. 6 Raman spectra of GO and PSNS@GO: (a) GO; (b) Sg-0; (c) Sg-1; (d) Sg-2; (e) Sg-3; (f) Sg-4; (g) ID/IG values of GO and PSNS@GO. | ||
Compared with GO, the ID/IG value of Sg-0 is significantly lower, and the value is 0.99. It is possibly due to that part of the oxygen-containing functional groups on the GO sheets are eliminated during the drying of the Sg-0.40 As can be seen in Fig. 6(g), when the GO is assembled with SNS or PSNS, the ID/IG value of the sample is lower than that of the GO sample without further treatment, and the ID/IG value of the sample Sg-2 is the lowest with a value of only 0.77. This indicates that the number of defects on GO sheets is decreasing, which may be due to the consumption of oxygen-containing functional groups on GO sheets by silica hydroxyl groups on the surface of silica nanospheres, and more importantly, the benzene ring on the surface of PSNS increases the concentration of sp2 hybridized carbon. The ID/IG value of Sg-1 is lower than that of Sg-0 because the surface of Sg-1 contains a large number of hydroxyl groups that can consume more oxygen-containing groups, resulting in a weaker D band intensity. The ID/IG value of Sg-2 is slightly lower than that of Sg-1 because the surface of Sg-2 is grafted with phenyl functional groups, thus increasing the concentration of sp2 hybridized carbon, resulting in an increase in the G band intensity. With the increase of phenyl content on the surface of PSNS, the ID/IG values of Sg-3 and Sg-4 gradually increased, which may be due to the weakening of G band intensity caused by GO wrapping of PSNS.
In order to investigate effect of phenyl groups from PSNS on assembling structure and compactiveness of dried GO film samples, namely Sg-1 and Sg-2 dry films were selected and studied by micro-CT and the results are shown in Fig. 7. Fig. 7(a) is the as prepared dry film samples of Sg-1 and Sg-2. As can be seen for sample Sg-1, there is a dense layer of white precipitant at the bottom, which is possibly agglomeration of SNS without phenyl groups that not adsorbed on the surface of GO during the drying process. This is in line with Fig. 4(a) where little SNS can be identified on the milled powder surface. Without phenyl groups, colloidal silica is difficult to assemble on GO lateral surface. In contrast, dry film sample of Sg-2 on Fig. 7(a) showed that SNS with phenyl groups were dispersed homogeneously, and there was no obvious agglomeration. A block of certain size was taken from the dried film for micro-CT testing, and the results are shown in Fig. 7(b) and (c). Both CT images were processed using the same parameters. The red part of the figure shows how dense the material in the GO block is, and the blue and purple parts represent materials of lower density in the sample. Fig. 7(b) is the micro-CT diagram of Sg-1, from which it can be seen that there are large red areas distributed on the top surface and bottom surface of Sg-1, which means that there is a layer of denser substances distributed on Sg-1. This is precisely because SNS that not adsorbed on the surface of GO is agglomerated during the drying process. It shows that SNS (S-1) are difficult to adsorb a large number of GO sheets only through silicon hydroxyl interaction, and the results are also consistent with Fig. 7(a). The red areas of the top surface and bottom surface in Fig. 7(c) are small and uniformly dispersed, indicating that PSNS (S-2) can be uniformly assembled onto GO and little precipitation occurred. It can also be seen from the figure that the blue and purple area of the cross section in dry film Sg-2 is larger than the area of the cross section in Sg-1. A fluffy structure can be found on the top surface. The density at the bottom of the film is much lower than dry film Sg-1. These evidences indicate that after drying, Sg-2 dry film has a much looser spatial structure than Sg-1. Therefore, the introduction of phenyl groups can effectively improve the assembly efficiency of silica nanospheres and GO.
 | ||
| Fig. 7 The topological structures of block samples (a) tested samples; (b) Sg-1; (c) Sg-2. | ||
13C solid-state NMR of GO and various PSNS@GO samples were measured to further investigate the interactions between GO and PSNS to provide more information on the assembly of PSNS on GO sheets, and the results are displayed in Fig. 8. According to SEM results, severe agglomeration occurred for samples Sg-3 and Sg-4, and there was no obvious distribution variance of the two. Therefore, 13C solid-state NMR test did not include sample Sg-4. C–O–C bonds and C–OH bonds, which belong to GO, appear at 60 ppm and 70 ppm, respectively, and sp2 hybridization emerges at 131 ppm.43–45 In this study, only the benzene rings in PTES and GO could reflect sp2 hybridization from 13C solid-state NMR for all used materials. Although there was residual solvent on the GO sheets, the following TG result shows residual solvent, including water and ethanol, below 5%, and the residual solvent would have little impact on the C–O–C and C–OH signals. As can be seen from the 13C solid-state NMR, the intensity of C–O–C and C–OH was similar for all samples, but there was a difference in sp2 hybridized carbon. The sp2 hybridized carbon intensity of Sg-1 was the same as GO, suggesting that mixture of S-1 and GO has little impact on the carbon atom signals. The sp2 hybridized carbon intensity of Sg-2 was higher than Sg-1, and Sg-3 was the highest. To quantify such trend, the corresponding peaks attributed to carbons from C–O bonds and sp2 hybridized carbon rings were integrated. The integral from C–O bond intensity was represented as S1, and the one from sp2 hybridized carbon rings was represented as S2. The ratio of S2 to S1, an index for sp2 carbon, is a normalized value indicating the amount of carbon atoms from sp2 hybridized carbon rings, as shown in Fig. 8(b). The index of GO and Sg-1 were 0.328 and 0.343, respectively. With the increased usage of PTES during the synthesis, the index increased to 0.528 for Sg-2 and to 0.882 for Sg-3, respectively. The whole assembly process did not involve C–O bonds, so the intensity of C–O–C bonds and C–OH bonds had no obvious change for all samples. Since PTES was not used in S-1, there were no extra benzene rings introduced in the mixture of S-1 and GO, so the intensity of Sg-1 was the same as GO. In this case, very few SNS was dispersed on the GO sheets. For S-2, PTES at 5 vol% was used. Due to the π–π interaction between benzene rings on PSNS and GO, PSNS containing phenyl groups could successfully assemble on the GO surface and stay stable, as confirmed by the SEM result (Fig. 4(b)), and a stronger intensity of sp2 hybridized carbon from the 13C solid-state NMR spectrum of Sg-2. Further increasing the usage of PTES led to more benzene rings being grafted on PSNS, and more solid particles appeared on the GO sheets by π–π interaction, revealed in Fig. 4(c), which resulted in the strongest sp2 hybridized carbon intensity for Sg-3.
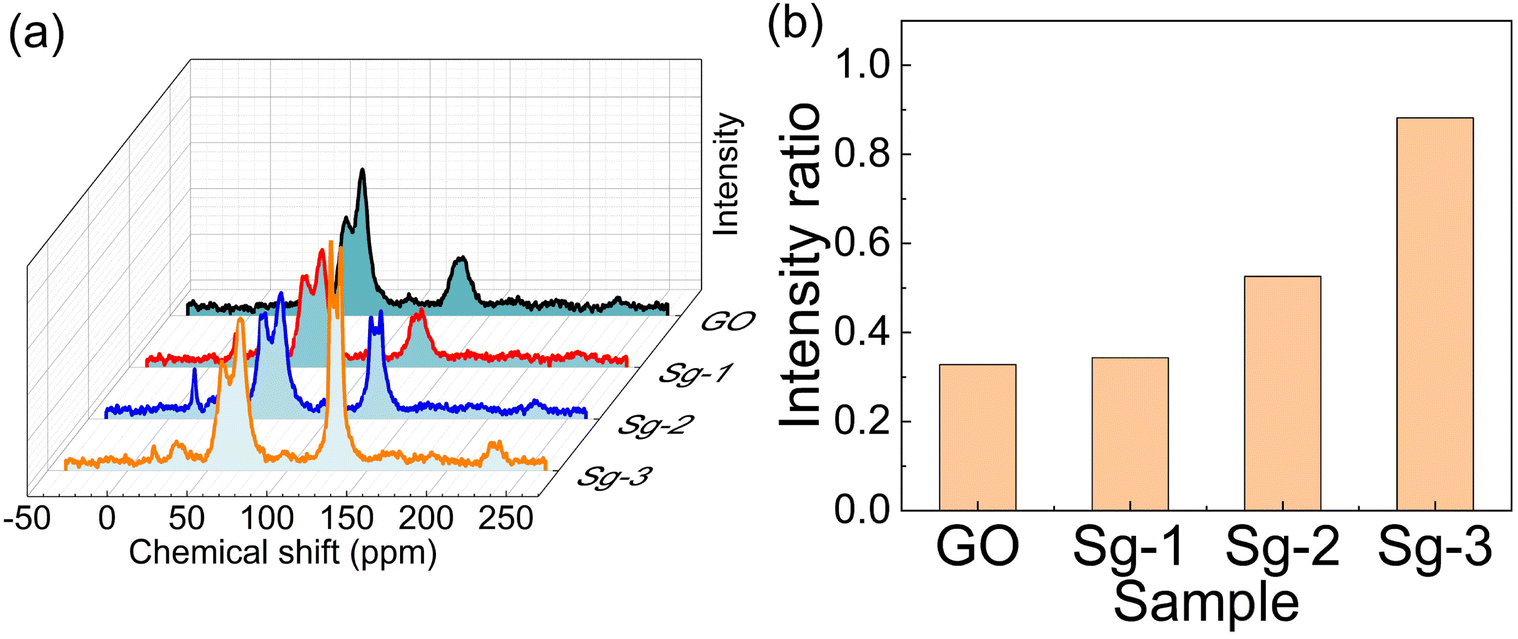 | ||
| Fig. 8 (a) 13C solid-state NMR of GO, Sg-1, Sg-2 and Sg-3; (b) the ratio of sp2 of C to C–O bond intensity. | ||
Fig. 9 shows the FTIR spectra of synthesized samples of S-2 and Sg-2 along with pure GO. The peaks located at 3435 cm−1, 1720 cm−1, 1612 cm−1, and 1230 cm−1 in the GO spectrum indicate the presence of –OH, C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O, CC, and C–O–C bonds,46 respectively. It suggests the existence of hydroxyl, carboxyl, and epoxide groups on the surface of GO. The spectrum of the Sg-2 has several new peaks that are not present in the GO spectrum, and the appearance of new peaks at 954 cm−1, 800 cm−1, 668 cm−1, and 694 cm−1 indicates the formation of Si–OH, Si–O–Si, and Si–O–C, which is further evidence of the presence of silane on the GO surface. The appearance of new peaks at 740 cm−1 and 697 cm−1 further demonstrates the presence of phenyl on the GO surface, which also provides strong evidence for the adsorption of phenyl functional groups onto GO via π–π interactions.
O, CC, and C–O–C bonds,46 respectively. It suggests the existence of hydroxyl, carboxyl, and epoxide groups on the surface of GO. The spectrum of the Sg-2 has several new peaks that are not present in the GO spectrum, and the appearance of new peaks at 954 cm−1, 800 cm−1, 668 cm−1, and 694 cm−1 indicates the formation of Si–OH, Si–O–Si, and Si–O–C, which is further evidence of the presence of silane on the GO surface. The appearance of new peaks at 740 cm−1 and 697 cm−1 further demonstrates the presence of phenyl on the GO surface, which also provides strong evidence for the adsorption of phenyl functional groups onto GO via π–π interactions.
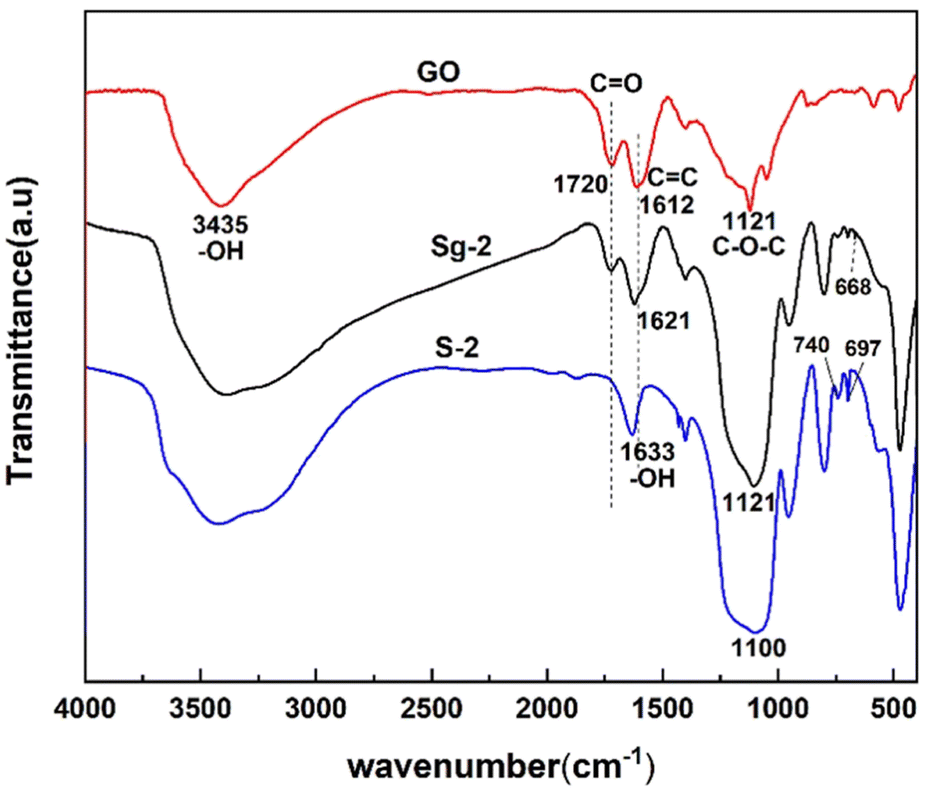 | ||
| Fig. 9 Fourier-transformed infrared spectra of GO, PSNS and PSNS@GO. | ||
In order to investigate the thermal stability as well as oxidation resistance of the samples, TG analysis experiments were performed in air atmosphere. Fig. 10 shows the TG and DTG for GO and PSNS@GO. As can be seen from the figure, GO lost 8.2% of its weight at temperature lower than 150 °C, which was due to the dissipation of the solvent adsorbed on the surface of GO.47 Increasing the temperature from 150 °C to 350 °C, a weight loss of 29.5% occurred in GO, which was attributed to the thermal decomposition of oxygen-containing functional groups such as hydroxyl, carbonyl, and carboxylic acid groups.48,49 Finally, in the temperature range from 450 °C to 600 °C, a 52.9% weight loss occurred due to the decomposition of the CC bond of the main structure of GO.50–53 It can also be seen in Fig. 10 that the weight losses of Sg-1, Sg-2, Sg-3, and Sg-4 are divided into three main stages. The first stage occurred in the temperature range from 40 °C to 150 °C, which was related to the evaporation of the solvent adsorbed on the sample surface. There is no significant difference in the weight losses of Sg-1, Sg-2, Sg-3 and Sg-4 in this temperature range, with weight losses of 4.2%, 3.5%, 3.6%, and 3.5%, respectively. The second stage of weight loss occurred from 150 °C to 350 °C, which was mainly caused by the decomposition of the unconsumed oxygen-containing functional groups on GO. In this temperature range, the sample Sg-1 showed the highest weight loss of 18.6%. This is due to that only a small amount of silica nanospheres in Sg-1 are adsorbed onto GO, which has the highest weight loss compared to Sg-2, Sg-3, and Sg-4, which have less consumption of oxygen-containing groups on GO. The most significant weight loss occurred in the temperature range of 350-600 °C, which was mainly owned to the decomposition of the phenyl functional groups on the surface of PSNS and the CC bond of the main structure of GO. Finally, the residual weight of Sg-1 and Sg-2 stabilized at 600 °C, and the weight losses from 350 °C to 600 °C were 31.3% and 29.5%, respectively. The residual weight of sample Sg-3 and sample Sg-4 was stable at 630 °C and 650 °C, and the weight losses were 28.9% and 30.8%, respectively. With the increase of the phenyl content on the surface of the PSNS, the temperature, at which the weight of the sample became stable in this section, becomes higher. This is possibly because for the PSNS (S-2, S-3, and S-4) on Sg-2, Sg-3, and Sg-4, π–π stacking effect occurred between phenyl group and the sp2 carbon skeleton of the GO, which makes the part less likely to react with oxygen and burn out more difficult. When the phenyl modifier content reaches 20% (Sg-4), the weight loss is increasing compared to the sample Sg-3. This is because the excessive phenyl modifier self-condensed to generate oligomers, doped in the middle of PSNS@GO, which reduces the thermal stability of the sample. From the above results, it can be seen that the weight loss of GO modified by silica nanospheres is significantly reduced, and the thermal stability and oxidation resistance are significantly improved, which are attributed to the excellent thermal stability and oxidation resistance of phenyl modified silica nanospheres.54
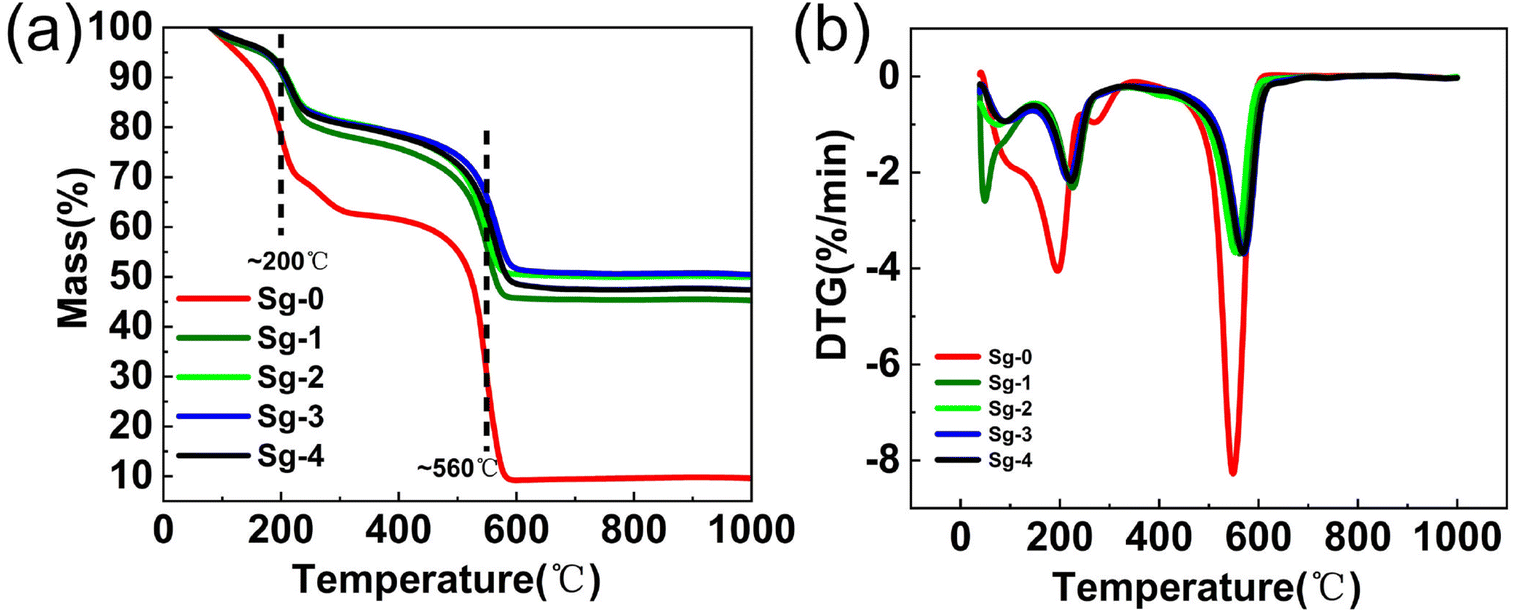 | ||
| Fig. 10 (a) TG and (b) DTG curves of PSNS@GO. | ||
3.3 Dispersion and redispersion of PSNS@GO in ethanol
To investigate the effect of non-covalent modification on the dispersibility of GO powders, PSNS@GO was ultrasonically dispersed in ethanol to observe the settling conditions. Fig. 11(a) shows the digital images taken after standing still for 30 min, 60 min, 120 min, 360 min, and 480 min. From left to right, the samples are Sg-0, Sg-1, Sg-2, Sg-3, and Sg-4, respectively. The settlement rate (SR, defined in Section 2.4) of samples as a function of standing time is shown in Fig. 11(c). It can be seen that Sg-0, Sg-1, Sg-3 and Sg-4 settle faster, and the SR values reached 60% to 80%, while that of Sg-2 was only 11% after 480 min.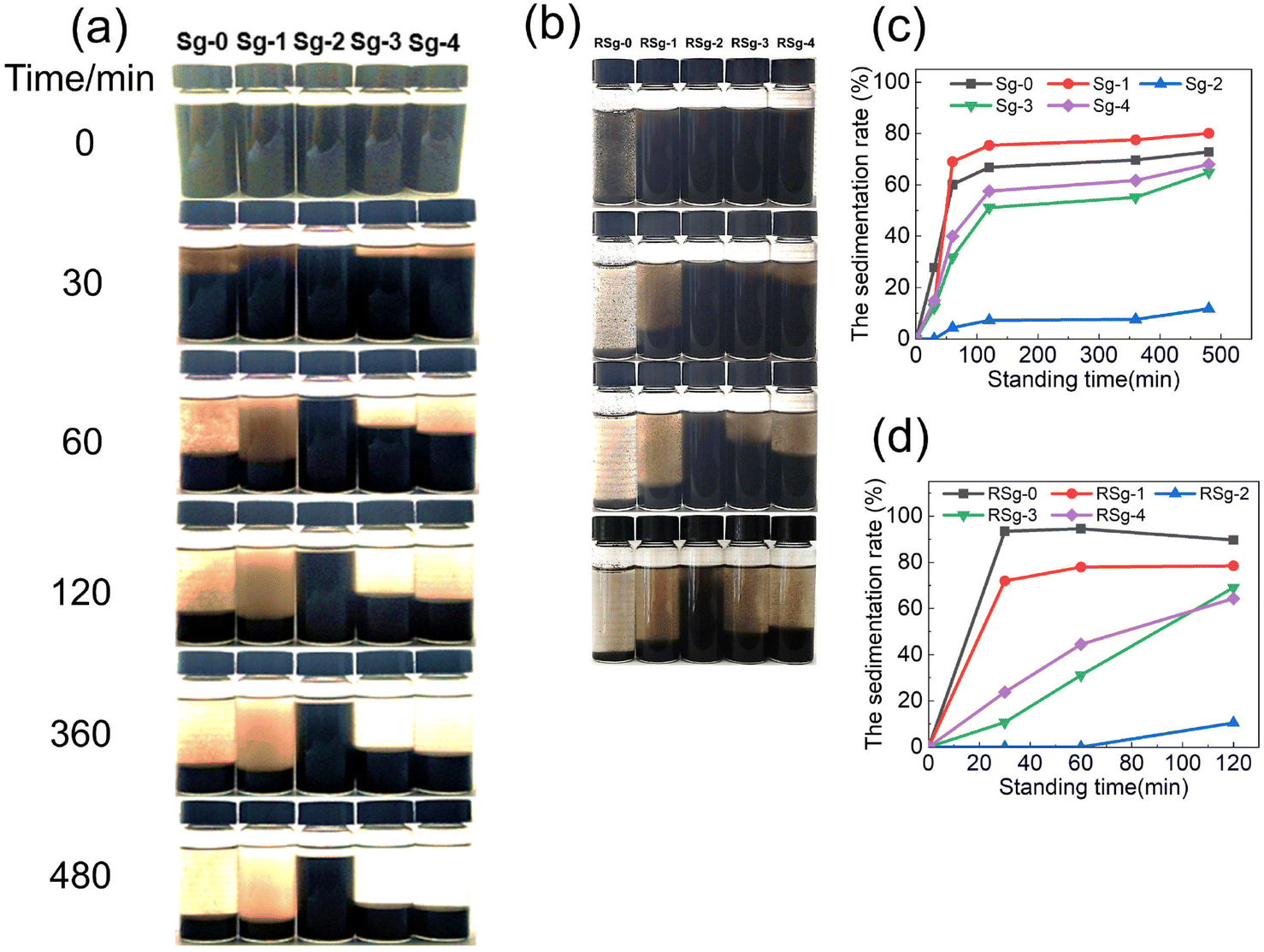 | ||
| Fig. 11 (a) Dispersion of 10 mg mL−1 PSNS@GO in ethanol (b) Redispersion of 10 mg mL−1 PSNS@GO in ethanol (c) the SR values with the standing time of first dispersion (d) the SR values with the standing time of redispersion. | ||
Within 60 min, most of the GO sheets settled in Sg-0, and the SR value was 60%. From the upper layer of Sg-0, it can be seen that a large amount of agglomerates was about to settle. The SR values of Sg-1 and Sg-0 in 480 min are both higher than 70%. It can be seen from the upper layer of Sg-1 that a large number of SNS without phenyl remained in it and did not improve the dispersion stability of GO.
The upper liquid of Sg-2, Sg-3, and Sg-4 is clear and transparent without obvious particles, which indicates that the PSNS are successfully adsorbed to the GO sheets. For PSNS@GO sample Sg-2, PSNS was modified by 5 vol% PTES, which settled the slowest. Within 480 min, most of the solid particles in Sg-2 can be stably dispersed in ethanol acting like colloidal suspension. When PSNS was modified by 10 vol% PTES in Sg-3, the SR of solid particles is higher than that of Sg-2, but lower than that of Sg-0 and Sg-1. The settling trend of Sg-4 is similar to Sg-3, but the SR is higher than Sg-3 at the same settling time. It can be seen that GO sheets loaded with PSNS of appropriate phenyl functional group content can greatly improve the dispersibility of GO. However, when the phenyl content is too high, PSNS will aggravate the agglomeration of GO nanosheets. This is consistent with the result of SEM images of PSNS@GO.
In order to verify the effect of PSNS on GO redispersibility, the first dispersed suspension samples were dried at 80 °C, milled into powder and added into ethanol to examine the SR values. Fig. 11(b) shows the digital images taken after holding still for 30 min, 60 min, and 120 min. Fig. 11(d) shows the SR of the redispersion of samples against standing time. As shown in Fig. 11(b), it can be found that the pure GO agglomerates in a large amount after drying and cannot be dispersed again. The PSNS@GO can still be redispersed. For PSNS@GO, the dispersion stability of Sg-2 is still the best, the SR value at 120 min is only 10%, while the SR values of Sg-1, Sg-3, and Sg-4 are 78%, 69%, and 64%, respectively, which is much higher than that of Sg-2. Therefore, the PSNS modified by 5 vol% PTES effectively improved the redispersibility of GO powder and enabled the preparation of redispersible GO powder.
3.4 Dispersion mechanism of PSNS@GO in ethanol
The mechanism of action of PSNS on the first dispersion and redispersion of GO in ethanol is shown in Fig. 12. Schematic diagrams of Sg-0, Sg-1, Sg-2, and Sg-3/Sg-4 first dispersion in ethanol, dry film formation, and redispersion of milled film powder in ethanol are shown in Fig. 12(a). During the first dispersion in ethanol, due to poor affinity between ethanol and GO, GO sheets cannot disperse in ethanol and settling of Sg-0 is the fastest. Although there is SNS in Sg-1, the interaction between SNS and GO is weak. Only a small amount of SNS is assembled on GO sheets, most of the SNS is in a free state in ethanol. The function of dispersion stability for SNS doesn't work, and the GO sheets in Sg-1 are still in an agglomerated state. For Sg-2, benzene rings have been successfully grafted on SNS and form PSNS, and the π–π stacking appeared between the phenyl group on PSNS and the GO carbon six-membered ring, PSNS is more easily adsorbed to GO sheets and is uniformly dispersed on GO. PSNS can enlarge the space between GO sheets and allow ethanol molecules to enter the GO sheets, allowing the sample to be stably dispersed in ethanol. For Sg-3/Sg-4, crosslinking between PSNS occurs due to the excessive phenyl content in PSNS, and its adsorption on GO sheets will also be excessive. Their settlement will be accelerated due to their more gravity brought by assembled excessive PSNS. Therefore, the dispersion effect in ethanol will be weakened.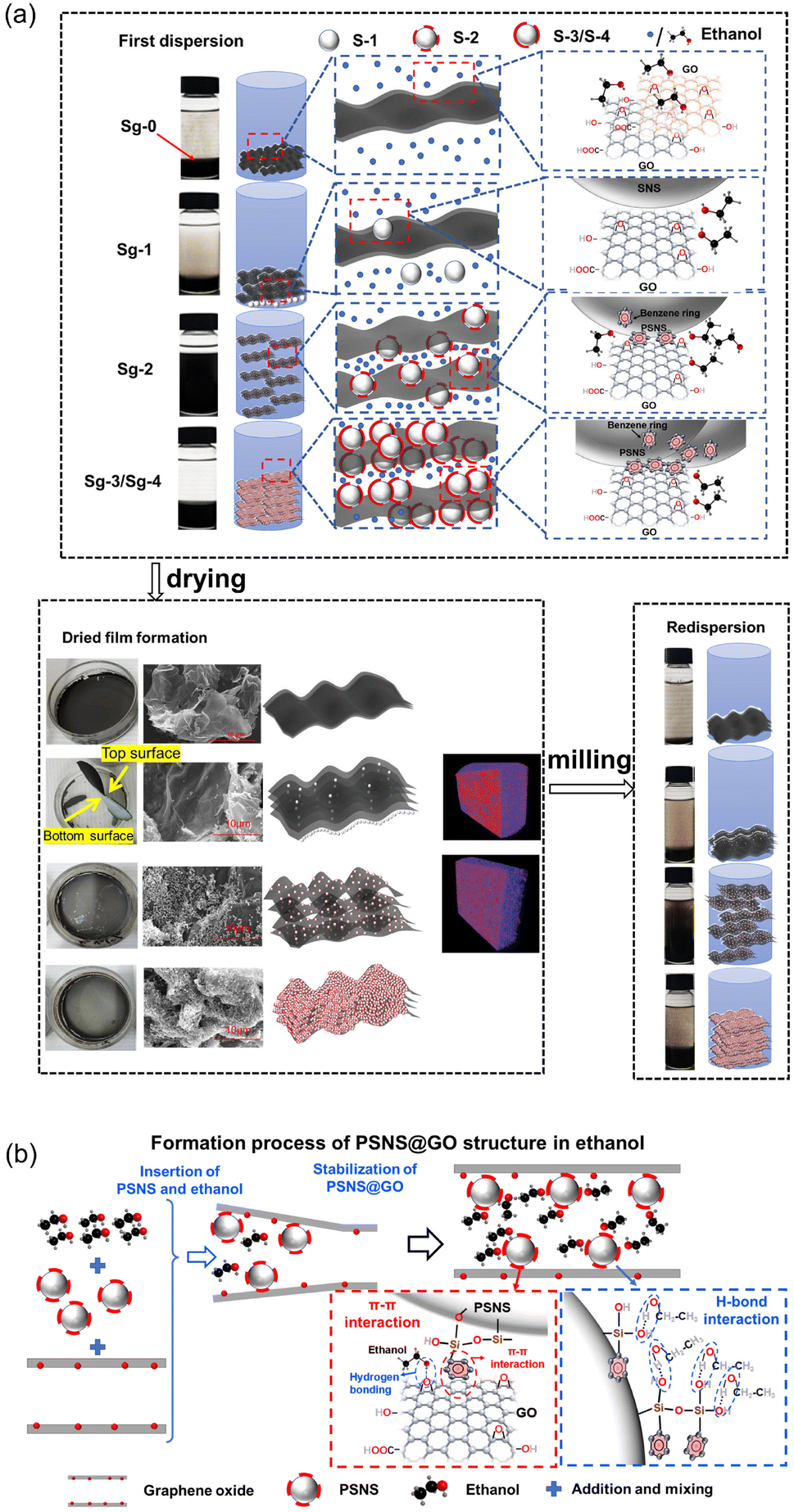 | ||
| Fig. 12 (a) Frist dispersion and redispersion mechanism of PSNS@GO in ethanol, and (b) formation process of PSNS@GO structure in ethanol. | ||
During drying and film formation process, Sg-0 film is in a homogeneous state, and there is a dense stacking in Sg-0. Because there is a very little connection between GO and SNS, and settlement of free SNS in Sg-1 suspension. A layer of white precipitated substance form at the bottom of the Sg-1 film, which is also displayed in micro-CT result. Sg-2 produces a relatively loose structure for the reason of wonderful assembly of PSNS on GO sheets. Sg-3/Sg-4 will still agglomerate due to the excessive phenyl content in PSNS.
For redispersion, when several groups of samples are redispersed, the samples will undergo a certain agglomeration. And Sg-2 can still maintain good redispersibility, while the redispersion effect of Sg-0, Sg-1 and Sg-3/Sg-4 suspension samples is relatively poor. Therefore, when PSNS with 5 vol% PTES, the preparation of redispersible GO powder can be achieved.
A detailed formation process of PSNS@GO structure in ethanol is shown in Fig. 12(b), for a better understanding of the above procedure. Ethanol only forms a monolayer attached on GO surface by the hydrogen bonds, and it's very hard for ethanol to pass through GO sheets.23 When PSNS is added, it firstly inserts and expands the space between GO sheet edges due to PSNS steric hindrance. The following PSNS and ethanol could insert deeper via the enlarged passage, and meanwhile, PSNS assemble on the GO sheets. PSNS and GO can stably connect with each other via supramolecular interaction, which is realized by π–π interaction between the benzene rings on GO sheets and PSNS. On the other hand, there are plenty of silicon hydroxyl groups on the assembled PSNS. The hydroxyl groups of ethanol can connect with them, and passages for ethanol are formed. The affinity of GO with PSNS and ethanol is improved by the introduction of silicon hydroxyl groups. The dispersion stability of GO in ethanol is promoted.
4 Conclusions
In this study, PSNS was prepared by sol–gel using TEOS and PTES, and PSNS@GO was successfully assembled by supramolecular interaction and physical intercalation between PSNS and GO. The results showed that the surface modification of SNS with 5 vol% PTES could obtain stabilizers capable of making GO achieve dispersible and redispersible stability in ethanol. In the ethanol solution of PSNS@GO, it could be stably dispersed for 480 min without significant precipitation. For redispersion, there was no significant precipitation in 120 min. The introduction of benzene rings on the GO surface can improve the redispersibility of GO in ethanol, and realize the preparation of redispersible GO powder, which provides a feasible method for the large-scale application of GO reduction to prepare graphene.Author contributions
Jian Huang: conceptualization, validation, resources, writing, funding acquisition, and project administration. Qian Zhang: conceptualization, methodology, software, formal analysis, investigation, data curation, and writing. Zhengcai Yang: conceptualization, methodology, investigation, and data curation. Hailong Hu: methodology, software, and data curation. Mesfin Manuka: investigation. Yuting Zhao: investigation. Xin Wang: methodology and supervision. Wufeng Wang: visualization. Rong Yang: supervision. Shouwei Jian: validation and methodology. Hongbo Tang: validation and supervision. Xiangguo Li: resources and methodology. Yang Lv: resources and investigation. Pei Tang: resources and supervision. Baoguo Ma: validation and supervision. All authors proofread, commented on, and approved the final manuscript for submission.Conflicts of interest
There are no conflicts to declare.Acknowledgements
Financial supports from the “14th Five-Year” National Key Research & Develop Program of China (2022YFC3902705), the National Natural Science Foundation of China (52272024), Guangdong Basic and Applied Basic Research Foundation (2022A1515011335), Fund of National Dam Safety Research Center (CX2022B07), the CRSRI Open Research Program (CKWV2021877/KY), and Hubei Three Gorges Laboratory (SC211018) are gratefully acknowledged. The authors would like to thank the State Key Laboratory and Center for Materials Research, Wuhan University of Technology, for the material characterization.References
- H. Zhao, R. Ding, X. Zhao, Y. Li, L. Qu, H. Pei, L. Yildirimer, Z. Wu and W. Zhang, Drug Discovery Today, 2017, 22, 1302–1317 CrossRef CAS PubMed.
- P. Govindaraj, A. Sokolova, N. Salim, S. Juodkazis, F. K. Fuss, B. Fox and N. Hameed, Composites, Part B, 2021, 226, 32 CrossRef.
- E. H. Cho, M. J. Kim, H. Sohn, W. H. Shin, J. Y. Won, Y. Kim, C. Kwak, C. S. Lee and Y. S. Woo, Nanoscale, 2018, 10, 628–638 RSC.
- X. Z. Xu and Y. L. Hsieh, Nanoscale, 2019, 11, 11719–11729 RSC.
- R. Ikram, B. M. Jan, P. B. Nagy and T. Szabo, Nanotechnol. Rev., 2023, 12, 45 Search PubMed.
- D. C. T. Nguyen, K. Y. Cho and W. C. Oh, RSC Adv., 2017, 7, 29284–29294 RSC.
- J. Wang, F. Ma and M. Sun, RSC Adv., 2017, 7, 16801–16822 RSC.
- R. Ikram, B. M. Jan and W. Ahmad, J. Mater. Res. Technol., 2020, 9, 15924–15951 CrossRef CAS.
- B. Dehghanzad, M. K. R. Aghjeh, O. Rafeie, A. Tavakoli and A. J. Oskooie, RSC Adv., 2016, 6, 3578–3585 RSC.
- S. Park, J. An, J. R. Potts, A. Velamakanni, S. Murali and R. S. Ruoff, Carbon, 2011, 49, 3019–3023 CrossRef CAS.
- P. Ren, D. Yan, Z. Li, X. Ji and T. Chen, Nanotechnology, 2011, 22, 055705 CrossRef PubMed.
- H. J. Shin, K. K. Kim, A. Benayad, S. M. Yoon, H. K. Park, I. S. Jung, M. H. Jin, H. K. Jeong, J. M. Kim, J. Y. Choi and Y. H. Lee, Adv. Funct. Mater., 2009, 19, 1987–1992 CrossRef CAS.
- S. F. Pei, J. P. Zhao, J. H. Du, W. C. Ren and H. M. Cheng, Carbon, 2010, 48, 4466–4474 CrossRef CAS.
- E. B. Nursanto, A. Nugroho, S. A. Hong, S. J. Kim, K. Y. Chung and J. Kim, Green Chem., 2011, 13, 2714–2718 RSC.
- M. Seo, D. Yoon, K. S. Hwang, J. W. Kang and J. Kim, Carbon, 2013, 64, 207–218 CrossRef CAS.
- J. S. Kim, C. J. Yu, K. C. Ri, S. H. Choe and J. C. Ri, J. Phys. Chem. C, 2019, 123, 8932–8942 CrossRef CAS.
- C. Gong, M. Acik, R. M. Abolfath, Y. Chabal and K. Cho, J. Phys. Chem. C, 2012, 116, 9969–9979 CrossRef CAS.
- B. Chen, L. Jiang, J. Wu, A. L. Hu, X. Y. Lu and Q. Jiang, ChemNanoMat, 2019, 5, 1317–1323 CrossRef CAS.
- F. Asghar, B. Shakoor, S. Fatima, S. Munir, H. Razzaq, S. Naheed and I. S. Butler, RSC Adv., 2022, 12, 11750–11768 RSC.
- V. V. Neklyudov, N. R. Khafizov, I. A. Sedov and A. M. Dimiev, Phys. Chem. Chem. Phys., 2017, 19, 17000–17008 RSC.
- Y. Shin, M. F. N. Taufique, R. Devanathan, E. C. Cutsforth, J. Lee, W. Liu, L. S. Fifield and D. W. Gotthold, Sci. Rep., 2019, 9, 11 CrossRef PubMed.
- J. Zhu, C. M. Andres, J. D. Xu, A. Ramamoorthy, T. Tsotsis and N. A. Kotov, ACS Nano, 2012, 6, 8357–8365 CrossRef CAS.
- A. V. Talyzin, T. Hausmaninger, S. J. You and T. Szabo, Nanoscale, 2014, 6, 272–281 RSC.
- W. Tong, D. D. Cui, Z. J. Xu and X. N. Yang, J. Phys. Chem. C, 2021, 125, 24692–24701 CrossRef CAS.
- S. X. Zheng, Q. S. Tu, M. N. Wang, J. J. Urban and B. X. Mi, ACS Nano, 2020, 14, 6013–6023 CrossRef CAS PubMed.
- C. Sainz-Urruela, S. Vera-Lopez, M. P. San Andres and A. M. Diez-Pascual, J. Mol. Liq., 2022, 357, 13 CrossRef.
- H. Wang, S. G. Bi, Y. S. Ye, Y. Xue, X. L. Xie and Y. W. Mai, Nanoscale, 2015, 7, 3548–3557 RSC.
- X. Qi, K. Y. Pu, H. Li, X. Zhou, S. Wu, Q. L. Fan, B. Liu, F. Boey, W. Huang and H. Zhang, Angew. Chem., Int. Ed., 2010, 49, 9426–9429 CrossRef CAS PubMed.
- S. P. Zhang and H. O. Song, New J. Chem., 2012, 36, 1733–1738 RSC.
- R. B. Cui, C. Zhang, J. Y. Zhang, W. Xue and Z. L. Hou, J. Alloys Compd., 2020, 834, 7 Search PubMed.
- Z. Y. Lu, D. S. Hou, M. Hanif, W. B. Hao, Z. J. Li and G. X. Sun, Cem. Concr. Compos., 2018, 94, 33–42 CrossRef CAS.
- X. H. Zhu, X. J. Kang, K. Yang and C. H. Yang, Constr. Build. Mater., 2017, 132, 290–295 CrossRef CAS.
- Z. K. Tu, J. Wang, C. J. Yu, H. W. Xiao, T. Jiang, Y. K. Yang, D. A. Shi, Y. W. Mai and R. K. Y. Li, Compos. Sci. Technol., 2016, 134, 49–56 CrossRef CAS.
- W. Stöber, A. Fink and E. Bohn, J. Colloid Interface Sci., 1968, 26, 62–69 CrossRef.
- B. G. Ma, Q. Jiang, J. Huang, Z. Z. Zhi, M. Q. Sun, Z. H. Lv, S. W. Jian and H. B. Tan, J. Sol-Gel Sci. Technol., 2017, 83, 582–589 CrossRef CAS.
- A. M. El-Rafei, Ceram. Int., 2022, 48, 32185–32195 CrossRef CAS.
- L. Y. Chen, J. Zheng and R. W. Fu, Carbon, 2020, 170, 658–665 CrossRef CAS.
- Z. L. He, J. K. Xu, L. Zhang, H. Y. Ren and S. Y. Fu, Composites, Part B, 2019, 170, 31–40 CrossRef CAS.
- D. M. Zhu, H. Gao, X. M. Zhang, T. Y. Yang, L. Li, G. Z. Yin, X. H. Li, C. Nicklin, X. Y. Gao, Z. Li, L. Yi and X. L. Li, Carbon, 2015, 94, 775–780 CrossRef CAS.
- H. P. Gong, W. M. Hua, Y. H. Yue and Z. Gao, Appl. Surf. Sci., 2017, 397, 44–48 CrossRef CAS.
- X. Y. Ma, B. Zhou, W. Gao, Y. N. Qu, L. L. Wang, Z. C. Wang and Y. C. Zhu, Powder Technol., 2012, 217, 497–501 CrossRef CAS.
- J. Yoo, H. S. Kim, S. Y. Park, S. Kwon, J. Lee, J. Koo and Y. S. Seo, Ultrason. Sonochem., 2020, 64, 10 CrossRef PubMed.
- G. Smith and J. Yard, Science, 2008, 321, 1812–1815 CrossRef CAS PubMed.
- M. A. Vieira, G. R. Goncalves, D. F. Cipriano, M. A. Schettino, E. A. Silva, A. G. Cunha, F. G. Emmerich and J. C. C. Freitas, Carbon, 2016, 98, 496–503 CrossRef CAS.
- S. Eissa, J. N'Diaye, P. Brisebois, R. Izquierdo, A. C. Tavares and M. Siaj, Sci. Rep., 2020, 10, 12 CrossRef PubMed.
- L. Jingzhong, C. Shuping, L. Yanan and Z. Bijing, J. Saudi Chem. Soc., 2022, 101560 Search PubMed.
- R. Ikram, B. M. Jan and W. Ahmad, J. Mater. Res. Technol., 2020, 9, 11587–11610 CrossRef CAS.
- A. Peyravi, F. Ahmadijokani, M. Arjmand and Z. Hashisho, J. Hazard. Mater., 2022, 433, 12 CrossRef PubMed.
- M. C. Hsiao, C. C. M. Ma, J. C. Chiang, K. K. Ho, T. Y. Chou, X. F. Xie, C. H. Tsai, L. H. Chang and C. K. Hsieh, Nanoscale, 2013, 5, 5863–5871 RSC.
- N. Justh, B. Berke, K. Laszlo and I. M. Szilagyi, J. Therm. Anal. Calorim., 2018, 131, 2267–2272 CrossRef CAS.
- J. F. Li, M. Gao, Y. D. Zheng, Y. P. Guan and D. Q. Yi, Macromol. Mater. Eng., 2020, 305, 14 Search PubMed.
- K. K. H. De Silva, H. H. Huang and M. Yoshimura, Appl. Surf. Sci., 2018, 447, 338–346 CrossRef CAS.
- J. P. Rourke, P. A. Pandey, J. J. Moore, M. Bates, I. A. Kinloch, R. J. Young and N. R. Wilson, Angew. Chem., Int. Ed., 2011, 50, 3173–3177 CrossRef CAS PubMed.
- W. Jiang, C. Sun, Y. Zhang, Z. Xie, J. Zhou, J. Kang, Y. Cao and M. Xiang, Polym. Test., 2023, 118, 107912 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2023 |