
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Pyrrole alkaloids from the fruiting bodies of edible mushroom Lentinula edodes†
Zhen-Zhu Zhao ,
Fei Zhang,
Bao-Yu Ji,
Ning Zhou,
Hui Chen,
Yan-Jun Sun,
Wei-Sheng Feng* and
Xiao-Ke Zheng*
,
Fei Zhang,
Bao-Yu Ji,
Ning Zhou,
Hui Chen,
Yan-Jun Sun,
Wei-Sheng Feng* and
Xiao-Ke Zheng*
School of Pharmacy, Henan University of Chinese Medicine, Zhengzhou 450046, China. E-mail: fwsh@hactcm.edu.cn; zhengxk.2006@163.com
First published on 16th June 2023
Abstract
Nine pyrrole alkaloid derivatives, including four new ones (1–4), were isolated from the wild mushroom Lentinula edodes for the first time. Their chemical structures were determined using UV-Vis spectroscopy, IR spectroscopy, MS, NMR spectroscopy, and single-crystal X-ray diffraction techniques. Compound 1, a previously unreported bicylo-pyrrole aldehyde homologue, was found to be a major component, approximately 8.2 μg g −1 in the dry powder of L. edodes. Compound 1 showed cytotoxicity against SMMC-772 (IC50 15.8 μM) without any cytotoxic effect on LO2, a normal hepatic cell line; compounds 1 and 2 displayed weak immunosuppressive activities by inhibiting the proliferation of induced T cells; compound 3 showed inhibition activity on the proliferation of HaCaT cell line (IC50 25.4 μM) and weak antioxidant activity at a concentration of 50 μM.
Introduction
Edible mushrooms are a well-known healthy food for their unique nutritional properties, which have low calories but are rich in nutrients, such as vitamins and minerals. The mushroom Lentinula edodes, commonly known as shiitake, is cultivated for both its culinary and medicinal qualities, which has long been utilized as an edible mushroom in East Asia, and its uses were first documented in an ancient Chinese book from the second century, titled Shen Nong Ben Cao Jing (The Divine Farmer's Materia Medica). Lentinan, a high molecular weight neutral polysaccharide purified from a hot water extract of L. edodes in 1969,1 has been approved as an anti-tumor drug. Subsequently, a series of research was carried out to illustrate the mechanism of lentinan and more active polysaccharides and proteins were found.2–5 However, unlike many progresses on biomacromolecule, only a few studies have been conducted to clarify the secondary metabolites (SMs) of the fruiting bodies of L. edodes, resulting in the isolation of some active compounds, such as eritadenine and polyacetylene molecules.6 Therefore, to complete the possible utilization of L. edodes from the perspective of SMs, we studied SMs in this mushroom leading to the isolation of nine pyrrole alkaloids.Alkaloids are a large group consisting of diverse subgroups of natural products that are most extensively studied in plants. Pyrrole is of special interest in developing new bioactive drugs owing to their anticancer, antimicrobial, antiviral, antimalarial, antitubercular, anti-inflammatory, and enzyme-inhibiting properties,7–9 e.g., the biggest-selling drug of all time, atorvastatin, a HMG-CoA reductase inhibitor, is just a pyrrole. Indeed, before 2002, diverse pyrrole alkaloids were known mostly from vegetal or marine origin. In mushrooms, the discovery of sciodole from Tricholoma sciodes,10 an alkaloid containing both pyrrole and indole moieties in its structure, led to the beginning of isolating pyrroles from mushrooms. Subsequently, a series of pyrroles were identified from different mushrooms, however, none of them were from L. edodes.8,11
Herein, we report the isolation and structural elucidation of compounds 1–9 (Fig. 1). Moreover, the potential activities of the isolated compounds 1–5 were evaluated, including inhibitory effects against five human cancer cell lines, a keratinocyte cell line, and T/B cells, as well as free radical scavenging activity on 2,2-diphenyl-1-picrylhydrazyl (DPPH).
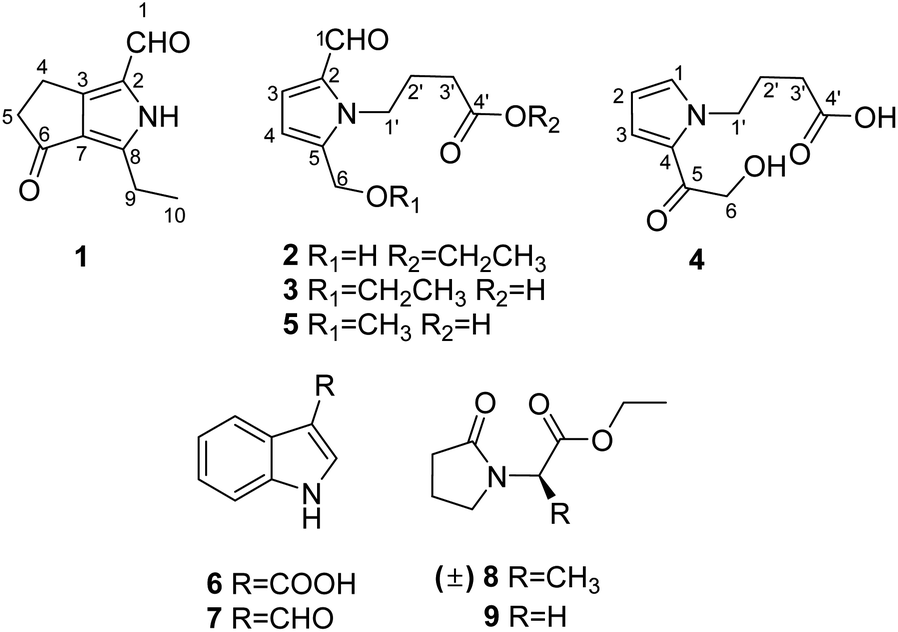 | ||
| Fig. 1 Structures of compounds 1–9. | ||
Results and discussion
Compound 1 was obtained as colourless crystals. The molecular formula C10H11NO2 with six sites of unsaturation was established by HRESIMS ion at m/z 200.0680 [M + Na]+ (calcd for C10H11NO2Na, 200.0687). Purified compound 1 showed UV absorption with two absorption maximums at 240 nm and 295 nm. The IR spectrum of 1 revealed the presence of amino (3212 cm−1) and two UV-conjugated carbonyl (1691 and 1651 cm−1) functionalities. The 1H NMR spectrum (Table 1) just revealed 10 protons clearly corresponding to an aldehyde hydrogen [δH 9.46 (s)], a methyl [δH 1.30 (t, J = 7.6 Hz)], and three methylenes [δH 2.80 (q, J = 7.6 Hz); 2.88 (m); 3.14 (m)]. The 13C NMR and DEPT spectra (Table 1) resolved 10 carbon resonances that were ascribed to a methyl (δC 13.4), three methylenes (δC 20.6, 21.4, 43.0), a conjugated aldehyde (δC 178.4), one pair of tetrasubstituted double bounds (δC 125.8, 127.5, 143.5, 155.3), and a conjugated ketone (δC 201.5). After the structure assignment using the 1D NMR data, compound 1 was deduced to possess a bicyclic system for the remaining two degrees of unsaturation. All proton resonances were assigned to their respective carbons via the HSQC spectrum. The planner structure of compound 1 comprising two rings, A and B, was successfully established as follows (Fig. 2). The HMBC correlations from H-1 to C-2, from H3-10 to C-8/C-9, and from H2-9 to C-7 confirmed the presence of the B ring, which constructed a motif of 3-ethyl-pyrrole-1-carbaldehyde. In addition, A ring, a cyclopentenone, was also constructed using the HMBC correlations from H2-4 to C-2/C-3/C-5, and from H2-5 to C-3/C-6/C-7. The fusion of rings A and B was identified at C-3 and C-7 based on the HMBC experiment (Fig. 2). Therefore, the structure of compound 1 was established as 3-ethyl-4-oxo-2,4,5,6-tetrahydrocyclopenta[c]pyrrole-1-carbaldehyde, as shown in Fig. 1, which was further proved by X-ray single crystal experiments (Fig. 3). A comprehensive literature research showed that the architecture of 1 is simple but quite novel.| No. | δC | δH (J in Hz) |
|---|---|---|
| 1 | 178.4, CH | 9.46, s |
| 2 | 125.8, C | |
| 3 | 155.3, C | |
| 4 | 20.6, CH2 | 3.14, m |
| 5 | 43.0, CH2 | 2.88, m |
| 6 | 201.5, C | |
| 7 | 127.5, C | |
| 8 | 143.5, C | |
| 9 | 21.4, CH2 | 2.80, q (7.6) |
| 10 | 13.4, CH3 | 1.30, t (7.6) |
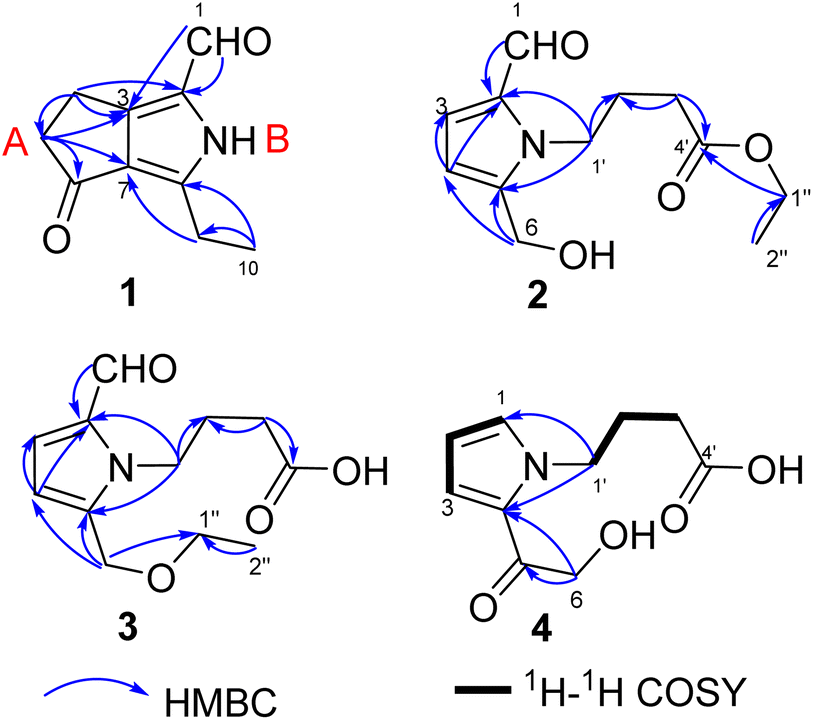 | ||
| Fig. 2 Key 2D NMR correlations of compounds 1–4. | ||
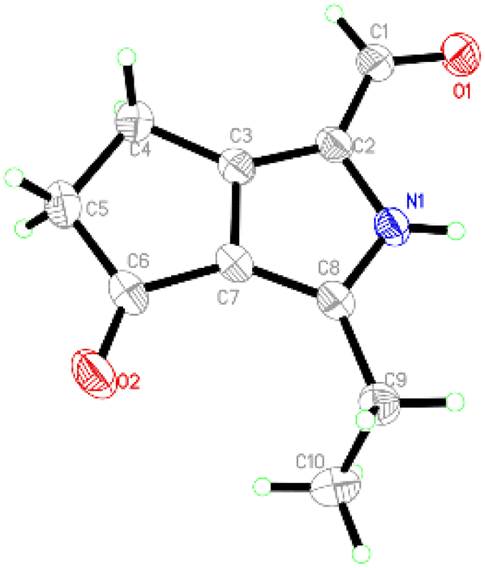 | ||
| Fig. 3 ORTEP drawing of 1. | ||
Compound 2 was obtained as a colourless oil with a molecular formula of C12H17NO4, determined by HRESIMS ion at m/z 262.1046 [M + Na]+ (calcd for C12H17NO4Na, 262.1050). The UV absorption spectrum of compound 2 showed an absorption maximum at 295 nm and a shoulder at 260 nm. The 1D NMR spectra indicated the presence of a pyrrole ring by displaying a set of two mutual coupled protons [δH 6.99 (1H, d, J = 4.0 Hz), H-3; 6.27 (1H, d, J = 4.0 Hz), H-4] and four olefinic carbon signals (δC 133.5, C-2; 126.4, CH-3; 111.5, CH-4; 144.6, C-5) (Table 2).12 Additionally, an aldehyde [δH/C 9.42 (1H, s)/180.9, CH-1], a hydroxymethyl group [δH/C 4.64 (2H, s)/56.4, CH2-6], and three methylene signals [δH/C 2.04 (2H, tt, J = 7.4, 7.4 Hz)/27.5, CH2-2′; 2.36 (2H, t, J = 7.4 Hz)/32.0, CH2-3′; δH/C 4.40 (2H, t, J = 7.4 Hz)/45.7, CH2-1′] as well as a carbonyl signal (δC 174.7, C-4′) were also observed in the 1D NMR and HSQC spectra. The above NMR data of 2 showed a close resemblance to those of compound 5,12 while the appearance of the signals for an ethoxyl group [δH/C 1.25 (3H, t, J = 7.1 Hz)/14.5; δH/C 4.12 (2H, q, J = 7.1 Hz)/61.7] indicated that compound 2 (Table 2) was an ethylated derivative of 4-[2-formyl-5-hydroxymethyl)-1H-pyrrol-1-yl]butanoic acid (5). Then, the HMBC correlations from H2-1′′ to C-4′ assigned this ethoxyl group to C-4′ (Fig. 2). Therefore, the structure of 2 was determined as ethyl 4-[2-formyl-5-(hydroxymethyl)-1H-pyrrol-1-yl]butanoate (Fig. 1).
| No. | 2 | 3 | 4 | |||
|---|---|---|---|---|---|---|
| δC | δH (J in Hz) | δC | δH (J in Hz) | δC | δH (J in Hz) | |
| 1 | 180.9, CH | 9.42, s | 181.1, CH | 9.44, s | 132.9, CH | 7.13, dd (2.1,1.2) |
| 2 | 133.5, C | 133.7, C | 109.9, CH2 | 6.18, dd (4.1,2.1) | ||
| 3 | 126.4, CH | 6.99, d (4.0) | 126.0, CH | 6.99, d (4.0) | 120.8, CH | 7.09, dd (4.1,1.2) |
| 4 | 111.5, CH | 6.27, d (4.0) | 112.7, CH | 6.28, d (4.0) | 127.9, C | |
| 5 | 144.6, C | 141.4, C | 190.1, C | |||
| 6 | 56.4, CH2 | 4.64, s | 64.5, CH2 | 4.54, s | 65.4, CH2 | 4.68, s |
| 1′ | 45.7, CH2 | 4.40, t (7.4) | 46.0, CH2 | 4.37, t (7.7) | 50.0, CH2 | 4.41, t (6.7) |
| 2′ | 27.5, CH2 | 2.04, tt (7.4, 7.4) | 27.7, CH2 | 2.01, tt (7.7, 7.3) | 29.0, CH2 | 2.00, m |
| 3′ | 32.0, CH2 | 2.36, t (7.4) | 32.0, CH2 | 2.33, t (7.3) | 34.5, CH2 | 2.17, m |
| 4′ | 174.7, C | 176.9, C | Unobserved C | |||
 |
61.7, CH2 | 4.12, q (7.1) | 66.9, CH2 | 3.55, q (7.0) | ||
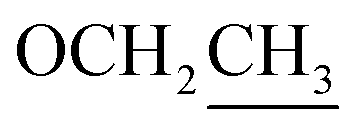 |
14.5, CH3 | 1.25, t (7.1) | 15.4, CH3 | 1.20, t (7.0) | ||
Compounds 3 and 2 are isomers by showing the same molecular formula, C12H17NO4, while a further comparison of the 1D NMR data between 3 and 2 (Table 2) indicated that compound 3 was also a derivative of 4-[2-formyl-5-(hydroxymethyl)-1H-pyrrol-1-yl]butanoic acid. However, compound 3 is an etherified derivative at HOCH2-6 for the obvious differences in chemical shifts of the ethoxyl and hydroxymethyl groups in the 13C NMR spectra [3: OCH2CH3 (δC 66.9, 15.4), OCH2-6 (δC 64.5); 2: OCH2CH3 (δC 61.7, 14.5), OCH2-6 (δC 56.4)], which was also supported by HMBC correlations from H2-6 to C-1′′ (Fig. 2). Finally, the structure of 3 was defined as 4-[2-formyl-5-(ethoxymethyl)-1H-pyrrol-1-yl]butanoic acid (Fig. 1).
Compound 4 was isolated as a colourless oil, and its molecular formula was C10H13NO4 deduced by the HRESIMS ion at m/z 234.0739 [M + Na]+ (calcd for C10H13NO4Na, 234.0737). The IR spectrum of 4 revealed the presence of a hydroxyl (3368, cm−1) and one carbonyl (1677 and 1645 cm−1) functionalities. Similar to the structures of compounds 3 and 5, compound 4 conserved the framework of (1H-pyrrol-1-yl)butanoic acid due to the resembling chemical shifts and coupling constants (Table 2). Whereas, instead of the obvious aldehyde signal, the newly appeared signal of pyrrole hydrogen [δH 7.13 (1H, dd, J = 2.1, 1.2 Hz)] suggested that CHO-1 was degraded in 4, which was consistent with 1H–1H COSY and HMBC spectra (Fig. 2). In addition, HMBC correlations from H2-6 [δH 4.68 (s)] to C-5 (δC 190.1) and C-4 (δC 127.9) successfully assigned the side chain –COCH2OH to C-4. Thus, the structure of 4 is 4-[5-(2-hydroxymethyl-1-carbonyl)-1H-pyrrol-1-yl]butanoic acid (Fig. 1).
By analysis and comparison of their NMR data with that documented in the literature, the known compounds 5–9 were identified as 4-[2-formyl-5-(hydroxymethyl)-1H-pyrrol-1-yl]butanoic acid (5),12 indole-3-acetic acid B (6),13 indole-3-carboxaldehyde (7),14 ethyl (±)2-(2-oxopyrrolidin-1-yl)propanoate (8),15 and ethyl 2-(2-oxopyrrolidin-1-yl)acetate (9)16 (Fig. 1), respectively. Since compounds 8 and 9 are uncomman natural products, their 1D NMR spectra are also listed in the ESI.†
As compounds 1–4 are new compounds, and compound 5 is a derivative of 1–4, they were chosen to test for their inhibitory effects against five human cancer cell lines, a keratinocyte cell line, and T/B cells, as well as DPPH-free radical scavenging activity. Among them, compound 1 exhibited inhibitory activity against SMMC-7721 without any cytotoxic effect on normal hepatocyte, LO2 (Table 3); compounds 1 and 2 showed weak anti-proliferation of induced T cells (Table 4); compound 3 displayed activities against proliferation of HaCaT cell line and scavenging of the DPPH free radical (Tables 3 and 5).
| Samples | C (μM) | Antioxidant rate (%) |
|---|---|---|
| Trolox | 25 | 94.8 ± 0.0 |
| 3 | 50 | 12.9 ± 2.4 |
Conclusions
In summary, four new pyrrole alkaloid derivatives (1–4) and five known ones (5–9) with various bioactivities were isolated from the wild edible mushroom L. edodes by alcohol (95%) extraction followed by ethyl acetate fractionation. Compound 1, bearing a novel cyclopenta[c]pyrrole-1-carbaldehyde scaffold, could inhibit the proliferation of SMMC-7721 without any cytotoxic effect on normal hepatocytes, showing the value for further research. Undoubtedly, this work not only enriches the ingredients of L. edodes, which is beneficial for the public's knowledge of it, but also provides solid evidence for L. edodes used as a functional food.Experimental section
General experimental procedures
Optical rotation studies were performed on a JASCO P-1020 digital polarimeter (Horiba, Kyoto, Japan). UV spectra were recorded on a Shimadzu UV-2401PC UV-visible recording spectrophotometer (Shimadzu, Kyoto, Japan). IR spectra were obtained on a Thermo Scientific Nicolet iS10 FT-IR Spectrometer (Thermo, Waltham, MA, USA). 1D and 2D NMR spectra were obtained on a Bruker Avance III 600 MHz spectrometer (Bruker Corporation, Karlsruhe, Germany) instrument. HRESIMS were recorded on an Agilent 6200 Q-TOF MS system (Agilent Technologies, Santa Clara, CA, USA). Single crystal X-ray diffraction was performed on a Bruker D8 Quest (Karlsruhe, Germany). The melting point was measured on an X-4 microscopic melting point meter (Yuhua Instrument Co., Ltd, Gongyi, China). Sephadex LH-20 (Amersham Biosciences, Uppsala, Sweden) and silica gel (Qingdao Haiyang Chemical Co., Ltd) were used for column chromatography (CC). Medium pressure liquid chromatography (MPLC) was performed on a Büchi Sepacore System equipped with a pump manager C-615, a pump modules C-605, a fraction collector C-660 (Büchi Labortechnik AG, Flawil, Switzerland), and a column packed with Chromatorex C-18 (dimensions 450 mm × i.d. 14 mm, particle size: 40–75 μm, Fuji Silysia Chemical Ltd., Kasugai, Japan). Preparative high-performance liquid chromatography (prep. HPLC) was performed on an Agilent 1260 liquid chromatography system equipped with Zorbax SB-C18 columns (particle size 5 μm, dimensions 150 mm × i.d. 9.4 mm or 150 mm × i.d. 21.2 mm, flow rate 7 or 15 mL min−1, respectively) and a DAD detector (Agilent Technologies, Santa Clara, CA, USA).Fungal material
Fruiting bodies of the wild mushroom L. edodes were purchased from Lijiang City, Yunnan Province, China in 2016, and were identified by the molecular evidence of ITS gene fragment (GenBank accession no. OQ680131), whose BLAST search result showed that the sequence was similar (>99%) to the sequence of L. edodes. A voucher specimen (no. HZYXG01) was deposited at the School of Pharmacy, Henan University of Chinese Medicine.Extraction and isolation
The dried fruiting bodies of L. edodes (3.5 kg) were soaked in 20 L EtOH (95%) at room temperature for three days (×4). Then, a total of 80 L solvents were concentrated till about 5 L under reduced pressure, which was further extracted by EtOAc (5 L × 3). The removal of all solvents gave an EtOAc layer (120 g). The crude extract was separated over silica gel flash CC, eluted with a gradient system of petroleum ether-acetone, to produce seven fractions, A to G. Fraction E (24 g) was divided on MPLC, eluted with CH3OH–H2O (v/v, from 20/80 to 0/100), yielding 14 subfractions, E1–E14. Subsequently, fractions E4 (CH3OH), E5 (acetone), and E6 (acetone) were subjected to a Sephadex LH-20 unit each affording 11 (E4a–E4k), 9 (E5a–E5i) and 12 fractions (E6a–E6l).Compounds 1 (28.7 mg, tR = 6.0 min) from fraction E4c, 4 (2.2 mg, tR = 9.5 min) from fraction E4d, 6 (1.6 mg, tR = 14.1 min) from fraction E4j, and 7 (2.8 mg, tR = 14.3 min) from fraction E4j were purified by prep. HPLC (CH3CN/H2O: 25/75 to 50/50, 25 min, flow rate 15 mL min−1). Compounds 2 (5.5 mg, tR = 10.0 min, CH3CN/H2O: 15/85 to 30/70) from fraction E5c, and 3 (2.6 mg, tR = 11.8 min, CH3CN/H2O: 15/85 to 35/65) from fraction E6f were obtained by prep. HPLC (20 min, flow rate 7 mL min−1).
Fraction E4a was subjected to a Sephadex LH-20 (acetone) unit affording four minor fractions (E4a1–E4a4). Compounds 5 (3.2 mg, tR = 14.6 min) from fraction E4a3, 8 from fraction E4a3 (3.1 mg, tR = 8.1 min), and 9 (4.4 mg, tR = 5.4 min) from fraction E4a4 were obtained by prep. HPLC (CH3CN/H2O: 35/65 to 60/40, 25 min, flow rate 15 mL min−1).
3-Ethyl-4-oxo-2,4,5,6-tetrahydrocyclopenta[c]pyrrole-1-carbaldehyde (1): colourless block crystals; mp 140.2–148.2 °C; UV (MeOH) λmax nm (log![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) ε): 195 (5.17), 240 (5.04), 295 (5.03); IR (MeOH) νmax cm−1: 3212,1691, 1651, 1576, 1520, 1466, 1291, 1025, 875; 1H NMR (600 MHz CD3OD) data see Table 1; 13C NMR (150 MHz CD3OD) data see Table 1; HRESIMS m/z 200.0680 [M + Na]+ (calcd for C10H11NO2Na, 200.0687), and m/z 178.0860 [M + H]+ (calcd for C10H12NO2, 178.0868).
ε): 195 (5.17), 240 (5.04), 295 (5.03); IR (MeOH) νmax cm−1: 3212,1691, 1651, 1576, 1520, 1466, 1291, 1025, 875; 1H NMR (600 MHz CD3OD) data see Table 1; 13C NMR (150 MHz CD3OD) data see Table 1; HRESIMS m/z 200.0680 [M + Na]+ (calcd for C10H11NO2Na, 200.0687), and m/z 178.0860 [M + H]+ (calcd for C10H12NO2, 178.0868).
Single crystal X-ray diffraction data for 1: a block-like specimen of C10H11NO2, approximate dimensions 0.180 mm × 0.330 mm × 0.370 mm, was used for the X-ray crystallographic analysis on the Bruker D8 Quest instrument. The integration of the data using a monoclinic unit cell yielded a total of 14437 reflections to a maximum θ angle of 74.55° (0.80 Å resolution), of which 1838 were independent (average redundancy 7.855, completeness = 98.9%, Rint = 2.99%, Rsig = 2.11%) and 1775 (96.57%) were greater than 2σ(F2). The final cell constants of a = 6.7030(4) Å, b = 9.0452(6) Å, c = 15.0090(9) Å, β = 93.234(2)°, volume = 908.55(10) Å3, T = 273(2) K. Data were corrected for absorption effects using the multi-scan method (SADABS). The structure was solved and refined using the Bruker SHELXTL Software Package, using the space group P121/n1, with Z = 4, μ(CuKα) = 1.54178. The final anisotropic full-matrix least-squares refinement on F2 with 119 variables converged at R1 = 3.97%, for the observed data and wR2 = 11.07% for all data. The goodness-of-fit was 1.056. These data can be obtained free of charge via https://www.ccdc.cam.ac.uk (CCDC no. 2251050).
4-[2-Formyl-5-(hydroxymethyl)-1H-pyrrol-1-yl]butanoate (2): a colourless oil; UV (MeOH) λmax nm (logε): 195 (4.71), 295 (4.92); IR (MeOH) νmax cm−1: 3382, 1732, 1659, 1379, 1186, 1032, 783; 1H NMR (600 MHz CD3OD) data see Table 2; 13C NMR (150 MHz CD3OD) data see Table 2. HRESIMS m/z HRESIMS m/z 262.1046 [M + Na]+ (calcd for C12H17NO4Na 262.1050).
4-[2-Formyl-5-(ethoxymethyl)-1H-pyrrol-1-yl]butanoic acid (3): a colourless oil; UV (MeOH) λmax nm (logε): 195 (4.87), 290 (4.93); IR (MeOH) νmax cm−1: 3416, 1658, 1645, 1555, 1411, 1175, 1088, 782; 1H NMR (600 MHz CD3OD) data see Table 2; 13C NMR (150 MHz CD3OD) data see Table 2; HRESIMS m/z 262.1055 [M + Na]+ (calcd for C12H17NO4Na, 262.1050).
4-[5-(2-Hydroxymethyl-1-carbonyl)-1H-pyrrol-1-yl]butanoic acid (4): a colourless oil, UV (MeOH) λmax nm (logε): 195 (4.04), 295 (2.86); IR (MeOH) νmax cm−1: 3368, 1677, 1645, 1570, 1425, 1114, 1087. 1H NMR (600 MHz CD3OD) data see Table 2; 13C NMR (150 MHz CD3OD) data see Table 2; HRESIMS m/z 234.0739 [M + Na]+ (calcd for C10H13NO4Na 234.0737).
Ethyl (±)2-(2-oxopyrrolidin-1-yl)propanoate (8): a colourless oil; 13C NMR data measured in CD3OD δC 14.5 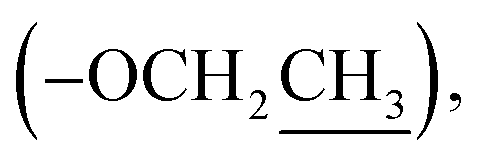 14.9 (CH3-3′), 19.1 (CH2-3), 31.9 (CH2-2), 45.4 (CH2-4), 51.2 (CH-1′), 62.5
14.9 (CH3-3′), 19.1 (CH2-3), 31.9 (CH2-2), 45.4 (CH2-4), 51.2 (CH-1′), 62.5 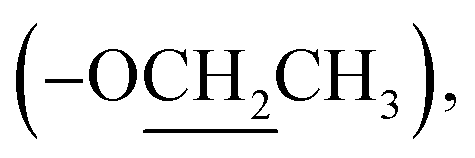 172.6 (C-2′), 178.0 (C-1).
172.6 (C-2′), 178.0 (C-1).
Ethyl 2-(2-oxopyrrolidin-1-yl)acetate (9): a colourless oil; 13C NMR data measured in CD3OD δC 14.4 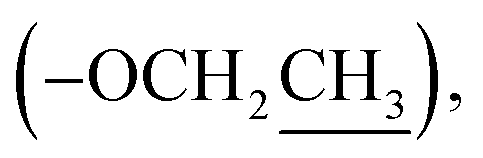 18.9 (CH2-3), 31.4 (CH2-2), 45.0 (CH2-4), 49.3 (CH-1′), 62.5
18.9 (CH2-3), 31.4 (CH2-2), 45.0 (CH2-4), 49.3 (CH-1′), 62.5 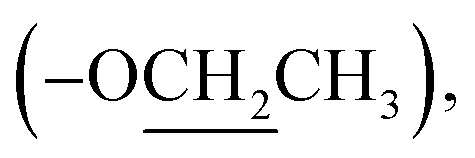 170.2 (C-2′), 178.5 (C-1).
170.2 (C-2′), 178.5 (C-1).
Biological assays
![[X with combining macron]](https://www.rsc.org/images/entities/i_char_0058_0304.gif) ± s. One-way ANOVA was used to compare the differences between groups. LSD or Dunnett's T3 test was used for pair comparison according to standard deviation values. P < 0.05 or P < 0.01 were considered statistically significant.
± s. One-way ANOVA was used to compare the differences between groups. LSD or Dunnett's T3 test was used for pair comparison according to standard deviation values. P < 0.05 or P < 0.01 were considered statistically significant.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was financially supported by the National Natural Science Foundation of China (No. 82003607), the Scientific and Technological Key Project in Henan Province (No. 232102310426), and Open Project of Henan University (No. 20200108).Notes and references
- T. Sasaki and N. Takasuka, Carbohydr. Res., 1976, 47(1), 99–104, DOI:10.1016/S0008-6215(00)83552-1.
- T. C. Finimundy, A. J. P. Dillon, J. A. P. Henriques and M. R. Ely, Food Nutr. Sci., 2014, 5, 1095–1105, DOI:10.4236/fns.2014.512119.
- X. F. Xu, H. D. Yan, J. Tang, J. Chen and X. W. Zhang, Crit. Rev. Food Sci. Nutr., 2014, 54, 474–487, DOI:10.1080/10408398.2011.587616.
- I. Sivanesan, M. Muthu, J. Gopal and J. W. Oh, Molecules, 2022, 27(13), 4090, DOI:10.3390/molecules27134090.
- Y. Gao, A. A. Padhiar, J. Wang, W. Zhang, M. Zhong, B. Liu, Z. Kang, X. Wang, X. Li and M. Huang, Gene, 2018, 642, 212–219, DOI:10.1016/j.gene.2017.10.080.
- E. Fukushima-Sakuno, J. Antibiot., 2020, 73, 687–696, DOI:10.1038/s41429-020-0354-x.
- K. Seipp, L. Geske and T. Opatz, Mar. Drugs, 2021, 19, 514, DOI:10.3390/md19090514.
- J. G. Zorrilla and A. Evidente, Biomolecules, 2022, 12, 1025, DOI:10.3390/biom12081025.
- S. S. Gholap, Eur. J. Med. Chem., 2016, 110, 13–31, DOI:10.1016/j.ejmech.2015.12.017.
- O. Sterner, Nat. Prod. Lett., 1994, 4, 9–14, DOI:10.1080/10575639408043885.
- J. A. Homer and J. Sperry, J. Nat. Prod., 2017, 80(7), 2178–2187, DOI:10.1021/acs.jnatprod.7b00390.
- T. Sakamoto, A. Nishida, N. Wada, Y. Nakamura, S. Sato, T. Konishi and S. Matsugo, Molecules, 2020, 25(21), 4879, DOI:10.3390/molecules25214879.
- S. Simon and J. Petrášek, Plant Sci., 2011, 180, 454–460, DOI:10.1016/j.plantsci.2010.12.007.
- N. Arnold, G. Palfner, J. Schmidt, C. Kuhnt and J. Becerra, J. Chil. Chem. Soc., 2012, 57, 1333–1335, DOI:10.4067/S0717-97072012000300029.
- K. D. Sarma, J. Zhang, Y. Huang and J. G. Davidson, Eur. J. Org. Chem., 2006, 16, 3730–3737, DOI:10.1002/ejoc.200600153.
- I. Y. Malya, J. Wu, E. Harada, M. Toda, C. N. D’Alessandro-Gabazza, T. Yasuma, E. C. Gabazza, J. H. Choi, H. Hirai and H. Kawagishi, Biosci., Biotechnol., Biochem., 2020, 84(7), 1332–1338, DOI:10.1080/09168451.2020.1743170.
- W. Jaidee, W. Maneerat, R. J. Andersen, B. O. Patrick, S. G. Pyne and S. Laphookhieo, Fitoterapia, 2018, 130, 219–224, DOI:10.1016/j.fitote.2018.09.004.
Footnote |
| † Electronic supplementary information (ESI) available: The 1D & 2D NMR, MS, and crystallographic data of compounds 1–9. CCDC 2251050. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d3ra02672h |
| This journal is © The Royal Society of Chemistry 2023 |