
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceDesign, synthesis and antitumor activity of Ascaphin-8 derived stapled peptides based on halogen–sulfhydryl click chemical reactions†
Xianglong Kong‡
a,
Nan Zhang‡ b,
Huaxing Shenb,
Nan Wangb,
Wei Cong*b,
Chao Liu*b and
Hong-gang Hu*ab
b,
Huaxing Shenb,
Nan Wangb,
Wei Cong*b,
Chao Liu*b and
Hong-gang Hu*ab
aSchool of Pharmacy, Weifang Medical University, Weifang 261053, PR China. E-mail: hhu66@shu.edu.cn; kongxi.anglong2008@163.com
bSchool of Medicine, Shanghai University, Shanghai 200444, China. E-mail: viviencong@shu.edu.cn; liuchao518@shu.edu.cn; huaxingshen@126.com; zhangnan@shu.edu.cn
First published on 4th July 2023
Abstract
Ascaphin-8 (GFKDLLKGAAKALVKTVLF-NH2), isolated from the norepinephrine-stimulated skin secretion of the North American-tailed frog Ascaphus truei, is a C-terminal α-helical antimicrobial peptide with potential antitumor activity. However, linear peptides are difficult to be applied directly as drugs because of their inherent defects, such as low hydrolytic enzyme tolerance and poor structural stability. In this study, we designed and synthesized a series of stapled peptides based on Ascaphin-8 via thiol-halogen click chemistry. Most of the stapled peptide derivatives showed enhanced antitumor activity. Among them, A8-2-o and A8-4-Dp had the most improved structural stability, stronger hydrolytic enzyme tolerance and highest biological activity. This research may provide a reference for the stapled modification of other similar natural antimicrobial peptides.
1 Introduction
Cancer is the general term for a large group of malignant neoplasms that are characterized by uncontrolled proliferation, transformation, and migration to other parts of the body.1 In 2020, there were approximately 19.3 million new cancer cases and nearly 10 million cancer deaths globally. Surpassing lung cancer, breast cancer has become the most common cancer among female patients with an estimated 2.3 million new cases (11.7%), followed by lung cancer (11.4%), colorectal cancer (10.0%), prostate cancer (7.3%), and gastric cancer (5.6%).2 The current methods of treating cancer include surgery and chemotherapy. Common chemotherapy drugs include mitomycin,3 doxorubicin,4 and 5-fluorouracil.5 Because of their poor selectivity, chemotherapy drugs can cause damage to adjacent normal cells.6,7 Therefore, the development of new antitumor drugs is still a research hotspot in the field of medicinal chemistry.The peptide drugs have promising properties for the development of new antitumor drugs. Peptides are composed of a series of ordered amino acids, which lie between biomolecules and small-molecule drugs.8 Compared with biomolecules, peptides have less antigenicity.9 Furthermore, compared with small-molecule drugs, peptides have a larger surface area and can target a wide range of protein–protein interactions.10
Ascaphin-8 (GFKDLLKGAAKALVKTVLF-NH2), isolated from the norepinephrine-stimulated skin secretion of the North American-tailed frog Ascaphus truei, is a C-terminal α-helical antimicrobial peptide with 19 amino acid residues.11 A previous study indicated that Ascaphin-8 can inhibit the growth of Candida albicans, Escherichia coli, and Staphylococcus aureus and can lyse human red blood cells.12 In addition, Ascaphin-8 has potential antitumor activity and an inhibitory effect on the growth of human HepG2 liver cancer–derived cells.13 Therefore, Ascaphin-8 can be used as a potential antitumor compound. However, it is difficult for linear peptides to maintain a stable α-helical structure in an aqueous solution.14 In addition, they are easily hydrolyzed by proteases in vivo and have poor membrane penetration.15
To address the deficiencies of linear peptides, chemical structure modification is considered as a means of solution. It was reported that peptide stapling strategy could stabilize linear peptides' conformation by introducing unnatural amino acids into the peptide backbone and cross-linking their side chains.16 Compared with linear peptides, stapled peptides usually have a stronger ability to resist hydrolytic enzymes and are better able to penetrate membranes.17 In previous work, the stapled structure obtained by the click reaction between sulfhydryl group and rigid connection bridge can effectively improve the α-helicity of the linear peptide.18 For example, Muppidi et al. used 4,4′-dibromomethylbiphenyl (Bph) as a linker to perform a click reaction with a cysteine side chain to obtain a thiomethylbiphenyl stapled peptide, significant enhancement in cell permeability was achieved without apparent cytotoxicity, along with modest increases in helicity and bioactivity.19 Herein, we report a series of Ascaphin-8 based stapled peptides constructed by halogen-sulfhydryl click chemical reactions. And their physical and chemical properties and antitumor bioactivity were also evaluated.
2 Materials and methods
2.1 Materials
The amino acids were purchased from GL Biochem (Shanghai) Ltd. Acetonitrile, dichloromethane (DCM) and N,N-dimethylformamide (DMF) were purchased from Sinopharm Chemical Reagent Co., Ltd. Trifluoroacetic acid (TFA), 1-hydroxybenzotriazole (HOBT), NaHCO3, N,N-diisopropylcarbodiimide (DIC), triisopropylsilane (TIPS), trifluoroethanol (TFE) and dimethylsulfoxide (DMSO) were bought from GL Biotech, J&K Scientific, Sinopharm Chemical Co., Ltd. Rink amide MBHA resin (loading = 0.32 mmol g−1) was obtained from Tianjin Nankai Synthetic Technology Co., Ltd.2.2 HPLC parameters
The analytical HPLC equipment used was a Shimadzu HPLC system (Shimadzu, Japan), including pump and column oven. The instrument was analyzed using an analytical column (Grace Vydac “Protein & Peptide C18”, 5 μm particle size, 250 × 4.6 mm, flow rate 1.0 mL min−1). Semi-preparative HPLC was also performed using a Shimadzu HPLC system with a semi-preparative column (Grace Vydac “Protein & Peptide C18”, 5 μm particle size, 250 × 4.6 mm, flow rate 10.0 mL min−1). The same mobile phase system was used for both analytical HPLC and semi-preparative HPLC, which both consisted of solution A and solution B. Solution A was an acetonitrile solution containing 0.1% trifluoroacetic acid. Solution B was an aqueous solution containing 0.1% trifluoroacetic acid. Analytical and semi-preparative HPLC were performed at 214 nm and 254 nm absorbance. For analytical HPLC, the gradients were as follows: water/acetonitrile: 90-0-40 min with a column temperature of 40 °C. For semi-preparative HPLC, the gradients were as follows: 0–5 min, 0–20% A; 5–20 min, 20–50% A; 20–40 min, 50–100% A. The column temperature was 40 °C.2.3 Peptide synthesis
The 500 mg rink amide MBHA resin was soaked with DCM (5 mL) for 10 min. Then the resin was treated with 20% piperidine/DMF to remove the Fmoc protection group. The resin was washed with DMF (3 times), DCM (3 times) and DMF (3 times) at room temperature. To couple natural amino acids, Fmoc-AA-OH (3 eq.), DIC (3 eq.), HOBT (3 eq.), and DMF (6 mL) were mixed for 1 min and added to the resin at 65 °C. After 2 h, the resin was washed with DMF (3 times), DCM (3 times) and DMF (3 times). The steps of deprotection, washing, coupling and washing were repeated until all amino acid residues were successfully assembled. The peptide-coupled resin was treated with a 20% piperidine/DMF solution to remove the Fmoc group from the N-terminus. The peptide-coupled resin was then washed with DMF (3 times), DCM (3 times) and DMF (3 times). The resin was treated with cocktail B regent (TFA/TIPS/H2O = 95/2.5/2.5, v/v/v) for 2 h at room temperature to cleave the peptide from its resin. After cleavage was completed, the peptides were precipitated with 40 ml of ice ether and centrifuged at 3000 rpm (four times) to obtain crude peptides. The supernatant was poured out, and the crude peptide was air-dried and purified by RP-HPLC and lyophilized to obtain the purified peptide. The purified peptide was dissolved in saturated NaHCO3 solution, and small molecules (1.1 eq.) were dissolved in acetonitrile. Then the peptide solution and small molecule solution were mixed in equal volume and reacted at room temperature for 1 h. The product could be easily purified to more than 95% purity.2.4 Circular dichroism
CD spectra were obtained in the range of 190–280 nm using a JASCO J-1500 spectro-polarimeter (Hachioji, Tokyo, Japan). Peptides were solubilized using water and trifluoroethanol (v/v = 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1) to obtain a final concentration of 50 μM peptide solution. Six repeated scans were accumulated to constitute the final average spectrum. After baseline correction, the observed ellipticity (mdeg) was converted to mean ellipticity (deg cm2 dmol−1) using the relation [Θ] = 100θ/(lcn). The α helicity was determined according to the assumed two-state model.20
:1) to obtain a final concentration of 50 μM peptide solution. Six repeated scans were accumulated to constitute the final average spectrum. After baseline correction, the observed ellipticity (mdeg) was converted to mean ellipticity (deg cm2 dmol−1) using the relation [Θ] = 100θ/(lcn). The α helicity was determined according to the assumed two-state model.20
2.5 Blood compatibility assay
Ascaphin-8 and its modifier were mixed with water and dissolved with 10% DMSO to prepare a 10 mM aqueous solution. 200 μL red blood cell solution was added to each well on plate 96, with equal volume of diluted gradient drug solution added to each well, resulting in a final concentration of 100, 50, 25, 12.5, 6.25, 3.125, 0 μM. 2 μL 10% Triton was mixed with red blood cell solution to complete hemolysis as positive control. 2 μL PBS was mixed with red blood cell solution as in the blank group. Cells were incubated in a 37 °C incubator for 1 hour, centrifuged at 1000 rpm for 10 minutes, 100 μL supernatant was transferred to a new 96 well plate. Absorption value was assessed with a Cytation 5 Imaging reader (Bio-Tek. Vermont, USA) at 450 nm.2.6 Cell culture and cell proliferation assay
The human colon cancer cell line (HCT116), human breast cancer cell line (MCF-7) and glioma cell line (U87) were purchased from the National Collection of Authenticated Cell Cultures (Shanghai, China). All cell lines were stored in Dulbecco's modified Eagle's medium (DMEM), which contained 10% fetal bovine serum (FBS) and 1% penicillin-streptomycin (PS), and were cultured at 37 °C in a humid environment containing 5% CO2. The cell counting kit (CCK8) was used to test the viability of tumor cells. A certain number of cells (3 × 103 cells per well) were inoculated into a 96-well plate and cultured in a 37 °C, 5% CO2 incubator. 24 hours later, cells were cultured with or without a series of peptide concentrations (0.78, 1.56, 3.125, 6.25, 12.5 and 25 μM) for another 48 hours. Then change the cell culture medium into serum-free medium and add CCK-8 reagent with 10 μL per well. The 96-well plate was incubated at 37 °C for 1 hour. The optical density (OD) was read by the imaging reader CYTATION 5 (Bio-Tek, Vermont, USA) at a wavelength of 450 nm. The half maximal inhibitory concentration (IC50) was calculated by GraphPad Prism v9.0 (San Diego, California, USA).2.7 Peptides stability assay
2 mg peptides (Ascaphin-8, A8-2-o, and A8-4-Dp) were mixed with a certain amount of DMSO to prepare a storage solution with a concentration of 1 nM. A certain amount of trypsin was dissolved in a phosphate buffer solution containing 1 mM calcium chloride (50 mM, pH = 7.4) until the concentration of trypsin was 0.5 ng μL−1. 1950 μL phosphate buffer solution containing trypsin and 50 μL of peptides storage solution undergoes enzymatic hydrolysis reaction in a 2 ml microcentrifuge tube. Take 50 μL of the reaction liquid at time points 0, 2, 4, 6, 8, 10, and 12 h respectively with 20 μL of hydrochloric acid (1 M) quench trypsin activity. The residual amounts of peptides at different time points were determined by HPLC analysis.2.8 Cell migration and invasion assays
2.9 Determination of cell apoptosis
MCF-7 cells were inoculated into a 6-well plate with 2 × 104 cells per well and cultured in a high sugar medium. 24 h later, the original culture medium was replaced with a concentration of 10 μM containing Ascaphin-8, A8-2-o, and A8-4-Dp and cultured for another 48 hours. About 1 × 106 cells were collected after centrifugally wash with pre-cooled PBS. The cells were resuspended by 500 μL 1 × binding buffer in new tubes. 5 μL annexin V-FITC and 10 μL PI were added and incubated at room temperature in dark for 5 minutes. The cells were observed by flow cytometry: annexin V-FITC was detected through FITC detection channel (Ex = 488 nm; Em = 530 nm) and PI was detected through PI detection channel (Ex = 535 nm; Em = 615 nm).2.10 Cell viability assays
MCF-7 cells with a density of 5 × 103 cells per well were inoculated in a PerkinElmer Cell Carrier Ultra 96 well microplate and cultured in a 37 °C incubator for 24 hours. Then Ascaphin-8, A8-2-o, and A8-4-Dp were added at a final concentration of 10 μM and incubated with cells for another 24 hours. According to the manufacturer's protocol, 2 μL calcein-AM storage solution, 3 μL PI storage liquid and 1 mL PBS mixed as a working liquid with the concentration was 2 μM and 4.5 μM respectively. 50 μL working liquid was added per well and incubated at 37 °C for 15 min. In the final analysis, the images were captured by a high content screening and analysis system.3 Result and discussion
3.1 Design and synthesis of peptides
To begin with, we noticed that it was important to avoid modifying key residues with biological activity. Based on previous research, 3-Lys, 7-Lys, 11-Lys, and 15-Lys were important residues in maintaining the biological activity of Ascaphin-8.21 Hence, these residues remained intact when designing derived peptides of Ascaphin-8. Next, based on the i, i + 4 strategy, we designed A8-n-o/m/p using o-substituted, m-substituted and p-substituted benzene bridges. In addition, A8-4-Dp was designed based on the i, i + 7 strategy by biphenyl structure (Fig. 1). As presented in Scheme 1, the stapled peptides were synthesized from the Rink amide AM resin (loading = 0.32 mmol g−1) by using HOBt/DIC as the coupling reagent to obtain on-resin peptide 1. Following Fmoc deprotection given. Subsequently, reagent B (trifluoroacetate/triisopropylsilane/water = 95:2.5:2.5, v/v/v) was used for acid cleavage to obtain peptide 3. Peptide 3 was then subjected to a halogen–sulfhydryl click reaction with 1,4-bis(bromomethyl)benzene to obtain crude stapled peptide 4. Further purification of the crude peptide was performed by reversed-phase high-performance liquid chromatography to provide target product with yields of 40–50%. The molecular weight of the peptide was verified by high resolution mass spectrometer (HR-MS), which was consistent with the theoretical molecular weight (ESI†).
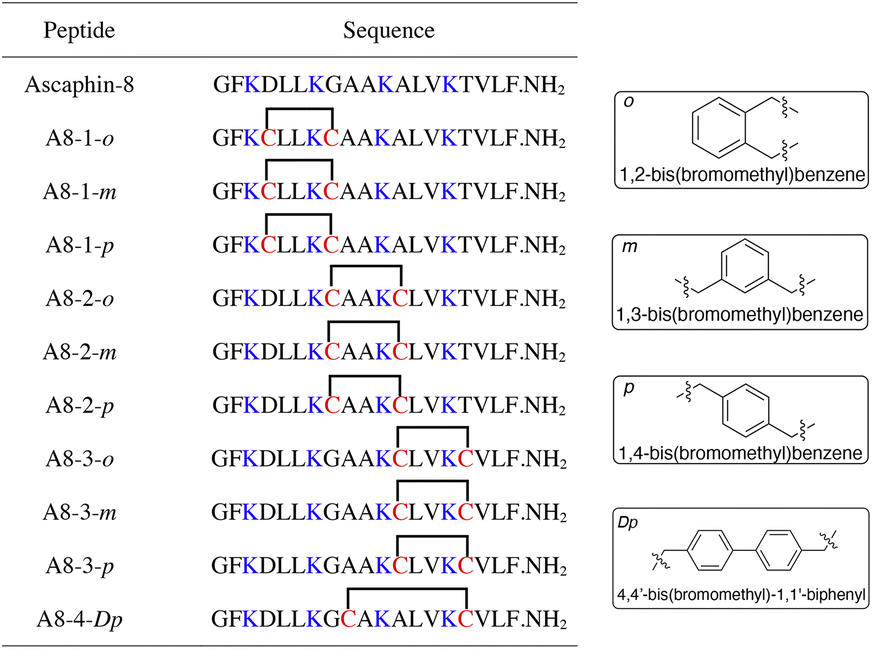 | ||
| Fig. 1 Structures of Ascaphin-8 and stapled derivatives. The key residues are colored blue. The Cys residues are colored red and two Cys were cross-linked by dibromo substituted benzene. | ||
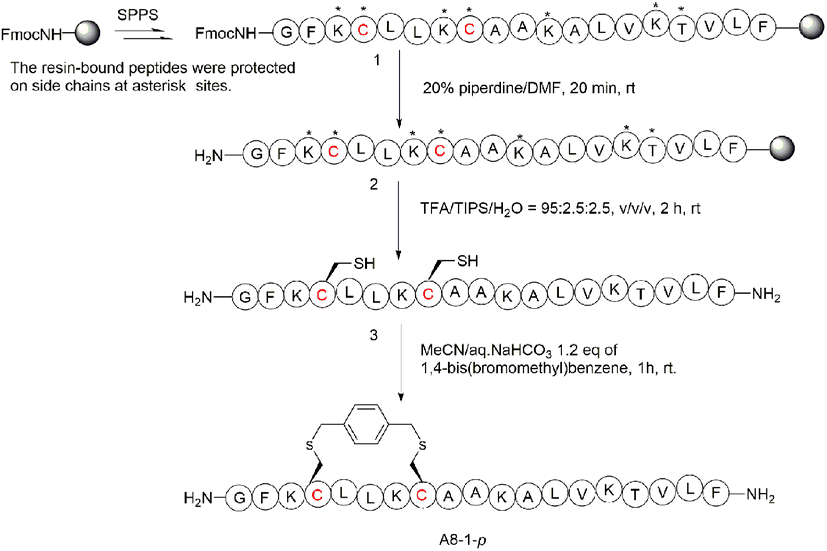 | ||
| Scheme 1 Synthesis route of A8-1-p. The following protecting groups for amino acids side chains were used: tert-butoxycarbonyl (Boc; for Lys), tert-butyl ester (OtBu, for Asp) and tert-butyl (tBu; for Thr). | ||
3.2 Helicity analysis of peptides
The secondary structure of Ascaphin-8 and its derived peptides was measured by circular dichroism chromatography. Circular dichroism analysis indicated that the α-helicity of Ascaphin-8 was 41% and that of the stapled peptides ranged from 18% to 71% (Fig. 2, Table 1). The results indicated that stapled peptide strategy can effectively improve the helicity level of the peptide if the side chain of cysteine was located in an appropriate position when compared with the linear peptide. Besides, compared with o/m/p three types of stapling bridge, the 1,2-bis(bromomethyl)benzene provided higher Secondary structural stability, suggesting a more compact spatial structure is more favorable to the helical structure formation of Ascaphin-8.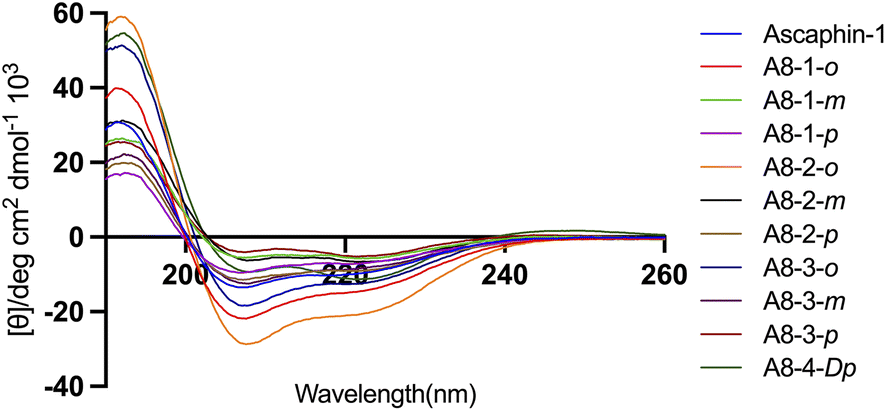 | ||
| Fig. 2 CD spectra of Ascaphin-8 and its derived peptide. | ||
| Peptide | Helicity (%) | Chromatographic purity (%) | Retention time (min) | Experimental molecular mass (m/z) | Theoretical molecular mass |
|---|---|---|---|---|---|
| Ascaphin-8 | 33 | >97 | 16.67 | [M + 2H]2+: 1009.91 | 2017.25 |
| [M + 3H]3+: 673.76 | |||||
| [M + 4H]4+: 505.56 | |||||
| A8-1-o | 49 | >97 | 18.45 | [M + 2H]2+: 1078.80 | 2153.26 |
| [M + 3H]3+: 719.35 | |||||
| [M + 4H]4+: 539.90 | |||||
| A8-1-m | 20 | >97 | 17.68 | [M + 2H]2+: 1078.35 | 2153.26 |
| [M + 3H]3+: 719.40 | |||||
| [M + 4H]4+: 539.90 | |||||
| A8-1-p | 24 | >97 | 17.45 | [M + 2H]2+: 1078.50 | 2153.26 |
| [M + 3H]3+: 719.45 | |||||
| [M + 4H]4+: 540.10 | |||||
| A8-2-o | 71 | >97 | 17.93 | [M + 2H]2+: 1100.31 | 2197.24 |
| [M + 3H]3+: 734.01 | |||||
| [M + 4H]4+: 550.86 | |||||
| A8-2-m | 23 | >97 | 18.50 | [M + 2H]2+: 1100.46 | 2197.24 |
| [M + 3H]3+: 734.01 | |||||
| [M + 4H]4+: 550.66 | |||||
| A8-2-p | 31 | >97 | 17.51 | [M + 2H]2+: 1100.40 | 2197.24 |
| [M + 3H]3+: 733.90 | |||||
| [M + 4H]4+: 550.90 | |||||
| A8-3-o | 42 | >97 | 18.98 | [M + 2H]2+: 1079.00 | 2154.20 |
| [M + 3H]3+: 719.60 | |||||
| [M + 4H]4+: 539.85 | |||||
| A8-3-m | 29 | >97 | 17.91 | [M + 2H]2+: 1078.50 | 2154.20 |
| [M + 3H]3+: 719.55 | |||||
| [M + 4H]4+: 540.00 | |||||
| A8-3-p | 18 | >97 | 17.43 | [M + 2H]2+: 1078.40 | 2154.20 |
| [M + 3H]3+: 719.50 | |||||
| [M + 4H]4+: 539.80 | |||||
| A8-4-Dp | 39 | >97 | 19.40 | [M + 2H]2+: 1116.25 | 2230.24 |
| [M + 3H]3+: 744.80 | |||||
| [M + 4H]4+: 558.80 |
3.3 Stapled peptide significantly inhibits tumor proliferation and migration
As presented in Fig. 3A, the vast majority of the peptides didn't trigger significant red blood cell rupture and hemolysis at concentrations below 25 μM. When the drug concentration reached 100 μM, only three peptides triggered more severe hemolytic events. These results indicate that Ascaphin-8 and its derivatives have good biocompatibility and safety. We expected Ascaphin-8 and its stapled peptide to have excellent antitumor activity. In this section, we used the Cell Counting Kit-8 reagent to test whether the peptides had the desired antitumor activity against three different cancer cells (human breast adenocarcinoma cell MCF-7, human colon cancer cell HCT116, and human glioblastoma cell U87). Of all the peptides tested, five modified peptides had lower half-maximal inhibitory concentration (IC50) values than the prototype peptide (Table 2). The IC50 of A8-2-o, A8-2-m and A8-4-Dp, which are great potential drug candidates for antitumor peptides, were significantly lower than that of Ascaphin-8.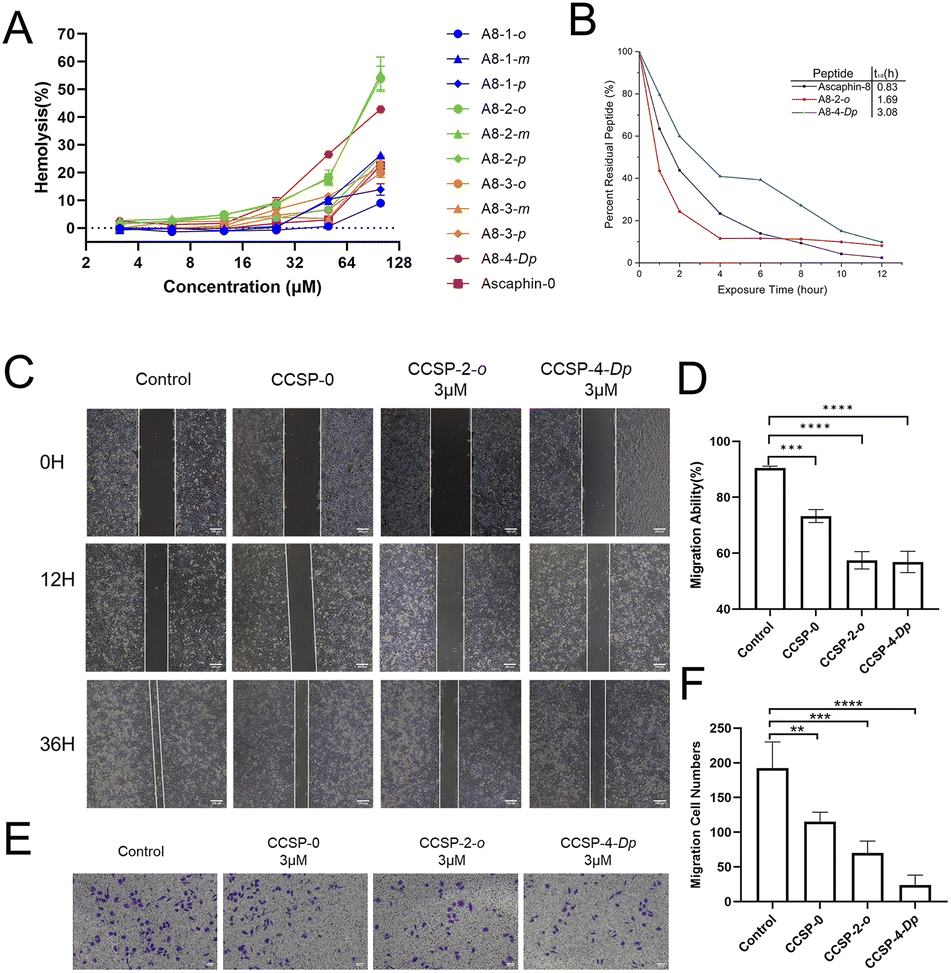 | ||
| Fig. 3 (A) Detection of haemolysis of rabbit erythrocytes by aromatic thioether staple peptide. (B) Detection of anti-enzymatic hydrolysis ability of Ascaphin-8, A8-2-o and A8-4-Dp. (C) Effects of Ascaphin-8, A8-2-o, and A8-4-Dp on lateral migration and growth of MCF-7 cells. (D) Calculation of crack area reduction. (E) Effects of Ascaphin-8, A8-2-o, and A8-4-Dp on vertical migration capacity of MCF-7 cells, (F) calculation of cell numbers that passed through the polycarbonate membrane. Scale: 200 μm (data are expressed as mean ± standard deviation; n = 3; compared with the control group, **p < 0.01, ***p < 0.001, ****p < 0.0001). | ||
| Peptide | IC50 (μM) | ||
|---|---|---|---|
| HCT116 | MCF-7 | U87 | |
| Ascaphin-8 | 18.83 | 9.33 | 9.25 |
| A8-1-o | 15.68 | 11.98 | 16.03 |
| A8-1-m | 20.88 | 22.90 | 16.73 |
| A8-1-p | 11.77 | 10.26 | 18.24 |
| A8-2-o | 4.78 | 3.79 | 4.63 |
| A8-2-m | 5.69 | 4.90 | 4.89 |
| A8-2-p | 10.77 | 9.27 | 12.03 |
| A8-3-o | 7.68 | 6.03 | 9.28 |
| A8-3-m | 9.21 | 7.80 | 8.69 |
| A8-3-p | 11.15 | 11.69 | 14.40 |
| A8-4-Dp | 3.03 | 2.99 | 3.63 |
It was reported that increased helicity and hydrophobicity are key factors in antimicrobial peptides' biological activity.22–25 Comparing protype peptide and A8-1-o to A8-2-m, we found that the antitumor activity of stapled antimicrobial peptides was not strictly linearly related to the degree of helicity, suggesting the increase in α-helicity may be one of the factors leading to an increase in its biological activity, other mechanisms such as their ability to selectively interact with ion channels may also significant for their antitumor activity. On the other hand, A8-4-Dp showed relatively low helicity but strong antitumor activity, a plausible explanation is that the biphenyl structure provided a higher degree of hydrophobicity, which may be more important than the increased degree of helicity.26
Anti–enzymatic hydrolysis ability is an important indicator of peptide drugs. Hence, enhancement of the enzymatic characterization of peptides is of great significance for peptide drugs. Fig. 3B illustrates that the half-lives (t1/2) of A8-2-o and A8-4-Dp were significantly longer than those of the control group, with A8-4-Dp having a longer half-life of 3.08 hours. These results demonstrated that the stapling modification with suitable sites can significantly enhance the enzymatic stability of linear peptides.
Fig. 3C illustrates the migration of tumor cells into the scratched area. The cells in the control group grew rapidly toward the center of the crack, and the crack gradually “closed” over time. The drug group did not grow as fast as the control group. In particular, the growth rate of cancer cells was significantly slower, and cell migration capacity was reduced when A8-2-o and A8-4-Dp were used to intervene in the cells (Fig. 3D). Fig. 3E and F illustrates the effect of stapled peptides on the migration capacity of tumor cells in the transwell migration assay. Compared with the control group, cells cultured with A8-2-o and A8-4-Dp rarely migrated to the other side of the polycarbonate membrane. These results suggest that A8-2-o and A8-4-Dp may inhibit the migration and invasion of MCF-7 cells.
3.4 A8-2-o and A8-4-Dp induce tumor cell apoptosis
To quantitatively evaluate the apoptotic effects of A8-2-o and A8-4-Dp on tumor cells, an annexin V-FITC apoptosis assay was performed to determine the percentage of cell apoptosis. As presented in Fig. 4A, MCF-7 cells were treated with 10 μM of A8-2-o and A8-4-Dp solutions. The number of apoptotic cells was significantly higher than that of MCF-7 cells cultured with the prototype peptide Ascaphin-8 and normal medium. Although tumor cells undergo apoptosis, the integrity of the cell membrane is destroyed. In calcein-acetyl methyl (AM)/propidium iodide (PI) staining of cells in different states, as presented in Fig. 4B, calcein-AM exhibits green fluorescence after degradation in living cells, mainly concentrated in the control and Ascaphin-8 groups. However, when A8-2-o and A8-4-Dp solutions (10 μM) were used to culture MCF-7 cells, a large amount of red fluorescence from the PI–DNA combination emerged. Therefore, it is suggested that A8-2-o and A8-4-Dp may induce apoptosis of tumor cells.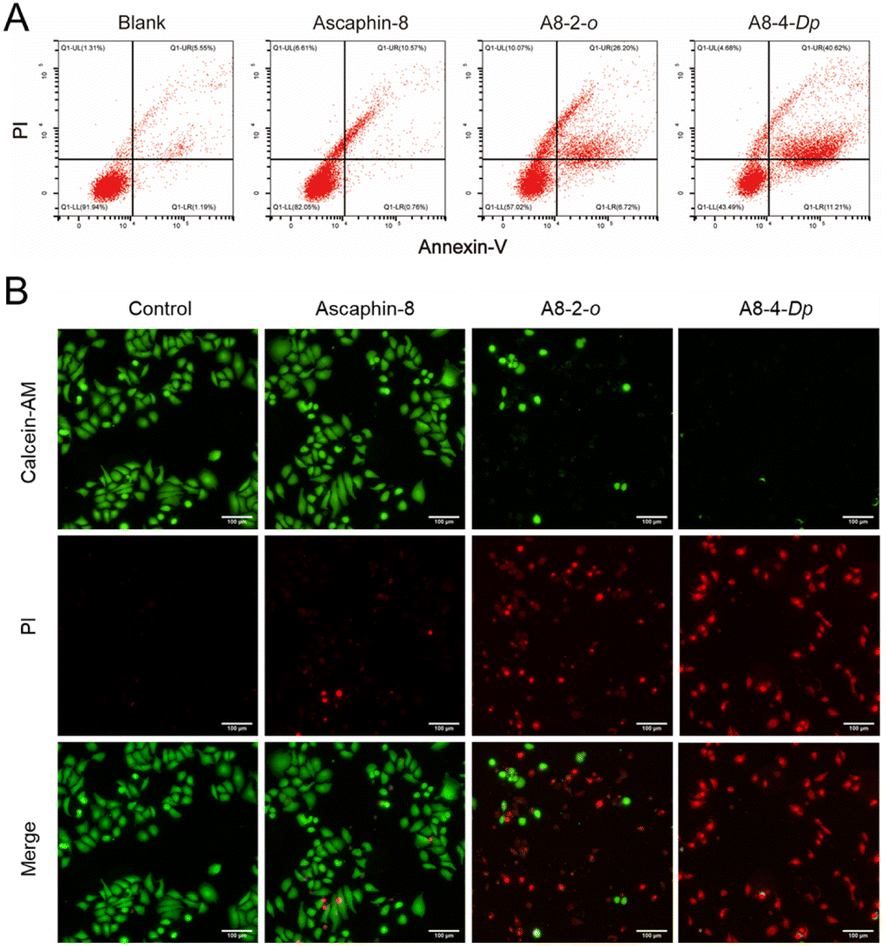 | ||
| Fig. 4 (A) Detection of apoptosis effects of Ascaphin-8, A8-2-o, and A8-4-Dp (10 μM) on MCF-7 cells. (B) Fluorescent images of life-and-death staining of MCF-7 cells treated with Ascaphin-8, A8-2-o and A8-4-Dp. | ||
4 Conclusions
In conclusion, a novel series of stapled Ascaphin-8 derivatives were designed and successfully synthesized. The CD and protease stability study suggested that the aromatization stapling strategy could improve the helicity and protease stability of the prototype linear peptide when suitable stapling bridge and sites were applied. In vitro antitumor inhibition tests confirm that A8-2-o and A8-4-Dp exhibit obviously stronger antitumor activity compared to the prototype peptide.However, we found that the antitumor activity of stapled antimicrobial peptides was not strictly linearly related to the degree of helicity, suggesting other mechanisms may also significant for their antitumor activity. An among them, A8-4-Dp showed relatively low helicity but strong antitumor activity, also indicating that higher degree of hydrophobicity may be more important than the increased degree of helicity26 for Ascaphin-8. All in all, this work not only provides a successful example of AMPs aromatization stapling structure optimization. More importantly, the promising lead compounds A8-2-o, A8-2-o and A8-4-Dp are expected to be studied more deeply as novel cancer therapeutics. Further molecular tracer research and related biological validation using fluorescent molecular labelling strategies are ongoing, and new findings will be published in due course.
Author contributions
CL, WC, and H-GH: conceptualization and design of the study. NW and NZ: synthesis of the compounds. X-LK and H-XS: performance of the pharmacological tests. XL-K: statistical analysis of the data. H-GH: writing and revising of the manuscript. All authors contributed to the article and approved the submitted version.Conflicts of interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.Acknowledgements
We thank LetPub (https://www.letpub.com) for its linguistic assistance during the preparation of this manuscript. This work was funded by the Science and Technology Commission of Shanghai Municipality (22140900300, 22140900302, 22140900303).References
- L. Zhang, B. Z. Zhai, Y. J. Wu and Y. Wang, Drug Delivery, 2023, 30, 1–18 CrossRef CAS PubMed.
- H. Sung, J. Ferlay, R. L. Siegel, M. Laversanne, I. Soerjomataram, A. Jemal and F. Bray, Ca-Cancer J. Clin., 2021, 71, 209–249 CrossRef PubMed.
- Y. T. Li and W. H. Chang, J. Chin. Med. Assoc., 2023, 86, 256 CrossRef PubMed.
- M. F. El-Behairy, W. H. Abd-Allah, M. M. Khalifa, M. S. Nafie, M. A. Saleh, M. S. Abdel-Maksoud, T. Al-Warhi, W. M. Eldehna and A. A. Al-Karmalawy, J. Enzyme Inhib. Med. Chem., 2023, 38, 2157825 CrossRef PubMed.
- A. H. Liao, Y. A. Lee, D. L. Lin, H. C. Chuang, J. K. Wang, C. E. Chang, H. T. Li, T. Y. Wu, C. P. Shih, C. H. Wang and Y. H. Chu, Drug Delivery, 2023, 30, 1–13 CrossRef CAS PubMed.
- D. D. Shi, J. A. Guo, H. I. Hoffman, J. Su, M. Mino-Kenudson, J. L. Barth, J. M. Schenkel, J. S. Loeffler, H. A. Shih, T. S. Hong, J. Y. Wo, A. J. Aguirre, T. Jacks, L. Zheng, P. Y. Wen, T. C. Wang and W. L. Hwang, Lancet Oncol., 2022, 23, e62–e74 CrossRef CAS PubMed.
- H. Gharwan and H. Groninger, Nat. Rev. Clin. Oncol., 2016, 13, 209–227 CrossRef CAS PubMed.
- A. Henninot, J. C. Collins and J. M. Nuss, J. Med. Chem., 2018, 61, 1382–1414 CrossRef CAS PubMed.
- L. Wang, N. Wang, W. Zhang, X. Cheng, Z. Yan, G. Shao, X. Wang, R. Wang and C. Fu, Signal Transduction Targeted Ther., 2022, 7, 48 CrossRef CAS PubMed.
- I. Petta, S. Lievens, C. Libert, J. Tavernier and K. De Bosscher, Mol. Ther., 2016, 24, 707–718 CrossRef CAS PubMed.
- N. Kamech, D. Vukicevic, A. Ladram, C. Piesse, J. Vasseur, V. Bojovic, J. Simunic and D. Juretic, J. Chem. Inf. Model., 2012, 52, 3341–3351 CrossRef CAS PubMed.
- J. M. Conlon, J. Kolodziejek and N. Nowotny, Biochim. Biophys. Acta, 2009, 1788, 1556–1563 CrossRef CAS PubMed.
- J. M. Conlon, S. Galadari, H. Raza and E. Condamine, Chem. Biol. Drug Des., 2008, 72, 58–64 CrossRef CAS PubMed.
- S. Kannan, P. G. A. Aronica, S. Ng, D. T. Gek Lian, Y. Frosi, S. Chee, J. Shimin, T. Y. Yuen, A. Sadruddin, H. Y. K. Kaan, A. Chandramohan, J. H. Wong, Y. S. Tan, Z. W. Chang, F. J. Ferrer-Gago, P. Arumugam, Y. Han, S. Chen, L. Renia, C. J. Brown, C. W. Johannes, B. Henry, D. P. Lane, T. K. Sawyer, C. S. Verma and A. W. Partridge, Chem. Sci., 2020, 11, 5577–5591 RSC.
- Y. Huan, Q. Kong, H. Mou and H. Yi, Front. Microbiol., 2020, 11, 582779 CrossRef PubMed.
- N. Wang, G. Xie, C. Liu, W. Cong, S. He, Y. Li, L. Fan and H. G. Hu, Front. Chem., 2020, 8, 616147 CrossRef CAS PubMed.
- M. Moiola, M. G. Memeo and P. Quadrelli, Molecules, 2019, 24, 3654 CrossRef CAS PubMed.
- L. D. Walensky and G. H. Bird, J. Med. Chem., 2014, 57, 6275–6288 CrossRef CAS PubMed.
- A. Muppidi, Z. Wang, X. Li, J. Chen and Q. Lin, Chem. Commun., 2011, 47, 9396–9398 RSC.
- C. A. Rohl and R. L. Baldwin, Methods Enzymol., 1998, 295, 1–26 CAS.
- M. Klein, Expert Opin. Drug Discovery, 2017, 12, 1117–1125 CrossRef CAS PubMed.
- T. T. Dinh, D. H. Kim, H. X. Luong, B. J. Lee and Y. W. Kim, Bioorg. Med. Chem. Lett., 2015, 25, 4016–4019 CrossRef CAS PubMed.
- H. X. Luong, D. H. Kim, B. J. Lee and Y. W. Kim, Arch. Pharmacal Res., 2017, 40, 1414–1419 CrossRef CAS PubMed.
- D. Migon, D. Neubauer and W. Kamysz, Protein J., 2018, 37, 2–12 CrossRef CAS PubMed.
- R. Mourtada, H. D. Herce, D. J. Yin, J. A. Moroco, T. E. Wales, J. R. Engen and L. D. Walensky, Nat. Biotechnol., 2019, 37, 1186–1197 CrossRef CAS PubMed.
- P. Gkeka and L. Sarkisov, J. Phys. Chem. B, 2010, 114, 826–839 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: The sequence, HPLC and MS data of the stapled peptides. See DOI: https://doi.org/10.1039/d3ra02743k |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2023 |