
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicencePseudo-multicomponent reactions
Julio C. Flores-Reyes
a,
Vanesa del C. Cotlame-Salinas
a,
Ilich A. Ibarra
 *b,
Eduardo González-Zamora
*a and
Alejandro Islas-Jácome
*a
*b,
Eduardo González-Zamora
*a and
Alejandro Islas-Jácome
*a
aDepartamento de Química, Universidad Autónoma Metropolitana-Iztapalapa, Av. Ferrocarril San Rafael Atlixco 186, Col. Leyes de Reforma 1A Sección, Iztapalapa, Ciudad de México C.P. 09310, Mexico. E-mail: egz@xanum.uam.mx; aij@xanum.uam.mx
bLaboratorio de Fisicoquímica y Reactividad de Superficies, (LaFReS), Instituto de Investigaciones en Materiales, Universidad Nacional Autónoma de México, Circuito Exterior S/N, CU, Coyoacán, Ciudad de México, Mexico. E-mail: argel@unam.mx
First published on 30th May 2023
Abstract
Classical multicomponent reactions (MCRs) are domino-type one-pot processes in which three or more different reactants are combined sequentially in the same reactor to synthesize compounds containing all or almost all atoms coming from the reactants. Besides, pseudo-MCRs are also domino-type one-pot processes involving combinations of at least three reactants but in which at least one of them takes part in two or more reaction steps. In consequence, the products synthesized through pseudo-MCRs contain also all or almost all atoms but coming from two or more identical reactants. Thus, pseudo-MCRs differ from classical MCRs because the first ones appear to involve an assembly of a higher number of different components than those that are being truly assembled. However, pseudo-MCRs are also useful synthetic tools to generate libraries of complex compounds in few experimental steps, and although the repeated reactants may make them appear less diverse than classical MCRs, this can be offset by the higher number of reactants that can participate in this type of reaction. Overall, there are two types of pseudo-MCRs. The first are those in which the duplicated reagents participate in different steps of the corresponding reaction mechanism. The second kind of pseudo-MCRs are those in which one or more components react simultaneously with a main reagent containing two or more identical functional groups. These latter are known as repetitive pseudo-MCRs. Thus, the aim of the present review is to cover for the first time selected works mainly published in the last two decades about pseudo-MCRs and their repetitive versions toward the synthesis of novel, complex, and highly symmetrical molecules, often including their interesting applications in various fields of science and technology. The manuscript has been categorized considering the number of reagents participating in the corresponding pseudo-MCRs, aiming to give readers novel insights for their future investigations.
 Julio C. Flores-Reyes | Julio César Flores-Reyes was born in Puebla, México, in 1989. He received his Bsc in biochemical engineering in 2013 from Universidad Autónoma Metropolitana – Iztapalapa (UAM-I). Then, he joined Professor Eduardo González-Zamora's research group in 2018 at UAM-I, where he got his MSc in chemistry in 2020 working on the synthesis of polytetrazole-based ligands toward new MOFs. He is currently enrolled as a PhD student working on multicomponent reactions, optical properties of complex polyheterocycles, and the ESIPT effect. |
 Vanesa del C. Cotlame-Salinas | Vanesa del Carmen Cotlame-Salinas was born in Veracruz, Mexico in 1995. She received her BSc in Industrial Chemistry from the Universidad Veracruzana (UV) in 2019 and obtained her MSc degree in Chemistry from Universidad Autónoma Metropolitana – Iztapalapa (UAM-I) Mexico under the guidance of Professor Eduardo Gonzalez-Zamora in 2022 working on the synthesis of new MOFs to capture CO2 and other gases with greenhouse effect, and in pseudo-multicomponent reactions. She is currently Professor of Chemistry at the Instituto Tecnológico Superior de Zongolica (ITSZ). |
 Ilich A. Ibarra | Ilich A. Ibarra was born in Mexico City in 1981. He completed his BSc at UAM-I, Mexico in 2005. Later, in 2010, he obtained his PhD in Chemistry under the supervision of Professor Martin Schröder at Nottingham University (UK). He then took a postdoctoral position, 2010–2012, at The University of Texas at Austin (USA) under the supervision of Professor Simon Humphrey. Then, in 2013 he was awarded a Wenner-Gren researcher position at Stockholm University (Sweden) under the supervision of Professor Xiaodong Zou. In 2014, he moved to UNAM (IIM, Mexico), working as an assistant professor. In 2017, he was promoted to associate professor. |
 Eduardo González-Zamora | Eduardo González-Zamora was born in 1961 in Mexico City. He obtained his PhD in 1998 from Paris XI University in France under the supervision of Professor R. Beugelmans. After one year of a post-doctoral position with Professor R. Cruz at the Instituto de Química (UNAM, Mexico), he joined the Institut de Chimie des Substances Naturelles (CNRS, France) in 2000 as a post-doctoral fellow with Professor J. Zhu. In 2011, he moved to UCLA (USA) as a visiting Professor in the M.A. Garcia-Garibay group. His research interests include synthesis via multicomponent reactions, MOF chemistry, and total synthesis. |
 Alejandro Islas-Jácome | Alejandro Islas-Jácome was born in Veracruz, México in 1981. He got his BSc in 2005 and PhD in 2011, both in chemistry and under the guidance of Professor Eduardo González-Zamora, in UAM-I, México. Then, he took up a post-doctoral position in the Rocío Gámez-Montaño research group from 2012 to 2016 at the Universidad de Guanajuato, México. He has been a full-time professor since 2017 at the Departamento de Química, UAM-I, México. His research interests include the synthesis of polyheterocycles via multicomponent reactions, click chemistry, reaction mechanisms, optical properties, as well as medicinal chemistry. |
1. Introduction
Synthetic organic chemistry is a rapidly growing field due to the constant research behind new methodologies that minimize the negative environmental impact. All this can be achieved with the guidance of the 12 principles of green chemistry,1 such as atom economy, less hazardous synthesis, energy efficiency, benign solvents, reduced derivatives, and by-products, among others. Multicomponent reactions (MCRs) are powerful tools leading modern synthetic organic chemistry, which exactly face the environmental challenges with many advantages like straightforward design, and processes requiring less time, resources, and efforts. In these kinds of reactions, all reagents are added sequentially in a single vessel (one-pot procedures) to assemble complex products with high structural diversity via a series of chemical transformations without needing solvent shifts, extra workups, or purifications in each reaction step.2Since the first MCR reported by Strecker in the mid-19th century,3 the impact of MCR-based strategies has been increasing in the last two decades compared to classical techniques, which is reflected in the large amount of publications describing the synthesis of new and complex organic molecules like heterocycles and polyheterocycles, multifunctional peptides, and natural products,4 that lead to the creation of extensive chemical libraries with potential biological activity, especially desirable for pharmaceutical5 and agrochemical6 industries, as well as the design of novel materials.7 For these reasons, there are many publications about MCRs in books,8 book chapters,9 recent reviews,10 and also in other sources. However, to date, there is no review about pseudo-MCRs.
Pseudo-MCRs (pretended, indented to be, or false MCRs) involve the sequential combination of two or more identical reactants with at least one different reactant in the same reactor. Indeed, pseudo-MCRs are domino-type one-pot process, equal to the classical MCRs, but the stoichiometry for one or more reactants is duplicated, triplicated, or more. It is worth noting that pseudo-MCRs have some limitations in comparison to classical MCRs such as less structural diversification and a relatively low degree of functional flexibility, which are offset by the high molecular symmetry that can be achieved with them.11 Pseudo-MCRs are divided into two major groups: (1) pseudo-MCRs (Fig. 1a), in which the duplicated, triplicated, or more, reactants participate in different steps of the reaction mechanism, and (2) repetitive pseudo-MCRs (Fig. 1b), in which a main reagent contains two or more identical functional groups to react simultaneously with also two or more reagents, just depending on the number of functional groups that are present in the main reactant. Fig. 1 shows a schematic example of a pseudo-4CR (in representation of any pseudo-MCR), and a repetitive pseudo-5CR, also on behalf of any repetitive pseudo-MCR.
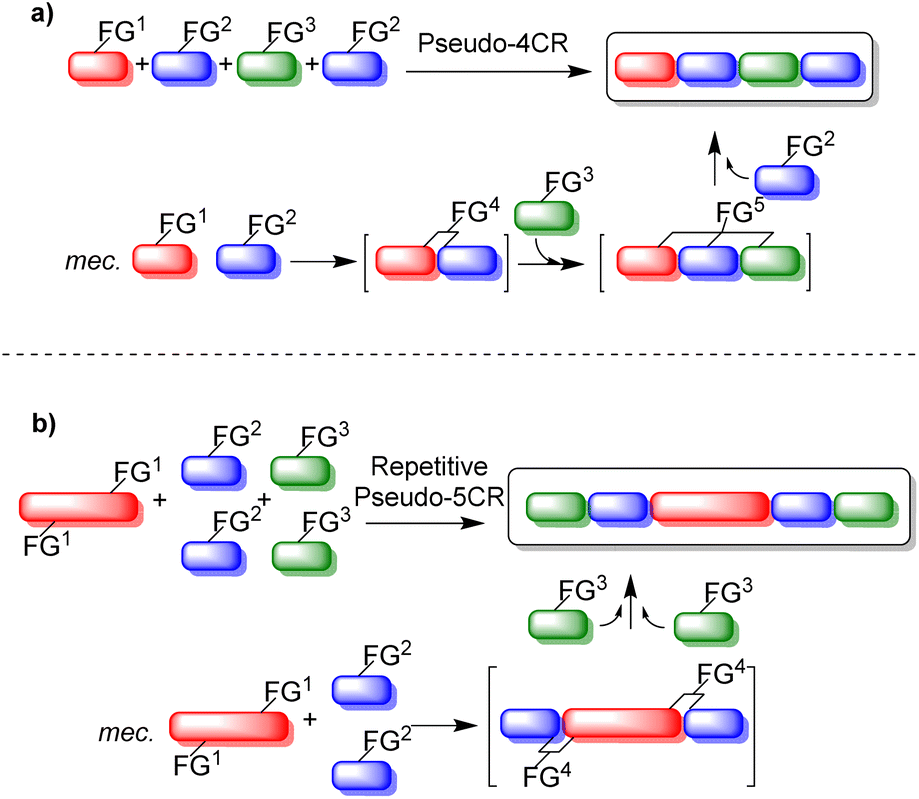 | ||
| Fig. 1 Schematic representation of (a) pseudo-MCRs, and (b) repetitive pseudo-MCRs. | ||
Pseudo-MCRs have led to a variety of compounds with interesting biological properties, especially desirable in pharmaceutical industry. For example, compound 1 exhibited anticancer activity on a panel of human cell lines,12a while compound 2 showed antitubercular activity.12b Moreover, other compounds synthesized through pseudo-MCRs were tested due to their potential applications in materials science. For instance, the polyheterocycle 3 exhibited moderate anticorrosive activity,12c and the hexasubstituted anilines 4 showed a high fluorescence emission (Fig. 2).12d
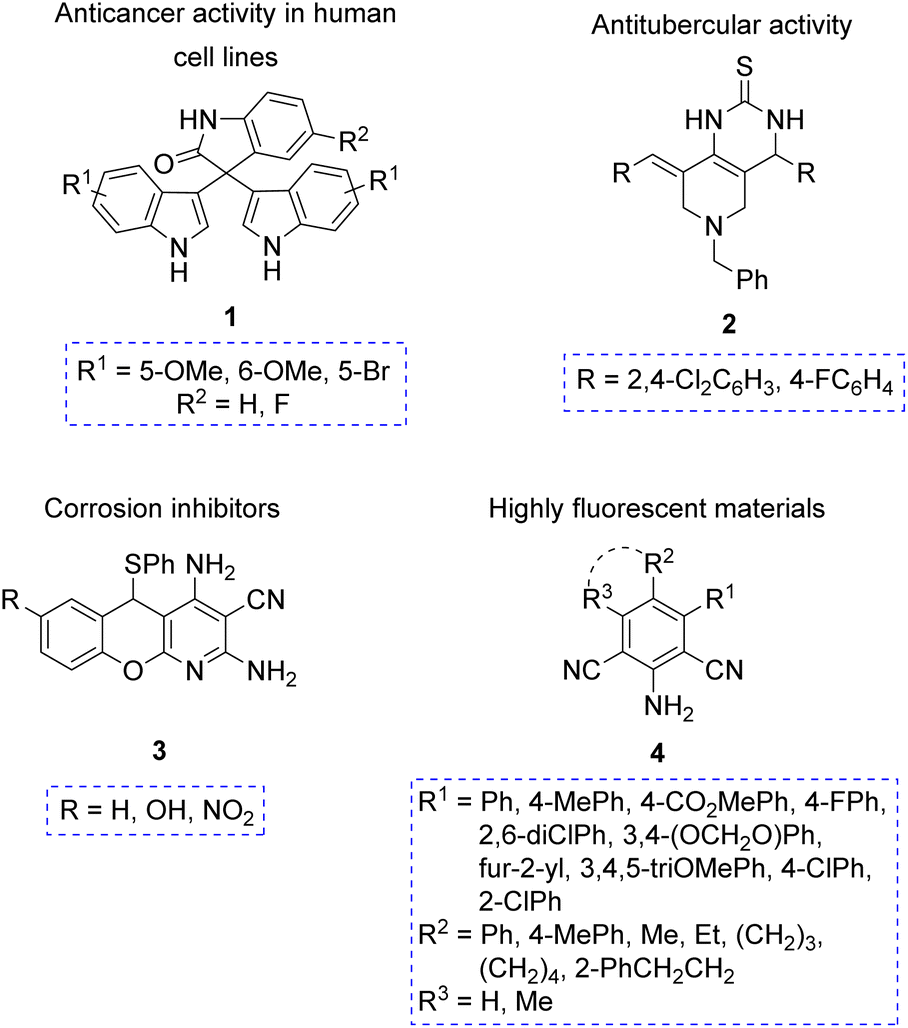 | ||
| Fig. 2 Molecules synthesized via pseudo-MCRs or repetitive pseudo-MCRs, and their applications. | ||
There are many research programs worldwide in which pseudo-MCRs (or their repetitive versions) play a central role. Thus, the present review covers selected works on the topic, mainly published in the last decade, and focusing on the reaction mechanisms. These were selected based on the number of participating reactants and the complexity of the products. The paper is classified by the number of reagents involved in the corresponding discussed one-pot procedure. It is worthy to note that a colour-key is used to highlight the ‘repeated’ reagents.
2. Pseudo-three component reactions
2.1 Knoevenagel condensation/Michael addition
A highly diverse strategy employed in pseudo-3CRs involves a Knoevenagel condensation coupled to a Michael addition. Brahmachari13 reported the synthesis of many functionalized bis-lawsone derivatives using two equivalents of lawsone 5 (2-hydroxy-1,4-naphthoquinone), one equivalent of aromatic aldehydes 6 and sulfamic acid as organo-catalyst, under mild reaction conditions. A reaction mechanism is proposed (Scheme 1a): the aldehydes 6, previously activated by sulfamic acid, react with a molecule of lawsone 5 via a Knoevenagel condensation forming the intermediates 7, which produce the alkenes 8 after dehydration. Then, 8 react with a second molecule of lawsone 5 via a Michael addition, producing the intermediates 9, which tautomerize towards the final bis-lawsones 10. Over the years, it has been known that bis-lawsones have numerous biological activities as well as cosmetics applications. Khan and co-workers14 reported a novel domino pseudo-MCR to synthesize thiopyrano{2,3-b:6,5-b′}bis(thiocromene)-12,14(13H)-diones 11 involving a Knoevenagel condensation/Michael-type addition, followed by intramolecular cyclization and loss of H2S (Scheme 1b). Using the same method, Naime-Jamal and Ghahremanzadeh15 synthetized the 3,3′-bis-substituted-2-oxindoles 12 using cyclohexyl isocyanide (Scheme 1b). Similar techniques were employed to synthesize series of furo[3,2-c]coumarins (using I2/K2S2O8/NA2CO3 as catalyst),16 10-(4-hydroxy-6-methyl-2-oxo-2H-pyran-3-yl)-3-methyl-1H,10H-pyrano[4,3-b]chromen-1-ones (using H3PMo12O40 as catalyst),17 tricyclic spiro-dihydrofurans18 and spirocyclopropanes.19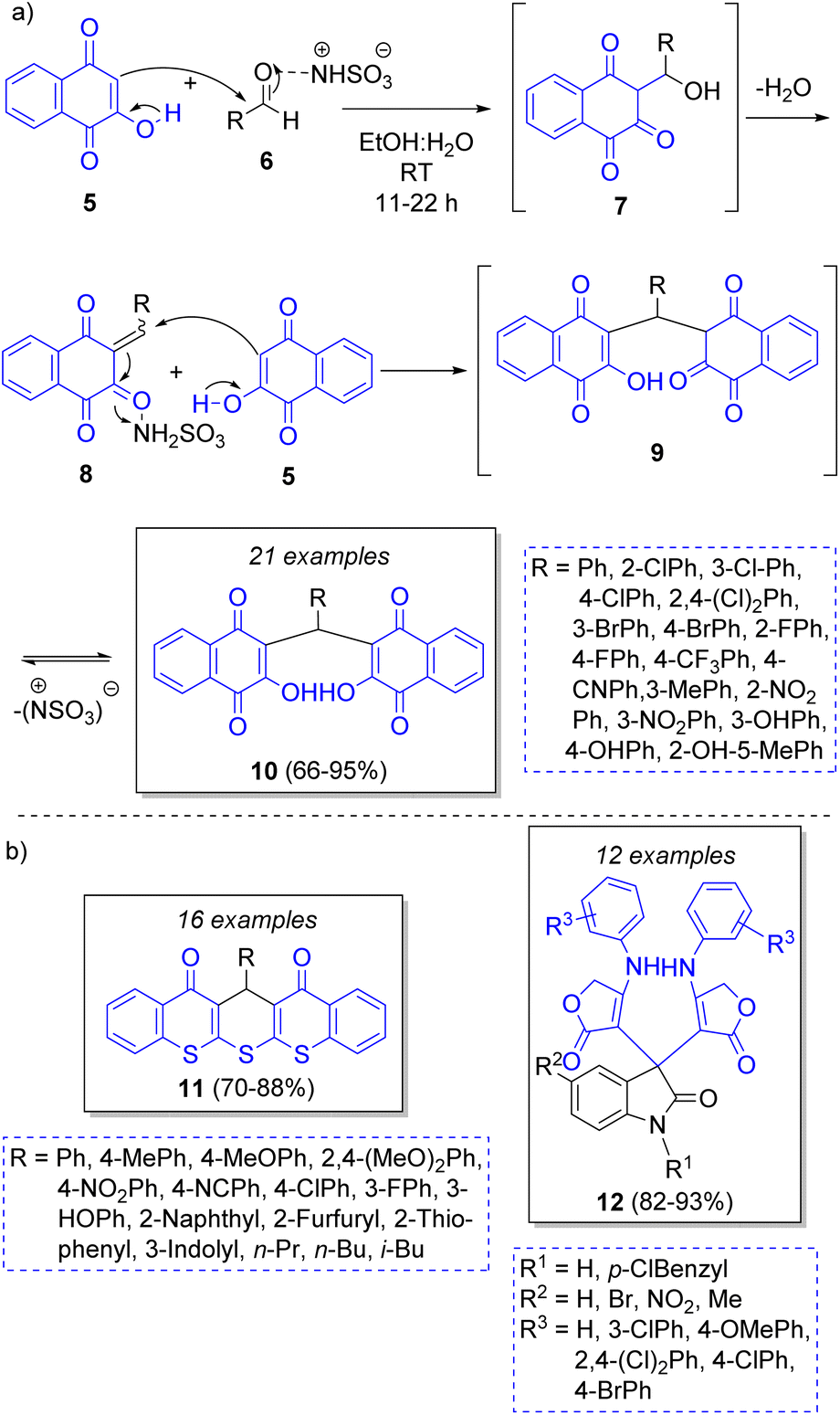 | ||
| Scheme 1 (a) Synthesis of bis-lowsone derivatives via Knoevenagel condensation/Michael addition. (b) Synthesized thiopyrano{2,3-b:6,5-b′}bis(thiocromene)-12,14(13H)-diones and 3,3′-bis-substituted-2-oxindoles. | ||
Following this approach, in 2019 Rimaz and co-workers20 reported a moderate to high-yielding synthesis of a series of many pyrano[2,3-d:6,5-d′]dipyrimidines using two equivalents of 1-ethyl-2-thioxodihydropyrimidine-4,6(1H,5H)-dione (13), an equivalent of arylglyoxal monohydrates 15, and 1,4-diazabicyclo[2.2.2]octane (DABCO) and ZrOCl2·8H2O as green catalysts. The Scheme 2 illustrates the reaction catalyzed by ZrOCl2·8H2O. The mechanism begins with a regioselective Knoevenagel condensation between thioxidihydropyrimidine 14 and arylglyoxals 17 activated by ZrOCl2·8H2O to form the intermediates 18. Then, a Michael addition of another molecule of 14 to 18 affords the intermediates 19. Finally, an intramolecular cyclization followed by a lactam-lactim tautomerization leads to the final products 22.21
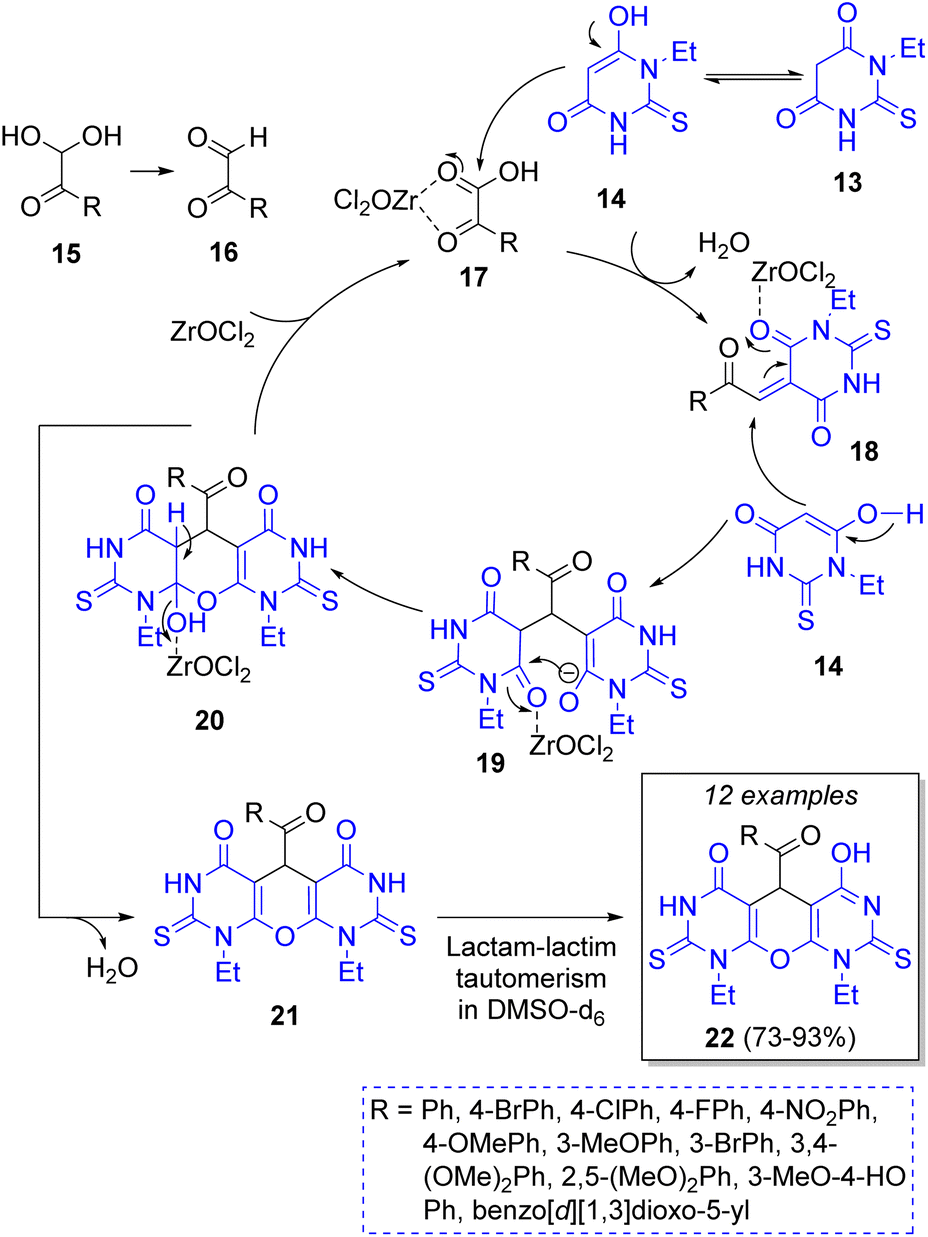 | ||
| Scheme 2 Synthesis of pyrano[2,3-d:6,5-d′]dipyrimidines. | ||
2.2 Others
In 2012 Yang and co-workers22 described the synthesis of pyranocoumarins 30 via a microwave-assisted pseudo-3CR starting from the 4-methylquinoline (23) and two equivalents of coumarins 24 in acetic anhydride as solvent. The proposed reaction mechanism begins with an acetylation of 23 by acetic anhydride to generate the intermediates 25, which add electrophilically to coumarins 24 to produce the alcoholates 27, after leaving one Ac2O molecule. This new intermediates react with another molecule of Ac2O to generate 28, which react with a second molecule of 24 to afford the zwitterions 29, which after an intramolecular cyclization followed by dehydration, furnish the final products 30 (Scheme 3). These products were found to be sensible to UV light, and thus, the aim behind their syntheses was to use them as molecular switches.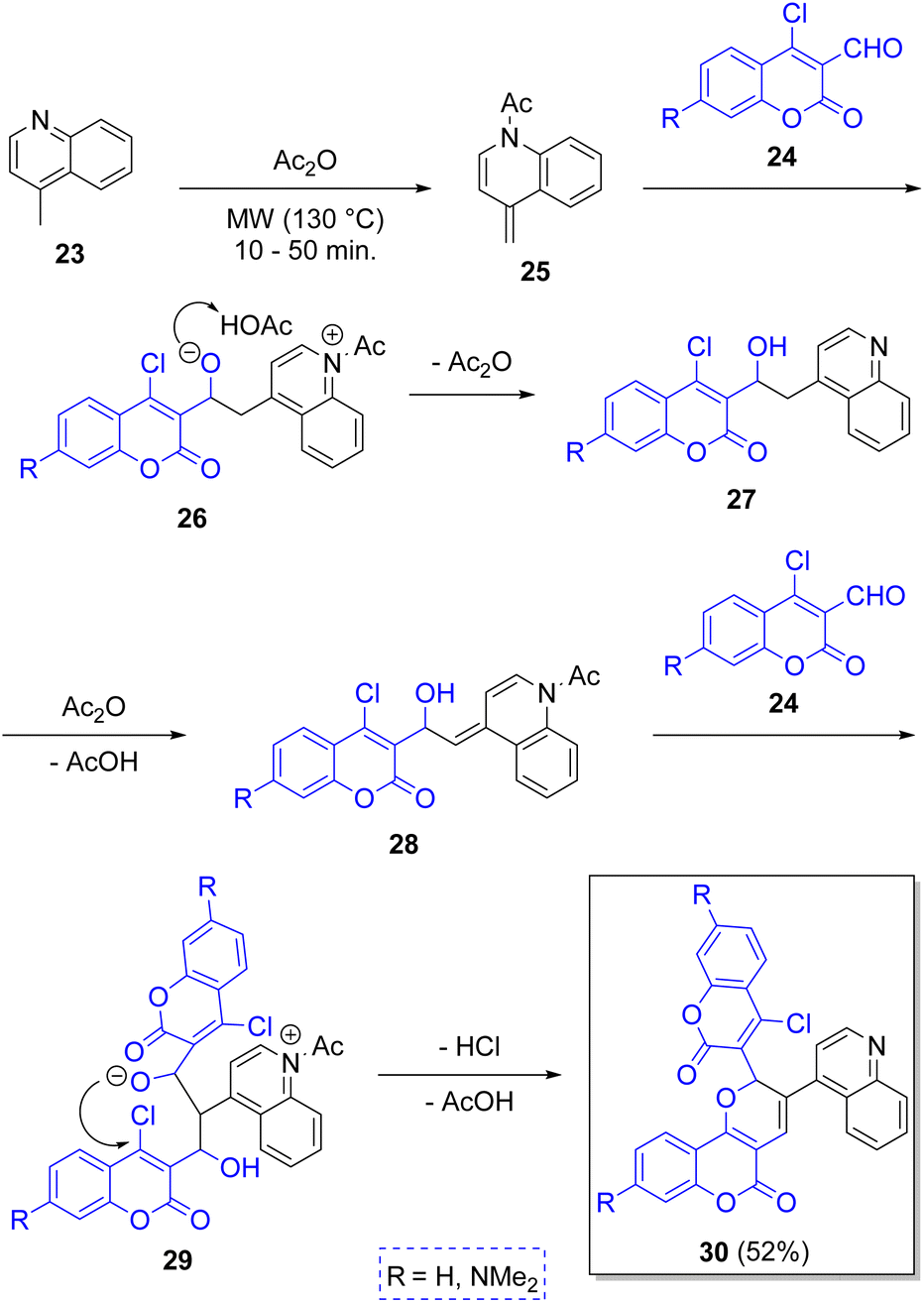 | ||
| Scheme 3 Synthesis of pyranocoumarins via a pseudo-3CR. | ||
In 2014, Rao and co-workers23 described a practical high-yielding domino MCR for the synthesis of hexa-substituted 1,4-dihydropyridines 37 (1,4-DHPs) by a reaction between a variety of aromatic aldehydes 31, nitroketene-N,S-acetals 32 (2 equiv.), and 2-aminopyridine as a catalyst. As Scheme 4 depicts, the reaction mechanism begins with the formation of iminium ion 33 from the condensation of aromatic aldehyde 31 with 2-aminopyridine (2-AP) 33. Then, it reacts with a molecule of N-methyl-S-methyl nitroethylene (NMSM, 32) forming the intermediate 34, which adopts the tautomeric form of 35. Then, intermediate 35 reacts with another molecule of 32 to give 36. The final product 37 results from an intramolecular elimination of methanethiol (MeSH). Encouraged by these results, the authors designed and synthesized a small library of 1,4-DPHs, functionalizing the C-4 and C-2 positions of benzaldehyde with electron-withdrawing groups (EWGs) and electron-releasing groups ERGs. 1,4-DHPs have important biological activities because of their similarity with the coenzymes NADH (nicotinamide adenine dinucleotide) and NADPH (nicotinamide adenine dinucleotide phosphate).
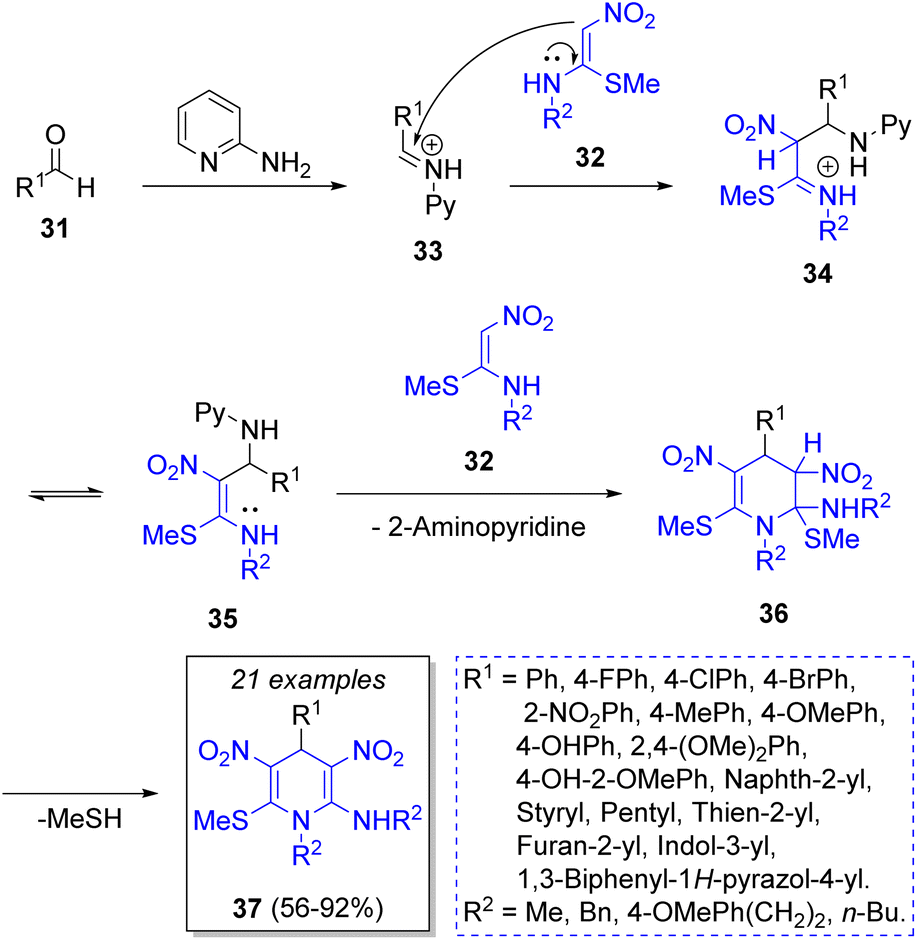 | ||
| Scheme 4 Synthesis of hexasubstituted 1,4-DHPs. | ||
Festa and Voskressensky24 reported an efficient synthesis of many 3- and 2-substituted pyrido[2,3-b]indolizines by a reaction between two equivalents of N-(cyanomethyl)pyridinium (38) and vinamidinium salts 39 or enaminones 40. The reaction mechanism showed in the Scheme 5 begins with the base-promoted dimerization of pyridinium salt 41, which then undergoes an intramolecular cyclization to form the intermediate 42. Then, elimination of pyridinium hydrochloride forms the aromatic compound aminoindolizine 43, which condenses with 1,3-dielectrophiles to give the pyridoindolizines 44 or 45. The optical properties of synthesized indolizines were investigated, showing a strong visible emission ranging from blue to green regions. The products have promising structures for preparing materials to fabricate OLED devices.
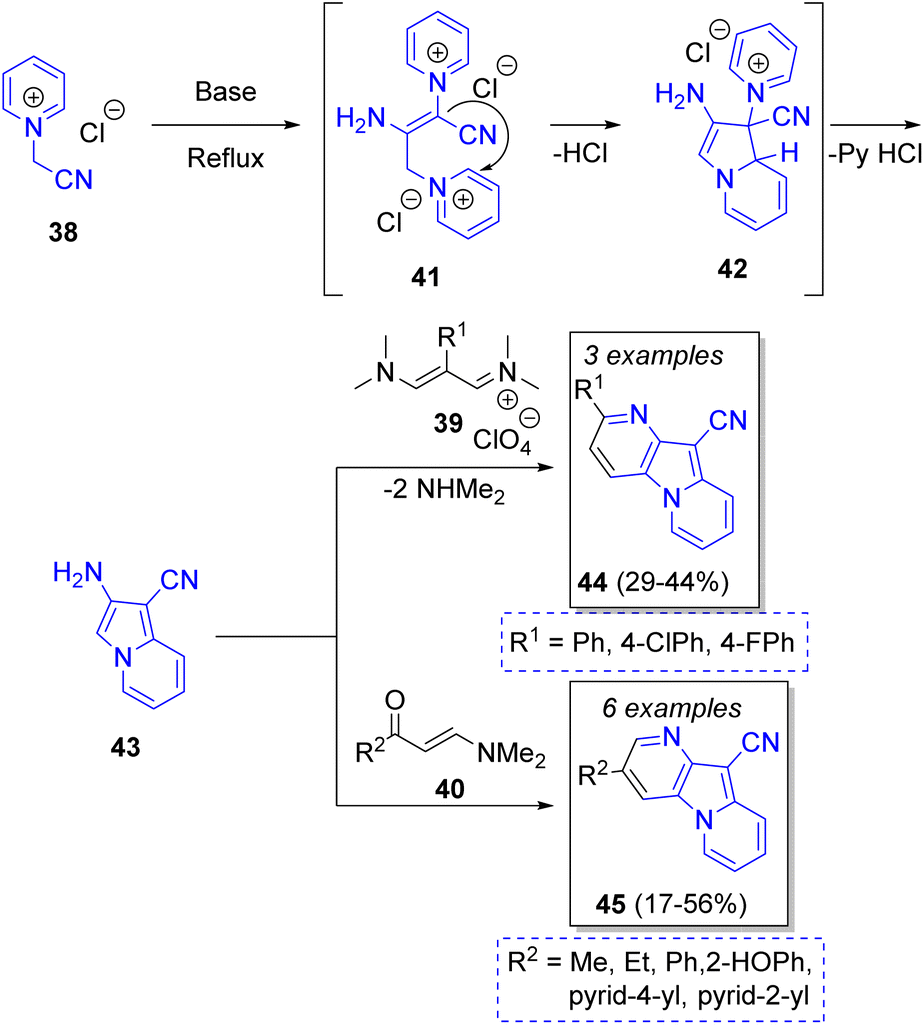 | ||
| Scheme 5 Synthesis of pyridoindolizines. | ||
A collection of 1,6-dihydroazaazulenes was achieved by Cortes-García and co-workers25 via a pseudo-3CR strategy using an acid-catalyzed cyclization of pyrrolyl-enones. The reaction comprises but-3-en-2-one (2 equiv., 46), functionalized pyrroles 47 and ionic liquid as catalyst (1-butyl-3-methylpyridinuim tetrafluoroborate, BMPy-BF4). The suggested mechanism is depicted in Scheme 6. Initially, the butenone 46 reacts with pyrroles 47 via Michael addition to produce the intermediates 48, which then undergo an 1,2 addition of a second molecule of 46 to produce the corresponding allylic alcohols 49, followed by a dehydration to form allylic carbocation intermediates 50. Upon electrophilic addition of 51 to the double bond of the enones and a subsequent deprotonation, the desired products 52 are formed.
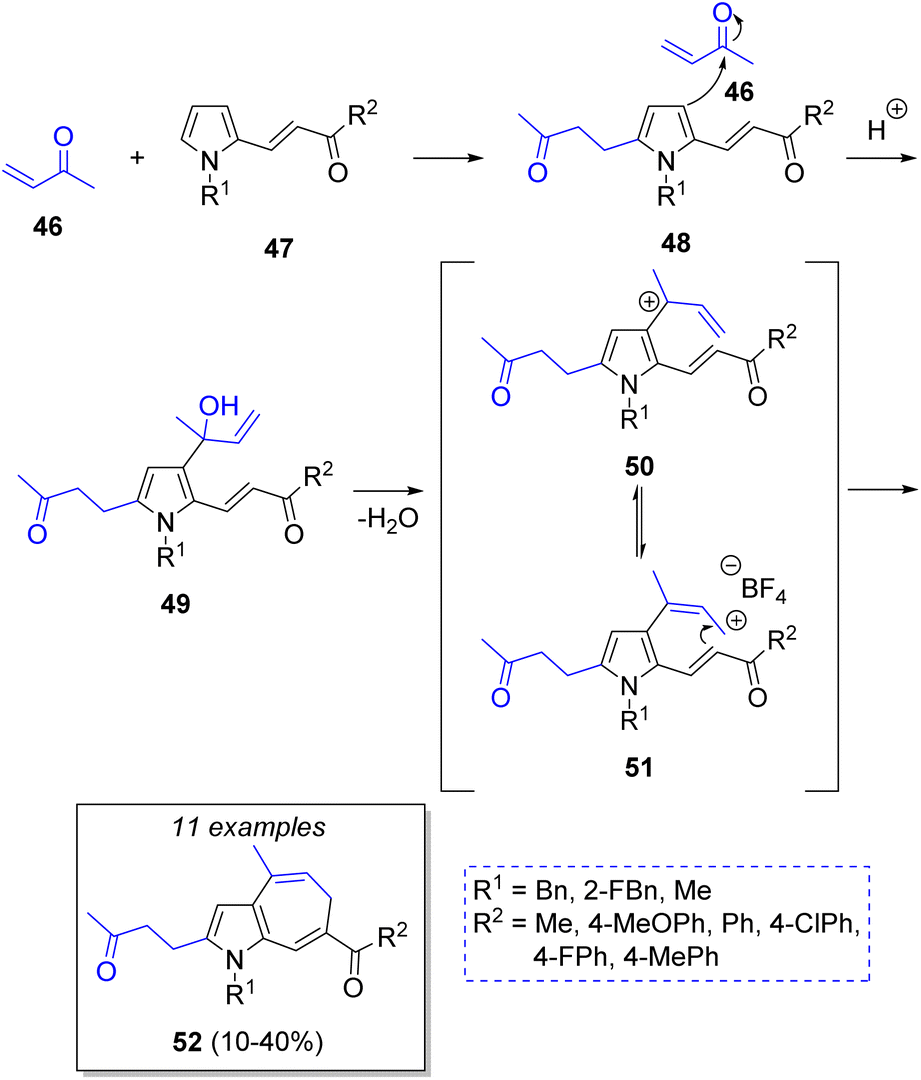 | ||
| Scheme 6 Synthesis of 1,6-dihydroazaazulenes via a pseudo-3CR. | ||
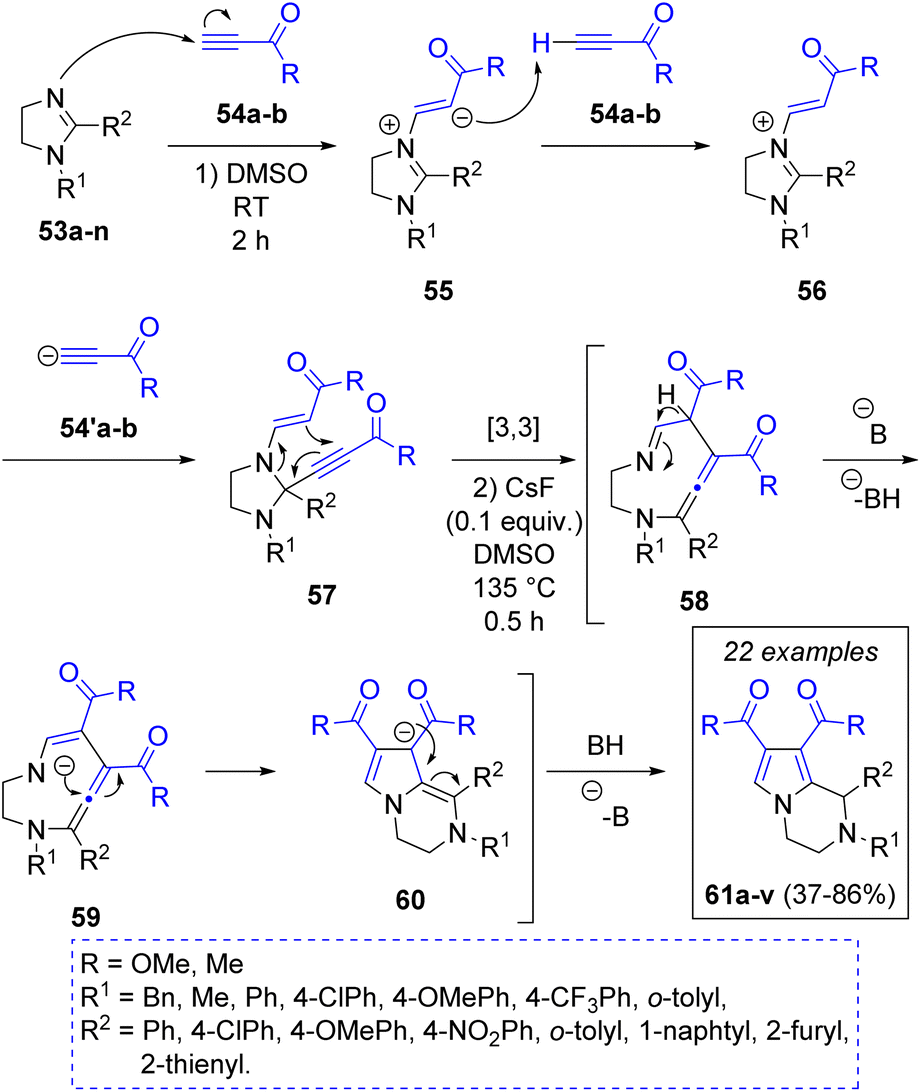
In 2022 N. E. Golantsov and L. V. Voskressensky26 reported a base catalyzed one-pot synthesis of 1,2,3,4-tetrahydropyrrolo[1,2-a]pyrazines via a pseudo three-component reaction between imidazolines 53a–n and two equivalents of electron-deficient terminal alkynes (methyl propiolate 54a or acetylacetylene 54b), obtaining the products 61a–v in moderate to excellent yields. To explain the transformation of imidazolines to pyrazines, the authors proposed a reaction mechanism (Scheme 7). The first step is a Michael addition of imidazolines to one molecule of terminal alkynes to form the zwitterions 55, which in turn deprotonate the second molecule of alkyne, producing the anions 54′a–b, which attack position 2 of the imidazolium ions 56 producing the key adducts 57. Under heating conditions, this adduct undergoes a [3,3]-sigmatropic rearrangement leading to the formation of a 9-membered intermediates 58, which are deprotonated by the base at their imino α-position, producing 59, which prompt a transannular cyclization leading to the formation of the target 1,2,3,4-tetrahydropyrrolo[1,2-a]pyrazines 61a–v after protonation of 60.
 | ||
| Scheme 7 Synthesis of 1,2,3,4-tetrahydropyrrolo[1,2-a]pyrazines. | ||
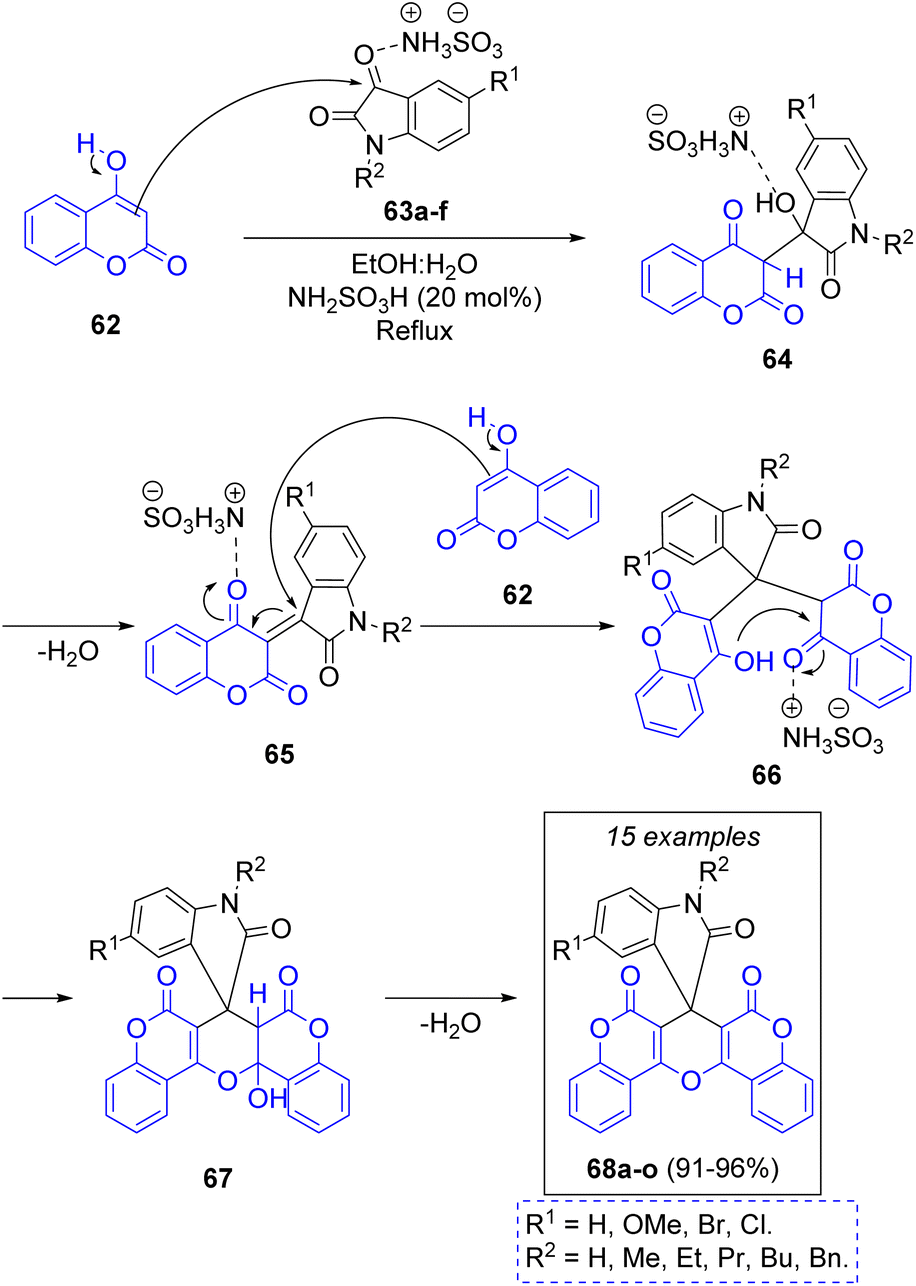
In 2022 C. Mukhopadhyay and co-workers reported the synthesis of a library of substituted spirooxindolopyrans via a green one-pot pseudo-3CR between substituted isatins 63a–f and two equivalents of 4-hydroxycoumarin (62) using a mixture of ethanol and water as solvent system, and sulfamic acid as catalyst. To explain the formation of the target products, the authors propose that the first step of the mechanism (Scheme 8) is the activation of the isatin's C-3 by sulfamic acid, prompting a nucleophilic addition of coumarin to form the intermediates 64, which after loss of a water molecule produces the key intermediates 65. These ones are attacked by other coumarin molecules to form 66, which undergo an intramolecular ring closure to generate the cycloadducts 67. Elimination of another water molecule generates the target compounds 68a–o. This remarkable methodology produced a compound library in excellent yields, and to further demonstrate the robustness of their methodology, the authors performed the synthesis of a single product in a multigram scale, obtaining the target molecule in an excellent yield of 94%.27
 | ||
| Scheme 8 Synthesis of spirooxindolopyrans. | ||
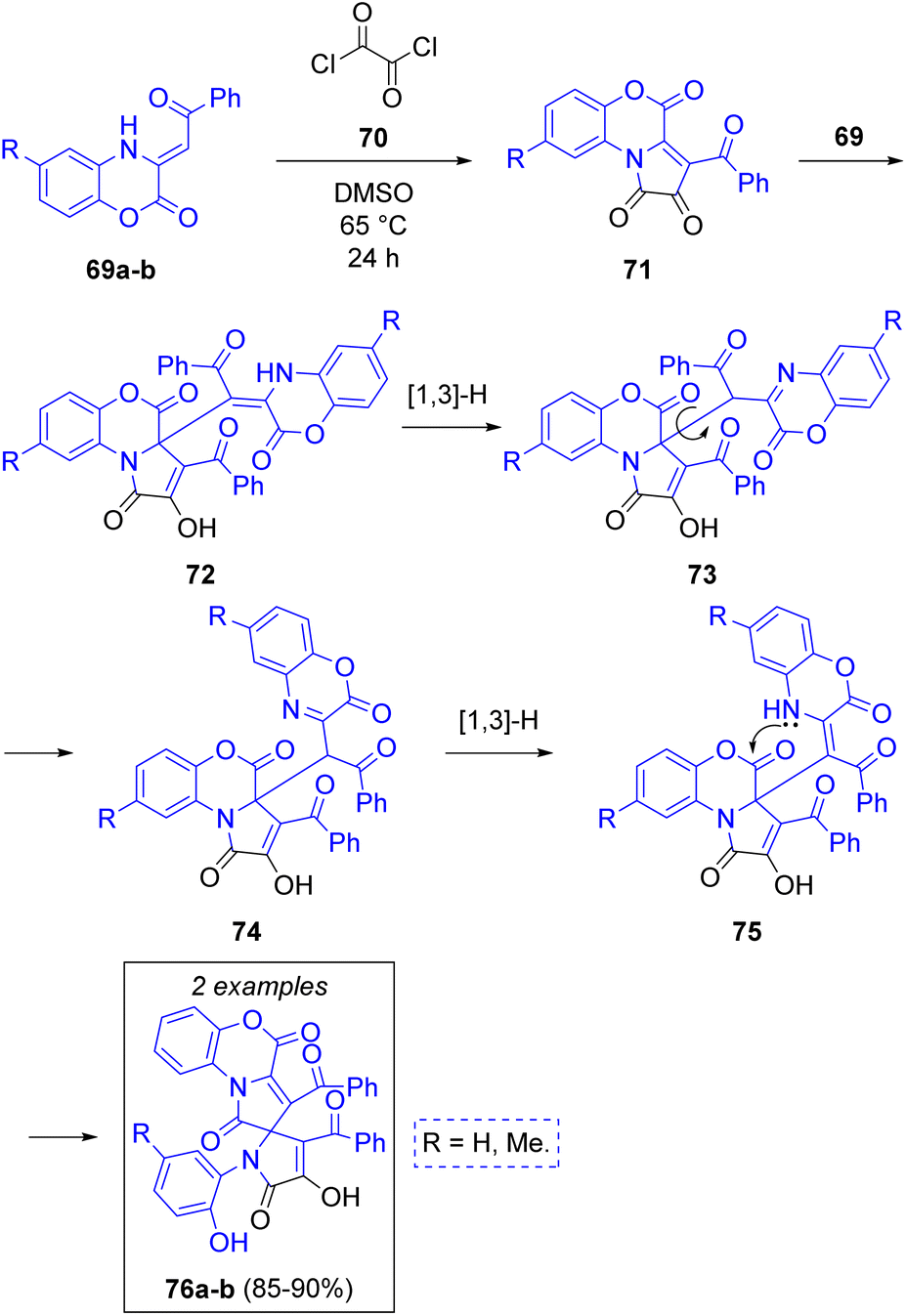
A. N. Maslivets and co-workers28 reported the synthesis of fused pyrrolobenzoxazines via a pseudo-3CR. The first step is a reaction between one equivalent of 3-benzoylmethylidene-3,4-dihydro-2H-1,4-benzoxazin-2-ones 69a–b with oxalyl chloride (70) to produce 3-aroylpyrrolo[2,1-c][1,4]benzoxazine-1,2,4-triones (71), which are attacked by another molecule of 69 to produce the intermediates 72. Next, tautomerization of 72 via [1,3]-H shift followed by rotation of the recently added benzoxazine fragment (73) to a less hindered position (74) and another [1,3]-H shift allows for an intramolecular ring closure via lactamization (75) with subsequent cleavage of the lactone rings to produce the spirocyclic target compounds 76a–b in excellent yield (Scheme 9).
 | ||
| Scheme 9 Synthesis of 3′-dibenzoyl-4-hydroxy-1-(2-hydroxyaryl)-1′H,4′H-spiro[pyrrole-2,2′-pyrrolo[2,1-c][1,4]benzoxazine]-1′,4′,5(1H)-triones. | ||
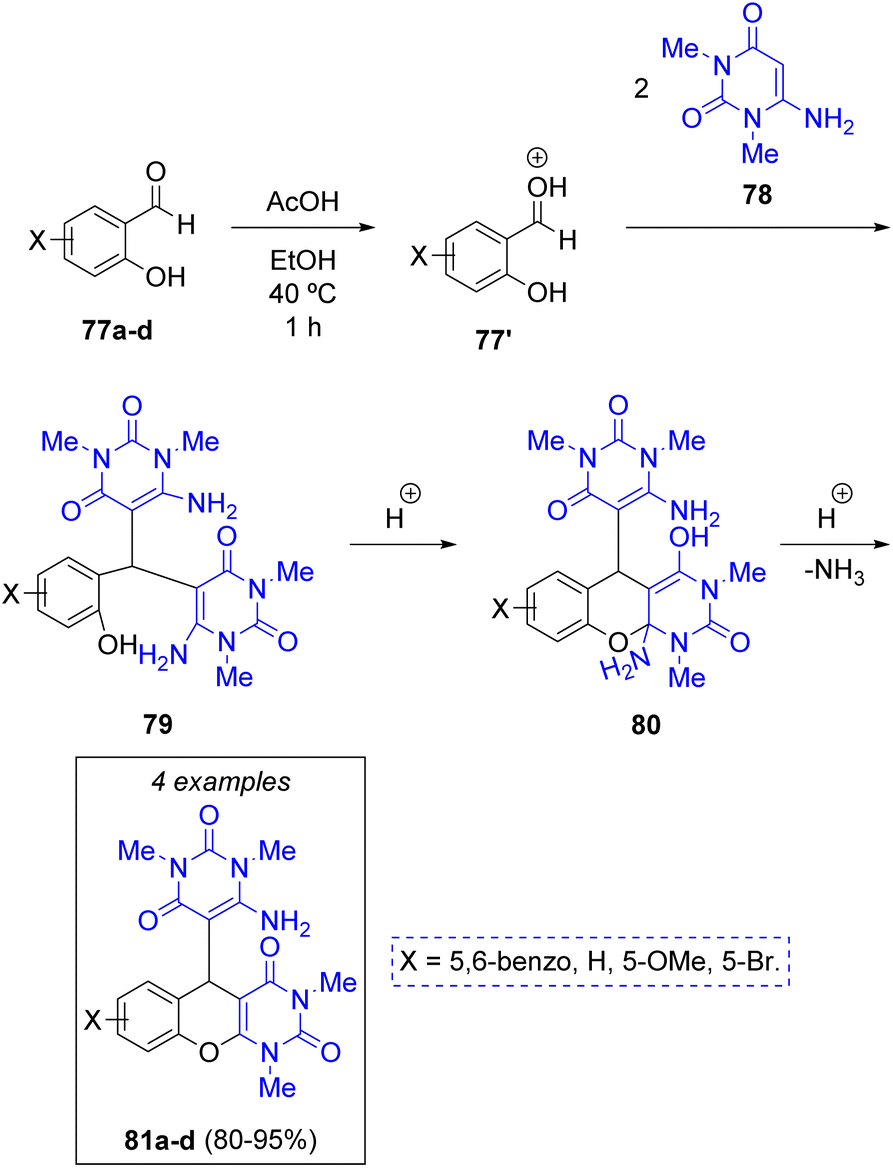
F. Farzaneh and co-workers29 synthesized four new fused pyranopyrimidinediones with antibacterial activity via a green, acetic acid catalyzed pseudo-3CR between substituted salicylaldehydes 77a–d and two equivalents of 6-amino-1,3-dimethyluracil (78), a derivative of the nucleobase uracil. The authors suggest that the first step towards the formation of the target molecule is a protonation of salicylaldehydes 77 by acetic acid, followed by a double condensation with the uracil derivatives to produce intermediates 79. Next, an intramolecular cyclization via Michael addition of the hydroxy group from salicylaldehyde unto the enamine moiety generates intermediates 80, which upon elimination of an ammonia molecule assisted by the acid catalyst, furnished the target products 81a–d in excellent yields (Scheme 10).
 | ||
| Scheme 10 Synthesis of fused pyranopyrimidinediones. | ||
3. Pseudo-four component reactions
3.1 Knoevenagel condensation/Michael addition
Knoevenagel condensation/Michael addition is also a common strategy used in pseudo-4CRs. For instance, Elinson and co-workers30 achieved a feasible and stereoselective synthesis of a spiroindole-3,1′-naphthalene tetracyclic system using the isatins 82, cyclic ketones 83a–h, two equivalents of malononitrile (84) and triethylamine as catalyst. The proposed reaction mechanism is illustrated in the Scheme 11. Initially, a Knoevenagel condensation takes place between isatins 82 and malononitrile (84) to generate the intermediates 85. Simultaneously, another malononitrile molecule (84) reacts with the cyclic ketones 83 to afford 86. Then, intermediates 86 are deprotonated by triethylamine, and then react with isatilidenemalononitriles 85 to afford 87. An intramolecular cyclization produces the anions 88 and finally, a proton abstraction from another malononitrile molecule provides the final products 90a–p in good to excellent yields.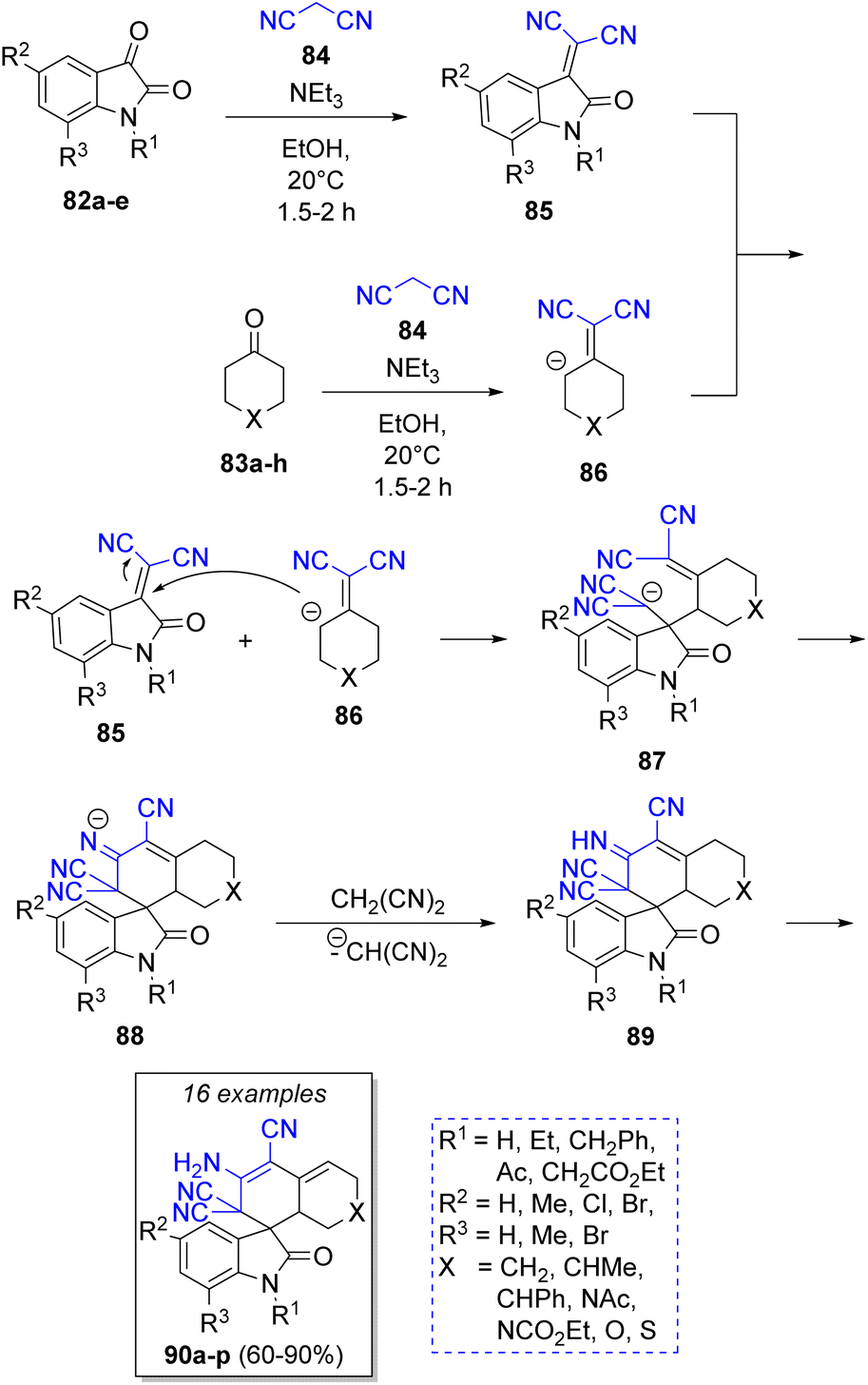 | ||
| Scheme 11 Synthesis of spiroindole-3,1′-naphthalene tetracyclic systems. | ||
Abaee and co-workers31 reported the synthesis of a collection of dicyanoanilines fused to a dithiane ring via a facile and efficient reaction among aldehydes 91a–j, 1,3-dithia-5-one (92), two equivalents of malononitrile (93) and triethylamine as catalyst. The proposed mechanism is depicted in Scheme 12. First, two parallel Knoevenagel condensations take place between aldehyde 91 and 92 with a molecule of malononitrile 93, respectively, to form the corresponding intermediate olefins 94 and 95 under basic conditions. Both intermediates then react via a Michael addition to afford the tetracyano intermediates 96. An intra-molecular cyclization forms intermediates 97. The aromatic final products 99a–j are obtained after a double bond rearrangement and the loss of one HCN molecule from 98. Diverse compounds such as 4-[2-(dicyanomethylene)cyclic or heterocyclic]-2-amino-4H-chromenes 100a–f32 dicyanoanilines,33 2-amino-4-(aryl)-5,6,7,8,9,10-hexahydro-benzo[a]cyclo-octene-1,3-dicarbonitriles,34 hexahydrobenzo[8]-annulene35 have been prepared using similar methodologies.
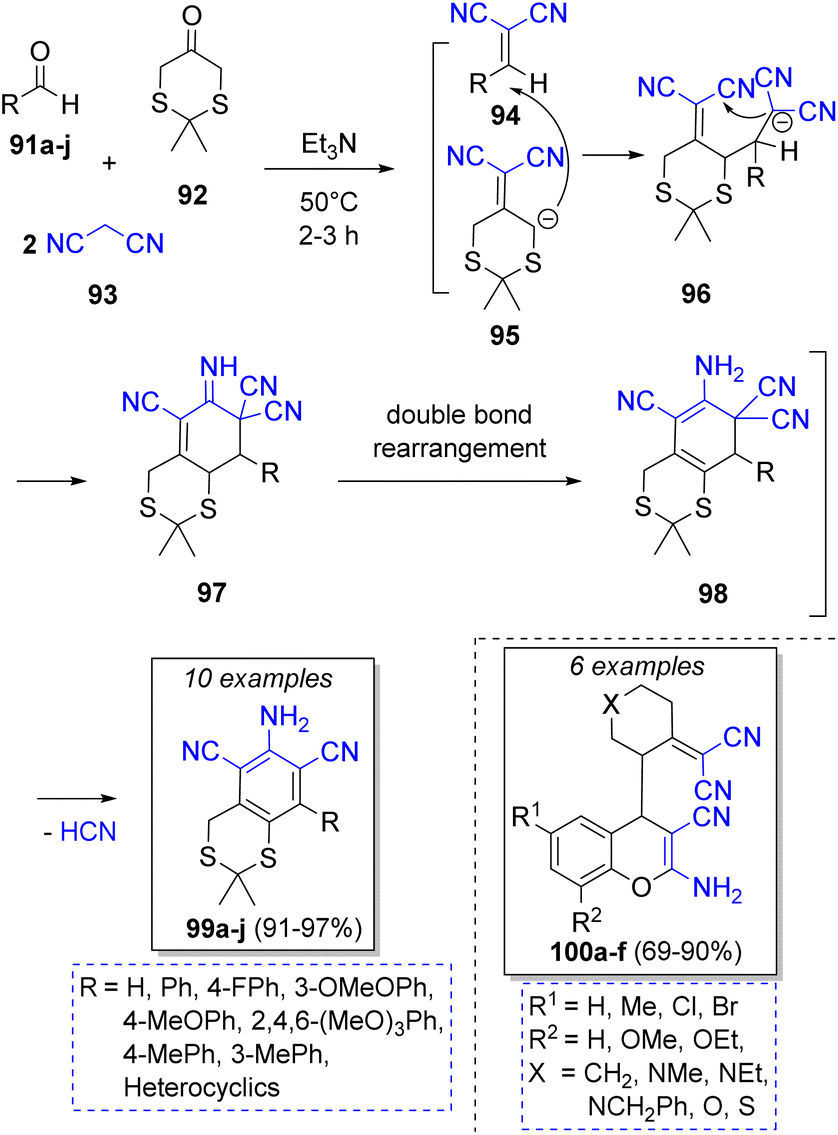 | ||
| Scheme 12 Synthesis of dicyanoanilines fused to heterocyclic dithiane ring. | ||
Ryzhkov and co-workers36 reported a pseudo-4CR approach for the synthesis of 5H-chromeno[2,3-b]pyridines via a Knoevenagel condensation/Pinner reaction/Michael addition/Pinner reaction strategy. The suggested mechanism is illustrated in the Scheme 13a. Initially, malononitrile anion (102′) (previously deprotonated by pyridine) reacts with salicylaldehydes 101 via a Knoevenagel condensation to form 103, followed by an intramolecular Pinner cyclization to form 105. Then, intermediates 105 react with 5-methyl-2,4-dihydro-3H-pyrazol-3-one anions 106 via Michael addition to produce 107. The regeneration of malononitrile anion produces the compounds 108. Then, the nitrile group of 108 is attacked by the malononitrile anion to form anions 109, which are better stabilized in 110. An intramolecular Pinner cyclization produces 111, which after tautomerization and protonation produce the final products 113a–k. A similar strategy was employed by Vereshchagin and co-workers.37 The synthesis of 5-C-substituted 2,4-diamino-5H-chromeno[2,3-b]pyridine-3-carbonitriles 114a–l was accomplished by the reaction of salicylaldehydes, two equivalents of malononitrile, 1,3-cyclohexanediones and triethylamine as catalyst (Scheme 13b).
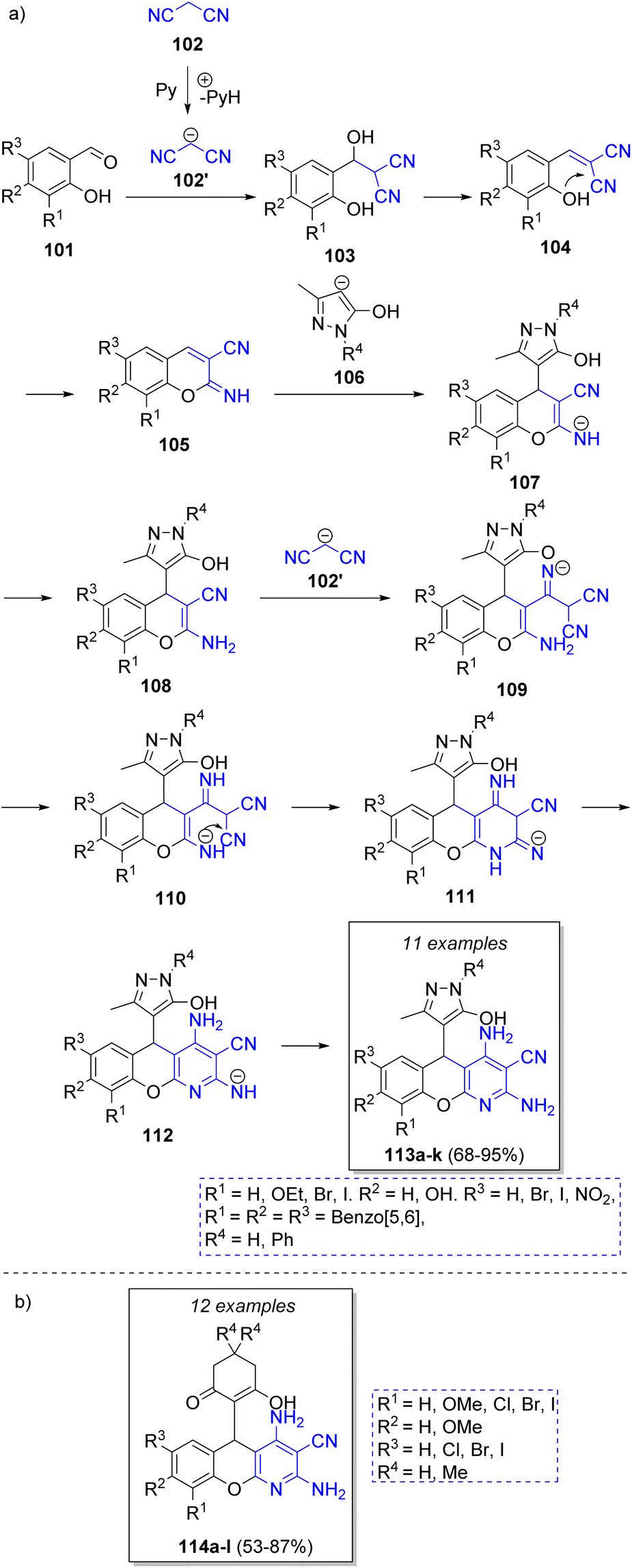 | ||
| Scheme 13 (a) Synthesis of 5H-chromeno[2,3-b]pyridines via pseudo-4CR. (b) Chromeno-pyridines synthesized via a pseudo-4CR. | ||
3.2 Others
An efficient method for the synthesis of benzopyranpyri-midines was developed by Bazgir and co-workers38 in 2013 via a Knoevenagel condensation/Pinner reaction, using two equivalents of salicylaldehydes 115a–d, malononitrile (116) and a variety of amines 119a–c with zirconyl chloride (ZrOCl2·8H2O) as catalyst. The proposed reaction mechanism is depicted in Scheme 14. A Knoevenagel condensation between aldehydes 115 and malononitrile (116) forms 117, followed by a Pinner reaction to form intermediates 118. Then, amines 119 react with intermediates 118 to form diimines 120. These intermediates react with another molecule of 115 to form 121, which form the final products 122a–j after a 1,5-hydrogen shift. The authors have also explored the use of lithium perchlorate as catalyst, obtaining similar yields.39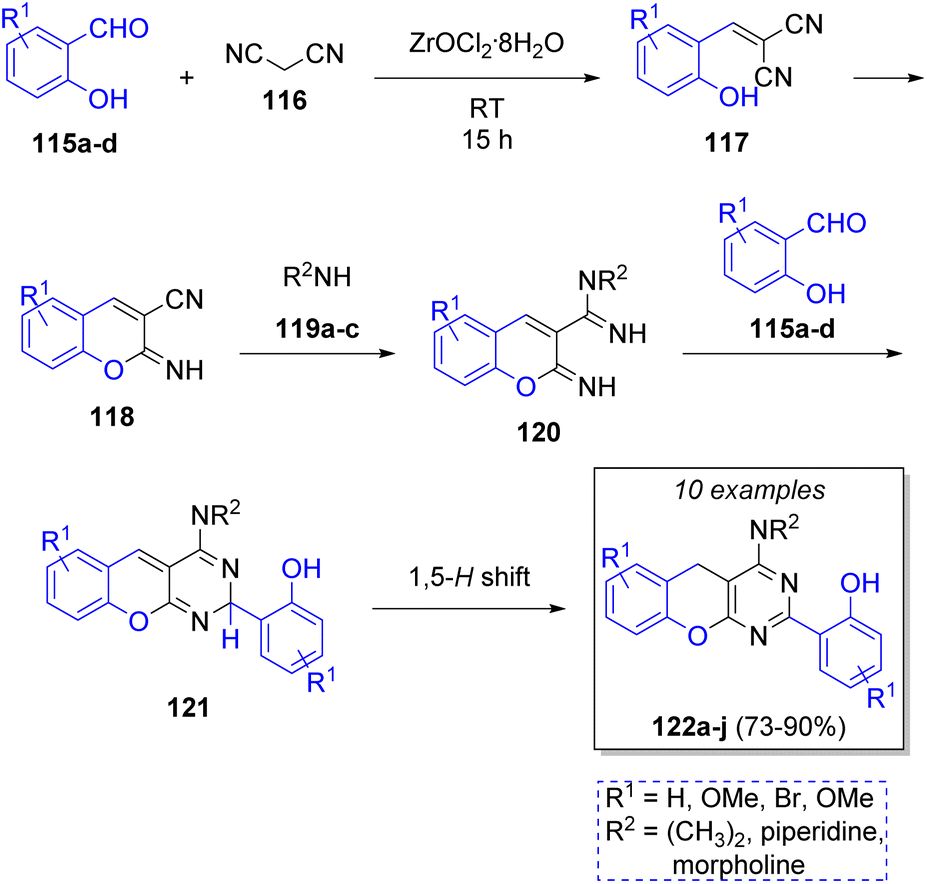 | ||
| Scheme 14 ZrOCl2-catalyzed synthesis of fused benzopyranopymidines. | ||
In 2017, Li and co-workers40 reported a highly efficient method to obtain 2,4,6-triphenylpyridines using aromatic aldehydes 123, two equivalents of aromatic ketones 124, ammonium acetate 125 and cerium(IV)carboxymethylcellulose (CMC-CeIV) as catalyst. The proposed catalytic cycle for this transformation is illustrated in Scheme 15a. Initially, CMC-CeIV promotes the formation of the enol tautomers of the complex 126, which react with 128 via a nucleophilic addition to form the α,β-unsaturated compounds 129. These intermediates perform a Michael addition with a second molecule of enol 127 to form the 1,5-diketone intermediates 130, which then react with ammonium acetate (125) to form intermediates 131, followed by an intramolecular cyclization promoted by CMC-CeIV, and tautomerization to form the dihydropyridines 133. Upon air oxidation of the last intermediates, the formation of the desired products 134a–t is completed. Another collection of 2,4,6-triarylpyridines 135a–p was prepared in the presence of salicylic acid as catalyst (Scheme 15b).41
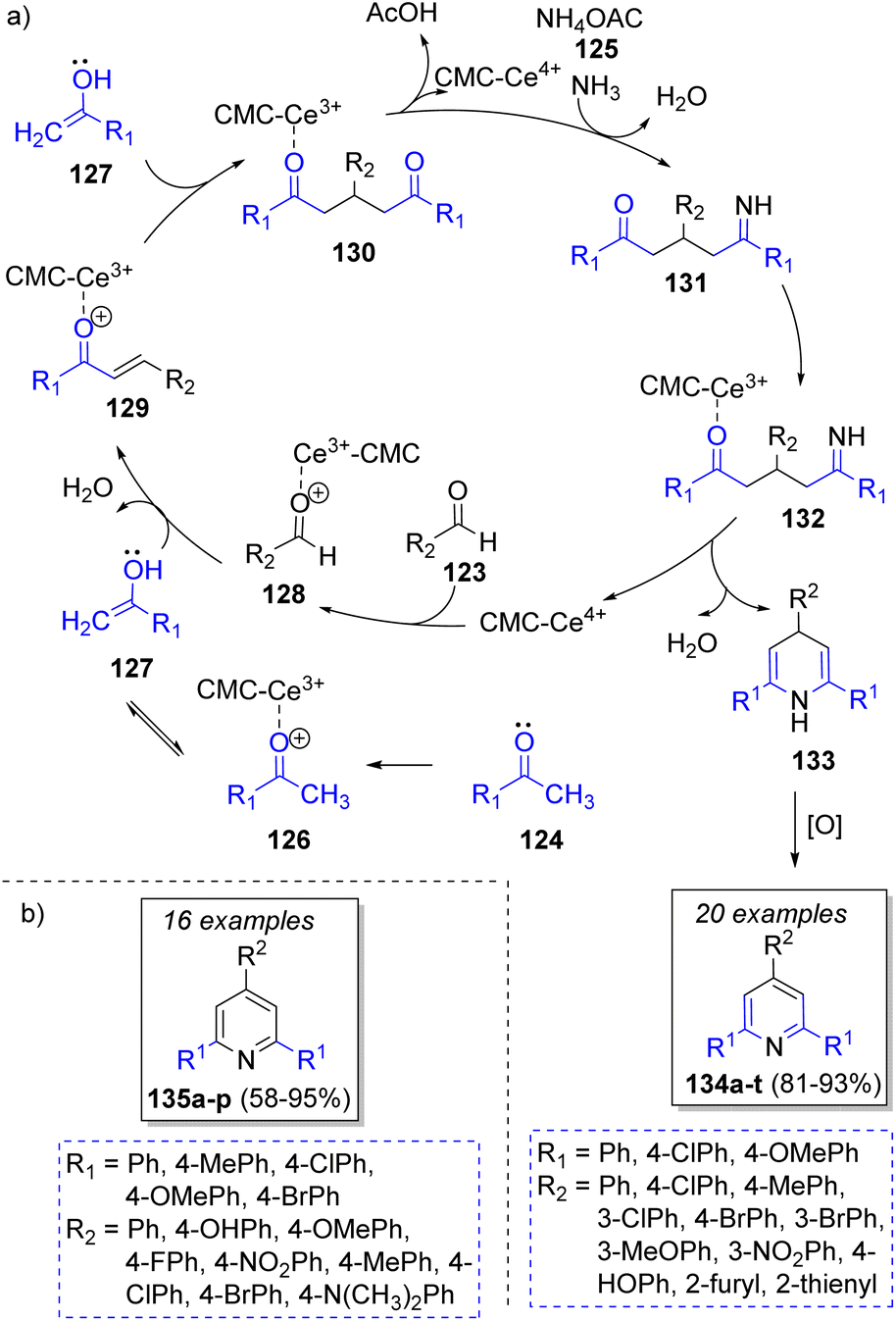 | ||
| Scheme 15 (a) Reaction mechanism for the synthesis of 2,4,6-triphenylpyridines. (b) Synthesized compounds via a pseudo-4CR. | ||
In 2004, Shaabani and co-workers42 performed a pseudo-4CR-based method to obtain some highly functionalized pyrroles from commonly available precursors. The reaction involved two equivalents of unhindered isocyanides 136a–b, one equivalent of dialkyl acetylenedicarboxylates 137a–b and one equivalent of five-membered cyclic imides (i.e. succinimide or maleimide) 142a–b to give 1′-alkyl, aryl-5′alkyl or arylamino-2,5-dioxo-2,3,4,5-tetrahydro- or 2,5-dihydro-1′H-[1,2′]bipyrrole-3′,4′-dicarboxylic acid dialkyl esters 144a–f. The suggested reaction mechanism is illustrated in Scheme 16. First, the intermediates 138 are generated by the reaction between isocyanides 136 and alkynes 137. Then, the addition of another molecule of isocyanides 136 produces the bis-ketenimine intermediates 141, which react with 142 to afford 143. Finally, an intramolecular cyclization forms the products 144a–f in moderate yields.
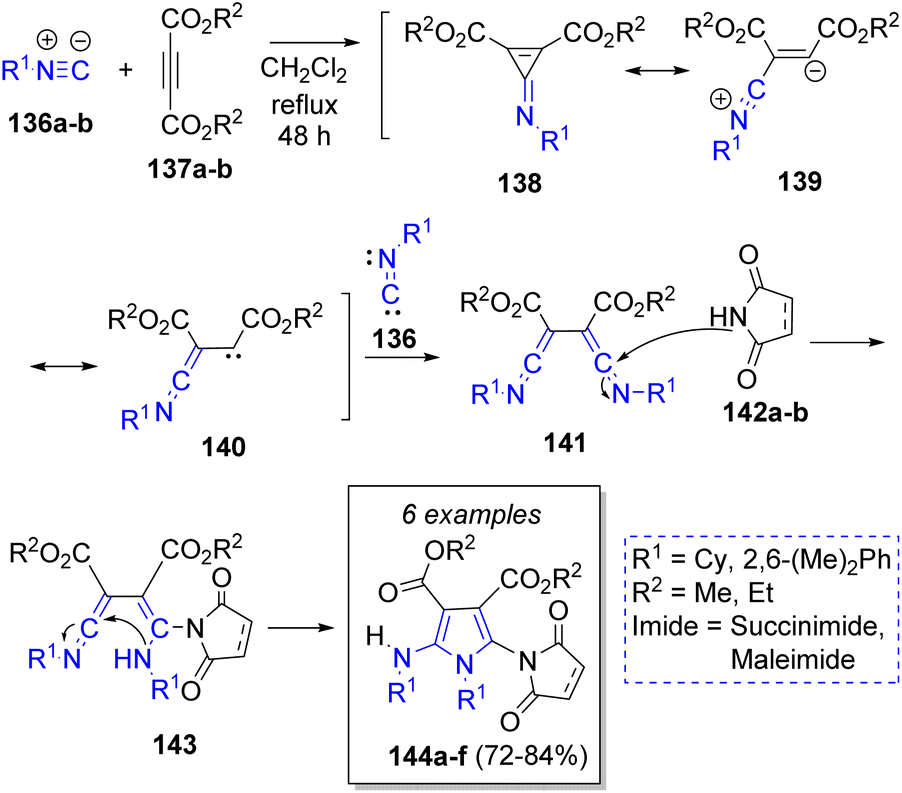 | ||
| Scheme 16 Synthesis of penta-substituted pyrroles. | ||
In the same year, Shaabani and co-workers43 also described a regioselective Biginelli-like pseudo-MCR of σ-symmetric spiro heterobicyclic rings extended to para-substituted aldehydes, including two equivalents of aldehydes 145a–d (para-substituted benzaldehydes with EWGs), an equivalent of urea (146), and a cyclic β-diester or β-diamides (Meldrum's acid or barbituric acid) 147. According to the authors, two probable reaction pathways were proposed, as illustrated in Scheme 17. The first starts with the nucleophilic addition of urea 146 to aldehydes 145 to afford the N-acylimines 148. Then, Meldrum's acids 147 react with 148 via Michael addition to afford 150. Then, addition of a second molecule of aldehydes 145 provided the intermediates 151, which after a cyclization gives the final products 152a–l. In the second pathway, aldehydes 145 react with cyclic β-diester or β-diamides 147 via Knoevenagel condensation to afford the intermediates 149, a Michael addition of urea 146 gives the intermediates 150, which react with another molecule of aldehydes 145 to afford 151. A final cyclization also provides the spiro heterobicyclic products 152a–l.
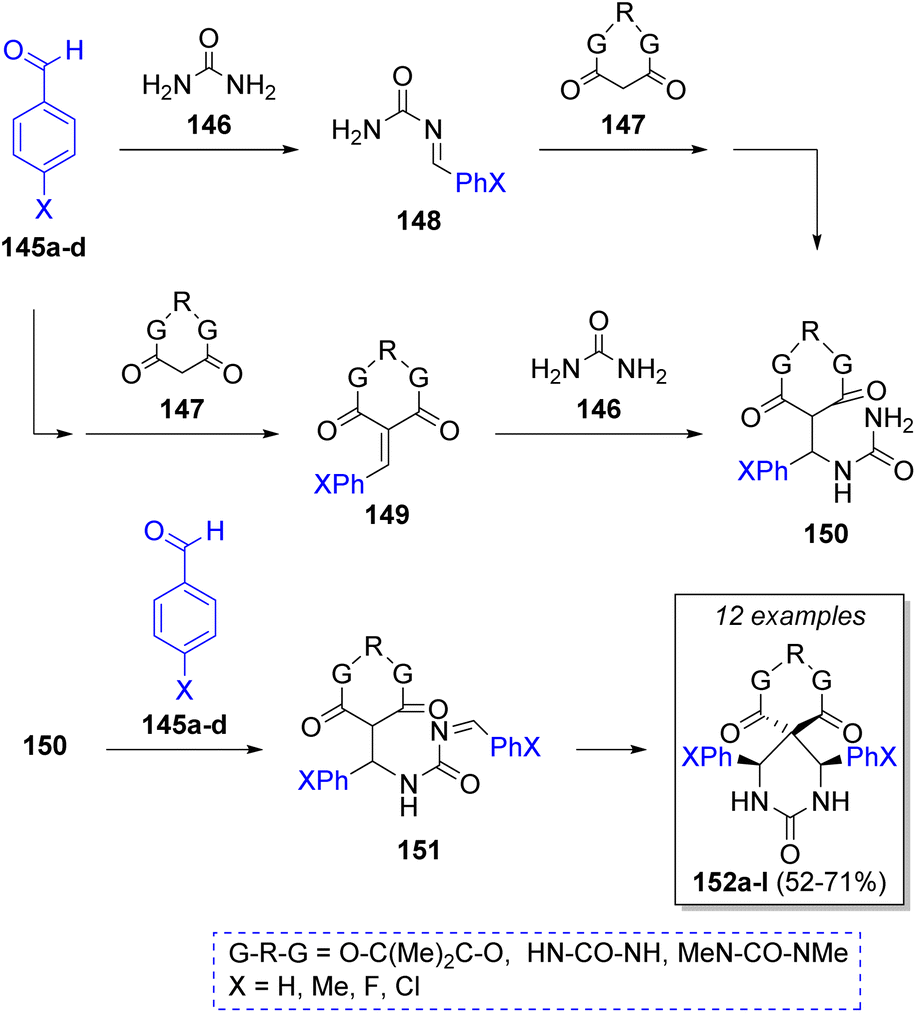 | ||
| Scheme 17 Synthesis of spiro-heterobicyclic rings. | ||
The synthesis of some dienes was described by Nair and co-workers44 in 2005. The synthetic approach included the use of one equivalent of the N-heterocyclic carbene 1,3-di-tert-butylimidazole-2-ylidenes 153, two equivalents of dimethyl acetylenedicarboxylates (DMAD, 154) and one equivalent of aromatic aldehydes 156a–g. The reaction mechanism was suggested as illustrated in Scheme 18. At first, the dipolar intermediates 155 are produced from the reaction between the carbenes 153 and DMAD 154, which then react with the aldehydes 156 to form the alkoxide intermediates 157. Then, the addition of another molecule of DMAD 154 lead to the zwitterionic intermediates 158. A proton abstraction provides the final products 159a–g.
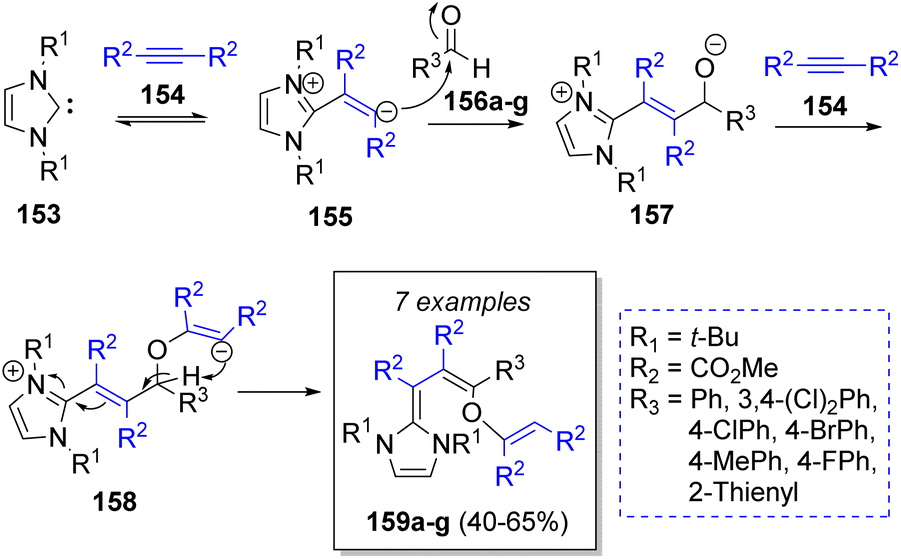 | ||
| Scheme 18 Synthesis of diene derivatives using N-heterocyclic carbenes. | ||
In 2008, Shaabani and co-workers45 described an efficient pseudo MCR condensation to afford 1-aminoimidazol[5,1-a]isoquinolinium salts, which involved the reaction of one equivalent of isoquinoline (160), two equivalents of isocyanides 162a–e and sulfonic 161a–d or bromic acids. The suggested mechanism is illustrated in Scheme 19. Initially, isoquinoline (160) is protonated by sulfonic acids 161, which then reacts with isocyanides 162a–e to form intermediates 163, which are trapped by second isocyanide molecules 162a–e to form 164. A hydrogen shift followed by intramolecular cyclization affords the intermediates 166. The reaction is completed after tautomerization of 166 to form the ionic products 167a–i.
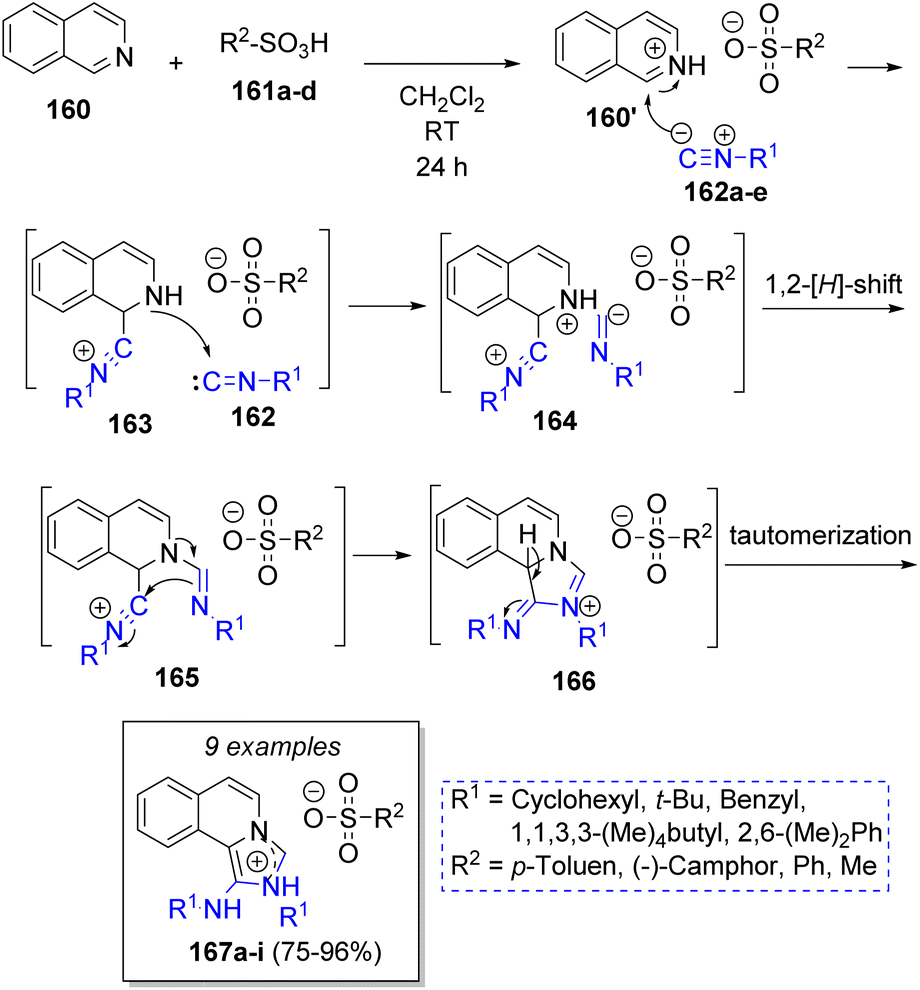 | ||
| Scheme 19 Synthesis of 1-aminoimidazol[5,1-a]isoquinolinium salts. | ||
Shaabani and co-workers46 also developed a novel good to high yielding technique for the synthesis of tetrahydrodiisoindolo-quinoxalines and tetrahydrobenzodiisoindolo-quinoxalines via the reaction between aromatic 1,2-diamines 168a–g, 2-formylbenzoic acid 169, and isocyanides 175a–d. The proposed mechanism is showed in Scheme 20. First, diimine intermediates 170 are formed by the condensation between 168a–g and two molecules of 169. Then, 174 is formed by 4n + 2 electrocyclic reaction followed by a [1,5] hydrogen shift and release of a water molecule via lactamization. Then, nucleophilic addition of isocyanides 175a–d to the imine, followed by release of another water molecule gives rise to the second indole ring in 177. The addition of one of the released water molecules unto the nitrilium ions 177 forms intermediates 178. Finally, a tautomerization affords either 179a–j or 180a–c.
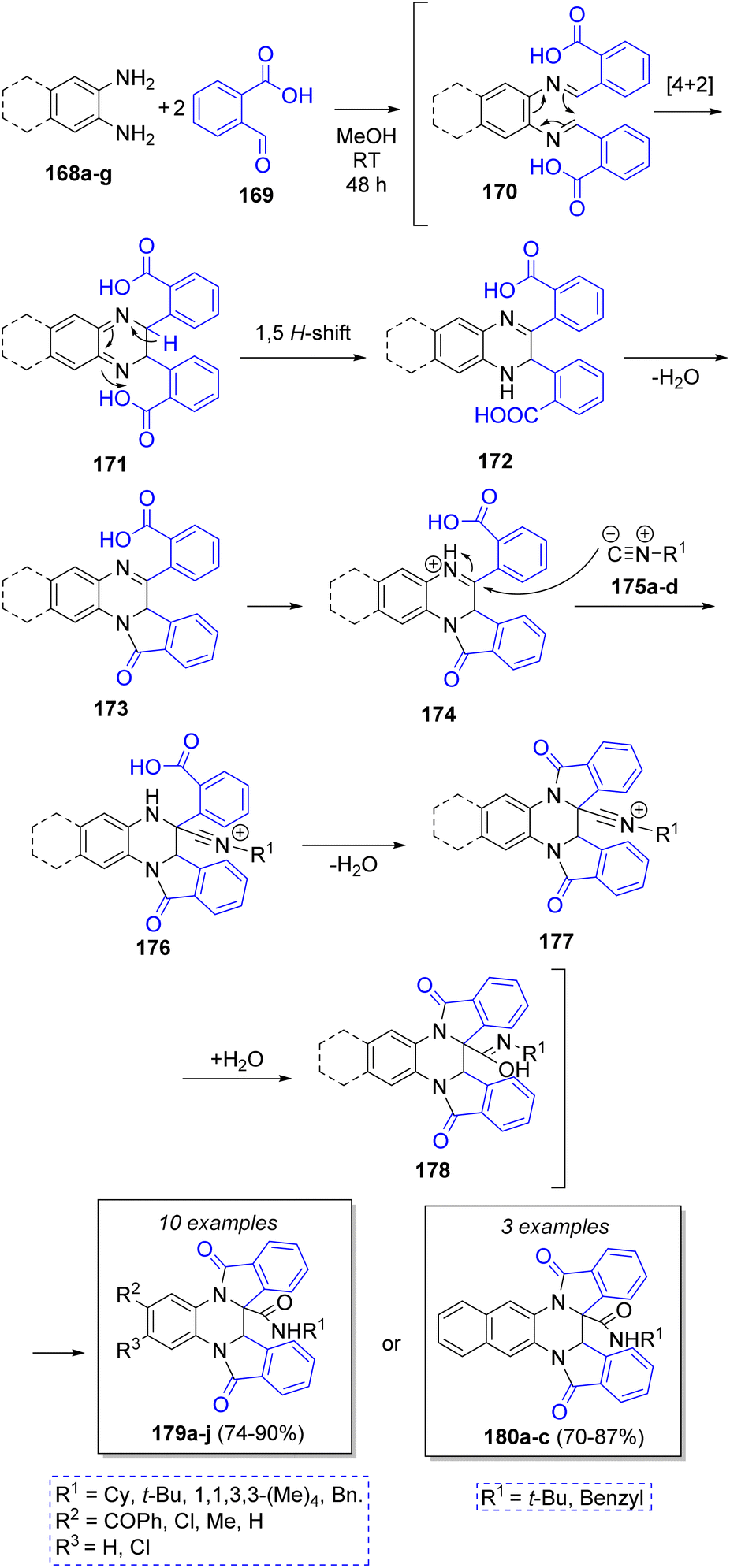 | ||
| Scheme 20 Synthesis of tetrahydrodiisoindoloquinoxalines and tetrahydrobenzodiisoindoloquinoxalines. | ||
A series of 4-amino-3,5-dicyano-6-arylphthalates catalyzed by triethylamine were successfully synthetized by Mohammadi and co-workers47 in 2013. The synthesis was achieved in satisfactory yields from the reaction of different benzaldehydes 181a–d, two equivalents of malononitrile (182), and dialkyl acetylenedicarboxylates 184a–c, in the presence of triethylamine as catalyst. A suggested mechanism is illustrated in Scheme 21. The reaction plausibly starts with a Michael addition of triethylamine to acetylenic esters 184a–c to form zwitterions 185, which react with the previously in situ formed arylidenemalononitriles 183 to afford the adducts 186, which after a [1,3]-proton shift the new zwitterions 187 are formed. Elimination of Et3N affords the electro-deficient 1,3-butadienes 188, which are attacked by another molecule of malononitrile 182 to give the intermediates 189, which after an intramolecular cyclization afford 190. The desired products 193a–j are formed after a [1,3]-proton shift (191), followed by HCN elimination (192), and finally by an imine isomerization.
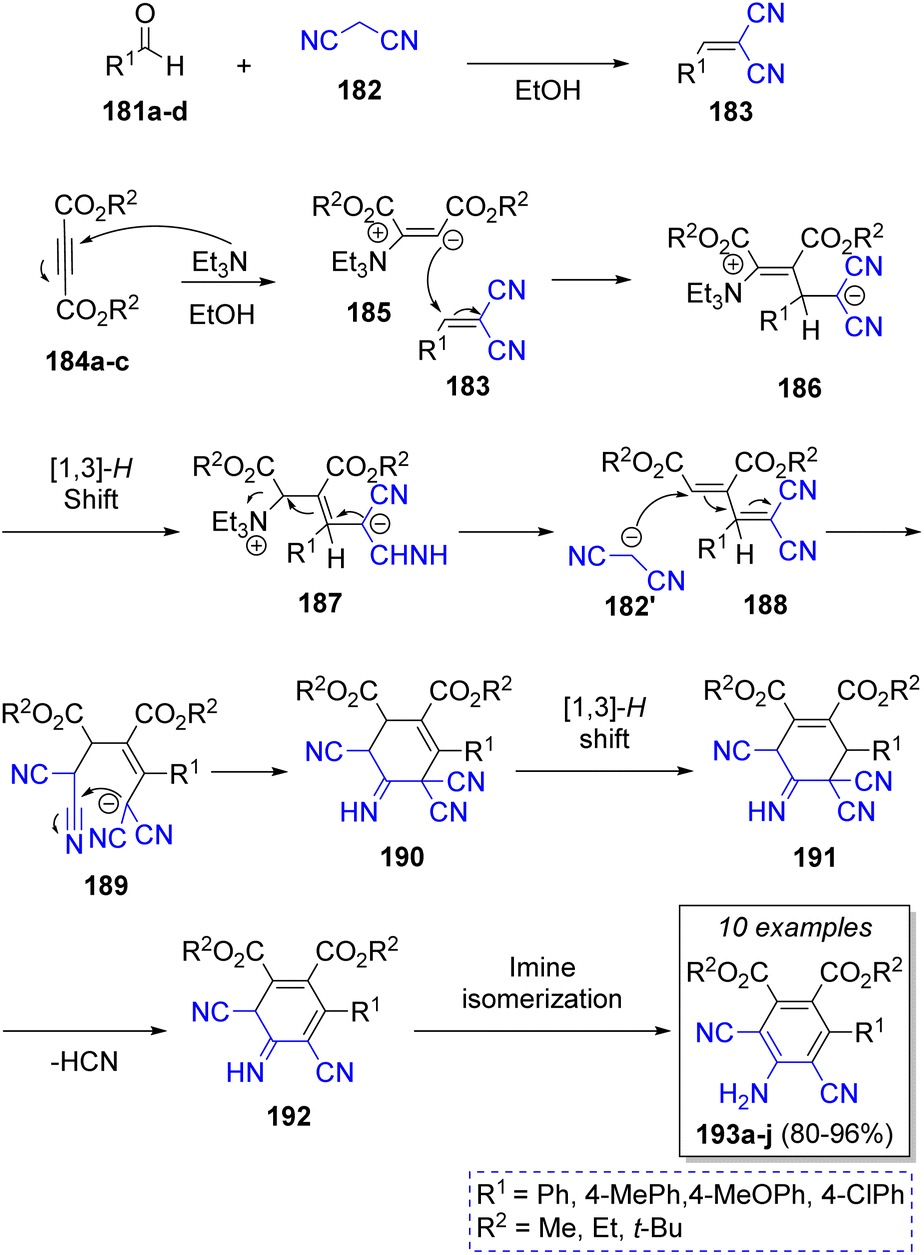 | ||
| Scheme 21 Synthesis of 4-amino-3,5-dicyano-6-arylphthalates. | ||
Khan48 reported a regioselective Yb(OTf)3-catalyzed synthesis of tri-substituted 3,4-dihydrothiochromeno[3,2-e][1,3]thiazin-5(2H)-ones via reaction between two equivalents of aldehydes 194a–k, 4-hydroxydithiocoumarin (195), and aliphatic primary amines 197a–c. A plausible mechanism for this reaction was suggested as illustrated in Scheme 22. Accordingly, adducts 194′ are formed by the reaction of aldehydes 194 and ytterbium triflate, which then react with hydroxydithiocoumarin 195 to produce the compounds 196 via a Knoevenagel condensation. Simultaneously, the other adducts 194′ react with the amines 197 to form imines 198. Furthermore, imines 198 form dienophiles 198′ when activated by ytterbium triflate. Finally, hetero-Diels–Alder cyclization between dienes 196 and dienophiles 198′ gives the desired products 199a–o.
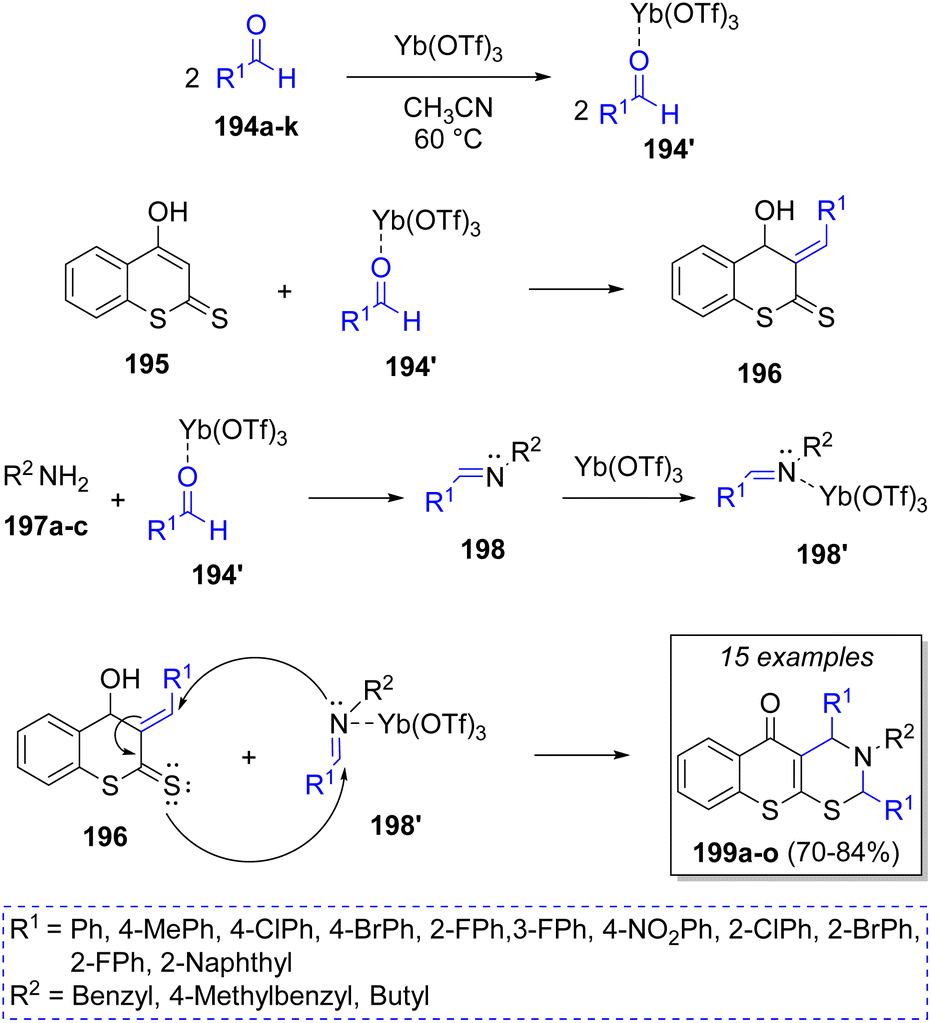 | ||
| Scheme 22 Synthesis of di- and tri-substituted 3,4-dihydrothiochromeno[3,2-e][1,3]thiazin-5(2H)-ones. | ||
Sanaeishoar49 in 2015 reported a sophisticated strategy to synthesize functionalized imidazo-[1,2-a]pyridines via the reaction of 2-aminopyridines 200a–b, aromatic aldehydes 201a–h and phenacyl bromides 204a–b, using DABCO (202) as catalyst under solvent-free conditions. The proposed reaction mechanism is depicted in the Scheme 23. First, the reaction between 2-aminopyridines 200 and aldehydes 201 in the presence of DABCO (202), produces the imine-type intermediates 203. A second molecule of 2-aminopyridines 200 react with phenacyl bromides 204 via SN2 to afford the intermediates 205, which undergo an intramolecular cycloaddition to form the 2-aryl-imidazo[1,2-a]pyridines 206. Finally, the products 207a–l are generated via a nucleophilic attack of intermediates 206 on the imine intermediates 203.
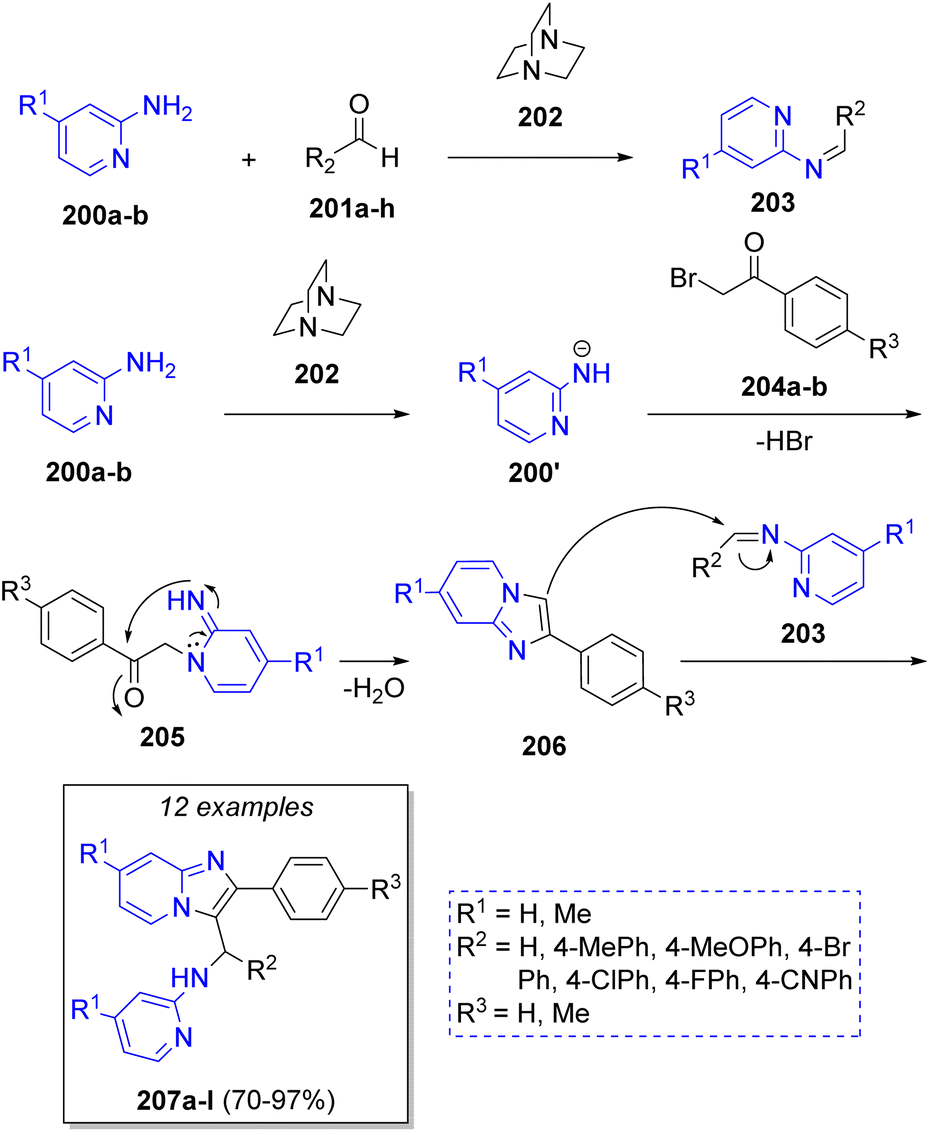 | ||
| Scheme 23 Synthesis of functionalized imidazo-[1,2-a]pyridines. | ||
D. S. Sharada50 published an environmentally friendly silica gel-promoted pseudo-4CR via a cascade process to synthesize first the α-aminoamidines 214 and then, an in situ iron(III) chloride-catalyzed cyclization towards the substituted 2H-indazoles 215a–j. The authors employed the 2-azidobenzaldehydes 208a–c, two equivalents of anilines 209a–g and alkyl isocyanides 211a–b to produce the corresponding α-aminoamidines in 68 to 82% overall yields. A plausible reaction mechanism for this transformation is proposed in the Scheme 24. Accordingly, the reaction between 208 and 209 produces imine intermediates 210. Then, the weakly Brønsted acidic nature of silica gel acted as activator of the imines 210 to promote the nucleophilic attack by the isocyanides 211 producing the intermediates 212, which are attacked by a second molecule of anilines 209, generating the intermediates 213. These compounds undergo an isomerization process affording the corresponding α-aminoamidines 214.
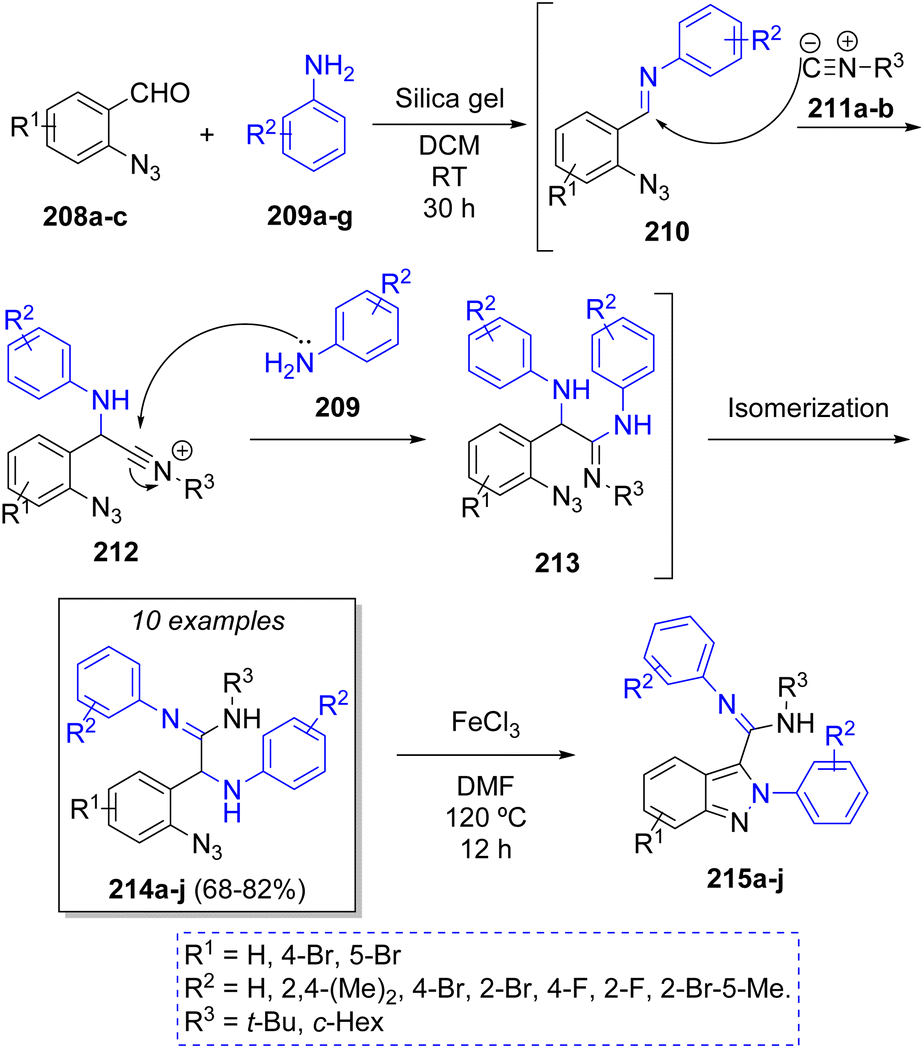 | ||
| Scheme 24 Synthesis of α-amino amidines and its cyclization towards 2H-indazoles. | ||
Sharma51 reported the synthesis of bis-amides via an organocatalytic oxidative pseudo-4CR. The reaction was carried out in a deep eutectic solvent (DES, choline chloride/urea mixture), which are considered as a greener alternative to organic solvents, and the oxidant (o-iodoxybenzoic acid) was soluble in such solvent system, which allowed the reaction to occur in homogeneous conditions. Thus, the authors utilized a variety of aryl and heterocyclic primary amines 216a–l, carboxylic acids 222a–h and alkyl isocyanides 223a–e (Scheme 25). The proposed reaction mechanism for the oxidative coupling of amines and the Ugi reaction begins when o-iodoxybenzoic acid 217 oxidizes the primary amines 216a–l generating the iminium ions 219 through the intermediates 218, after loss of water molecule. Then, 219 are hydrolyzed leading to the formation of aldehydes 220. The presence of benzoic acid catalyses the condensation between aldehydes 220 and other amine molecules 216a–l, producing the corresponding Schiff bases 221. These imines react with carboxylic acids 222a–h and isocyanides 223a–e affording the pseudo-Ugi-4CR products 224a–x.
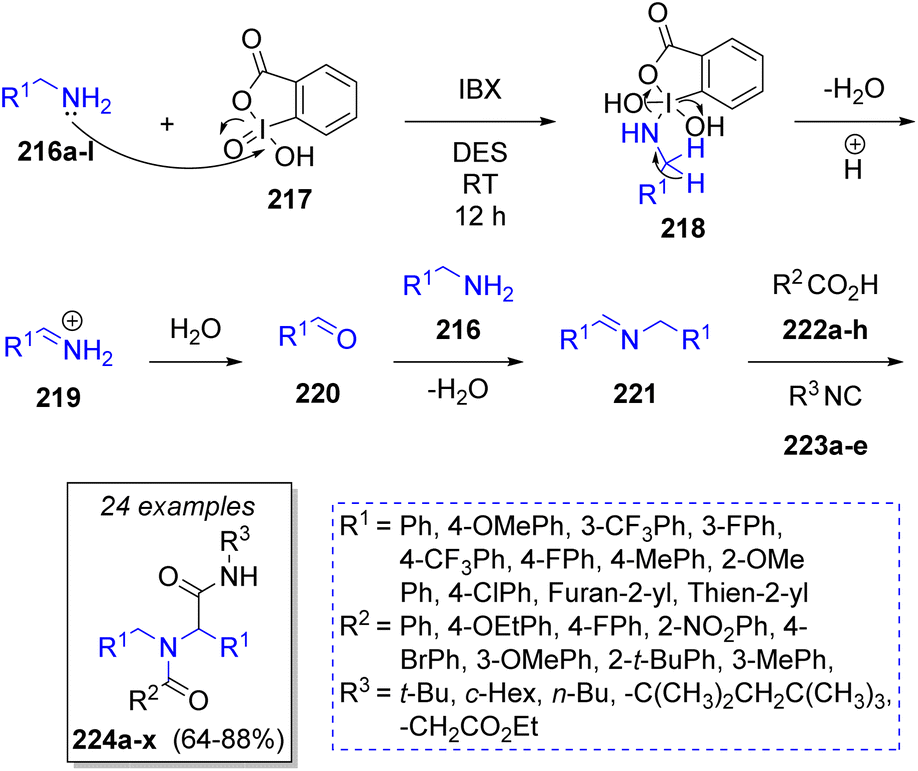 | ||
| Scheme 25 Synthesis of bis-amides via a hybrid oxidative pseudo-Ugi-4CR strategy. | ||
In 2018, Shaterian and Mohammadi52 reported a highly efficient visible light-induced pseudo-4CR synthesis of many new chromeno[4,3,2-de][1,6]naphthyridines by using two equivalents of malononitrile (225), aromatic aldehydes 226a–p and 2′-hydroxyacetephenones 230a–b. Scheme 26 illustrates the plausible reaction mechanism proposed by the authors. Initially, light irradiation induced malononitrile (225) to react with aldehydes 226 to form benzylidene malononitriles 227 followed by formation of radical intermediates 228. Then, these intermediates 228 induced the formation of malononitrile radical by abstracting a methylenic hydrogen to form the intermediates 229. The malononitrile radical 225′ reacts with enol intermediates 230′ to form 231. Later, intermediates 231 react with 229 to form 232, which upon tautomerization generates 233. Then, the elimination of malononitrile group produces chalcones 234. Intermediates 235 are formed via the reaction of chalcones 234 and a molecule of malononitrile (225), and after an intramolecular Pinner reaction 236 is formed. Subsequently, another molecule of malononitrile (225) reacts with 236 to form 237, further intramolecular cyclization and aromatization generates the desired products 238a–p.
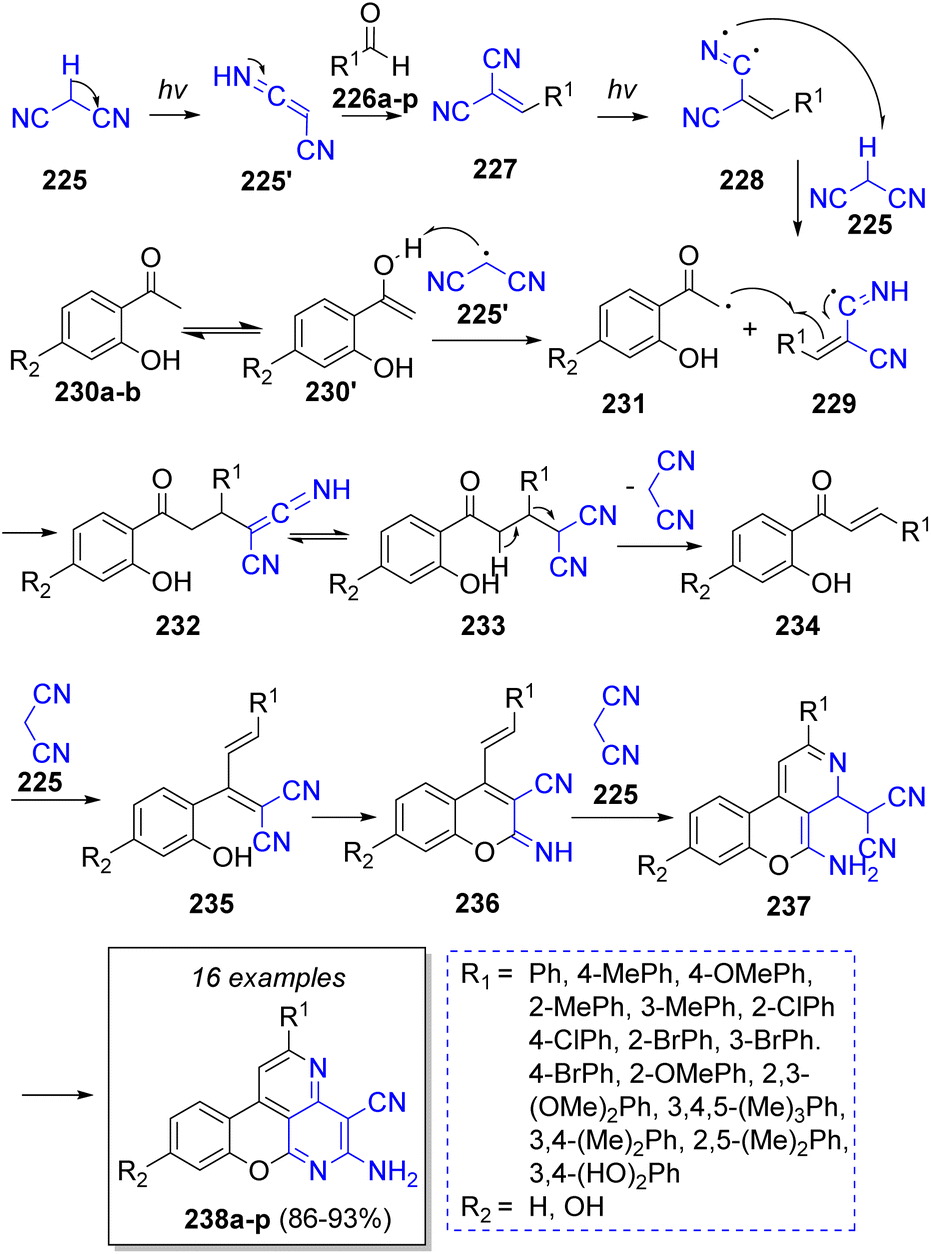 | ||
| Scheme 26 Synthesis of chromeno[4,3,2-de][1,6]naphthyridines. | ||
Gill and co-workers53 reported a highly efficient (up to 91%) pseudo-4CR for the synthesis of trisubstituted imidazoles using a cheap and recoverable ionic liquid as catalyst. Following the Scheme 27, the reaction initially proceeds via nucleophilic attack of ammonia (from NH4OAc, 240) to aromatic aldehydes 239a–v (previously activated by [DBUH+][Im−]) to form the hydroxylamines 241, which after loss of water produces the imine-type intermediates 242. Then, a second attack of ammonia forms the diamine intermediates 243, which react with a previously activated reagent 244 to give the intermediates 245. Upon loss of water, [DBUH+][Im−] and a 1,5-H shift, the trisubstituted imidazoless 247a–v are formed. One of the synthetized products demonstrated a potential cytotoxic activity against the human breast cancer cell line PC-3.
 | ||
| Scheme 27 Synthesis of trisubstituted imidazoles. | ||
A novel technique for the synthesis of spiro[diindenopyridine-indoline]triones was reported by Naimi-Jamal and co-workers,54 using a reusable heterogeneous nano-ordered mesoporous SO3H functionalized-silica (MCM-41-SO3H) catalyst. Following the proposed mechanism depicted in Scheme 28, isatins 248a–i are activated by the SO3H groups, which attack the enolic form 250 of 1,3-indanedione 249 to give intermediates 251. The final products 254a–y are afforded after the addition of anilines 252a–f, followed by a cyclization reaction.
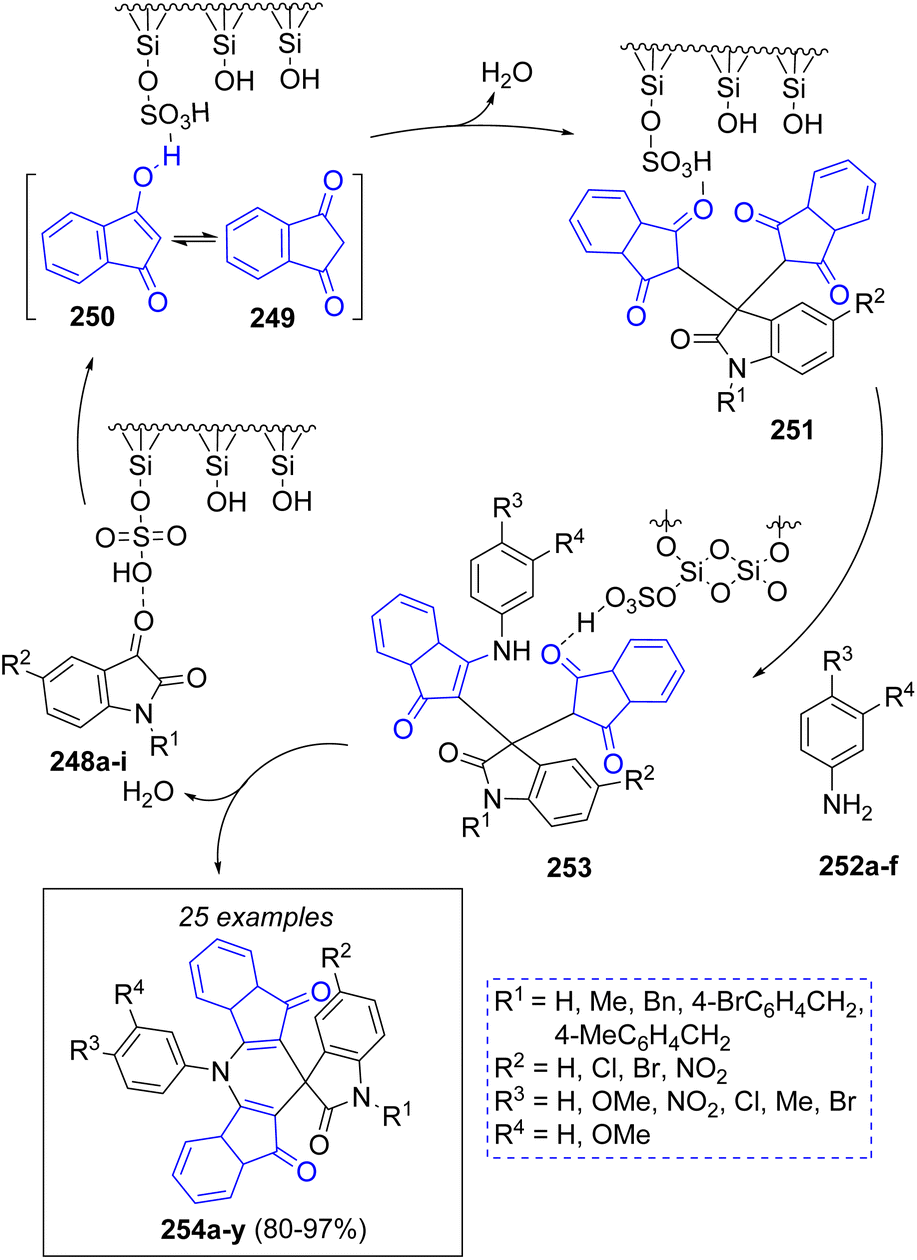 | ||
| Scheme 28 Synthesis of spiro[diindenopyridine-indoline]triones. | ||
4. Pseudo-five component reactions
4.1 Knoevenagel condensation/Michael addition
Safaei-Ghomi and co-workers55 reported an efficient strategy for the synthesis of 4,4′-(arylmethylene)bis(3-methyl-1H-pyrazol-5-ol) derivatives catalyzed by ZnAl2O4. This pseudo-5CR involves the reaction between two equivalents of ethyl acetoacetate (255), two equivalents of hydrazine hydrate (256) and aromatic aldehydes 258a–h. The process is shown in the Scheme 29a. Initially, hydrazine 256 reacts with ethyl acetoacetate 255 to form a pyrazole intermediate 257, which then undergoes a Knoevenagel condensation with the aldehydes 258a–h to form intermediates 259. Finally, another previously formed pyrazole 257 reacts with 259 to form the final products 260a–j.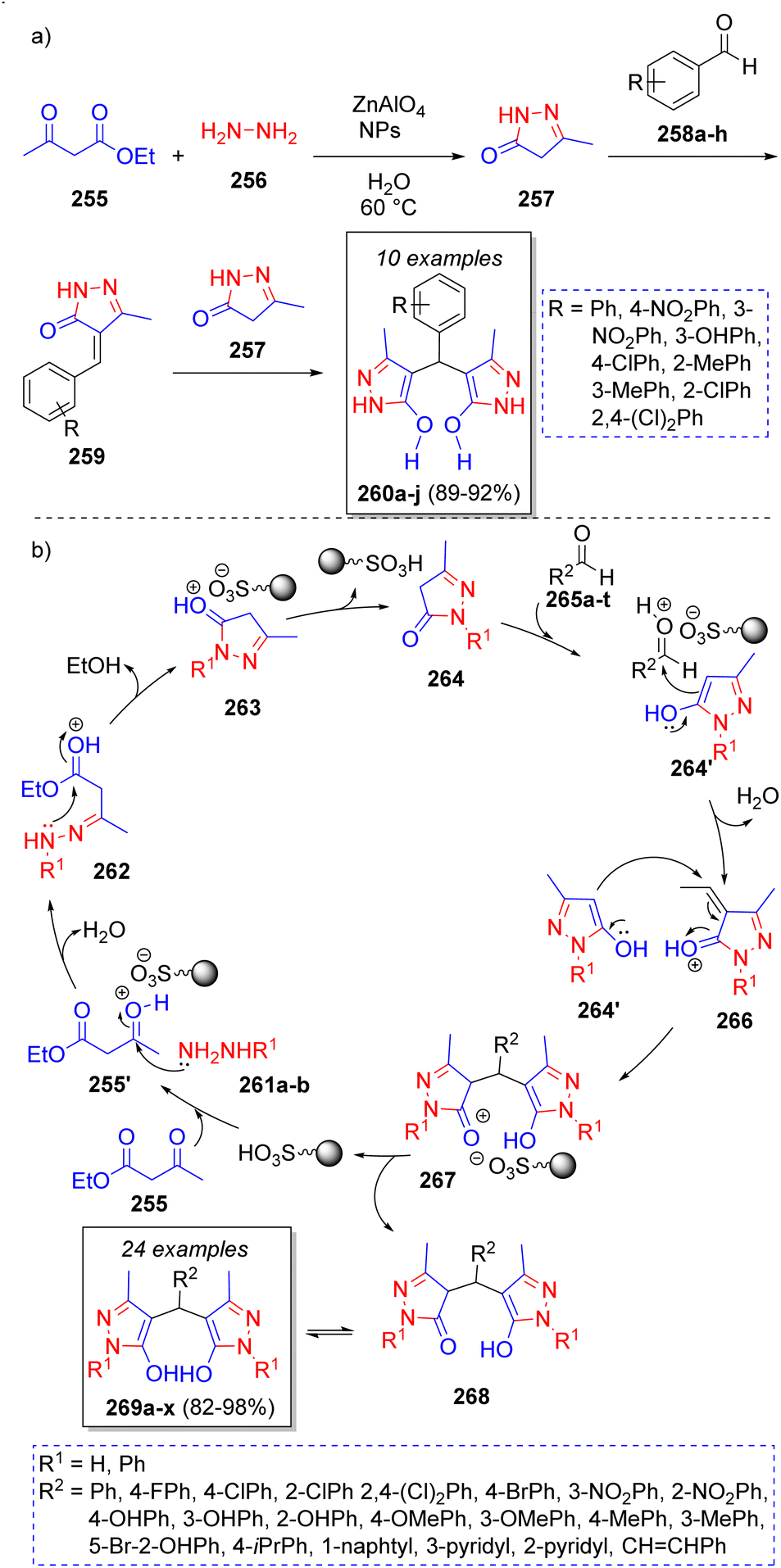 | ||
| Scheme 29 (a) Synthesis of 4,4′-(arylmethylene)bis(3-methyl-1H-pyrazol-5-ol)s. (b) Synthesis of 4,4′-(arylmethylene)bis(3-methyl-1H-pyrazol-5-ol)s. | ||
Later in 2017, Jahanshashi and Akhlaghinia56 reported a similar synthesis of a large collection of new 4,4′-(arylmethylene)bis(3-methyl-1H pyrazol-5-ol)s via a one-pot pseudo-5CR in the presence of sulfonated honeycomb coral (HC–SO3H) as green catalyst. Scheme 29b depicts the proposed reaction mechanism. Accordingly, pyrazolone intermediates 264 (and their tautomeric form 264′) are formed by the reaction between the protonated form of ethyl acetoacetate 255′ and phenylhydrazine/hydrazine 261a–b, followed by an intramolecular cyclization. Consequently, enol intermediates 264′ condense with aromatic aldehydes 265a–t, which are also activated by the acid catalyst, followed by a loss of water to afford 266. Finally, the desired products 269a–x are formed after a Michael addition of another 264′ molecules unto the intermediates 266 to form 268, which undergo a tautomeric proton shift.
A similar work to the previous two was recently reported by A. Kohzadian and co-workers57a for the pseudo-5CR synthesis of bis(pyrazolyl)methanes from the substituted benzaldehydes 274a–m and two equivalents of phenylhydrazine (271) and ethyl acetoacetate (270), respectively. This multicomponent reaction was catalyzed by the acidic ionic liquid (N,N-diethyl-N-sulfoethanaminium methane sulfonate [Et3N-SO3H][MeSO3]), which allowed the authors to obtain their target compounds in truly remarkable yields. Thus, a plausible reaction mechanism was proposed (Scheme 30): ethyl acetoacetate (270) is activated by the acidic catalyst facilitating a nucleophilic attack by a molecule of phenylhydrazine (271) producing the enamine 272, which cyclizes to produce the intermediate 273 after loss of an ethanol molecule followed by tautomerization. Pyrazole 273′ undergoes Knoevenagel condensation with the catalyst-activated benzaldehydes 274a–m, affording intermediates 275. Michael addition of another molecule of 276 affords intermediates 277 which tautomerize to the target compounds 278a–m. A similar strategy was recently utilized by N. Sarmasti and co-workers,57b with the use of a different catalyst (Fe3O4@SiO2/Si(OEt)(CH2)3NH/CC/EDA/Cu(OAc)2). A very interesting review about the syntheses of compounds based on the 4,4′-arylmethylene-bis-1H-pyrazol-5-ol scaffold was recently published by S. U. Deshmukh.57c
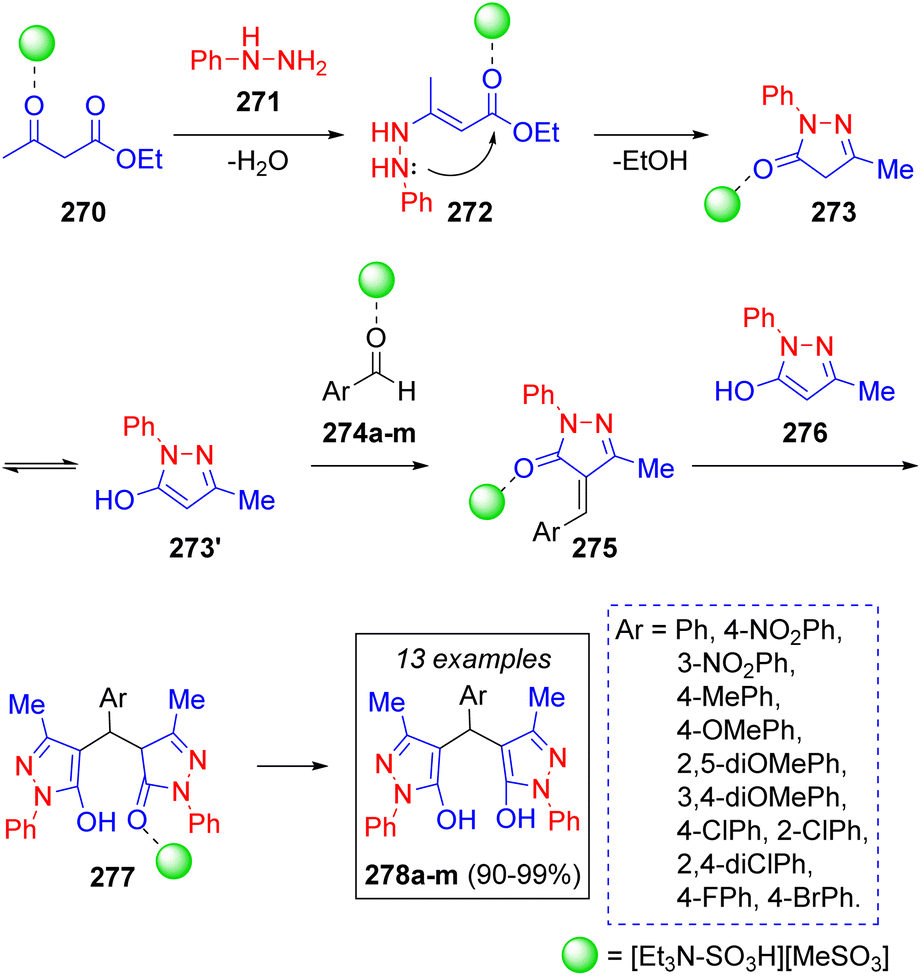 | ||
| Scheme 30 Synthesis of bis(pyrazolyl)methanes via an ionic liquid-catalyzed pseudo 5-CR. | ||
Following a similar reaction sequence as in the previous entries, M. Heravi and co-workers58 reported in 2022 a green synthesis of 5,5′-(arylmethylene)bis(4-hydroxythiazol-2(3H)-ones) 288a–q via a reaction between chloroacetic acid (280), thiourea (279) and aromatic aldehydes 285a–q, using Triton X-100, which is a non-ionic surfactant, as catalyst in water as reaction medium. The antibacterial activity of these molecules was evaluated against antibiotic-resistant bacterial strains, as well as their antifungal activity. To explain the formation of the products, a reaction mechanism was proposed (Scheme 31): the first 4-hydroxythiazol intermediate 284′ is synthesized via a series of reactions starting with a nucleophilic substitution between thiourea (279) and chloroacetic acid (280). This intermediate condenses with the aldehyde molecules 285a–q to produce 286, which are attacked by another molecule of 284′ to form 287, which tautomerize to the target compounds 288.
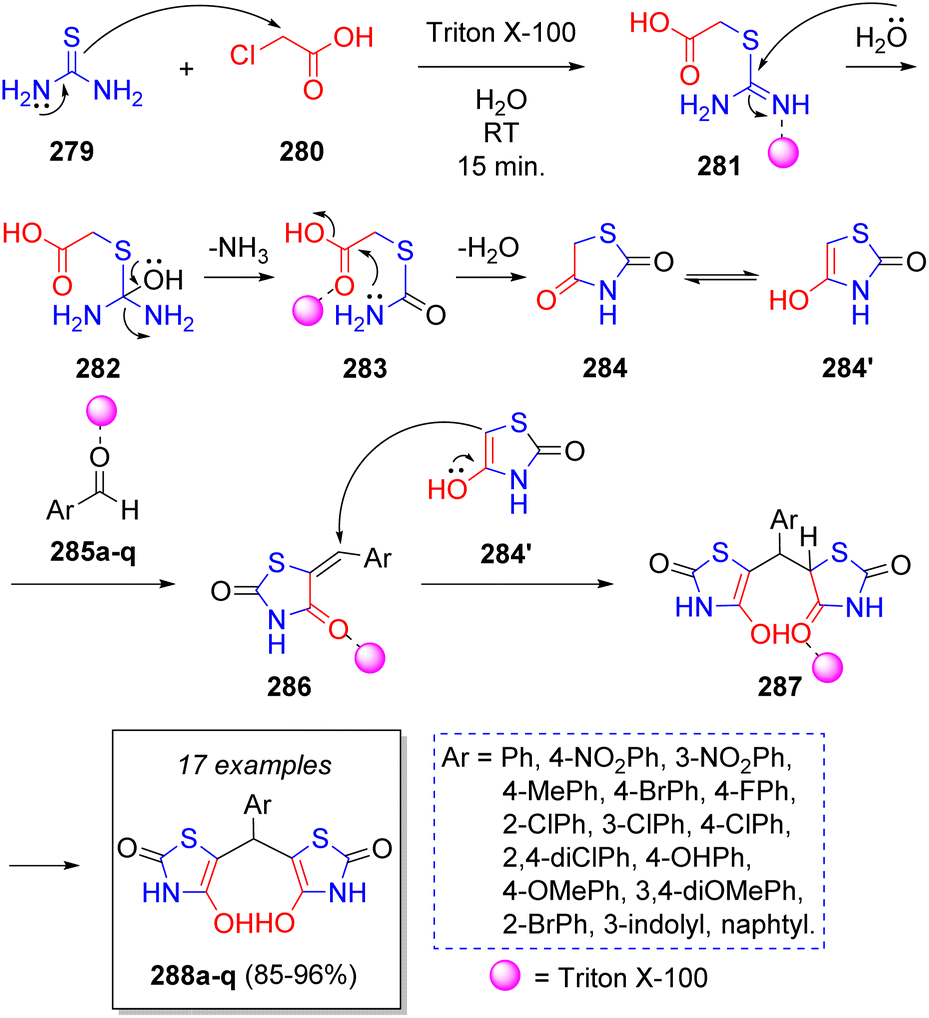 | ||
| Scheme 31 Synthesis of 5,5′-(arylmethylene)bis(4-hydroxythiazol-2(3H)-ones) via a pseudo-5CR. | ||
Das and co-workers,59 in 2014, synthesized a collection of [1,6]naphtyridines via a domino pseudo-5CR, involving the reaction between two equivalents of malononitrile (289), two equivalents of methyl ketones 290a–g, and phenols or thiols 298a–t on water as solvent. A reaction mechanism was proposed as illustrated in Scheme 32. Intermediates 292 are formed via a Knoevenagel condensation of malononitrile anion 289′ with ketones 290a–g promoted by triethylamine, followed by Michael addition of another previously formed intermediates 292 to afford 295 upon loss of a malononitrile fragment. Next, the attack of another malononitrile anion forms the cyclic intermediates 296, which then tautomerize to 297. After nucleophilic attack of phenols or thiols 298a–t on the electrophilic nitrile group and further aromatization, the final products 300a–b are formed.
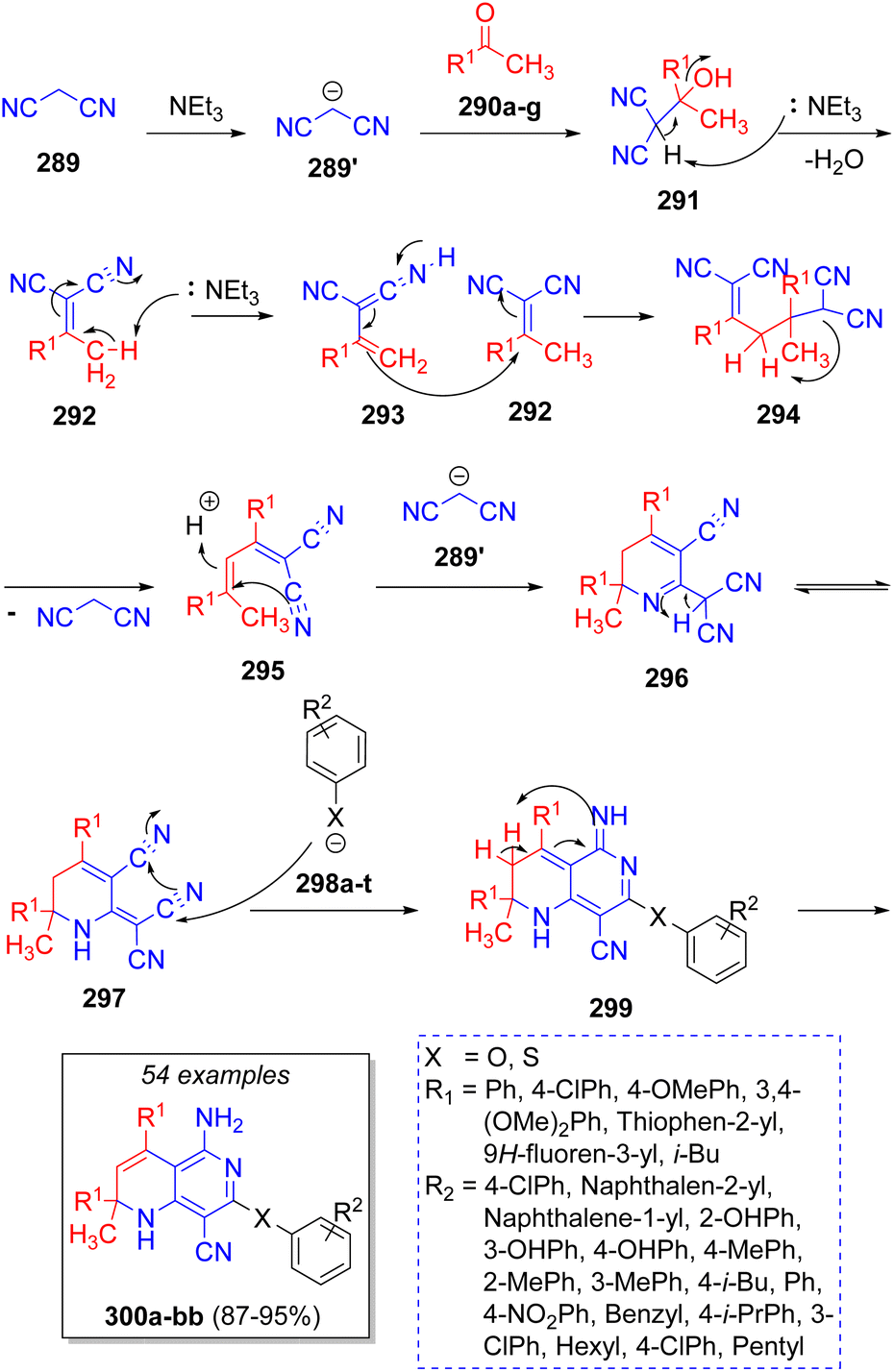 | ||
| Scheme 32 Synthesis of [1,6]naphtyridines. | ||
A collection of 4H-thiopyrans was successfully synthesized by Bodaghifard and co-workers60 in 2016. This pseudo-5CR involves a variety of aromatic aldehydes 301a–k, two equivalents of malononitrile (302), carbon disulfide (307), primary amines 308, and triethylamine as catalyst. A mechanistic pathway for the synthesis of 4H-thiopyran derivatives was suggested as illustrated in Scheme 33. Initially, Knoevenagel condensation of aldehydes 301 and malononitrile 302 generates arylidene malononitrile intermediates 303, which then react with anion 302′ via Michael addition to form intermediates 304. Simultaneously, intermediates 305 are formed from the addition of 308 to carbon disulfide (307), which then react with intermediates 304 to form 306. A hydrogen shift and loss of an isothiocyanate fragment leads to the formation of intermediates 309. Next, another H-shift induces an intramolecular cyclization by the attack of sulfur unto the cyano group. After a final H-shift the desired products 311a–l are formed.
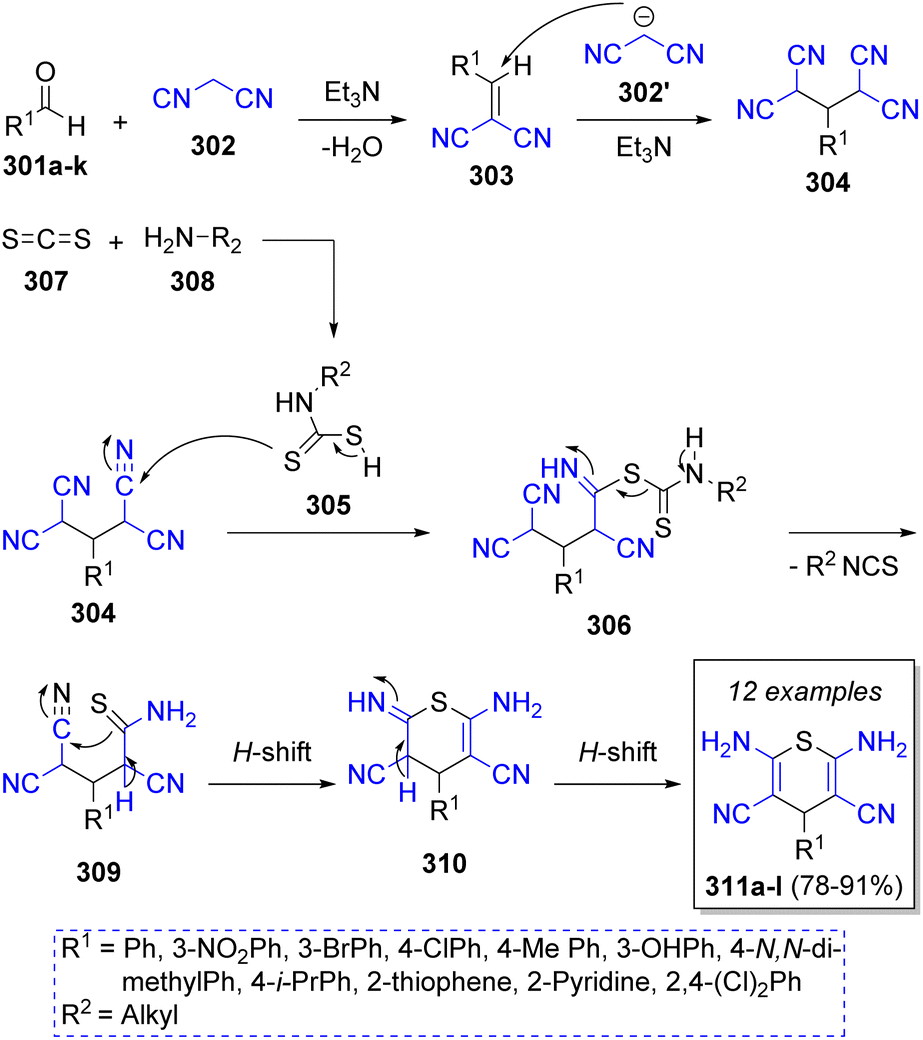 | ||
| Scheme 33 Synthesis of 4H-thiopyran derivatives. | ||
A method to synthesize tris-amides via a microwave-assisted pseudo-5CR was achieved in excellent yields by Hosseini-Tabatabaei and co-workers61 in 2017. This MCR included the use of Meldrum's acid (312), aryl aldehydes 313a–f, alkyl isocyanides 315a–b and two equivalents of aniline (317). The proposed mechanism for this reaction comprises an initial Knoevenagel condensation between Meldrum's acid 312 and aldehydes 313a–f to form the conjugated electron-deficient heterodienes 314, which react with isocyanides 315a–b via Michael addition to give aminolactone intermediates 316. Next, the reaction of aminolactones 316 with aniline 317 affords the vinylogous carbonates 318. Upon loss of acetone and a final ring-opening reaction with another molecule of aniline 317, the desired products 320a–k are formed (Scheme 34).
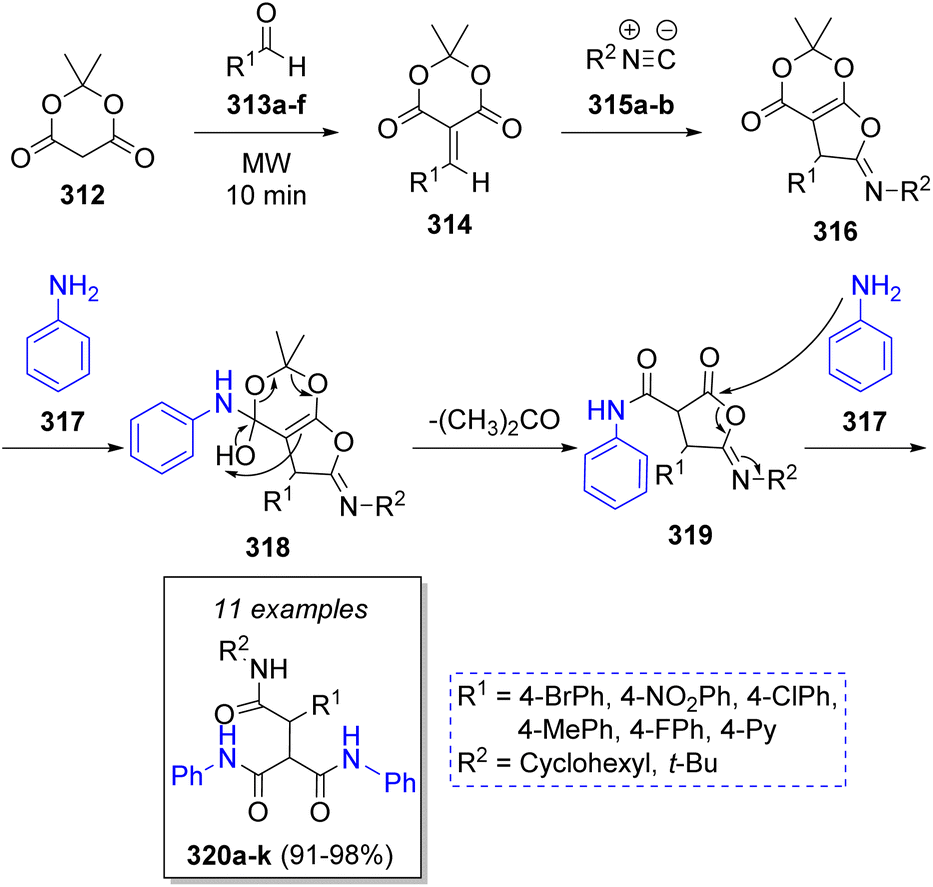 | ||
| Scheme 34 Synthesis of tris-amides via pseudo-MCRs. | ||
Dabiri and Salehi62 reported a series of triazolyl methoxyphenyl 1,8-decahydroacridine derivatives using aromatic propargylated aldehydes 321a–c, a variety of azides 322a–e, two equivalents of dimedone (324) and amines 328a–d in the presence of Cu(OAc)2/sodium ascorbate and 1-methylimidazolium trifluoroacetate ([Hmim]TFA) as catalyst. The proposed reaction mechanism for this transformation is illustrated in Scheme 35. Initially, aromatic propargylated aldehydes 321 react with azides 322 to form, via a click reaction, the intermediate 1,2,3-triazolyl methoxy benzaldehydes 323. Then, dimedone 324 reacts with intermediates 323 via a Knoevenagel condensation, with acidic ionic liquid (IL) acting as catalyst, to afford intermediates 326 after loss of water. Subsequently, a second molecule of dimedone 324′ reacts, via Michael addition, with 326 to afford 327. Lastly, a nucleophilic attack of amines 328a–d followed by intramolecular cyclization and loss of a water molecule affords the final products 331a–n.
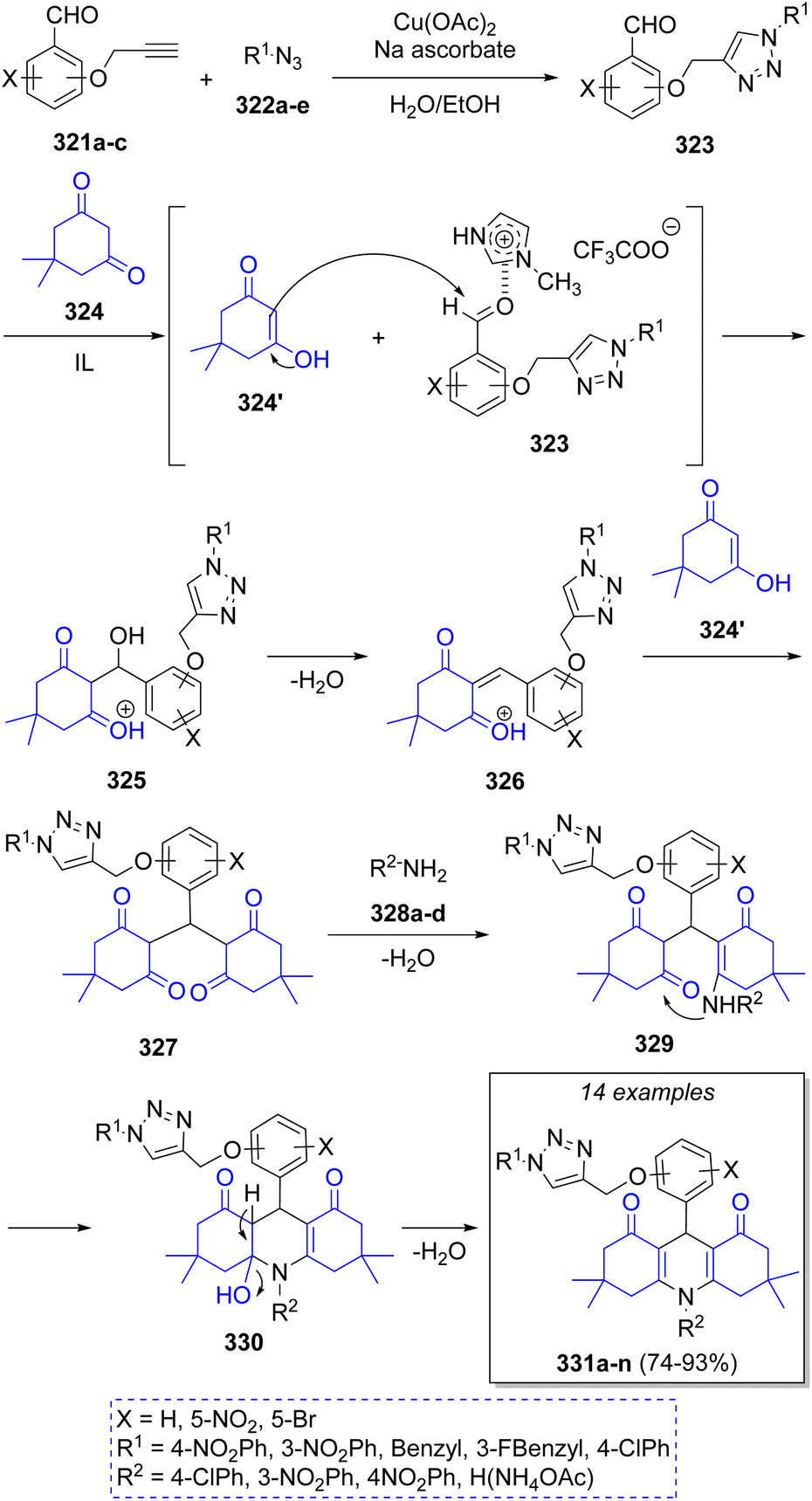 | ||
| Scheme 35 Synthesis of triazolyl methoxyphenyl 1,8-decahydroacridine derivatives. | ||
An efficient and high-yielding method to obtain highly functionalized [1,6]naphthyridines via pseudo-5CR was achieved by Khan and co-workers63 using two equivalents of arylmethyl ketones or alkyl methyl ketones 332a–j, two equivalents of malononitrile (333) and alkyl alcohols or thiols 337a–d, with sodium hydroxide as catalyst. The reaction was proposed to proceed as is depicted in the Scheme 36. An initial Knoevenagel condensation between methyl ketones 332a–j and malononitrile (333) forms the intermediates 334, followed by an auto-Michael addition of ylidenes 334, which after loss of one molecule of malononitrile give the intermediates 335. Upon a reaction with another malononitrile molecule, intermediates 336 are formed. Subsequently, alcohols or thiols 337a–d attack to form intermediates 338. A proton shift affords intermediates 339, which undergo an intramolecular cyclization to form 340. A final aromatization step affords the desired products 341a–k.
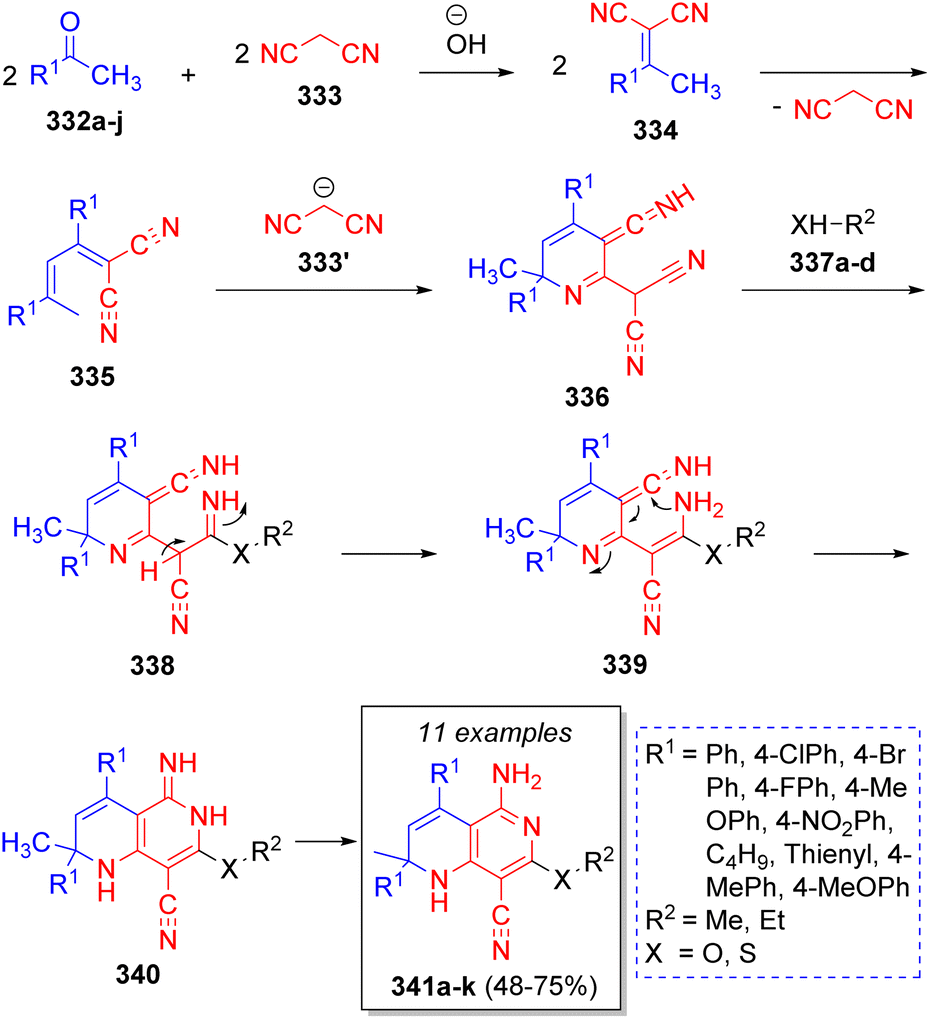 | ||
| Scheme 36 Synthesis of highly functionalized [1,6]-naphthyridines. | ||
A small library of highly functionalized 3-azabicyclo[3.3.1]nona-2.7-dienes was reported by Ismiyeva and co-workers64 via a stereoselective cascade pseudo-5CR, comprising the aromatic aldehydes 342a–e (2 equivalents), cyanoacetic esters 343a–b, malononitrile (345) and acetylacetone (347). The probable reaction mechanism is illustrated in Scheme 37. Initially, cyanoacetic esters 343a–b undergo a Knoevenagel condensation with 342a–e to produce intermediates 344. Simultaneously, the same happens with malononitrile 345 and another molecule of 342a–e to form arylidenemalononitrile-type intermediates 346. Next, 346 reacts with acetylacetone 347 to form 348, which upon further heterocyclization form 4H-pyrans 349. Then, an acyclic intermediates 350 result from OH-assisted nucleophilic cleavage of 349, followed by a Michael addition of anions 350 to the previously formed arylidenemalononitriles 344 and a carbocyclization to give 352. Upon an intramolecular cyclization, the final products 353a–h are obtained.
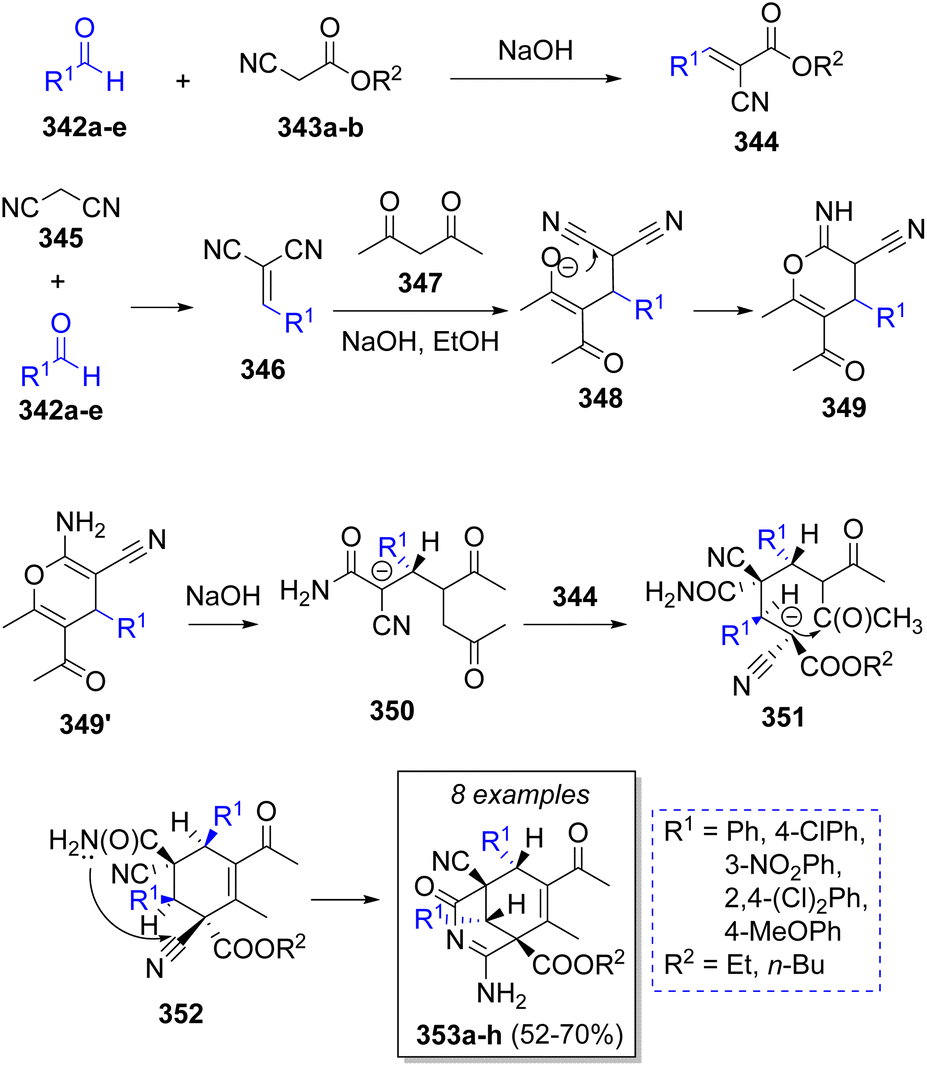 | ||
| Scheme 37 Synthesis of 3-azabicyclo[3.3.1]nona-2.7-dienes. | ||
4.2 Condensation/cyclization
In 2008, Shaabani and co-workers65 reported a high-yielding pseudo-5CR synthesis of 4,5,6,7-tetrahydro-1H-1,4-diazepine-5-carboxamides using 2,3-diaminomaleonitrile (DAMN, 354), two equivalents cyclic or acyclic ketones 355a–c, isocyanides 359a–d, and water 361 with p-toluenesulfonic acid as catalyst. As depicted in Scheme 38, the proposed reaction mechanism begins with two simultaneous condensations between DAMN 354 and acid activated ketones 355a–c to form the diimine intermediates 356, which upon an imine-enamine tautomerization followed by intramolecular cyclization affords iminium ion intermediates 358. Then, α-attack by the isocyanides 359a–d affords nitrilium ion intermediates 360. Finally, the desired products 363a–l are formed by water addition to nitrilium ions 360 and a final tautomerization process. Similarly, a series of bifunctional diazepine-tetrazoles 369a–p were synthesized by Shaabani and co-workers66 using 2,3-diaminomaleonitrile or aromatic diamines, ketones, trimethylsilylazide, isocyanides and p-toluenesulfonic acid as catalyst in methanol. A similar mechanism was proposed by the authors, upon formation of iminium ions 364, there are two possible pathways to form intermediates 368, pathway A involves two steps, first a nucleophilic attack of isocyanides 366 to iminium ions 364 to form 367, followed by a nucleophilic attack by the azide ion 365. The second pathway involved a one-step concerted reaction with the nucleophilic attack of azide ion 365 to isocyanides 366, followed by isocyanides 366 attack to iminiums 364. The last step consists of an intramolecular [3 + 2] cycloaddition to afford the final products 369a–p (Scheme 38).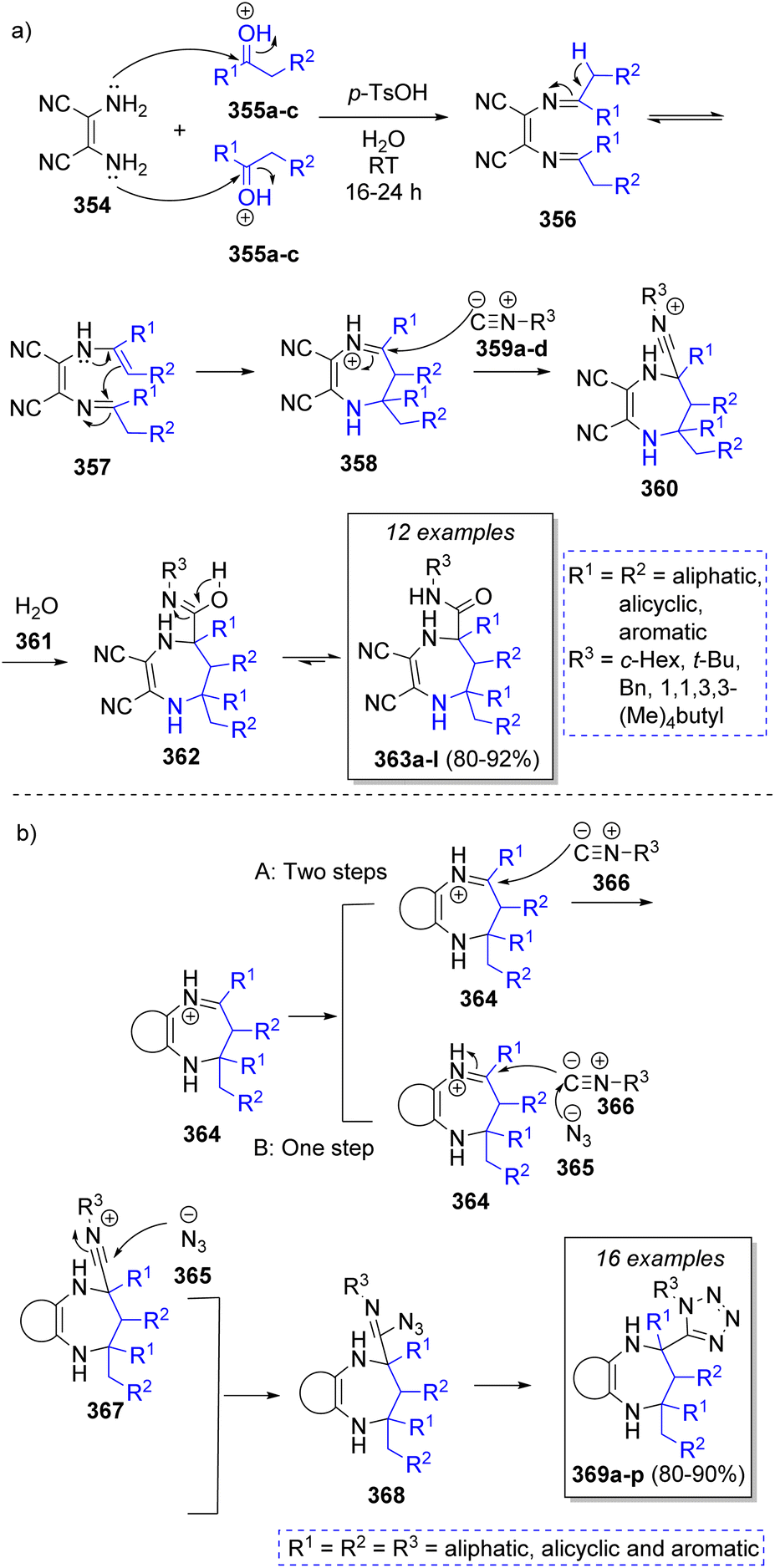 | ||
| Scheme 38 Synthesis of (a) 4,5,6,7-tetrahydro-1H-1,4-diazepine-5-carboxamides, and (b) 1H-tetrazolyl-benzo[b][1,4]diazepines. | ||
4.3 Knoevenagel condensation/[1,4] cycloaddition
In 2011, Teimouri and Akbari-Moghaddam67 reported an efficient method to obtain ferrocene-triamides from the reaction of ferrocenecarboxaldehyde (370), Meldrum's acid (371) with alkyl or aryl isocyanide 373a–h and two equivalents of different amines 376a–o in anhydrous dichloromethane as solvent. Scheme 39 illustrates the synthetic methodology. The first step is a Knoevenagel condensation of ferrocenecarboxaldehyde (370) with Meldrum's acid 371 to form the electro-deficient heterodiene 372. A [1 + 4] cycloaddition reaction with isocyanides 373a–h leads to iminolactone intermediates 374; after loss of acetone, 375 reacts with two molecules of anilines 376 to form the final products 377a–q.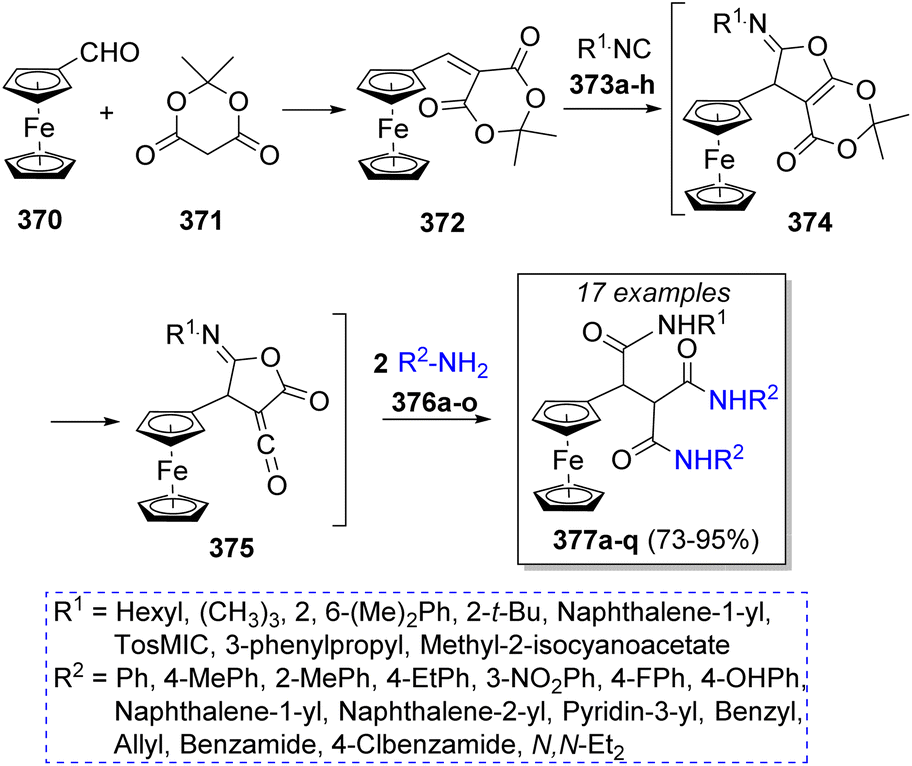 | ||
| Scheme 39 Synthesis of ferrocene-triamides. | ||
Likewise, Safaei-Ghomi and co-workers68 published a high-yielding method to obtain functionalized tricarboxamides using two equivalents of benzaldehydes 378a–f, Meldrum's acid (379), isocyanides 381a–b, and anilines 383a–f in the presence of catalytic amounts of copper(I) iodide nanoparticles. It is noteworthy that the catalyst proved to be recyclable as it was recovered and reused several times. The suggested reaction mechanism is depicted in Scheme 40. Initially, a Knoevenagel condensation between benzaldehydes 378a–f and Meldrum's acid 379 is catalyzed by CuI nanoparticles to afford intermediates 380, which then react with isocyanides 381a–b in a [1 + 4] cycloaddition to form iminolactones 382. Subsequently, iminolactones 382 react with an aniline molecule 383a–f to produce vinylogous carbonates 384. Ultimately, 384 is converted into the desired products 386a–r through the reaction with another molecule of amine 383a–f.
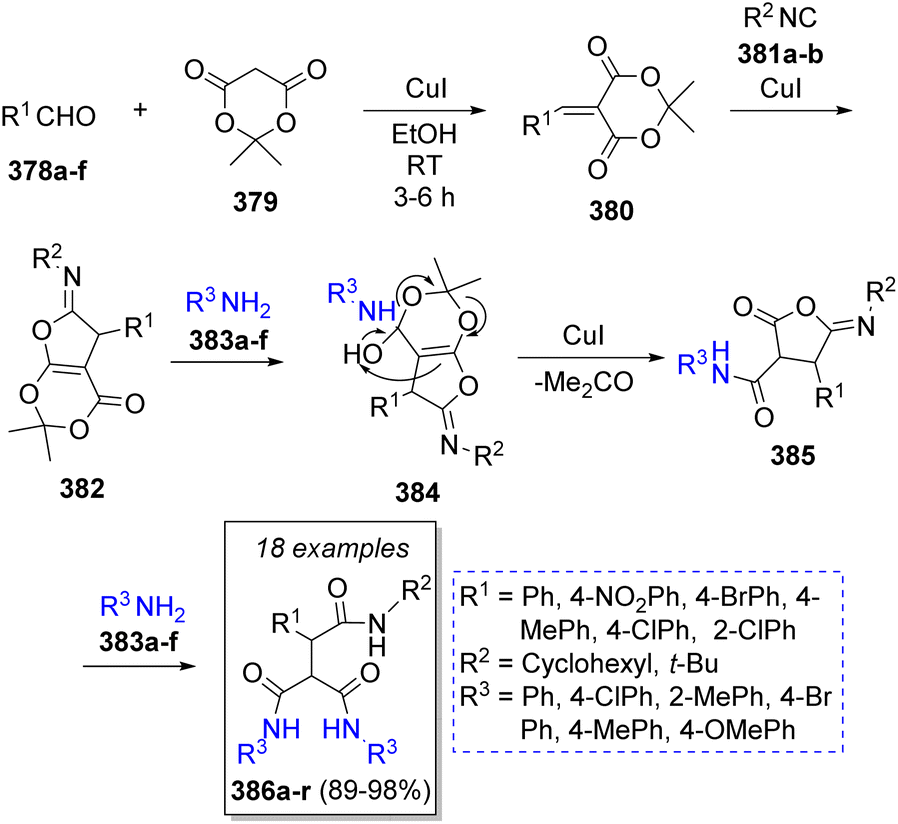 | ||
| Scheme 40 Synthesis of tricarboxamides. | ||
The synthesis of a library of indole and quinoline tricarboxamides was achieved by Shiri and Heravi69 using aromatic aldehydes (2-formylindole or 2-Cl-3-formyl quinolines) 387a–c, Meldrum's acid (388), isocyanides 390a–b and aromatic amines 392a–f (two equivalents) in dichloromethane as solvent. According to the suggested mechanism (Scheme 41), aldehydes 387a–c and Meldrum's acid (388) react via Knoevenagel condensation to afford imine intermediates 389 which then undergo a [1 + 4] cycloaddition with isocyanides 390a–b to give aminolactone intermediates 391. Then, nucleophilic addition of arylamines 392a–f to 391 with subsequent releasing of an acetone molecule forms the intermediates 393, followed by the addition of a second molecule of amines 392a–f at lactone carbonyl group to form the desired products 394a–g and 395a–m in excellent yields.
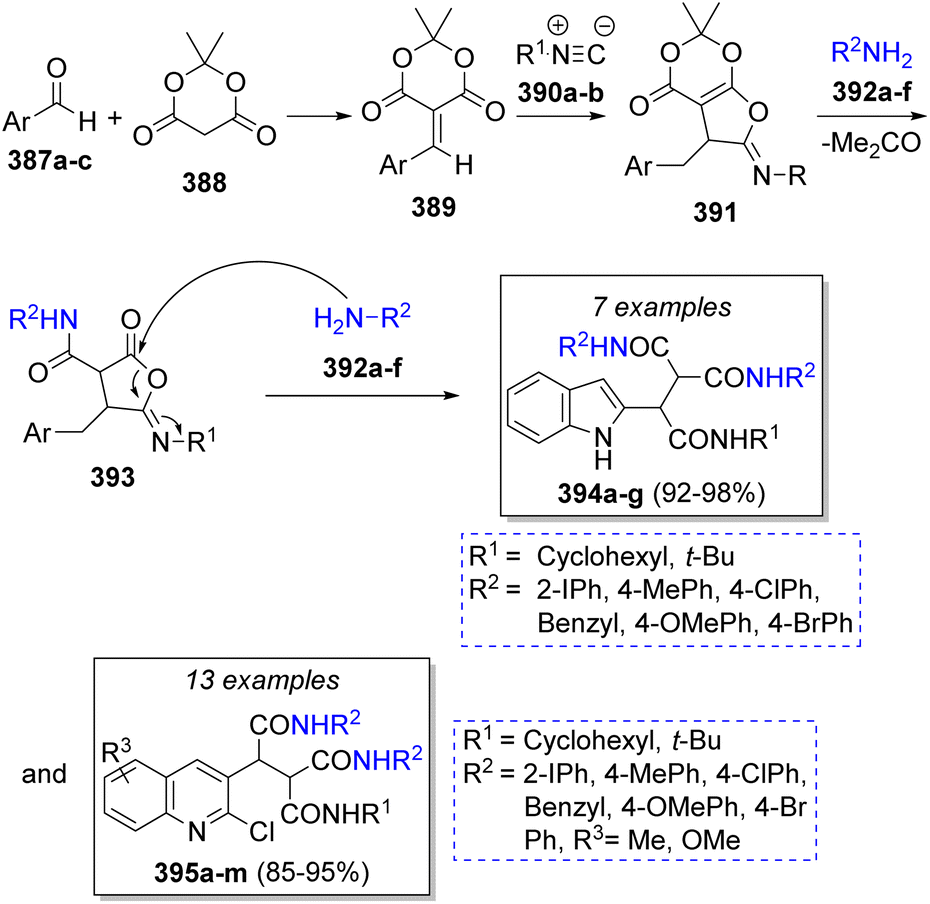 | ||
| Scheme 41 Synthesis of indole and quinoline tricarboxamides. | ||
In 2022 M. Mojtahedi and co-workers70 reported a novel pseudo-5CR synthesis of dicyanoanilines 409a–k by reacting different substituted aromatic aldehydes 396a–k with four equivalents of malononitrile (397) in the presence of triethylamine. To explain this transformation the authors proposed the following reaction mechanism: the first step is a Knoevenagel condensation of the aldehydes 396 with a molecule of malononitrile to produce the intermediates 398. Simultaneously, two molecules of malononitrile react to form dimer 399. Then, 398 and 399 undergo a cycloaddition to produce the intermediates 400, which after loss of an HCN molecule, followed by annulation and ring rearrangement generates 403, which reacts with another molecule of malononitrile followed by a ring expansion and contraction sequence and a decarboxylation to afford the target compounds 409a–k in good to excellent yields (Scheme 42).
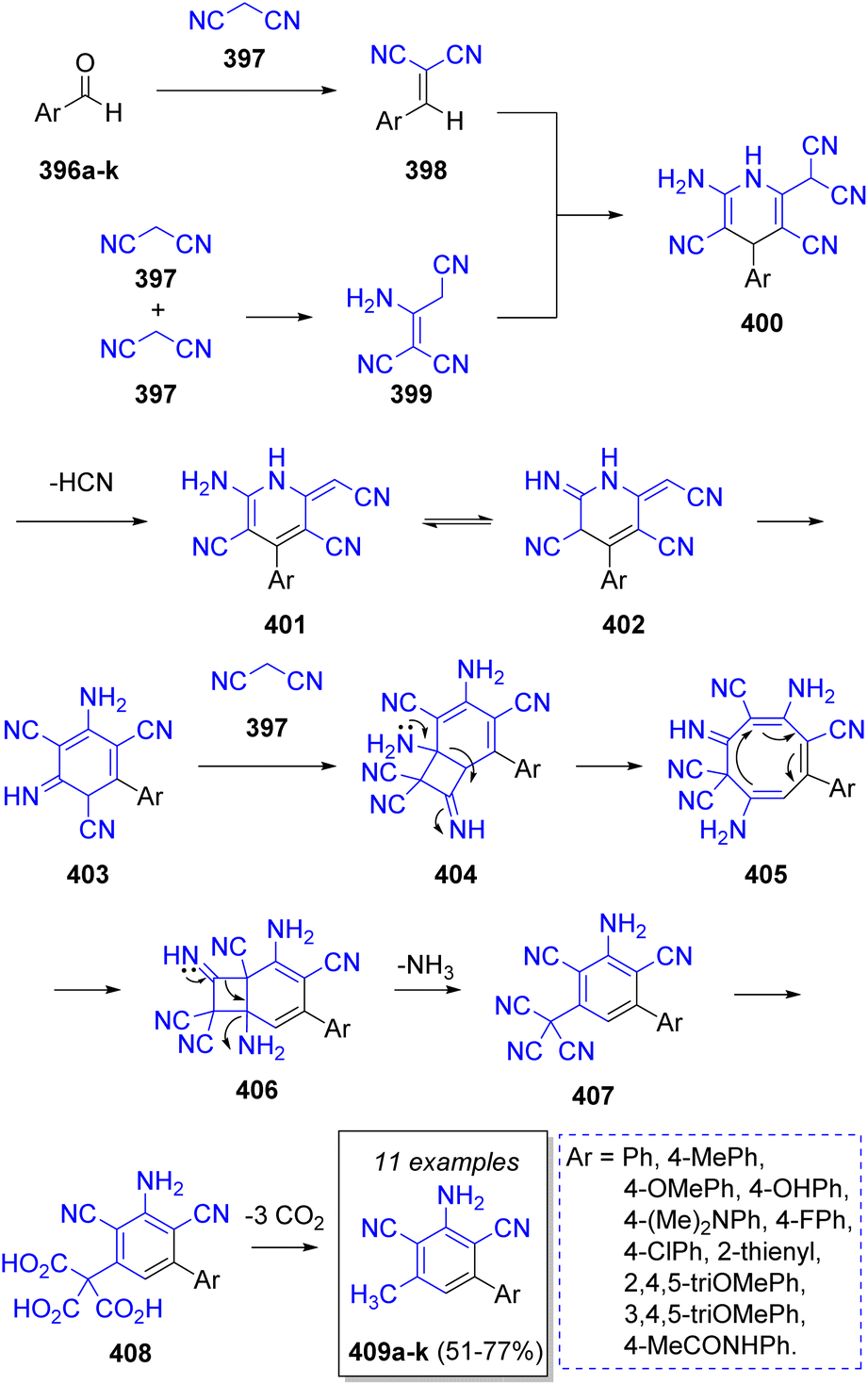 | ||
| Scheme 42 Synthesis of dicyanoanilines via a pseudo-5CR. | ||
4.4 Others
In 2012, Perumal and co-workers71 reported a domino pseudo-5CR for the synthesis of 11 examples of 5-aroryl-1,3-diarylhexahydropyrimidines involving the use of the (E)-3-(dimethylamino)-1-arylprop-2-en-1-ones 410a–e, formaldehyde (four equivalents, 413), and aromatic amines (2 equivalents) 411a–g. A reasonable reaction mechanism was suggested as depicted on Scheme 43. The reaction initially proceeds via Michael addition of amines 411a–g to 410a–e, the subsequent loss of the dimethylamine moiety affords intermediates 412. Simultaneously, formaldehyde 413 condenses with another molecule of amines 411a–g to form the corresponding imines 414. Then, a Mannich-type reaction occurs between 412 and 414 to produce the intermediates 415, which condense with a second molecule of formaldehyde to afford intermediates 416. Upon protonation and reduction, induced by the formic acid arising from the air oxidation of excess of formaldehyde, the final products 418a–q are formed.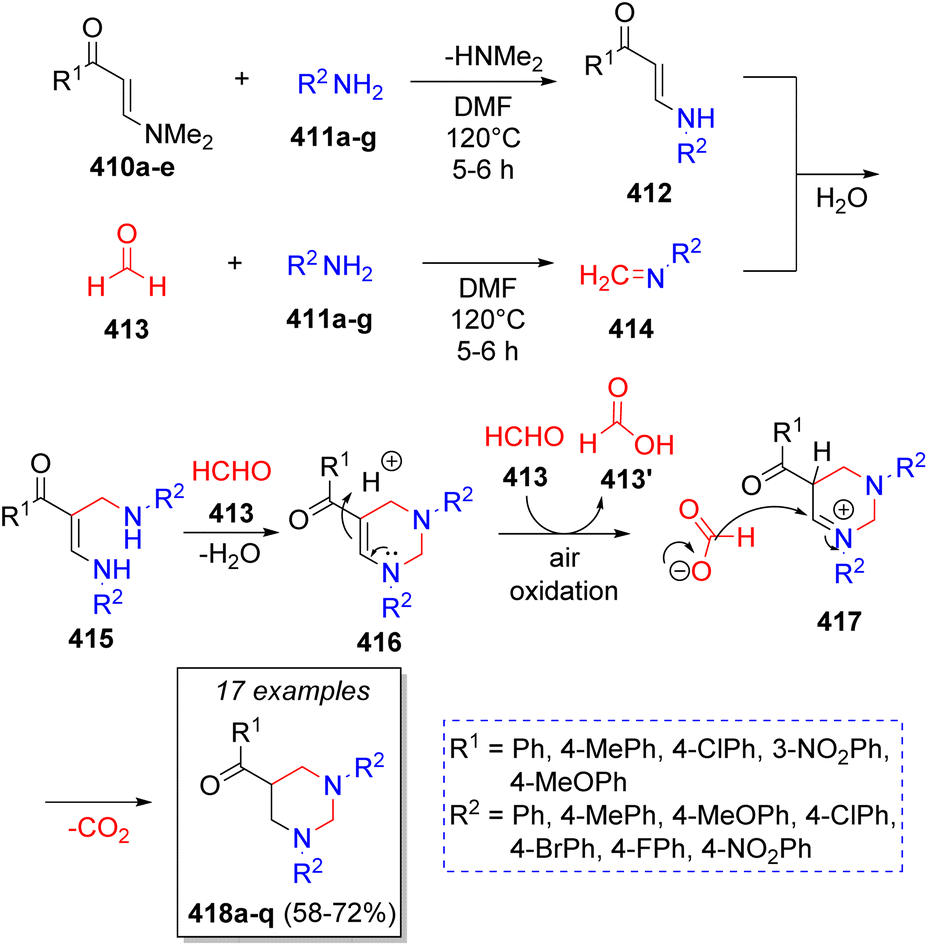 | ||
| Scheme 43 Synthesis of 5-aroryl-1,3-diarylhexahydropyrimidines. | ||
In the same year, a series of spirooxindoles were prepared from a pseudo-5CR by Alizadeh and co-workers,72 comprising the reaction of two equivalents of ammonia 419, 1,1-bis(methylthio)-2-nitroethylene (420), isatin (or derivatives) 422a–f and 1,3-dicarbonyl compounds 423a–b in water as solvent. A plausible reaction mechanism was proposed for this reaction as illustrated in Scheme 44. This pseudo MCR starts with two nucleophilic attacks of ammonia 419 on 1,1-bis(methylthio)-2-nitroethylene (420) which resulted in the formation of ketene diaminal 421. Later, a Knoevenagel condensation between isatins 422a–f, 1,3-dicarbonyl compounds 423a–b with p-toluenesulfonic acid (p-TSA) as catalyst forms intermediates 424. Intermediates 425 are formed after a Stork enamine alkylation between 421 and 424. Further intramolecular cyclization and loss of a water molecule leads to the formation of the final products 427a–f.
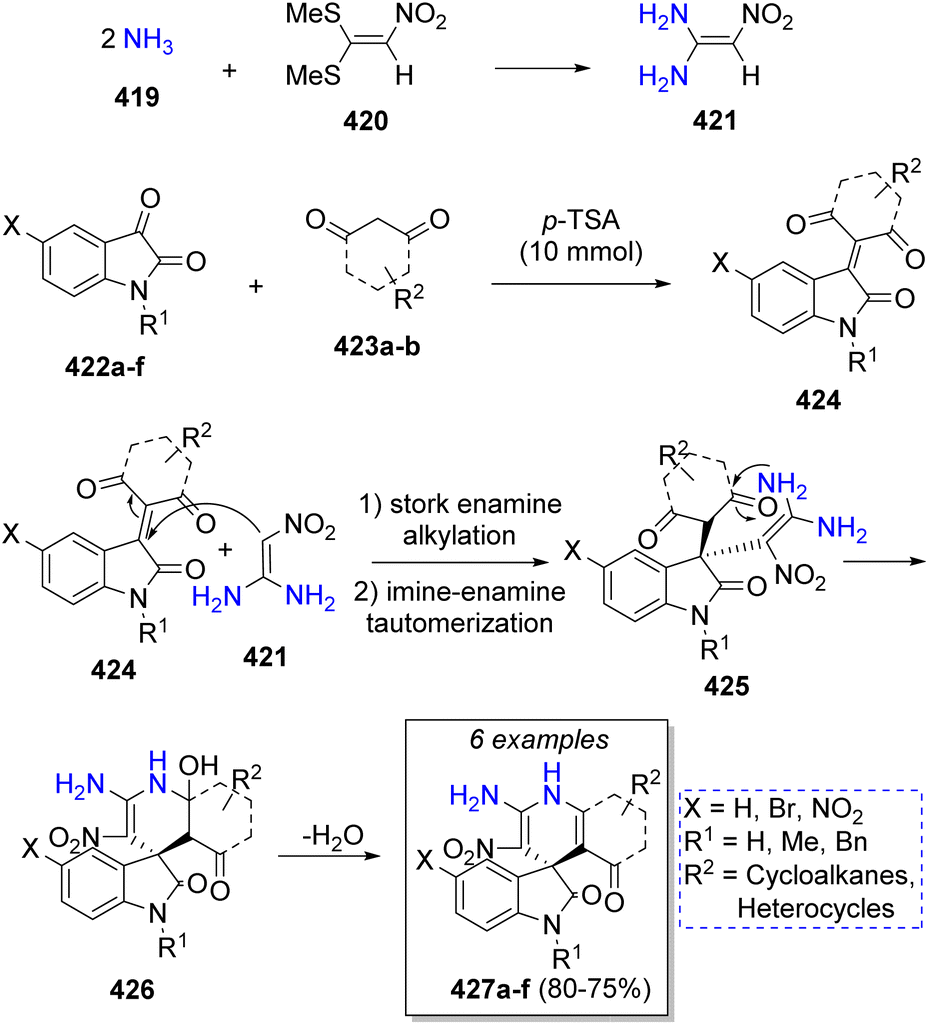 | ||
| Scheme 44 Synthesis of spirooxindoles. | ||
A series of 1,2,4-triazolo[1,5-a]pyridines were synthesized by Alizadeh and co-workers73 in 2013. Their syntheses involved the reaction between the benzylidene-hydrazines 428a–c, acetylene-dicarboxylates 429a–b, two equivalents of aromatic aldehydes 431a–d and malononitrile (432) in ethanol, with molecular iodine as catalyst. Scheme 45 shows the proposed mechanism for the reaction. It is likely that, initially, enaminone compounds 430 are formed via the reaction of benzylidenehydrazines 428a–c with dialkyl acetylenedicarboxylates 429a–b. Simultaneously, aldehydes 431a–d condense with malononitrile via a Knoevenagel reaction to form intermediates 433, which then react with the previously formed enaminones 430 to yield the intermediates 434. Upon iodine coordination to the imine group and intramolecular nucleophilic attack of amine group, intermediates 435 are formed, which after oxidation leads to the desired products 436a–h.
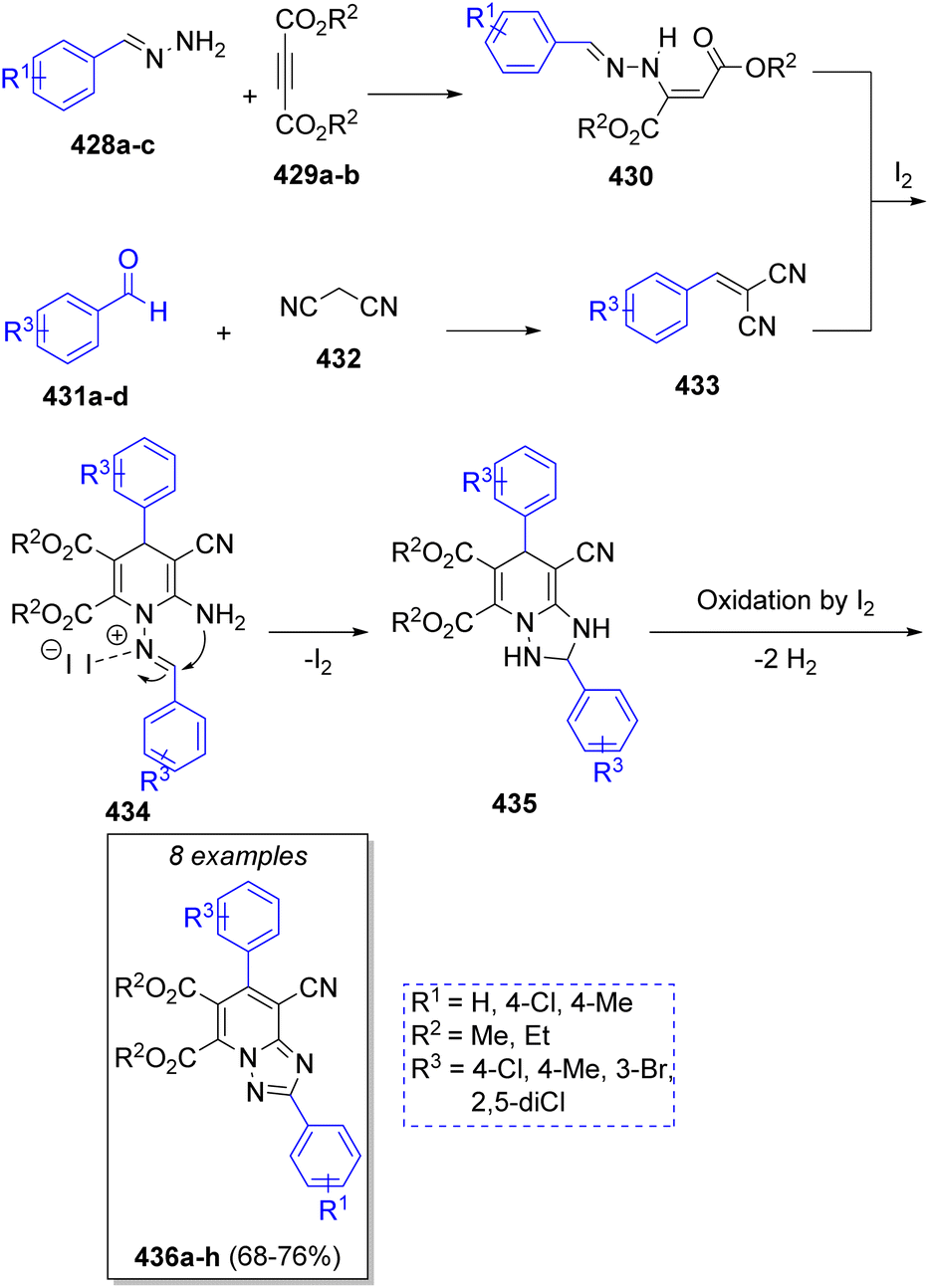 | ||
| Scheme 45 Synthesis of 1,2,4-triazolo[1,5-a]pyridines. | ||
A library of chiral tetrahydropyridines were obtained by Lin and co-workers74 via an organocatalytic asymmetric pseudo-5CR using two equivalents of aromatic amines 437a–c, β-ketoesters 438a–d, two equivalents of aromatic aldehydes 440a–h and SPINOL-derived phosphoric acid as catalyst. The reaction sequence is illustrated in the Scheme 46. Initially, the reaction is proposed to proceed via condensation of amines 437a–c with β-ketoesters 438a–d to give enamines 439, which further react with aldehydes 440a–h to form intermediates 441. These compounds undergo an imine-enamine tautomerization to afford the diene intermediates 442. Simultaneously, another molecule of aldehydes and amines react to form imines 443, which are activated via protonation of the amine and hydrogen bonding between enamines 442 and SPINOL-phosphoric acid to afford 444. Then, a pseudo-intramolecular 1,2-addition gives the iminium ions 445, which undergo an intramolecular-1,4-addition to afford intermediates 446. Finally, a tautomerization of these later ones leads to the final products 447a–l.
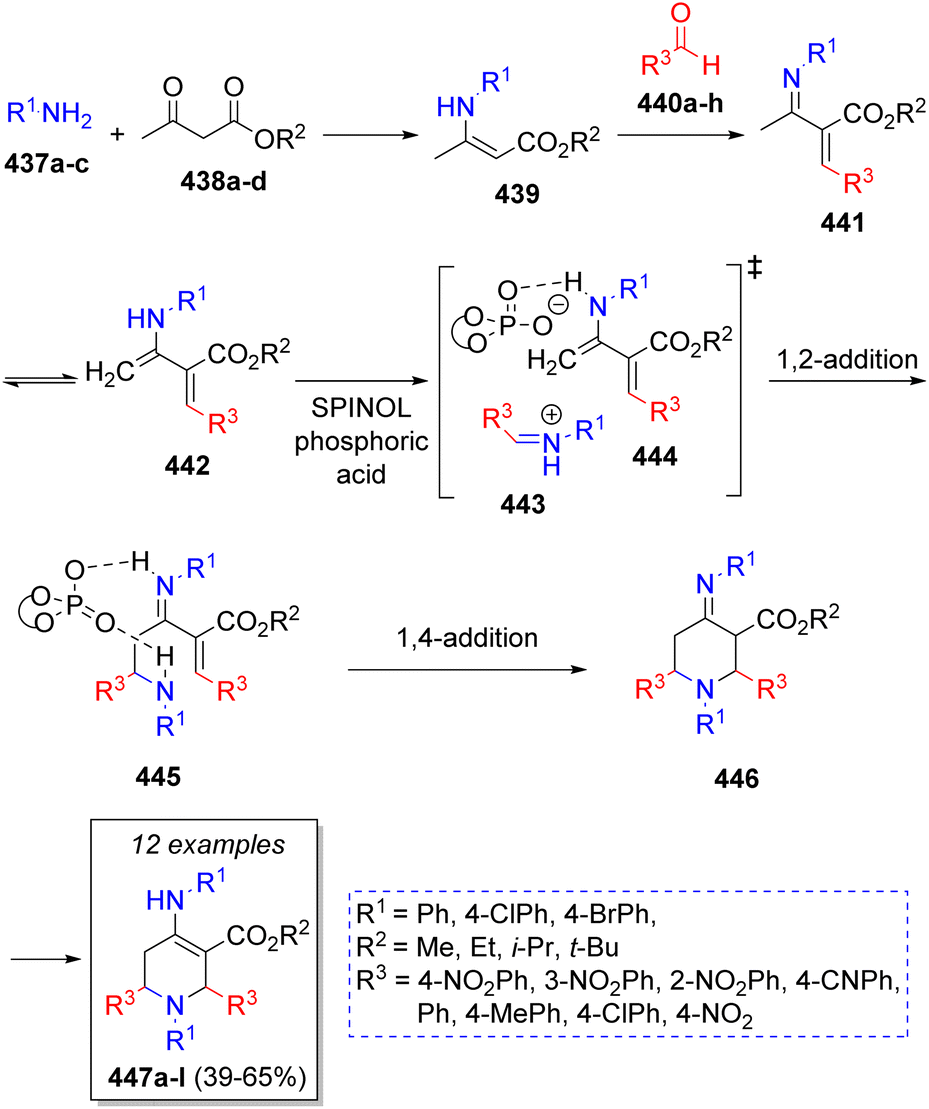 | ||
| Scheme 46 Synthesis of pentasubstituted-tetrahydropyridines. | ||
A high-yielding and diastereoselective synthesis of α-hydrazino tetrazole scaffolds were reported by Balalaie and co-workers75 via an Ugi-azide pseudo-5CR. The strategy involved the sequential combination of hydrazine hydrate (448), cyclic ketones 449a–e, trimethylsilyl azide (452) and isocyanides 450a–b. A plausible mechanism was proposed as depicted in Scheme 47. Apparently, diimines 451 are generated via the condensation of hydrazine 448 with cyclic ketones 449a–e. Next, nucleophilic attack of isocyanides 450a–b affords nitrilium ion intermediates 453, which are then attacked by the azide anion 452 from TMSN3 to produce the intermediates 454, which undergo a final intramolecular cyclization to afford the products 455a–h.
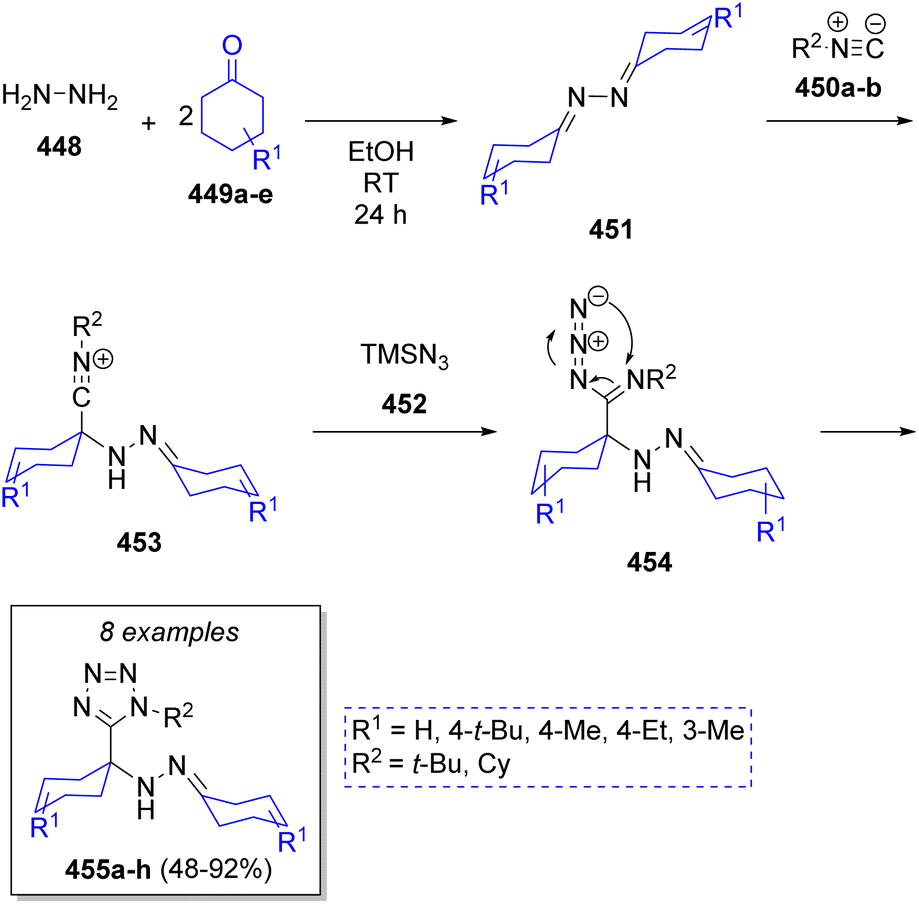 | ||
| Scheme 47 Synthesis of α-hydrazino tetrazoles via a pseudo-5CR. | ||
A synthesis of a series of N,N′-substituted-4-imidazolidinones was successfully accomplished by Ramirez and co-workers76 in 2018 via a pseudo-5CR using two equivalents of formaldehyde (456), primary amines 457a–k, isocyanides 459a–k, water 463 and trifluoroethanol as solvent. The proposed mechanism for this MCR is depicted in Scheme 48. This reaction is assumed to begin with the formation of the imines 458 from the condensation of formaldehyde (456) with amines 457a–k, followed by their protonation. Next, a reaction with isocyanides 459a–k leads to the formation of nitrilium ions, which are trapped by trifluoroethanol to give the imidates 460. Then, a second molecule of formaldehyde condenses with 460 to form intermediates 461, followed by an intramolecular cyclization to give 462. Finally, addition of a water molecule affords hemi-orthoamides 464, which after release of a solvent molecule generates the desired products 465a–m.
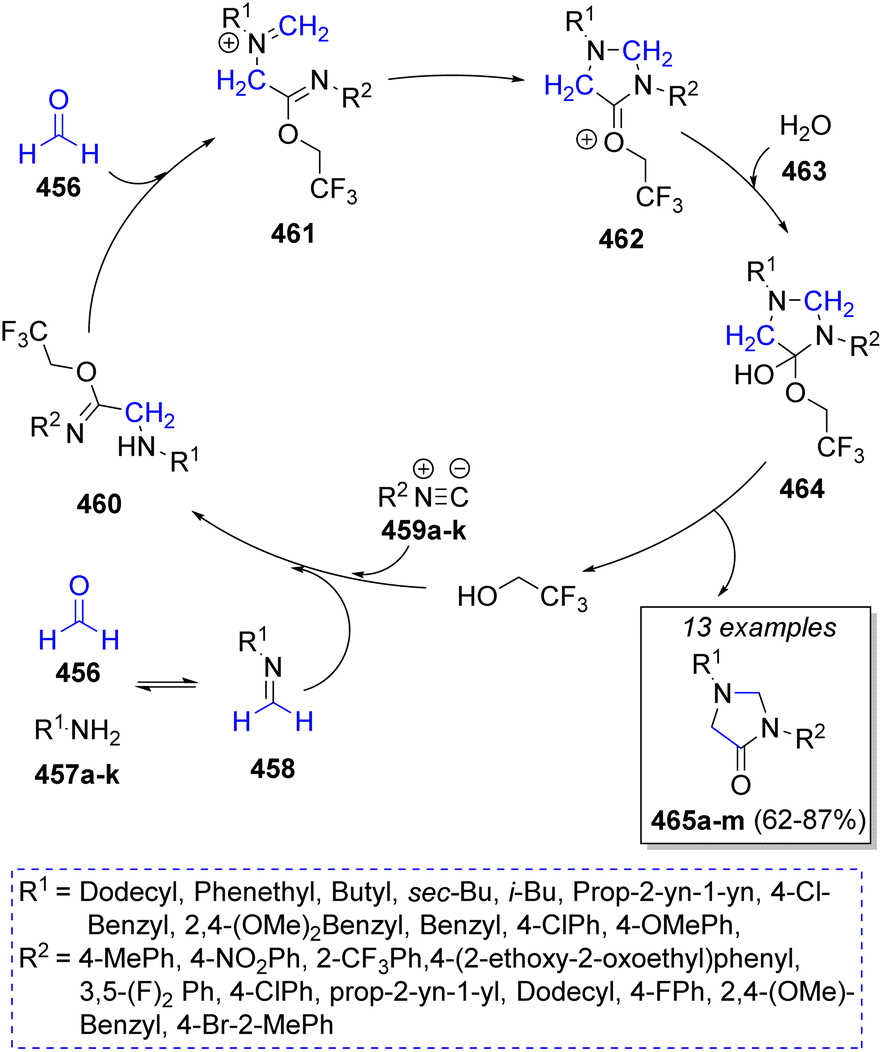 | ||
| Scheme 48 Synthesis of N,N′-substituted 4-imidazolidinones. | ||
In 2020, Ma and co-workers77 reported an acid-catalyzed synthesis of 2,4,6-triarylpyrimidines using a variety of methyl ketones 467a–h, two equivalents of aromatic aldehydes 466a–k, two equivalents of ammonium acetate (469) and trifluoromethanesulfonic acid (TfOH) as catalyst. Two plausible mechanisms were proposed as Scheme 49 depicts. In path A, α,β-unsaturated ketones 468 are formed via aldol condensation of aldehydes 466a–k and methyl ketones 467a–h in the presence of TfOH, which react with ammonium acetate (469) via 1,4-Michael addition, followed by the loss of AcOH to afford intermediates 470, which then condense with another molecule of ammonium acetate (469) to form the imine-type intermediates 471, which further condense with a second molecule of aldehydes 466 to afford 472 and upon a final intramolecular cyclization and oxidation, the products 475a–s are formed. On the other hand, via path B, intermediates 470 react with previously formed imines 476 (from the reaction between aldehydes 466a–k and NH4OAc (469)) to form the intermediates 473, which then undergo an intramolecular cyclization producing the intermediates 474, which upon air oxidation, generates the final products 475a–s.
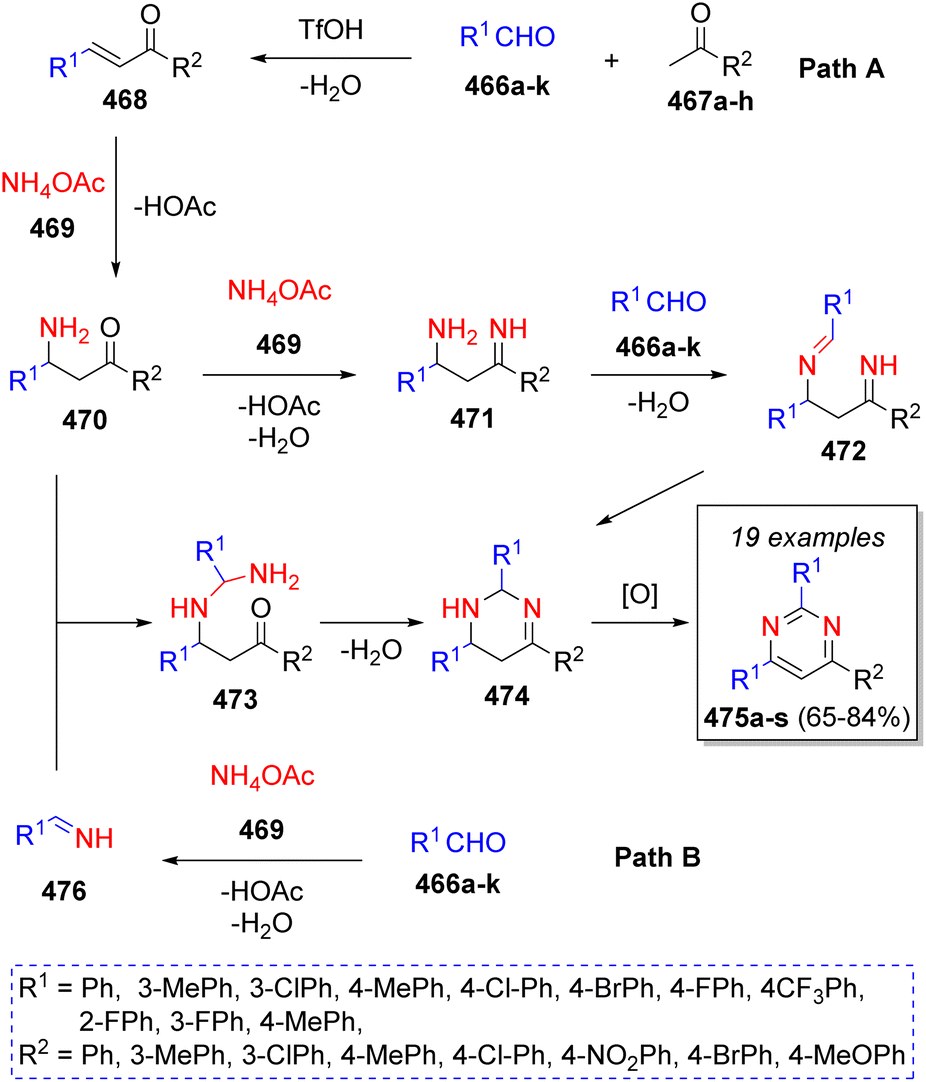 | ||
| Scheme 49 Synthesis of triarylpyrimidines via pseudo-5CR. | ||
Recently, in 2020 a series of 6,6′(arylmethylene)bis(benzo[a]phenazine-5-ol)s were synthesized by Olyaei and co-workers,78 using 2-hydroxynaphthalene-1,4-dione (477), o-phenylenediamine (478), aromatic aldehydes 480a–i, with p-toluensulfonic acid (p-TsOH) as catalyst and 2-aminopyridine (481) as co-catalyst, via a tandem pseudo-5CR. According to the proposed reaction mechanism illustrated in Scheme 50, 477 tautomerizes to 477′, which then reacts with o-phenylenediamine (478) to afford benzo[a]phenanzin-5-ol 479. Additionally, aromatic aldehydes 480a–i condense with 2-aminopyridine (481), in the presence of p-TsOH producing Schiff bases 482 as intermediates. Next, intermediate 479 reacts with intermediates 482 affording 483, which then tautomerize to compounds 483′. Upon loss of a 2-aminopyridine fragment, o-quinonemethide 484 is produced. The desired products 485a–i are formed after Michael addition of benzo[a]phenazine-5-ol 479 unto the 484 intermediates.
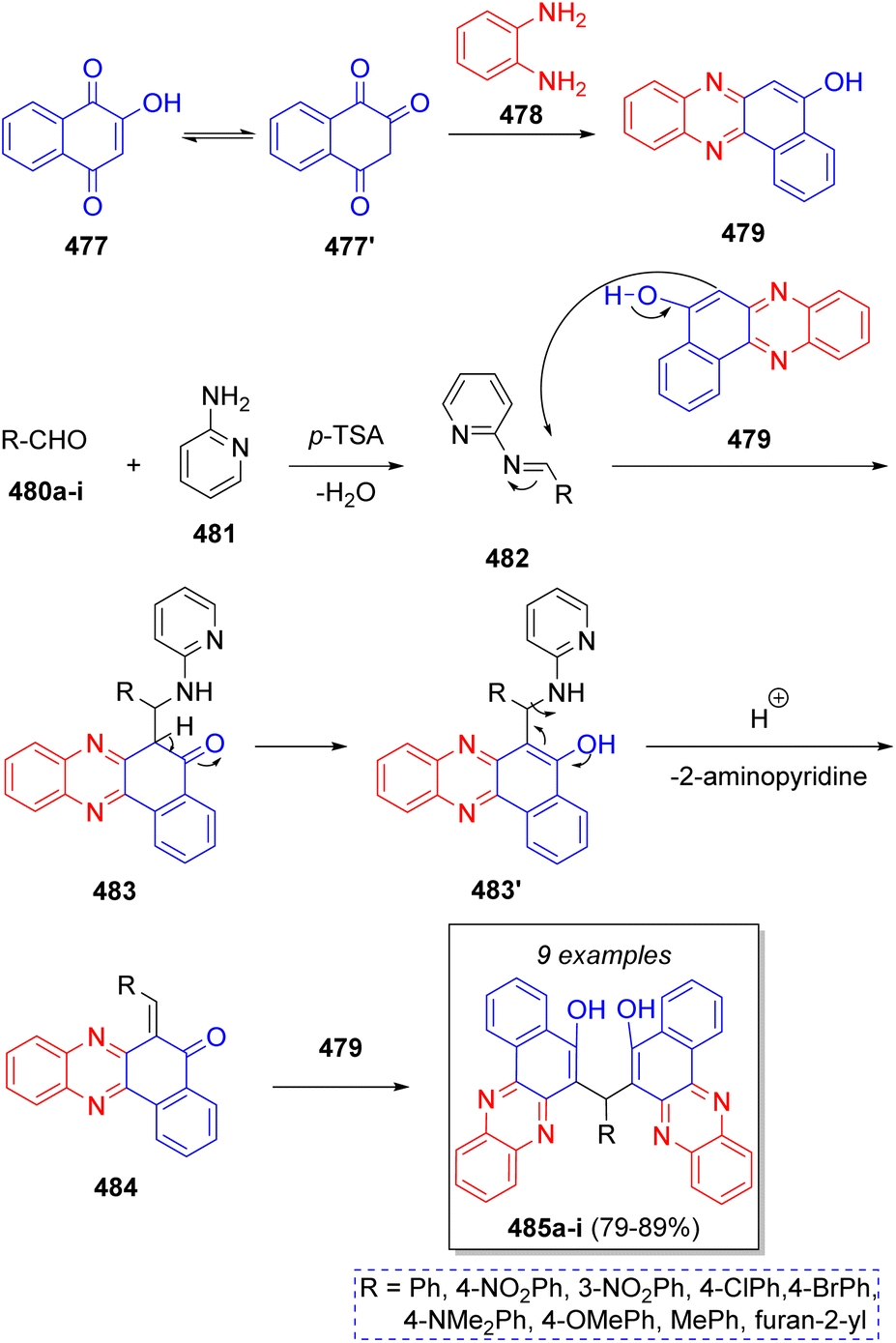 | ||
| Scheme 50 Synthesis of 6,6′(arylmethylene)-bis(benzo[a]phenazine-5-ol). | ||
5. Pseudo-six component reactions
5.1 Knoevenagel/Michael addition
In 2016, Mousavi and co-workers79 reported a simple and eco-friendly method to obtain bis-spiro piperidines using formaldehyde (486), dimedone (487), aromatic amines 493a–i and acetic acid as solvent and catalyst. The proposed mechanism for this pseudo-6CR is illustrated in the Scheme 51. Initially, formaldehyde (486) is protonated and dimedone (487) tautomerized by acetic acid. Subsequently, intermediate 488 and enol form of 487′ react via Knoevenagel condensation to afford intermediate 489, which after the loss of water, the α,β-unsaturated carbonyl compound 490 is produced. Next, intermediate 491 is formed from the Michael addition of another enol molecule 487′ to intermediate 490. On the other hand, the reaction between an activated aldehyde 492 and amines 493a–i produces imine intermediates 494 which then react with intermediate 491 via Mannich reaction to give intermediates 495. Further reaction with another activated formaldehyde 492 generates iminium ions 496. The final products 497a–i are formed after an intramolecular cyclization via Mannich reaction.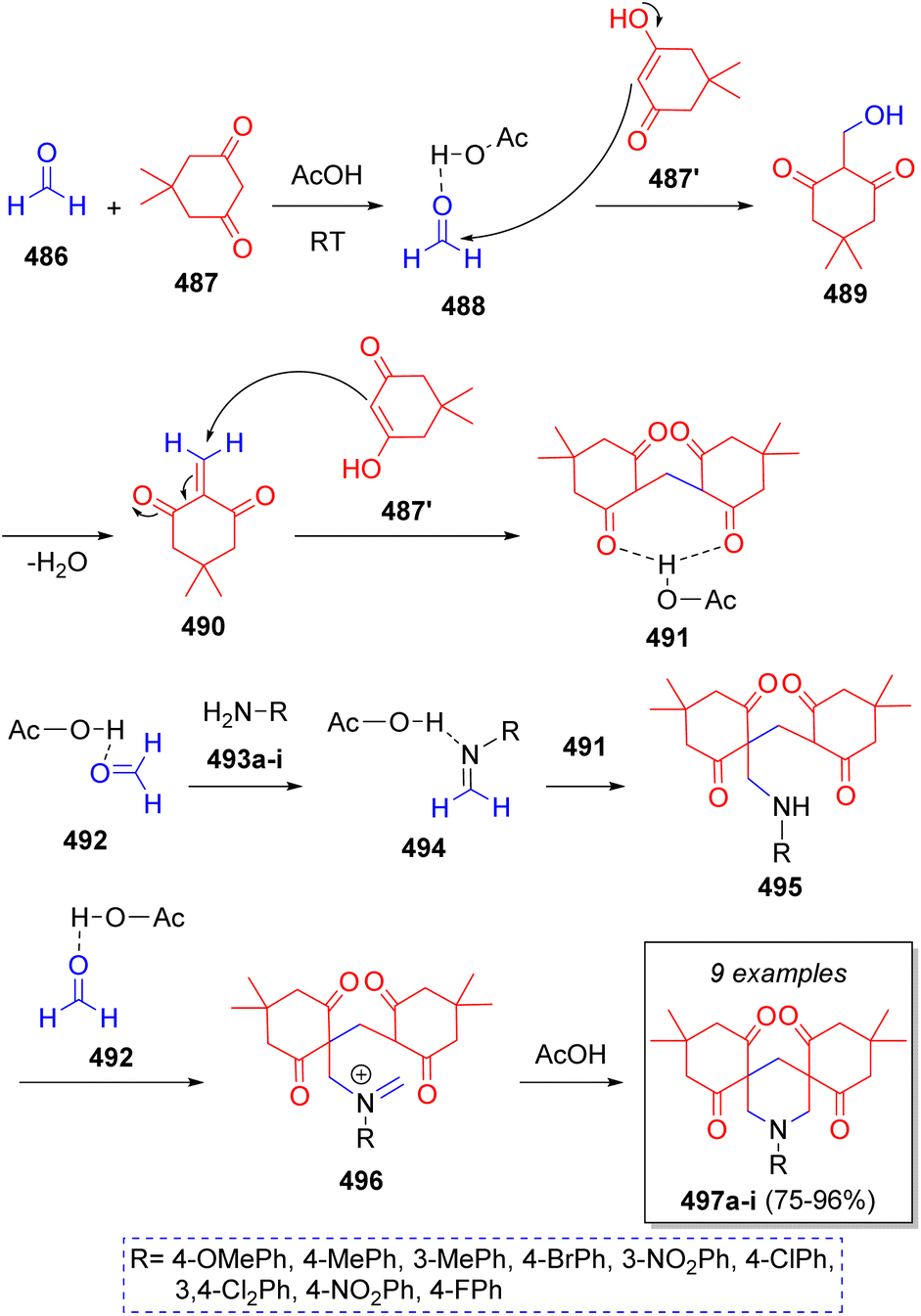 | ||
| Scheme 51 Synthesis of bis-spiro piperidines via a Mannich type pseudo-6CR. | ||
A stereoselective synthesis of cis,cis-2,4,6-triaryl-3,3,5,5-tetracyanopiperidines was achieved by Vereshchagin and co-workers.80 The cascade pseudo-6CR involved the use of two equivalents of malononitrile (498), three equivalents of aromatic aldehydes 499a–g, and ammonium acetate or aqueous ammonia 502, in methanol as solvent. The reaction sequence is depicted in the Scheme 52. Initially, ylidene-malononitriles 500 are formed from the reaction between malononitrile anion 498′ and aromatic aldehydes 499a–g via Knoevenagel condensation prompted by ammonium acetate. Michael addition of another molecule of malononitrile anion 498′ to 500 affords the 1,1,3,3-tetracyanopropene anions 501. Next, tetracyanoamides 503 are produced from the Mannich reaction of 501, aldehydes 499a–g and ammonia (from ammonium acetate). These intermediates then react with another molecule of aldehydes 499a–g which leads to Schiff bases 504, which upon further intramolecular cyclization afford the final cyclic amines 505a–g.
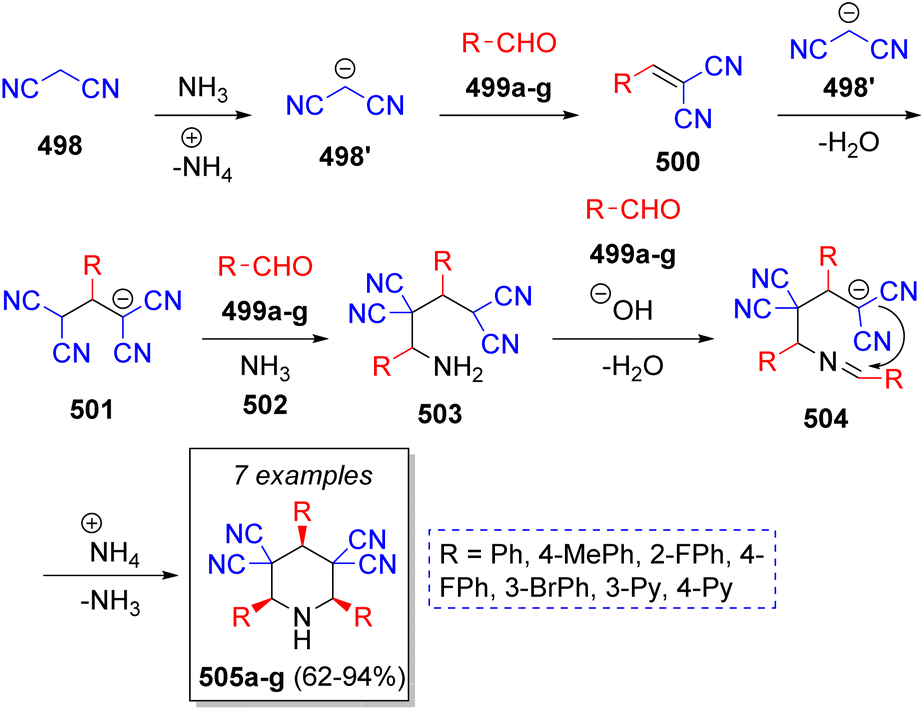 | ||
| Scheme 52 Synthesis of cis,cis-2,4,6-triaryl-3,3,5,5-tetracyanopiperidines. | ||
A collection of fused tetrahydrodipyrazolopyridines was synthesized by Safaei-Ghomi and co-workers81 via a highly efficient pseudo-6CR using ionic liquid supported on FeNi3 as nanocatalyst. This procedure involved the reaction between ethyl acetoacetate (506), hydrazine (507), aromatic aldehydes 509a–k, and ammonium acetate 512 as source of ammonia. A plausible reaction mechanism was suggested for the synthesis of the products as illustrated in Scheme 53a. The first step consists of a nucleophilic attack of hydrazine 507 on ethyl acetoacetate 506 to form the pyrazolone intermediate 508. This intermediate then reacts with activated aldehydes 509a–k via Knoevenagel condensation to form intermediates 510. Then, another molecule of pyrazolone 508 attacks intermediates 510 to afford 511. Afterwards, intermediates 511 are attacked by ammonia producing 513, which upon a subsequent intramolecular condensation, the desired products 514a–l are formed. Other collection of tetrahydrodipyrazolopyridines 515a–l (Scheme 53b) were synthetized by Tamaddon and Khorram82 using a similar strategy in water. This environmentally friendly procedure involved alkyl acetoacetates (two equivalents), hydrazine hydrate (two equivalents), ammonium carbonate and a variety of aromatic aldehydes.
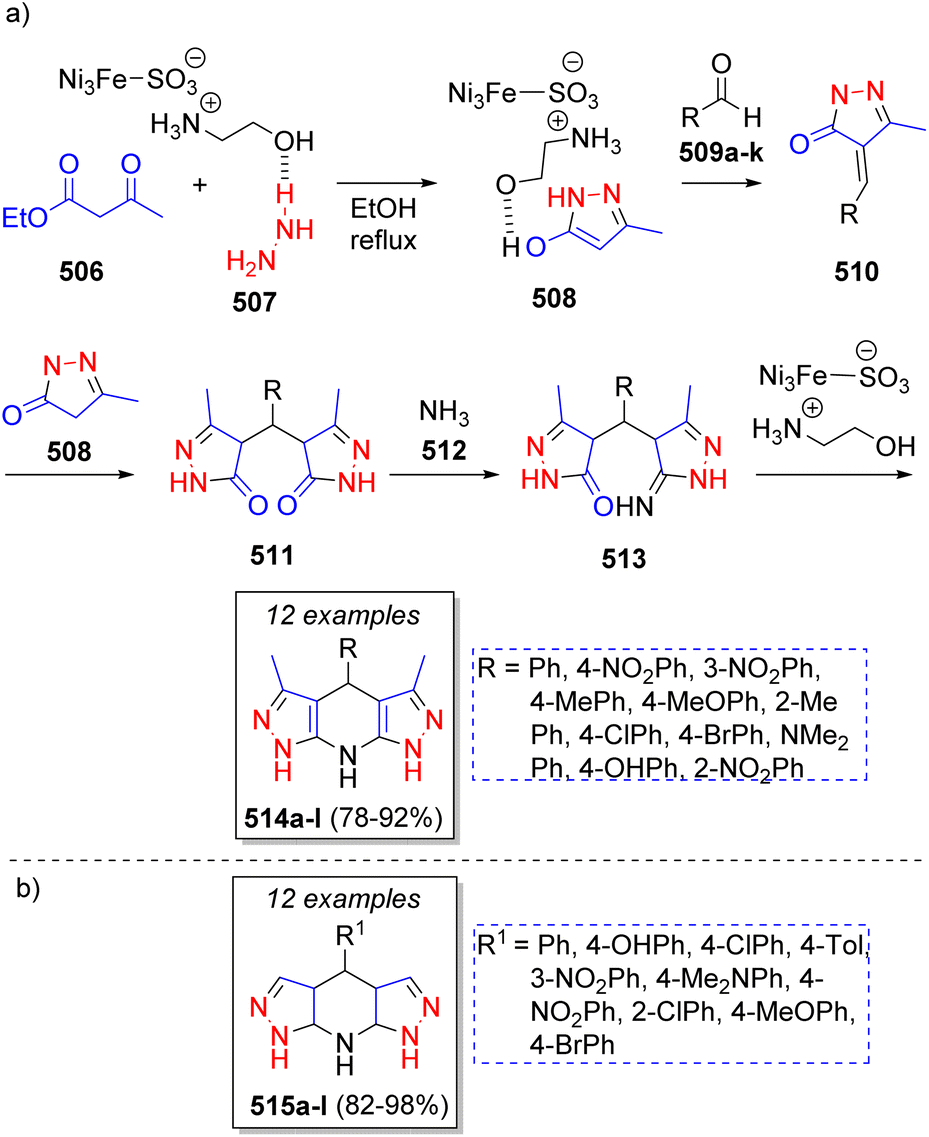 | ||
| Scheme 53 (a) Synthesis of tetrahydrodipyrazolo pyridines. (b) Library of tetrahydrodipyrazolopyridines reported by Tamaddon and Khorram. | ||
The synthesis of many novel substituted pyrazoles were reported in 2018 by Rezvanian and co-workers83 using aromatic aldehydes 516a–f, malononitrile (517), hydrazine (519), and nitro ketene dithioacetal (520). The mechanism for the pseudo-6CR is depicted on Scheme 54a. Initially, aldehydes 516a–f react with malononitrile 517 via Knoevenagel condensation to form intermediates 518. Next, the SN2 reaction of a 2.5![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 ratio of hydrazine (519) with nitro ketene dithioacetal (520) leads to the formation of 1,1-dihydrazino-2-nitroethylene (521) with the subsequent loss of two molecules of MeSH. The reaction of 521 with another molecule of aromatic aldehydes 516a–f affords intermediates 522. Subsequently, amine group of intermediates 522 attack the previously formed intermediates 518 producing 523. Finally, 523 undergo an intramolecular cyclization via nucleophilic attack of amino group onto the CN group to form 524, which then carry out a rapid imine-enamine tautomerization to afford the final products 525a–f.
:1 ratio of hydrazine (519) with nitro ketene dithioacetal (520) leads to the formation of 1,1-dihydrazino-2-nitroethylene (521) with the subsequent loss of two molecules of MeSH. The reaction of 521 with another molecule of aromatic aldehydes 516a–f affords intermediates 522. Subsequently, amine group of intermediates 522 attack the previously formed intermediates 518 producing 523. Finally, 523 undergo an intramolecular cyclization via nucleophilic attack of amino group onto the CN group to form 524, which then carry out a rapid imine-enamine tautomerization to afford the final products 525a–f.
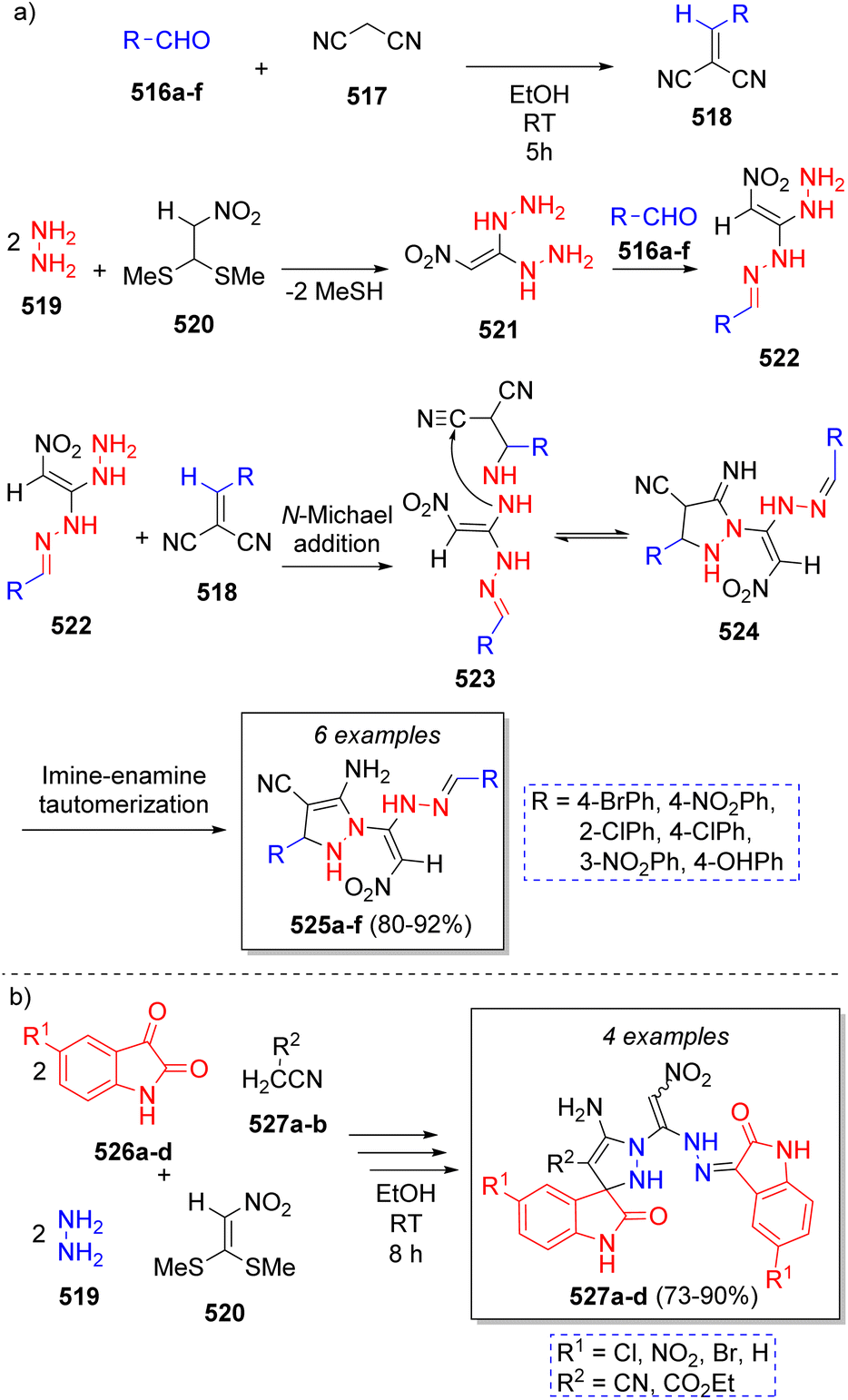 | ||
| Scheme 54 (a) Synthesis of substituted 1H-pyrazoles-4-carbonitriles. (b) Synthesis of highly functionalized spiropyrazolines. | ||
Using a similar reaction sequence, Rezvanian and Babashah84 reported the synthesis of highly functionalized spiropyrazolines via a pseudo six-component reaction comprising isatins 526a–d as the carbonylic component, active methylene compounds 527a–b, hydrazine hydrate (519) and nitro ketene dithioacetal (520), which afforded the products 527a–d as a mixture of two diastereomers in good to excellent yields (Scheme 54b).
5.2 Others
A series of tetrazolo-spiroquinazolinones were synthesized by Balalaie and co-workers85 via a one-pot stereoselective pseudo-6CR comprising isatoic anhydride (528), hydrazine (529), cyclic ketones 531a–e (2 equivalents), isocyanides 535a–b and trimethylsilyl azide (534). The proposed reaction mechanism is outlined in Scheme 55. It is proposed that initially, hydrazine (529) reacts into the carbonyl group of isatoic anhydride (528), followed by ring opening and CO2 elimination to afford the intermediate 530. Upon a double condensation with cyclic ketones 531a–e, hydrazones 532 are formed and upon protonation, the corresponding intermediates 533 are produced. Aminal intermediates 536 are generated by intramolecular nucleophilic attack of the amide nitrogen onto the imine moiety and axial nucleophilic attack of isocyanides 535a–b on the imine. These intermediates undergo a nucleophilic addition of the azide ion producing 537, followed by a final cyclization to generate the 1,5-disubstituted tetrazoles 538a–h.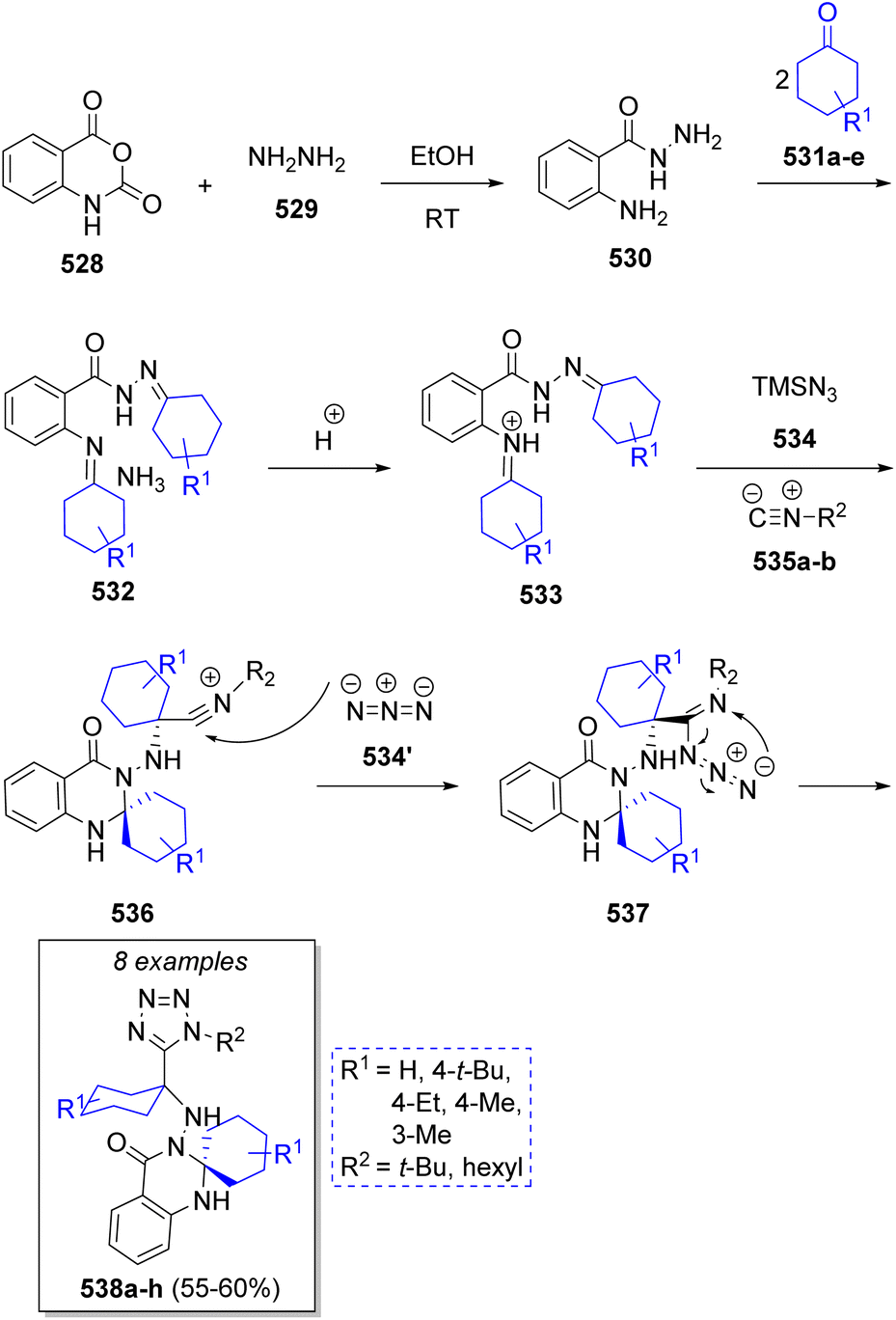 | ||
| Scheme 55 Synthesis of tetrazolospiroquinazolinones. | ||
6. Pseudo-seven component reactions and more
In 2010, a small collection of new phosphanylidene bis(2,5-dioxotetrahydro-1H-pyrrole-3-carboxylates) were prepared by Alizadeh and co-workers86 via a diastereoselective pseudo-7CR. The reaction involved two equivalents of dialkyl acetylenedicarboxylates (DAAD) 539a–b, triphenylphosphine (TPP, 540), two equivalents of isocyanides 543a–b and two molecules of water (545). As Scheme 56 depicts, the mechanism proceeds initially via the formation of zwitterions 541 by the addition of TPP to 539a–b, which then react with another molecule of DAAD 539a–b to produce intermediates 542. Then, 542 are protonated by TFA to generate the ion-paired intermediates 542′. Nucleophilic addition of isocyanides 543a–b produces nitrilium ion intermediates 544 to which a water molecule is added to produce 546. These intermediates undergo an intramolecular cyclization to afford ylides 547, followed by addition of isocyanides 543a–b and a water molecule to afford intermediates 548. Finally, a proton shift prompts another intramolecular cyclization, expelling an alcohol molecule, to provide the desired products 549a–e.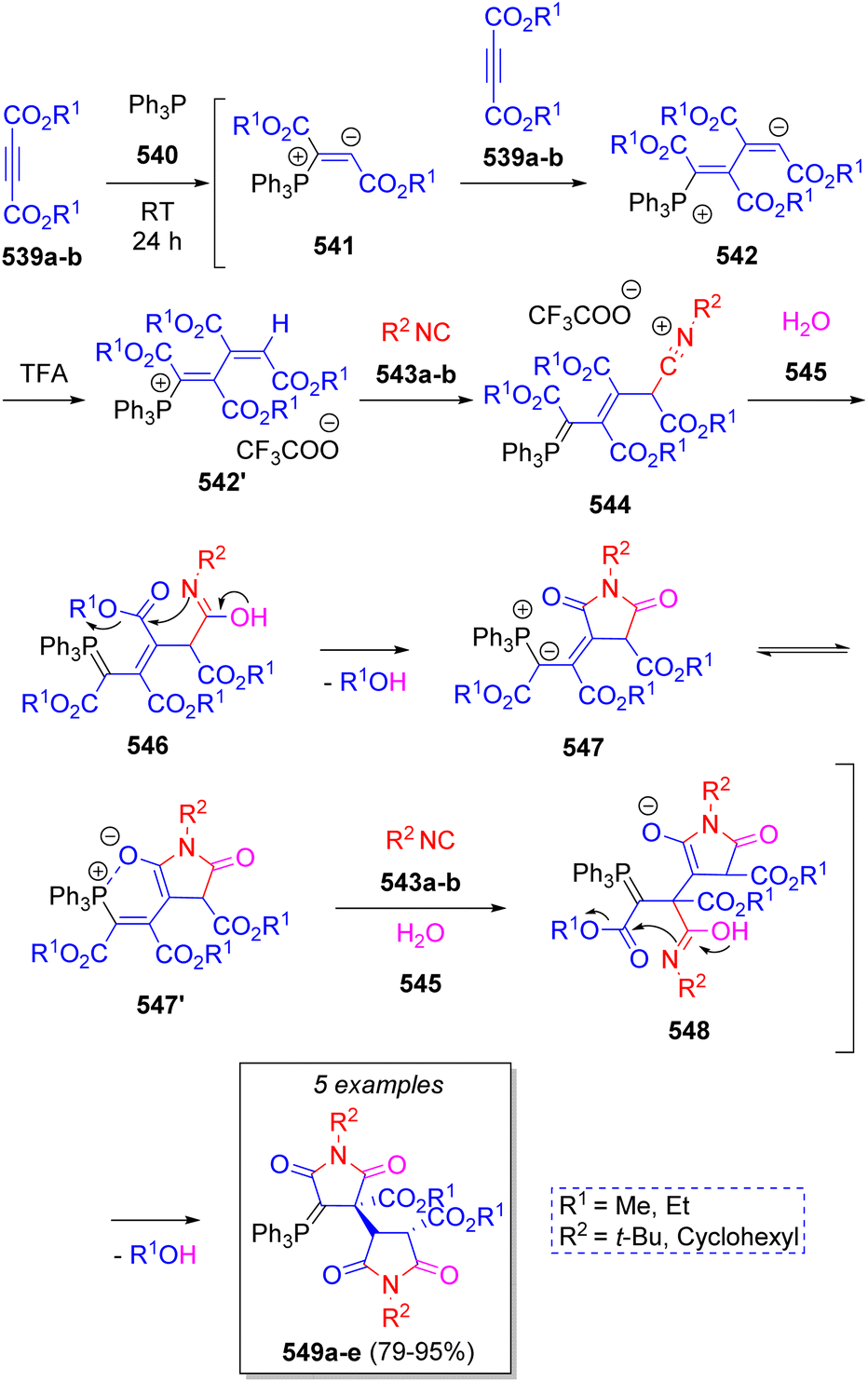 | ||
| Scheme 56 Synthesis of phosphanylidene bis(2,5-dioxotetrahydro-1H-pyrrole-3-carboxylates). | ||
In 2015, a series of bis-1,5-disubstituted-1H-tetrazoles were reported by Gámez-Montaño and co-workers via pseudo seven double Ugi-azide MCR, obtaining excellent yields (88–95%) at room temperature and slightly lower yields (80–91%) using MW conditions but in a reduced time.87 The reaction involved different amines 550a–c, two equivalents of a variety of aldehydes 551a–b, two equivalents of isocyanides 553a–j, and two equivalents of azide anion (555 from trimethylsilyl azide) in methanol. Scheme 57a illustrates a reasonable mechanism for the reaction. Initially, intermediates 552 are formed from condensation of amines 550a–c and aldehydes 551a–b to which isocyanides 553a–j are added to afford nitrilium ions 554. Next, azide anion 555 adds to 554 to give intermediates 556 which undergo an intramolecular cyclization to afford the mono-tetrazoles 557. These intermediates react again with aldehydes 551a–b to afford iminium ions 558, which react with isocyanides 553a–j to afford 559, and then a second molecule of azide 555 provides intermediate 560, which after an intramolecular cyclization leads to the final products 561a–e. Other collection of bis-1,5-disubstituted-1H-tetrazoles 562a–p via pseudo seven-component Ugi-azide reactions were reported in 2019 by Nenajdenko and co-workers88 using various amines, carbonyl compounds, isocyanides and TMS (as source of N3) in methanol at room temperature. Scheme 57b illustrates the compounds reported in this work.
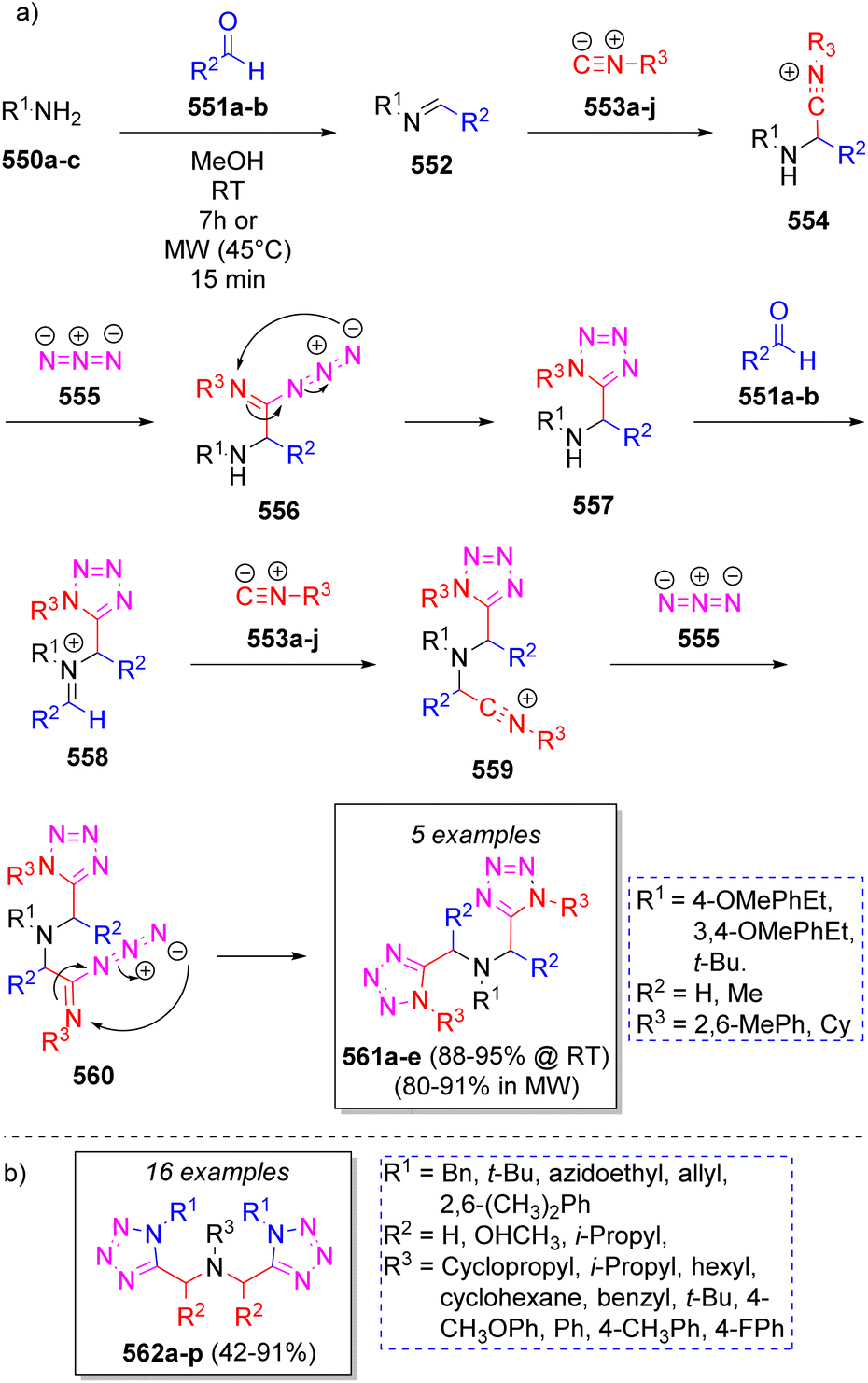 | ||
| Scheme 57 (a) Synthesis of bis-1,5-disubstituted-1H-tetrazoles. (b) Compound library synthesized by Nenajdenko and co-workers. | ||
In 2018, Hasaninejad and Mojikjalifen89 reported the synthesis of novel poly substituted pyrazolyl-1,2-diazepine scaffolds via a one-pot pseudo-7CR from condensation of 1,1-bis(methylthio)-2-nitroethylene (BMTNE) (563), two equivalents of hydrazine (564), two equivalents of aryl aldehydes 566a–l, and two equivalents of malononitrile (567) in EtOH as solvent. The synthetic strategy is depicted in the Scheme 58. Initially, a nucleophilic substitution of hydrazine 564 on methylsulfanyl groups of BMTNE (563) leads to the formation of intermediate 565. Now, two pathways (A and B) are possible. In pathway A, intermediates 570 are formed from the Schiff base formation between 565 and aryl aldehydes to form diimines 568, which then react with two molecules of malononitrile 567. In pathway B, intermediates 570 are formed from the Knoevenagel condensation of malononitrile with aryl aldehydes 566a–l to form intermediates 569 which then undergo Michael addition of 565. Next, intermediates 570 undergo an intermolecular cycloaddition to afford 571, followed by three imine-enamine tautomerizations to produce 572 and after a final air oxidation the desired products 573a–l are formed.
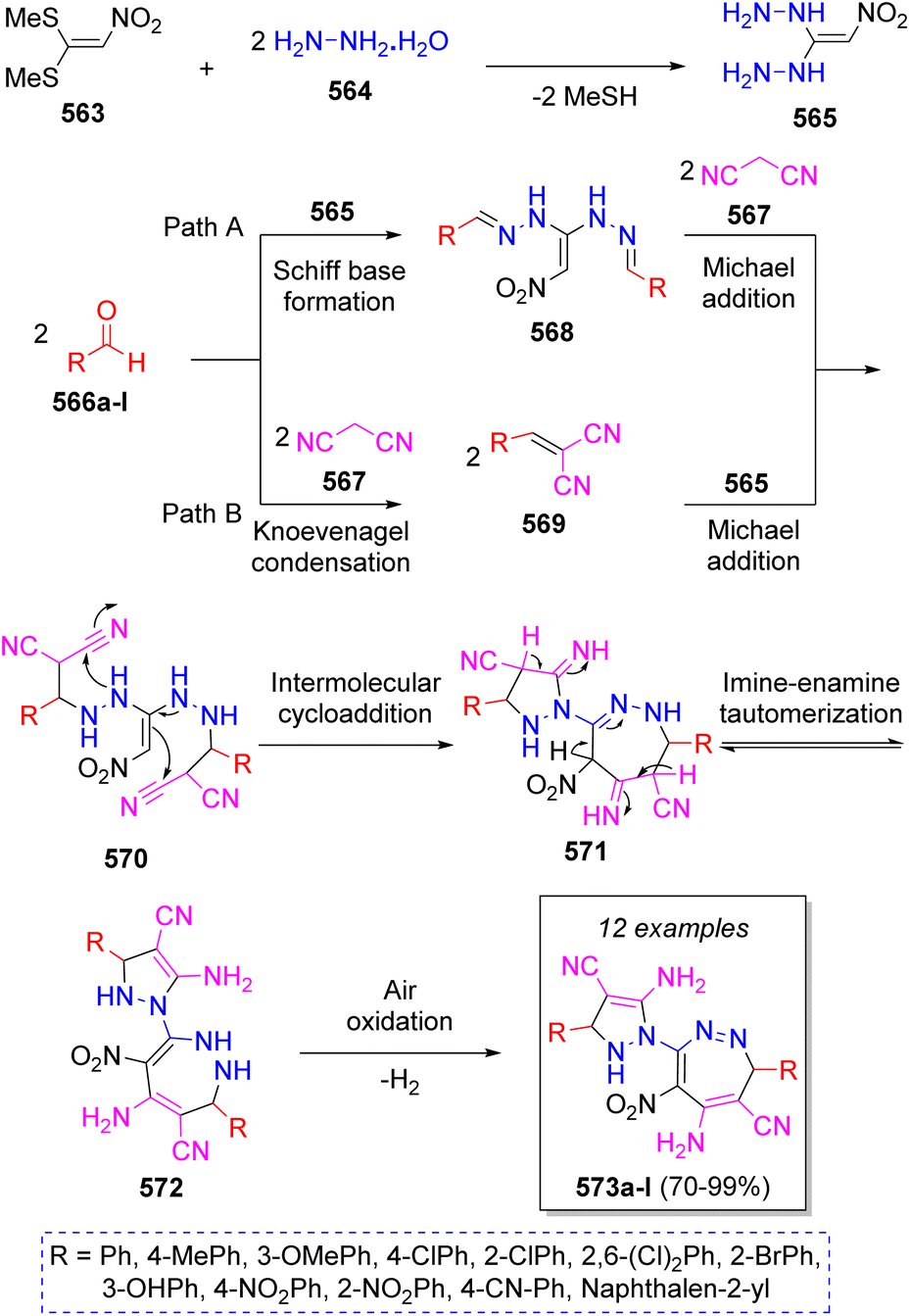 | ||
| Scheme 58 Synthesis of poly substituted pyrazolyl-1,2-diazepines. | ||
A collection of fully substituted furans containing pseudopeptide base were successfully prepared via an efficient pseudo-7CR by Shaabani and co-workers90 in 2020. The reaction comprises Meldrum's acid (574), benzaldehydes 575a–b, two different isocyanides 577a–b, and acetylenedicarboxylates 582a–b in a mixture of water and acetonitrile at 70 °C. According to Scheme 59, the proposed reaction proceeds via Knoevenagel condensation between aromatic aldehydes 575a–b and Meldrum's acid (574) to give intermediates 576, followed by a [1 + 4] cycloaddition of isocyanides 577a–b to produce iminolactones 578, which then react with water to produce 579. These intermediates lose an acetone molecule to produce ketenes 580, to which a water molecule is added producing 581. These compounds undergo decarboxylation to afford mono carboxylic acids 583. Simultaneously, the reaction between acetylenedicarboxylates 582a–b and isocyanides 577a–b produce a zwitterionic species which are protonated by 583 affording nitrilium ions 584 and carboxylate 583′. Both intermediates then react to produce 585, which undergo a Mumm rearrangement to generate 586. A [1 + 4] cycloaddition of isocyanides 577a–b with the newly formed 586 affords iminolactones 587 which tautomerize to provide the final products 588a–p.
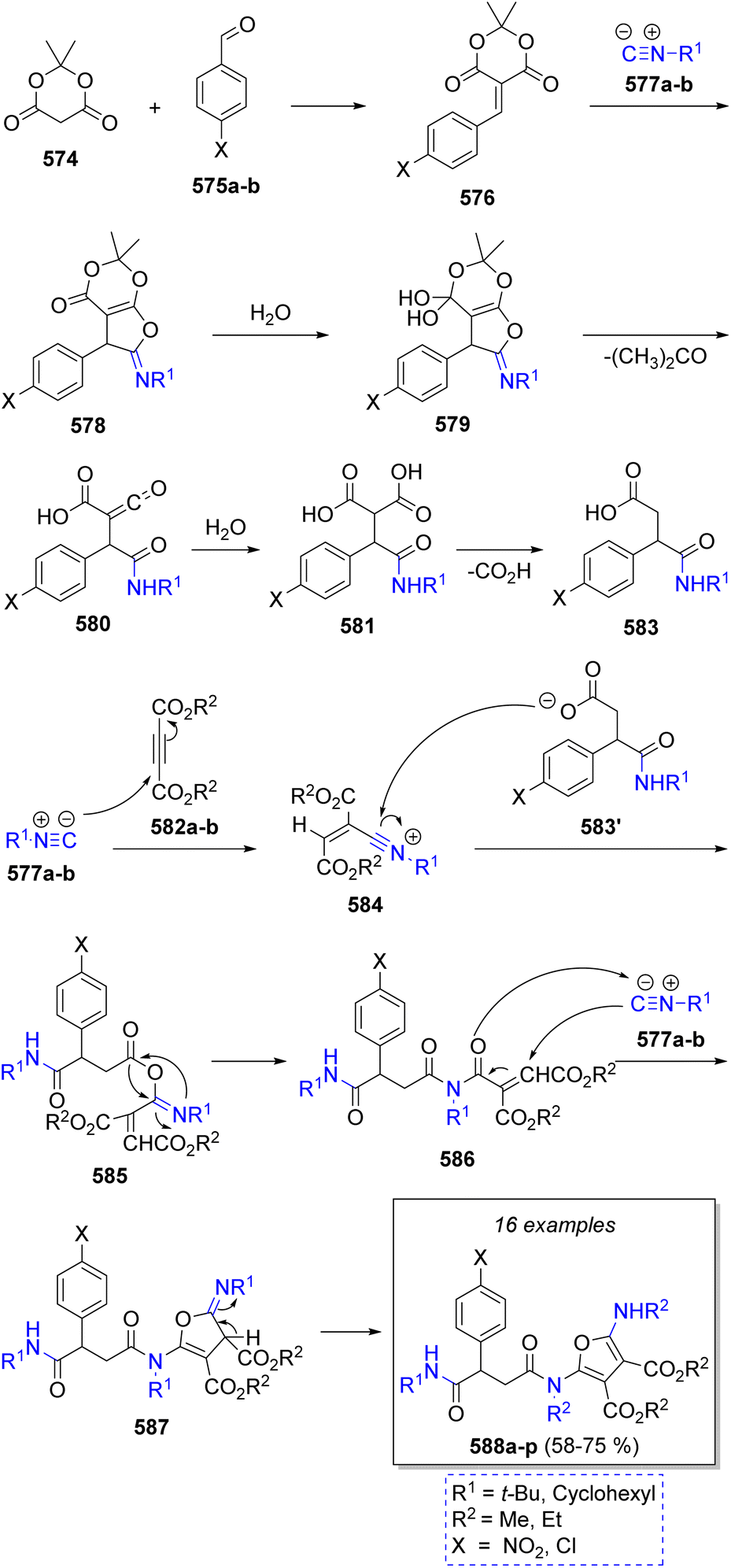 | ||
| Scheme 59 Synthesis of fully substituted furans containing pseudopeptide base. | ||
Yielzoleh and Nikoofar91 reported the synthesis of a collection of 7,7′-((aryl/alkyl)methylene)bis(N-cyclohexyl-2-(aryl/alkyl)-6-methyl-3H-imidazo[1,2-b]pyrazol-3-imines) via a one-pot pseudo-7CR using 3-amino-5-methylpyrazole (589, 2 equiv.), aldehydes 590a–f (3 equiv.) and cyclohexyl isocyanide (592, 2 equiv.) in ethanol as solvent. The suggested reaction mechanism is depicted in Scheme 60. Initially, 3-amino-4-methylpyrazole 589 reacts with aldehydes 590a–f (previously activated by SO3H-D-Leu@SiO2–Fe3O4) to produce imines 591 activated as the iminium salts by catalyst complexation. Next, iminium ions 591 are attacked by cyclohexyl isocyanide (592) to produce the intermediate nitrilium ions 593, which then undergo an intramolecular cyclization to form imines 594. Upon [1,3]-H shift, imidazopyrazoles 595 are formed, which then react with another molecule of aldehydes to produce 596 after releasing a water molecule. This is followed by a condensation of intermediates 595 and 596 to obtain the dimerized imidazopyrazoles 597. Upon [1,3]-H shift and oxidation, the final products 599a–f are obtained.
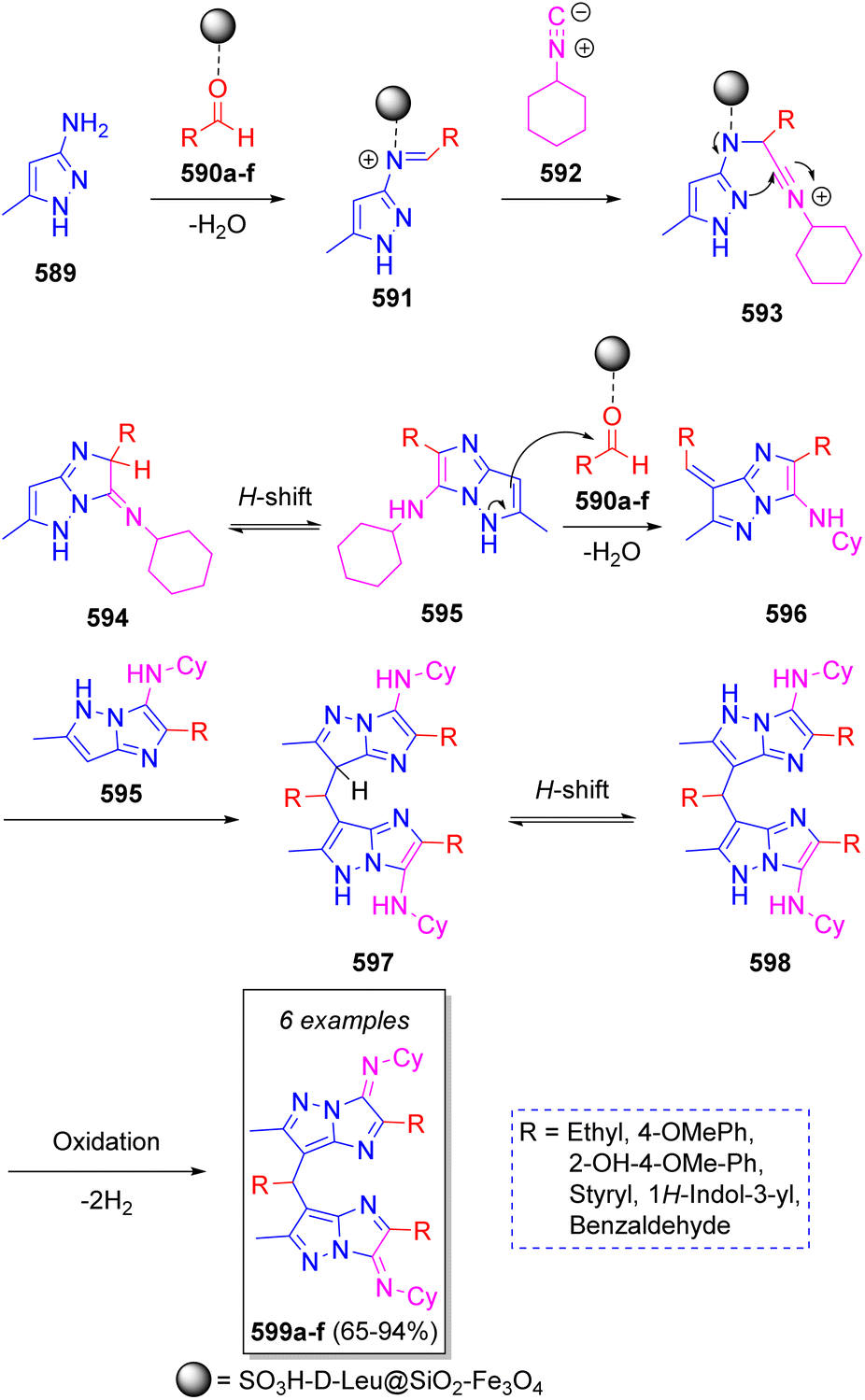 | ||
| Scheme 60 Synthesis of 7,7′-((aryl/alkyl)methylene)bis(N-cyclohexyl-2-(aryl/alkyl)-6-methyl-3H-imidazo[1,2-b]pyrazol-3-imines). | ||
In 2014, Hazeri and co-workers92 reported a diastereoselective synthesis of poly-substituted hydroquinolines via a one-pot domino pseudo-8CR, using three equivalents of Meldrum's acid (600), four equivalents of aromatic aldehydes 601a–h, aromatic amines 605a–e, in acetonitrile in the presence of trichloroacetic acid. A reaction mechanism was proposed as Scheme 61 depicts. Initially, the reaction between Meldrum's acid 600 and benzaldehydes 601a–h via Knoevenagel condensation affords benzylidenes of Meldrum's acid 602, which in acidic conditions are decomposed to 603 and acetone. Subsequently, acetone reacts with anilines 605a–e to produce imines 606 which further tautomerize to enamines 607, which react with the aldehydes to afford dienamines 609. These compounds act as a diene and undergo Diels–Alder cycloaddition with 602 to form intermediates 610. Finally, the Michael addition of the previously formed 603 produces 611, which tautomerize to their enamine-enol forms 612. Then, the reaction with aldehydes 601a–h affords intermediates 613, which after an intramolecular cyclization afford the desired products 614a–m. Notably, the product contains four stereocenters and 10 new bonds were generated in a single vessel operation. Other collections of poly-substituted hydroquinolines were reported using benzoic acid,93 Brønsted acid ionic liquid94 and phthalic acid95 as catalyst.
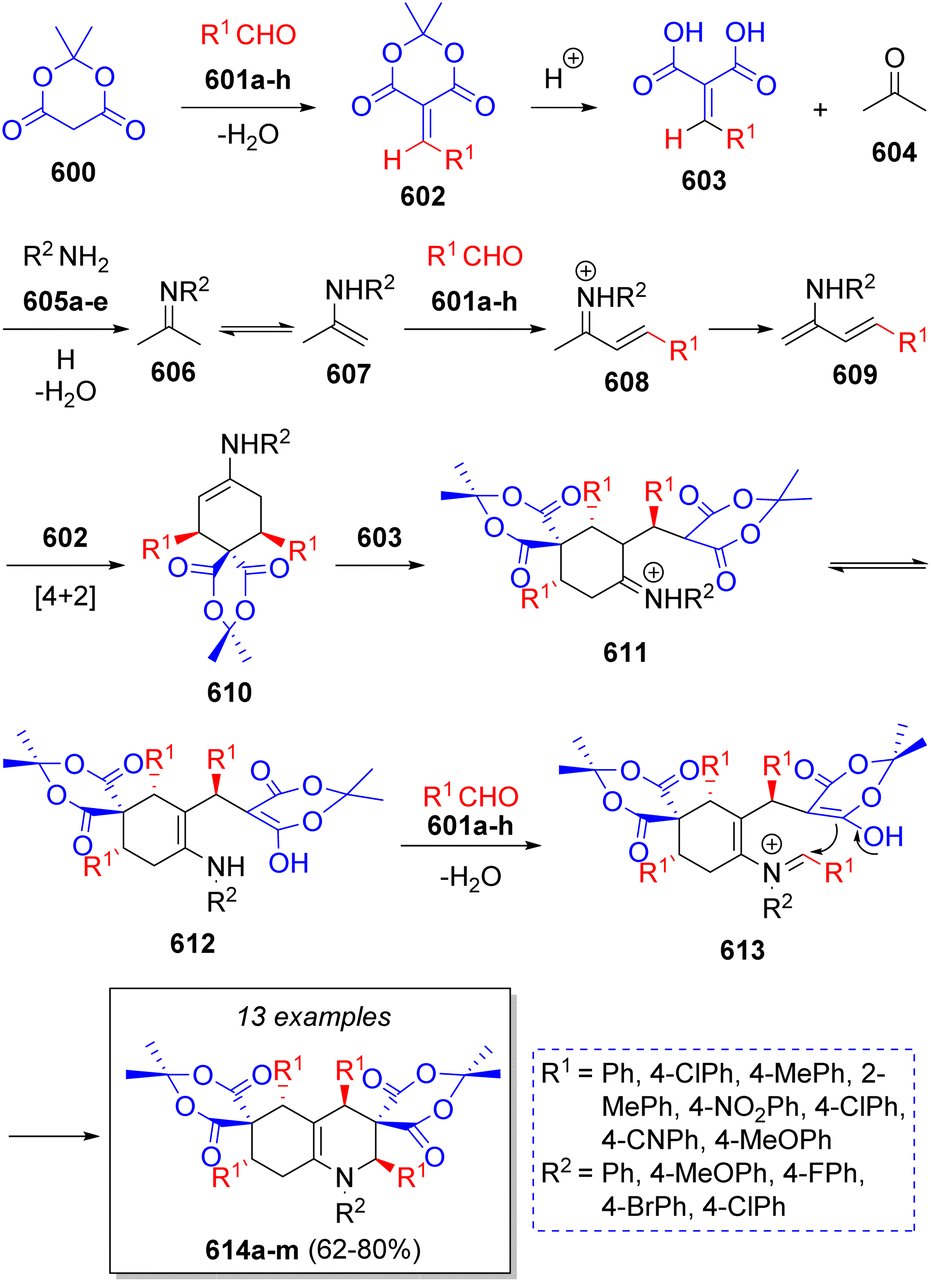 | ||
| Scheme 61 Synthesis of poly-substituted tetrahydroquinolines. | ||
In 2011, a small collection of zinc 1,5-disubstituted-1H-tetrazol-5-yl coordination complexes were prepared by Shaabani and co-workers96 via one-pot pseudo-9CR. The reaction plausibly proceeds via condensation of 1,3-dicarbonyls 615a–b and DMF-DMA (616) to produce intermediates 617, which then coordinate to Zn(II), followed by a Michael addition of isocyanides 619a–c to give the nitrilium ions 620. These intermediates coordinate with the azide anion 621 producing 622, which allow an intramolecular 1,5-cycloaddition to generate the intermediates 624, which further tautomerize to 625. Upon reaction with another molecule of 617, 619 and NaN3 (621), the complete complexes 626a–e are produced (Scheme 62).
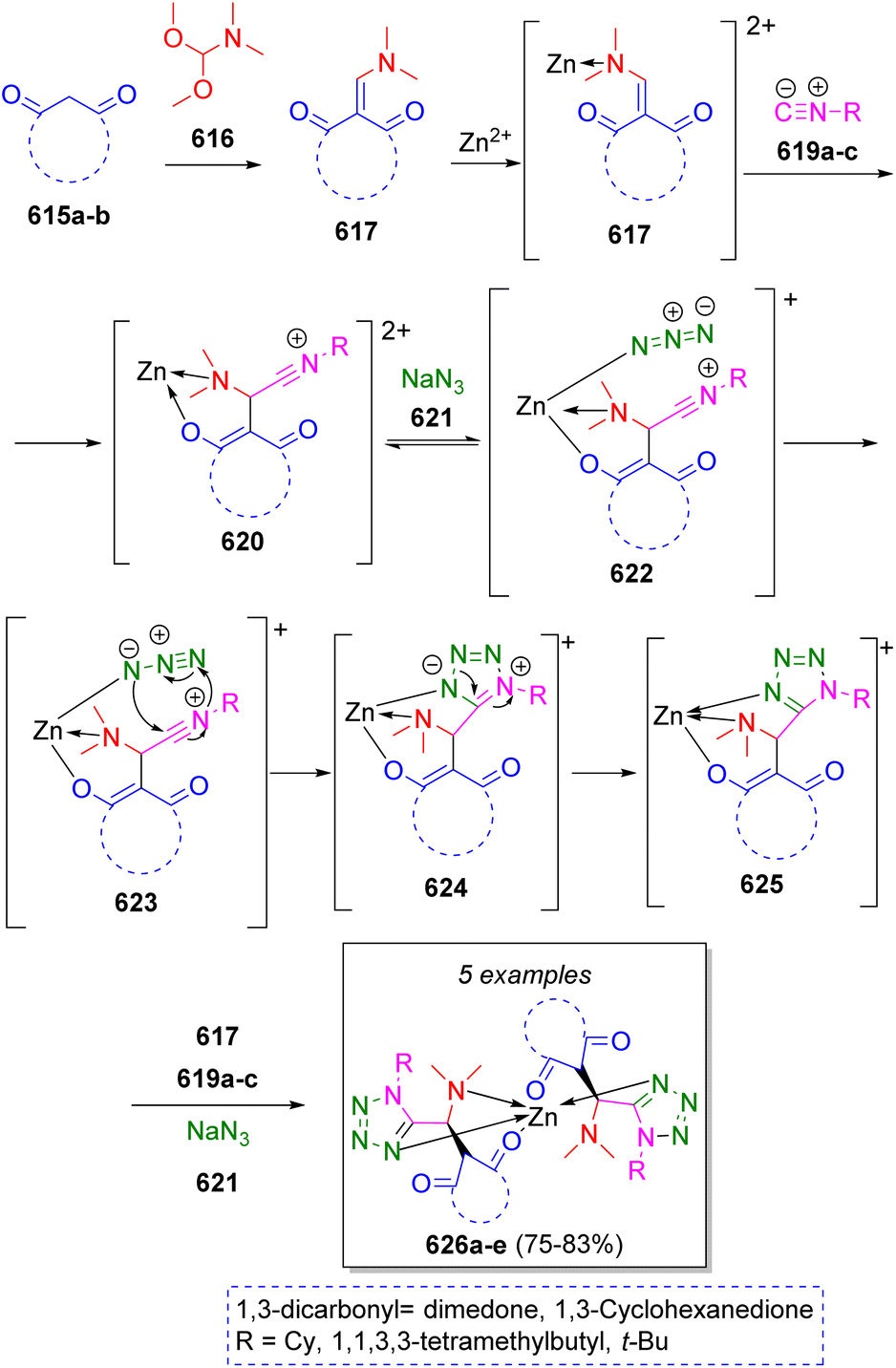 | ||
| Scheme 62 Synthesis of 1,5-disubstituted 1H-tetrazol-5-yl coordination complexes. | ||
7. Repetitive pseudo-MCRs
Repetitive pseudo-MCRs are a subgroup of pseudo-MCRs in which there is a bifunctional reactant participating in the reaction sequence. Most of the examples depicted in this section could be classified as pseudo five-component reactions as well, but the difference in the reaction mechanism leading to the products is enough to warrant its own separate classification. In this case, an MCR occurs simultaneously on both functional groups of the bifunctional reactant. As it will become apparent throughout this section, the products of repetitive pseudo-MCRs are highly symmetrical, which is of high interest for ligand design and can also lead to compounds with interesting optical properties due to the high π-conjugation that can be achieved.Shaabani and co-workers97 reported a method to obtain bis(4H-chromene)-3,4-dicarboxylates via the reaction of two equivalents of isocyanides 627a–f, two equivalents of acetylenedicarboxylates 628a–c and 2,5-dihydroxycyclohexa-2,5-diene-1,4-dione (630) in acetonitrile. The proposed reaction mechanism is illustrated in Scheme 63a. The vinylisonitrilium cations 629 are generated by the reaction between isocyanides 627a–f and alkynes 628a–c via Michael addition. Then, a double aldol condensation takes place between the nucleophilic 2,5-dihydroxycyclohexa-2,5-diene-1,4-dione (630) and two cations 629 producing the keteneimides 631, which via an intramolecular cyclization produce the final products 632a–j via a repetitive pseudo-5CR. During the same year, the same authors98 also reported a similar method to obtain highly functionalized benzo[g]- and dihydropyrano[2,3-g]chromenes via the reaction of two equivalents of aldehydes 633a–f, two equivalents of malononitrile (634) and 2,5-dihydroxycyclohexa-2,5-diene-1,4-dione (636), with triethylamine as catalyst. The suggested mechanism is depicted in Scheme 63b. Initially, a Knoevenagel condensation between aldehydes 633a–f and malononitriles (634) forms intermediates 635, which undergo a double aldol-type condensation with the 2,5-dihydroxycyclohexa-2,5-diene-1,4,-dione (636) producing the intermediates 637, followed by a cyclization to form 638, and a further imine/enamine tautomerization that result in the final products 639a–f.
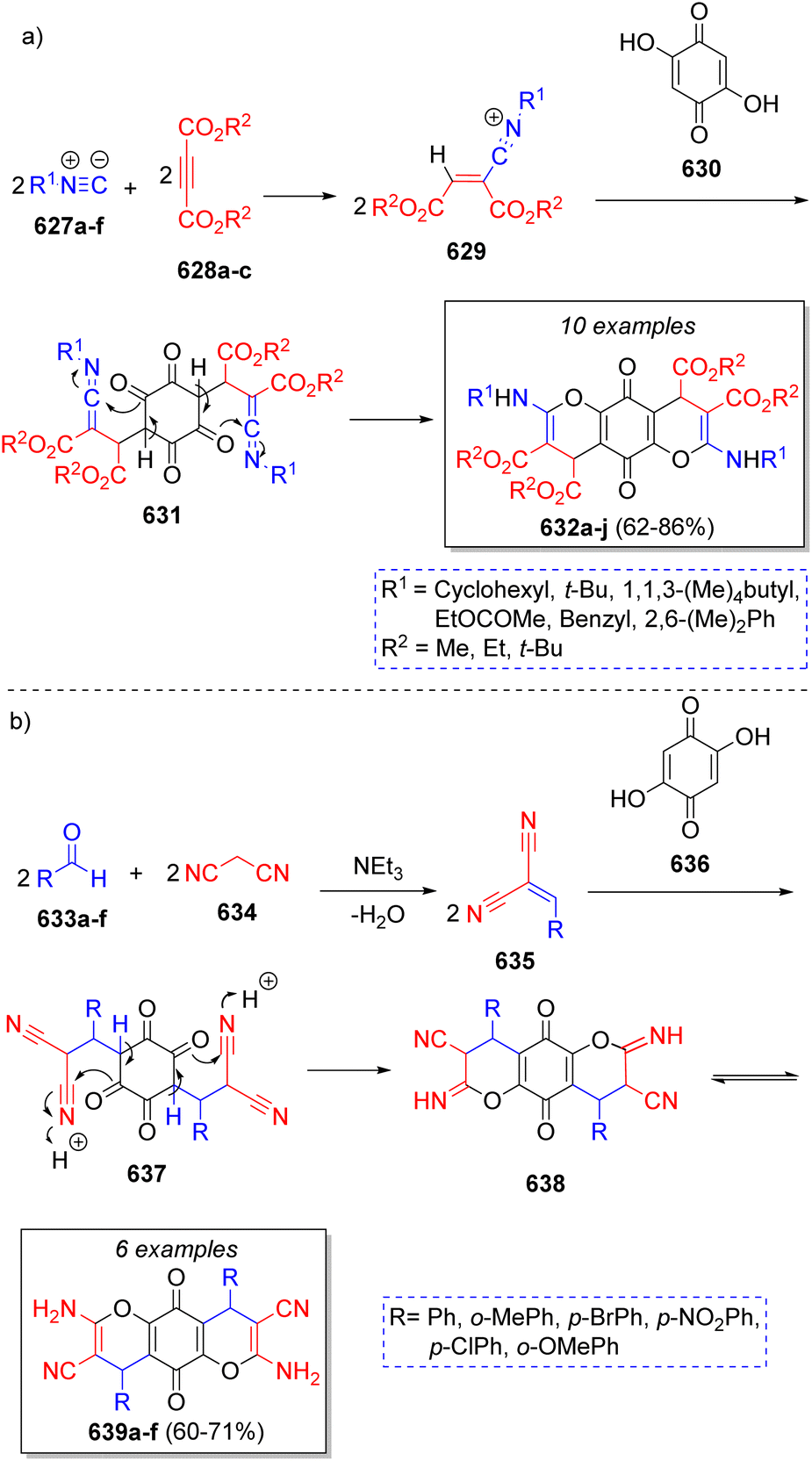 | ||
| Scheme 63 (a) Synthesis of bis(4H-chromene)-3,4-dicarboxylates. (b) Synthesis of highly functionalized benzo[g]- and dihydropyrano[2,3-g]chromenes. | ||
In 2010, Alizadeh and co-workers99 described the synthesis of some bis[2-(arylimino)-1,3-thiazolidin-4-ones] via the reaction of aliphatic diamines 640a–b, two equivalents of isothiocyanatobenzene (641) and two equivalents of dialkyl but-2-ynedioate 643a–b. Scheme 64 illustrates the synthetic methodology. Initially, aliphatic diamines react with two molecules of isothiocyanatobenzene (641) leading to the bis-thiourea intermediates 642, which in turn react with the dialkyl but-2-ynedioates 643a–b to produce the intermediates 644. The desired products 645a–f are obtained after intramolecular lactamization of intermediates 644.
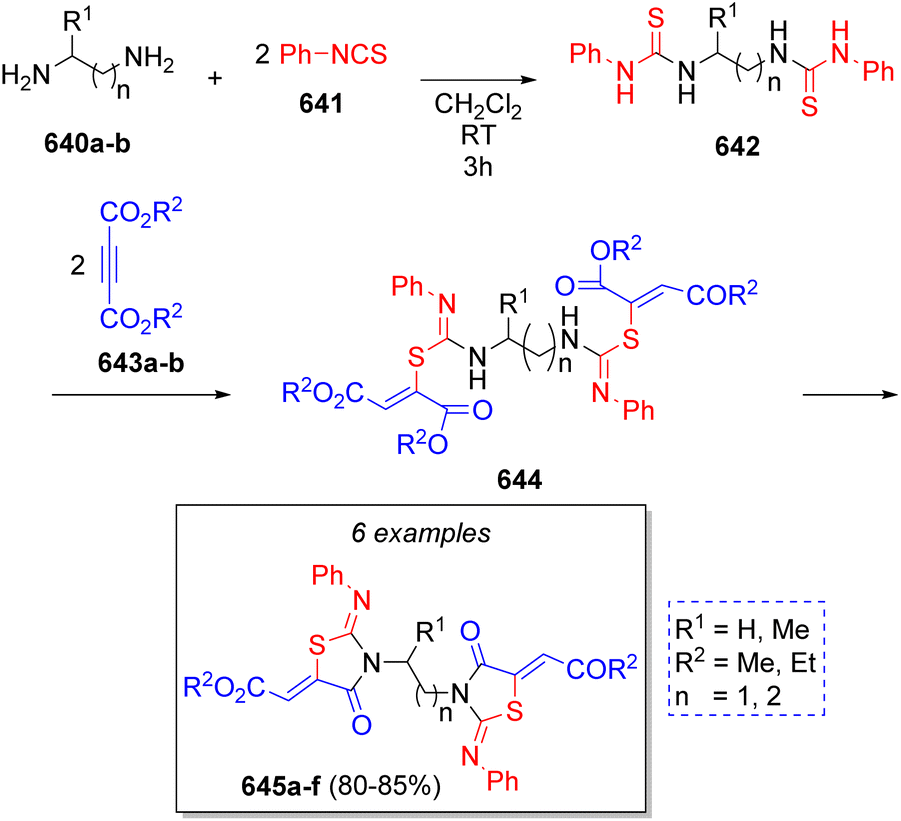 | ||
| Scheme 64 Synthesis of bis[2-(arylimino)-1,3-thiazolidin-4-ones]. | ||
In 2014, Ghahremanzadeh and co-workers100 reported a high yielding repetitive pseudo-5CR to obtain a collection of dispiro[furan-2,1′-naphthalene-4′,2′′-furan] compounds, using two equivalents of isocyanides 646a–e, two equivalents of acetylenedicarboxylates 647a–b and 2,3-dichloronaphthalene-1,4-dione (649). A suggested reaction mechanism for this transformation is shown in Scheme 65. Initially, isocyanides 646a–e and dialkyl acetylenedicarboxylates 647a–b react to form zwitterionic intermediates 648, which then react simultaneously with the 2,3-dichloronaphthalene-1,4-dione (649) to afford the dipolar intermediates 650. Next, these latter ones undergo a double intramolecular cyclization to form the final di-spiro compounds 651a–h.
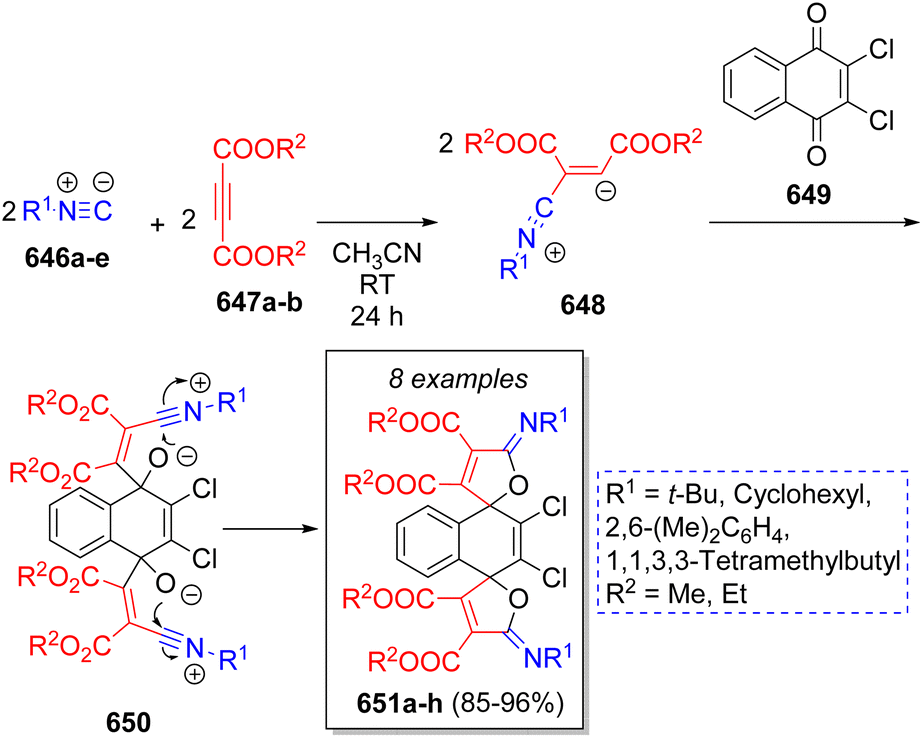 | ||
| Scheme 65 Synthesis of dispiro[furan-2,1′-naphthalene-4′,2′′-furan] compounds. | ||
In 2021, Abdelhamid and Elwahy101 achieved the synthesis of novel bis(14H-dibenzo[a,j]xanthenes), bis(pyrano[3,2-c:5,6-c′]dichromenedione) and bis(dihydrobenzo[a]-xanthenones) using p-toluenesulfonic acid as catalyst. The repetitive pseudo-5CR involved bis-aldehydes 652a–d and two equivalents of β-alcohols 653a–c. Scheme 66 illustrates the suggested reaction mechanism. Initially, bis-aldehydes 652a–d are activated by AcOH or p-TsOH, then undergoing a Knoevenagel condensation with β-alcohols 653a–c to produce 654, followed by a Michael addition of another molecule of 653a–c, producing the intermediates 655. Finally, an intramolecular dehydrative cyclization affords the final products 656a–d.
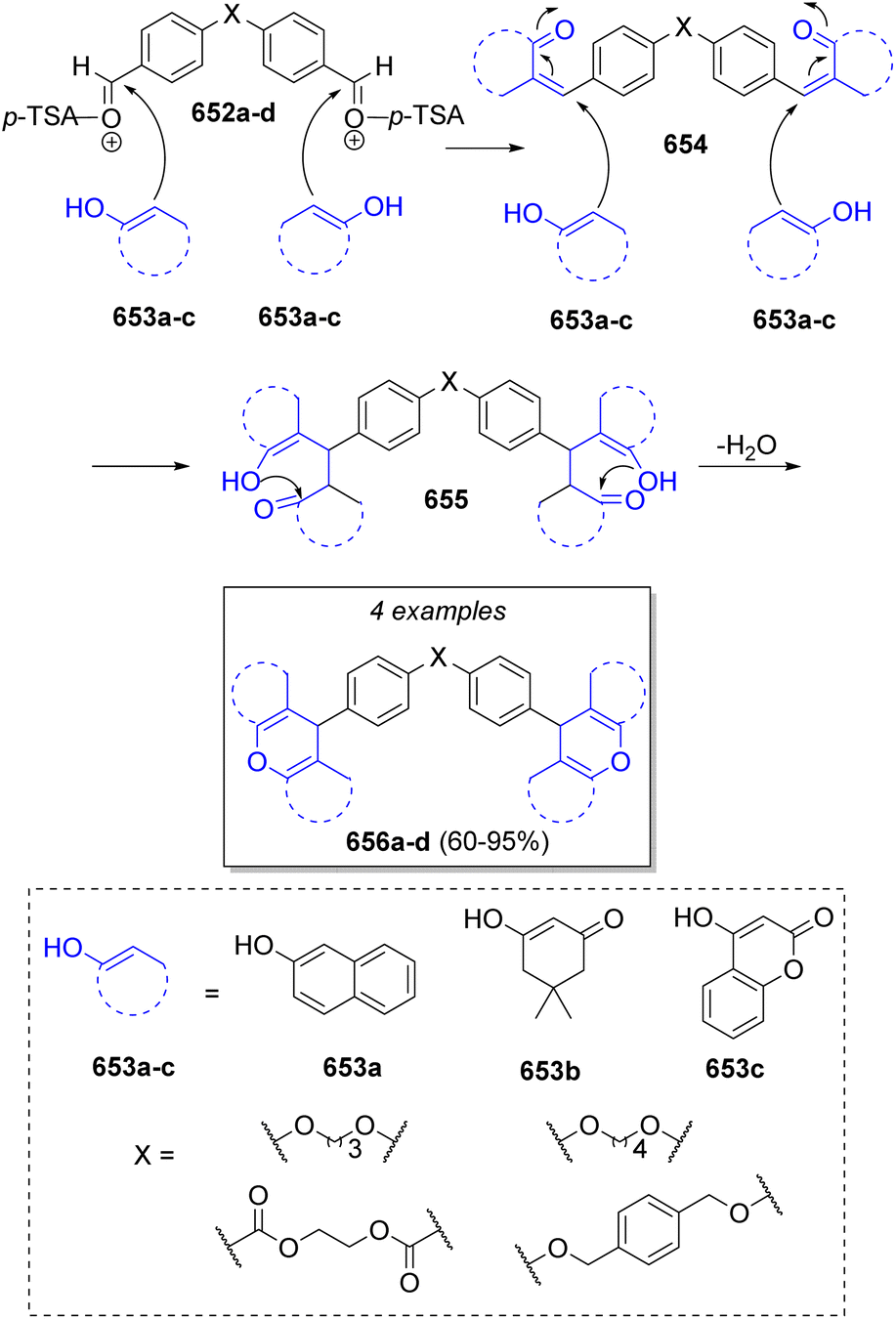 | ||
| Scheme 66 Synthesis of bis(14H-dibenzo[a,j]xanthenes), bis(pyrano[3,2-c:5,6-c′]dichromenedione) and bis(dihydrobenzo[a]-xanthenones). | ||
A small collection of indole-3-aminopropenylidene merocyanine dimers was synthesized by Müller and co-workers102 via a repetitive pseudo five-component reaction using 2 equivalents of N-tosyl 3-phenyl propynoyl ortho-bromo anilide (657), two equivalents of aryl alkynes 660a–c, and N,N′-(1,4-phenylenebis(methylene))dialkanamines 662a–b. According to the literature,103 the mechanism occurs via an intramolecular Pd-catalyzed coupling of propynoyl ortho-bromo anilides 657 to produce the key intermediates 658 and 659, which further couple with the aryl alkynes 660a–c to form alkynylidene indolones 661. These intermediates then react simultaneously with the bifunctional reactant N,N′-(1,4-phenylenebis(methylene))-dialkanamines 662a–b to produce 663a–d via a double nucleophilic addition (Scheme 67).
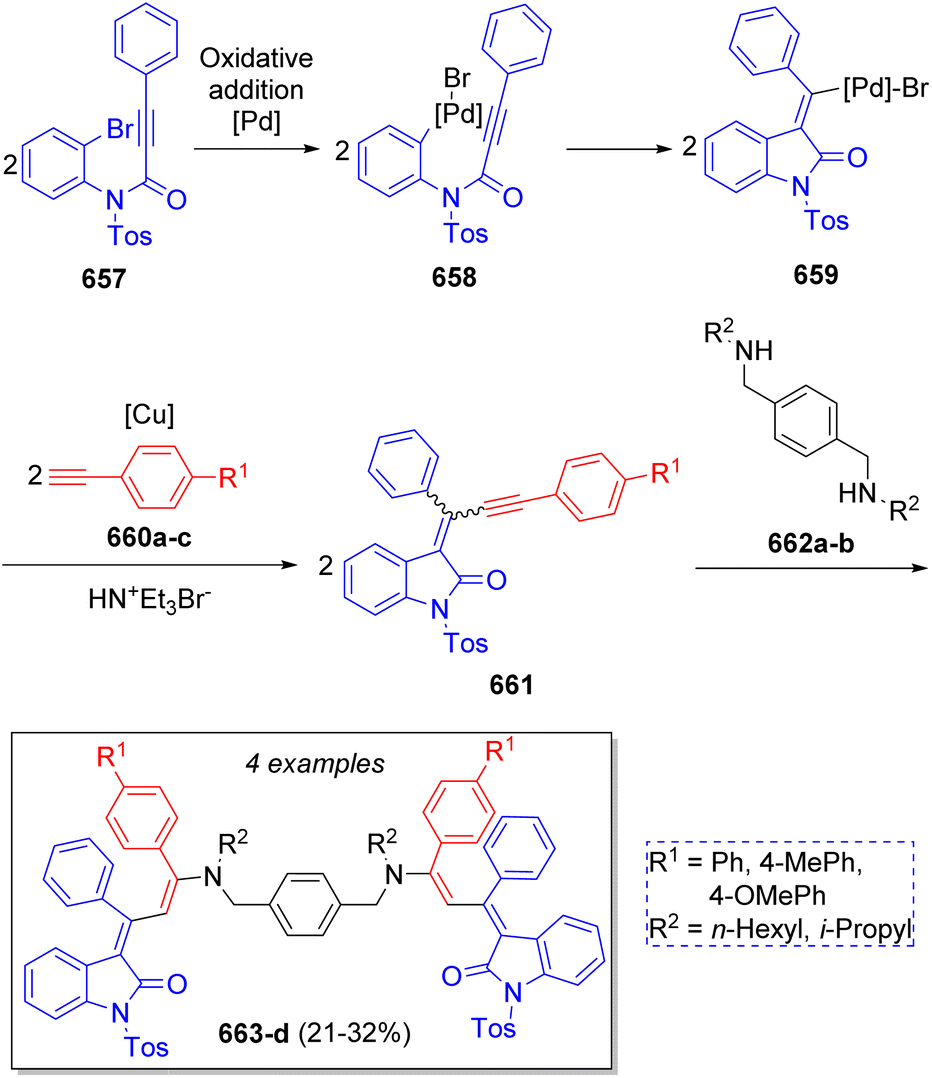 | ||
| Scheme 67 Synthesis of indole-3-aminopropenylidene merocyanine dimers. | ||
The synthesis of three novel bis-1-substituted-1H-tetrazoles were reported by Islas-Jácome and co-workers104 via repetitive-type pseudo-MCR heterocyclizations from symmetric dianilines 666a–c with two equivalents of trimethyl orthoformate (664), and sodium azide (669), respectively, in acetic acid as solvent. The proposed mechanism is illustrated in Scheme 68. Initially, protonation of the trimethylorthoester (664) by the acidic medium results in loss of a methanol molecule and formation of electrophile 665. Iminoester 670 is formed from the reaction between intermediate 665 and dianilines 666a–c after loss of another methanol molecule, followed by 1,3-dipolar cycloaddition with the azide ion 669 to form the corresponding bis-1-substituted 1H-tetrazoles 671a–c with subsequent loss of final methanol molecules.
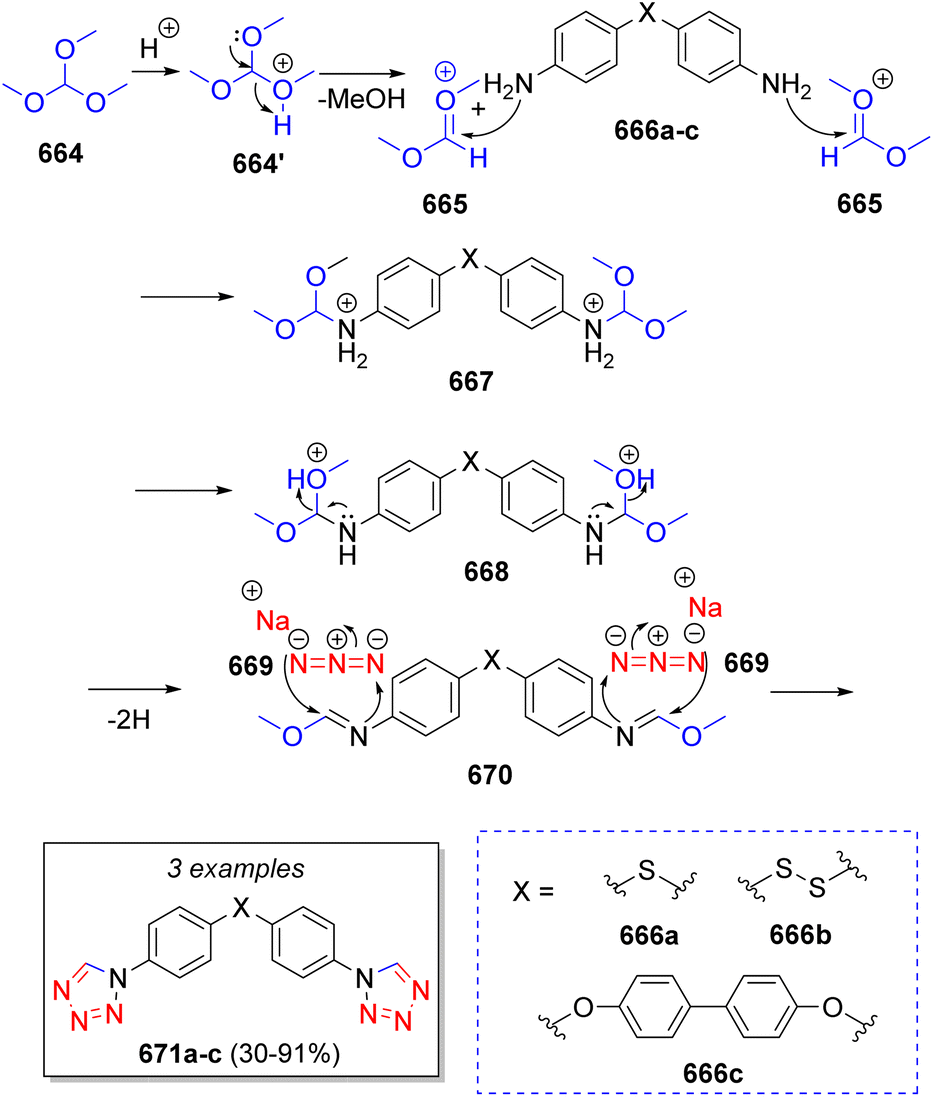 | ||
| Scheme 68 Synthesis of bis 1-substituted-1H-tetrazoles. | ||
A collection of substituted 1,4,8,11-tetrathiacyclotetradeca-5,12-dienes was obtained by Akhmetova and co-workers105 via a repetitive pseudo-6CR macroheterocyclization using malononitrile (672), aromatic aldehydes 673a–i, and 1,2-ethanedithiol (675) (in a 2:2:1 ratio), using triethylamine in ethanol as catalyst. The proposed mechanism is illustrated in Scheme 69. It its suggested that the reaction proceeds via Knoevenagel condensation between malononitrile (672) and aromatic aldehydes 673a–i to form intermediates 674. Subsequently, a reaction with two molecules of dithiol 675 to two molecules of intermediates 674 yield the symmetrical products 676a–i in moderate yields.
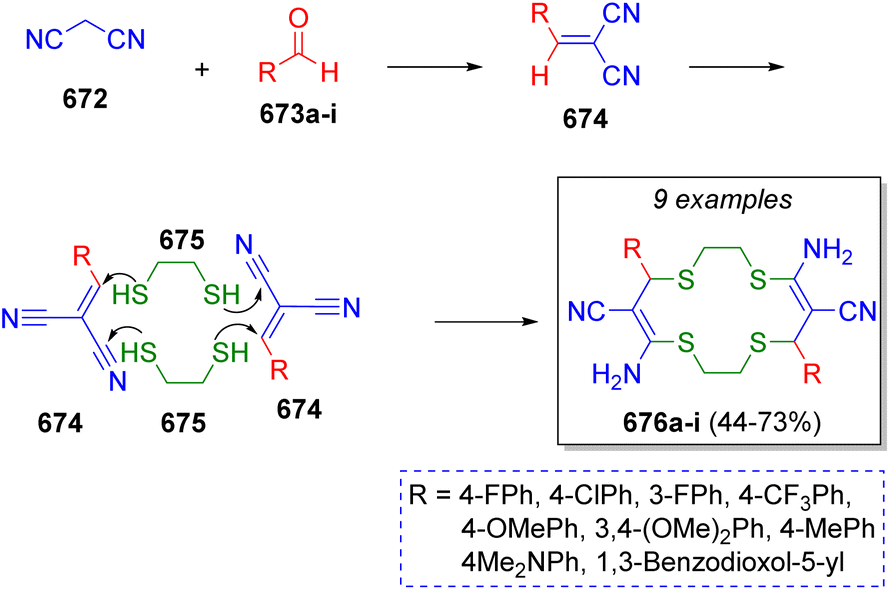 | ||
| Scheme 69 Synthesis of 1,4,8,11-tetrathiacyclotetradeca-5,12-dienes. | ||
Islas-Jácome and co-workers recently published the synthesis of bis-5-aminooxazoles linked through a ferrocene unit via a Sc(III)-catalyzed repetitive pseudo-5C Ugi–Zhu reaction. The central oxazole-ferrocene-oxazole motif makes this complex molecule a potential candidate to be an antimalarial agent, while the presence of the trifluoromethyl groups could further enhance its pharmacokinetics. To synthesize the target compound, 1,1′-ferrocenedicarboxaldehyde (677) was condensed with two equivalents of 4-(trifluoromethyl)benzylamine (678) to afford the bis-imine 679. This intermediate was activated towards nucleophilic addition with scandium(III) triflate, after which two equivalents of isocyanoacetamide 680 were added to afford the target ferrocene-linked bis-5-aminooxazole 681 (Scheme 70). The product was obtained in a one-pot fashion in under 2 hours of reaction time and in 73% yield, which is remarkable for such a complex molecule.106
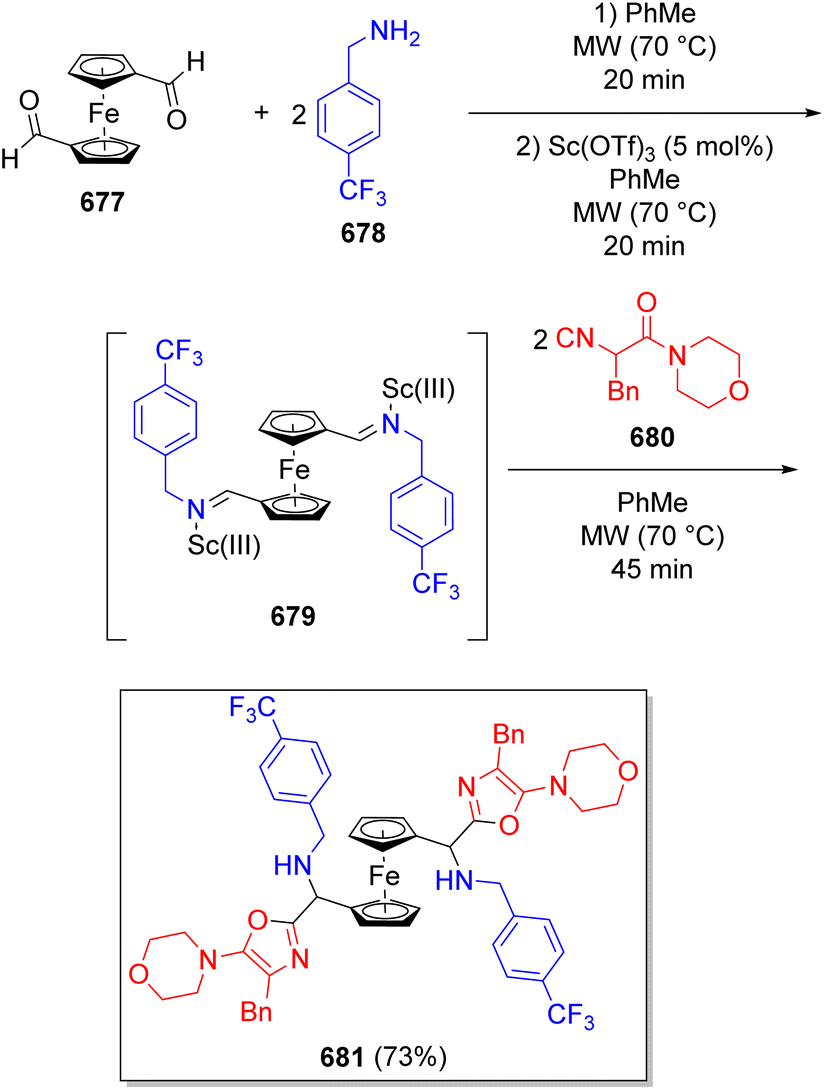 | ||
| Scheme 70 Synthesis of ferrocene-linked bis-5-aminooxazole via a repetitive pseudo-5C Ugi–Zhu reaction. | ||
M. Sadeghpour and co-workers107 recently reported the synthesis of a small compound library of bis-aminomethylnaphtols via a repetitive pseudo-5C Betti reaction, which is a modified Mannich-type reaction involving a diamine 682, two equivalents of different benzaldehydes 683 and two equivalents of 2-naphthol (685). Once the optimal reaction conditions were found by the authors, they synthesized a compound library using p-toluenesulfonic acid as organocatalyst, affording the target compounds in good to excellent yields. The authors also proposed a reaction mechanism: first, the bis-heterocyclic diamine component 682 undergoes a condensation with two equivalents of different benzaldehydes 683a–j in the presence of the acid catalyst to afford the bis-Schiff bases 684a–j. These intermediates react with two equivalents of 2-naphthol (685) to produce the intermediates 686a–j, and after a [1,3]-H shift the target products 687a–j are obtained (Scheme 71). It is worth noting that the products have potential use as ligands in coordination chemistry and were obtained in good yields in a one-pot procedure without solvent, making these conditions environmentally benign.
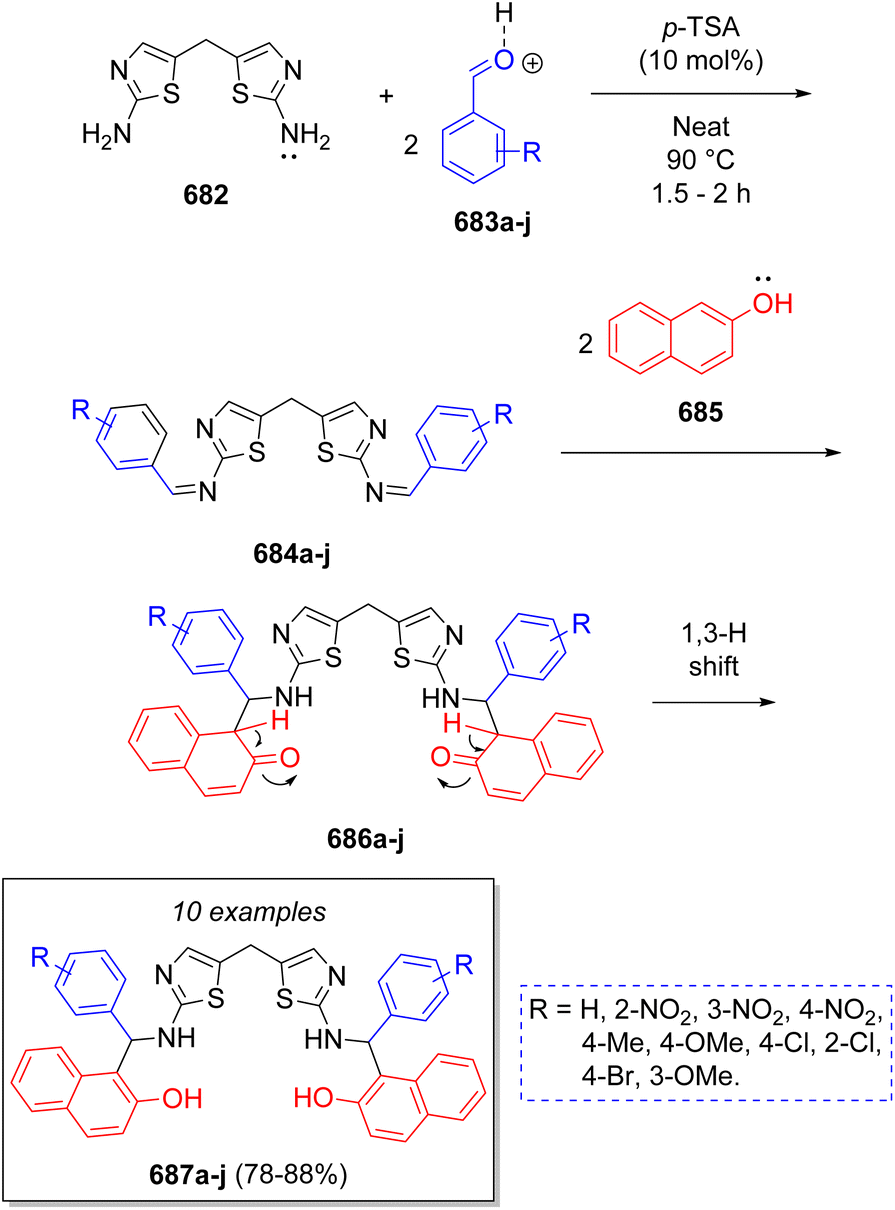 | ||
| Scheme 71 Synthesis of bis-aminomethylnaphtols via a repetitive-type pseudo-5C Betti condensation. | ||
8. Conclusions and outlook
Multicomponent reactions are very useful tools for rapidly generating molecular complexity and diversity in few experimental steps, especially when they are coupled with further processes such as post synthetic transformations, allowing access to a large variety of chemical libraries of heterocyclic and polyheterocyclic compounds with various potential applications, particularly in medicinal chemistry, agrochemical industry, optics, and innovative materials design. In this sense, it is important to generate bibliographic material that documents the development of new methodologies based on MCRs. With this review, it was intended to provide attention to a subclass of MCRs, namely, pseudo-MCRs, as well as proposing a simple classification for the most commonly encountered synthetic strategies when pseudo-MCRs are employed: pseudo-MCRs, which have one or more reactants in a stoichiometric ratio equal or higher than 2:1, and repetitive pseudo-MCRs, in which one of the reactants have two or more identical functional groups with react simultaneously with the reactants stoichiometrically in excess (also equal or higher than 2:1), often generating highly symmetrical products. In addition, we consider this review will provide new insights for further investigations behind new reactions and/or novel domino-type one-pot based strategies.
Author contributions
J. C. F.-R and V. C. C.-S. (writing – original draft), I. A. I. (writing – review & editing), E. G.-Z. (supervision), A. I.-J. (conceptualization & funding acquisition).Conflicts of interest
There are no conflicts to declare.Acknowledgements
J. C. F.-R. (886026) and V. C. C.-S. (892844) thank CONACyT for their scholarships. E. G.-Z. and I. A. I. specially thank U. Winnberg for the format/style revision. A. I.-J. acknowledges “Proyecto Apoyado por el Fondo Sectorial de Investigación para la Educación CONACyT-SEP CB-2017-2018 (A1-S-32582)”.Notes and references
- R. C. Cioc, E. Ruijter and R. V. A. Orru, Green Chem., 2014, 16, 2958 RSC
.
- J. C. Flores-Reyes, A. Islas-Jácome and E. González Zamora, Org. Chem. Front., 2021, 8, 5460 RSC
- A. Strecker, Liebigs Ann. Chem., 1850, 7, 27 CrossRef
-
(a) M. A. Fouad, H. Abdel-Hamid and M. S. Ayoup, RSC Adv., 2020, 10, 42644 RSC
-
(a) E. Ruijter and R. V. A. Orru, Drug Discovery Today: Technol., 2013, 10, e15 CrossRef PubMed
- C. Lamberth, Bioorg. Med. Chem., 2020, 28, 115471 CrossRef CAS PubMed
- B. T. Tuten and C. Barner-Kowollik, Macromol. Rapid Commun., 2020, 42, 2000495 CrossRef PubMed
-
(a) Multicomponent Reactions, ed. J. Zhu and H. Bienaymé, Wiley-VCH Verlag GmbH, Weinheim, 2005 Search PubMed
-
(a) A. Tuch and S. Wallé, in Handbook of Combinatorial Chemistry: Drugs, Catalysts, Materials, ed. K. C. Nicolaou, R. Hanko and W. Hartwig, Wiley-VCH Verlag GmbH, Weinheim, 1st edn, 2002, vol. 2, ch. 23, pp. 685–705 Search PubMed
-
(a) P. S. G. Nunes, H. D. A. Vidal and A. G. Corrêa, Org. Biomol. Chem., 2020, 18, 7751–7773 RSC
- J. C. Castillo, J. Quiroga, R. Abonia, J. Rodriguez and Y. Coquerel, J. Org. Chem., 2015, 80, 9767 CrossRef CAS PubMed
-
(a) A. Kamal, Y. V. V. Srikanth, M. N. A. Khan, T. B. Shaik and Md. Ashraf, Bioorg. Med. Chem. Lett., 2010, 20, 5229 CrossRef CAS PubMed
- G. Brahmachari, ACS Sustainable Chem. Eng., 2015, 3, 2058 CrossRef CAS
- K. Mahato, P. R. Bagdi and A. T. Khan, Synlett, 2014, 25, 2438 CrossRef CAS
- E. Ali, M. R. Naimi-Jamal, Z. Rashid and R. Ghahremanzadeh, Polycyclic Aromat. Compd., 2022, 42, 1157 CrossRef CAS
- Z. Zareai, M. Khoobi, A. Ramazani, A. Foroumadi, A. Souldozi, K. Slepokura, T. Lis and A. Shafiee, Tetrahedron, 2012, 68, 6721 CrossRef CAS
- L. Saher, M. Makhloufi-Chebli, L. Dermeche, S. Dermeche, B. Boutemeur-Khedis, C. Rabia, M. Hamdi and A. M. S. Silva, Tetrahedron, 2018, 74, 872–879 CrossRef CAS
- C. Yao, Y. Wang, T. Li, C. Yu, L. Li and C. Wang, Tetrahedron, 2013, 69, 10593 CrossRef CAS
- A. A. Mohammadi, S. Makarem, R. Ahdenov and N. A. Notash, Mol. Diversity, 2020, 24, 763 CrossRef CAS PubMed
- M. Rimaz, H. Mousavi, B. Khalili and L. Sarvari, J. Iran. Chem. Soc., 2019, 16, 1687 CrossRef CAS
- For an additional example see: M. Rimaz, H. Mousavi, M. Behnam, L. Sarvari and B. Khalili, Curr. Chem. Lett., 2017, 6, 55 CrossRef
- K.-T. Li, Y.-B. Lin and D.-Y. Yang, Org. Lett., 2012, 14, 1190 CrossRef CAS PubMed
- H. S. P. Rao and A. Parthiban, Org. Biomol. Chem., 2014, 12, 6223 RSC
- E. A. Sokolova, A. A. Festa, N. E. Golantsov, N. S. Lukonina, I. N. Ioffe, A. V. Varlamov and L. G. Voskressensky, Eur. J. Org. Chem., 2019, 40, 6770 CrossRef
- J. Valentín-Escalera, A. K. García-Dueñas, C. R. Solorio-Alvarado, C. Contreras-Celedón, C. J. Cortés-García and L. Chacón-García, Synlett, 2021, 32, 1461 CrossRef
- N. E. Golantsov, A. S. Golubenkova, A. A. Festa, A. V. Varlamov and L. G. Voskressensky, J. Org. Chem., 2022, 87, 3242 CrossRef CAS PubMed
- K. Mal and C. Mukhopadhyay, J. Mol. Struct., 2022, 1253, 132213 CrossRef CAS
- A. I. Kobelev, M. V. Dmitriev and A. N. Maslivets, Russ. J. Org. Chem., 2022, 58, 159 CrossRef CAS
- Z. Azarkamanzad, F. Farzaneh, M. Ghandi, M. M. Feizabadi and S. Jasemi, Russ. J. Org. Chem., 2022, 58, 372 CrossRef CAS
- M. N. Elinson, A. N. Vereshchagin, R. F. Nasybullin, S. I. Bobrovsky, A. I. Ilovaisky, V. M. Merkulova, I. S. Bushmarinov and M. P. Egorov, RSC Adv., 2015, 5, 50421 RSC
- M. S. Abaee, F. Ehteshami, S. Forghani, M. M. Mojtahedi and A. Hadizadeh, J. Iran. Chem. Soc., 2017, 14, 1151 CrossRef CAS
- M. N. Elinson, A. N. Vereshchagin, S. I. Bobrovsky, R. F. Nasybullin, A. I. Ilovaisky and V. M. Merkulova, C. R. Chim., 2016, 19, 293 CrossRef
- M. S. Abaee, S. Forghani, M. M. Mojtahedi and K. Harms, J. Sulfur Chem., 2016, 37, 683 CrossRef CAS
- S. Maharani and R. R. Kumar, Tetrahedron Lett., 2013, 54, 4800 CrossRef CAS
- S. M. Chinchkar, J. D. Patil, S. N. Korade, G. S. Gokavi, R. V. Shejawal and D. M. Pore, Lett. Org. Chem., 2017, 14, 403 CrossRef CAS
- F. V. Ryzhkov, M. N. Elinson, Y. E. Ryzhkova, A. N. Vereshchagin, V. A. Korolev and M. P. Egorov, J. Heterocycl. Chem., 2021, 58, 793 CrossRef CAS
- A. N. Vereshchagin, M. N. Elinson, Y. E. Anisina, F. V. Ryzhkov, R. A. Novikov and M. P. Egorov, ChemistrySelect, 2017, 2, 4593 CrossRef CAS
- H. R. Tavakoli, S. M. Moosavi and A. Bazgir, J. Korean Chem. Soc., 2013, 57, 260 CrossRef CAS
- R. Ghahremanzadeh, T. Amanpour and A. Bazgir, Tetrahedron Lett., 2010, 51, 4202 CrossRef CAS
- Y. Chen, T. Zhang, D. Wang, J. Zhou, Y. Zhang and Y. Li, J. Chem. Sci., 2017, 129, 421 CrossRef CAS
- M. Roozifar, N. Hazeri and H. F. Niya, J. Heterocycl. Chem., 2021, 58, 1117 CrossRef CAS
- A. Shaabani, M. B. Teimouri and S. Arab-Ameri, Tetrahedron Lett., 2004, 45, 8409 CrossRef CAS
- A. Shaabani, A. Bazgir and H. R. Bijanzadeh, Mol. Diversity, 2004, 8, 141 CrossRef CAS PubMed
- V. Nair, V. Sreekumar, S. Bindu and E. Suresh, Org. Lett., 2005, 7, 2297 CrossRef CAS PubMed
- A. Shaabani, E. Soleimani and H. R. Khavasi, J. Comb. Chem., 2008, 10, 442 CrossRef CAS PubMed
- A. Shaabani, F. Hajishaabanha, M. Mahyari, H. Mofakham and S. W. Ng, Tetrahedron, 2011, 67, 8360 CrossRef CAS
- B. Mohammadi, M. Shafieey, H. Kazemi and A. Ramazani, Chin. Chem. Lett., 2013, 24, 497 CrossRef CAS
- K. Mahato, P. R. Bagdi and A. T. Khan, RSC Adv., 2015, 5, 48104 RSC
- H. Sanaeishoar, R. Nazarpour and F. Mohave, RSC Adv., 2015, 5, 68571 RSC
- A. Sagar, V. N. Babu and D. S. Sharada, RSC Adv., 2015, 5, 29066 RSC
- K. Singh, A. Kaur, V. S. Mithu and S. Sharma, J. Org. Chem., 2017, 82, 5285 CrossRef CAS PubMed
- H. Mohammadi and H. R. Shaterian, ChemistrySelect, 2018, 3, 11059 CrossRef
- C. K. Jadhav, A. S. Nipate, A. V. Chate, P. M. Kamble, G. A. Kadam, V. S. Dofe, V. M. Khedkar and C. H. Gill, J. Chin. Chem. Soc., 2021, 68, 1067 CrossRef CAS
- M. Fathi, M. R. Naimi-Jamal, M. G. Dekamin, L. Panahi and O. M. Demchuk, Sci. Rep., 2021, 11, 4820 CrossRef CAS PubMed
- J. Safaei-Ghomi, B. Khojastehbakht-Koopaei and H. Shahbazi-Alavi, RSC Adv., 2014, 4, 46106 RSC
- R. Jahanshahi and B. Akhlaghinia, Chem. Pap., 2017, 71, 1351 CrossRef CAS
-
(a) A. Zare, A. Kohzadian, H. Filian, M. S. G. Nezhad and A. Karami, Res. Chem. Intermed., 2022, 48, 1631 CrossRef CAS
- S. Hemmati, M. R. P. Heravi, N. Nami and M. A. Khalilzadeh, Polycyclic Aromat. Compd., 2022, 43, 2540 CrossRef
- P. Das, T. Chaudhuri and C. Mukhopadhyay, ACS Comb. Sci., 2014, 16, 606 CrossRef CAS PubMed
- A. Mobinikhaledi, M. A. Bodaghifard and S. Asadbegi, Mol. Diversity, 2016, 20, 461 CrossRef CAS PubMed
- Z. Shirdel and M. R. Hosseini-Tabatabaei, Inorg. Nano-Met. Chem., 2017, 47, 810 CrossRef CAS
- M. Dabiri, P. Salehi, M. Bahramnejad, F. Sherafat and M. Bararjanian, Synth. Commun., 2012, 42, 3117 CrossRef CAS
- S. Sarkar, D. K. Das and A. T. Khan, Eur. J. Org. Chem., 2013, 30, 6823 CrossRef
- A. I. Ismiyev, V. V. Dotsenko, N. A. Aksenov, I. V. Aksenova and A. M. Magarramov, Russ. J. Gen. Chem., 2021, 91, 758 CrossRef CAS
- A. Shaabani, A. Maleki, H. Mofakham and J. Moghimi-Rad, J. Org. Chem., 2008, 73, 3925 CrossRef CAS PubMed
- H. Mofakham, A. Shaabani, S. Mousavifaraz, F. Hajishaabanha, S. Shaabani and S. W. Ng, Mol. Diversity, 2012, 16, 351 CrossRef CAS PubMed
- M. B. Teimouri and P. Akbari-Moghaddam, Tetrahedron, 2011, 67, 5928 CrossRef CAS
- A. Ziarati, J. Safaei-Ghomi and S. Rohani, Chin. Chem. Lett., 2013, 24, 195 CrossRef CAS
- M. Shiri, M. M. Heravi, V. Zadsirjan, A. N. Arani, S. A. Shintre and N. A. Koorbanally, Iran. J. Chem. Chem. Eng., 2018, 37, 101 CAS
- M. S. Abaee, A. Hatamifard, M. M. Mojtahedi, B. Notash and S. Naderi, Synth. Commun., 2022, 52, 346 CrossRef CAS
- S. Muthusaravanan, S. Perumal and A. I. Almansour, Tetrahedron Lett., 2012, 53, 1144 CrossRef CAS
- A. Alizadeh, A. Mikaeili and T. Firuzyar, Synthesis, 2012, 44, 1380 CrossRef CAS
- A. Alizadeh, V. Saberi and J. Mokhtari, Synlett, 2013, 24, 1825 CrossRef CAS
- X. Li, Y. Zhao, H. Qu, Z. Mao and X. Lin, Chem. Commun., 2013, 49, 1401 RSC
- A. Nikbakht, S. Ramezanpour, S. Balalaie and F. Rominger, Tetrahedron, 2015, 71, 6790 CrossRef CAS
- C. I. Attorresi, E. L. Bonifazi, J. A. Ramirez and G. F. Gola, Org. Biomol. Chem., 2018, 16, 8944 RSC
- Y. Ding, R. Ma, R. C. Hider and Y. Ma, Asian J. Org. Chem., 2020, 9, 242 CrossRef CAS
- A. Olyaei, A. Aghajanzadeh, E. Feizy and M. Sadeghpour, J. Chin. Chem. Soc., 2021, 68, 704 CrossRef CAS
- M. R. Mousavi, H. Gharari, M. T. Maghsoodlou and N. Hazeri, Res. Chem. Intermed., 2016, 42, 3875 CrossRef CAS
- A. N. Vereshchagin, K. A. Karpenko, M. N. Elinson, E. O. Dorofeeva, A. S. Goloveshkin and M. P. Egorov, Mendeleev Commun., 2018, 28, 384 CrossRef CAS
- J. Safaei-Ghomi, R. Sadeghzadeh and H. Shahbazi-Alavi, RSC Adv., 2016, 6, 33676 RSC
- F. Tamaddon and A. Khorram, Synlett, 2020, 31, 691 CrossRef CAS
- A. Rezvanian, M. Babashah and M. Anafcheh, Mol. Diversity, 2019, 23, 875 CrossRef CAS PubMed
- A. Rezvanian and M. Babashah, J. Heterocycl. Chem., 2019, 56, 1362 CrossRef CAS
- S. Ramezanpour, S. Balalaie and F. Rominger, Tetrahedron, 2016, 72, 6409 CrossRef CAS
- A. Alizadeh, S. Rostamnia and L.-G. Zhu, Tetrahedron Lett., 2010, 51, 4750 CrossRef CAS
- L. E. Cárdenas-Galindo, A. Islas-Jácome, K. M. Colmenero-Martínez, A. Martínez-Richa and R. Gámez-Montaño, Molecules, 2015, 20, 1519 CrossRef PubMed
- I. V. Kutovaya, D. P. Zarezin, O. I. Shmatova and V. G. Nenajdenko, Eur. J. Org. Chem., 2019, 24, 3908 CrossRef
- S. Mojikhalifeh and S. Hasaninejad, Org. Chem. Front., 2018, 5, 1516 RSC
- M. T. Nazeri, R. Mohammadian, H. Farhid, A. Shaabani and B. Notash, Tetrahedron Lett., 2020, 61, 151408 CrossRef CAS
- F. M. Yielzoleh and K. Nikoofar, Polycyclic Aromat. Compd., 2021, 42, 3884 Search PubMed
- N. Hazeri, M. Lashkari, S. García-Granda and L. Torre-Fernández, Aust. J. Chem., 2014, 67, 1656 CrossRef CAS
- S. Salahi, N. Hazeri, M. T. Maghsoodlou, S. García-Granda and L. Torre-Fernández, J. Chem. Res., 2014, 38, 383 CrossRef CAS
- S. Salahi, M. T. Maghsoodlou, N. Hazeri, M. Lashkari, N. A. Torbati, M. A. Kazemian, S. García-Granda and L. Torre-Fernández, J. Saudi Chem. Soc., 2016, 20, 349 CrossRef CAS
- M. Fatahpour, M. Lashkari, N. Hazeri, F. N. Sadeh and M. T. Maghsoodlou, Org. Prep. Proced. Int., 2019, 51, 576 CrossRef CAS
- A. Shaabani, M. Mahyari, M. Seyyedhamzeh, S. Keshipour and S. W. Ng, Tetrahedron Lett., 2011, 52, 4388 CrossRef CAS
- A. Shaabani, R. Ghadari, A. Sarvary and A. H. Rezayan, J. Org. Chem., 2009, 74, 4372 CrossRef CAS PubMed
- A. Shaabani, R. Ghadari, S. Ghasemi, M. Pedarpour, A. H. Rezayan, A. Sarvary and S. W. Ng, J. Comb. Chem., 2009, 11, 956 CrossRef CAS PubMed
- A. Alizadeh, Z. Noaparast, H. Sabahno and N. Zohreh, Helv. Chim. Acta, 2010, 93, 1401 CrossRef CAS
- H. R. Sadabad, A. Bazgir, M. Eskandari and R. Ghahremanzadeh, Monatsh. Chem., 2014, 145, 1851 CrossRef CAS
- A. F. Darweesh, S. K. Salama, I. A. Abdelhamid and A. H. M. Elwahy, J. Heterocycl. Chem., 2021, 58, 315 CrossRef CAS
- A.-L. Elsner, L. Biesen and T. J. J. Müller, Arkivoc, 2021, III, 53 Search PubMed
- M. Denißen, N. Nirmalananthan, T. Behnke, K. Hoffmann, U. Resch-Genger and T. J. J. Müller, Mater. Chem. Front., 2017, 1, 2013 RSC
-
(a) R. E. Blanco-Carapia, J. C. Flores-Reyes, Y. Medina-Martínez, P. Islas-Jácome, D. Pérez-Martínez, L. Lomas-Romero, I. A. Ibarra, A. Islas-Jácome and E. González-Zamora, Proceedings, 2019, 9, 32 Search PubMed
- N. S. Akhmadiev, E. S. Mescheryakova, V. R. Akhmetova and A. G. Ibragimov, RSC Adv., 2021, 11, 18768 RSC
- R. E. Blanco-Carapia, E. A. Aguilar-Rangel, A. Islas-Jácome and E. González-Zamora, Molbank, 2022, 2022, M1444 CrossRef CAS
- A. Olyaei, P. Kouhfar and M. Sadeghpour, ChemistrySelect, 2023, 8, e202204596 CrossRef CAS
| This journal is © The Royal Society of Chemistry 2023 |