
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceHierarchical carbon hollow nanospheres coupled with ultra-small molybdenum carbide as sulfiphilic sulfur hosts for lithium–sulfur batteries†
Huifa Shi*ab,
Jiakai Caoab,
Sa Hanab,
Weiyi Sunab,
Xiaoyang Zhua,
Guixia Lu*ef,
Hongbo Lana,
Huicong Yangd and
Shuzhang Niu *c
*c
aShandong Engineering Research Center for Additive Manufacturing, Qingdao University of Technology, Qingdao 266520, China. E-mail: shihuifa@qut.edu.cn
bKey Lab of Industrial Fluid Energy Conservation and Pollution Control (Qingdao University of Technology), Ministry of Education, Qingdao 266520, China
cCollege of New Materials and New Energies, Shenzhen Technology University, Shenzhen 518118, China. E-mail: niushuzhang@sztu.edu.cn
dShenyang National Laboratory for Materials Science, Institute of Metal Research, Chinese Academy of Sciences, Shenyang, 110016, China
eSchool of Civil Engineering, Qingdao University of Technology, Qingdao, Shandong 266520, China. E-mail: lgxia1988@163.com
fEngineering Research Center of Concrete Technology Under Marine Environment, Ministry of Education, Qingdao University of Technology, Qingdao, Shandong 266520, China
First published on 11th July 2023
Abstract
Lithium–sulfur (Li–S) batteries are an attractive candidate to replace the current state-of-the-art lithium-ion batteries due to their promising theoretical capacity of 1675 mA h g−1 and energy density of 2500 W h kg−1. However, the lithium polysulfide (LiPS) shuttle effect and the slow sulfur redox kinetics seriously decrease the utilization of sulfur and deteriorate battery performance. Here, hierarchical carbon hollow nanospheres containing intimately coupled molybdenum carbide nanocrystals were synthesized as a sulfiphilic sulfur host. The sufficient interior void space accommodates the sulfur and physically confines LiPSs, while the in situ introduced molybdenum carbide nanoparticles can chemically immobilize LiPSs and catalytically accelerate their redox transformations. As a result, the Li–S batteries with this synergistic effect achieve an excellent rate capability of 566 mA h g−1 at 2C and a long cycle stability over 300 cycles at 1C.
Introduction
Due to their mature processing technology and extensive application, lithium-ion batteries have played an important role in mobile energy storage since their commercialization.1 Especially in the last decade, the development of electric vehicles has further promoted the demand for lithium-ion batteries, and also put forward higher requirements for their energy density. However, the energy density of lithium-ion batteries is approaching the theoretical maximum, which is ultimately determined by the intercalation reaction mechanism. Among various alternatives, lithium–sulfur (Li–S) batteries based on a sulfur redox conversion mechanism have obvious advantages, including much lower cost, higher gravimetric/volumetric energy densities (2500 W h kg−1/2800 W h L−1) and environmental friendliness.2,3 Thus, they have attracted considerable research interest. In practice, however, several challenges seriously hinder the practical use of Li–S batteries, including poor ionic/electronic conductivity of sulfur and lithium sulfide (Li2S),4 shuttle behaviors of lithium polysulfides (LiPSs)5 and safety risk of Li metal anode.6 These problems greatly reduce the battery cycle life and sulfur utilization. In addition, the redox conversion of sulfur species is kinetically tardy, which not only further aggravates the shuttle effect, but also causes large amounts of deactivated “dead sulfur”.7,8Carbon materials are often used as sulfur hosts to tackle the above intrinsic shortcomings of the sulfur cathode, due to their diversified pore structure, high conductivity and high electrochemical stability.9–12 Among them, hollow carbon sphere is an earlier proposed sulfur host.13,14 Its large cavity can effectively encapsulate large amounts of sulfur and buffer its volume changes. Besides, the carbon shell acts as a physical barrier to prevent the direct contact between sulfur and bulk electrolyte, and further confine the produced LiPSs. However, the non-polar carbon surface has a poor affinity with the sulfur species.15–17 The dissolved LiPSs will still gradually diffuse out from the hollow cavity of carbon in the long cycle process, and cause the parasitic shuttle effect.18 Therefore, imparting sulfiphilic property to the carbon surface to enhance the interaction between carbon-based hosts and sulfur species is necessary. According to previous literature reports, polar transition metal compounds can chemically immobilize dissolved sulfur species and improve the stability of the sulfur cathode, such as metal oxides,19 metal carbides,20 metal phosphides,21 metal sulfides14,22 and metal nitrides.23,24 More importantly, some compounds show obvious catalytic activity on the sulfur redox conversion, which could accelerate the conversion of sulfur species and improve their utilization.25,26 Therefore, the introduction of highly dispersed and tightly coupled polar nanoparticles into the common carbon hollow sphere can improve the sulfur affinity of the carbon interface and greatly enhance the confinement and utilization of sulfur species.
Metal carbides have the advantages of high conductivity, high chemical stability and thermal stability, and have been used as catalysts in hydrogen evolution reaction,27 ammonia decomposition28 and formic acid oxidation.29 In the sulfur cathodes, theoretical calculation30 shows that metal carbides have moderate chemical bonding for LiPSs, and also can realize rapid LiPS transformation and diffusion. In our previous study,15 necklace-like molybdenum carbide (MoC) nanoparticles were used to modify the carbon fiber network to obtain a sulfiphilic 3D carbon collector. The fabricated sulfur cathode with a sulfur loading of 10 mg cm−2 delivered an ultra-high areal capacity of 12.3 mA h cm−2 at 0.1C. Nevertheless, the preparation process of metal carbides usually requires high temperature treatment. Due to inhomogeneous carburization and inevitable high-temperature aggregation, the obtained carbides tend to be large and irregular particles.31 The resulting limited active sulfiphilic sites greatly decreases their chemisorption and catalytic ability. Another concern is the interfacial bonding between the carbide catalyst and the carbon. Poor interfacial binding greatly deteriorates interfacial electron transfer, further affecting the catalytic activity. Therefore, in the Li–S battery application, there is an urgent need to develop a convenient method for preparing a carbon-based sulfur host with uniformly dispersed and tightly coupled carbide nanoparticles.
In this work, nitrogen-doped hollow carbon spheres strongly coupled with molybdenum carbide nanocrystals (MoC@N-HCS) were prepared by a simple metal chelate-assisted polymerization and heat treatment. When used as the sulfur host, this purposefully designed MoC@N-HCS has the following advantages: (1) the carbon shell is composed of a large number of carbon sheets, which increases the specific surface area and the active sites; (2) solid-state thermal reaction is a common but effective method to prepare carbon/carbide composites. The carbon produced by dopamine plays a role in limiting the growth of MoC, realizing its ultra-small size and high dispersion. The close coupling between MoC and conductive carbon enhances their interface electron transfer; (3) MoC nanocrystals have high conductivity and sulfur affinity, which can chemically adsorb LiPSs and catalyze their redox conversion. Combined with the hollow carbon structure, the shuttle effect can be effectively inhibited. As a result, the Li–S batteries with these intimately coupled MoC@N-HCS hosts show a reduced charge/discharge polarization, and deliver a decent rate capability of 566 mA h g−1 at 2C and long cycle stability over 300 cycles at 1C.
Experimental
Materials preparation
According to the experimental procedures reported in reference,3 300 mg ammonium molybdate tetrahydrate ((NH4)6Mo7O24·4H2O) was dissolved in 80 mL deionized water, and then 160 mL ethanol was added into the solution. Subsequently, 300 mg of dopamine hydrochloride (C8H11NO2·HCl) was added and stirred thoroughly until it was completely dissolved. Finally, 0.2 mL ammonium hydroxide (25–28%) was added and stirred at room temperature for 6 h. The stirred solution was centrifuged and washed twice with ethanol. The obtained precipitation was dried in an oven at 80 °C. The dried powder was placed in a tubular furnace for heat treatment at 700 °C for 2 h under the protection of Ar. The resulting sample is labeled as MoOx@N-HCS.MoOx@N-HCS was mixed with concentrated hydrochloric acid for 12 h to remove partial MoOx and increase the specific surface area and the porosity of the final sample. Subsequently, the powders were washed several times with deionized water to fully remove residual hydrochloric acid and dried in an oven at 80 °C. To obtain MoC@N-HCS, the powders were subjected to a heat treatment at 800 °C for 3 h in an atmosphere of 10% H2/Ar.
The preparation of comparison sample nitrogen-doped carbon sphere (N-CS) has the same preparation procedure, except that (NH4)6Mo7O24·4H2O was not added.
Preparation of MoC@N-HCS/S
The impregnation of sulfur was carried out using a melting-diffusion method. The MoC@N-HCS and sublimated sulfur powder were mixed in a mass ratio of 3![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :7, and then heated at 155 °C for 12 h to obtain the MoC@N-HCS/S composite.
:7, and then heated at 155 °C for 12 h to obtain the MoC@N-HCS/S composite.
LiPS adsorption test
The preparation of Li2S6 solution was as follows: 115 mg of lithium sulfide (Li2S) and 400 mg of sublimed sulfur were added in a mixed solvent of 1,2-dioxolane and dimethoxymethane (1:1, v%). The mixture was stirred at 60 °C until the powder was completely dissolved. Li2S6 solution with a concentration of 0.5 M was obtained.
20 mg MoC@N-HCS powder, conductive carbon Super P and reduced graphene oxide (rGO) were firstly added into different glass vials respectively. Then, the diluted Li2S6 (1 mM, 5 mL) was added and mixed with the powders. Digital photos were taken after adsorption for 12 h.
Materials characterization
The morphology and structure of the prepared materials were analyzed using scanning electron microscopes (SEM, FEI Nova NanoSEM 430, 15 kV) and transmission electron microscopes (TEM, Tecnai F20, 200 kV). The element distribution was obtained using the Energy Dispersive Spectrometer. XRD data were collected on a Rigaku diffractometer equipped with Cu Kα radiation at room temperature. The thermogravimetric analysis (TGA) curves were obtained using a NETZSCH STA 449 C thermo-balance. MoC content of MoC@N-HCS was tested in the air from room temperature to 1000 °C with a heating rate of 10 °C min−1, while the sulfur content in the MoC@N-HCS/S was determined in Ar from room temperature to 600 °C with a heating rate of 10 °C min−1. X-ray photoelectron spectroscopy (XPS) using an ESCALAB 250Xi (Thermo Fisher) instrument with a monochromated Al Kα radiation analysis was proceeded to analyze the elemental composition and valence.Electrochemical measurements
The cathode was prepared by mixing the MoC@N-HCS/S composite, Super-P, and polyvinylidene difluoride with a weight ratio of 80:10:10 in N-methylpyrrolidone solvent to form a slurry. Then the slurry was coated onto a carbon-coated aluminum foil using a doctor blade and dried at 60 °C under a vacuum. The mass loading of sulfur in the obtained cathode is ∼1 mg cm−2. Commercial polypropylene membrane (Celgard 2500) and lithium metal were used as the separator and the anode. The coin cell (CR2032) was assembled in a glove box to evaluate the electrochemical performance. 1 M bis(trifluoromethane) sulfonamide lithium salt (LiTFSI) dissolved in a mixture of 1,2-dioxolane and dimethoxymethane (1:1, v%) with adding 2 wt% LiNO3 was used as the electrolyte. Cyclic voltammetry (CV) experiment was carried out in the potential range of 1.7–2.8 V (versus Li/Li+) at a scanning rate of 0.1 mV s−1 using VSP-300 multichannel workstation. Electrochemical cycle stability and rate capability at various currents were performed using a LAND instrument.
Results and discussion
As shown in Fig. 1, MoC@N-HCS is synthesized through a simple metal chelate-assisted polymerization method. First, a metal chelation reaction between the Mo7O246− anion and dopamine hydrochloride happened in the water/ethanol solution, forming the chelates. Then, the mixed solvent produced water phase/organic phase micelles during the stirring process and the chelates polymerized along the micellar surface in a weakly alkaline environment. A hollow Mo-PDA spherical structure with the lamellar shell was formed (Fig. S1a and b†). This resulting precursor spheres were thermally treated at 700 °C to produce MoOx@N-HCS spheres. To adjust the content of MoC in the final product appropriately and increase the sulfur storage sites, partial MoOx was dissolved and removed using concentrated hydrochloric acid. After pickling, the samples were further heat treated in a reducing atmosphere (10% H2/Ar). MoOx reacted with carbon to produce MoC nanoparticles. Due to the in situ and confined reaction in the carbon matrix, the obtained MoC has an ultra-small size and a uniform distribution, leading to the high exposure of sulfiphilic sites. Additionally, the conductive carbon nanosheets in the shell and the nanocrystals have extremely intimate coupled interactions, realizing rapid electron migration between the interface. When used as a sulfur host, MoC@N-HCS hollow sphere can collaboratively achieve physical confinement, chemisorption, and catalytic conversion towards LiPSs. In order to verify the importance of metal chelate in the polymerization process, we polymerized and carbonized dopamine under the same condition only without the addition of (NH4)6Mo7O24·4H2O. The resulting product is the solid carbon spheres with a diameter of 200–300 nm (Fig. S3†), which is used as a comparison sample for subsequent electrochemical tests.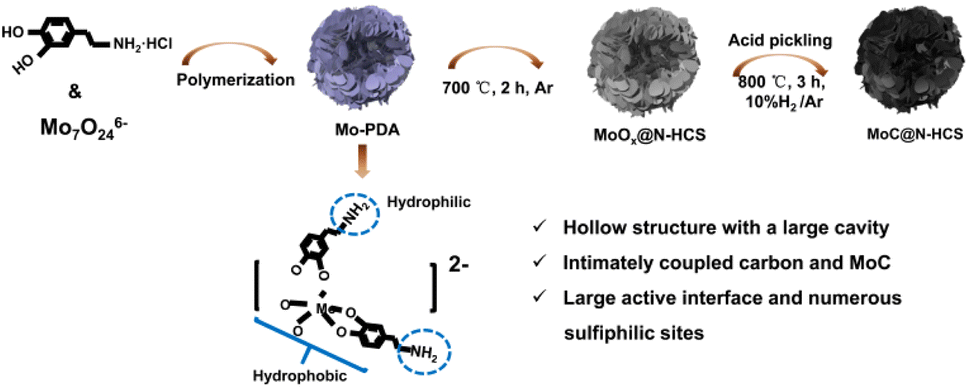 | ||
| Fig. 1 Schematic diagram of the synthesis steps for MoC@N-HCS. | ||
The phase and component contents of the final products were characterized and determined by XRD and TGA. As shown in Fig. 2a, except for the characteristic peak of carbon at 24.5°, several obvious peaks at 36.6, 39.3, 42.4, 61.7 and 74.4 are observed, corresponding to the (1 0 2), (1 0 3), (1 0 4), (1 1 0) and (1 1 6) planes of hexagonal η-MoC phase (JCPDS Card no. 89-4305).32 The TGA curve is shown in Fig. 2b. Before 320 °C, the increase in mass is attributed to the conversion of MoC to molybdenum oxide in the air. Above 320 °C, carbon materials are oxidized in air and the mass is rapidly reduced. At 450 °C, the residual mass fraction is 50.8 wt%. Therefore, the content of MoC in the sample is 38.1 wt%. The N2 adsorption isotherm (Fig. 2c) recorded on MoC@N-HCS resembles type I and IV isotherm and a H3 hysteresis loop, indicating the existence of adequate micropores and mesopores.33 The specific surface area of MoC@N-HCS based on the Brunauer–Emmett–Teller method was calculated to be 201 m2 g−1. The high specific surface area provides sufficient active sites for the adsorption of LiPSs. Fig. S4b† shows that the pore diameter of micropores and mesopores mainly concentrated in 1 nm and 5.6–12.9 nm.
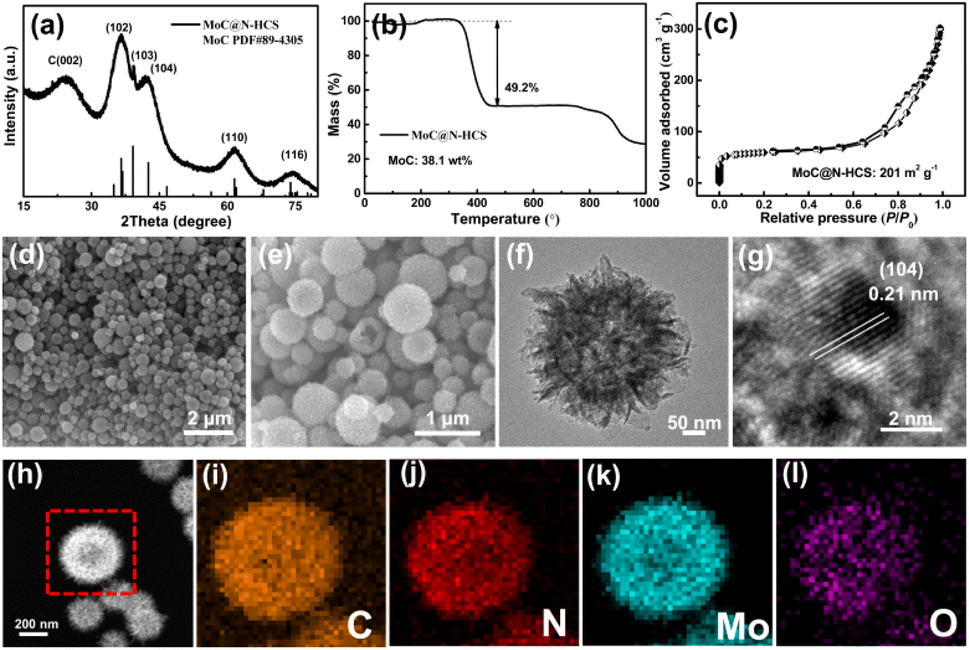 | ||
| Fig. 2 (a) XRD pattern and (b) TGA pattern of MoC@N-HCS. (c) N2 adsorption/desorption isotherm of MoC@N-HCS. (d and e) SEM images, (f) TEM and (g) HRTEM images of MoC@N-HCS. MoC@N-HCS STEM picture (h) and corresponding elemental mapping images of carbon (i), nitrogen (j), molybdenum (k) and oxygen (l). | ||
SEM images of the MoC/N-HCS show that the flower-like structure of Mo-PDA precursor was largely maintained after twice annealing (Fig. 2d, e and S1a†). The carbon shell consists of a large number of carbon sheets and the sphere size is 200–500 nm. A partially broken carbon sphere verifies the hollow structure, which provides a large interior void for accommodating sulfur and its discharge products in the charge and discharge process. TEM image (Fig. 2f) further shows the internal structure of the carbon sphere more clearly, including the hollow carbon cavities and the carbon shell composed of carbon sheets. More importantly, lots of molybdenum carbide nanoparticles are homogeneously incorporated into the carbon sheet. High-resolution transmission electron microscopy (HRTEM) image (Fig. 2g) confirms that the nanoparticle with high crystallinity has an ultra-small size of ∼2 nm. The lattice fringe with a spacing of 0.21 nm is attributed to the (104) crystal facet of the MoC. Fig. 2h–l further characterize the distribution of MoC on the surface of the carbon sphere. Elements carbon, nitrogen, oxygen and molybdenum are all consistent with the morphology of MoC@N-HCS hollow spheres in STEM images, indicating that MoC is evenly distributed in the entire spherical shell. The element oxygen comes from the amorphous oxide layer formed due to the oxidation of MoC in the air. The formed Mo–O–C like species may help to activate surface Mo sites for chemically binding dissolved LiPSs.21 The same conclusion can be obtained from the element line scanning of MoC@N-HCS (Fig. S2†).
The sulfur is impregnated into MoC@N-HCS by the classical melt-diffusion process, and the mass fraction of sulfur in the composite is 68.2 wt% (Fig. 3a). The microstructure of the MoC@N-HCS/S composite was observed. As shown in Fig. S4,† the composite still maintains the original flower-like shape of MoC@N-HCS, and there is no obvious sulfur agglomeration, indicating that all sulfur is fully diffused into the shell and hollow cavity of MoC@N-HCS. To further investigate the sulfur distribution within the as-synthesized MoC@N-HCS, EDS analysis (Fig. 3b–f) demonstrates that sulfur was homogeneously distributed within MoC@N-HCS. In this case, sulfur and MoC particles are evenly and fully combined. After sulfur loading, the specific surface area decreases to 3.4 m2 g−1 (Fig. S5a†), and the original micropores and mesoporous pores disappeared (Fig. S5b†), indicating that these pores were completely filled with sulfur.
 | ||
| Fig. 3 (a) TGA curve of MoC@N-HCS/S in Ar. (b) SEM images of the MoC@N-HCS/S composite, and the corresponding EDS images of carbon (c), nitrogen (d), molybdenum (e) and sulfur (f). | ||
The obtained MoC@N-HCS/S composite was mixed with Super P and binder to prepare sulfur cathode. The cyclic voltammetry (CV) curves are shown in Fig. 4a. During the cathodic scan from 2.8 to 1.7 V, two well-defined reduction peaks appear at 2.25 and 1.96 V, corresponding to the generation of long-chain LiPSs (Li2Sn, 4 ≤ n ≤ 8) and the reduction of long-chain LiPSs to Li2S2/Li2S, respectively. In the subsequent anodic scan, two oxidation peaks occurred at 2.39 and 2.45 V, which corresponds to the reversible oxidation of Li2S and the final conversion of long-chain LiPSs into sulfur.34,35 Notably, there are no obvious current or potential changes of these reduction/oxidation peaks in the first three scans and the obtained curves are highly overlapped, proving superior reversibility and stability of the cell.36 Cycle and rate performance MoC@N-HCS/S and N-CS/S are shown in Fig. 4b and c. The initial specific capacity of MoC@N-HCS/S at 0.3C reaches 985 mA h g−1, and the residual capacity after 50 cycles is 699 mA h g−1, both of which are much higher than those of N-CS/S. Meanwhile, MoC@N-HCS/S delivers discharge specific capacities of 1193, 908, 799, 657 and 566 mA h g−1 at 0.1, 0.3, 0.5, 1 and 2C, respectively. In contrast, the specific capacities of N-CS/S at the above discharge rate are only 721.8, 491.3, 416.5, 289.4, 184.9 mA h g−1. The excellent cycle and rate performance are attributed to two points. On the one hand, MoC@N-HCS has a hollow structure, which can effectively store sulfur and buffer the volume expansion during the charge–discharge process. On the other hand, the ultra-small molybdenum carbide nanoparticles enable the sulfiphilic surface of the carbon shell, realizing the effective strong chemical anchoring and fast redox conversion of LiPSs.
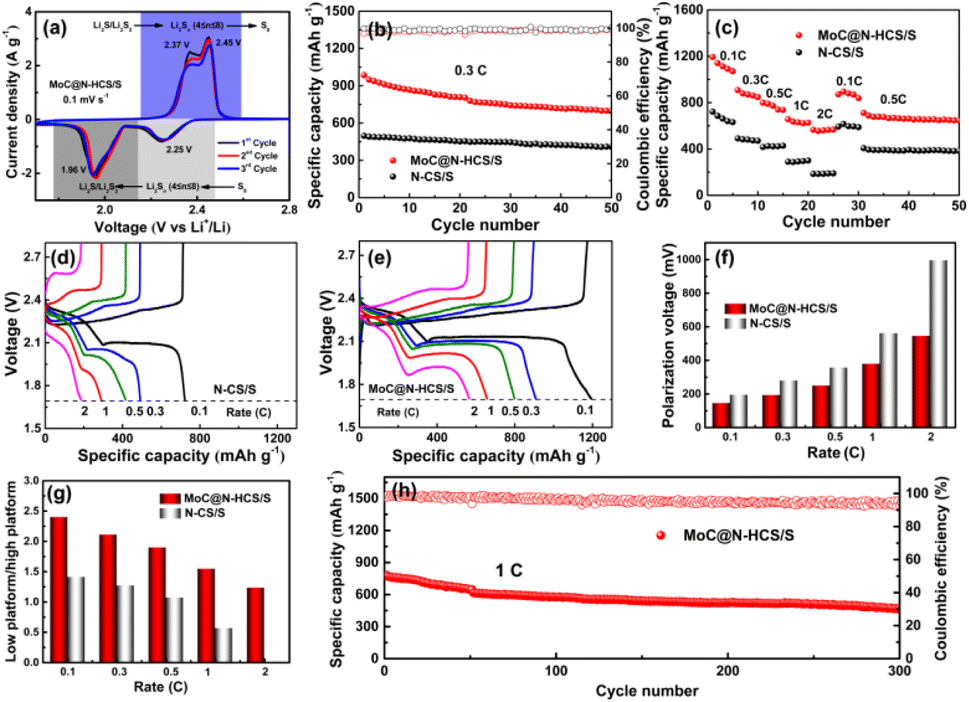 | ||
| Fig. 4 (a) CV curve of MoC@N-HCS/S at a scan speed of 0.1 mV s−1. Comparison of electrochemical properties of MoC@N-HCS/S and N-CS/S: (b) cycling stability at 0.3C and (c) rate performance. Charge and discharge curves of the cells with N-CS/S cathode (d) and MoC@N-HCS/S cathode (e) at different discharge rates. (f) Polarization voltages of charge and discharge at different discharge rates. (g) Capacity ratio of low voltage platform to high voltage platform. (h) Long cycle performance of MoC@N-HCS/S at 1C. | ||
Fig. 4d and e show the charge–discharge curves of N-CS/S and MoC@N-HCS/S at different rates. Results show that the corresponding discharge curves of MoC@N-HCS/S at all rates show two obvious platforms, while the second discharge platform of N-CS/S disappears at 2C. As shown in Fig. 4f, the polarization voltages of the charge–discharge curves are analyzed and compared. It is found that the polarization of MoC@N-HCS/S is significantly less than that of N-CS/S at all discharge rates. In addition, the discharge curve is divided into high-voltage platform capacity and low-voltage platform capacity. The ratio of low-voltage platform capacity to high-voltage platform capacity reflects whether the long-chain LiPSs generated in the first step can be further utilized effectively. As shown in Fig. 4g, the ratios of MoC@N-HCS/S cathode are higher than those of N-CS/S at all current densities. The above results indicate that the cavity structure not only stores sulfur, but also achieves physical confinement for sulfur and its discharge products. MoC nanoparticles incorporated in the shell can chemically adsorb dissolved LiPSs and catalyze their rapid and full conversion to Li2S2/Li2S. In contrast, N-CS has a non-hollow structure, which is lack of sufficient storage space and reaction sites for generated LiPSs during the discharge/charge process, leading to sluggish LiPS conversion and the serious shuttle phenomenon. Fig. 4h shows that the cell assembled with MoC@N-HCS/S cell exhibits good long-cycle stability. At 1C, the cell delivers an initial specific discharge capacity of 783 mA h g−1 and a remaining capacity of 472 mA h g−1 after 300 cycles.
MoC nanoparticles enhances the sulfur affinity of the carbon interface. To verify this, MoC@N-HCS was added to the Li2S6 solution for the chemical adsorption test. Nanocarbon materials commonly used in laboratories, rGO and Super P, are used as control samples. As shown in Fig. 5a–d, after resting for 12 h, the Li2S6 solution with MoC@N-HCS becomes almost completely colorless and transparent, while the solution with rGO or Super P is still light yellowish green. The corresponding UV-visible absorption spectroscopy (Fig. S6†) further proves that the LiPS concentration of the supernatant solutions with MoC@N-HCS decreases more. This result indicates that MoC@N-HCS has stronger chemical adsorption ability towards LiPSs, and more Li2S6 is chemically anchored to the surface of the MoC@N-HCS.37,38 To more deeply study the interaction between the embedded MoC nanoparticles and LiPSs, the samples before and after the adsorption of Li2S6 were tested by XPS. Fig. 5e shows the full spectrum of elements before LiPS adsorption, and the sample contains O 1s, N 1s, C 1s and Mo 3d peaks. After LiPS adsorption, the S 2p peak appears at 162.6 eV,39 which indicates that LiPSs are strongly adsorbed on the MoC surface. In Mo 3d fine spectrum (Fig. 5f), Mo–O and Mo–C bonds are mainly included. After the adsorption of LiPSs (Fig. 5g), Mo–S bonds appear at the positions of 229.9 eV and 233.9 eV, confirming the chemical interaction between MoC nanoparticles and Li2S6.40 Based on the above discussion results, the working principle diagram of MoC@N-HCS as a high efficient sulfur host is shown in Fig. 5h. Hollow carbon spheres have large cavities that provide storage space for active materials and also physically confine dissolved LiPSs. MoC nanocrystals closely coupled with the carbon sheets function as sulfiphilic sites, which chemical adsorb LiPSs, and catalyze their rapid redox transformation. More critically, the growth of MoC is limited by carbon substrate based on unique metal chelate-assisted polymerization and solid phase heat treatment method. Its high dispersion and ultra-small particle size further maximize its chemisorption and catalytic capability. Therefore, the resulting sulfiphilic hollow carbon spheres exhibit the above excellent electrochemical properties.
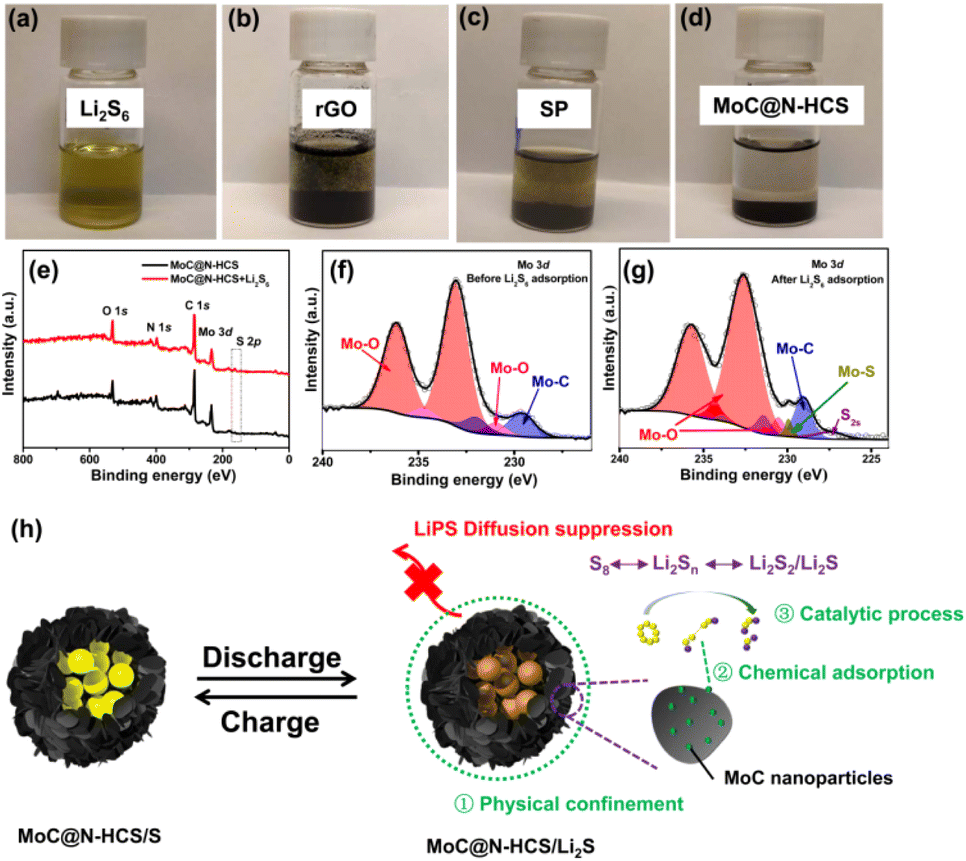 | ||
| Fig. 5 Digital images of the Li2S6 captured by different materials: (a) Li2S6 solution, (b) rGO + Li2S6 solution, (c) Super P + Li2S6 solution and (d) MoC@N-HCS + Li2S6 solution. (e) XPS full spectra of MoC@N-HCS before and after Li2S6 adsorption. Mo 3d fine spectra before (f) and after (g) Li2S6 adsorption. (h) A working principle diagram of MoC@N-HCS as a high efficient sulfur host. | ||
Conclusions
In this work, a hierarchical carbon hollow sphere with embedded ultra-small molybdenum carbide nanoparticles was prepared by a metal chelate-assisted polymerization method. The obtained MoC@N-HCS as the sulfur host shows multiple advantages. The large cavity provides sufficient space for sulfur storage and physically prevents the outward diffusion of discharge products. More importantly, conductive MoC nanoparticles with ultra-small size and uniform distribution are intimately coupled with the carbon, making carbon surface sulfiphilic. Dissolved LiPSs are chemically anchored onto these MoC sites and undergo a rapid redox transformation. Based on the synergistic coupling of the above multiple functions, Li–S cells with the MoC@N-HCS/S cathodes show excellent rate performance (566 mA h g−1, 2C) and cycle stability (300 cycles, 1C).Conflicts of interest
The author declares no conflict of interest.Acknowledgements
The work was supported by the Natural Science Foundation of Shandong Province, China (no. ZR2021QE098), Postdoctoral Science Foundation of China (no. 2022M712329), and Qingdao Institute of Collaborative Innovation (A2022-JG05).References
- P. G. Bruce, S. A. Freunberger, L. J. Hardwick and J. M. Tarascon, Nat. Mater., 2011, 11, 19–29 CrossRef PubMed.
- W. G. Chong, J.-Q. Huang, Z.-L. Xu, X. Qin, X. Wang and J.-K. Kim, Adv. Funct. Mater., 2017, 27, 1604815 CrossRef.
- Y. Yang, M. C. Luo, Y. Xing, S. T. Wang, W. Y. Zhang, F. Lv, Y. J. Li, Y. L. Zhang, W. Wang and S. J. Guo, Adv. Mater., 2018, 30, 1706085 CrossRef PubMed.
- J. L. Yang, P. Yang, D. Q. Cai, Z. Wang and H. J. Fan, Nano Lett., 2023, 23, 4000–4007 CrossRef CAS PubMed.
- V. P. Nguyen, J. S. Park, H. C. Shim, J. M. Yuk, J. H. Kim, D. Kim and S. M. Lee, Adv. Funct. Mater., 2023 DOI:10.1002/adfm.202303503.
- T. Kakimi, S. Miyakawa, S. Taminato, T. Saito, D. Mori and N. Imanishi, RSC Adv., 2023, 13, 9142–9153 RSC.
- C. Jiang, L. Li, Q. Jia, M. Tang, K. Fan, Y. Chen, C. Zhang, M. Mao, J. Ma, W. Hu and C. Wang, ACS Nano, 2022, 16, 9163–9171 CrossRef CAS PubMed.
- W. Hua, T. Shang, H. Li, Y. Sun, Y. Guo, J. Xia, C. Geng, Z. Hu, L. Peng, Z. Han, C. Zhang, W. Lv and Y. Wan, Nat. Catal., 2023, 6, 174–184 CrossRef CAS.
- H. Shi, W. Lv, C. Zhang, D.-W. Wang, G. Ling, Y. He, F. Kang and Q.-H. Yang, Adv. Funct. Mater., 2018, 28, 1800508 CrossRef.
- S. Feng, R. K. Singh, Y. Fu, Z. Li, Y. Wang, J. Bao, Z. Xu, G. Li, C. Anderson, L. Shi, Y. Lin, P. G. Khalifah, W. Wang, J. Liu, J. Xiao and D. Lu, Energy Environ. Sci., 2022, 15, 3842–3853 RSC.
- N. Kang, Y. Lin, L. Yang, D. Lu, J. Xiao, Y. Qi and M. Cai, Nat. Commun., 2019, 10, 4597 CrossRef PubMed.
- P. R. Adhikari, E. Lee, L. Smith, J. Kim, S. Shi and W. Choi, RSC Adv., 2023, 13, 9402–9412 RSC.
- N. Jayaprakash, J. Shen, S. S. Moganty, A. Corona and L. A. Archer, Angew. Chem., Int. Ed., 2011, 50, 5904–5908 CrossRef CAS PubMed.
- C. Ye, L. Zhang, C. X. Guo, D. D. Li, A. Vasileff, H. H. Wang and S. Z. Qiao, Adv. Funct. Mater., 2017, 27, 1702524 CrossRef.
- H. F. Shi, Z. H. Sun, W. Lv, S. G. Wang, Y. Shi, Y. B. Zhang, S. J. Xiao, H. C. Yang, Q. H. Yang and F. Li, J. Mater. Chem. A, 2019, 7, 11298–11304 RSC.
- L. Wang, X. Meng, X. Wang and M. Zhen, Small, 2023, 19, 2300089 CrossRef CAS PubMed.
- S. S. Yao, Y. P. He, M. Arslan, C. J. Zhang, X. Q. Shen, T. B. Li and S. B. Qin, New Carbon Mater., 2021, 36, 606–613 CrossRef CAS.
- Y. Wen, Z. Shen, J. Hui, H. Zhang and Q. Zhu, Adv. Energy Mater., 2023, 13, 2204345 CrossRef CAS.
- C. Zheng, S. Niu, W. Lv, G. Zhou, J. Li, S. Fan, Y. Deng, Z. Pan, B. Li, F. Kang and Q.-H. Yang, Nano Energy, 2017, 33, 306–312 CrossRef CAS.
- W. L. Cai, G. R. Li, K. L. Zhang, G. N. Xiao, C. Wang, K. F. Ye, Z. W. Chen, Y. C. Zhu and Y. T. Qian, Adv. Funct. Mater., 2018, 28, 1704865 CrossRef.
- Y. R. Zhong, L. C. Yin, P. He, W. Liu, Z. S. Wu and H. L. Wang, J. Am. Chem. Soc., 2018, 140, 1455–1459 CrossRef CAS PubMed.
- T. Chen, L. B. Ma, B. R. Cheng, R. P. Chen, Y. Hu, G. Y. Zhu, Y. R. Wang, J. Liang, Z. X. Tie, J. Liu and Z. Jin, Nano Energy, 2017, 38, 239–248 CrossRef CAS.
- Z. H. Li, Q. He, X. Xu, Y. Zhao, X. W. Liu, C. Zhou, D. Ai, L. X. Xia and L. Q. Mai, Adv. Mater., 2018, 30, 1804089 CrossRef PubMed.
- M. J. Zhang, Y. Lu, Z. J. Yue, M. M. Tang, X. K. Luo, C. Chen, T. Peng, X. M. Liu and Y. S. Luo, RSC Adv., 2023, 13, 9322–9332 RSC.
- J. He and A. Manthiram, Adv. Energy Mater., 2020, 10, 2002654 CrossRef CAS.
- D. Liu, C. Zhang, G. Zhou, W. Lv, G. Ling, L. Zhi and Q. H. Yang, Adv. Sci., 2018, 5, 1700270 CrossRef PubMed.
- R. Michalsky, Y. J. Zhang and A. A. Peterson, ACS Catal., 2014, 4, 1274–1278 CrossRef CAS.
- W. Q. Zheng, T. P. Cotter, P. Kaghazchi, T. Jacob, B. Frank, K. Schlichte, W. Zhang, D. S. Su, F. Schuth and R. Schlogl, J. Am. Chem. Soc., 2013, 135, 3458–3464 CrossRef CAS PubMed.
- C. Y. He, J. Z. Tao, Y. B. Ke and Y. F. Qiu, RSC Adv., 2015, 5, 66695–66703 RSC.
- F. Zhou, Z. Li, X. Luo, T. Wu, B. Jiang, L. L. Lu, H. B. Yao, M. Antonietti and S. H. Yu, Nano Lett., 2018, 18, 1035–1043 CrossRef CAS PubMed.
- Y. Chen, X. C. Gao, D. W. Su, C. Y. Wang and G. X. Wang, Trends Chem., 2020, 2, 1020–1033 CrossRef CAS.
- C. Y. Tang, H. Zhang, K. F. Xu, Q. L. Zhang, J. H. Liu, C. X. He, L. D. Fan and T. Asefa, J. Mater. Chem. A, 2019, 7, 18030–18038 RSC.
- M. Thommes, K. Kaneko, A. V. Neimark, J. P. Olivier, F. Rodriguez-Reinoso, J. Rouquerol and K. S. W. Sing, Pure Appl. Chem., 2015, 87, 1051–1069 CrossRef CAS.
- H. D. Shi, X. M. Ren, J. M. Lu, C. Dong, J. Liu, Q. H. Yang, J. Chen and Z. S. Wu, Adv. Energy Mater., 2020, 10, 2002271 CrossRef CAS.
- M. J. Shi, Z. Liu, S. Zhang, S. C. Liang, Y. T. Jiang, H. Bai, Z. M. Jiang, J. Chang, J. Feng, W. S. Chen, H. P. Yu, S. X. Liu, T. Wei and Z. J. Fan, Adv. Energy Mater., 2022, 12, 2103657 CrossRef CAS.
- R. Xiao, S. Yang, T. Yu, Z. H. Sun and F. Li, Batteries Supercaps, 2022, 5, 202100389 Search PubMed.
- D. Lu, X. Wang, Y. Hu, L. Yue, Z. Shao, W. Zhou, L. Chen, W. Wang and Y. Li, Adv. Funct. Mater., 2023, 33, 2212689 CrossRef CAS.
- C. Zhao, H. Liu, J. Liu, Y. Shi, S. Wang, Q. Tang, X. Zhu, H. Zhang and Y. Zhao, RSC Adv., 2023, 13, 8706–8717 RSC.
- X. Y. Zhou, L. Li, J. Yang, L. Xu and J. J. Tang, Chemelectrochem, 2020, 7, 3767–3775 CrossRef CAS.
- Y. Y. Li, S. S. Yao, C. J. Zhang, Y. P. He, Y. Q. Wang, Y. Z. Liang, X. Q. Shen, T. B. Li, S. B. Qin and W. Wen, Int. J. Energy Res., 2020, 44, 8388–8398 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3ra03167e |
| This journal is © The Royal Society of Chemistry 2023 |