
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceConvergent micro-wave assisted synthesis of quinazolinone and its precursor using the bio-sourced solvent pinane†
Antoine Richieu and
Philippe Bertrand*
Institut de Chimie des Milieux et Matériaux de Poitiers, UMR CNRS 7285, 4 rue Michel Brunet, TSA 51106, B27, 86073 Poitiers, France. E-mail: philippe.bertrand@univ-poitiers.fr
First published on 10th July 2023
Abstract
A general microwave synthesis of 4-oxo-3,4-dihydroquinazolin-2-yl propanoic acids and their diamide precursors from the corresponding substituted benzamide and succinic anhydride is described, using pinane as a sustainable solvent that favors the cyclization step. The conditions are some of the most simple and cost effective reported.
Heterocycles are common important building blocks in medicinal chemistry. Quinazolines or quinazolinones are regularly used in the synthesis of bioactive compounds to inhibit epigenetic targets,1 gamma-aminobutyric acid (GABA) receptors,2 phosphoinositide 3-kinase (PI3K) signalling3 and many other biological pathways.4 Some typical quinazolin(on)es approved for use in clinical applications are presented in Fig. 1, like erlotinib, an endothelial growth factor receptor (EGFR) inhibitor, and idelalisib, a PI3K inhibitor. Indeed the synthesis of quinazolin(on)e derivatives is of high interest. A conventional synthesis of quinazoline 4 (Scheme 1) starts with ortho-substituted anilines 1 being reacted with carbonyl derivatives 2 to give an amide 3.
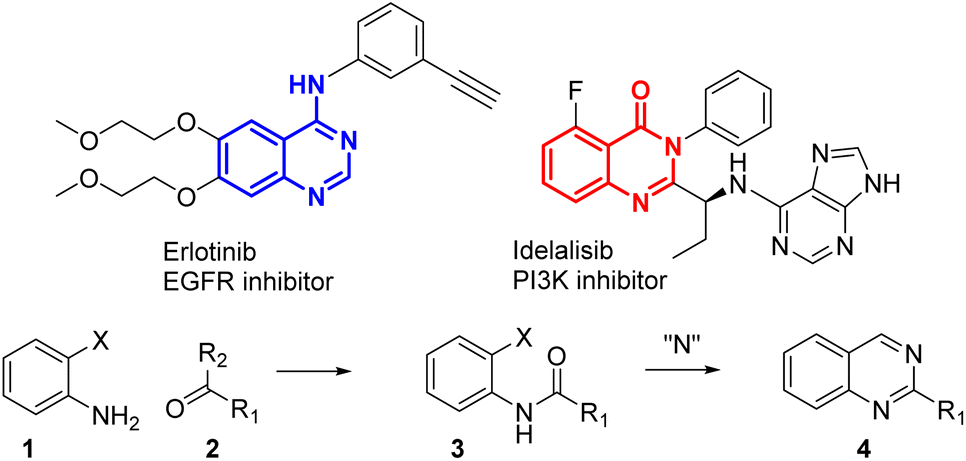 | ||
| Fig. 1 Two examples of quinazolin(on)e-based approved drugs. | ||
 | ||
| Scheme 1 Preparation of pinane and example for the synthese of quinazolinones 8. | ||
The final intramolecular cyclization to 4 needs a nitrogen atom that can come from the initial substituents (X, R2) or through an additional reagent. Many methods for synthesizing quinazolin(on)es are available and were reviewed.5
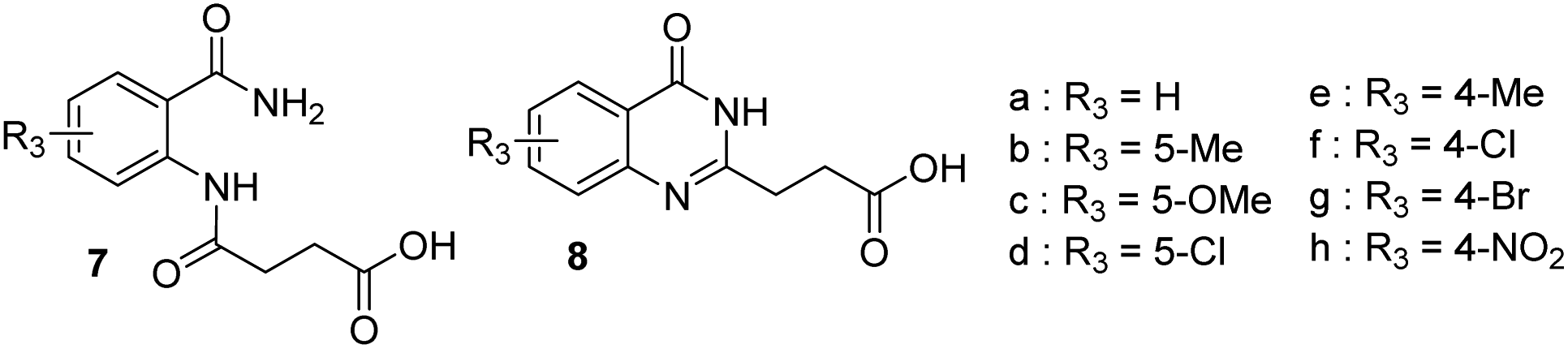
Our team being involved in sustainable chemistry to produce key building blocks for various applications, we studied the preparation of a series of quinazolinones with a greener approach in the frame of an ongoing medicinal project. The targeted compounds were quinazolin-2-ylpropanoic acid 8a (R = H, Scheme 2) and substituted versions (R =/= H) as intermediates toward compounds 9 (Scheme 2). A possible synthesis of compound 8a under thermal conditions requires two steps:6 (i) mixing 2-aminobenzamide 5a and succinic anhydride 6 in toluene with 3 h refluxing to afford a diamide 7a, (ii) cyclization to quinazolinone 8a by heating 7a in the presence of aqueous NaOH. Alternative thermal solutions exists for the second steps with NaOAc/acetic anhydride,7 or through benzooxazinone in formamide/ethanol.8 In the first step to prepare compounds 7 toluene is used and, as a fossil-based solvent, its replacement by an hydrocarbon like terpenes such as pinane (Scheme 2), obtained from renewable resources, could give a greener synthesis.9,10
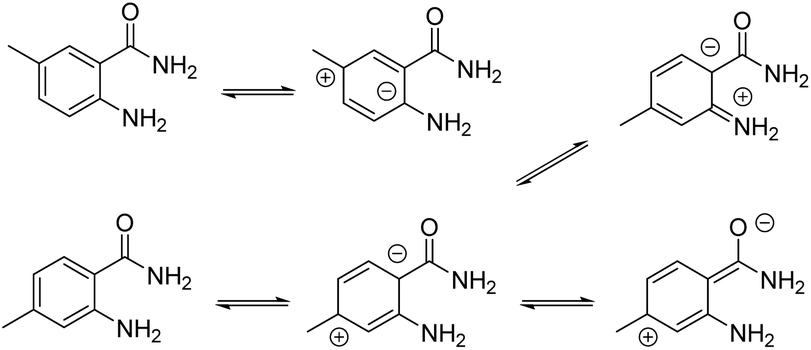 | ||
| Scheme 2 Possible explanation for substitution-dependent reactivity of benzamide. | ||
The starting materials 5 and 6 (Scheme 1) used for the first step of the preparation are all powders not soluble in toluene, except the anhydride 6 whose partial solubility in toluene probably favours the first step of the preparation. We though that keeping a suspension of these finely devised powders could give better results than performing the reaction neat. Terpenes like pinene, the major component of the abundant and cheap turpentine pine tree oil and produced in large quantities worldwide, are considered as substitutes for fossil-based hexane.11 The structure of pinene has some similarity with toluene, with an additional bridge. In this family of terpene the 2,6,6-trimethylbicyclo[3.1.1]heptane, called herein pinane (Scheme 1), was selected for it similarity to toluene and no potential side reactivity as it could happen with the double bond of the pinene. Pinane is obtained by the catalytic reduction of the pinene,12 with a price currently at 10 euros per kg, and around 30 euros per kg for toluene. As toluene, pinane makes an azeotrope with water (50/50, pEb 120 °C). Beside the possible uses of terpenes to prepare bioactive compounds, pinane was often used as solvent for oil extractions13 but to our knowledge nothing is described with pinane as a solvent for chemical reactions, whereas pinene is used either as reagent or acid scavenger.14
A regular way to transform heating under reflux for hours is to take advantage of microwave reactors. This strategy was already applied to the synthesis of various quinazolin(on)es, starting from various substrates.15,16 A reported green microwave-assisted synthesis of quinazolinone was performed in two steps.17 A preparation of the derivative 8a in particular in ethanol with acetic acid under microwave conditions in a domestic oven without temperature control was reported18 but the 1H NMR chemical shifts in the aromatic region are not supported by spectra and are different from those reported in another reference work.6 As the combination of microwaves and pinane alone as solvent for the preparation of compounds such 8a–g (Scheme 1) was never described we report below our results in preparing selectively the intermediates 7 and/or the expected derivatives 8 using thermal and microwaves conditions.
A series of quinazolynylpropanoic acid 8b–g (Scheme 2) was thus synthesized in two steps with the procedure used to prepare compound 8a (ref. 6) to have all the reference compounds and for comparison purposes with the microwave conditions. Substituted benzamides 5b–g were mixed with succinic anhydride in toluene and the mixture was refluxed for 3 hours in the case of 5-substituted benzamides. The less reactive 4-substituted benzamides 5e–g required longer heating (12 h for 4-bromo and 2 days for 4-nitro), to have a more complete reaction. After filtration the diamides 5b–g were heated in aqueous sodium hydroxide to perform the intramolecular cyclization leading to quinazolinones 8b–g. All the compounds were characterized by 1H and 13C NMR and HRMS. The yields obtained when preparing compounds 7 and 8 under thermal conditions are reported in Table 1.
|
|||||
|---|---|---|---|---|---|
| R= | Thermal yields | Isolated microwave yieldsc (from benzamide 5) | |||
| 5 → 7 | 7 → 8 | 5 × 7 → 8 | 7 | 8c | |
a Optimized conditions are: 110 °C, 10 min, 300 W, 0.8 mmol ml−1, ratio 5![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :6 = 1:2.b Optimized conditions are: 110 °C, 15 min, 300 W, 0.4 mmol ml−1, ratio 5:6 = 1:4.c 110 °C, 10 min, 300 W then 180 °C, 15 min, 300 W. n.d.: not done. :6 = 1:2.b Optimized conditions are: 110 °C, 15 min, 300 W, 0.4 mmol ml−1, ratio 5:6 = 1:4.c 110 °C, 10 min, 300 W then 180 °C, 15 min, 300 W. n.d.: not done. |
|||||
| a = H | 90 (ref. 6) | 91 (ref. 6) | 81.9 | 99a | 85 |
| b = 5-Me | 95 | 98.6 | 93.7 | 94a | 88 |
| c = 5-OMe | 98 | 91 | 89.2 | 99a | 98.8 |
| d = 5-Cl | 96 | 99.5 | 95.5 | 96a | 84.4 |
| e = 4-Me | 92 | 97.9 | 90.1 | 93b | 79 |
| f = 4-Cl | 94 | qt | 94.0 | n.d. | n.d. |
| g = 4-Br | 92.9 | 91.2 | 84.7 | 90b | 89.3 |
| h = 4-NO2 | 94 | 86 | 80.8 | n.d. | n.d. |
We next evaluate the transposition of the thermal conditions used for the preparation of the diamide 7 into microwaves conditions.
We selected the preparation of the diamide 7a as the test reaction. The equipment we used (Discover 2.0) has an algorithm to convert the thermal conditions to microwave ones. Converting thermal conditions with toluene at reflux for 3 hours gave initial experimental conditions of 6 minutes at 180 °C and 150 Watts with stirring. According to the thermal conditions the molar ratio of benzamide/anhydride to solvent was kept at 1:1 mmol in 1.25 ml. In a first reaction using a 1 cm diameter 10 ml reactor we observed, with the internal camera, that the stirring was not efficient, the magnetic bar size possible for this diameter being too small to be able to mix correctly the suspension in the pinane. The limitations observed with the 10 ml vial were solved by using a 35 ml one with a bigger magnetic bar. In these conditions the stirring of the suspension was slightly more efficient.
The visualization of the ongoing reaction with the camera indicated two key changes during the reaction: a first one when the heat reached 110–130 °C with the deposition of a grey solid sticking to the glass vessel and a second one when 150–180 °C was reached with a full melting of the solids that was maintained during the rest of the reaction. At the end of the reaction the mixture solidified with the pinane as the supernatant, allowing its easy recovery by filtration or a simple transfer into another flask. The solid was removed with methanol after careful trituration.
We were surprised to see that the 1H NMR spectrum of the crude reaction showed the quinazolinone 8a as the major product (Fig. S1 left line 2, ESI†). Other compounds observed were the starting benzamide 5 and another unknown compound we named 10 whose 1H NMR signals did not corresponded to the initial substrate or the intermediate diamide 7a. In the same conditions using toluene as the solvent a clean diamide 7a was obtained. In contrast to pinane that gave a stable plateau at 180 °C with discontinued irradiation, the heating of toluene was limited to 176–177 °C with almost constant irradiation. This observation suggested that toluene absorbed more energy than pinane lowering the reactivity of the diamide for the final cyclization. We decided to stop the reaction after 6 minutes at 176–177 °C to match the 180 °C plateau for pinane. This demonstrated that toluene is superior to pinane for the first step but is not able to perform the second cyclization step. The reaction times were then evaluated at 180 °C and 150 W (10 and 15 min, Fig. S1 left lines 3 and 4, ESI†), showing a minor impact on the final composition.
We then studied both the reaction time, power and temperature to find the best conditions to improve the preparation of the quinazolinone in pinane while reducing the formation of compound 10. 1H NMR spectra of the crude reactions were used to monitor the parameters. Typical NMR signals (Fig. S1 left, ESI†) were a multiplet and a triplet at 2.55 and 7.1 ppm for the intermediate 7a, two triplets and the doublet at 2.75, 2.85 and 8.1 ppm for the quinazolinone 8a and the overlapping signal at 7.55 ppm for the unknown compound 10. Increasing the time of reaction was useful to fully transform the starting benzamide but also increased the formation of the unknown compound. To limit the formation of 10 that increased after 10 min the temperature of the reaction was decreased to 150 °C for 10 min. In these conditions a mixture of intermediate 7a and quinazolinone 8a was obtained, along with other unidentified side-products including product 10. A temperature below 150 °C gave also mixtures but slightly less side products, and at 110 °C some unreacted benzamide 5a was observed.
A better approach to limit this side reactions leading to 10 was to ensure a rapid consumption of the benzamide 5a. This was achieved by two equivalents of the anhydride 6 relative to the benzamide 5a. Controlling the first step leading to compound 7a selectively could be of interest for preparing such compounds and we focused on a protocol to obtain clean compound 7a. In addition we thought critical to achieve high initial conversion prior to the second cyclization step leading to 8a. We selected 130 °C for the temperature to reach, this temperature being a compromise between the conversion to quinazolinone 8a and the formation of 10. A first method at 130 °C for 5 min at 300 W with a 1:2 ratio of 5a:6 gave results similar to a previous one (150 °C, 10 min, 150 W), e.g. a mixture of 7a and 8a but with less side products formed (Fig. S1 right, lines 1 and 2, ESI†). The reaction time was then decreased down to 4, 3 and 2 minutes, the best results being obtained for 3 minutes, yielding a clean diamide 7a with some remaining benzamide 5a (Fig. S1 right, line 3, ESI†). After a couple of optimizations we found that 110 °C, 10 min, 300 W, 600 rpm stirring and a 5a:6 ratio of 1:2 gave 100% conversion of the benzamide 5a to the diamide 7a with no side products, except the excess of succinic anhydride (Fig. S1 right, line 4, ESI†).
The method was then applied to 4- and 5-substituted benzamides 5 and give high conversions for all examples (Table 1).
However, if the conversion was 100% for all 5-substituted benzamide 5a–d, the conversion of 4-substituted benzamides 5e–g did not exceeded 95%. A possible explanation for these differences lies in the electronic effects where the doublet of the nitrogen atom in the 2-amino group did not participate in the mesomeric forms initiated from position 5, whereas it becomes possible with position 4 (Scheme 2), decreasing the reactivity of the doublet.
Better conditions were evaluated for a complete conversion of 4-substituted benzamides starting with 4-methylbenzamide as test molecule. Increasing the reaction time to 15 minutes did not improve the conversion. The other possibility was to increase the ratio of anhydride to 2.5, 3, 4 and 5 equivalents. The more reliable results were obtained for 4 equivalents. However, with the initial suspension, an early formation of blocks of solid in the mixture did not help to achieve completion of the reaction. To reduce this drawback we used a more diluted suspension by reducing the amount of substrate 50% for the same volume of pinane. The stirring was also made a little more efficient with a smaller magnetic bar and a pre-stirring for five minutes of the suspension outside the apparatus. In these conditions only a slight increase of the conversion was achieved up to 96% and 93% for 5e and 5f (methyl and bromo derivatives), respectively. A purification was necessary to isolate the corresponding diamides from the starting benzamides.
Having found the best conditions for the initial formation of diamides 7 we then tried to achieve a direct formation of quinazolinones 8. A second step was added to the method to reach 180 °C for 10 minutes. Using 8a the cyclization occurred with some remaining diamide 7 and the side product 10. Varying the reaction time for the second step between 5 and 25 minutes showed that 15 minutes was the best compromise (Fig. S2, line 3, ESI†). Increasing the temperature gave more compound 10 and below 180 °C more diamide 7 was recovered, with 160 °C giving low cyclization. The difficulties seen during the reaction were the lack of solubility and limited stirring of the suspension and with the use of two equivalents of anhydride, succinic acid was formed. The best conditions were finally pre-stirring of the succinic anhydride as fine powder in the pinane during 5 minutes in the reactor without microwave, followed by addition of the benzamide in one shot and stirring for 5 minutes more. The reactor was then placed in the microwave equipment with the following method: pre-stirring 1 min, then 110 °C, 10 min, 300 W, then 180 °C, 15 min, 300 W, with overall stirring at 900 rpm. After cooling the solid was broken down to small pieces and the pinane filtered off. The solid was then re-suspended in water and stirred until it formed a fine suspension in the reactor. The pH may be adjusted to 5–6 with saturated aqueous NaHCO3. A final filtration and washing with water gave the quinazolinone (84% for 8a). Experimental details are in footnotes.
We finally compared the result for the full sequence for preparing compound 8a in pinane with the same made in toluene. In toluene we again did not observed any cyclization, the major compound being still the benzamide 7a. As previously mentioned, a major difference between toluene and pinane as the solvent is the continuous irradiation with toluene to maintain the temperature just below 180 °C when in pinane 180 °C is readily obtained. Toluene has a dipolar moment (0.375 D (ref. 19)) surely superior to pinane (cyclohexane = 0 D) but their dielectric constant may be almost the same (toluene 2.4, hexane 1.9) as well as their low microwave absorbing capability. As a result we postulate that some part of the energy is spent in toluene to maintain its evaporation and that its condensation also consumes some energy. In pinane, with a higher boiling point, all the energy goes to the substrates, possibly explaining the observed results. Beside the temperature or absorbing effect, the mechanism of cyclization may involve proton mobility in the solvent, and pi-stacking may exist20 in toluene that could contribute to stabilize the intermediate 7,21 whereas the apolar pinane must have low interactions with the substrates that can react faster. It can be anticipated that other renewable hydrocarbons with high boiling point could be used for this reaction.
Recovery of the pinane from microwave reactions
In the synthesis of diamides 7, the recovery of the pinane can be made by prior dilution of the reaction mixture with petroleum ether (PE), filtration and washing with PE. The combined filtrates were filtered on a pad of silica and the colorless oil concentrated under vacuum to yield pinane with 1H NMR spectra identical to the reference. For quinazolinone synthesis a similar protocol can be applied with or without adding PE.Conclusions
Originally designed for preparing diamide 7, a practical and simple microwave-assisted synthesis of quinazolinones 8 or diamide intermediates 7 is proposed using a common protocol. The yields are similar to the reported thermal conditions.The originality of this work is the use of pinane as a renewable and recyclable solvent to mix the substrates. Pinane appeared to have a specific role in the reaction, favouring the second step of cyclization that give a direct access to our quinazolinones from 2-aminobenzamides. Although the pinane did not compete with water as a green solvent, the reaction is performed without the need for K2CO3 used in microwave aqueous conditions. The prepared compounds are intermediates in a medicinal project leading to N3-methyl quinazolinone 9 (Scheme 2) analogues as potentially improved ZnF-UBP inhibitors.6
Experimental section
Example of thermal synthesis of diamide 7d. In a flask equipped with a condenser, 2-amino-benzamide (10 mmol, 1 eq.) and succinic anhydride (1 g, 10 mmol, 1 eq.) were mixed in 12.5 ml of toluene. The suspension was vigorously refluxed for 3 h (>10 h for 4-substituted benzamides) and then cooled to RT. The resulting white solid was filtered, washed with Et2O and dried.Example of microwave synthesis of diamide 7a. In a tubular 35 ml vial equipped with a magnetic stirrer, 2-amino-benzamide (500 mg, 3.67 mmol, 1 eq.) and succinic anhydride (734.5 mg, 7.34 mmol, 2 eq.) were mixed in 4.6 ml of pinane. The vial was closed with a pressure cap and put on the auto-sampler system of the microwave apparatus. After insertion in the reaction hole, a pre-stirring of 1 minute was followed by irradiation (300 W) up to 110 °C maintained for 10 minutes and wit stirring at 600 rpm. The resulting brown solid was poured into a round flask with methanol and after concentration under vacuum a brown solid was obtained analyzed by 1H NMR to measure the conversion. The purification was made as previously by washing with Et2O and dried to yield solids with NMR spectra identical to those obtained by thermal reactions.
Example of thermal synthesis of quinazolinone 8d. In a flask equipped with a condenser, compound 7d (9.2 mmol, 1 equiv.) was dissolved in an aqueous 2 N NaOH solution (15 ml, 3.3 equiv.). The resulting solution was refluxed for 2 h and then cooled to RT. The pH was carefully adjusted to pH 5–6 with aqueous 3 N HCl solution under vigorous stirring. The resulting white powder was filtered, washed with water and then dried to afford the expected quinazolinone 8d.
Example of microwave synthesis of quinazolinone 8a. In a tubular 35 ml vial equipped with a magnetic stirrer, succinic anhydride finely powdered (734.5 mg, 7.34 mmol, 2 eq.) was suspended in 4.6 ml of pinane. After 5 min stirring 2-amino-benzamide 5a (500 mg, 3.67 mmol, 1 eq.) was added, followed by 5 min stirring. The vial was then placed in the microwave oven with pre-stirring 1 minute, followed by 110 °C, 10 min, 300 W, then 180 °C, 15 min, 300 W, all at 900 rpm. After cooling the supernatant pinane is filtered on a glass filter 4, and the solid washed with PE. The solid is suspended in water (5 ml) and after stirring is filtered again and dried to give 680 mg (85%) of ivory solid with NMR spectra identical to those obtained by thermal reactions. The pinane can be purified by rapid filtration on a pad of silica.
4-((2-Carbamoyl-phenyl)amino)-4-oxobutanoic acid 7a. Prepared from benzamide 5a (500 mg, 3.67 mmol). Microwave: 499 mg, 99%.
4-((2-Carbamoyl-4-methylphenyl)amino)-4-oxobutanoic acid 7b. Prepared from 5b (600 mg, 4 mmol). Thermal: 945 mg, 95%. Microwave: 94%.
4-((2-Carbamoyl-4-methoxyphenyl)amino)-4-oxobutanoic acid 7c. Prepared from 5c (2.49 g, 15 mmol). Thermal: 3.92 g, 98%. Microwave 99%.
4-((2-Carbamoyl-4-chlorophenyl)amino)-4-oxobutanoic acid 7d. Prepared from 5d (1.7 g, 10 mmol). Thermal: 2.6 g, 96%. Microwave: 96%.
4-((2-Carbamoyl-5-methylphenyl)amino)-4-oxobutanoic acid 7e. Prepared from 5e (1.0 g, 10 mmol or 550 mg, 3.67 mmol). Thermal: 2.3 g, 92%. Microwave: 93%.
4-((2-Carbamoyl-5-chlorophenyl)amino)-4-oxobutanoic acid 7f. Prepared from 5f (1.7 g, 10 mmol). 2.54 g, 94%.
4-((2-Carbamoyl-5-bromophenyl)amino)-4-oxobutanoic acid 7g. Prepared from 5g (3.67 mmol). Thermal: 1.07 g, 92.9%. Microwave: 1.04 g, 90%.
4-((2-Carbamoyl-5-nitrophenyl)amino)-4-oxobutanoic acid 7h. Prepared from 5h (544 mg, 3 mmol). 2.54 g, 94%.
3-(6-Methyl-4-oxo-3,4-dihydroquinazolin-2-yl)propanoic acid 8b. Thermal: prepared from 7b (874 mg, 3.5 mmol). 0.80 g, 98.6%. Microwave: prepared from 5b, 3.67 mmol, 85%.
3-(6-Methoxy-4-oxo-3,4-dihydroquinazolin-2-yl)propanoic acid 8c. Thermal: prepared from 7c (3.6 g, 13.5 mmol). 3.07 g, 91%. Microwave: prepared from 5c, 3.67 mmol, 900 mg, 98.8%.
3-(6-Chloro-4-oxo-3,4-dihydroquinazolin-2-yl)propanoic acid 8d. Thermal: prepared from 7d (2.48 g, 9.2 mmol). 2.31 g, 99.5%.
3-(7-Methyl-4-oxo-3,4-dihydroquinazolin-2-yl)propanoic acid 8e. Thermal: prepared from 7e (2.2 g, 8.8 mmol). 2.0 g, 97.9%. Microwave: prepared from 5e, 3.67 mmol, 79%.
3-(7-Chloro-4-oxo-3,4-dihydroquinazolin-2-yl)propanoic acid 8f. Thermal: prepared from 7f (2.44 g, 9 mmol). 2.28 g, quant.
3-(7-Bromo-4-oxo-3,4-dihydroquinazolin-2-yl)propanoic acid 8g. Thermal: prepared from 7g (0.5 g, 1.59 mmol). 430 mg, 91.2%. Microwave: prepared from 5g (3.67 mmol), 89.3%.
3-(7-Nitro-4-oxo-3,4-dihydroquinazolin-2-yl)propanoic acid 8h. Thermal: prepared from 7h (655 mg, 2.3 mmol), 527 mg, 86%.
Author contributions
AR did the thermal and microwaves synthesis. PB wrote the manuscript. All authors have given approval to the final version of the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
Authors thank Ligue Contre le Cancer Comité Charentes Maritimes, Fond National Suisse (FNS) for funding AR postdoc, CNRS and University of Poitiers for the Equipment Program, Dr Ayman Karam (SARL SICO-CHEM), Dr Antoine Piccirilli (La Fabrique Végétale) and Christel Lafont (POC SARL) for providing pinane.References
- Q. Liu, B. Zhang, Y. Wang, X. Wang and S. Gou, Eur. J. Med. Chem., 2022, 229, 114058 CrossRef CAS PubMed.
- R. D. Amrutkar and M. S. Ranawat, Med. Chem. (Sharjah, United Arab Emirates), 2021, 17, 453 CAS.
- Z. A. Wani, et al., Food Chem. Toxicol., 2016, 87, 1 CrossRef CAS PubMed.
- A. M. Alsibaee, H. M. Al-Yousef and H. S. Al-Salem, Molecules, 2023, 28(3), 978 CrossRef CAS PubMed.
- M. Faisal and A. Saeed, Front. Chem., 2021, 8, 594717 CrossRef PubMed.
- R. Ferreira de Freitas, et al., J. Med. Chem., 2018, 61, 4517 CrossRef CAS PubMed.
- H. Bregman, J. L. Buchanan, N. Chakka, E. F. Dimauro, H. Gunaydin, P. A. Guzman, Z. Hua, X. Huang, WO2014036022, 2014.
- G. A. Khodarahmi, M. Shamshiri and F. Hassanzadeh, Res. Pharm. Sci., 2012, 7, 119 CAS.
- J. H. Clark and S. J. Tavener, Org. Process Res. Dev., 2007, 11, 149 CrossRef CAS.
- N. Winterton, Clean Technol. Environ. Policy, 2021, 23, 2499 CrossRef PubMed.
- R. Ciriminna, M. Lomeli-Rodriguez, P. Demma Carà, J. A. Lopez-Sanchez and M. Pagliaro, Chem. Commun., 2014, 50, 15288 RSC.
- J. N. M. Fanta, et al., C. R. Chim., 2018, 5972, 987 Search PubMed.
- E. Yara-Varón, et al., Green Chem., 2016, 18, 6596 RSC.
- B. Li, et al., Biomater. Sci., 2019, 7, 3675 RSC.
- L. Mohammadkhani and M. M. Heravi, Front. Chem., 2020, 8, 580086 CrossRef CAS PubMed.
- T. Besson and E. Chosson, Comb. Chem. High Throughput Screening, 2007, 10, 903 CrossRef CAS PubMed.
- Y. Kabri, A. Gellis and P. Vanelle, Green Chem., 2009, 11, 201 RSC.
- T. O. Olomola, D. A. Akinpelu and C. A. Obafemi, J. Pharm. Res., 2013, 6, 633 CAS.
- JACS, CRC Handbook of Chemistry and Physics, J. Am. Chem. Soc., 2009, 131, 2009–2010, p. 12862, 90th ed Search PubMed.
- G. A. DiLabio and E. R. Johnson, J. Am. Chem. Soc., 2007, 129, 6199 CrossRef CAS PubMed.
- G. Platzer, et al., Angew. Chem., Int. Ed. Engl., 2020, 59, 14861 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental details, 1H and 13C NMR of all compounds. See DOI: https://doi.org/10.1039/d3ra03702a |
| This journal is © The Royal Society of Chemistry 2023 |