
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSurface-strain-enhanced oxygen dissociation on gold catalysts†
Tianqi Gao‡
,
Yongli Shen‡,
Lin Gu,
Zhaocheng Zhang,
Wenjuan Yuan * and
Wei Xi*
* and
Wei Xi*
Center for Electron Microscopy, Tianjin Key Laboratory of Advanced Functional Porous Materials, Institute for New Energy Materials and Low-Carbon Technologies, School of Materials Science and Engineering, Tianjin University of Technology, Tianjin 300384, China. E-mail: yuanwj@email.tjut.edu.cn; weiandna1234@163.com
First published on 26th July 2023
Abstract
The excellent low-temperature oxidation performance and stability of nanogold catalysts have attracted significant interest. However, the main active source of the low-temperature oxidation of gold remains to be determined. In situ electron microscopy and mass spectrometry results show that nitrogen is oxidized, and the catalyst surface undergoes reconstruction during the process. Strain analysis of the catalyst surface and first-principles calculations show that the tensile strain of the catalyst surface affects the oxidation performance of gold catalysts by enhancing the adsorption ability and dissociation of O2. The newly formed active oxygen atoms on the gold surface act as active sites in the nitrogen oxidation reaction, significantly enhancing the oxidation ability of gold catalysts. This study provides evidence for the dissociation mechanism of oxygen on the gold surface and new design concepts for improving the oxidation activity of gold catalysts and nitrogen activation.
Introduction
Gold was previously considered an unsuitable catalytic material owing to its chemical stability. However, in 1987, Nobumasa et al.1 reported that gold nanoparticles exhibit excellent catalytic oxidation activity toward CO.2 Since then, the catalytic performance of more than thirty annuities has been extensively studied.3 Gold exhibits good catalytic activity in various catalytic reactions,2,4 such as CO oxidation,5,6 acetylene hydrogenation,7,8 methanol oxidation,9,10 methane pyrolysis,11 glucose oxidation,12,13 and other small-molecule heterogeneous catalytic reactions.14 Gold catalysts exhibit excellent CO oxidation activity even at a low temperature of −70 °C.15 This property renders them an excellent reference system for studying the characteristics of oxidation reaction catalysts under mild conditions.16–19 Therefore, the significant potential of gold catalysts in mild oxidation reactions necessitates the exploration of methods that can further improve their oxidation performance.In practical oxidation reactions, various factors can affect the catalytic activity of gold, and understanding the mechanism of heterogeneous catalysis at the microscale level remains a complex issue. The size of the gold catalyst,20,21 valence state of gold,22,23 surface curvature of gold,24–27 and gold carrier interactions21,28,29 are important factors that affect the catalytic oxidation activity of gold. Among the numerous influencing factors, oxygen or oxygen species adsorbed on the catalyst surface play significant roles in multiple oxidation reactions.30–32 For example, in the selective oxidation of alcohols, oxygen molecules adsorbed and activated on the surface of gold catalysts dissociate into active oxygen atoms, O*, which interact with alcohol molecules to form alkoxy intermediates and initiate the reaction cycle. In CO oxidation33,34 and volatile organic compound oxidation,35,36 the co-adsorption of reactants and oxygen at oxide interfaces or surface defect sites37 is an essential step.38,39 A study on alcohol oxidation reaction showed that gold catalysts exhibit better selective oxidation activity after being subjected to ozone treatment.40 Although the precise origin of the oxidation activity of gold catalysts remains varied in studies, previous studies have emphasized the crucial role of the active O species on the catalyst surface in the low-temperature oxidation of gold.26,31,41,42 Using gases with minimal impact on oxygen dissociation as reactants helps to better understand this process. The adsorption capacity of nitrogen on the gold surface is relatively weak,43 and nitrogen compounds have rich valence states and high bond energy,44 making N2 an ideal molecular probe.45–49 Currently, nitrogen fixation under relatively mild conditions still requires the synergistic activation of nitrogen–nitrogen triple bonds under various conditions. Therefore, studying the interactions between gold catalysts and various nitrogen oxides is beneficial for distinguishing the role of oxygen species on the surface of gold in the oxidation ability of gold catalysts and for nitrogen activation methods.
In this study, we investigated the effect of surface strain on nitrogen oxidation reactions (NORs) on gold catalysts. Nanoporous gold (NPG), whose environment has abundant stress that eliminates the influence of the catalyst support, is used as a model catalyst. Mass spectrometry (MS) results showed that the gold catalyst oxidized nitrogen to N2O. Significant changes were observed in the catalyst morphology during nitrogen oxidation. Additionally, strain analysis was employed to determine the surface strain, which was mainly tensile, of ligaments with different sizes. Density functional theory (DFT) calculations showed that the tensile strain reduced the activation energy of oxygen dissociation, whereas the presence of surface O atoms significantly promoted the activation of N2 molecules.
Experimental
Materials synthesis
NPG was prepared by dealloying Au–Ag alloy films (12-Karats, Gerstendörder) in concentrated HNO3 (65 wt%, Sinopharm Chemical Reagent Co., Ltd). To obtain NPG with a low silver content and ligament sizes of ∼10 (sample 1#), 15 (sample 2#), and 20 (sample 3#) nm, the Au–Ag alloy films were floated on the surface of 6, 8, and 10 mol L−1 HNO3 solutions under an applied potential (0.2 V, 240 s). Before further use, all NPG samples were rinsed several times in ultrapure water and floated on the surface of ultrapure water.Compositional and morphological characterization
High-resolution transmission electron microscopy (HRTEM, Talos F200, FEI) was employed to obtain morphological and crystallographic information on the NPG substrates. The scanning transmission electron microscopy (STEM) mode was used to determine the morphology and composition of the NPG samples. High-quality HRTEM images were collected using an electron microscope equipped with a spherical aberration-corrected transmission electron microscopy (AC-TEM) system (Themis Z, FEI). The X-ray photoelectron spectroscopy (XPS) spectra of the NPG-Pt samples were measured using an X-ray photoelectron spectrometer (ESCALAB 250Xi, Thermo Fisher Scientific) with Al-Kα radiation.In situ gas phase catalysis experiment
The NPG samples were loaded onto gas-phase chips and mounted on a gas-phase sample holder (Climate S3+, DENSsolutions). After gas tightness testing, the sample was analyzed using a high-resolution field-emission transmission electron microscope (Talos F200X, FEI) connected to a heating control system (Digiheater) and gas mass spectrometer (GSD 320, Omnistar Thermostat). Before the experiment, Ar was used to flush the reaction and gas supply systems. During the experiment, the actual pressure in the reaction chamber was 998.5 mbar, the gas flow rate was 0.26 mL min−1, and the temperature error was <5%. Mixed gases with different proportions of nitrogen and oxygen were used, and the temperature was raised by 10 °C each time after the MS signal and TEM morphology behavior stabilized. The changes in the morphology and lattice of the samples were recorded in situ using a charge-coupled device camera during the experiment. A mass spectrometer was used to detect the composition of the outgoing gas during the entire process by a Faraday detector.First-principles calculations
A five-layered face-centered cubic (FCC)-Au(111)-p(2√3 × 3)R30 structure was constructed to represent the pristine Au(111) model catalyst, and the crystal cell parameters of Au(111) in the x- and y-directions were amplified to 105% (Au(111)+5%). During the calculations, the two Au layers at the bottom were fixed to their bulk positions for pristine Au(111), whereas only the z-component was fixed for Au(111)+5%. All calculations in this study were performed using the Gaussian plane-wave method implemented in the CP2K Quickstep module.50 The generalized gradient approximation and Perdew–Burke–Ernzerhof functional51 were used for the calculations in this study. The van der Waals correction proposed by Grimme (DFT-D3(BJ) method)52 was also considered. The DZVP-MOLOPT-SR-GTH basis set with the Goedecker (Teter) pseudopotential was used for each atom in the calculations. The transition states were located using a two-step approach.53 First, the climbing nudged elastic band method was used to optimize the energies for a series of structures along the reaction path. The highest-energy structure was then used as the input for the dimer method to locate the transition state. For all geometric optimization and transition state search calculations performed in this study, the maximum force, maximum geometry change, root mean square (RMS) force, and RMS geometry change for atoms were set to 4.5 × 10−4, 3 × 10−3, 3 × 10−4, and 1.5 × 10−3, respectively, and the criterion for self-consistent field convergence was 1 × 10−6. An energy cutoff of 400 Ry was used for all calculations. The k-point mesh for the calculation was set to 5 × 4 × 1. All independent gradient models based on Hirshfeld partition (IGMH) analysis54 were performed using Multiwfn.55Results and discussion
Characterization of NPG
We prepared NPG samples with a ligament size of 9 nm using the dealloying method and subsequently characterized them. The bi-continuous ligament structure of NPG with uniform ligament and pore sizes (Fig. 1a) and the TEM bright-field image exhibit many positive and negative curvature surfaces. The HRTEM image and fast Fourier transform (FFT) images (Fig. 1b) show that the crystal structure of the NPG ligament is the same as that of the gold crystal, and the residual Ag atoms do not affect the crystal structure of gold. The high-angle annular dark-field (HAADF) image (Fig. 1c) and the corresponding energy-dispersive X-ray spectroscopy (EDX) mapping show a uniform distribution of residual Ag atoms. Multiple measurements in different regions show that the concentration of residual Ag is less than 1 at% and that of Au is >99 at%. To eliminate the influence of adsorbents on the surface of the NPG thin films, the samples were characterized by XPS after Ar plasma cleaning. Fig. 1d shows the electronic structures of Ag 3d5/2 in the NPG catalysts, indicating that Ag mainly exists in the metallic form on the NPG surface.56 The double-peak spectra (Fig. S1†) of Au 4f7/2 (84 eV), Au 4f5/2 (87.6 eV), Ag 3d3/2 (367.8 eV), and Ag 3d5/2 (373.8 eV) indicate that a negligible amount of oxide is present on the NPG surface after the Ar plasma treatment.57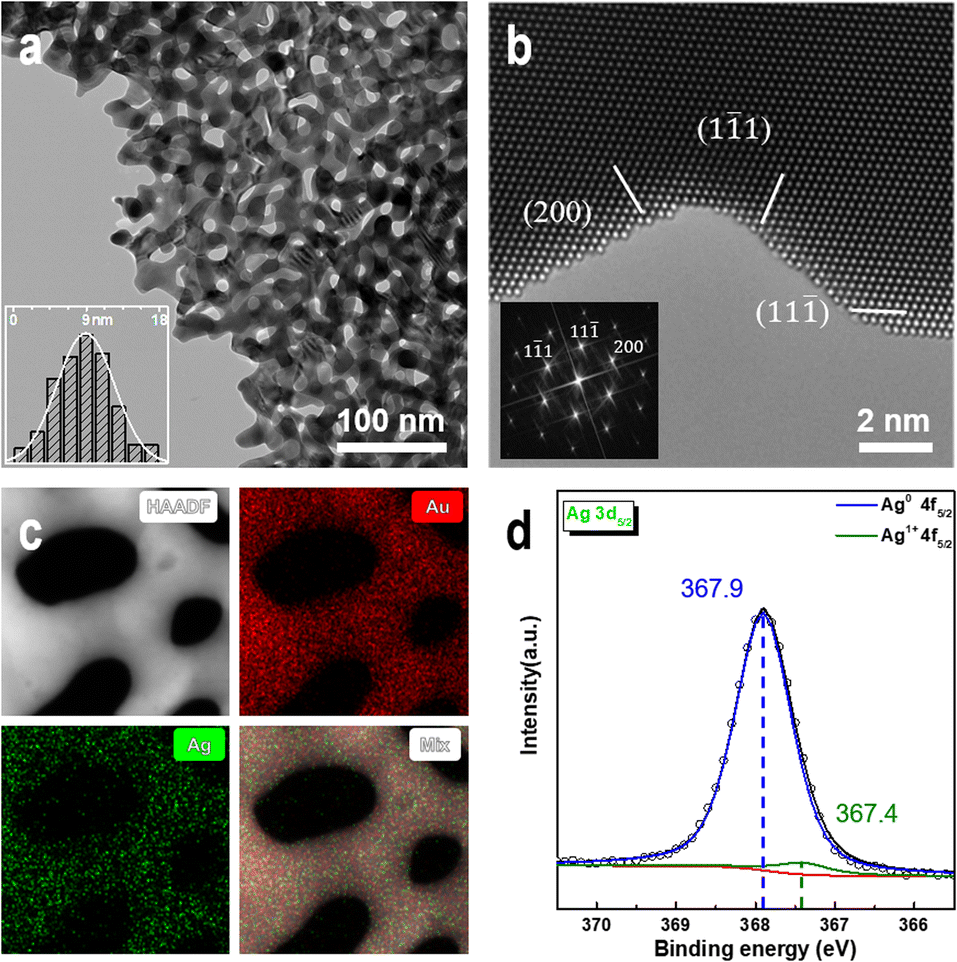 | ||
| Fig. 1 Characterization of NPG samples. (a) Low-power TEM image of NPG. The inset shows the size distribution of NPG ligaments. (b) HRTEM image of NPG. The inset is a FFT image of NPG. (c) HAADF image of NPG and its corresponding elemental distribution in the region. (d) Ag 3d5/2 XPS spectra of NPG. | ||
Fig. 2a–c shows that at 200 °C, the ligament of NPG ruptures when injected with a mixture of nitrogen and oxygen gas, and the surface morphology of the intact ligament also undergoes significant reconstruction. Additionally, the contrast change of the ligament shows a shift in the crystal orientation of the NPG ligament. The ligament rupture phenomenon is also observed outside the continuous electron beam irradiation area (Fig. S2†). However, before introducing the mixed gas at the same temperature, no significant changes were observed in the morphology of NPG in a pure argon, nitrogen, oxygen atmosphere and vacuum environment (Fig. S3†), and the ligaments of NPG were flattened after a long-term reaction (Fig. S4†).
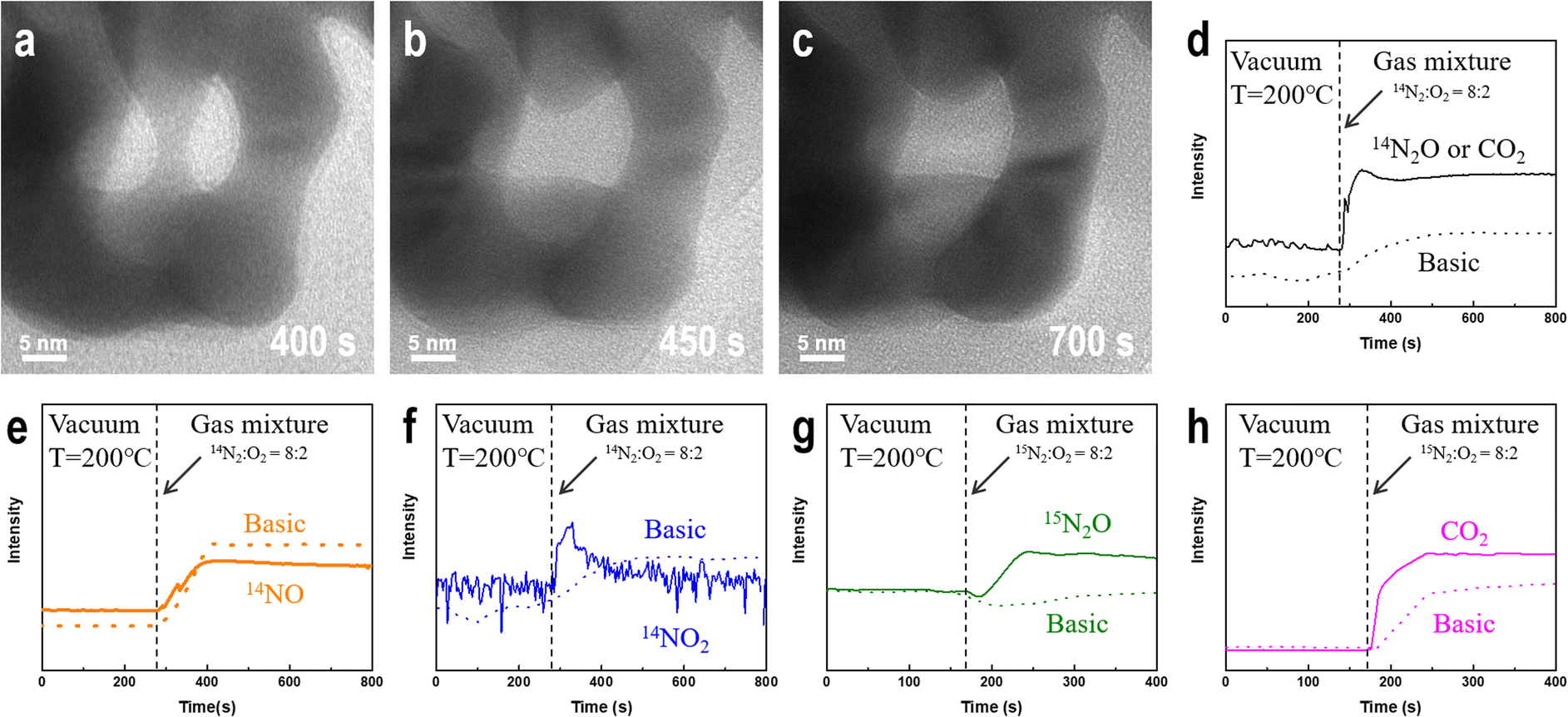 | ||
| Fig. 2 In situ changes in NPG morphology and MS data observed in gas-phase TEM experiments. (a)–(c) Morphological changes of NPG after introducing a nitrogen–oxygen mixture at 200 °C. (d)–(f) Changes in the signal intensities of NOx species in the effluent gas as determined by MS. (g) Changes in the signal intensity of 15N2O in the outflow gas as determined by MS. (h) Changes in the signal intensity of CO2 in the outflow gas as determined by MS. | ||
Analysis of the reaction products
To determine whether NPG causes nitrogen oxidation in oxygen atmosphere, we used a gas mass spectrometer to monitor the composition of the outflow gas. Because nitrogen exists in multiple valence states in nitrogen oxides, we monitored the changes in the intensities associated with three nitrogen-containing compounds, N2O, NO, and NO2, before and after introducing a nitrogen–oxygen gas mixture (Fig. 2d–f). The results were compared with those of a blank control under the same background conditions. After introducing a mixture of nitrogen and oxygen, as shown in Fig. 2d, the possible presence of the N(I) compound (N2O) is deduced. However, the mass spectrometer could not distinguish gas molecules with the same molecular weight, such as N2O and CO2. The intensities of the N(II) (NO) and N(IV) (NO2) compounds in the middle of the profile are significantly lower than those in the blank control group after the introduction of the nitrogen–oxygen mixture gas, as shown in Fig. 2e and f. Comparing Fig. 2e and f, nitrogen is not oxidized to NO and NO2 owing to the easy conversion of NO into NO2 under oxygen atmosphere. To verify that gold oxidizes nitrogen to N2O, we used the isotope 15N2 instead of ordinary nitrogen and repeated the experiment using the same sample (1#) under the same conditions. Fig. 2g shows that the intensity of the 15N2O signal is significantly higher than that in the blank control after introducing the reaction gas, confirming that nitrogen undergoes a NOR in the presence of NPG, and the nitrogen atoms in the N2O product originate from the introduced gas rather than the possible residual nitrogen-containing compounds in the sample and reactor. Furthermore, small amounts of CO2 (Fig. 2h) in the outflow gas can be detected.Factors affecting the degree of oxygen dissociation
To determine the factors affecting the degree of nitrogen oxidation, we conducted in situ experiments considering different parameters, including electron beam irradiation, the oxygen content, temperature, and the NPG ligament size. Subsequently, we summarized the criteria for evaluating their impact on the strength of nitrogen-containing compounds.First, the electron beam irradiation was shielded using a beam blank in the electron microscopy equipment to confirm that the electron beam exhibited no significant effects on the catalytic reaction. The MS spectrum showed no significant change in the intensity of the 15N2O signal (Fig. S5†). Subsequently, while the temperature and pressure remained constant, the proportion of oxygen in the mixed gas was increased continuously. As shown in Fig. 3a, the intensity of the detected 15N2O signal increases with increasing oxygen content. Moreover, at low oxygen concentrations (<60%), the 15N2O signal intensity shows a stepwise increase, reaching its maximum value when the oxygen content reaches 90%. The intensity of the 15N2O product signal shown in Fig. 3b continuously increases with an increase in the oxygen content from 30% to 40% rather than increasing with mutations. These results indicate that oxygen plays an essential role in gold-catalyzed nitrogen oxidation.
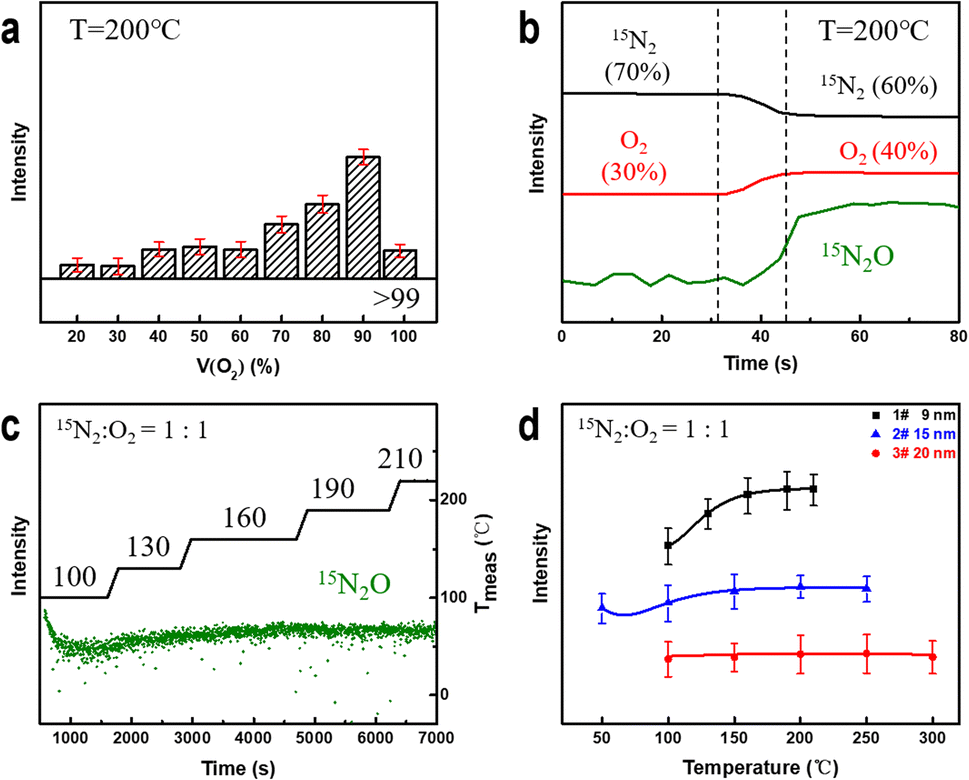 | ||
Fig. 3 Changes in the N2O signal intensity measured by MS under different conditions. (a) and (b) Continuous variation in the N2O signal intensity at different oxygen contents. (c) Changes in the N2O signal intensity as a function of time at different temperatures when the gas ratio is 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1, Tmeas is the actual measured temperature value. (d) Changes in the N2O signal intensity of samples with differing ligament sizes during heating. :1, Tmeas is the actual measured temperature value. (d) Changes in the N2O signal intensity of samples with differing ligament sizes during heating. | ||
Next, we studied the effect of temperature on the oxidation reaction at 50% oxygen content. As shown in Fig. 3c, the 15N2O signal intensity decreases and gradually stabilizes after introducing a nitrogen–oxygen mixture at 100 °C for 100 s. Subsequently, the signal strength of the product increases with increasing reaction temperature and stabilizes at approximately 190 °C. Coarsening of ligaments (Fig. S6†) may be the reason for no further elevation above 190 °C. This indicates that NPG can oxidize nitrogen at lower temperatures, and increasing the temperature can improve the NOR. To explore this further, we prepared NPG samples 2# (Fig. S7†) and 3# (Fig. S8†) with ligament sizes of ∼15 and ∼20 nm, respectively, and residual Ag atom percentages of less than 1 at%. At 50% oxygen, the reaction temperature was gradually increased until the MS signal intensity of the product no longer increased significantly. As shown in Fig. 3d, the 15N2O signal of the NPG samples with smaller ligament sizes is stronger and more sensitive to temperature changes. The larger the ligament size, the weaker the N2O signal of the samples (2# and 3#) under the same reaction conditions. The experimental results indicate that NPG samples with smaller ligaments exhibit better oxidation activity, which is consistent with current research findings obtained for NPG in heterogeneous catalytic reactions.58,59 The variation in the surface structure of NPG samples with small and large ligaments accounts for their distinct NOR activities.
To investigate the effect of size on the surface structure of the NPG ligaments, we collected HRTEM images of ligaments with differing sizes using AC-TEM and performed atomic-scale strain analysis.60 Fig. 4a shows the presence of numerous exposed (111) crystal surfaces on the ligaments. Fig. 4b reveals unevenly distributed strain on the surface of the ligaments in the 〈111〉 axial direction. The strain analysis shows that the tensile strain in certain areas (yellow) is higher than 5%. The degree of strain in the radial direction 〈112〉 is significantly smaller than that in the axial direction, with uniform distributions of the tensile and compressive strains. Comparing the strain distribution results for different ligament sizes (Fig. S9†), the axial strain is significantly higher than the radial strain, and the surface strain of small-sized ligaments is higher than that of large-sized ligaments. The internal strain of NPG arises from the process of dealloying, during which gold atoms cannot rapidly fill the voids left by the large-scale rapid detachment of silver atoms (Fig. S10†), forming a double connected structure with complex internal stress. These findings are consistent with the local strain law for nanoporous metals.61–63
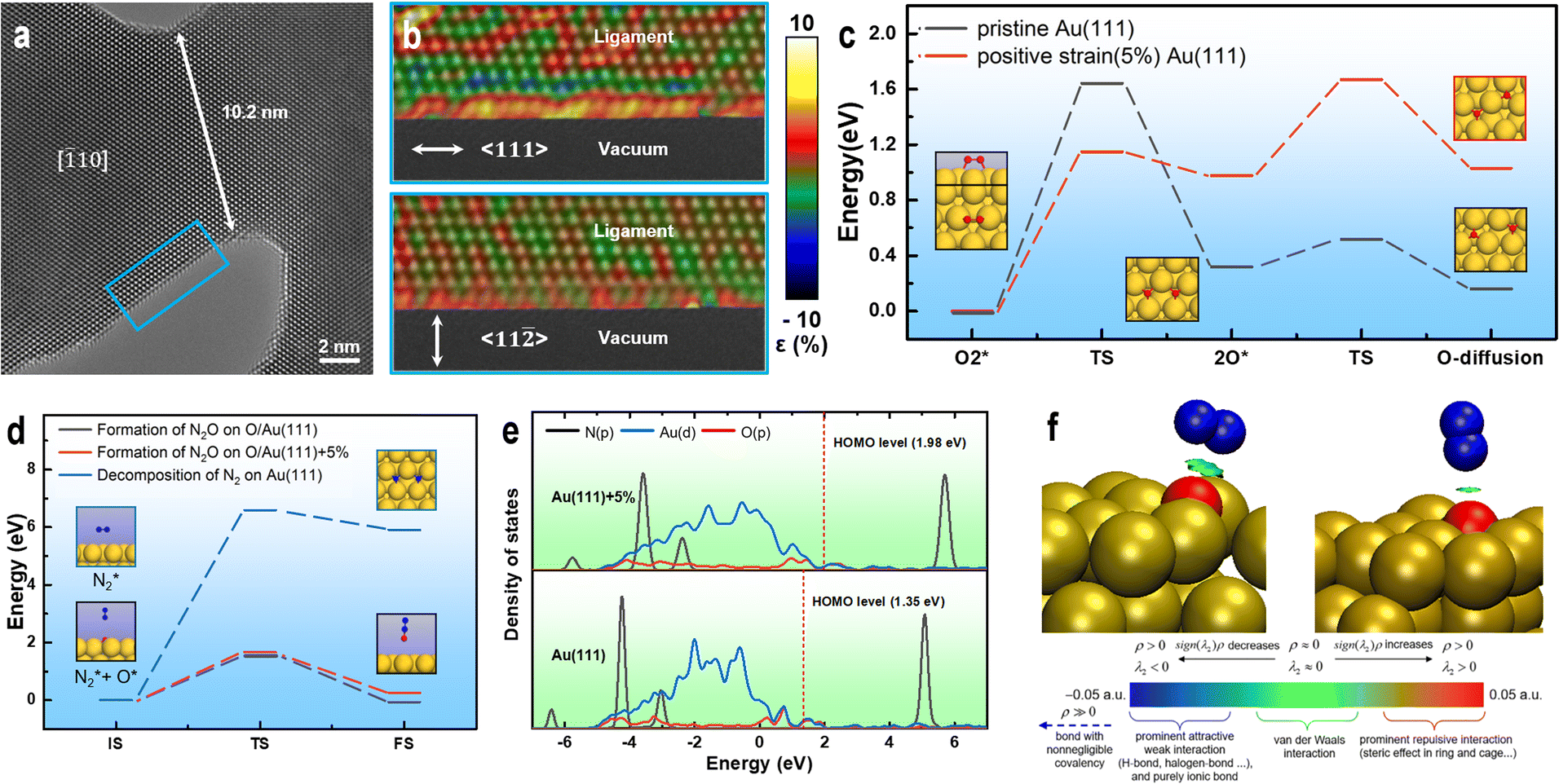 | ||
| Fig. 4 Surface strain analysis and theoretical calculation of ligaments. (a) Strain distribution in the blue area of the HRTEM image. (b) Segment of the NPG ligament with a size of ∼11 nm. (c) Minimum energy path (MEP) for the decomposition of O2 and O atom diffusion on Au(111) and Au(111)+5%. (d) MEP for the decomposition of N2 on Au(111) O/Au(111) and O/Au(111)+5%. (e) DOS of the N2–O/Au(111) and N2–O/Au(111)+5% adsorption structures. (f) IGMH analysis of the interactions between N2 and O/Au(111) and O/Au(111)+5%. Standard interpretation of the coloring method of the mapped function sign(λ2)ρ in IGMH is based on ref. 5 (dark blue sphere: N, red sphere: O, and yellow sphere: Au). | ||
Various factors indicate that tensile strain enhances the oxidation activity of gold catalysts. Therefore, it is necessary to investigate the effect of strain on the nitrogen and oxygen interaction on the gold catalyst surface. First, the activation of O2 on the surface of the catalysts was analyzed. The computational results show that positive-strain Au(111) can facilitate O2 decomposition. Additionally, the activation energy required for O2 decomposition on Au (111)+5% is lower (0.49 eV) than that on pristine Au(111) (Fig. 4c). The newly formed O atoms on both pristine Au(111) and Au(111)+5% easily diffuses to the adjacent stable adsorption sites on the catalyst surface (Fig. 4c), providing active oxygen atoms for subsequent oxidation reactions. For N2 adsorption, the computational results show that the vertical distances between N2 and the possible active centers (O atom adsorbed on Au) on the surfaces of Au(111) and Au(111)+5% are 3.34 and 2.77 Å, respectively, indicating that the interaction between N2 and the catalyst may be weak. The density of state (DOS) calculations show no significant overlap between the p orbitals of N2 and p orbitals of O in the N2 adsorption structures on both catalysts (Fig. 4e). IGMH analysis of the molecular density48 reveals only van der Waals interactions between N2 and O in the N2 adsorption structures (Fig. 4f). However, the adsorption strength of N2 on O/Au(111)+5% is slightly higher than that on O/Au(111). The activation energy required for the decomposition of N2 on the surfaces of the O/Au(111) and O/Au(111)+5% catalysts is significantly lower than that on the pristine Au(111) catalyst (Fig. 4d). Therefore, the active O atoms on the Au surface are speculated to play a crucial role in the activation of N2. The calculation results show that the difficulty of N2O generation on the surfaces of the two catalysts (O/Au(111) and O/Au(111)+5%) is not closely related to the adsorption strength of N2, but only depends on the availability of highly active O atoms on the catalyst surface (Fig. 4d).
Conclusions
This study demonstrated that the presence of strain enhances the ability of gold catalysts to oxidize nitrogen. Specifically, changes in the surface strain of gold catalysts significantly enhance the adsorption capacity of oxygen molecules on the Au(111) crystal surface and reduce the activation energy required to dissociate O2 into oxygen atoms. The interaction between these active oxygen atoms and nitrogen molecules is stronger than the adsorption of nitrogen on the gold surface, which plays an essential role in the NOR on the gold surface. These findings suggest that strain engineering can enhance the gold oxidation reaction, and precise control over the catalyst surface strain can be effective for designing high-oxidation-activity catalysts.Author contributions
T. G. and L. G. performed the main experiments. T. G., Z. Z. and W. Y. analyzed the data of the TEM experiments. Y. S. performed the theoretical calculations. W. Y. and W. X. co-supervised the project. All authors took part in analyzing the data and writing the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was financially supported by the National Natural Science Foundation of China [grant number 51701143] and the Tianjin Municipal Science and Technology Commission [grant number 18JCYBJC86900].References
- M. Haruta, T. Kobayashi, H. Sano and Y. Nobumasa, Chem. Lett., 1987, 16, 405 CrossRef.
- R. Ciriminna, E. Falletta, C. D. Pina, J. H. Teles and M. Pagliaro, Angew. Chem., Int. Ed., 2016, 55, 14210 CrossRef CAS PubMed.
- M. Okumura, T. Fujitani, J. H. Huang and T. Ishida, ACS Catal., 2015, 5, 4699 CrossRef CAS.
- Z. J. Guan, J. J. Li, F. Hu and Q. M. Wang, Angew. Chem., Int. Ed., 2022, 61, e202209725 CAS.
- C. Xu, X. Xu, J. Su and Y. Ding, J. Catal., 2007, 252, 243–248 CrossRef CAS.
- F. Kettemann, S. Witte, A. Birnbaum, B. Paul, G. Clavel, N. Pinna, K. Rademann, R. Kraehnert and J. Polte, ACS Catal., 2017, 7, 8247 CrossRef CAS.
- H. Chen, Z. Li, Z. Qin, H. J. Kim, H. Abroshan and G. Li, ACS Appl. Nano Mater., 2019, 2, 2999 CrossRef CAS.
- A. C. Gluhoi, J. W. Bakker and B. E. Nieuwenhuys, Catal. Today, 2010, 154, 13 CrossRef CAS.
- Z. Borkowska, A. Tymosiak-Zielinska and R. Nowakowski, Electrochim. Acta, 2004, 49, 2613 CrossRef CAS.
- L. C. Wang, M. L. Personick, S. Karakalos, R. Fushimi, C. M. Friend and R. J. Madix, J. Catal., 2016, 344, 778 CrossRef CAS.
- W. Xi, K. Wang, Y. Shen, M. Ge, Z. Deng, Y. Zhao, Q. Cao, Y. Ding, G. Hu and J. Luo, Nat. Commun., 2020, 11, 1919 CrossRef CAS PubMed.
- P. Beltrame, M. Comotti, C. D. Pina and M. Rossi, Appl. Catal., A, 2006, 297, 1 CrossRef CAS.
- H. D. Toit and M. D. Lorenzo, Sens. Actuators, B, 2014, 192, 725 CrossRef.
- I. McCue, E. Benn, B. Gaskey and J. Erlebacher, Annu. Rev. Mater. Res., 2016, 46, 263 CrossRef CAS.
- M. Haruta, Angew. Chem., Int. Ed., 2014, 53, 52 CrossRef CAS PubMed.
- C. Xu, J. Su, X. Xu, P. Liu, H. Zhao, F. Tian and Y. Ding, J. Am. Chem. Soc., 2007, 129, 42 CrossRef CAS PubMed.
- S. Witzel, A. S. K. Hashmi and J. Xie, Chem. Rev., 2021, 121, 8868 CrossRef CAS.
- M. Rudolph and A. S. K. Hashmi, Chem. Soc. Rev., 2012, 41, 2448 RSC.
- X. K. Wan, J. Q. Wang, Z. A. Nan and Q. M. Wang, Sci. Adv., 2017, 3, 1701823 CrossRef PubMed.
- M. Luna, A. Cruceira, A. Diaz, J. M. Gatica and M. J. Mosquera, Environ. Technol. Innovation, 2023, 30, 103070 CrossRef CAS.
- Y. Lykhach, S. M. Kozlov, T. Skala, A. Tovt, V. Stetsovych, N. Tsud, F. Dvorak, V. Johanek, A. Neitzel, J. Mysliveček, S. Fabris, V. Matolín, K. M. Neyman and J. Libuda, Nat. Mater., 2016, 15, 284 CrossRef CAS PubMed.
- D. W. Wang, R. Cai, S. Sharma, J. Jirak, S. K. Thummanapelli, N. G. Akhmedov, H. Zhang, X. Liu, J. L. Petersen and X. Shi, J. Am. Chem. Soc., 2012, 134, 9012 CrossRef CAS.
- A. Ade, E. Cerrada, M. Contel, M. Laguna, P. Merino and T. Tejero, J. Organomet. Chem., 2004, 689, 1788 CrossRef CAS.
- T. Fujita, P. F. Guan, K. Mckenna, X. Lang, A. Hirata, L. Zhang, T. Tokunaga, S. Arai, Y. Yamamoto, N. Tanaka, Y. Ishikawa, N. Asao, Y. Yamamoto, J. Erlebacher and M. Chen, Nat. Mater., 2012, 11, 775 CrossRef CAS PubMed.
- S. Satapathy, J. Mohanta and S. Si, ChemistrySelect, 2016, 1, 4940 CrossRef.
- Z. Lin, H. Hirao, C. Sun and X. Zhang, Phys. Chem. Chem. Phys., 2020, 22, 14458 RSC.
- X. Cao, C. J. Li, Y. Lu, B. Zhang, Y. Wu, Q. Liu, J. Wu, J. Teng, W. Yan and Y. Huang, J. Catal., 2019, 377, 389 CrossRef CAS.
- K. Sakamoto, S. Masuda, S. Takano and T. Tsukuda, ACS Catal., 2023, 13, 3263 CrossRef CAS.
- Y. Y. Li, Y. S. Zhang, K. Qian and W. Huang, ACS Catal., 2022, 12, 1268 CrossRef CAS.
- Q. Y. Li, J. H. Zhou, D. D. Li and Z. Ao, J. Mater. Chem. A, 2022, 10, 9025 RSC.
- L. L. Huang, W. Liu, J. Hu and X. Xing, J. Phys. Chem. A, 2022, 126, 5594 CrossRef CAS PubMed.
- T. A. Baker, X. Y. Liu and C. M. Friend, Phys. Chem. Chem. Phys., 2011, 13, 34 RSC.
- J. D. Lee, J. B. Miller, A. V. Shneidman, L. Sun, J. F. Weaver, J. Aizenberg, J. Biener, J. A. Boscoboinik, A. C. Foucher, A. I. Frenkel and J. E. van der Hoeven, Chem. Rev., 2022, 122, 8758 CrossRef CAS PubMed.
- Y. G. Wang, D. H. Mei, V. A. Glezakou, J. Li and R. Rousseau, Nat. Commun., 2015, 6, 6511 CrossRef CAS PubMed.
- E. Iro, H. Ariga-Miwa and T. Sasaki, Catalysts, 2022, 12, 1365 CrossRef CAS.
- S. Chu, E. Wang, F. Feng, C. Zhang, J. Jiang, Q. Zhang, F. Wang, L. Bing, G. Wang and D. Han, Catalysts, 2022, 12, 1543 CrossRef CAS.
- T. A. Baker, C. M. Friend and E. Kaxiras, J. Phys. Chem. C, 2009, 113, 3232 CrossRef CAS.
- K. Sun, Chin. J. Catal., 2016, 37, 1608 CrossRef CAS.
- Y. Shen, X. Yang, Y. Wang, Y. Zhang, H. Zhu, L. Gao and M. Jia, Appl. Catal., B, 2008, 79, 142 CrossRef CAS.
- M. L. Personick, B. Zugic, M. M. Biener, J. Beiner, R. J. Madix and C. M. Friend, ACS Catal., 2015, 5, 4237 CrossRef CAS.
- T. Fujitani, I. Nakamura and M. Haruta, Catal. Lett., 2014, 144, 1475 CrossRef CAS.
- M. G. Sensoy and M. M. Montemore, J. Phys. Chem. C, 2020, 124, 8843 CrossRef CAS.
- A. R. Davies, H. Azim and T. G. Wright, Mol. Phys., 2023, 121, 24 CrossRef.
- J. G. Chen, R. M. Crooks, L. C. Seefeldt, K. L. Bren, R. M. Bullock, M. Y. Darensbourg, P. L. Holland, B. Hoffman, M. J. Janik, A. K. Jones, M. G. Kanatzidis, P. King, K. M. Lancaster, S. V. Lymar, P. Pfromm, W. F. Schneider and R. R. Schrock, Science, 2018, 360, 7 Search PubMed.
- Z. F. Nie, L. L. Zhang, X. Ding, M. Y. Cong, F. F. Xu, L. H. Ma, M. X. Guo, M. Z. Li and L. X. Zhang, Adv. Mater., 2022, 34, 8 Search PubMed.
- Y. Yu, C. H. Wang, Y. F. Yu, Y. M. Huang, C. B. Liu, S. Y. Lu and B. Zhang, J. Mater. Chem. A, 2020, 8, 19623 RSC.
- A. Pashkova, B. O. Burek and J. Z. Bloh, Catal. Sci. Technol., 2022, 12, 2755 RSC.
- X. R. Zhang, R. Shi, Z. H. Li, J. Q. Zhao, H. N. Huang, C. Zhou and T. R. Zhang, Adv. Energy Mater., 2022, 12, 7 Search PubMed.
- C. X. Zhang, Y. M. Huang, B. H. Zhao, Y. Yu, Y. F. Yu and B. Zhang, J. Catal., 2022, 409, 70 CrossRef CAS.
- T. D. Kühne, M. Iannuzzi, M. D. Ben, V. V. Rybkin, P. Seewald, F. Stein, T. Laino, R. Z. Khaliullin, O. Schütt, F. Schiffmann and D. Golze, J. Chem. Phys., 2020, 152, 194103 CrossRef PubMed.
- Y. Zhang and W. Yang, Phys. Rev. Lett., 1998, 80, 890 CrossRef CAS.
- S. Grimme, S. Ehrlich and L. Goerigk, J. Comput. Chem., 2011, 32, 1456 CrossRef CAS PubMed.
- T. Ricciardulli, S. Gorthy, J. S. Adams, C. Thompson, A. M. Karim, M. Neurock and D. W. Flaherty, J. Am. Chem. Soc., 2021, 143, 5445 CrossRef CAS PubMed.
- T. Lu and Q. X. Chen, J. Comput. Chem., 2022, 43, 539 CrossRef CAS PubMed.
- T. Lu and F. W. Chen, J. Comput. Chem., 2012, 33, 580 CrossRef CAS PubMed.
- M. Graf, M. Haensch, J. Carstens, G. Wittstock and J. Weissmüller, Nanoscale, 2017, 9, 17839 RSC.
- A. R. S. Olaya, F. Kühling, C. Mahr, B. Zandersons, A. Rosenauer, J. Weissmüller and G. Wittstock, ACS Catal., 2022, 12, 4415 CrossRef.
- T. Fujita, T. Tokunaga, L. Zhang, D. Li, L. Chen, S. Arai, Y. Yamamoto, A. Hirata, N. Tanaka, Y. Ding and M. Chen, Nano Lett., 2014, 14, 1172 CrossRef CAS PubMed.
- A. Y. Chen, J. W. Wang, Y. Wang, Y. Q. Jia, J. F. Gu, X. F. Xie and D. Pan, Electrochim. Acta, 2015, 153, 552 CrossRef CAS.
- P. L. Galindo, S. Kret, A. M. Sanchez, J. Y. Laval, A. Yanez, J. Pizarro, E. Guerrero, T. Ben and S. I. Molina, Ultramicroscopy, 2007, 107, 1186 CrossRef CAS PubMed.
- C. Mahr, K. Müller-Caspary, M. Graf, A. Lackmann, T. Grieb, M. Schowalter, F. F. Krause, T. Mehrtens, A. Wittstock, J. Weissmüller and A. Rosenauer, Mater. Res. Lett., 2018, 6, 84 CrossRef CAS.
- E. J. Schofield, B. Ingham, A. Turnbull, M. F. Toney and M. P. Ryan, Appl. Phys. Lett., 2008, 92, 043118 CrossRef.
- J. Weissmüller, H. L. Duan and D. Farkas, Acta Mater., 2010, 58, 1 CrossRef.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3ra03781a |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2023 |