
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Synthesis of cyclic carbonates from CO2 cycloaddition to bio-based epoxides and glycerol: an overview of recent development
Muhammad Usman ab,
Abdul Rehman*ab,
Faisal Saleemab,
Aumber Abbasc,
Valentine C. Ezeb and
Adam Harveyb
ab,
Abdul Rehman*ab,
Faisal Saleemab,
Aumber Abbasc,
Valentine C. Ezeb and
Adam Harveyb
aDepartment of Chemical and Polymer Engineering, University of Engineering and Technology Lahore, Faisalabad Campus, Pakistan. E-mail: a.rehman2@uet.edu.pk
bSchool of Engineering, Newcastle University, Newcastle Upon Tyne, NE1 7RU, UK
cSongshan Lake Materials Laboratory, University Innovation Park, Dongguan, 523808, China
First published on 26th July 2023
Abstract
Anthropogenic carbon dioxide (CO2) emissions contribute significantly to global warming and deplete fossil carbon resources, prompting a shift to bio-based raw materials. The two main technologies for reducing CO2 emissions are capturing and either storing or utilizing it. However, while capture and storage have high reduction potential, they lack economic feasibility. Conversely, by utilizing the CO2 captured from streams and air to produce valuable products, it can become an asset and curb greenhouse gas effects. CO2 is a challenging C1-building block due to its high kinetic inertness and thermodynamic stability, requiring high temperature and pressure conditions and a reactive catalytic system. Nonetheless, cyclic carbonate production by reacting epoxides and CO2 is a promising green and sustainable chemistry reaction, with enormous potential applications as an electrolyte in lithium-ion batteries, a green solvent, and a monomer in polycarbonate production. This review focuses on the most recent developments in the synthesis of cyclic carbonates from glycerol and bio-based epoxides, as well as efficient methods for chemically transforming CO2 using flow chemistry and novel reactor designs.
1. Introduction
A terpene is a hydrocarbon consisting of at least two isoprene units (C5H8) and is odourless. Five carbon atoms with double bonds make up an isoprene molecule, also referred to as an isoprene unit. Isoprene itself, a gaseous hydrocarbon, is emitted by leaves as a natural by-product of plant metabolism. Monoterpenes (C10H16) contain two isoprene units (Fig. 1), while sesquiterpenes have three, diterpenes have four, triterpenes have six, and tetraterpenes have eight isoprene units.1 The name of a terpene always ends with –ene. The structure of terpenes determines whether they are cyclic or acyclic, with cyclic terpenes forming a ring and acyclic terpenes being linear. Most terpenes are monoterpenes, which can be categorized into groups based on their structure. Terpenes have low molecular weight, which enables them to evaporate quickly. Terpenes are sometimes confused with terpenoids, but the key difference is that terpenes do not contain oxygen while terpenoids do.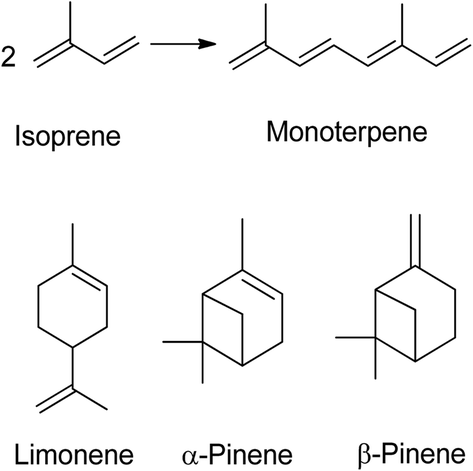 | ||
| Fig. 1 Two isoprene units give monoterpene, the chemical structure of limonene and pinene.1 | ||
Monoterpenes are volatile and commonly used for flavouring and perfumes due to their strong odour. They are also lipophilic, allowing them to easily penetrate cell membranes, and are known to have antispasmodic properties.2–4 Cannabis contains approximately 200 terpenes, which is only a small fraction of the 20![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 terpenes known to exist in plants.5 Among these, limonene and pinene are major terpenes.6 Limonene, a colourless liquid, has a boiling point of 175 °C and is comprised of two isomers: D-limonene and L-limonene. D-limonene is the primary component of essential oils found in citrus fruit rinds, such as oranges, lemons, mandarins, grapefruits, and limes. On the other hand, L-limonene is emitted as a key component of volatile oils by oaks and pines.7 Orange peels contain around 97% of limonene,8 and with around 110 million tons of citrus fruits produced annually, this waste can be a source of essential oil extraction. Various methods, such as solvent extraction, steam distillation, and water distillation extraction, have been employed to extract the oil from orange peels. Steam distillation has been found to produce the maximum yield of oil at 4.40%, followed by water distillation extraction at 3.47% and solvent extraction at 2.53%.9
000 terpenes known to exist in plants.5 Among these, limonene and pinene are major terpenes.6 Limonene, a colourless liquid, has a boiling point of 175 °C and is comprised of two isomers: D-limonene and L-limonene. D-limonene is the primary component of essential oils found in citrus fruit rinds, such as oranges, lemons, mandarins, grapefruits, and limes. On the other hand, L-limonene is emitted as a key component of volatile oils by oaks and pines.7 Orange peels contain around 97% of limonene,8 and with around 110 million tons of citrus fruits produced annually, this waste can be a source of essential oil extraction. Various methods, such as solvent extraction, steam distillation, and water distillation extraction, have been employed to extract the oil from orange peels. Steam distillation has been found to produce the maximum yield of oil at 4.40%, followed by water distillation extraction at 3.47% and solvent extraction at 2.53%.9
D-Limonene is widely manufactured globally, with approximately 300 orange oil distillation plants operating worldwide to produce technical-grade (95%) and food-grade (96%) D-limonene from orange oil.7 The market for D-limonene was estimated to be worth USD 473.72 million in 2021, and it is anticipated to increase to USD 694.59 million between 2022 and 2029, increasing at a compound annual growth rate (CAGR) of 4.90%.10 Limonene is mainly used in the flavour and fragrance industry, as a solvent, and for making polymers and adhesives.11 It was first used in 2008 to obtain fats and oils, replacing toxic solvents like n-hexane, effectively eliminating toxicity and environmental impact. D-Limonene is also an excellent cleansing agent, with a high KB (74) and density of 0.84, which allows it to hold a large amount of dirt before becoming ineffective. A compound's KB value is a measure of its ability to hold dirt.7 Additionally, L-limonene and D-limonene are among the 44 sustainable solvents developed by GlaxoSmithKline (GSK).12
In Kraft wood pulping, turpentine oil is produced as an energy recovery oil. The increasing demand for chemical products containing artificial flavours, fragrances, and solvents is driving the demand for turpentine oil, which is also a promising alternative fuel for various industrial applications.13 Turpentine oil is currently the primary source of pinene, with an average yield of 0.3–1 kg per tonne of pulp production. Worldwide, the pulping industry produces an estimated 3.5 × 105 Mt per year of turpentine oil, with α-pinene accounting for 70% of the total monoterpene content.14,15 α-Pinene is the dominant form of pinene, found in pine trees and cannabis, and responsible for the characteristic pine scent. The majority of applications use α-pinene. Pine-sol, a cleaning product, contains this terpene. α-Pinene has significant potential as an engine biofuel, as it is comparable to rocket fuel in terms of volumetric heating.16
Glycerol is a versatile chemical that may be processed through numerous paths to creating a variety of products with value-added, such as reforming, dehydration, esterification, selective oxidation, hydrogenolysis, and acetylation.17 However, a direct reaction of glycerol and CO2 is an attractive route that effectively combines two waste products to produce glycerol carbonate. The production of glycerol is a by-product of the biodiesel or green diesel industry, where approximately 10 wt% of glycerol is obtained from every kg of biodiesel, and around 70% of glycerol is produced in this manner.18 The biodiesel market was estimated to be worth USD 40.6 billion in 2021 and is anticipated to increase by 4.4% to USD 52.7 billion by 2027.19 With the expected increase in biodiesel production, there will be a surplus of glycerol that can be utilized as a low-cost chemical feedstock to produce glycerol carbonates and glycerol acetates (also known as acetins). Acetins are non-natural, synthetic products that can be obtained either through traditional chemical conversion or new biological conversion. Chemical conversions involve toxic solvents and reactants (such as acetic acid and acetic anhydride) and require harsh temperature and pressure conditions, making them non-green and polluting. In contrast, biological processes for acetin production are considered green as they use enzymes or microbes as biocatalysts. Acetins have various applications, such as plasticizers, explosives, emulsifiers, food additives, and fuel additives.
Both terpenes and glycerol offer pathways for the conversion of low-value compounds into high-value chemicals with diverse applications. Terpenes, such as limonene, can be coupled with CO2 to produce terpene-based cyclic carbonates, while glycerol can be transformed into glycerol carbonate (GC). Glycerol carbonate (GC) is a liquid with a high water solubility, high boiling point, and low toxicity. It has potential applications as a solvent, as a surfactant, and as an electrolyte in Li-ion batteries.20,21
1.1 CO2 and its utilisation
In 2020, global CO2 emissions reached 34.8 Gt, resulting in a reported temperature increase of 1.16 °C relative to 1920.22 In April 2022, global monthly mean CO2 emissions were at a record high of 418.39 ppm, up from 415.82 ppm in April 2021.23 It is projected that by 2050, 5.6 Gt per year of CO2 will need to be captured and stored, equivalent to 40 Mt year of CO2 today.24 However, currently, only 100 Kt of cyclic carbonates are produced annually, consuming 40 Kt of direct CO2 each year.25,26 Approximately 35 Mt of CO2 is released each year, with less than half being emitted by large point sources that can be captured, stored, and utilized.27 To address this global CO2 challenge, a multifaceted approach is necessary, which includes Carbon Capture and Storage (CCS) primarily focusing on capturing and storing, Carbon Capture and Utilization (CCU) focusing on utilization after capturing, energy transition to non-fossil fuels such as solar, wind, and tidal, aiming to improve energy efficiency and minimizing heat loss.25There are now competitive technologies such as solar and wind that can replace fossil fuels.28 However, renewable energy resources are intermittent, posing a significant concern.29 To reduce greenhouse gas (GHG) emissions, efforts are being made to replace fossil fuel-based power plants and synthesis gas generators with biomass-based processes. Nevertheless, bio-based fuel requires being used as a combined mixed fuel due to its low energy density. Thus, conventional power generation may need to be employed to meet peak demands. Moreover, most chemical and fuel production currently rely on fossil fuels as feedstocks, necessitating technologies to reduce negative emissions associated with these processes.30
1.2 Methods of CO2 utilisation
Two main technologies that have been deployed for CO2 emission reduction are CCS and CCU. CCS captures and stores CO2 in large quantities, while CCU methods valorize waste and renewable CO2 as inputs for added-value products.31 Both technologies involve CO2 capture, compression, and storage or utilization using catalytic technologies. However, CCS consumes a lot of energy and provides a negative return on investment due to the associated cost of capturing and storing, whereas CCU consumes less energy and provides a net positive ROI because the product sold after manufacturing gives a profit.32 Climate change mitigation is the primary objective of CCS, whereas resource security is the main objective of CCU. The focus of current research is on developing CO2 utilization methods that consume less energy.CCS targets large source points or ambient air to capture a large amount of CO2, while chemical production quantities vary regionally in CCU, limiting the CO2 demand. CCS deployment currently has few economic incentives, while incentives are already provided for CCU to a variety of actors. By minimizing the cost of CO2 capture, CO2 can become an appreciating asset rather than a liability.33 However, it's important to note that CO2 stored in CCS is intended to last more than a thousand years, while CCU stores CO2 in the product only temporarily, which is released at the end of its life cycle back into the atmosphere. CSS is more mature and well-established, while CCU is less familiar and in the early development stages, but the latter is regarded as more positive than the former due to the associated risk of storage and transport with the former technology.34
CSS, which captures and compresses CO2, costs 80% of the total cost associated with the process. Research and development efforts must aim to reduce these costs. Currently, technologies such as pre-combustion, post-combustion, and oxy-fuelling can capture CO2 from cement, steel, iron, and biogas sweetening processes.35 Once captured and separated, CO2 should be transported cost-effectively through pipelines to the injection station. To reduce corrosion and surplus costs, impurities and moisture must be removed. CO2 must be compressed into supercritical form and monitored after injection to prevent it from seeping out and remaining confined.36,37 Long-term sequestration is preferred in active or depleted gas reservoirs and deep aquifers.38 According to the Intergovernmental Panel on Climate Change (IPCC) and Special Report on Emission Scenarios (SRES), CCS may reduce global CO2 emissions by 17% by 2050 and is one of the least expensive strategies for doing so in the near future.39,40
CO2 is a challenging C1-building block because of its high kinetic inertness and thermodynamic stability, with a standard heat of formation of ΔHf = −394 kJ mol−1 and a standard Gibbs energy of ΔGf = −395 kJ mol−1.41 To react effectively to epoxide and CO2, a catalyst is needed. Metal-containing catalysts can create cyclic carbonate and polycarbonate depending on the nucleophile species, metal species, nucleophile-to-metal ratio, and reaction conditions, but metal-free catalysts are often more selective towards cyclic carbonates. Optimization is necessary to maximize production. Two broad categories of CO2 utilization processes exist physical and chemical. To be physically utilised, CO2 molecules must maintain their identity, either as a distinct entity or suspended in a solution. These processes, which include carbonated drinks, fire extinguishers, dry ice, enhanced oil recovery (EOR), enhanced gas recovery (EGR), and enhanced geothermal systems, are based on the physical properties of CO2. Some physical utilization processes, such as EOR and EGR, have the potential to store large volumes of CO2 and are called sequestration processes.42 Chemical utilization involves CO2 being converted into a new compound. The chemical potential of CO2 can be utilized to produce fuels and chemicals. This article focuses on chemical utilization because of its limitless potential to abate CO2. Several prominent chemical utilization pathways, such as those summarized in Table 1, are available to convert CO2 chemically into useful products, with varying production rates, technology readiness levels (TRL), the specific mass (CO2 utilization in ton per ton of product), and CO2 uptake potential.43–47
| Product | Current production (Mt per year) | Technology readiness level (TRL) | Specific mass (CO2 utilization in ton perton of product) | CO2 uptake (MtCO2 per year) | Reference |
|---|---|---|---|---|---|
| Urea | 180 | 9 | 0.74 | 132.3 | 43 |
| Salicylic acid | 0.17 | 9 | 0.32 | 0.054 | 44 |
| Methanol | 65 | 9 | 1.37 | 89.24 | 44 |
| Polycarbonate | 5 | 9 | 0.17 | 0.87 | 43 |
| Polyurethane | 15 | 9 | 0.30 | 4.5 | 45 |
| Ethylene carbonate | 0.2 | 8 | 0.50 | 0.099 | 45 |
| Algae | 35 | 7 | 1.8 | 63 | 46 |
| Calcium carbonate | 113.9 | 7 | 0.44 | 113.9 | 45 |
| Methane | 1100–1500 | 7 | 2.75 | 3050–4150 | 44 |
| Propylene carbonate | 0.2 | 7 | 0.43 | 0.086 | 45 |
| Syngas | 359 | 6 | 1.47 | 526.55 | 47 |
| Sodium carbonate | 62 | 6 | 0.42 | 25.73 | 44 |
| Dimethyl carbonate | 1.60 | 5 | 1.47 | 2.35 | 45 |
| Magnesium carbonate | 20.50 | 4 | 0.26 | 5.35 | 44 |
| Acetic acid | 10.25 | 3 | 0.73 | 7.51 | 44 |
| Acrylic acid | 5.85 | 3 | 0.61 | 3.57 | 44 |
| Formaldehyde | 21 | 3 | 1.45 | 30.45 | 45 |
| Dimethyl ether | 11.4 | 3 | 1.91 | 21.79 | 45 |
| Ethanol | 80 | 2 | 1.91 | 152.88 | 44 |
Technologies for producing urea, salicylic acid, methane, methanol, ethylene carbonate, polyurethanes, and polycarbonates have a technology readiness level (TRL) of 9, indicating that they are well-established and mature. Other technologies listed are currently maturing and developing, while some are still immature and in the early stages of development. The CO2 uptake potentials mentioned do not include emissions associated with processing. However, the reduction that can be achieved if environmentally benign processes are developed and deployed for utilization is an overview of the overall reduction. Currently, approximately 220 Mt per year of CO2 is utilized for producing different chemicals, excluding methane, syngas, calcium carbonate, sodium carbonate, ethanol, etc. Urea production consumes the majority of CO2, followed by salicylic acid, cyclic carbonate, and polycarbonate. The remaining CO2 is used for methanol, ethanol, etc. Additionally, around 20 Mt per year of its usage as a technical fluid must be added to this amount. It is estimated that today, the chemical industry consumes 92% of the approximately 240 Mt per year of CO2 that is consumed.48
Over the next decade, a growing synthetic technology industry may increase the rate at which CO2 is utilized to 350 Mt per year. By 2040, we could see a surge in the consumption of CO2 for chemicals, rising to around 1000 Mt per year, potentially making a considerable difference.49
1.3 Industrial processes of CO2 utilizations
The first mature method for CO2 chemical utilization is urea production, which consumes 0.735 to 0.75 tons of CO2 per ton of urea and is highly exothermic (Scheme 1).28,37 The global urea market has reached 180 Mt per year in 2021, with a predicted growth of 0.8% between 2022 and 2027.50 A novel process has been proposed to produce urea from power plants and electricity simultaneously by using CO2 in the flue gas as a reactant for urea production, with 1 ton of CO2 utilization producing 1.68 tons of urea.51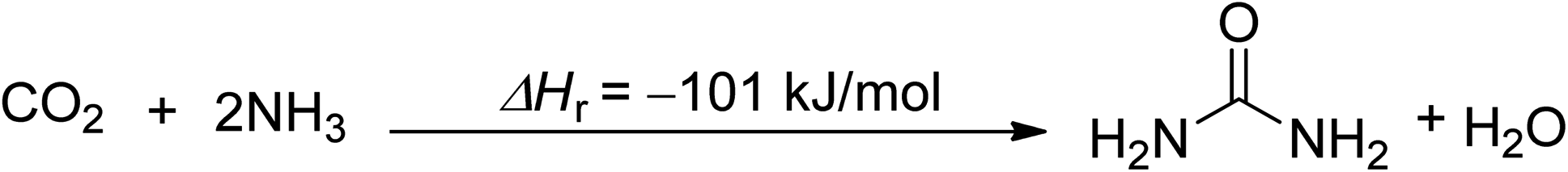 | ||
| Scheme 1 Synthesis of urea (reproduced from ref. 26 with permission from Wiley-VCH copyright 2021). | ||
Salicylic acid synthesis is a second example of efficient CO2 utilisation (Scheme 2).52 The commercialization of this process dates back to the 19th century, and salicylic acid production currently consumes 29 Kt of CO2 directly, with a total production of 90 Kt per year.27,53
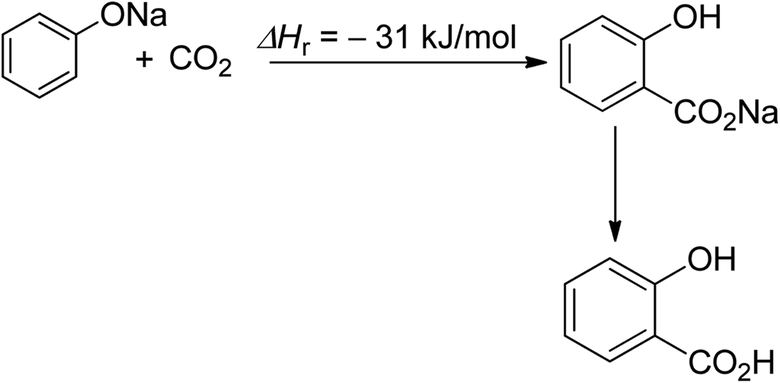 | ||
| Scheme 2 Synthesis of salicylic acid (reproduced from ref. 26 with permission from Wiley-VCH copyright 2021). | ||
The production of carbon-neutral fuels like methanol and methane is the third efficient chemical use of CO2 (Scheme 3).54,55 These fuels have zero CO2 emissions when combusted under certain conditions, and their potential scale is almost limitless, as they can replace liquid and gaseous fossil fuels. Methane and methanol production has the potential to reduce 2.75 and 1.373 tons of CO2 per ton of product produced, respectively.44 Hydrogen is the main reactant needed for the feasibility of methanol and methane production, and methane can also be used for the production of carbon black and rubber.56
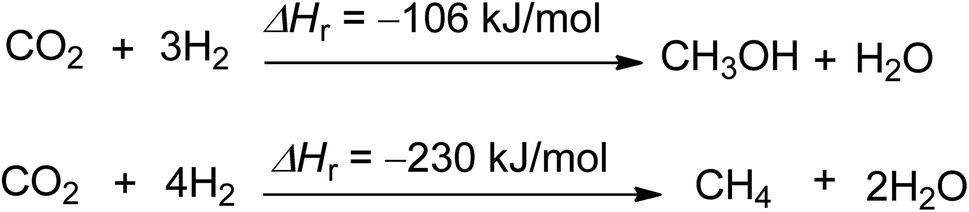 | ||
| Scheme 3 Synthesis of methanol and methane using CO2 and hydrogen (reproduced from ref. 26 with permission from Wiley-VCH copyright 2021). | ||
The production of polycarbonates is the fourth illustration of efficient CO2 utilisation, which uses renewable, economical, and non-toxic CO2 instead of the poisonous phosgene route (Scheme 4).57,58 Similarly, the fifth example of effective CO2 utilization is the production of polyurethanes (PU),59 which have dominated coatings, adhesives, sealants, and elastomers for the past three to four decades and can be tailored for different applications (Scheme 4).60–62 The current production rate of polycarbonates and polyurethanes is 5 and 15 Mt per year, respectively.45,47
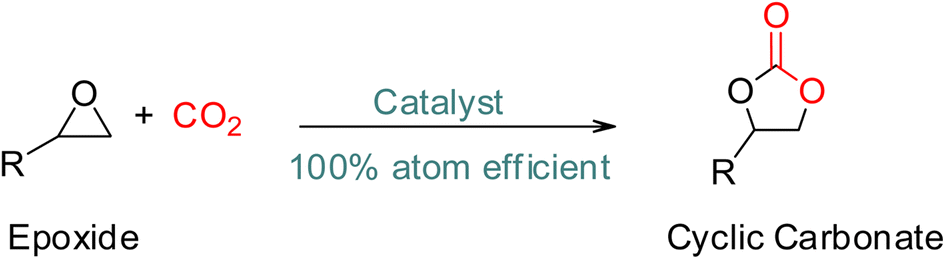 | ||
| Scheme 4 Synthesis of cyclic carbonates from epoxide and CO2.32 | ||
Other developing CO2 utilization technologies include dimethyl carbonate (DMC), dimethyl ether (DME), ethanol and algae. Ethanol is produced conventionally by fermenting sugars or corn, and it has the potential to be used as a zero-emission fuel, but the process design and low catalyst activity remain challenges.61,62 DME is an alternative green fuel that results in fewer GHGs and can coproduce electricity, while DMC has potential as a methylating agent but faces catalyst selection issues (Table 1).63,64 Algae cultivation has great potential as a biomass source for green fuel production and can absorb CO2 and convert it to oxygen for water purification.65 Fig. 2 shows examples of mature CO2 utilization technologies and applications of cyclic carbonates.
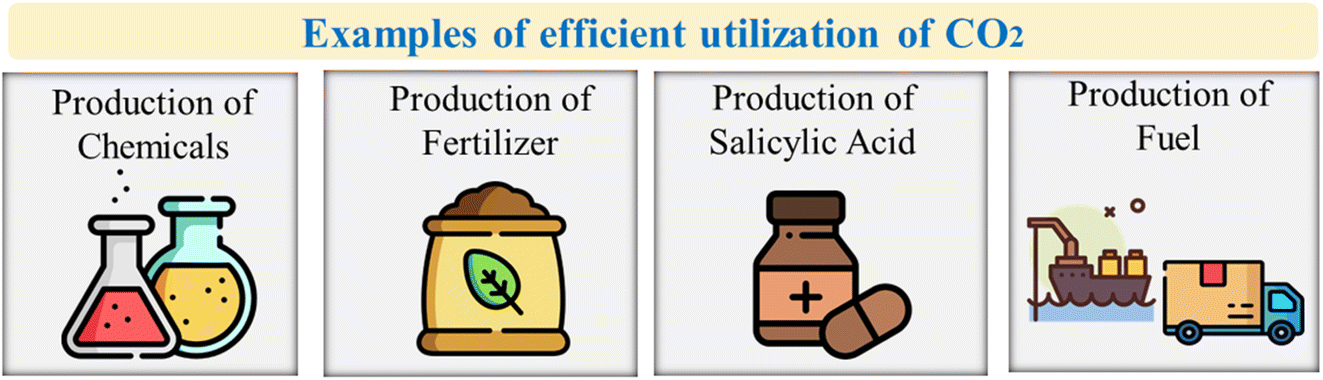 | ||
| Fig. 2 Examples of effective utilization of CO2 and applications of cyclic carbonates.26 | ||
2. Cyclic carbonates
The production of five-membered cyclic carbonates through the reaction of epoxides and CO2 is a commercially viable green and sustainable chemistry reaction (Scheme 4).32 This process uses CO2 as a readily available, non-toxic, renewable reactant, which is one of its key advantages. Additionally, it is a 100% atom-efficient reaction as the product is comprised of all reactants, with no need for a solvent. The reaction is also thermodynamically favourable, as epoxides have greater free energy than CO2, which offsets the required thermodynamic stability of CO2.66 Therefore, cyclic carbonates production from CO2 and epoxides is a promising and environmentally friendly approach to chemical synthesis.Cyclic carbonates are considered green solvents, with propylene carbonate (PC), ethylene carbonate (EC), and glycerol carbonate (GC) being the most widely used (Fig. 3.).66 The physiochemical properties of cyclic carbonates are listed in Table 2.66–73 EC and PC are the most commonly used globally.74,75
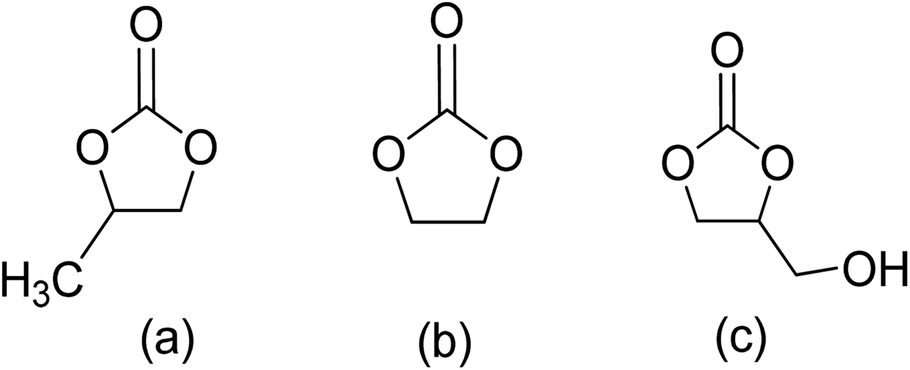 | ||
| Fig. 3 Cyclic carbonates: (a) propylene carbonate (PC), (b) ethylene carbonate (EC), (c) glycerol carbonate (GC).66 | ||
| Cyclic carbonates | Propylene carbonate (PC) | Ethylene carbonate (EC) | Glycerol carbonate (GC) |
|---|---|---|---|
| Formula | C4H6O3 | C4H6O3 | C4H6O3 |
| CAS number | 108-32-7 | 96-49-1 | 931-40-8 |
| IUPAC name | 4-Methyl-1,3-dioxolan-2-one | 1,3-Dioxolan-2-one | 4-Hydroxymethyl-1,3-dioxolan-2-one |
| Molecular mass, [g mol−1] | 102 | 88 | 118 |
| Specific gravity [g cm−3] | 1.20 (20 °C) | 1.32 (40 °C) | 1.40 (25 °C) |
| Boiling point, Tb (°C) | 242 | 248 | 354 |
| Freezing point, Tf (°C) | −49 | 36 | −69 |
| Flash point (°C) | 135 | 145 | >204 |
| Vapour pressure [kPa] | 0.131 (50 °C) | 0.003 (25 °C) | n.a |
| Viscosity, (cP or mPa s at 25 °C) | 2.53 | 1.93 | 85.40 |
| Solubility (at 20 °C) (mg kg−1) | Very soluble | Soluble | Miscible |
| Hazards | Induces severe eye irritation | A prolonged or repeated exposure can cause serious eye irritation, and cause injury to the kidneys if swallowed | Ingestion or skin absorption can be harmful |
2.1 Applications of cyclic carbonates
Propylene carbonate (PC) is a highly desirable polar aprotic solvent due to its high dipole moment, high dielectric constant, odorlessness, colourlessness, and low viscosity.68,76 Additionally, it remains liquid across a wide temperature range and is biodegradable, hydrolyzing in air and water. PC is also non-toxic, non-flammable, and corrosion-resistant, making it an excellent solvent from a health and safety standpoint.66,68,77 PC ranks highly on the GSK solvent suitability ranking, making it a green solvent. However, it has a high boiling point, which requires energy-intensive distillation for separation, and therefore, it may not be considered a green solvent under all circumstances.78 Despite this, PC can replace harmful polar aprotic solvents affordably and sustainably. EC is not used as a solvent due to its solid nature, so it is combined with other compounds to overcome this issue. Several other cyclic carbonates, such as 1,2-limonene carbonate (1,2-LC) and styrene carbonate, have the same restraint.66 Glycerol carbonate (GC) has similar properties and applications, but its use as a solvent is limited by its high viscosity.77Due to their dielectric characteristics, cyclic carbonates make excellent electrolytes for Li-ion batteries.79 Li-ion batteries have become popular due to their use in portable devices.80 Among Li-based battery electrolytes, EC and PC are commonly used as solvents. Lithium salts can dissolve in cyclic carbonate, providing the high dielectric constant needed for conductive electrolytes.25 PC and EC have various uses such as cleaning, paint stripping, degreasing, and the production of paints and coatings, lubricants, dyes, cosmetics, and personal care products.74,75 Cyclic carbonates can also be employed as monomers in the production of polymers like polycarbonate and polyurethanes without isocyanate.59,81 Cyclic carbonates are more effective than urea or salicylic acid at sequestering CO2 in electronic devices, which typically have a lifespan measured in years. Electric cars may have a lifespan of decades, making cyclic carbonates even more effective at sequestering CO2.26 Fig. 4 represents the applications of cyclic carbonates.
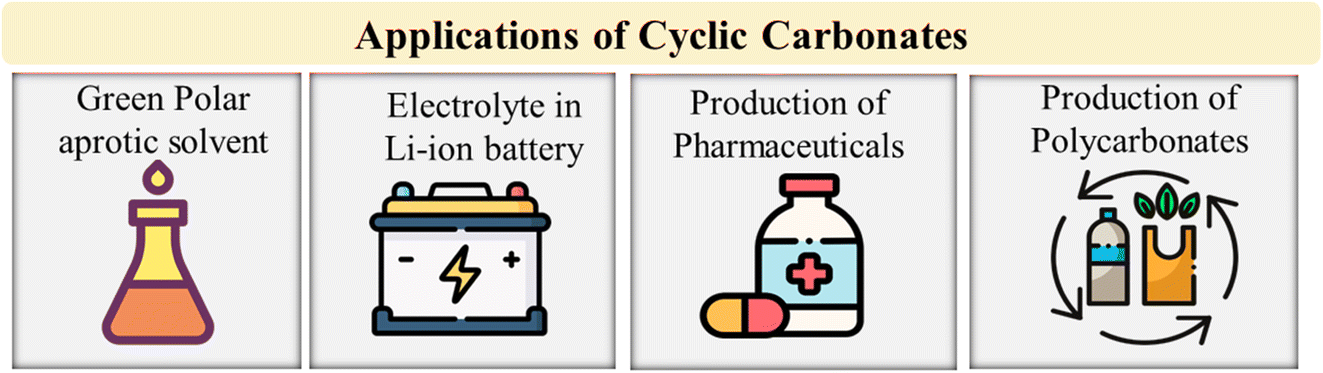 | ||
| Fig. 4 Applications of cyclic carbonates.32 | ||
2.2 Terpene-based cyclic carbonates
The diverse structure of terpenes makes them an excellent source of complex cyclic carbonates, as opposed to other bio-based feedstocks. The use of waste CO2 as a coupling agent after oxidation is a straightforward process. Recently, the synthesis of cyclic carbonates from terpenes has gained prominence in research. While past efforts focused on preparing five- and six-membered cyclic carbonates, there is now a growing interest in developing more complex compounds.82 It is thermodynamically efficient to react CO2 with compounds that have high free energy. In nature, fir cone oil and citrus fruits produce cyclic terpenes like limonene (both isomers). (R)-(+)-Limonene can be cost-effectively obtained from orange peel waste. The presence of several double bonds allows the epoxidation of 1,2-limonene oxide (1,2-LO), 8,9-limonene oxide (8,9-LO), and limonene dioxide (LDO). Coupling these compounds with CO2 results in the formation of corresponding carbonates (Fig. 5).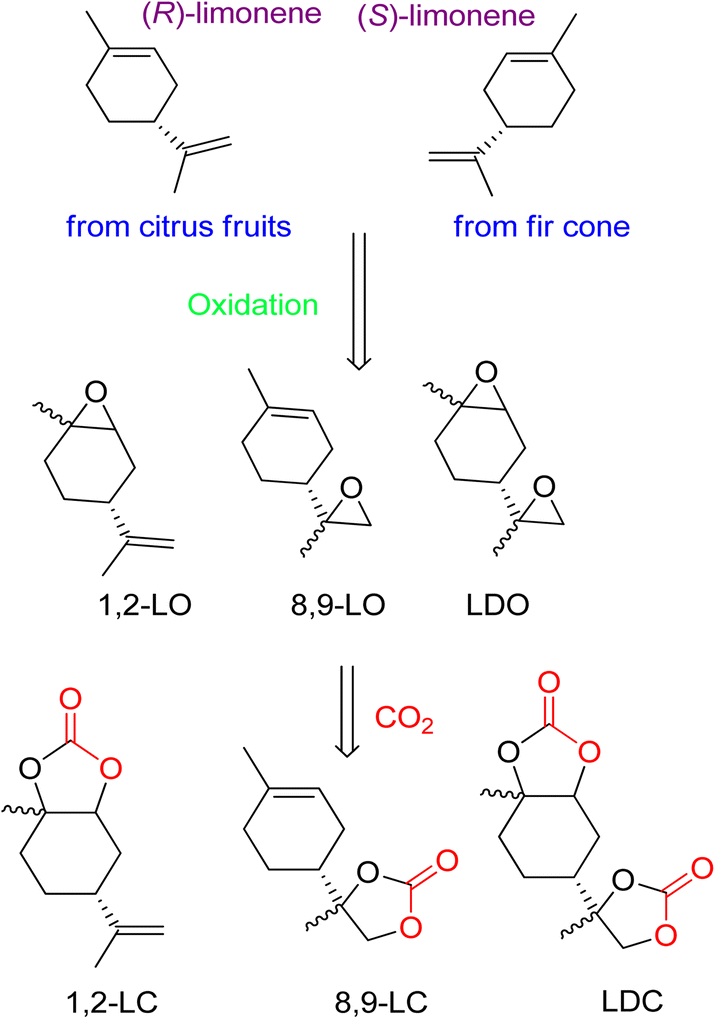 | ||
| Fig. 5 Structure of both (R)-(+)-limonene enantiomers, oxidation to afford limonene-based oxides and reaction with CO2 to obtain corresponding cyclic carbonates, reproduced from (reproduced from ref. 82 with permission from RSC copyright 2021). | ||
Extensive studies have been conducted on 1,2-LO due to its wide availability. However, when comparing sterically demanding 1,2-LO with other epoxides, the former exhibits lower reactivity during coupling. As a result, a longer reaction time (16–66 h) is required at higher temperatures and pressures (75–100 °C, 10–50 bar). Fig. 6 summarises metal-based catalytic systems that have been reported for the coupling of 1,2-LO and CO2.83–87
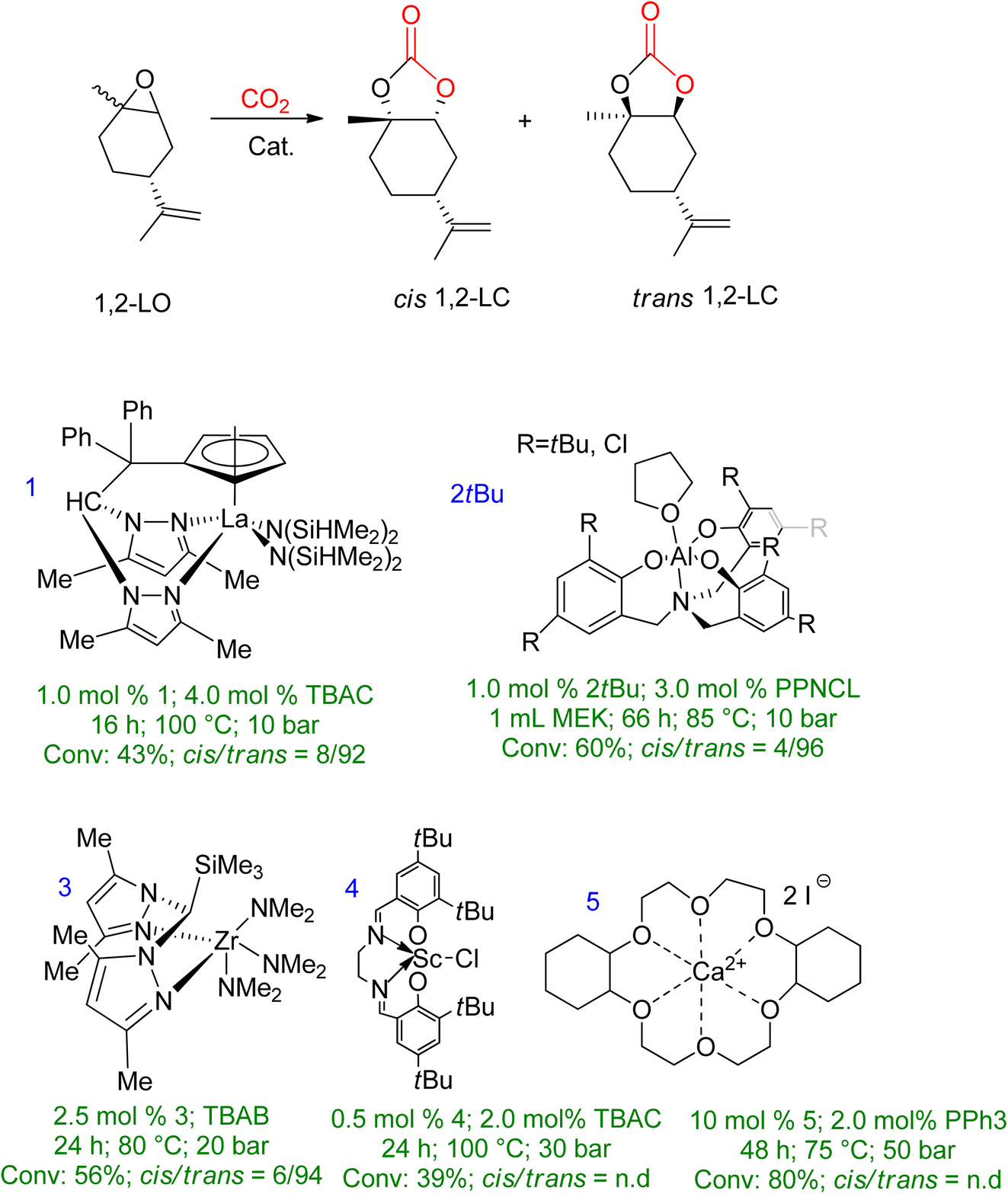 | ||
| Fig. 6 A metal-based catalytic system reported for the synthesis of 1,2-LC, along with comparisons of the reaction conditions and chemistry of the products (n.d. = no determination) (reproduced from ref. 82 with permission from RSC copyright 2021). | ||
Several catalyst systems, including 1, 2tBu, and 3, have been reported to selectively form trans-1,2-LC, leading to different reactivity between the cis and trans isomers of 1,2-LO.83,84,86 In 2016, efficient conversion of various terpene-based substrates was achieved using 2R/PPNCL, which produced LDO with good yield. However, the synthesis of LDC was unsuccessful due to the formation of polyether (PE) side products. The reaction conditions for LDO synthesis were 1.0 mol% 2R, 3.0 mol% PPNCL, 1.0 mL MEK, 66 h, 85 °C, p(CO2) = 1 MPa.84 Another study reported the formation of LDC with a concentration of up to 78% in the presence of 10.0 mol% 5/2.0 mol% PPh3 catalyst, after 48 h at 75 °C and 50 bar.85
Tetrabutylammonium halide (TBAX) is an efficient and effective catalyst for synthesizing 5-ring cyclic carbonates.88 Therefore, a similar reaction was carried out using TBAX as a catalyst for the reaction between 1,2-LO and CO2 with the substrate being ct-LO [43:57].89 The reaction conditions were extreme with high temperature, pressure, and reaction time (100 °C, 3 MPa, 20 h) and the catalyst consisted of 10 mol% ct-LO without any solvent. Tetrabutylammonium chloride (TBAC) had a 51% conversion rate of cis–trans-LO, with the individual conversion of trans-LO and cis-LO being 76% and 19%, respectively. However, the conversion rate and NMR yield decreased when tetrabutylammonium bromide (TBAB) and tetrabutylammonium iodide (TBAI) were used as activating catalysts. The order of catalytic activity increment was reported as TBAX > TBAB > TBAI < TBAC.
The research group continued their investigation using TBAC as the catalyst model to study the effect of reaction conditions. The typical conditions used were a 10 mol% catalyst based on cis–trans-LO, 20 h reaction time, 100 °C temperature, and 3 MPa of CO2 pressure. The study focused on varying reaction times, CO2 pressure, and the amount of TBAC. Prolonging the reaction time did not improve the conversion of cis–trans-LO, as 48 h and 72 h resulted in 65% and 69% yields, respectively. The next step was to vary the CO2 pressure while keeping all other parameters constant. The results showed that only increasing the pressure by 5 MPa increased the individual yield/conversion to cis-LO/trans-LO to 27:85. Finally, increasing the amount of TBAC increased trans-LO yield, while cis-LO conversion remained low.
A detailed kinetic study of the coupling reaction between 1,2-LO and CO2 in the presence of TBAC was recently reported, which found that the trans-isomer exhibits greater reactivity. The reaction was determined to be a first-order reaction with respect to all reactants, and the Eyring equation was used to determine the thermodynamic parameters of conversion. The entropy value was found to be 60.6 kJ mol−1 and the enthalpy value was −103.6 J (mol K)−1.90 In another recent study by Mikšovsky et al.,91 tetrabutylammonium-based halides and 1-ethyl-3-methyl imidazolium halides were used as homogeneous catalysts, while a supported ionic liquid was used as a heterogeneous catalyst for the synthesis of cyclic carbonates from limonene epoxides. It was found that imidazolium-based liquids have less catalytic activity in comparison or are even inactive when LO (cis/trans-mixture) is used as a substrate. TBAC proved to be the most selective and highest-yielding catalyst for converting the diastereomeric mixture (cis/trans = 44/56). Silica without immobilization, TBAC, and TBAB showed lower yield, while in the case of imidazolium-based catalysts TBAI yield increased, but the overall selectivity was still low. The same order of catalytic activity was seen for supported ionic liquid phases and ammonium-based ionic liquids (ILs) physisorbed on silica as it was for homogeneous mode.
Mülhaupt's research group used LDC to prepare poly-hydroxy urethanes (Scheme 5).92 For the synthesis of LDC on a one-kilogram scale, TBAB was used, and the reaction parameters were optimized to allow for 3 mol% TBAB usage in less than 50 h to achieve complete LDO conversion. The resulting product was a brownish oil, which can be directly used to prepare non-isocyanate polyurethanes (NIPUs). Several by-products were identified after analyzing the reaction. LDC was cooled and crystallised to create a pure product, resulting in a 3:1 combination (cis-to-trans ratio). X-ray analysis proved that the trans-LDC produced by further crystallisation was 100% pure.93
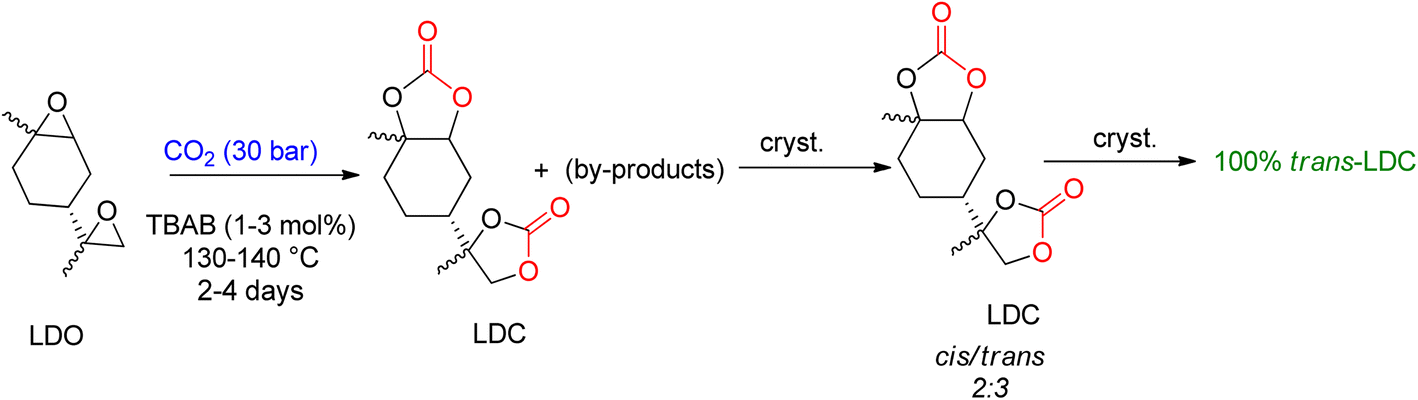 | ||
| Scheme 5 Formation of limonene di-carbonate (LDC) by the reaction of limonene di-oxide (LDO) and CO2 catalyzed by TBAB and subsequent crystallization to obtain trans-LDC (reproduced from ref. 82 with permission from RSC copyright 2021). | ||
Through TBAB-mediated reaction, 8,9-LC was formed by reacting CO2 with 8,9-LO. To synthesize the desired carbonates, the less hindered terminal alkene of (R)-(+)-limonene was selectively epoxidized (Scheme 6). Perchloric acid was used as the oxidant, and bulky polyoxometalates (POM) were used as the catalyst. Compared to 1,2-LO, 8,9-LC is easier to produce due to less steric hindrance, reaching maximum conversion in only 2.5 h under comparable reaction conditions. Additionally, the two diastereomer-isomers obtained showed the same reactivity during coupling with CO2, which is in contrast to 1,2-LO, where trans-1,2-LO was found to be more reactive.94
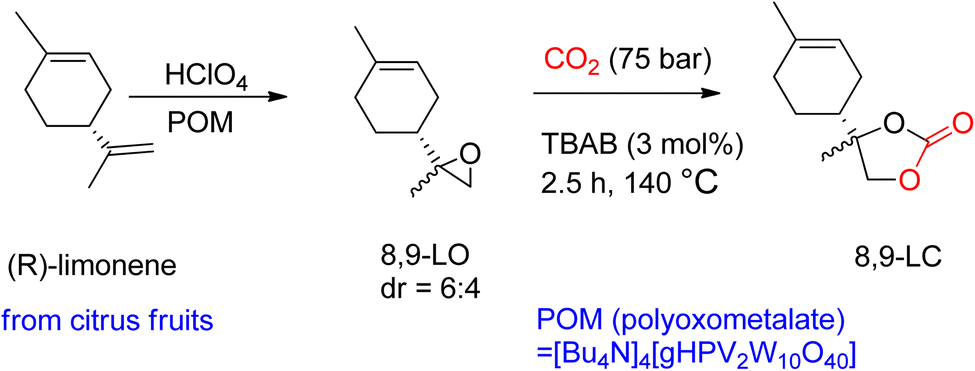 | ||
| Scheme 6 Highly selective (R)-(+)-limonene epoxidation to obtain 8,9-limonene oxide (8,9-LO) and subsequent reaction with CO2 to obtain 8,9-LC by TBAB-catalysed reaction (reproduced from ref. 82 with permission from RSC copyright 2021). | ||
2.3 Glycerol carbonate
Glycerol carbonate (GC), also referred to as 4-hydroxymethyl-1,3-dioxolan-2-one, is a bio-derived 5-membered ring carbonate synthesized from glycerol. It is one of the few newly commercialized bio-derived products from biomass.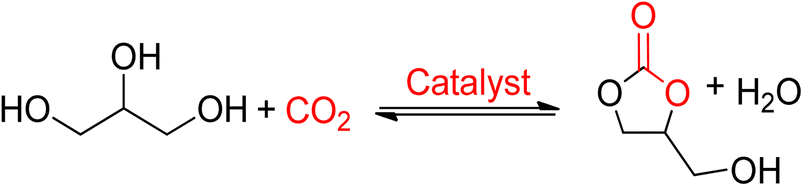 | ||
| Scheme 7 Glycerol carbonate (GC) obtained from glycerol and CO2 (reproduced from ref. 82 with permission from RSC copyright 2021). | ||
Vieville et al. reported the first attempt to synthesise GC from glycerol and CO2 in 1998.96 The reaction did not occur under supercritical CO2 conditions. The first successful production of GC was reported by Aresta et al. in 2006 using Sn-based catalysts under 5 MPa and 180 °C of pressure and temperature, respectively.97 A maximum yield of 5.7% was achieved without the use of a solvent, and molecular sieves were used for the removal of in situ water. During the mechanistic study, it was proposed that the Sn-based catalyst and CO2 could be inserted into glycerol by forming the active Sn-glycerate intermediate. George et al. intensified this process in 2009 using MeOH as a medium, increasing the yield of GC to 35%, as shown in Scheme 8.98 Methanol acted as a solvent and was involved in the reaction mechanism as well. Metal oxide/complexes, supported metal catalysts, modified zeolites, or hydrotalcite have been reported as catalytic systems. For GC synthesis, most heterogeneous catalysts require CO2 at high pressures and temperatures above 40 bar and 150 °C, respectively.
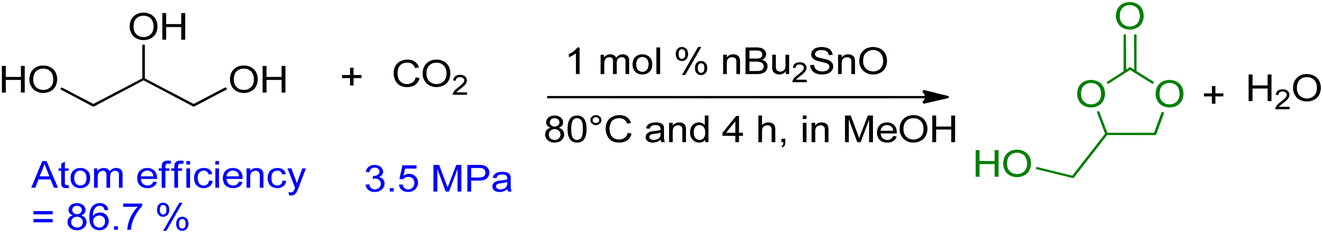 | ||
| Scheme 8 Improved carbonation of glycerol using MeOH as solvent.98 | ||
The dehydrating reagent for the removal of water was more efficacious than using molecular sieves. However, in some cases, a decrease in chemo selectivity towards GC was noted. The most used dehydrating agents for this reaction are acetonitrile, adiponitrile, and 2-cyanopyridine, as shown in Scheme 10. In most studies, acetonitrile is the first choice because it is cheap, but upon reaction with the water molecule, it generates acetamide in situ. The produced acetamide reacts with another water molecule to produce acetic acid, which decreases selectivity by forming mono- and di-acetins. McGregor et al.18 used adiponitrile as a dehydrating agent to remove water and found that it degrades to ammonia upon reacting with glycerol, resulting in a mixture of two regio-isomers of 4-HMO (4-(hydroxymethyl)oxazolidine-2-one). In 2016, Liu et al.99 employed 2-cyanopyridine as a highly effective dehydrating reagent. By using 3 equivalents of 2-cyanopyridine with a CeO2 catalyst in DMF at 4 MPa of CO2 and 150 °C, GC yields can increase to 79%. Recent research by Zhang et al.100 has shown the benefits of employing CaC2 as a dehydration agent for GC production. In their work, CaC2 was combined with Zn(OTf)2/phen (1,10-phenanthroline) in N-methyl-2-pyrrolidone (NMP), and the reaction was carried out at 50 bar CO2 pressure at 180 °C for 24 h to get 88% GC. In their experiment, CaC2 in combination with Zn(OTf)2/phen (1,10-phenanthroline) in NMP (N-methyl-2-pyrrolidone) at 50 bar CO2 pressure and 180 °C for 24 h yielded 88% GC. Su et al.101 reported a year later that GC can also be produced using 2-cyanopyridine at 15 MPa of CO2 and 180 °C without the use of a metal catalyst, with a 19% yield (Scheme 9). FTIR analysis confirmed the formation of a 5-membered ring via the activation of CO2 by 2-cyanopyridine, followed by a reaction with glycerol to form GC. The formation of this heterocycle was also supported by theoretical analysis, and glycerol was subsequently transesterified by it. Notably, there was no substantial GC production when using acetonitrile, 4-cyanopyridine, or 3-cyanopyridine (Table 3).
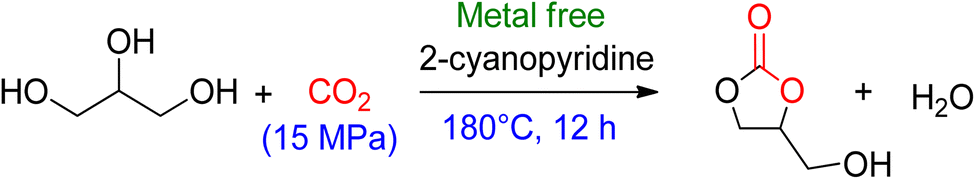 | ||
| Scheme 9 Metal-free formation of GC using 2-cyanopyridine (reproduced from ref. 82 with permission from RSC copyright 2021). | ||
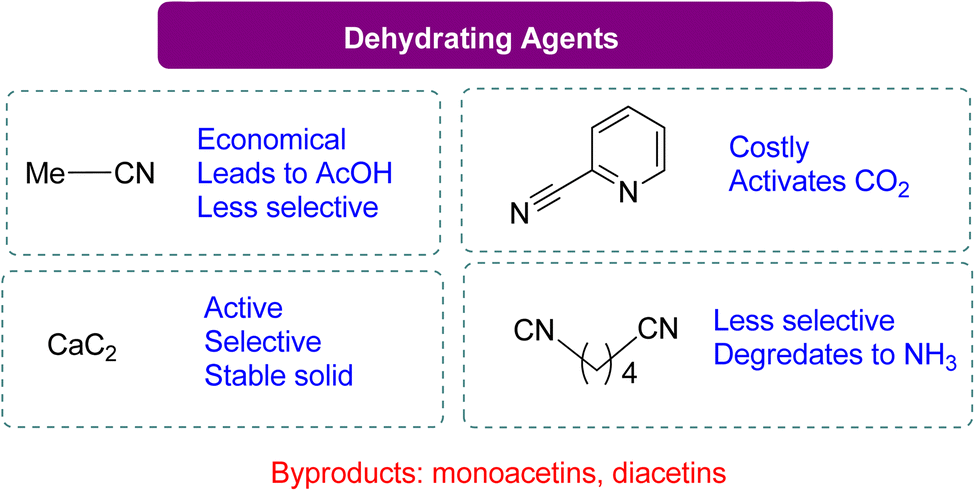 | ||
| Scheme 10 Comparison of different dehydrating agents in the formation of GC from glycerol and CO2 (ref. 18, 21, 99 and 100) (reproduced from ref. 82 with permission from RSC copyright 2021). | ||
| System | Catalyst | Dehydrating agent | Pressure (MPa) | T (°C) | Time req. | Yielda/conversionb | Other reported | Reference |
|---|---|---|---|---|---|---|---|---|
| a Yield.b Conversion. | ||||||||
| Homogeneous | n-Bu2Sn (OMe)2 | Molecular sieves | 5 | 180 | 5.7a | No solvent | 97 | |
| Homogeneous | nBu2SnO | 13X-soda zeolites | 3.5 | 80 | 4 h | 35a | MeOH as medium | 98 |
| Heterogeneous | La2O3 | Adiponitrile | 4.5 | 160 | 18 h | 58b | 18 | |
| Heterogeneous | CeO2 | 2-Cyanopyridine | 4 | 150 | 33.3a/38b | 99 | ||
| Heterogeneous | Zn (OTf)2/phen (1,10-phenanthroline) | CaC2 | 50 bars | 180 | 24 h | 88a | In NMP (N-methyl-2-pyrrolidone) | 100 |
| Metal-free | No | 2-Cyanopyridine | 15 | 180 | 18.7a/31.1b | 101 | ||
 | ||
| Scheme 11 General reaction of glycerol with CO2 in the presence of a coupling agent, reproduced from (reproduced from ref. 82 with permission from RSC copyright 2021). | ||
| System | Catalyst | Coupling agents | P (MPa) | T (°C) | Time req. | Yield% | Reference |
|---|---|---|---|---|---|---|---|
| Heterogeneous | KI |  |
2.0 | 115 | 1.5 h | 77 | 102 |
| Heterogeneous | P-DVB-(vIm-BuBr) |  |
2.0 | 100 | 4 h | 81 | 103 |
| Heterogeneous | Ag2CO3/xanthophores |  |
1.0 | 80 | 12 h | 82 | 104 |
| Heterogeneous | Silver sulfadiazine and Et4NBr | 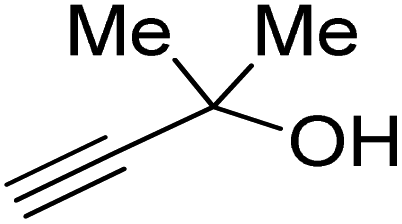 |
0.1 | 80 | 24 h | 56 | 105 |
| Heterogeneous | Amidine-CO2 adduct/MTBD | 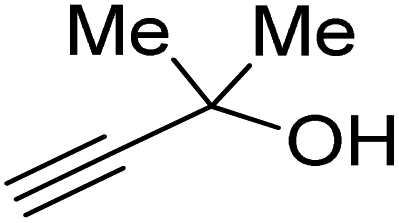 |
0.1 | 25 | 24 h | 85 | 106 |
| Heterogeneous | DBU (8-diazabicyclo[5.4.0]undec-7-ene), DMF | 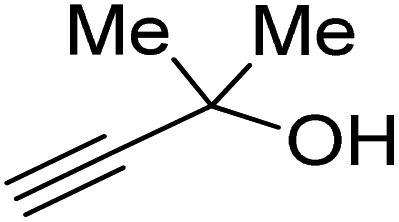 |
3.0 | 120 | 10 h | 97 | 107 |
| Heterogeneous | DBU (8-diazabicyclo[5.4.0]undec-7-ene), BmimPF6 | CH2Br2 | 1.0 | 70 | 18 h | 86 | 108 |
| Heterogeneous | Guanidine catalyst | n-BuBr | 0.1 | 50 | 4 h | 74 | 109 |
Propargylic alcohol (dimethyl ethynyl carbinol) has been used as a coupling reagent along with PO. He et al.104 utilized Ag2CO3/xanthaphos catalyst in MeCN with 1 MPa CO2 pressure at 80 °C to produce a good yield of GC in one pot approach using CH3CN as a solvent (entry 3, Table 4). Li et al.105 improved the process by using silver sulfadiazine and the Et4NBr synergistic system without any solvent (entry 4, Table 4), but the yield of GC decreased. Silver sulfadiazine activated the hydroxyl groups simultaneously in propargylic alcohols (or vicinal diols), resulting in an excellent performance. The Lu and Liu groups reported GC preparation by organo-catalytic approach. Zhou et al.106 used N-hetrocyclic carbene-CO2 adducts which formed an intermediate and further transesterification took place with glycerol catalyzed by MTBD (methyl-1,5,7-triazabicyclo [4.4.0] dec-5-ene) to achieve an 85% yield of GC at room temperature and 0.1 MPa (entry 5, Table 4). Han et al.107 reported a three-component reaction using DBU (8-diazabicyclo [5.4.0] under-7-ene) to promote both steps, resulting in 97% yield of GC at 120 °C and 3 MPa in 10 h (entry 6, Table 4). Alkyl halides can also be used as effective reactants, but they generate halogen-containing waste, making the method unsustainable. Jang et al.108 reported the use of DBU, BmimPF6, and CH2Br2 for direct coupling of various alcohols, including glycerol, without using any Mitsunobu-type reagents (entry 7, Table 4). At 70 °C and 1 MPa, it was possible to achieve 86% yield. Mihara et al.109 used tert-butyl tetramethyl guanidine and n-BuBr as an additional component at 50 °C and 0.1 MPa CO2 pressure employing NMP (N-methyl pyrrolidone) as a solvent to obtain GC in 74% yield (entry 8, Table 4). The authors suggest that the ionic liquid is effective in improving CO2 solubility, glycidol (Gly) may react with an NHC carbene formed that can capture/activate CO2.
Rintjema et al.112 discovered that the hydroxyl group on Gly, which can be used to create transitory hemi-carbonate species, can be used to activate CO2. Halide-free processing is made possible by this activation, which causes the oxirane ring to be opened by an intramolecular nucleophile under mild conditions and without the requirement for an external nucleophilic addition. It has been determined the exact process by which the oxirane ring of Gly is opened intramolecularly by the production of hemi-carbonate. Additionally, metal and halide-free production of GC using a packed bed reactor under flow conditions has been reported.113 Several catalytical systems, including heterogeneous, homogenous, and organo-metallic, have been reported to produce GC from Gly. However, not all catalytic systems are superior to simple TBAB.
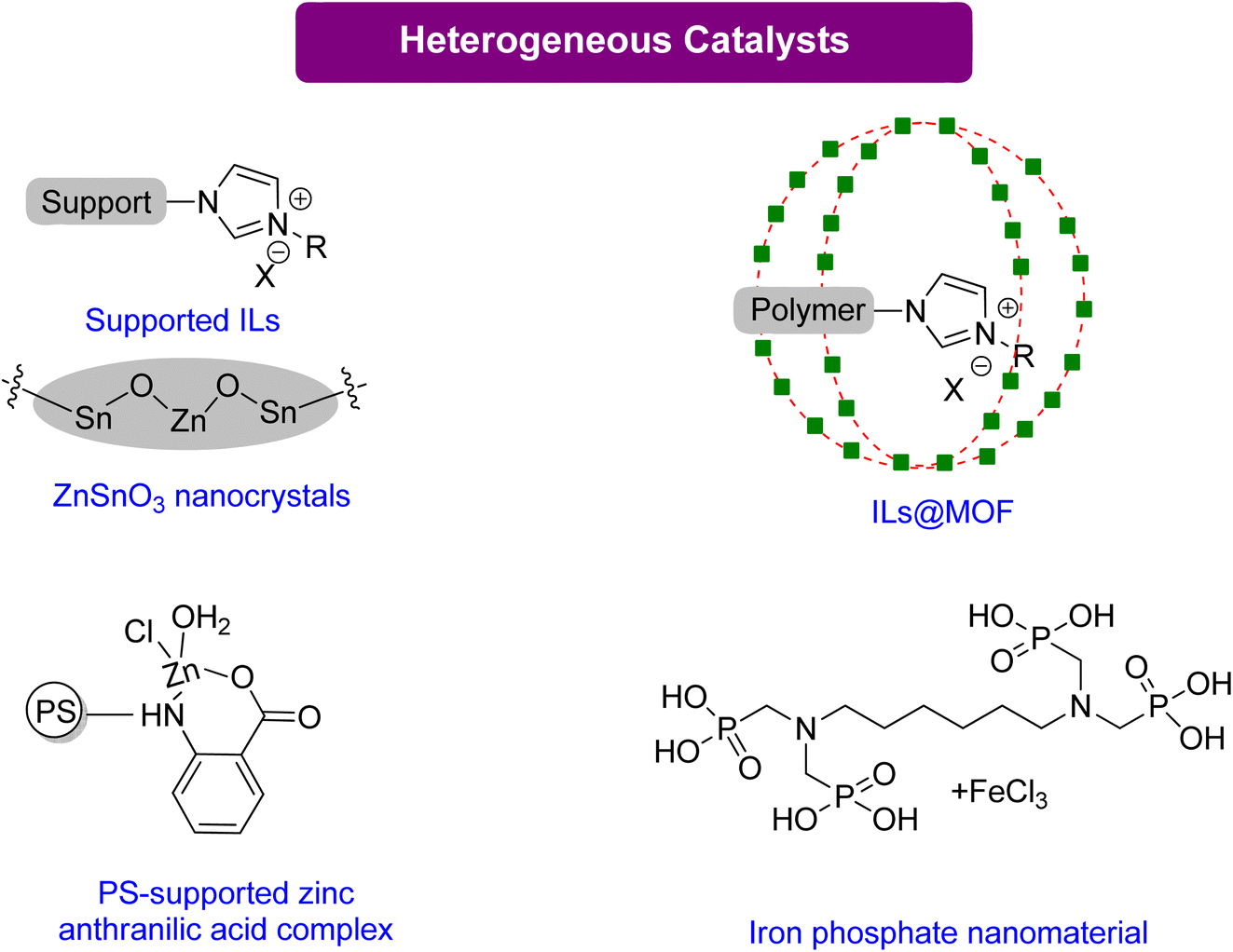 | ||
| Scheme 12 Highly efficient heterogeneous catalysts for the synthesis of GC from Gly,120–123 (reproduced from ref. 82 with permission from RSC copyright 2021). | ||
Multifunctional components integrated into composite materials can enhance the activity for specific uses. Jiang et al.120 reported an efficient composite catalyst, polyILs@MIL-101, by confining imidazolium-based polyionic liquids (polyILs) into a metal–organic framework (MOF) material MIL-101 via in situ polymerizations of encapsulated monomers. This material has CO2-capturing capability and is a good catalyst without any co-catalyst under normal atmospheric pressure. With high surface area and hierarchical pores, PolyILs@MIL-101 is a promising catalyst material. The superior catalytic activity of polyILs@MIL-101, in comparison to either polyILs or MIL-101, is due to the synergy between the CO2 capturing capability, the Lewis base site in polyILs, and the Lewis acid site in the MOF. Only a few reports have explored the synergistic effects of CO2 capture and conversion. The results of this study demonstrate the necessity of enriching CO2 using a heterogeneous catalyst to assist in its conversion. Moreover, this paper proposes a strategy for designing practical catalysts that could be used in the future to directly capture and convert CO2 from flue gases.
Ghosh et al.121 reported using iron-phosphonate hybrid organic–inorganic nanoparticles HPFP-1(NP) as a nano-catalyst for synthesizing organic carbonate at mild conditions and CO2 under atmospheric pressure. This catalyst demonstrated excellent recyclability and reusability. In addition, a polystyrene–zinc–anthra complex was found to be an efficient catalyst, achieving 89% conversion to GC in 5 h.122 While both HPFP and PS-Zn require TBAB as a co-catalyst, zinc stannate nanocrystals do not need any co-catalyst. These nanocrystals could also be recovered and reused without losing their activity in the presence of PEG-600 as a green solvent, showing the potential of nano-catalysts as an environmentally friendly solution.123 The summary of the homogenous catalytical system reported for GC production from Gly is given in Table 5.
| Catalyst | Pressure | Temperature (°C) | Time req. | Conversiona/yieldb | Other reported | Reference |
|---|---|---|---|---|---|---|
| a Conversion.b Yield. | ||||||
| PolyILs@MIL-101 | 1.5 bar | 70 | 24 h | >99%a | 2 mL acetonitrile | 120 |
| PS-Zn-anthra complex | 1 atm | 25 | 4 h | 89%b | Bu4NBr, TON/TOF = 141/28 | 122 |
| Zinc-stannate nanocrystals | 1 atm | 80 | 10 h | 91%b | PEG-600 as a green solvent | 123 |
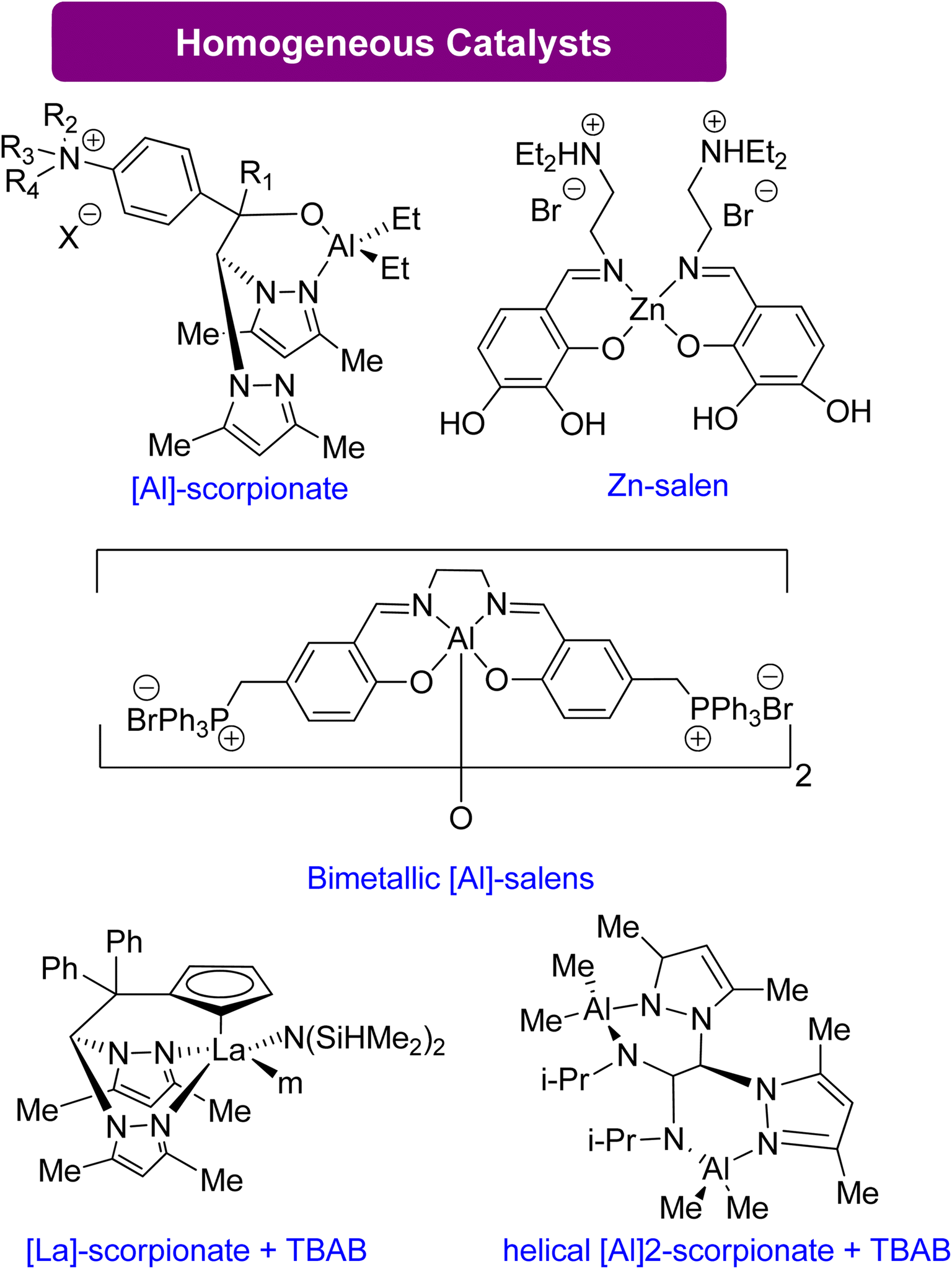 | ||
| Scheme 13 Synthesis of GC from Gly using highly effective homogeneous catalysts83,124–129 (reproduced from ref. 82 with permission from RSC copyright 2021). | ||
| Catalyst | Loading | Pressure (MPa) | Temperature (°C) | Time Req. | Co-catalyst | Reference |
|---|---|---|---|---|---|---|
| Aluminium scorpionate complexes | 0.25–0.5 mol% | — | 70–85 | — | No co-catalyst | 124–126 |
| Bifunctional zinc salen-like complex | 0.3 mol% | 0.1 | 100 | — | No co-catalyst | 127 |
| Bimetallic aluminium salen complexes | — | 0.1 | 27 | 3 h | No co-catalyst | 128 |
| Heteroscorpionate lanthanum complex | 0.05 mol% | 1.0 | 70 | 4 h | Catalytic amount of halide as co-catalyst | 83 |
| Bimetallic helical aluminium complex | 0.5 mol% | 1.0 | 50 | 8 h | Catalytic amount of halide as co-catalyst | 129 |
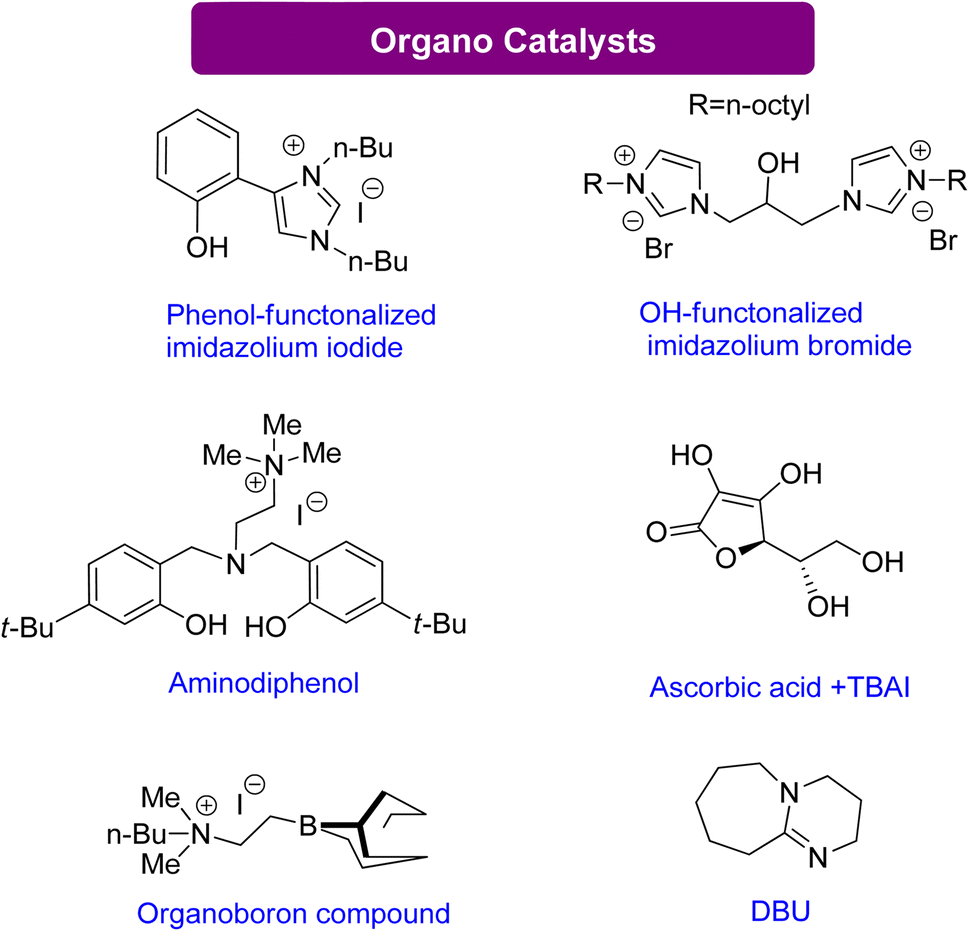 | ||
| Scheme 14 Efficient organo catalysts reported for the conversion of Gly into GC130–135 (reproduced from ref. 82 with permission from RSC copyright 2021). | ||
Sopeña et al.135 concluded that DBU can enhance GC production from Gly and other epoxy alcohols by creating Hemi-carbonate intermediates in mild conditions. Furthermore, a new organocatalytic process has been discovered to couple tri- and tetra-substituted epoxides with CO2, allowing for the synthesis of cyclic carbonates previously difficult to produce under mild reaction conditions. This discovery is advantageous for industrial applications. By controlling the substrate conversion of CO2, new heterocyclic scaffolds can be created with enhanced synthetic potential.
3. New methods for the synthesis of cyclic carbonates
3.1 Synthesis of cyclic carbonates in flow reactors
The production of cyclic carbonates from epoxides and CO2 comprises a gas–liquid process in which mass is transferred from the gas phase to the liquid phase, followed by a catalytic cycloaddition reaction.136 The rate of CO2 mass transfer has been identified as a critical factor for controlling the reaction rate in batch reactors.137 An effective reactor design is required to attain high rates of heat and mass transfer. However, traditional batch reactors are less efficient, costly, and less secure. In contrast, continuous flow systems have several advantages, including lower reaction temperatures, shorter residence times, and ease of industrial scaling.138 As a result, continuous flow reactors can address many of the drawbacks of conventional batch reactors. Moreover, the design of the reactor significantly affects the gas–liquid reaction rates.139 Recent advancements in flow chemistry have enabled the chemical transformation and incorporation of CO2 into valuable organic compounds. These advances have made CO2 attractive to both industry and academia.140–143 Batch transformations using CO2 as a C1 synthon have rapidly developed,144–147 while CO2 reactions in continuous flow systems have become increasingly popular. Compared to conventional batch reactors, flow reactors provide several advantages. Firstly, they have a large surface-to-volume ratio, enabling effective mixing of different phases and precise temperature control. This rapid heat and mass transfer can greatly increase the effectiveness of reactions. Secondly, flow reactors can be much safer due to their excellent thermal management and low material hold-up, facilitating transformations that are difficult to perform in batch reactors, streamlining multistep processes, and optimizing reactions.148–150 Additionally, small flow reactors can produce large quantities of product because of their numbering-up methodology. Thus, lab research can be applied feasibly in the industrial setting within a short period.151Although flow chemistry offers several advantages for CO2 transformation, its application is still a challenge. The low reactivity of CO2 makes many batch reactions slow, and different flow patterns can occur due to the reactor geometry and operating conditions.152 These patterns, such as annular flow, bubble flow, slug flow and churn flow, directly affect the experimental results. Thus, continuous processes require different operating parameters than batch processes, which must be adjusted accordingly. Additionally, CO2 fixation under flow conditions poses other challenges, such as product separation, catalyst recycling, and solvent and/or waste compatibility during multistep reactions. Innovative approaches have been reported to solve these issues, but further developments are necessary, including the development of new concepts, more efficient catalysts, and novel equipment. Process intensification has exciting possibilities and flow chemistry should be viewed as a complement to batch chemistry rather than as its competitor. Research and development efforts are needed for commercialization, and high-potential applications of process intensification include biological CO2 transformation, catalytic CO2 transformation, electrochemical CO2 transformation, and CO2 transformation using plasma technology.153 These advancements can significantly reduce the feasibility gap between newly developed sustainable technologies and previous technologies. Flow platforms are commonly equipped with the equipment listed in Fig. 7.
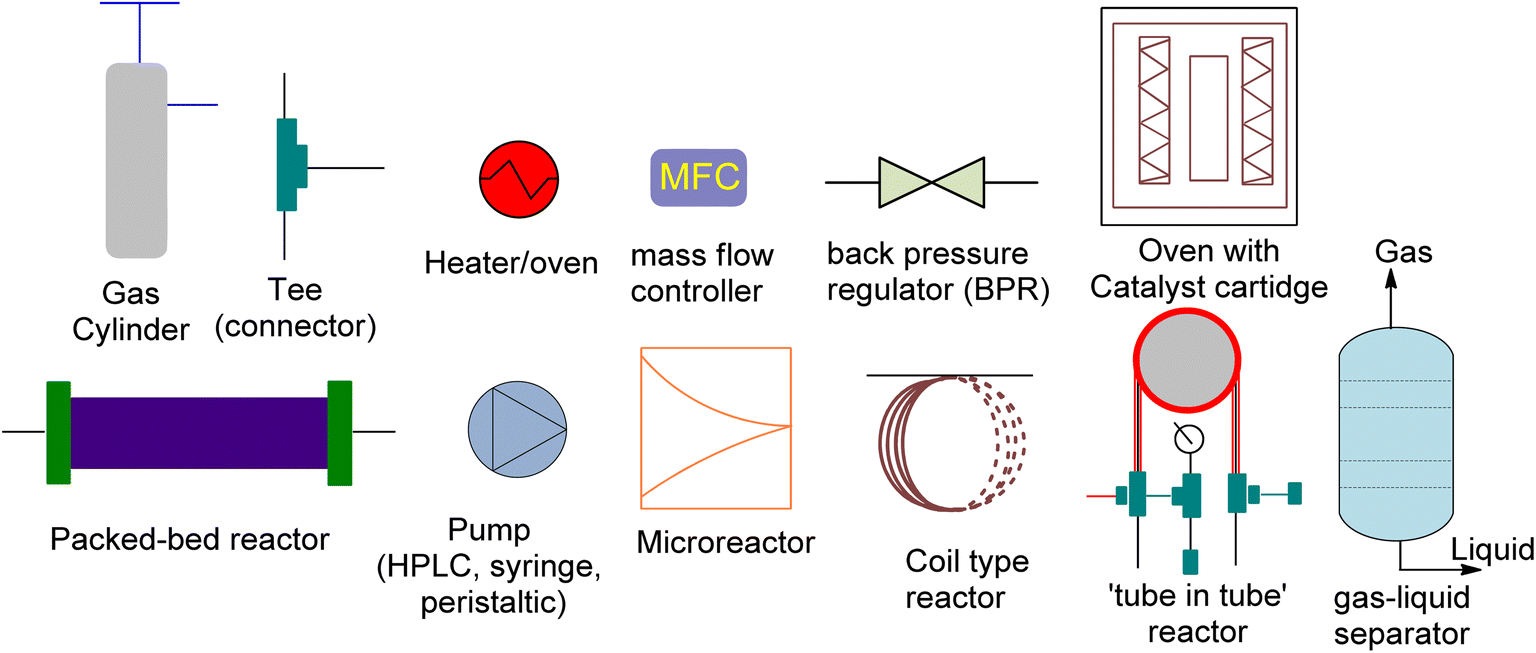 | ||
| Fig. 7 The schematic diagram for the representation of reaction equipment (reproduced from ref. 154 with permission from Wiley-VCH Verlag copyright 2021). | ||
Ionic liquids have become a popular catalysis method in recent years due to their stability, non-flammability, recyclability, and tailoring ease.137,155 The cycloaddition of CO2 and propylene oxide (PO) to create PC was reported on by Takahashi et al. in 2006.156 They used a packed-bed reactor with an effective organic–inorganic hybrid catalyst. Compared to homogeneously used onium-salts, immobilized phosphonium-halides increased catalytic activity significantly. The inorganic-acid support also synergistically enhanced the organic component (Fig. 8). SiO2–C3H6–P (n-Bu)3 I had a pseudo-first-order rate constant 300 times greater than P (n-Bu)4I, normalized to phosphorus atoms. The fixed bed flow reactor used 10 g of SiO2–C3H6–P(n-Bu)3Br as a catalyst, with CO2 and PO flow rates of 0.1 and 0.2 mL min−1, respectively. Under 10 MPa, the temperature increased from 90 to 160 °C, yielding 80% in over 1000 h with 99% selectivity. One phosphonium group can produce more than 11500 molecules of PC. Using packed-bed reactors and supported catalysts, this ground-breaking study found that the cycloaddition of CO2 to PC could be accomplished with remarkable efficiency.
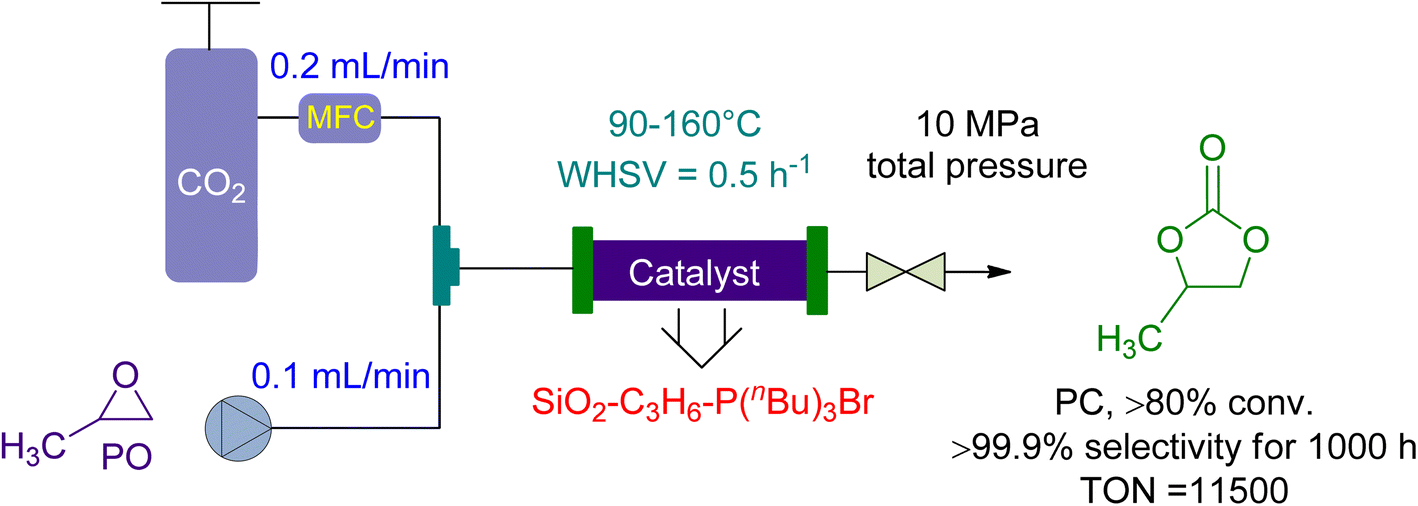 | ||
| Fig. 8 The catalytic coupling between Propylene oxide and CO2 catalysed by SiO2–C3H6–P(n-Bu)3Br (reproduced from ref. 156 with permission from RSC copyright 2017). | ||
Zhang et al.157 examined the utilisation of a packed-bed reactor for the cycloaddition of epichlorohydrin in 2015. They used coconut shell activated carbon (CSAC) anchored ionic liquids as a catalyst and no solvent (Fig. 9). The epichlorohydrin flowrate was maintained at 1.0 mL min−1 and the CO2 flowrate at 50 mL min−1. The reaction was carried out at 1.4 MPa and 140 °C, resulting in an 82% yield in 5 h, with a liquid hourly space velocity (LHSV) of 6 h−1.
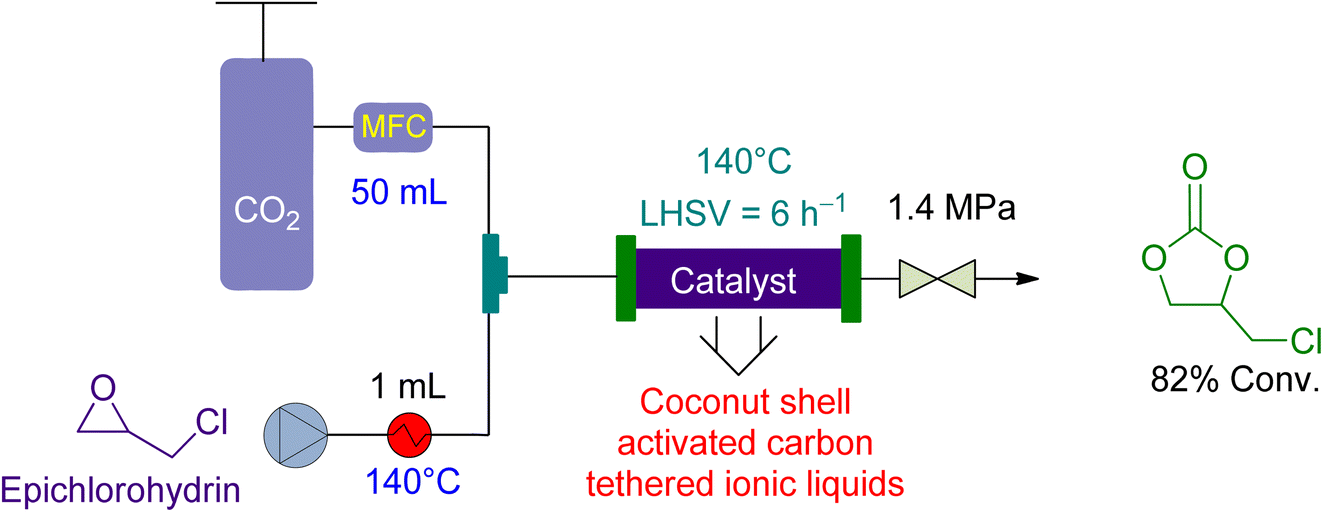 | ||
| Fig. 9 The catalytic coupling of epoxide and CO2 using coconut shell activated carbon (CSAC) tethered ionic liquid as a catalyst.157 | ||
In 2017, Wang et al.158 utilized a fixed-bed reactor to perform the cycloaddition of CO2 to PO to PC using newly synthesized PSIL(IMD) Imidazolium-based polymer-supported ionic liquids as a catalyst, without the use of a solvent. A total of 0.3 g of the catalyst was used in the experiment (Fig. 10). The gas hourly space velocity (GHSV) of the PO was kept constant at 4000 h−1 while the flow rate was kept at 15 μL min−1, under a 2 MPa pressure. With a liquid hourly space velocity (LHSV) of 6 h−1, a yield of 41 to 45% was attained in 130 hours. This system demonstrated good activity and was a significant milestone in the field.
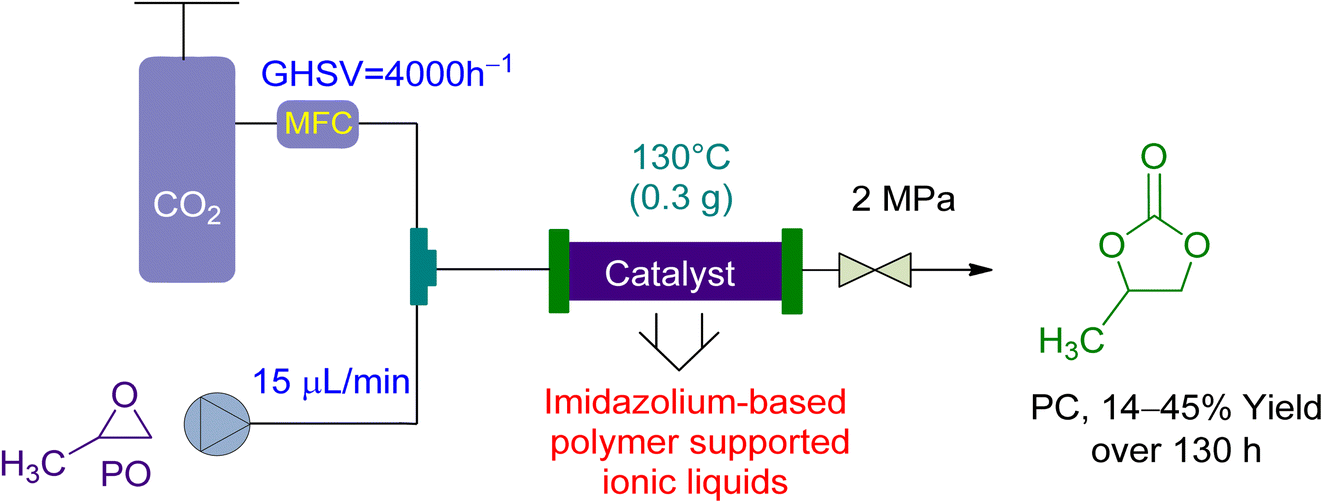 | ||
| Fig. 10 The catalytic coupling between Propylene oxide and CO2 catalysed by Polymer supported ionic liquid (PSIL) (reproduced from ref. 158 with permission from RSC copyright 2017). | ||
For CO2 cycloaddition to epoxides in a continuous flow reactor, Valverde et al.159 looked into the use of multifunctional polymers based on ionic liquids and Rose Bengal (RB) fragments (Fig. 11). A highly effective catalyst was produced under flow conditions by modifying the RB characteristics by changing the chemical nature of the supporting liquid. The substrate and CO2 flowed at rates of 5 and 50 μL min−1, respectively. At 140 bar and 150 °C, the system achieved 53% conversion over 10 days, with no leaching and no decrease in catalytic activity.
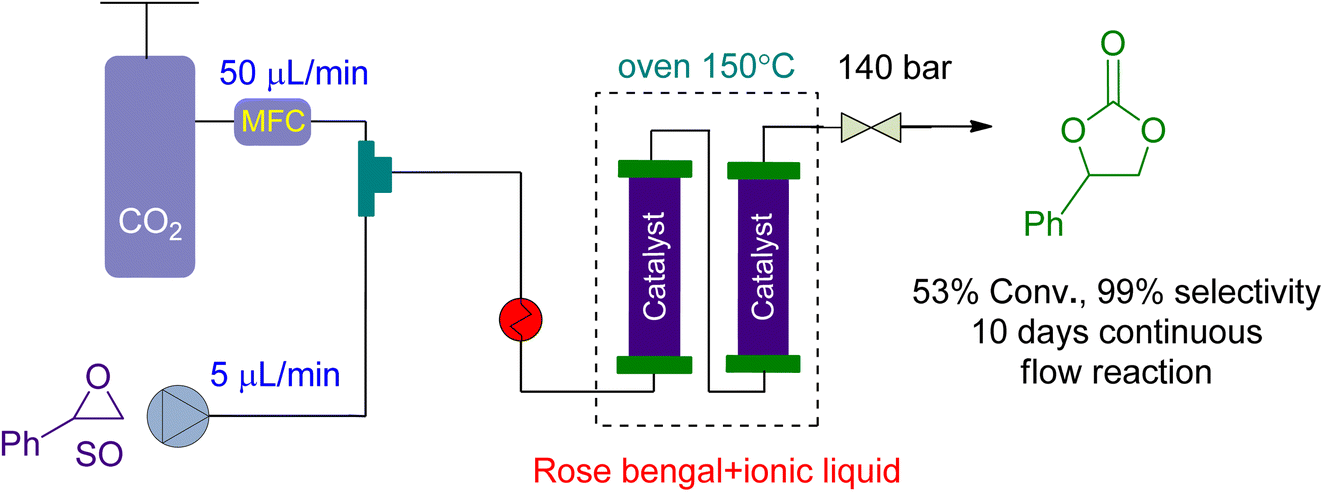 | ||
| Fig. 11 The catalytic coupling between styrene oxide and CO2 catalysed by Rose Bengal (RB) based polymer (reproduced from ref. 159 with permission from ACS, copyright 2021). | ||
In 2021, Yin et al.160 investigated the use of a green DBU-based IL catalyst (DBU@SBA) for CO2 fixation and PC synthesis in a new packed bed reactor design (Fig. 12). The reactor had a substrate flow rate of 0.1 mL min−1 and CO2 flowrate of 5 mL min−1. At 2 MPa and 90 °C, the reaction was conducted, yielding 57.1% conversion after 2 h and 16.86% after 24 h of continuous operation. However, the catalytic activity was lost after 26 h, resulting in only a 4% yield.
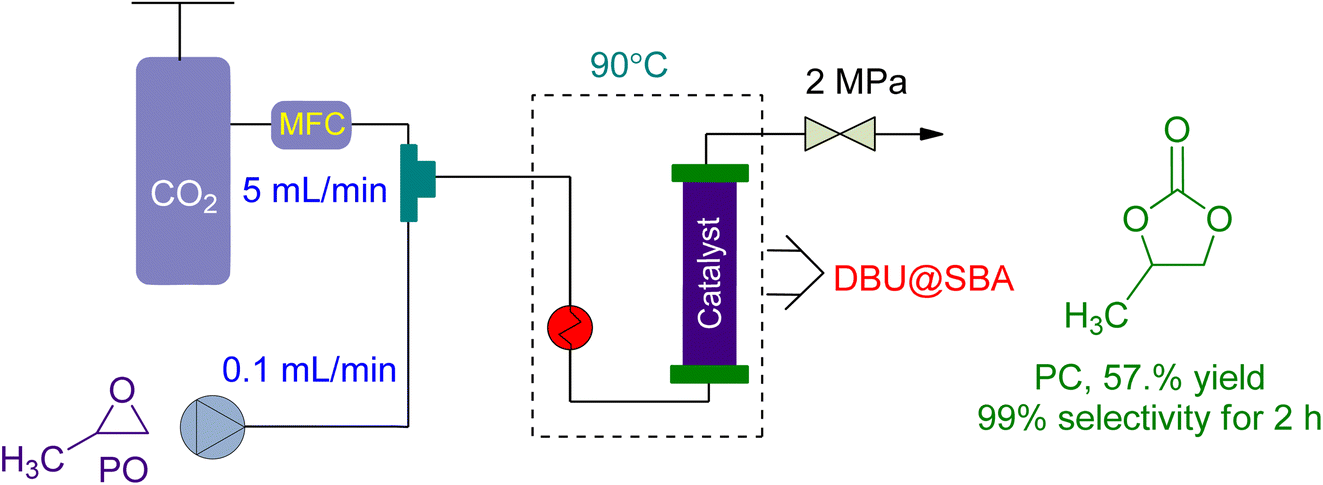 | ||
| Fig. 12 The coupling of propylene oxide with CO2 catalysed by DBU@SBA-15 (reproduced from ref. 160 with permission from Elsevier copyright 2021). | ||
Bui et al.161 reported the use of a vertical-fixed bed reactor for the cycloaddition of CO2 to epichlorohydrin and 1,2 butylene oxide using economical Mesoporous melamine-formaldehyde resins (MMFR) as a heterogeneous catalyst (Fig. 13).
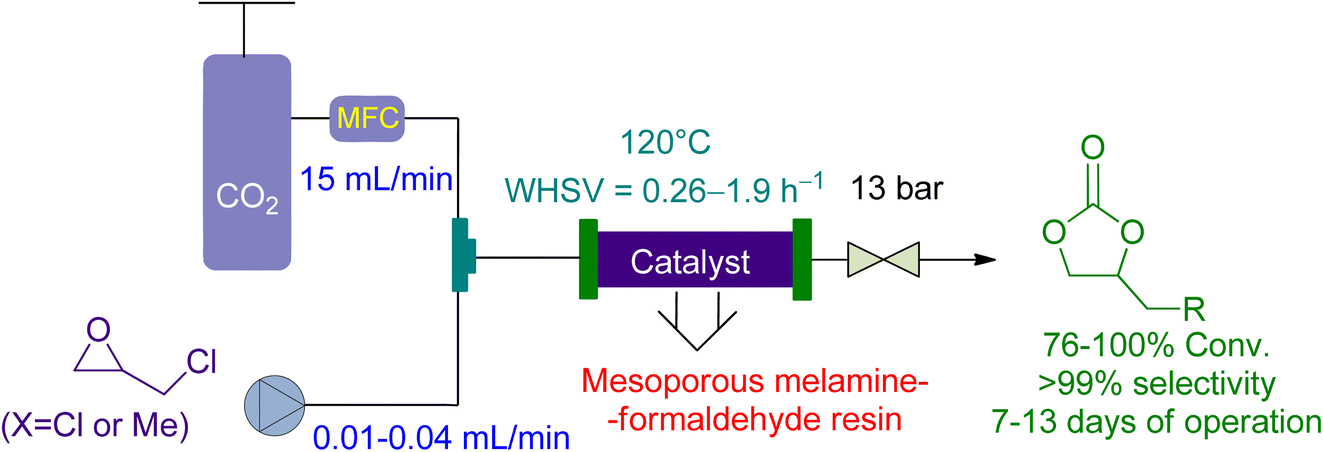 | ||
| Fig. 13 Mesoporous melamine-formaldehyde resins (MFFR) catalysed epoxide-CO2 coupling (reproduced from ref. 161 with permission from ACS, copyright 2020). | ||
3.2 Synthesis of cyclic carbonates in microflow reactors
New types of flow reactors have been developed to improve the efficiency of production. Microreactors are ideal for two-phase gas–liquid processes due to their controllability and large specific surface area. Moreover, they offer opportunities for homogeneous catalysis for CO2-based chemical reactions.162,163 Zhao et al.136 investigated the use of a microreactor for the cycloaddition of PO to PC using a hydroxyl functionalized ionic liquid (HETBAB) as a catalyst. The high surface-to-volume ratio of the microreactor resulted in a significant improvement in heat and mass transfer compared to a conventional CSTR (Fig. 14). The reaction achieved a yield of 99.8% in just 14 seconds at 180 °C and 3.0 MPa.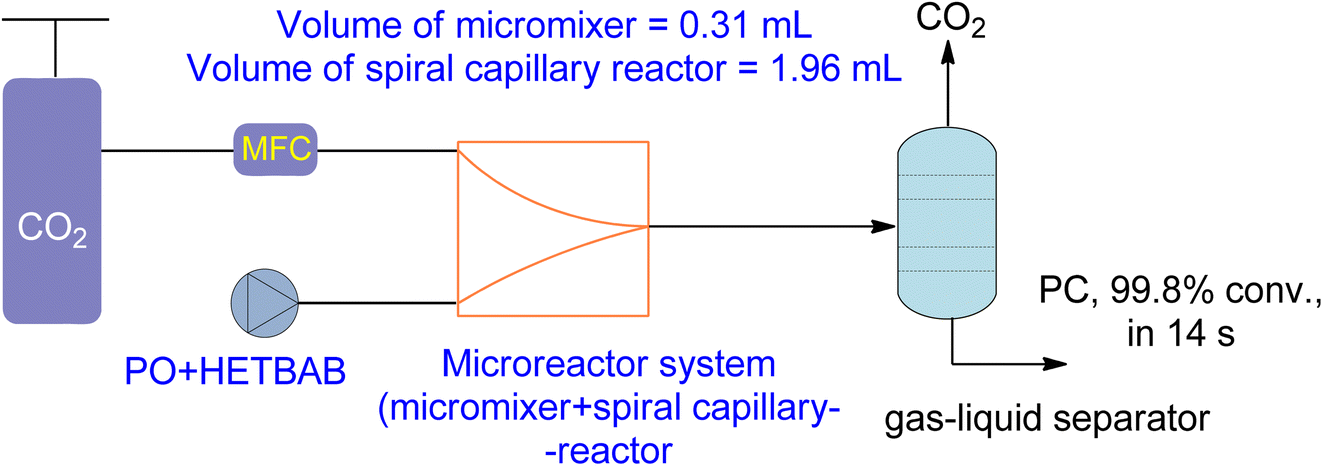 | ||
| Fig. 14 2-Hydroxyl-ethyl-tributyl ammonium bromide (HETBAB) catalysed reaction in micro reaction system including micromixer and capillary reactor (reproduced from ref. 136 with permission from RSC copyright 2013). | ||
Li et al.164 reported the use of a microreactor followed by a delayed reactor for the cycloaddition of CO2 to various epoxides using a binary catalytic system (Fig. 15). The flowrate of epoxide and CO2 was maintained at 0.3–1.2 mL min−1 and 195.2–770 mL min−1, respectively, achieving 90 to 99% conversion in less than 100 seconds with TOF ranging from 6800 to 14700 under 2.0 MPa and 150 °C. Compared to a conventional stirred reactor, the microreactor reaction exhibited significantly higher TOF values, a higher and stable reaction rate, and negligible effect by the CO2/epoxide molar ratio. Furthermore, the “Electrophile–Nucleophile” synergistic effect in the microreactor could be enhanced to further improve the catalytic activity.
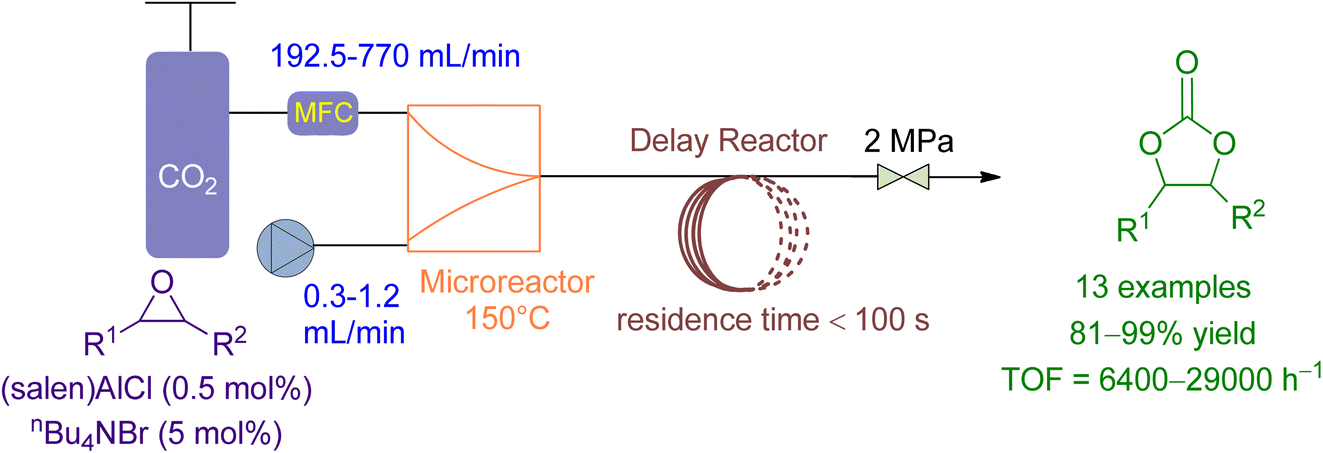 | ||
| Fig. 15 Coupling reaction of CO2 in the presence of (Salen) AlCl (reproduced from ref. 164 with permission from RSC copyright 2018). | ||
In 2021, Wu et al. 138 stated the use of a micro reaction system in a continuous flow setup for CO2 fixation, using 1-butyl-3-methylimidazolium bromide ([BMIM]Br) and H2O as catalyst and co-catalyst, respectively (Fig. 16). At 3 MPa and 140 °C, they achieved 90 to 99.8% conversion with a residence time of just 166 seconds. The molar ratio of [BMIM]Br/H2O/PO was set at 0.14/0.25/1, and the CO2/PO molar ratio was 1.4. Additionally, the recycling performance of [BMIM]Br catalyst was examined, yielding over 86% even after being recycled and repurposed 5 times without losing selectivity.
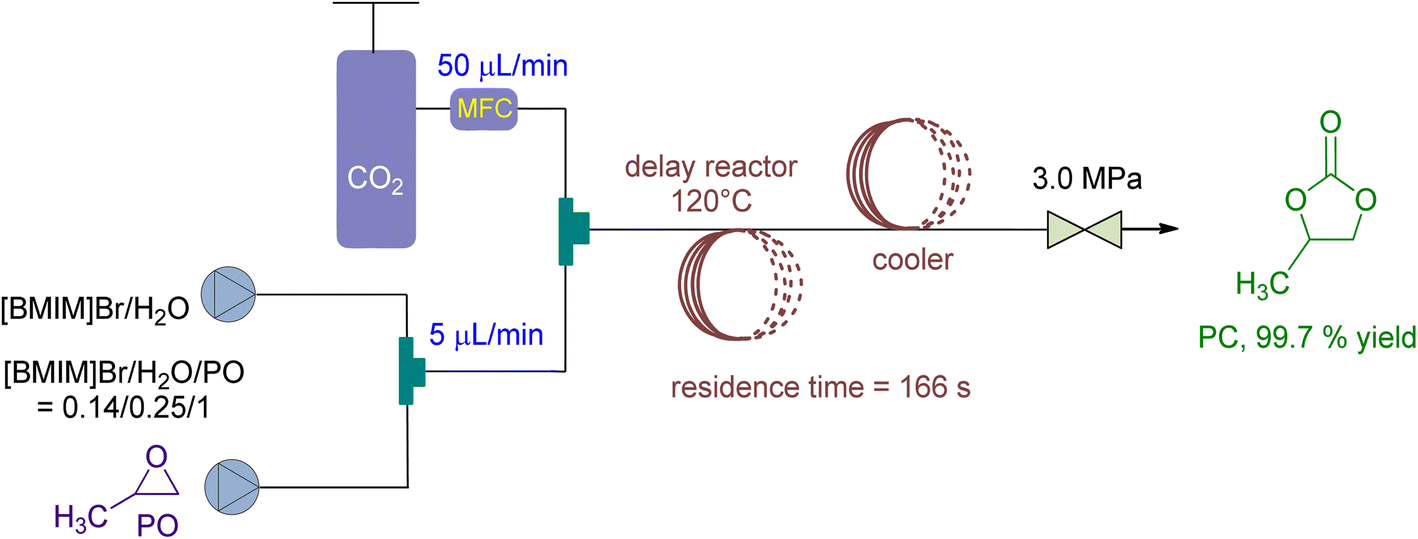 | ||
| Fig. 16 Propylene oxide and CO2 coupling are coupled by [BMIM]Br (reproduced from ref. 138 with permission from Elsevier copyright 2021). | ||
In 2021, Rigo et al.165 used a micro-fluidic reactor to perform cycloaddition to epoxides with a binary system NaBr/diethylene glycol (DEG) as a catalytic system, using DEG as a reaction medium (Fig. 17). The flow rate of epoxide and CO2 was kept at 0.1 mL min−1 and 1.0 mL min−1, respectively. At 120 bar and 220 °C, they obtained a conversion of 91 to 99% for a range of cyclic carbonates.
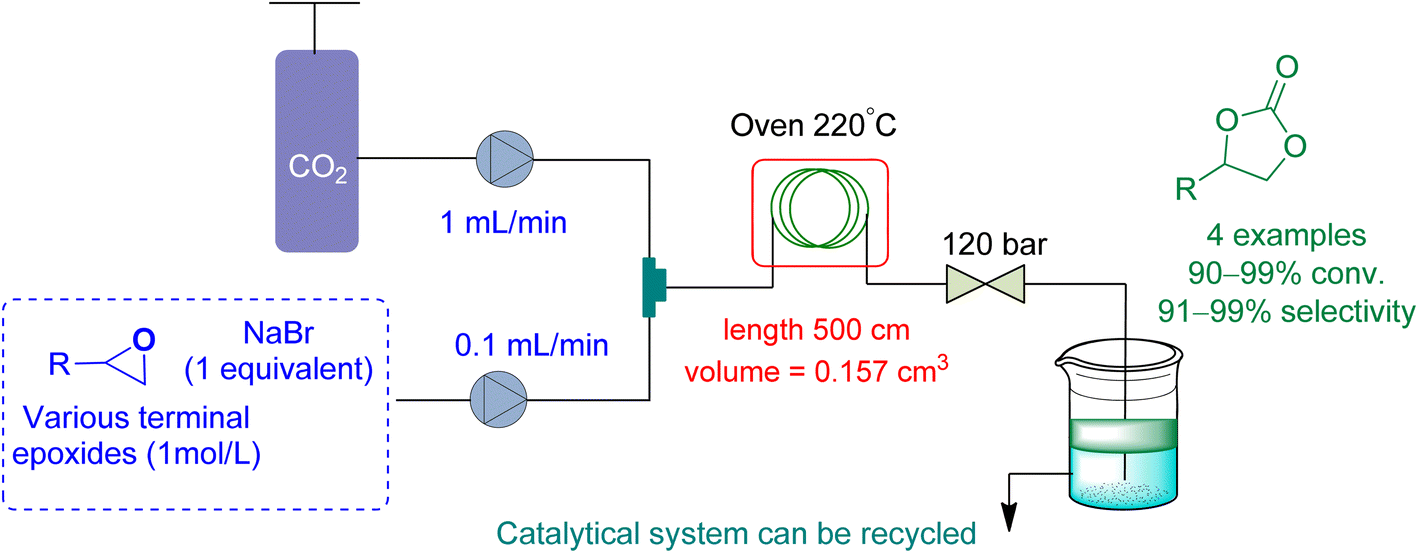 | ||
| Fig. 17 Propylene oxide and CO2 are coupled using diethylene glycol/NaBr system catalysis (reproduced from ref. 165 with permission from Wiley-VCH copyright 2021). | ||
Tube-in-tube reactors are gaining attention as an intelligent process for gas–liquid transformations. In 2018, Rehman et al.166 studied the CO2 cycloaddition of styrene oxide (SO) using a ZnBr2/TBAB catalytic system in a gas–liquid continuous flow reactor of this type. They achieved 100% conversion at 6 bar and 120 °C in 45 min (Fig. 18). The kinetic study showed that it was a first-order reaction with respect to the reactants. The decrease of activation energy by 23 kJ mol−1 using ZnBr2 as a co-catalyst highlighted the importance of synergy.
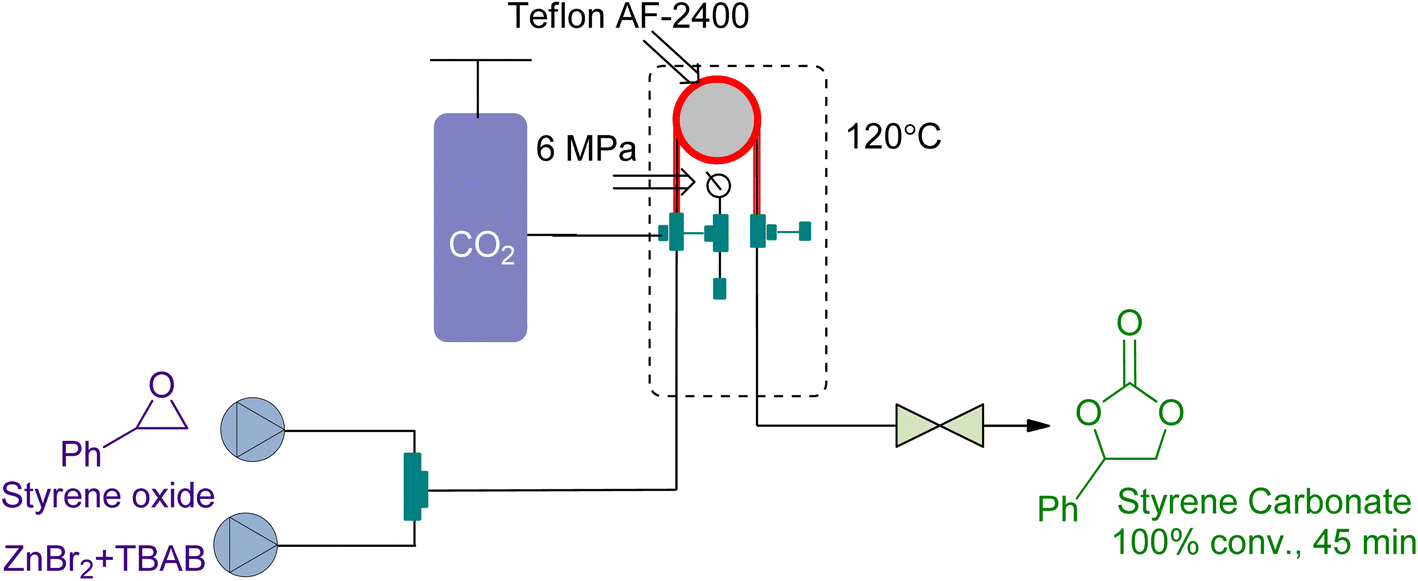 | ||
| Fig. 18 Styrene oxide and CO2 coupling using a synergistic system of ZnBr2/TBAB (reproduced from ref. 166 with permission from Elsevier, copyright 2018). | ||
In 2021, Zanda et al.113 used a tube-in-tube reactor followed by a packed bed reactor in series for the preparation of GC from Gly. They used 1.59 g of heterogeneous TBD @ Merrifield as a catalyst and MEK as a solvent (Fig. 19). In 48 h, 99% conversion was attained while the reaction was being conducted at 70 °C and 3 bar of CO2 pressure.
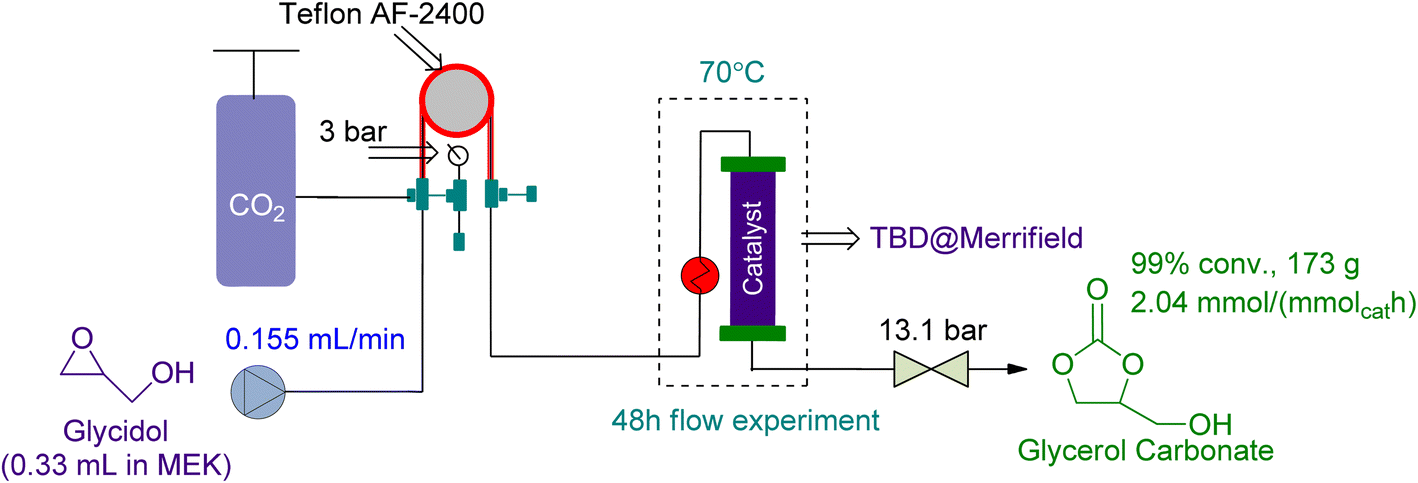 | ||
| Fig. 19 Coupling reaction in a tube-in-tube reactor using TBD@Merrifield as a catalyst (reproduced from ref. 113 with permission ACS from copyright 2021). | ||
3.3 Synthesis of limonene carbonate using a flow reactor
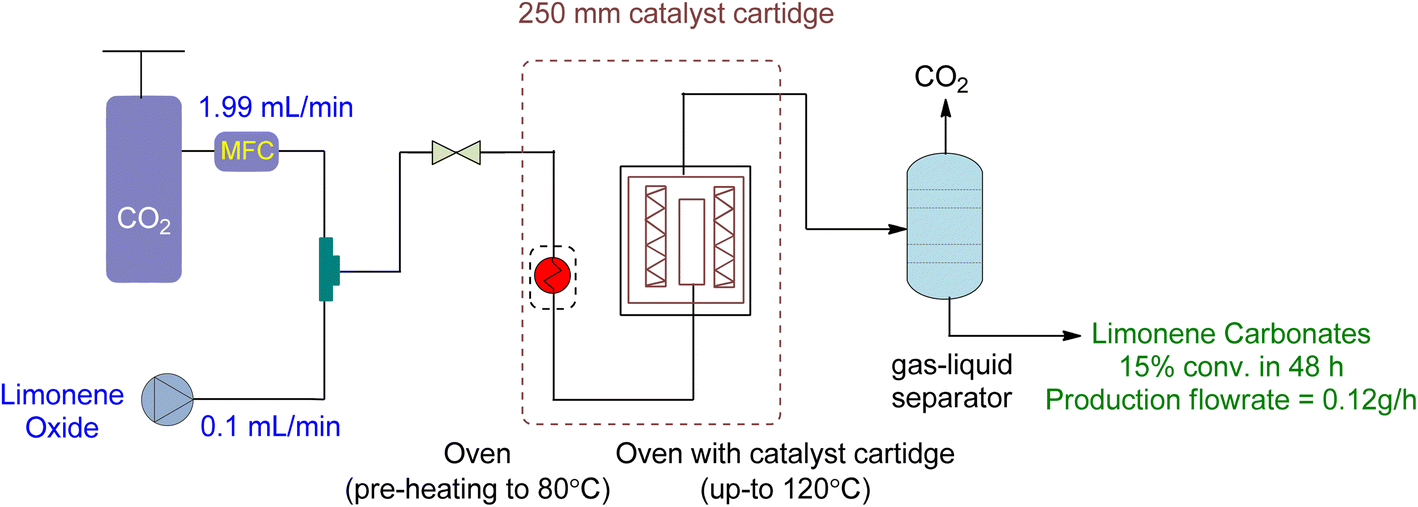
A novel continuous flow method for converting 1,2-LO and LDO to limonene cyclic carbonates was recently reported.91 Supercritical CO2 was the only reagent and solvent used in the experiment, and easily producible heterogeneous supported ionic liquid phase (SILP) catalysts were applied. ILs are excellent candidates for use as reaction media in catalytic processes due to the high solubility of supercritical CO2 in them. SILP materials have a high surface area that is beneficial for catalytic processes and are made up of a thin layer of ionic liquid on a supporting material, such as mesoporous silica. This enables the researchers to overcome mass transfer limitations caused by short diffusion lengths in the thin film.The setup for continuous flow reaction is shown in Fig. 20. Both scCO2 and substrate were mixed and sent to the preheater when a temperature greater than 80 °C was required, followed by a second heating unit to heat the catalyst cartridge to 150 °C. The catalyst input for two different cartridge lengths (150 and 250 mm) was 1.34 and 2.22 g of SILP material, respectively. Back-pressure regulators were used to enabling reactions to occur at different pressures. The final product was collected after passing through a gas–liquid separator, containing carbonates and unreacted starting material. Optimization was performed on the temperature, pressure, catalyst loading, and flow rates of CO2 and substrates. The reaction required a 2–3 h preliminary lead time under the low flow rate of 1,2-LO of 0.01 mL min−1. The SILP catalysts used were easily producible heterogeneous and best-performing supported ionic liquid phase (SILP) catalysts (Fig. 21).
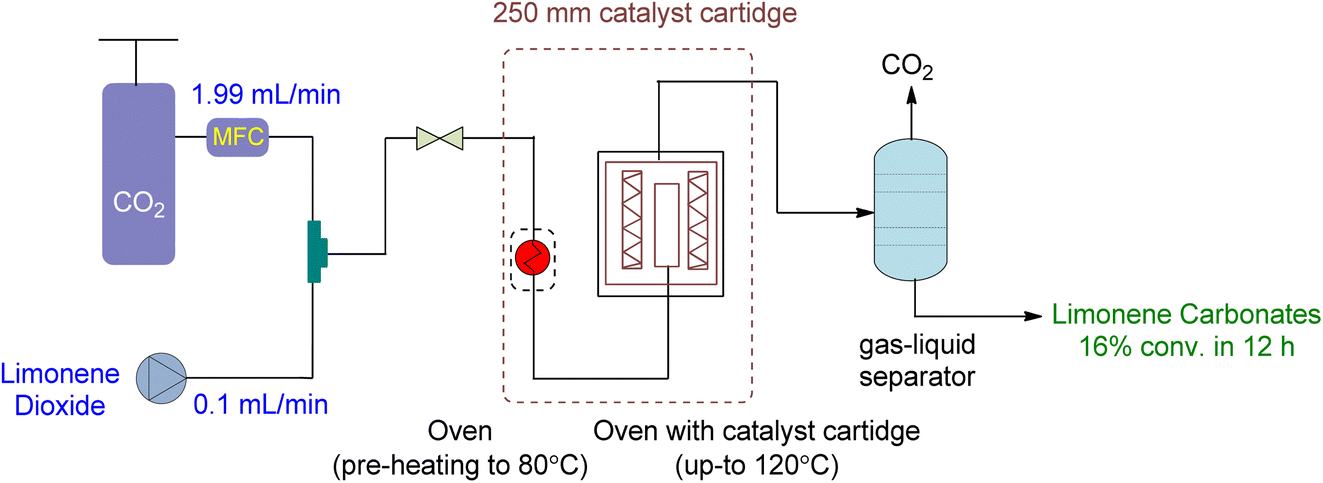 | ||
| Fig. 20 Supported Ionic liquid (SLIP) catalysed heterogeneous reaction of limonene oxide and CO2 (reproduced from ref. 91 with permission from ACS, copyright 2022). | ||
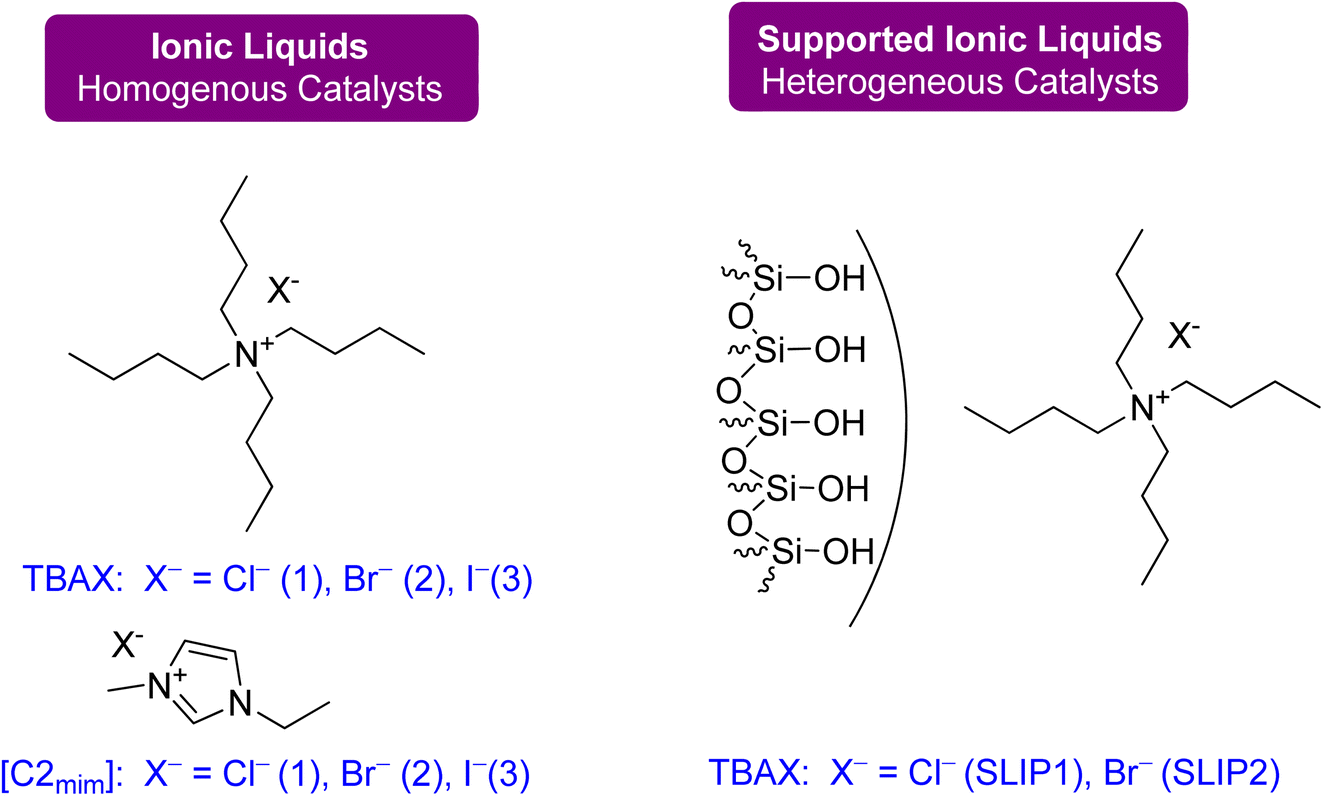 | ||
| Fig. 21 Catalysts: ammonium- and imidazolium-based ionic liquids were used as homogeneous catalysts, and SILPs were applied as heterogeneous catalysts for flow synthesis (reproduced from ref. 91 with permission from ACS, copyright 2022). | ||
Various factors were studied to optimize the continuous flow process for the conversion of 1,2-LO and LDO to limonene cyclic carbonates. Firstly, an increase in temperature from 80 to 120 °C increased yield. However, at 150 °C, both degradation and Hoffmann elimination of TBAC 1 to tributylamine caused a decline in yield over time. Secondly, to prevent the leaching of the catalyst from its supporting material during prolonged industrial use, the corresponding pressure of 6 to 20 MPa was studied at 120 °C. It was observed that 12% leaching occurred at 6 MPa, while no leaching was observed at 15 and 20 MPa. However, at 20 MPa, product formation declined. Thirdly, a catalyst cartridge of 250 mm (residence time of 125 s) was used instead of 150 mm (residence time of 75 s), and the increased input of SILP to 2.22 g resulted in a 60% yield increment. Fourthly, catalyst loading of 20 to 40 wt% was studied, and it was found that a 30 wt% loading instead of 20 wt% increased the yield, while a 40 wt% loading was unsuitable and overburdened the pressure. Lastly, the flow rates of scCO2 and substrate (LO-cis/trans) were studied. The flow rate of CO2 between 1.99 and 2.49 mL min−1 was found to be optimum, while a flow rate of 0.99 mL min−1 caused choking and a flow rate of 3.99 mL min−1 caused leaching. Similarly, a higher flow rate of (LO-cis/trans) 0.02 L min−1 instead of 0.01 mL min−1 resulted in leaching.
Using a heterogeneous catalyst optimized under specific conditions (48 h reaction time, 15 MPa pressure, 120 °C temperature, 2.22 g of SLIP 1 catalyst input, 0.01 mL min−1 of LO-cis/trans substrate flow rate, 1.99 mL min−1 of CO2 flow rate, and a 250 mm catalyst cartridge), a maximum yield of 22% and an overall yield of 16% were achieved. This resulted in a novel production flow rate of 0.12 g h−1, which had not been reported before. To further optimize the process, future experiments could focus on using only the more reactive cis isomer for the chemical fixation of CO2 under flow conditions.
Diastereomeric mixtures of epoxy carbonate and bicarbonate can be formed by converting LDO, as shown in Fig. 22. Epoxy carbonate, being less sterically hindered, yields higher yields than bicarbonate. To optimize the reaction, a heterogeneous catalyst was used under specific conditions (12 h, 20 MPa, 120 °C, 2.22 g of SLIP 1, 0.01 mL min−1 of LO-cis/trans, 1.99 mL min−1 of CO2, 250 mm catalyst cartridge), resulting in a maximum yield of 27% for epoxy carbonate and 14% for bicarbonate, with an overall yield of 16%. Table 7 provides a summary of cyclic carbonates synthesis under flow conditions, showcasing the various types of reactors and their respective reaction conditions.
 | ||
| Fig. 22 Supported Ionic liquid (SLIP) catalysed heterogeneous reaction of LDO and CO2 (reproduced from ref. 91 with permission from ACS, copyright 2022). | ||
| Substrate/epoxide | Epoxide flow rate | CO2 flow rate | Reactor type | Reaction/reactor conditions (T, P) | Catalyst req. | Conversion + time | Selectivity | Other reported | References |
|---|---|---|---|---|---|---|---|---|---|
| Propylene oxide (PO) | 0.1 mL min−1 | 0.2 mL min−1 | Fixed bed flow reactor | 10 MPa, 90–160 °C | 10 g SiO2– C3H6–P(n-Bu)3Br (8.5 mmol phosphorus) | 80% in 1000 h | 99.9% | WHSV = 0.5 per h, TON = 11500, synergistic organic-inorganic catalyst |
156 |
| Epichlorohydrin | 1.0 mL min−1 | 50.0 mL min−1 | Packed bed reactor | 1.4 MPa, 140 °C | Coconut shell-activated carbon (CSAC) tethered Bmim-COOH/Br | 82% in 50 h | n.a | Liquid hourly space velocity (LHSV) = 6 per h | 157 |
| Propylene oxide (PO) | 15 μL min−1 | Fixed bed flow reactor | 2 MPa, 130 °C | PSIL (IMD) imidazolium-based polymer-supported ionic liquids | 41–45% in 130 h | n.a | GHSV 4000 per h | 158 | |
| Styrene oxide (SO) | 5 μL min−1 | 50 μL min−1 | Continuous flow reactor | 140 bar, 150 °C | Rose bengal supported ionic liquid-like phases (RB-SILLP) | 53% in 10 days | 99% | Productivity of 1.129 | 159 |
| Propylene oxide (PO) | 0.1 mL min−1 | 5.0 mL min−1 | Packed bed reactor | 2 MPa, 90 °C | DBU@SBA-15 (100 mg) | 57.1% for 2 h continuous reaction/16.86% for 24 h | 99% | 160 | |
| Epichlorohydrin and 1,2 butylene oxide | 0.01–0.04 mL min−1 | 15.0 mL min−1 | Vertical fixed-bed reactor packed with the polymeric MMFR (continuous flow) | 13 bar, 120 °C | Mesoporous melamine-formaldehyde resins MMFR (1.95 g, ≤100 μm) or MMFR 250 (1.5 g, ≤100 μm) | 76–100% conversion in 7 to 13 days | 99.9% | Weight hourly space velocity (WHSV) of 0.26–1.9 h−1 | 161 |
| Propylene oxide (PO) | n.a | n.a | Microreactor | 3.5 MPa, 180 °C | HETBAB | 99.8% in 14 s | n.a | 136 | |
 |
0.3–1.2 mL min−1 | 192.5–770 mL min−1 | Microreactor followed by a delayed reactor | 2 MPa and 150 °C | Binary (salen) AlCl/nBu4NBr (TBAB) system | 90–99% with a residence time of less than 100 s | 99% | Residence time 100 s, TOF = 6800 to 14700 h−1 |
164 |
| Propylene oxide (PO) | n.a | n.a | Continuous-flow micro-reaction system | 3 MPa, 140 °C | 1-Butyl-3-methylimidazolium bromide ([BMIM]Br)/H2O | 99.8% | n.a | Residence time 166 s, molar ratio of [BMIM]Br/H2O/PO to be 0.14/0.25/1, the molar ratio of CO2/PO to be 1.4 | 138 |
| Terminal epoxide (R = nC4H9) | 0.1 mL min−1 | 1 mL min−1 | Microfluidic reactor | 120 bar and 220 °C | NaBr/diethylene glycol (DEG) | 91 to 99% | 92 to 99% | Reaction medium (DEG) | 165 |
| Styrene oxide | n.a | n.a | “Tube-in-tube” gas–liquid continuous flow reactor | 6 bar and 120 °C | Tetrabutylammonium bromide (TBAB)/ZnBr2 homogenous system | 100% in 45 min | n.a | Activation energy decrement 55–32 kJ mol−1 = 23 kJ mol−1 | 166 |
| Glycidol (Gly) | 0.155 mL min−1 | n.a | “Tube-in-tube” reactor followed by a packed bed reactor | 3 (bar CO2 local pressure) | TBD@Merrifield (catalyst loading of 1.59 g/1.48 mmol) | 99% in 48 h | n.a | MEK solvent | 113 |
| LO-(cis/trans) | 0.1 mL min−1 | 1.99 mL min−1 | Continuous flow reaction | 15 MPa, 120 °C | Supported ionic liquid SLIP 1 (2.22 g) (Fig. 21) | 22% (max) and 15% (overall) in 48 h | n.a | 250 mm catalyst cartridge, production flow rate of 0.12 g h−1 | 91 |
| Limonene dioxide (LDO) | 0.1 mL min−1 | 1.99 mL min−1 | Continuous flow reaction | 20 MPa, 120 °C | Supported ionic liquid SLIP 1 (2.22 g) (Fig. 21) | 27% (epoxy carbonate) 14% (bis-carbonate), and 16% (overall) in 12 h | n.a | 250 mm catalyst cartridge | 91 |
4. Summary
This review summarizes the latest developments in using CO2 to produce cyclic carbonates from bio-based precursors. Future research should focus on proposing catalytical systems that can reduce the temperature and pressure requirements for CO2 and epoxide reactions. The selection of product routes based on CCU is complex, and economic and environmental assessments of each route are necessary. Novel designs for integrating Capture and Utilization at the same station are anticipated, and research should aim to improve financial and energy penalties. Ethanol, methanol, and syngas production have high potential, but research and development efforts are needed to make them feasible. Processes that require hydrogen will need a sustainable or green method of hydrogen production for commercial viability. Process Intensification tools such as intensified equipment and processes can improve mass and heat-transfer rates. Over 50% of processes are laboratory-based, so industrialization efforts are necessary. Recent advances in flow chemistry have enabled the use of CO2 as a C1 synthon and packed-bed and microreactors have shown promise for coupling CO2 and epoxides. Further research should focus on developing robust catalytic systems, novel reactor designs, and efficient synthesis techniques. With the growing number of publications in this field, a bright future is forecasted, and this review provides a starting point for further advancements.Conflicts of interest
There are no conflicts to declare.References
- M. E. Mosquera, et al., Terpenes and Terpenoids: Building Blocks to Produce Biopolymers, Sustainable Chem., 2021, 2(3), 467–492 CrossRef CAS.
- S. Perveen, Introductory chapter: terpenes and terpenoids, in Terpenes and Terpenoids-Recent Advances, IntechOpen. 2021 Search PubMed.
- E. M. Thurman, Analysis of terpenes in hemp (Cannabis sativa) by gas chromatography/mass spectrometry: Isomer identification analysis, in Comprehensive Analytical Chemistry, Elsevier, 2020, pp. 197–233 Search PubMed.
- J. Buckle, Basic Plant Taxonomy, Chemistry, Extraction, Biosynthesis, and Analysis (Chapter 3), Clinical aromatherapy. Essential Oils in Practice, 2003, pp. 38–75 Search PubMed.
- [cited 2022 August, 26], available from, https://www.truelabscannabis.com/blog/terpene-boiling-points.
- V. C. Eze, et al., Synthesis of cyclic α-pinane carbonate–a potential monomer for bio-based polymers, RSC Adv., 2022, 12(27), 17454–17465 RSC.
- R. Ciriminna, et al., Limonene: a versatile chemical of the bioeconomy, Chem. Commun., 2014, 50(97), 15288–15296 RSC.
- N. Mercy, B. Nithyalakshmi and L. Aadhithiya, Extraction of orange oil by improved steam distillation and its characterization Studies, Int. J. Eng. Technol., Manage., Appl. Sci., 2015, 3(2), 1–8 Search PubMed.
- S. O. Giwa, M. Muhammad and A. Giwa, Utilizing orange peels for essential oil production, J. Eng. Appl. Sci., 2018, 13, 17–27 CAS.
- Global D-limonene Market – Industry Trends and Forecast to 2029, [cited 2022 August, 11], available from, https://www.databridgemarketresearch.com/reports/global-d-limonene-market Search PubMed.
- A. Thomas and Y. Bessiere, Nat. Prod. Rep., 1989, 6(3), 291–309 RSC.
- C. M. Alder, et al., Updating and further expanding GSK's solvent sustainability guide, Green Chem., 2016, 18(13), 3879–3890 RSC.
- Tuepentine Oil Market Size, Share and Industry Analysis, [cited 2022 August 11], available from: https://www.fortunebusinessinsights.com/turpentine-oil-market-103803 Search PubMed.
- M. Colonna, et al., Synthesis and radiocarbon evidence of terephthalate polyesters completely prepared from renewable resources, Green Chem., 2011, 13(9), 2543–2548 RSC.
- A. Gandini, The irruption of polymers from renewable resources on the scene of macromolecular science and technology, Green Chem., 2011, 13(5), 1061–1083 RSC.
- Pinene Terpene: Strains, Effects, Dosage, & More, [cited 2022 August 26], available from: https://finestlabs.com/pinene-terpene/ Search PubMed.
- C. Hu, et al., Lanthanum nanocluster/ZIF-8 for boosting catalytic CO 2/glycerol conversion using MgCO 3 as a dehydrating agent, J. Mater. Chem. A, 2021, 9(11), 7048–7058 RSC.
- N. Razali, M. Conte and J. McGregor, The role of impurities in the La2O3 catalysed carboxylation of crude glycerol, Catal. Lett., 2019, 149(5), 1403–1414 CrossRef CAS.
- I. Group, Biodiesel Market: Global Industry Trends, Share, Size, Growth, Opportunity and Forecast 2022-2027, [cited 2022 09 November], available from, https://www.imarcgroup.com/biodiesel-market Search PubMed.
- B. Zada, M. Kwon and S.-W. Kim, Current Trends in Acetins Production: Green versus Non-Green Synthesis, Molecules, 2022, 27(7), 2255 CrossRef CAS PubMed.
- N. Razali and J. McGregor, Improving product yield in the direct carboxylation of glycerol with CO2 through the tailored selection of dehydrating agents, Catalysts, 2021, 11(1), 138 CrossRef CAS.
- IPCC, [cited 2022 August, 11], available from, https://www.co2.earth/global-co2-emissions.
- NOAA, National oceanic and atmospheric administration, [cited 2022 August 11], available from, https://gml.noaa.gov/ccgg/trends/global.html.
- A. Townsend and A. Gillespie, Scaling up the CCS market to deliver net-zero emissions, Thought Leadership Piece, Global CCS Institute, 2020 Search PubMed.
- A. J. Kamphuis, F. Picchioni and P. P. Pescarmona, CO2-fixation into cyclic and polymeric carbonates: principles and applications, Green Chem., 2019, 21(3), 406–448 RSC.
- A. W. Kleij, M. North, and A. Urakawa, CO2 Catalysis, Wiley Online Library, 2017, pp. 1036–1038 Search PubMed.
- M. Aresta and A. Dibenedetto, Carbon Recycling Through CO2-Conversion for Stepping Toward a Cyclic-C Economy. A Perspective, Front. Energy Res., 2020, 8, 159 CrossRef.
- A. Arif, et al., Optimal selection of integrated electricity generation systems for the power sector with low greenhouse gas (GHG) emissions, Energies, 2020, 13(17), 4571 CrossRef CAS.
- G. Notton, et al., Intermittent and stochastic character of renewable energy sources: Consequences, cost of intermittence and benefit of forecasting, Renewable Sustainable Energy Rev., 2018, 87, 96–105 CrossRef.
- M. Yousaf, et al., Carbon dioxide utilization: A critical review from multiscale perspective, Energy Sci. Eng., 2022, 10(12), 4890–4923 CrossRef CAS.
- P. Markewitz, et al., Worldwide innovations in the development of carbon capture technologies and the utilization of CO 2, Energy Environ. Sci., 2012, 5(6), 7281–7305 RSC.
- A. Rehman, et al., Recent advances in the synthesis of cyclic carbonates via CO2 cycloaddition to epoxides, J. Environ. Chem. Eng., 2021, 9(2), 105113 CrossRef CAS.
- T. Bruhn, H. Naims and B. Olfe-Kräutlein, Separating the debate on CO2 utilisation from carbon capture and storage, Environ. Sci. Policy, 2016, 60, 38–43 CrossRef CAS.
- K. Arning, et al., Same or different? Insights on public perception and acceptance of carbon capture and storage or utilization in Germany, Energy Policy, 2019, 125, 235–249 CrossRef.
- D. Y. Leung, G. Caramanna and M. M. Maroto-Valer, An overview of current status of carbon dioxide capture and storage technologies, Renewable Sustainable Energy Rev., 2014, 39, 426–443 CrossRef CAS.
- M. E. Boot-Handford, et al., Carbon capture and storage update, Energy Environ. Sci., 2014, 7(1), 130–189 RSC.
- A. Raza, et al., Significant aspects of carbon capture and storage–A review, Petroleum, 2019, 5(4), 335–340 CrossRef.
- S. Bachu, Screening and ranking of sedimentary basins for sequestration of CO2 in geological media in response to climate change, Environ. Geol., 2003, 44(3), 277–289 CrossRef CAS.
- E. J. Novek, et al., Low-temperature carbon capture using aqueous ammonia and organic solvents, Environ. Sci. Technol. Lett., 2016, 3(8), 291–296 CrossRef CAS.
- B. Metz, et al., IPCC Special Report on Carbon Dioxide Capture and Storage, Cambridge University Press, Cambridge, 2005 Search PubMed.
- NIST Database, [cited 2022 August, 11], available from, https://webbook.nist.gov/chemistry/name-ser/ Search PubMed.
- M. Andrei, et al., Enhanced Oil Recovery with CO2 Capture and Sequestration, 2010 Search PubMed.
- M. Aresta, A. Dibenedetto and E. Quaranta, State of the art and perspectives in catalytic processes for CO2 conversion into chemicals and fuels: The distinctive contribution of chemical catalysis and biotechnology, J. Catal., 2016, 343, 2–45 CrossRef CAS.
- R. Chauvy, et al., Selecting emerging CO2 utilization products for short-to mid-term deployment, Appl. Energy, 2019, 236, 662–680 CrossRef CAS.
- M. Aresta, A. Dibenedetto and A. Angelini, The changing paradigm in CO2 utilization, J. CO2 Util., 2013, 3, 65–73 CrossRef.
- X. Zhang, Microalgae Removal of CO2 from Flue Gas, IEA Clean Coal Centre, UK, 2015 Search PubMed.
- R. Hands, Market Drivers for the Syngas Industry, Gasification and Syngas Technology Council, 2017 Search PubMed.
- M. Aresta, A. Dibenedetto, and L.-N. He, Analysis of demand for captured CO2 and products from CO2 conversion. November, A Report Exclusively for Members of the Carbon Dioxide Capture & Conversion (CO2 CC) Program of The Catalyst Group Resources (TCGR), 2012 Search PubMed.
- M. Aresta, F. Nocito, and A. Dibenedetto, What catalysis can do for boosting CO2 utilization, in Advances in Catalysis, Elsevier, 2018, pp. 49–111 Search PubMed.
- Global Urea Market Outlook, [cited 2022 August, 11], available from, https://www.expertmarketresearch.com/reports/urea-market Search PubMed.
- E. Koohestanian, et al., A Novel Process for CO2 Capture from the Flue Gases to Produce Urea and Ammonia. 2018 Search PubMed.
- M. Aresta and A. Dibenedetto, The Contribution of the Utilization Option to Reducing the CO2 Atmospheric Loading: Research Needed to Overcome Existing Barriers for a Full Exploitation of the Potential of the CO2 Use, Elsevier, 2004 Search PubMed.
- C. Federsel, R. Jackstell and M. Beller, Moderne Katalysatoren zur Hydrierung von Kohlendioxid, Angew. Chem., 2010, 122(36), 6392–6395 CrossRef.
- G. A. Olah, Beyond oil and gas: the methanol economy, Angew. Chem., Int. Ed., 2005, 44(18), 2636–2639 CrossRef CAS PubMed.
- M. Yousaf, et al., Techno-economic analysis of integrated hydrogen and methanol production process by CO2 hydrogenation, Int. J. Greenhouse Gas Control, 2022, 115, 103615 CrossRef CAS.
- C. Miaohua, Agricultural ecology and varied functional synthetical uses of methane, Journal of Guangxi Teachers Education University, 1996, 1(2), 101–105 Search PubMed.
- B. M. Bhanage and M. Arai, Transformation and Utilization of Carbon Dioxide, Springer, 2014 Search PubMed.
- A. Chapman, et al., Adding value to power station captured CO2: tolerant Zn and Mg homogeneous catalysts for polycarbonate polyol production, ACS Catal., 2015, 5(3), 1581–1588 CrossRef CAS.
- G. Laugel, et al., Homogeneous and heterogeneous catalysis for the synthesis of cyclic and polymeric carbonates from CO2 and epoxides: a mechanistic overview, Adv. Chem. Lett., 2013, 1(3), 195–214 CrossRef CAS.
- A. Das and P. Mahanwar, A brief discussion on advances in polyurethane applications, Adv. Ind. Eng. Polym. Res., 2020, 3(3), 93–101 Search PubMed.
- L. Olsson and B. Hahn-Hägerdal, Fermentation of lignocellulosic hydrolysates for ethanol production, Enzyme Microb. Technol., 1996, 18(5), 312–331 CrossRef CAS.
- M. E. Dias De Oliveira, B. E. Vaughan and E. J. Rykiel, Ethanol as fuel: energy, carbon dioxide balances, and ecological footprint, BioScience, 2005, 55(7), 593–602 CrossRef.
- C. Arcoumanis, et al., The potential of di-methyl ether (DME) as an alternative fuel for compression-ignition engines: A review, Fuel, 2008, 87(7), 1014–1030 CrossRef CAS.
- W. Deng, et al., A review on transesterification of propylene carbonate and methanol for dimethyl carbonate synthesis, Carbon Resour. Convers., 2019, 2(3), 198–212 CrossRef CAS.
- Y. Liang, Producing liquid transportation fuels from heterotrophic microalgae, Appl. Energy, 2013, 104, 860–868 CrossRef CAS.
- P. P. Pescarmona, Cyclic carbonates synthesised from CO2: Applications, challenges and recent research trends, Curr. Opin. Green Sustainable Chem., 2021, 29, 100457 CrossRef CAS.
- M. North, F. Pizzato and P. Villuendas, Organocatalytic, asymmetric aldol reactions with a sustainable catalyst in a green solvent, ChemSusChem, 2009, 2(9), 862–865 CrossRef CAS PubMed.
- M. O. Sonnati, et al., Glycerol carbonate as a versatile building block for tomorrow: synthesis, reactivity, properties and applications, Green Chem., 2013, 15(2), 283–306 RSC.
- D. R. Lide, Handbook of Organic Solvents, CRC Press, Boca Raton, 1995 Search PubMed.
- G. Wypych, Databook of Green Solvents, Elsevier, 2019 Search PubMed.
- J. Garche and K. Brandt, Electrochemical Power Sources: Fundamentals, Systems, and Applications: Li-Battery Safety, Elsevier, 2018 Search PubMed.
- P. De Caro, et al., Recent progress in synthesis of glycerol carbonate and evaluation of its plasticizing properties, Front. Chem., 2019, 7, 308 CrossRef CAS PubMed.
- M. Szori, et al., Glycerol carbonate as a fuel additive for a sustainable future, Sustainable Energy Fuels, 2018, 2(10), 2171–2178 RSC.
- Propylene Carbonate Market, [cited 2022 August, 11], available from, https://www.marketresearchfuture.com/reports/propylenecarbonate-market-8089 Search PubMed.
- Ethylene Carbonate Market by Application, [cited 2022 August, 11], available from: https://www.marketsandmarkets.com/Market-Reports/ethylene-carbonate-market-229766138.html Search PubMed.
- J. S Bello Forero, et al., Propylene carbonate in organic synthesis: Exploring its potential as a green solvent, Curr. Org. Synth., 2016, 13(6), 834–846 CrossRef.
- S. Christy, et al., Recent progress in the synthesis and applications of glycerol carbonate, Curr. Opin. Green Sustainable Chem., 2018, 14, 99–107 CrossRef.
- D. Prat, et al., CHEM21 selection guide of classical-and less classical-solvents, Green Chem., 2016, 18(1), 288–296 RSC.
- V. Etacheri, et al., Challenges in the development of advanced Li-ion batteries: a review, Energy Environ. Sci., 2011, 4(9), 3243–3262 RSC.
- V. Aravindan, et al., Lithium-ion conducting electrolyte salts for lithium batteries, Chem. - Eur. J., 2011, 17(51), 14326–14346 CrossRef CAS PubMed.
- J. W. Comerford, et al., Sustainable metal-based catalysts for the synthesis of cyclic carbonates containing five-membered rings, Green Chem., 2015, 17(4), 1966–1987 RSC.
- V. Aomchad, et al., Recent progress in the catalytic transformation of carbon dioxide into biosourced organic carbonates, Green Chem., 2021, 23(3), 1077–1113 RSC.
- J. Martinez, et al., An efficient and versatile lanthanum heteroscorpionate catalyst for carbon dioxide fixation into cyclic carbonates, ChemSusChem, 2017, 10(14), 2886–2890 CrossRef CAS PubMed.
- G. Fiorani, et al., Catalytic coupling of carbon dioxide with terpene scaffolds: access to challenging bio-based organic carbonates, ChemSusChem, 2016, 9(11), 1304–1311 CrossRef CAS PubMed.
- L. Longwitz, et al., Calcium-based catalytic system for the synthesis of bio-derived cyclic carbonates under mild conditions, ACS Catal., 2018, 8(1), 665–672 CrossRef CAS.
- J. Fernández-Baeza, et al., NNC-Scorpionate Zirconium-Based Bicomponent Systems for the Efficient CO2 Fixation into a Variety of Cyclic Carbonates, Inorg. Chem., 2020, 59(17), 12422–12430 CrossRef.
- V. Aomchad, et al., Exploring the potential of group III salen complexes for the conversion of CO2 under ambient conditions, Catal. Today, 2021, 375, 324–334 CrossRef CAS.
- S. Y. Park, et al., Synthesis of poly [(2-oxo-1, 3-dioxolane-4-yl) methyl methacrylate-co-ethyl acrylate] by incorporation of carbon dioxide into epoxide polymer and the miscibility behavior of its blends with poly (methyl methacrylate) or poly (vinyl chloride), J. Polym. Sci., Part A: Polym. Chem., 2001, 39(9), 1472–1480 CrossRef CAS.
- H. Morikawa, et al., Two diastereomers of d-limonene-derived cyclic carbonates from d-limonene oxide and carbon dioxide with a tetrabutylammonium chloride catalyst, Bull. Chem. Soc. Jpn., 2018, 91(1), 92–94 CrossRef CAS.
- A. Rehman, et al., Highly selective, sustainable synthesis of limonene cyclic carbonate from bio-based limonene oxide and CO2: A kinetic study, J. CO2 Util., 2019, 29, 126–133 CrossRef CAS.
- P. Mikšovsky, et al., Continuous Formation of Limonene Carbonates in Supercritical Carbon Dioxide, Org. Process Res. Dev., 2022, 26(10), 2799–2810 CrossRef PubMed.
- M. Bähr, A. Bitto and R. Mülhaupt, Cyclic limonene dicarbonate as a new monomer for non-isocyanate oligo-and polyurethanes (NIPU) based upon terpenes, Green Chem., 2012, 14(5), 1447–1454 RSC.
- V. Schimpf, et al., High purity limonene dicarbonate as versatile building block for sustainable non-isocyanate polyhydroxyurethane thermosets and thermoplastics, Macromolecules, 2017, 50(3), 944–955 CrossRef CAS.
- K. Kamata, et al., Efficient Epoxidation of Electron-Deficient Alkenes with Hydrogen Peroxide Catalyzed by [γ-PW10O38V2 (μ-OH) 2] 3−, Chem. - Eur. J., 2011, 17(27), 7549–7559 CrossRef CAS PubMed.
- J. Li and T. Wang, Chemical equilibrium of glycerol carbonate synthesis from glycerol, J. Chem. Thermodyn., 2011, 43(5), 731–736 CrossRef CAS.
- C. Vieville, et al., Synthesis of glycerol carbonate by direct carbonatation of glycerol in supercritical CO2 in the presence of zeolites and ion exchange resins, Catal. Lett., 1998, 56(4), 245–247 CrossRef CAS.
- M. Aresta, et al., A study on the carboxylation of glycerol to glycerol carbonate with carbon dioxide: the role of the catalyst, solvent and reaction conditions, J. Mol. Catal. A: Chem., 2006, 257(1–2), 149–153 CrossRef CAS.
- J. George, et al., Methanol assisted selective formation of 1, 2-glycerol carbonate from glycerol and carbon dioxide using nBu2SnO as a catalyst, J. Mol. Catal. A: Chem., 2009, 304(1–2), 1–7 CrossRef CAS.
- J. Liu, et al., Glycerol carbonylation with CO2 to glycerol carbonate over CeO2 catalyst and the influence of CeO2 preparation methods and reaction parameters, Appl. Catal., A, 2016, 513, 9–18 CrossRef CAS.
- Q. Zhang, et al., Calcium carbide as a dehydrating agent for the synthesis of carbamates, glycerol carbonate, and cyclic carbonates from carbon dioxide, Green Chem., 2020, 22(13), 4231–4239 RSC.
- X. Su, et al., Metal-free catalytic conversion of CO 2 and glycerol to glycerol carbonate, Green Chem., 2017, 19(7), 1775–1781 RSC.
- J. Ma, et al., One-pot conversion of CO 2 and glycerol to value-added products using propylene oxide as the coupling agent, Green Chem., 2012, 14(6), 1743–1748 RSC.
- X. Song, et al., Functionalized DVB-based polymer catalysts for glycerol and CO2 catalytic conversion, J. CO2 Util., 2018, 28, 326–334 CrossRef CAS.
- Z.-H. Zhou, Q.-W. Song and L.-N. He, Silver (I)-promoted cascade reaction of propargylic alcohols, carbon dioxide, and vicinal diols: Thermodynamically favorable route to cyclic carbonates, ACS Omega, 2017, 2(1), 337–345 CrossRef CAS PubMed.
- J.-Y. Li, et al., Cascade strategy for atmospheric pressure CO2 fixation to cyclic carbonates via silver sulfadiazine and Et4NBr synergistic catalysis, ACS Sustainable Chem. Eng., 2019, 7(3), 3378–3388 CrossRef CAS.
- H. Zhou, et al., Highly regio-and stereoselective synthesis of cyclic carbonates from biomass-derived polyols via organocatalytic cascade reaction, Green Chem., 2019, 21(23), 6335–6341 RSC.
- L.-H. Han, et al., Thermodynamic favorable CO2 conversion via vicinal diols and propargylic alcohols: A metal-free catalytic method, Chin. Chem. Lett., 2020, 31(2), 341–344 CrossRef CAS.
- Y. N. Lim, C. Lee and H. Y. Jang, Metal-free synthesis of cyclic and acyclic carbonates from CO2 and alcohols, Eur. J. Org. Chem., 2014, 2014(9), 1823–1826 CrossRef CAS.
- M. Mihara, et al., Selective Synthesis of Carbonates from Glycerol, CO2, and Alkyl Halides Using tert-Butyltetramethylguanidine, Synlett, 2018, 29(13), 1759–1764 CrossRef CAS.
- D. Cespi, et al., A simplified early stage assessment of process intensification: glycidol as a value-added product from epichlorohydrin industry wastes, Green Chem., 2016, 18(16), 4559–4570 RSC.
- F. Della Monica, et al., Glycidol: an hydroxyl-containing epoxide playing the double role of substrate and catalyst for CO2 cycloaddition reactions, ChemSusChem, 2016, 9(24), 3457–3464 CrossRef CAS PubMed.
- J. Rintjema, et al., Substrate-Controlled Product Divergence: Conversion of CO2 into Heterocyclic Products, Angew. Chem., 2016, 128(12), 4040–4044 CrossRef.
- N. Zanda, et al., Organocatalytic and halide-free synthesis of glycerol carbonate under continuous flow, ACS Sustainable Chem. Eng., 2021, 9(12), 4391–4397 CrossRef CAS.
- S. M. Sadeghzadeh, A heteropolyacid-based ionic liquid immobilized onto fibrous nano-silica as an efficient catalyst for the synthesis of cyclic carbonate from carbon dioxide and epoxides, Green Chem., 2015, 17(5), 3059–3066 RSC.
- S. M. Sadeghzadeh, A heteropolyacid-based ionic liquid immobilized onto Fe3O4/SiO2/Salen/Mn as an environmentally friendly catalyst for synthesis of cyclic carbonate, Res. Chem. Intermed., 2016, 42(3), 2317–2328 CrossRef CAS.
- A. H. Jadhav, et al., Effect of anion type of imidazolium based polymer supported ionic liquids on the solvent free synthesis of cycloaddition of CO2 into epoxide, Catal. Today, 2016, 265, 56–67 CrossRef CAS.
- T. Jin, et al., Novel and effective strategy of dual bis (trifluoromethylsulfonyl) imide imidazolium ionic liquid immobilized on periodic mesoporous organosilica for greener cycloaddition of carbon dioxide to epoxides, New J. Chem., 2019, 43(6), 2583–2590 RSC.
- P. Zhang and R. Zhiani, Synthesis of ionic liquids as novel nanocatalysts for fixation of carbon dioxide with epoxides by using a carbon dioxide balloon, Catal. Lett., 2020, 150(8), 2254–2266 CrossRef CAS.
- Y. Zhang, et al., Two-in-one: Construction of hydroxyl and imidazolium-bifunctionalized ionic networks in one-pot toward synergistic catalytic CO 2 fixation, Chem. Commun., 2020, 56(22), 3309–3312 RSC.
- M. Ding and H.-L. Jiang, Incorporation of imidazolium-based poly (ionic liquid) s into a metal–organic framework for CO2 capture and conversion, ACS Catal., 2018, 8(4), 3194–3201 CrossRef CAS.
- S. Ghosh, et al., Porous iron-phosphonate nanomaterial as an efficient catalyst for the CO2 fixation at atmospheric pressure and esterification of biomass-derived levulinic acid, Catal. Today, 2018, 309, 253–262 CrossRef CAS.
- S. Ghosh, et al., Use of PS-Zn-anthra complex as an efficient heterogeneous recyclable catalyst for carbon dioxide fixation reaction at atmospheric pressure and synthesis of dicoumarols under greener pathway, J. Organomet. Chem., 2018, 866, 1–12 CrossRef CAS.
- S. Roy, et al., CO 2 fixation at atmospheric pressure: porous ZnSnO 3 nanocrystals as a highly efficient catalyst for the synthesis of cyclic carbonates, RSC Adv., 2016, 6(37), 31153–31160 RSC.
- J. Martínez, et al., One-Component Aluminum (heteroscorpionate) Catalysts for the Formation of Cyclic Carbonates from Epoxides and Carbon Dioxide, ChemSusChem, 2017, 10(6), 1175–1185 CrossRef.
- F. de la Cruz-Martinez, et al., Bifunctional aluminum catalysts for the chemical fixation of carbon dioxide into cyclic carbonates, ACS Sustainable Chem. Eng., 2018, 6(4), 5322–5332 CrossRef CAS.
- J. Martínez, et al., Influence of the counterion on the synthesis of cyclic carbonates catalyzed by bifunctional aluminum complexes, Inorg. Chem., 2019, 58(5), 3396–3408 CrossRef.
- X.-D. Lang, Y.-C. Yu and L.-N. He, Zn-salen complexes with multiple hydrogen bonding donor and protic ammonium bromide: Bifunctional catalysts for CO2 fixation with epoxides at atmospheric pressure, J. Mol. Catal. A: Chem., 2016, 420, 208–215 CrossRef CAS.
- M. North, P. Villuendas and C. Young, Inter-and intramolecular phosphonium salt cocatalysis in cyclic carbonate synthesis catalysed by a bimetallic aluminium (salen) complex, Tetrahedron Lett., 2012, 53(22), 2736–2740 CrossRef CAS.
- M. Navarro, et al., Bimetallic scorpionate-based helical organoaluminum complexes for efficient carbon dioxide fixation into a variety of cyclic carbonates, Catal. Sci. Technol., 2020, 10(10), 3265–3278 RSC.
- M. H. Anthofer, et al., Hydroxy-functionalized imidazolium bromides as catalysts for the cycloaddition of CO2 and epoxides to cyclic carbonates, ChemCatChem, 2015, 7(1), 94–98 CrossRef CAS.
- J. A. Castro-Osma, et al., Development of hydroxy-containing imidazole organocatalysts for CO 2 fixation into cyclic carbonates, Catal. Sci. Technol., 2018, 8(7), 1981–1987 RSC.
- M. Hong, et al., Scorpionate catalysts for coupling CO2 and epoxides to cyclic carbonates: A rational design approach for organocatalysts, J. Org. Chem., 2018, 83(16), 9370–9380 CrossRef CAS PubMed.
- N. Yadav, et al., Versatile functionalization of polymer nanoparticles with carbonate groups via hydroxyurethane linkages, Polym. Chem., 2019, 10(26), 3571–3584 RSC.
- Y. Y. Zhang, et al., Scalable, Durable, and Recyclable Metal-Free Catalysts for Highly Efficient Conversion of CO2 to Cyclic Carbonates, Angew. Chem., 2020, 132(51), 23491–23498 CrossRef.
- S. Sopeña, et al., Organocatalyzed domino [3+ 2] cycloaddition/payne-type rearrangement using carbon dioxide and epoxy alcohols, Angew. Chem., 2018, 130(35), 11373–11377 CrossRef.
- Y. Zhao, Highly efficient synthesis of cyclic carbonate with CO2 catalyzed by ionic liquid in a microreactor, Green Chem., 2013, 15(2), 446–452 RSC.
- I. S. Metcalfe, M. North and P. Villuendas, Influence of reactor design on cyclic carbonate synthesis catalysed by a bimetallic aluminium (salen) complex, J. CO2 Util., 2013, 2, 24–28 CrossRef CAS.
- Y. Wu, et al., Efficient fixation of CO2 into propylene carbonate with [BMIM] Br in a continuous-flow microreaction system, Green Energy Environ., 2021, 6(2), 291–297 CrossRef CAS.
- D. Lokhat, et al., Gas–liquid mass transfer in a falling film microreactor: Effect of reactor orientation on liquid-side mass transfer coefficient, Chem. Eng. Sci., 2016, 155, 38–44 CrossRef CAS.
- F. M. Akwi and P. Watts, Continuous flow chemistry: where are we now? Recent applications, challenges and limitations, Chem. Commun., 2018, 54(99), 13894–13928 RSC.
- M. B. Plutschack, et al., The hitchhiker's guide to flow chemistry∥, Chem. Rev., 2017, 117(18), 11796–11893 CrossRef CAS.
- M. Guidi, P. H. Seeberger and K. Gilmore, How to approach flow chemistry, Chem. Soc. Rev., 2020, 49(24), 8910–8932 RSC.
- M. Trojanowicz, Flow chemistry in contemporary chemical sciences: A real variety of its applications, Molecules, 2020, 25(6), 1434 CrossRef CAS PubMed.
- H. Zhang, et al., An efficient ternary catalyst ZnBr2/K2CO3/[Bmim] Br for chemical fixation of CO2 into cyclic carbonates at ambient conditions, J. CO2 Util., 2016, 14, 76–82 CrossRef CAS.
- A.-L. Girard, et al., Insights on recyclable catalytic system composed of task-specific ionic liquids for the chemical fixation of carbon dioxide, Green Chem., 2014, 16(5), 2815–2825 RSC.
- J. Sun, et al., Hydroxyl-functionalized ionic liquid: a novel efficient catalyst for chemical fixation of CO (2) to cyclic carbonate, Tetrahedron Lett., 2008, 49, 3588–3591 CrossRef CAS.
- B.-H. Xu, et al., Fixation of CO 2 into cyclic carbonates catalyzed by ionic liquids: a multi-scale approach, Green Chem., 2015, 17(1), 108–122 RSC.
- J. Wegner, S. Ceylan and A. Kirschning, Flow chemistry–a key enabling technology for (multistep) organic synthesis, Adv. Synth. Catal., 2012, 354(1), 17–57 CrossRef CAS.
- B. Gutmann, D. Cantillo and C. O. Kappe, Continuous-flow technology—a tool for the safe manufacturing of active pharmaceutical ingredients, Angew. Chem., Int. Ed., 2015, 54(23), 6688–6728 CrossRef CAS PubMed.
- M. Movsisyan, et al., Taming hazardous chemistry by continuous flow technology, Chem. Soc. Rev., 2016, 45(18), 4892–4928 RSC.
- I. Rossetti and M. Compagnoni, Chemical reaction engineering, process design and scale-up issues at the frontier of synthesis: Flow chemistry, Chem. Eng. J., 2016, 296, 56–70 CrossRef CAS.
- C. Mallia and I. Baxendale, Org. Process Res. Dev., 2016, 20(2), 327–360 CrossRef CAS.
- A. Adamu, F. Russo-Abegão and K. Boodhoo, Process intensification technologies for CO2 capture and conversion–a review, BMC Chem. Eng., 2020, 2(1), 1–18 Search PubMed.
- H. Seo, L. V. Nguyen and T. F. Jamison, Using carbon dioxide as a building block in continuous flow synthesis, Adv. Synth. Catal., 2019, 361(2), 247–264 CrossRef CAS.
- M. J. Earle, et al., The distillation and volatility of ionic liquids, Nature, 2006, 439(7078), 831–834 CrossRef CAS PubMed.
- T. Takahashi, et al., Synergistic hybrid catalyst for cyclic carbonate synthesis: Remarkable acceleration caused by immobilization of homogeneous catalyst on silica, Chem. Commun., 2006,(15), 1664–1666 RSC.
- Y. Zhang, et al., Coconut shell activated carbon tethered ionic liquids for continuous cycloaddition of CO2 to epichlorohydrin in packed bed reactor, Catal. Commun., 2015, 68, 73–76 CrossRef CAS.
- T. Wang, et al., Highly recyclable polymer supported ionic liquids as efficient heterogeneous catalysts for batch and flow conversion of CO 2 to cyclic carbonates, RSC Adv., 2017, 7(5), 2836–2841 RSC.
- D. Valverde, et al., Multifunctional polymers based on ionic liquid and rose bengal fragments for the conversion of CO2 to carbonates, ACS Sustainable Chem. Eng., 2021, 9(5), 2309–2318 CrossRef CAS.
- J. Sun, Z. Li and J. Yin, Continuous flow synthesis of propylene carbonate using DBU-based ionic liquid in a packed bed reactor, J. CO2 Util., 2021, 53, 101723 CrossRef CAS.
- T. Q. Bui, et al., Mesoporous melamine-formaldehyde resins as efficient heterogeneous catalysts for continuous synthesis of cyclic carbonates from epoxides and gaseous CO2, ACS Sustainable Chem. Eng., 2020, 8(34), 12852–12869 CrossRef CAS.
- J. Deng, et al., Microreaction technology for synthetic chemistry, Chin. J. Chem., 2019, 37(2), 161–170 CrossRef CAS.
- P. T. Baraldi and V. Hessel, Micro Reactor and Flow Chemistry for Industrial Applications in Drug Discovery and Development, 2012 Search PubMed.
- M.-R. Li, et al., Highly efficient conversion of CO2 to cyclic carbonates with a binary catalyst system in a microreactor: intensification of “electrophile–nucleophile” synergistic effect, RSC Adv., 2018, 8(68), 39182–39186 RSC.
- D. Rigo, et al., Diethylene Glycol/NaBr Catalyzed CO2 Insertion into Terminal Epoxides: From Batch to Continuous Flow, ChemCatChem, 2021, 13(8), 2005–2016 CrossRef CAS.
- A. Rehman, et al., Kinetic investigations of styrene carbonate synthesis from styrene oxide and CO2 using a continuous flow tube-in-tube gas–liquid reactor, J. CO2 Util., 2018, 24, 341–349 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2023 |