DOI:
10.1039/D3RA03828A
(Paper)
RSC Adv., 2023,
13, 29568-29583
Structure-directing role of CH⋯X (X = C, N, S, Cl) interactions in three ionic cobalt complexes: X-ray investigation and DFT study using QTAIM Vr predictor to eliminate the effect of pure Coulombic forces†
Received
8th June 2023
, Accepted 22nd September 2023
First published on 9th October 2023
Abstract
Three cobalt complexes, namely [CoIII(HL1)2(N3)2]ClO4 (1), [CoIII(L2)(HL2)(N3)]ClO4·1.5H2O (2), and [CoIII(L3)(HL3)(NCS)]2 [CoIICl2(NCS)2] (3), where HL1 = 2-(3-(dimethylamino)propyliminomethyl)-6-methoxyphenol, HL2 = 2-(2-(dimethylamino)ethyliminomethyl)-4,6-dichlorophenol, and HL3 = 2-(2-(dimethylamino)ethyliminomethyl)-6-methoxyphenol, as potential tridentate N2O-donor Schiff base ligands, were synthesized and characterized using elemental analysis, IR and UV-vis spectroscopy, and single-crystal X-ray diffraction studies. All three were found to be monomeric ionic complexes. Complex 1 crystallizes in the orthorhombic space group Pbcn, whereas both complexes 2 and 3 crystallize in triclinic space groups, P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) . Further, 1 and 2 are cationic complexes of octahedral cobalt(III) with perchlorate anions, whereas complex 3 contains a cationic part of octahedral cobalt(III) and an anionic part of tetrahedral cobalt(II). Hydrogen-bonding interactions involving aromatic and aliphatic CH bonds as H-bond donors and the pseudo-halide co-ligands as H-bond acceptors were established, which are important aspects governing the X-ray packing. These interactions were analyzed theoretically using the quantum theory of atoms in molecules (QTAIM) and non-covalent interaction plot (NCI plot) analyses. Moreover, energy decomposition analysis (EDA) was performed to analyze the stabilization of the complexes in terms of the electrostatic, dispersion, and correlation forces.
. Further, 1 and 2 are cationic complexes of octahedral cobalt(III) with perchlorate anions, whereas complex 3 contains a cationic part of octahedral cobalt(III) and an anionic part of tetrahedral cobalt(II). Hydrogen-bonding interactions involving aromatic and aliphatic CH bonds as H-bond donors and the pseudo-halide co-ligands as H-bond acceptors were established, which are important aspects governing the X-ray packing. These interactions were analyzed theoretically using the quantum theory of atoms in molecules (QTAIM) and non-covalent interaction plot (NCI plot) analyses. Moreover, energy decomposition analysis (EDA) was performed to analyze the stabilization of the complexes in terms of the electrostatic, dispersion, and correlation forces.
Introduction
Schiff bases have long been widely used as versatile ligands for the synthesis of different transition and non-transition metal complexes with potential application in bioinorganic chemistry, opto-electronics and magnetism, catalysis, separation and encapsulation, hydrometallurgy, drug designing, the transport and activation of small molecules, and others.1–11 Among them, potential tridentate N2O donor Schiff bases are very popular for their mesmerizing ability to form both mononuclear facial and meridional isomers of bis-ligand complexes as well as varieties of poly-nuclear complexes by exploiting the bridging efficiency of their phenolate oxygen atoms along with many other type of complexes.12–24 They have also been used as mono-/bi-dentate ligands for keeping other donor centers pendant.25,26 Focusing on cobalt(III) complexes of N, O donor Schiff bases, many studies have been reported in the literature regarding their bio-mimetic catalytic activity, such as phenoxazinone synthase mimicking and catechol oxidase mimicking activity.27–31 It is, however, true that cobalt has not been found in the active site structure of any metallo-oxidase yet, probably because of the inert character of cobalt(III), but this inert character of cobalt(III) attracts the interests of coordination chemists to synthesize bio-mimetic catalysts with cobalt(III), so that the intermediate species formed in the catalytic cycle could be identified easily and the mechanistic pathway of a catalytic reaction could be figured out.29 The exploration of opto-electronic properties of cobalt(III) complexes of Schiff base ligands is also a growing research topic owing to the low cost and good availability of the metal sources and the structural flexibility and stability of the ligands.32 The utilization of cobalt(III) complexes to model photosynthesis systems, where they have been used for either the oxidation of water to O2 or reduction of H+ to H2 or both, has also been reported in the literature.33–38
On the other hand, to construct an interesting supramolecular architecture by modulating various non-covalent interactions is an appealing research topic today.39–41 A large number of supramolecular systems based on transition metal complexes have been used as probes, sensors, and photonic devices in last couple of years.42–48 Several non-covalent intermolecular forces, such as hydrogen bonding, π–π stacking, cation-π, C–H⋯π interactions, halogen bonding, and ion pairing, have been used by crystal engineers to direct the pattern of numerous supramolecular assemblies.49–64 An important role is also played by these non-covalent interactions in drug–receptor interactions, crystal engineering, enzyme inhibition, protein folding, etc.65–69 The relevance of several non-covalent interactions has therefore been analyzed by means of theoretical and experimental investigations.70,71
In the present work, we synthesized three cobalt(III) complexes, namely [CoIII(HL1)2(N3)2]ClO4 (1), [CoIII(L2)(HL2)(N3)]ClO4·1.5H2O (2), and [CoIII(L3)(HL3)(NCS)]2 [CoIICl2(NCS)2] (3), with tridentate N2O donor Schiff base ligands, namely HL1 = 2-(3-(dimethylamino)propyliminomethyl)-6-methoxyphenol, HL2 = 2-(2-(dimethylamino)ethyliminomethyl)-4,6-dichlorophenol, and HL3 = 2-(2-(dimethylamino)ethyliminomethyl)-6-methoxyphenol. Both 1 and 2 are cationic complexes, whereas both cationic and anionic complexes are present in 3. The CH⋯X contacts (X = C, N, S, Cl) make a strong contribution to the total Hirshfeld surface and have a predominant role in the crystal packing of complexes 1–3 (see Scheme 1). Complexes 1 and 2 form self-assembled dimers, where the azide co-ligands interact with several aliphatic CH bonds of the Schiff base ligand. In the case of 2, additional CH⋯π interactions are also established that further stabilize the assemblies. Complex 3 forms a tetrameric assembly, where the [CoCl2(SCN)2]− anion is surrounded by three counter-cations and a network of CH⋯N,S,C interactions are established. The interaction energies were evaluated here using the quantum theory of atoms in molecules (QTAIM) and the potential energy density (Vr) predictor. This method is very useful as the interactions can be established between charged systems. Conventional procedures based on the supramolecular approach would lead to repulsive cation⋯cation interactions in 1 and 2 or to very large and attractive interactions for the anion⋯cation interactions in 3. The evaluation of the CH⋯X (X = C, N, S, Cl) contacts using the QTAIM Vr predictor is also free from the effect of pure Coulombic forces.
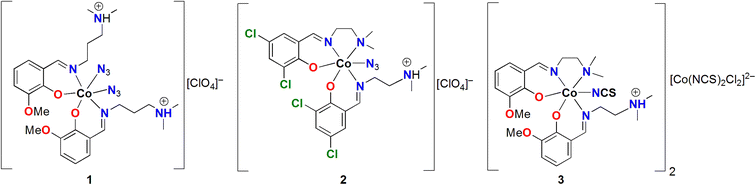 |
| | Scheme 1 Molecular diagrams complexes 1–3. | |
Experimental section
Materials
All the starting materials and solvents were commercially available, reagent grade, and used as purchased from Sigma-Aldrich without further purification.
Synthesis of the ligands
Synthesis of 2-(3-(dimethylamino)propyliminomethyl)-6-methoxyphenol (HL1). The Schiff base ligand HL1 was prepared by refluxing N,N-dimethyl-1,3-diaminopropane (∼1 mmol, 0.1 mL) with 3-methoxysalicylaldehyde (∼1 mmol, 0.160 g) in methanol (20 mL) for ca 1 h. The ligand was not isolated and the methanol solution was used directly for the preparation of complex 1.
Synthesis of 2-(2-(dimethylamino)ethyliminomethyl)-4,6-dichlorophenol (HL2). The Schiff base ligand HL2 was prepared by refluxing a methanol solution (20 mL) of N,N-dimethyl-1,2-diaminoethane (∼1 mmol, 0.1 mL) with 3,5-dichlorosalicylaldehyde (∼1 mmol, 0.190 g) in methanol (20 mL) for ca 1 h. The ligand was not isolated and the methanol solution was used directly for the preparation of complex 2.
Synthesis of 2-(2-(dimethylamino)ethyliminomethyl)-6-methoxyphenol (HL3). The Schiff base ligand HL3 was synthesized in a similar method to that of HL2, except that 3-methoxysalicylaldehyde (∼1 mmol, 0.160 g) was used instead of 3,5-dichlorosalicylaldehyde. The ligand was not isolated and was used directly for the preparation of complex 3.
Synthesis of the complexes
Synthesis of [CoIII(HL1)2(N3)2]ClO4 (1). A methanol solution (5 mL) of cobalt(II) perchlorate hexahydrate (1 mmol, 0.365 g) was added to the methanol solution of the ligand HL1, followed by the addition of a methanol solution (5 mL) of sodium azide (1 mmol, 0.065 g) with constant stirring. The stirring was continued for an additional 2 h. Dark brown diffraction quality single crystals were obtained after a few days slow evaporation of the solution in an open atmosphere.Yield: 0.50 g {70%}. Anal. Calc. for C26H40ClCoN10O8 (FW 717.08): C, 43.55; H, 5.90; N, 19.53%; found: C, 43.53; H, 5.87; N, 19.54%. IR (KBr, cm−1): 1623 (νC![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N), 2012, 2023 (νN3), 3232, 3293 (νN–H), 2968–2806 (νCH). UV-vis, λmax (nm), [εmax (L mol−1 cm−1)] (CH3CN), 246 nm [5.1 × 104], 426 nm [5.06 × 103], 708 nm [1.69 × 103]. Magnetic moment: diamagnetic. ESI-MS (positive ion mode, CH3CN) m/z: 529.06 [Co(L1)2]+. 1H NMR (DMSO-d6) (ppm) δ: 8.03 (s, 2H, benzylic CH), 6.90 (d, 4H, H atom, attached to the nitrogen of +NHMe2 group, and aromatic CH), 6.79 (dd, J = 1.5 Hz, 2H, aromatic CH), 6.43 (dd, J = 7.5 Hz, 2H, aromatic CH), 3.85–3.75 (m, 10H, –OCH3 and N–CH2), 3.18–1.24 (m, 20H, methyl protons of +NHMe2 groups and methylene protons).
N), 2012, 2023 (νN3), 3232, 3293 (νN–H), 2968–2806 (νCH). UV-vis, λmax (nm), [εmax (L mol−1 cm−1)] (CH3CN), 246 nm [5.1 × 104], 426 nm [5.06 × 103], 708 nm [1.69 × 103]. Magnetic moment: diamagnetic. ESI-MS (positive ion mode, CH3CN) m/z: 529.06 [Co(L1)2]+. 1H NMR (DMSO-d6) (ppm) δ: 8.03 (s, 2H, benzylic CH), 6.90 (d, 4H, H atom, attached to the nitrogen of +NHMe2 group, and aromatic CH), 6.79 (dd, J = 1.5 Hz, 2H, aromatic CH), 6.43 (dd, J = 7.5 Hz, 2H, aromatic CH), 3.85–3.75 (m, 10H, –OCH3 and N–CH2), 3.18–1.24 (m, 20H, methyl protons of +NHMe2 groups and methylene protons).
Synthesis of [CoIII(L2)(HL2)(N3)]ClO4·1.5H2O (2). Complex 2 was prepared in a similar method to that of complex 1 except that HL2 was used instead of HL1. Diffraction quality dark brown single crystals were obtained after a few days slow evaporation of the solution in an open atmosphere.Yield: 0.78 g {52%}. Anal. Calc. for C44H58Cl10Co2N14O15 (FW 1495.40): C, 35.34; H, 3.91; N, 13.11%; found: C, 35.33; H, 3.90; N, 13.13%. IR (KBr, cm−1): 1637 (νCN), 2022 (νN3), 3292 (νN–H), 2968–2806 (νCH). UV-vis, λmax (nm), [εmax (L mol−1 cm−1)] (CH3CN), 314 nm [1.40 × 104], 404 nm [6.7 × 103], 638 nm [1.21 × 102]. Magnetic moment: diamagnetic. ESI-MS (positive ion mode, CH3CN) m/z: 578.81 [Co(L2)2]+. 1H NMR (DMSO-d6) (ppm) δ: 8.50 (s, 2H, benzylic CH), 7.47 (s, J = 1.5 Hz, 2H, aromatic CH), 7.40 (s, 3H, aromatic CH and H atom attached to N of +NHMe2 group), 3.18 (t, J = 7.1 Hz, 6H, methylene protons), 2.91–2.05 (J = 7.1 Hz, 8H, methylene protons and methyl protons of +NHMe2 group), 1.57–1.24 (s, 6H, methyl protons of NMe2).
Synthesis of [CoIII(L3)(HL3)(NCS)]2 [CoIICl2(NCS)2] (3). A methanol solution (5 mL) of cobalt(II) chloride tetrahydrate (∼3 mmol, 0.510 g) was then added to the methanol solution of the ligand HL3, followed by the addition of a methanol solution (5 mL) of sodium thiocyanate (∼4 mmol, 0.330 g) with constant stirring. The stirring was continued for an additional 2 h. Dark brown single crystals suitable for X-ray diffraction were obtained after a few days slow evaporation of the solution in an open atmosphere.Yield: 0.75 g {55%}. Anal. Calc. for C52H70Cl2Co3N12O8S4 (FW 1365.17): C, 45.68; H, 5.16; N, 12.29%; found: C, 45.66; H, 5.14; N, 12.30%. IR (KBr, cm−1): 1647 (νCN), 2023, 2065 (νNCS), 2968–2806 (νCH). UV-vis, λmax (nm), [εmax (L mol−1 cm−1)] (CH3CN), 388 nm [1.28 × 104], 572 nm [1.29 × 103], 654 nm [1.27 × 103]. Magnetic moment: μ = 3.78 B.M. ESI-MS (positive ion mode, CH3CN) m/z: 500.99 [Co(L3)2]+, 501.99 [Co(L3)(HL3)]+.
Physical measurements
Elemental analyses (carbon, hydrogen, and nitrogen) were performed using a PerkinElmer 240C elemental analyzer. IR spectra in KBr (4500–500 cm−1) were recorded with a PerkinElmer RX-1 FTIR spectrophotometer. Electronic spectra in acetonitrile were recorded on a UV-vis spectrofluorometer, SHIMADZU (UV-1900i). The magnetic susceptibility measurements were done with an EG&PAR vibrating sample magnetometer, model 155, at room temperature, with diamagnetic corrections made using Pascal's constants. Effective magnetic moments were calculated using the formula μeff = 2.828(χMT)1/2, where χM is the corrected molar susceptibility. Electrospray ionization mass spectra were recorded on a Waters Xevo G2 Q-TOF instrument. 1H NMR spectra were recorded using a BRUKER 400 MHz NMR spectrometer in DMSO-d6 solvent, with MestReNova software used for plotting the NMR data.
X-ray crystallography
Suitable single crystals of all three complexes were used for data collection using a ‘Bruker D8 QUEST area detector’ diffractometer equipped with graphite-monochromated Mo Kα radiation (λ = 0.71073 Å) at 110 K. The molecular structures were solved by direct methods and refined by full-matrix least squares on F2, using the SHELX-18/1 package.72 Anisotropic thermal parameters were used for the refinement of the non-hydrogen atoms. Hydrogen atoms attached to oxygen atoms were located by different Fourier maps and were kept at fixed positions. All other hydrogen atoms were placed in their geometrically idealized positions and constrained to ride on their parent atoms. Multi-scan empirical absorption corrections were applied to the data using the program SADABS.73 A summary of the crystallographic data and refinement details of all three complexes are given in Table 1. Selected bond lengths are listed in Tables 2 and 3, while selected bond angles are listed in Tables 4 and 5.
Table 1 Crystal data and refinement details for complexes 1, 2, and 3
| Complex |
1 |
2 |
3 |
| Formula |
C26H42ClCoN10O8 |
C44H58Cl10Co2N14O15 |
C52H70Cl2Co3N12O8S4 |
| Formula weight |
717.08 |
1495.40 |
1367.13 |
| Temperature (K) |
273(2) |
273(2) |
273(2) |
| Crystal system |
Orthorhombic |
Triclinic |
Triclinic |
| Space group |
Pbcn |
P |
P |
| a (Å) |
12.8143(7) |
8.2780(6) |
10.3465(4) |
| b (Å) |
17.6436(10) |
13.6175(9) |
15.4956(6) |
| c (Å) |
14.6798(9) |
15.2737(10) |
20.7242(9) |
| α |
(90) |
68.354(2) |
105.535(1) |
| β |
(90) |
82.668(2) |
101.074(1) |
| γ |
(90) |
86.839(2) |
97.799(1) |
| Z |
4 |
1 |
2 |
| dcalc (g cm−3) |
1.435 |
1.564 |
1.474 |
| μ (mm−1) |
0.659 |
1.015 |
1.081 |
| F(000) |
1504 |
764 |
1418 |
| Total reflections |
106![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 239 239 |
44389 |
91950 |
| Unique reflections |
2957 |
5680 |
10987 |
| Observed data [I > 2σ(I)] |
2735 |
5065 |
9723 |
| No. of parameters |
231 |
399 |
762 |
| R(int) |
0.048 |
0.039 |
0.048 |
| R1, wR2 (all data) |
0.0752, 0.2322 |
0.0553, 0.1484 |
0.0527, 0.1143 |
| R1, wR2 [I > 2σ(I)] |
0.0716, 0.2265 |
0.0498, 0.1420 |
0.0465, 0.1099 |
Table 2 Selected bond lengths (Å) in the coordination of cobalt(III) in the cationic units of complexes 1, 2, and 3
| Complex |
1 |
2 |
3 |
| (For Cation A) |
(For Cation B) |
| Co(1)-N(1) |
— |
2.042(3) |
2.037(3) |
2.038(3) |
| Co(1)-N(2) |
1.954(4) |
1.895(3) |
1.900(3) |
1.898(2) |
| Co(1)-N(3) |
1.967(4) |
1.942(3) |
1.927(3) |
1.942(3) |
| Co(1)-N(4) |
— |
1.962(3) |
1.951(3) |
1.953(3) |
| Co(1)-O(1) |
1.899(2) |
1.888(3) |
1.924(2) |
1.908(2) |
| Co(1)-O(2) |
— |
1.910(2) |
1.882(2) |
1.881(2) |
Table 3 Selected bond lengths (Å) in the coordination sphere of cobalt(II) in the anionic unit of complex 3
| Co(2)-N(6) |
1.977(4) |
| Co(2)-N(7) |
2.034(5) |
| Co(2)-Cl(1) |
2.2845(11) |
| Co(2)-Cl(2) |
2.2600(15) |
Table 4 Selected bond angles (°) in the coordination of cobalt(III) in the cationic units of complexes 1, 2, and 3
| Complexa |
1 |
2 |
3 |
| (For cation A) |
(For cation B) |
| Symmetry transformation* = 1 − x, y, 3/2 − z. |
| N(1)-Co(1)-N(2) |
— |
85.15(13) |
85.22(12) |
86.21(12) |
| N(1)-Co(1)-N(3) |
— |
88.72(13) |
90.43(12) |
92.34(12) |
| N(1)-Co(1)-N(4) |
— |
94.76(11) |
92.77(12) |
93.64(12) |
| N(2)-Co(1)-N(2*) |
178.96(13) |
— |
— |
— |
| N(2)-Co(1)-N(3*) |
87.53(17) |
— |
— |
— |
| N(2)-Co(1)-N(3) |
93.21(17) |
89.90(12) |
86.20(13) |
84.40(12) |
| N(2)-Co(1)-N(4) |
— |
177.86(12) |
177.19(11) |
178.00(11) |
| N(3)-Co(1)-N(3*) |
90.46(17) |
— |
— |
— |
| N(3)-Co(1)-N(4) |
— |
92.24(12) |
95.80(12) |
97.60(12) |
| O(1)-Co(1)-O(1*) |
90.05(10) |
— |
— |
— |
| O(1)-Co(1)-O(2) |
— |
89.37(11) |
88.90(9) |
90.91(9) |
| O(1)-Co(1)-N(1) |
— |
178.27(11) |
178.23(11) |
177.77(11) |
| O(1)-Co(1)-N(2) |
92.25(12) |
94.91(12) |
95.53(10) |
95.94(10) |
| O(1)-Co(1)-N(2*) |
87.01(12) |
— |
— |
— |
| O(1)-Co(1)-N(3) |
174.54(16) |
89.56(12) |
88.03(11) |
88.48(11) |
| O(1)-Co(1)-N(3*) |
90.01(14) |
— |
— |
— |
| O(1)-Co(1)-N(4) |
— |
85.25(11) |
86.53(10) |
84.20(10) |
| O(2)-Co(1)-N(1) |
— |
92.35(11) |
92.78(11) |
88.74(11) |
| O(2)-Co(1)-N(2) |
— |
84.03(12) |
83.44(11) |
83.37(10) |
| O(2)-Co(1)-N(3) |
— |
173.73(12) |
168.85(11) |
167.62(11) |
| O(2)-Co(1)-N(4) |
— |
93.83(11) |
94.71(10) |
94.64(10) |
Table 5 Selected bond angles (°) in the coordination sphere of cobalt(II) in the anionic unit of complex 3
| N(6)-Co(2)-N(7) |
104.62(18) |
| Cl(1)-Co(2)-Cl(2) |
110.84(5) |
| Cl(1)-Co(2)-N(6) |
109.16(12) |
| Cl(1)-Co(2)-N(7) |
107.90(12) |
| Cl(2)-Co(2)-N(6) |
111.21(14) |
| Cl(2)-Co(2)-N(7) |
112.85(14) |
Hirshfeld surface analysis
Hirshfeld surfaces74–76 and the associated two-dimensional (2D) fingerprint77–79 plots were calculated using Crystal Explorer,80 with bond lengths to hydrogen atoms set to the standard values.81 The Hirshfeld surface is unique for a fixed CIF.82
Theoretical methods
The calculations reported in this manuscript were performed using the Turbomole 7.2 program83 and the BP86-D3/def2-TZVP level of theory.84,85 The X-ray geometries were used instead of fully optimized geometries because we wanted to evaluate the CH⋯C,N,S,Cl as they stand in the solid state. The NCI plot86 reduced density gradient (RGD) isosurfaces were used to characterize the non-covalent interactions since combining both methods can reveal more about the non-covalent interactions in real space. The cubes needed to generate the NCI plot surfaces were computed at the same level of theory using the wave functions generated by means of the Turbomole 7.2 program. The NCI plot index isosurfaces corresponded to both favorable and unfavorable interactions, as differentiated by the sign of the second density Hessian eigenvalue and defined by the isosurface color. The QTAIM analysis87 and the cube files from the wave functions were computed at the same level of theory by means of the MULTIWFN program88 and represented using VMD software.89
Results and discussion
Synthesis
3-Methoxysalicylaldehyde was refluxed in methanol with N,N-dimethyl-1,3-diaminopropane to form compartmental Schiff base ligand, HL1, following the literature method.90–92 This Schiff base upon reaction with cobalt(II) perchlorate hexahydrate and sodium azide produced the mononuclear complex, 1. The formation process of this complex is shown in Scheme 2.
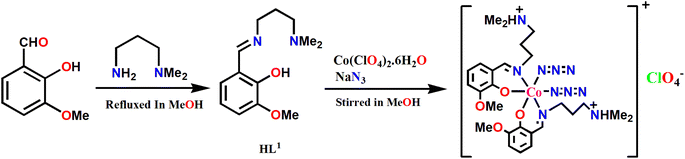 |
| | Scheme 2 Synthetic route to complex 1. | |
On the other hand, 3,5-dichlorosalicylaldehyde and 3-methoxysalicylaldehyde were refluxed in methanol with N,N-dimethyl-1,2-diaminoethane to form the compartmental Schiff base ligands HL2 and HL3 respectively following the literature method. HL2 upon reaction with cobalt(II) perchlorate hexahydrate and azide gave mononuclear complex 2. Similarly, HL3 upon reaction with cobalt(II) chloride tetrahydrate and sodium thiocyanate gave mononuclear complex 3. Here, cobalt(II) was first oxidized to cobalt(III) by areal oxygen in the presence of the Schiff base ligand producing a strong crystal field, as has also been observed in similar complexes.27,93,94 The formation of the complexes is shown in Scheme 3.
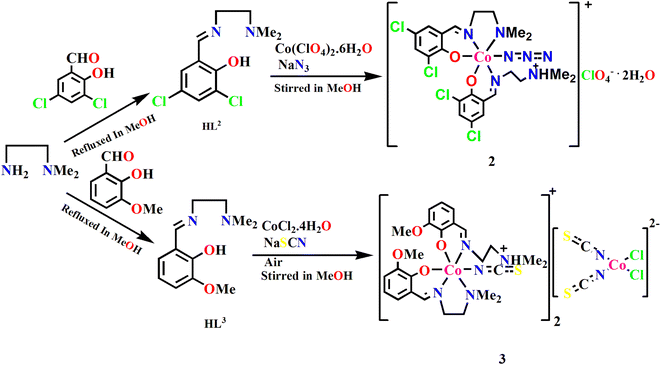 |
| | Scheme 3 Synthetic routes to complexes 2 and 3. | |
Description of the structures
[CoIII(HL1)2(N3)2]ClO4 (1). Complex 1 crystallizes in the orthorhombic space group, Pbcn. A perspective view of complex 1 with the selective atom numbering scheme is shown in Fig. 1. The structure of the complex consists of a hexa-coordinated cobalt(III) in a distorted octahedral geometry. A non-coordinated perchlorate is also present. The Schiff base ligand (HL1) is trapped in its zwitterionic form. The cobalt(III) center is coordinated by two imine nitrogen atoms, N(2) and N(2)*, and two phenolate oxygen atoms, O(1) and O(1)*, of the Schiff base (where, the symmetry transformation* = 1−x, y, 3/2−z). The fifth and sixth coordination sites of cobalt(III) are occupied by two nitrogen atom, N(3) and N(3)*, of two terminal azides to complete its octahedral geometry. The azides occupy cis positions. Each azide is quasi-linear with the N–N–N angle being 170.1(6)°, as expected.27,95 The bond lengths and bond angles of the complex are comparable with previously reported octahedral cobalt(III) Schiff base complexes.92,95
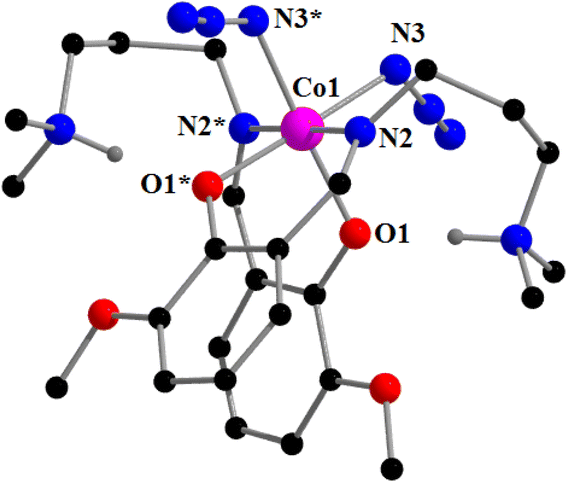 |
| | Fig. 1 Perspective view of complex 1 with the selective atom numbering scheme. Perchlorate anion is not shown for clarity. Only two hydrogen atoms (attached with two amine nitrogen atoms) are shown. Symmetry transformation* = 1 − x, y, 3/2 − z. | |
The hydrogen atoms, H(3B), attached to the carbon atom C(3), and H(4A), attached to the carbon atom, C(4), of one molecule are involved in intermolecular hydrogen bonding interactions, with the azide nitrogen atoms N(4A)a and N(5A)a, respectively, {symmetry transformation, a = 1 − x, 1 − y, 1 − z } of a neighboring molecule, thus forming a self-assembled dimer (Fig. 2). The details of these interactions are listed in Table 6.
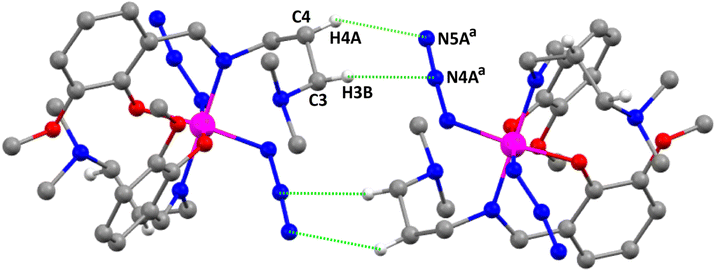 |
| | Fig. 2 Perspective view of the hydrogen bonding interactions in complex 1. Only the relevant hydrogen atoms are shown for clarity. Symmetry transformation, a = 1 − x, 1 − y, 1 − z. | |
Table 6 List of non-covalent interactions studied in the complexesa
| Complex |
Atoms involved (D–H⋯A) |
Interaction |
Distance D–H (Å) |
Distance H⋯A (Å) |
Distance D⋯A (Å) |
Angle D–H⋯A (°) |
| Symmetry transformation, a = 1 − x, 1 − y, 1 − z; b = 1 − x, 1 − y, −z and c = 1 + x, y, z. Cg(1)b is the centroid of the aromatic ring, R(1)b [C(6)b-C(7)b-C(8)b-C(9)b-C(10)b-C(11)b]. |
| 1 |
C(3)-H(3B)⋯N(4A)a |
CH⋯N |
0.971(4) |
3.081(5) |
3.568(7) |
145.7(3) |
| C(4)-H(4A)⋯N(5A)a |
CH⋯N |
0.969(5) |
2.693(7) |
3.537(8) |
112.6(3) |
| 2 |
C(12)-H(12C)⋯N(7)b |
CH⋯N |
0.960(5) |
2.587(4) |
3.489(6) |
156.7(3) |
| C(13)-H(13B)⋯Cg(1)b |
CH⋯π |
0.960(7) |
3.321 |
3.694 |
105.44 |
| 3 |
C(2)-H(2A2)⋯S(3) |
CH⋯S |
0.960(4) |
2.956(1) |
3.602(4) |
125.7(3) |
| C(2)-H(2A3)c⋯S(2) |
CH⋯S |
0.960(3) |
2.972(1) |
3.915(3) |
167.6(3) |
| C(3)-H(3A2)c⋯Cl(1) |
CH⋯Cl |
0.970(5) |
2.871(1) |
3.742(5) |
149.8(2) |
| C(15)-H(15A)⋯Cl(1) |
CH⋯Cl |
0.970(5) |
2.737(1) |
3.649(5) |
156.8(3) |
| C(17)-H(17A)⋯Cl(1) |
CH⋯Cl |
0.930(3) |
2.867(1) |
3.734(3) |
155.7(2) |
| C(13)-H(13D)⋯S(3) |
CH⋯S |
0.960(1) |
3.032(2) |
3.93(1) |
157.4(6) |
| C(14)-H(14F)⋯S(2) |
CH⋯S |
0.960(1) |
2.888(1) |
3.65(1) |
136.8(7) |
[CoIII(L2)(HL2)(N3)]ClO4·1.5H2O (2). Complex 2 crystallizes in the triclinic space group, P. A perspective view of complex 2 with the selective atom numbering scheme is shown in Fig. 3. The structure of the complex consists of a hexa-coordinated cobalt(III) in a distorted octahedral geometry. One molecule of the Schiff base ligand (HL1) is trapped in its zwitterionic form, while the other is trapped in its anionic form. The cobalt(III) center in this complex is meridionally coordinated by one amine nitrogen atom, N(1), one imine nitrogen atom, N(2), and one phenolate oxygen atom, O(1) of a deprotonated Schiff base, and one imine nitrogen atom, N(4), and one phenolate oxygen atom, O(2), of the zwitterionic Schiff base. An azide nitrogen atom, N(3), occupies the sixth coordination site of cobalt(III) to complete its distorted octahedral geometry. The cobalt(III)–Nimine distance {1.895(3) Å} is shorter than the cobalt(III)–Namine distance {2.043(3) Å}, as has also been observed in similar systems90,92,96,97 The terminal azide is quasi-linear with the N–N–N angle being 175.9 (4)°, as expected.27,95 The saturated five membered chelate ring, Co(1)-N(1)-C(3)-C(4)-N(2), represents a half chair conformation (Fig. 4) with puckering parameters98 Q = 0.407(4) Å, φ(2) = 61.4(5)°.
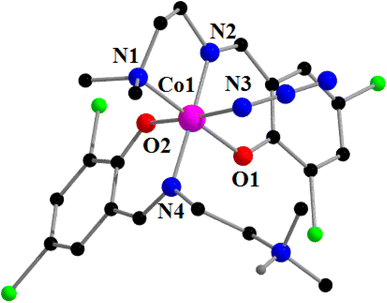 |
| | Fig. 3 Perspective view of complex 2 with the selective atom numbering scheme. Perchlorate anion and lattice water molecules are not shown for clarity. Only one hydrogen atom (attached with the amine nitrogen atom) is shown. | |
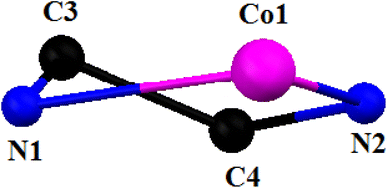 |
| | Fig. 4 Half chair conformation of complex 2 with the selective atom numbering scheme. | |
The hydrogen atom, H(12C), attached with the carbon atom, C(12), is involved in intermolecular CH⋯N with the azide nitrogen atom, N(7)b, of a symmetry related {symmetry transformation, b = 1 − x, 1 − y, −z} neighboring molecule, also forming a self-assembled dimer. This dimer is further stabilized by CH⋯π interactions between the hydrogen atom, H(13A), attached to the carbon atom, C(13), and the centroid, Cg(1)b, of the aromatic ring, R(1)b [C(6)b-C(7)b-C(8)b-C(9)b-C(10)b-C(11)b]. The dimer is shown in Fig. 5. The relevant CH⋯N and CH⋯π interactions observed in complex 2 are listed in Table 6.
 |
| | Fig. 5 Perspective view of the hydrogen bonding interactions in complex 2. Only the relevant hydrogen atoms are shown for clarity. Symmetry transformation, b (b = 1 − x, 1 − y, −z). | |
[CoIII(L3)(HL3)(NCS)]2 [CoIICl2(NCS)2] (3). Complex 3 crystallizes in the triclinic space group, P. The asymmetric unit contains two crystallographically independent cationic units with the identical chemical formula [CoIII(L3)(HL3)(NCS)]+ (may be designated as unit A and unit B) and one anionic unit, [CoIICl2(NCS)2]. A perspective view of one cationic unit (unit A) with the selective atom numbering scheme is shown in Fig. 5. The other cationic unit (unit B) has a more or less similar structure as that of unit A (Fig. S1 in the ESI†). A perspective view of the anionic unit of complex 3 is shown in Fig. 6. The structures of both the cationic and anionic units are described below.
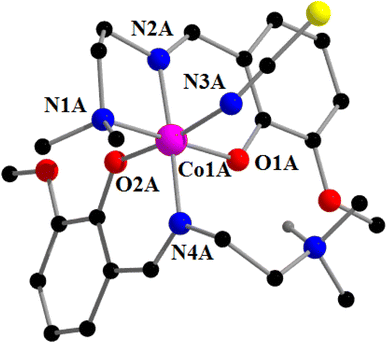 |
| | Fig. 6 Perspective view of the cationic part (unit A) of complex 3 with the selective atom numbering scheme. Only one hydrogen atom (attached with the amine nitrogen atom) is shown. | |
The cobalt(III) centers are more or less octahedral in both cationic units (unit A or unit B), being bonded by one amine nitrogen atom, N(1), one imine nitrogen atom, N(2), and one phenolate oxygen atom, O(1), of the deprotonated Schiff base in a meridional fashion, one imine nitrogen atom, N(4), and one phenolate oxygen atom, O(2), of the zwitterionic Schiff base, and one thiocyanate nitrogen atom, N(3). The cobalt(III)–Nimine distances are shorter than the cobalt(III)–Namine distances in both units, as has also been observed in similar systems.96,97 In each cationic unit, the thiocyanate is almost linear, as evident from the N–C–S angle {179.1(3)° in unit A and 178.7(4)° in unit B}, as has also been observed in related systems.27,99 The saturated five membered chelate rings, Co(1)-N(1)-C(3)-C(4)-N(2), represent envelope conformations with the puckering parameters98 Q = 0.427(4) Å, φ(2) = 79.0(4)° for unit A and Q = 0.427(4) Å, φ(2) = 254.3(4)° for unit B. Fig. 7 shows the envelope configuration of the ring in unit A.
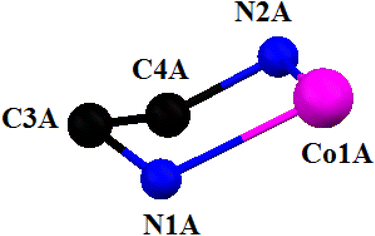 |
| | Fig. 7 Envelope conformation of the cationic part (unit A) of complex 3 with the selective atom numbering scheme. | |
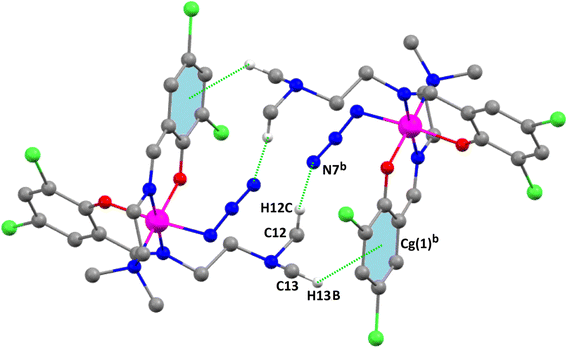
The anionic part of the complex consists of a tetra-coordinated distorted tetrahedral cobalt(II), being coordinated by two thiocyanate nitrogen atoms, N(6) and N(7), and two chloride ions, Cl(1) and Cl(2) (Fig. 8). The thiocyanates are quasi-linear with the N–C–S angles being 178.0 (4) ° and 177.6 (6)°, as expected.100
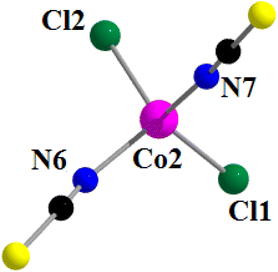 |
| | Fig. 8 Perspective view of the anionic part of complex 3 with the selective atom numbering scheme. | |
The thiocyanate sulfur atom, S(3), of the anionic unit forms bifurcated intermolecular hydrogen bonds with the hydrogen atoms H(2A2) {attached to carbon atom C(2)} of the cationic unit A and H(13D) {attached to carbon atom C(13)} of the cationic unit B. Another thiocyanate sulfur atom, S(2), of the anionic [CoIICl2(NCS)2]2− unit is also involved in intermolecular bifurcated hydrogen bonds with the hydrogen atoms, H(2A3)c {attached to carbon atom C(2)c} {Symmetry transformation, c = 1 + x, y, z}, of a neighboring cationic unit A and H(14F) {attached to carbon atom C(14)} of the cationic unit B, forming a 3D tetrameric assembly. The chlorine atom, Cl(1), of the anionic unit is involved in trifurcated hydrogen bonding with the hydrogen atoms H(15A), attached to C(15), H(17A), attached to C(17), and H(3A2)c, attached to C(3)c to further stabilize the tetrameric H-bonded assembly {Symmetry transformation, c = 1 + x, y, z} of the two cationic subunits A, as shown in Fig. 9.
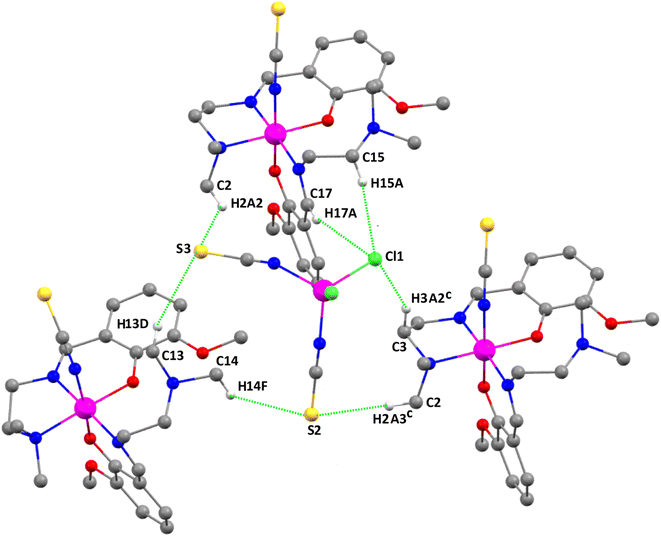 |
| | Fig. 9 Perspective view of the tetrameric assembly of complex 3. Only the relevant hydrogen atoms are shown for clarity. Symmetry transformation, c (c = 1 + x, y, z). | |
Hirshfeld surface analysis
The Hirshfeld surfaces of all three complexes were mapped over dnorm (range ∼0.1 Å to 1.5 Å), shape index, and curvedness (Fig. S6, ESI†). Red spots on the Hirshfeld surfaces denote the dominant interactions. The intermolecular interactions appear as distinct spikes in the 2D fingerprint plot showing the different spikes with their corresponding interactions. The dominant interactions in complex 1 corresponded to C⋯H/H⋯C (9.6%), O⋯H/H⋯O (21.1%), and N⋯H/H⋯N (24.4%) contacts. The proportion of C⋯H/H⋯C, O⋯H/H⋯O, N⋯H/H⋯N, and Cl⋯H/H⋯Cl interactions in complex 2 comprised 15.4%, 17.1%, 8.2%, and 31.7% of the Hirshfeld surface, respectively. Again the interactions in complex 3 comprised 16.2%, 10.2%, 17.2%, 6.6%, and 2.7% of the Hirshfeld surface as shown by the proportion of C⋯H/H⋯C, Cl⋯H/H⋯Cl, S⋯H/H⋯S, O⋯H/H⋯O, and N⋯H/H⋯N respectively. The 2D fingerprint plots of complexes 1, 2, and 3 are shown in Fig. 10–12, respectively.
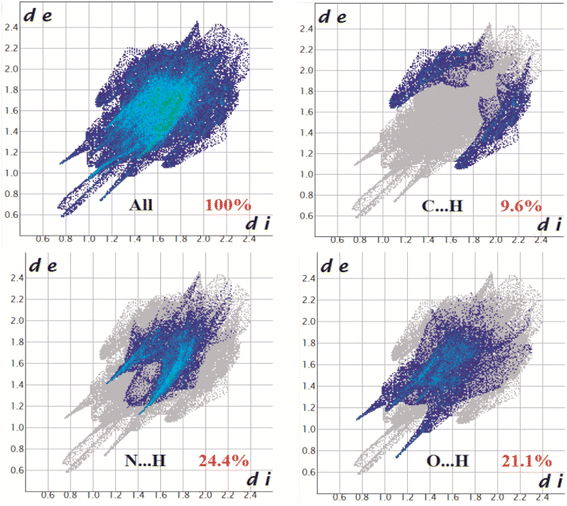 |
| | Fig. 10 Fingerprint plot of complex 1: Full and resolved into C⋯H/H⋯C, N⋯H/H⋯N, and O⋯H/H⋯O contacts contributing to the total Hirshfeld surface area. | |
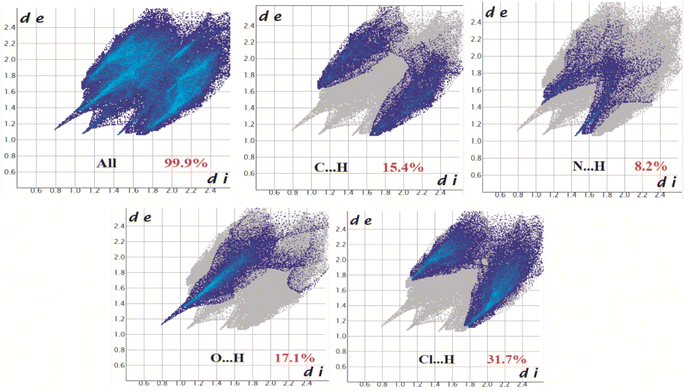 |
| | Fig. 11 Fingerprint plot of complex 2: Full and resolved into C⋯H/H⋯C, N⋯H/H⋯N, O⋯H/H⋯O, and Cl⋯H/H⋯Cl contacts contributing to the total Hirshfeld surface area. | |
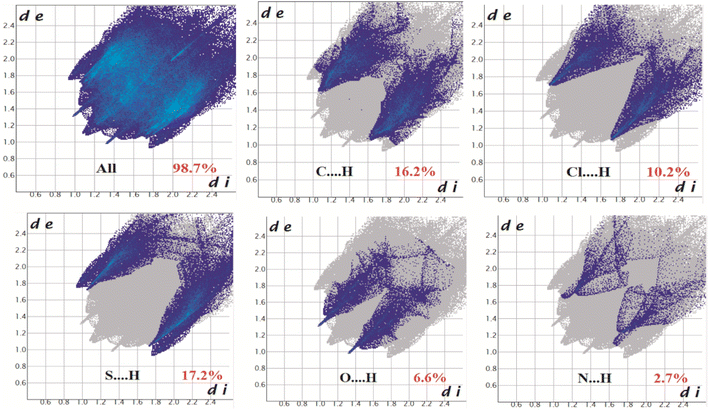 |
| | Fig. 12 Fingerprint plot of complex 3: Full and resolved into C⋯H/H⋯C, Cl⋯H/H⋯Cl S⋯H/H⋯S, O⋯H/H⋯O, and N⋯H/H⋯N contacts contributing to the total Hirshfeld surface area. | |
Theoretical study on the supramolecular interaction
As commented in the previous section, the CH⋯X contacts (X = C, N, S, Cl) make a strong contribution to the total Hirshfeld surface and have a predominant role in the X-ray packing of complexes 1–3 (see Fig. 2, 5 and 9). In the case of complex 2, additional CH⋯π interactions are established that further stabilize the assemblies. The CH⋯N distances range from 2.55 to 3.06 Å.
Fig. 9 shows a tetrameric assembly of complex 3, where the [CoCl2(SCN)2]− anion is surrounded by three counter-cations, where a network of CH⋯N,S,C interactions are established with distances ranging from 2.74 to 3.03 Å. These long distances (also observed in the self-assembled dimers of 1 and 2), along with the modest ability of C–H groups as H-bond donors, anticipate that each individual contact is weak. However, an additive number of contacts can lead to a significant stabilization of the system. This is the main purpose of the present theoretical study. The interaction energies were evaluated using the quantum theory of atoms in molecules and the potential energy density predictor. This method is very useful in systems like these reported herein where the interactions are established between charged systems. Conventional procedures based on the supramolecular approach would lead to repulsive cation⋯cation interactions in complexes 1 and 2 or to very large and attractive interactions for the anion⋯cation interactions in complex 3. The evaluation of the CH⋯X (X = C, N, S, Cl) contacts using the QTAIM Vr predictor is free from the effect of pure Coulombic forces.
Fig. 13 shows the combined QTAIM/NCI plot characterization of the self-assembled dimers of complexes 1 and 2. Each H-bond is characterized by a bond critical point (CP, red sphere) and bond path (orange line) connecting the H atom to the N atom. Moreover, a green RDG isosurface also appears upon dimerization, coincident with the location of the bond CPs. The green color of the isosurface reveals the CH⋯N contacts are weakly attractive. In the case of the dimer of complex 2, the QTAIM/NCI plot analysis also confirmed the existence of the two symmetrically equivalent CH⋯π interactions, characterized by a bond CP, bond path, and extended green RDG isosurface that connects the CH bond to one C-atom of the aromatic ring. The interaction energy of the four H-bonds corresponding to the dimer of complex 1 is −2.96 kcal mol−1, thus confirming the weak nature of each individual contact. The H-bond contribution in the dimer of complex 2 is similar (−3.51 kcal mol−1) to the dimerization energy of complex 1. The dimer is further reinforced with the CH⋯π interactions, which are slightly weaker (−2.13 kcal mol−1) than the H-bonds.
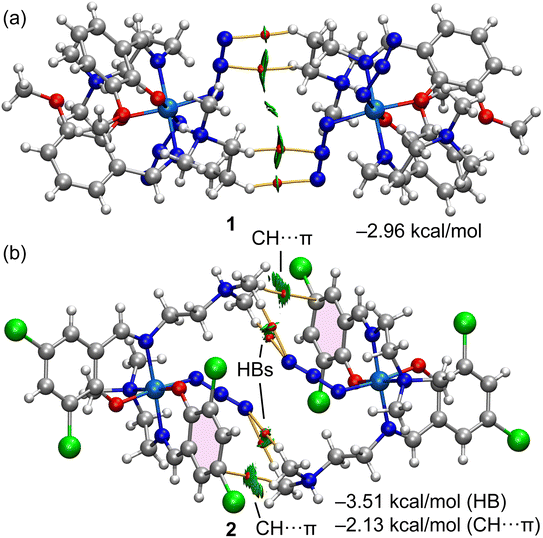 |
| | Fig. 13 QTAIM (bond CPs in red and bond path as orange lines) and NCI plot (RDG = 0.5, ρcut-off = 0.04 a.u., color scale −0.035 a.u. ≤ (signλ2)ρ ≤ 0.035 a.u.) for the self-assembled dimers of complexes 1 (a) and 2 (b). The interaction energies calculated using the Vr energy predictor are indicated in the figure. Only intermolecular interactions are represented. | |
Finally, Fig. 14 shows the combined QTAIM/NCI plot analysis of three heterodimers (ion pairs) extracted from the tetrameric assembly represented in Fig. 9. In the case of the dimer denoted as “A”, the cationic part interacts with both SCN arms of the anion. The QTAIM/NCI plot analysis revealed a total of five CH⋯S contacts with an interaction energy of −3.29 kcal mol−1, in line with the H-bonding energies of the self-assembled dimers. In the dimer denoted as “B” (Fig. 14b), the cationic part interacts with one Cl and one SCN ligand of the anionic counterpart. A total of three CH⋯Cl, two CH⋯N, and two CH⋯S contacts were revealed by the QTAIM/NCI plot analysis. The total interaction energy for this binding mode is moderately strong (−6.19 kcal mol−1) due to this intricate network of H-bonds, confirming its importance in the solid-state architecture of complex 3. The third dimer analyzed, denoted as “C” in Fig. 14c, presented a total of five contacts with a total interaction energy of −3.53 kcal mol−1, similar to heterodimer “A” and those of complexes 1 and 2.
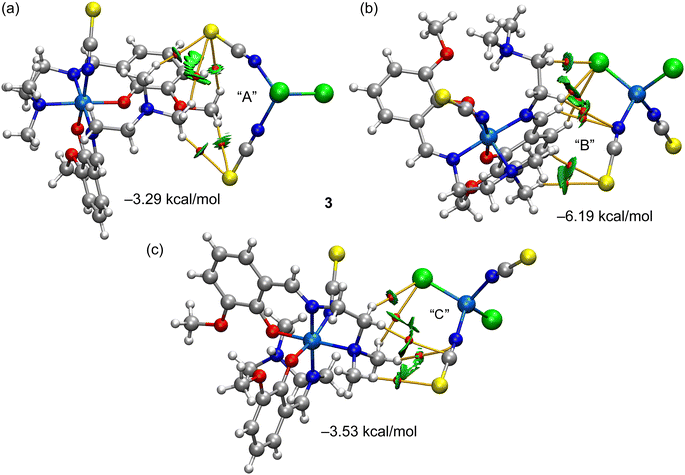 |
| | Fig. 14 (a–c) QTAIM (bond CPs in red and bond path as orange lines) and NCI plot (RDG = 0.5, ρcut-off = 0.04 a.u., color scale −0.035 a.u. ≤ (signλ2)ρ ≤ 0.035 a.u.) for the three dimers of complex 3. The interaction energies calculated using the Vr energy predictor are indicated in the figure. Only intermolecular interactions are represented. | |
The significant contribution of the CH⋯X (X = S, Cl, N) in complexes 1–3 agreed well with the Hirshfeld surface analysis since these type of contacts comprise most of the HS surface.
As previously indicated, the significance of CH⋯X contacts in the solid-state structures of complexes 1–3 was established without considering potent electrostatic influences. In order to comprehensively account for the role of Coulombic forces in stabilizing these complexes, we employed energy decomposition analysis for complexes 1 and 2 utilizing the Kitaura–Morokuma methodology.101
We assessed the relative contributions of the electrostatic, dispersion, orbital, and correlation terms to the stability of complexes 1 and 2. Our findings, as illustrated in Fig. 15, underscore the paramount importance of the electrostatic term (depicted by blue bars), accounting for substantial stabilization energies of −44.70 kcal mol−1 and −45.53 kcal mol−1 for complexes 1 and 2, respectively. Minor disparities are apparent in the exchange repulsion (depicted in red bars), dispersion (illustrated in green bars), and correlation (represented by violet bars) components, exhibiting resemblances across both complexes. Notably, a pronounced discrepancy of the surfaces in the orbital contribution was noted, significantly more pronounced in complex 1 than in complex 2. Consequently, this disparity yielded a notably higher total stabilization energy (portrayed in pink bars) in complex 1. This variance likely stems from the closer proximity of the perchlorate anion to the Schiff base cation in complex 1, culminating in shorter CH⋯O interactions (with the shortest CH⋯O distance measuring 2.24 Å in complex 1, in contrast to 2.46 Å in complex 2). It is well-established that orbital effects are acutely sensitive to distance, with larger distances resulting in diminished orbital overlap. This interpretation finds further support in the Hirshfeld surface analysis, where the CH⋯O contribution was registered at a higher 21.1% in complex 1 compared to 17.1% in complex 2.
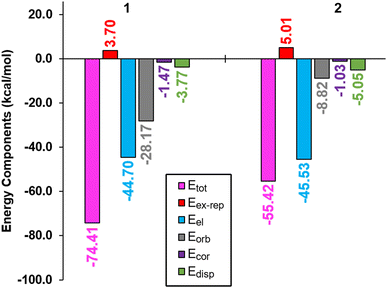 |
| | Fig. 15 Representation of the total (Etot, pink), exchange repulsion (Eex-rep, red), electrostatic (Eel, blue), orbital (Eorb, gray), correlation (Ecor, violet), and dispersion (Edisp, green) terms for the ion-pair complexes 1 (left) and 2 (right). | |
IR, electronic spectroscopy, ESI-MS, and 1H-NMR studies
In the IR spectra of all three complexes, distinct bands due to azomethine (CN) stretching vibrations appeared in the region of 1623–1647 cm−1.102–105 The sharp absorption bands located at 2015 and 2023 cm−1 (in 1) and 2022 cm−1 (in 2) indicated the presence of terminal azide (for 1 and 2), while those at 2023 and 2065 cm−1 (in 3) indicated the presence of N-bonded thiocyanate (for 3),106,107 respectively, which are also evident from crystal structure determination. The sharp bands at 3293 and 3232 cm−1 indicated N–H stretching vibrations in the IR spectrum of complex 1 and the sharp bands at 3292 cm−1 also indicated N–H stretching vibrations in the IR spectrum of complex 2 as well.108–110 The characteristic absorption band for non-coordinated perchlorate anion appeared at 1076 cm−1 in the IR spectra of complexes 1 and 2. The bands in the range of 3006–2806 cm−1 were due to alkyl C–H bond stretching vibrations, as customarily noticed in the IR spectra of all three complexes.94,111 The IR spectra of all the complexes are given in Fig. S2–S4 (ESI).† These assignments were in good agreement with the theoretical calculations, as detailed in Table 7.
Table 7 Theoretical in parenthesis and experimental values (in cm−1) for representative IR bands for complexes 1–3
| Complexes |
(CN) Schiff base |
N3/SCN |
NH stretching |
| 1 |
1625 (1619) |
2023 (2018) |
3293 (3281) and 3232 (3225) |
| 2 |
1641 (1638) |
2022 (2016) |
3292 (3285) |
| 3 |
1648 (1637) |
2023 (2025) & 2065 (2070) |
— |
Electronic spectra of the complexes were recorded in acetonitrile medium at room temperature in the range of 200–800 nm. In the high-energy region, complex 1 showed intense absorption bands around 243–275 nm, corresponding to π–π* transitions.28,112,113 Broad absorption bands at 314 nm (for 2) and 388 nm (for 3) were also observed, which were consistent with the LMCT band.114–116 The electronic absorption spectra of all three complexes in acetonitrile medium showed d–d transition bands around 426 nm (for 1), 404 nm (for 2), and 654 nm (for 3), as expected for a low spin cobalt(III) complex in an octahedral geometry.117,118 The weak band around 708 nm (for 1), 638 nm (for 2), and 572 nm (for 3) may be assigned as one of the two expected transitions for any low spin cobalt(III) complex in an octahedral geometry (1A1g → 1T1g and 1A1g → 1T2g). The UV spectrum of complex 3 is given in Fig. S5 (ESI).†
The molecular ion peaks in acetonitrile solution appeared at m/z = 529.0591 for complex 1 (theoretical m/z = 529.55 for [Co(L1)2]+), 578.81 for complex 2 (theoretical m/z = 579.21 for [Co(L2)2]+), and 500.99 (theoretical m/z = 501.49 for [Co(L3)2]+) and 501.99 ([Co(L3)(HL3)]+, theoretical m/z = 502.49) for complex 3. The mass spectra of the complexes are shown in Fig. S7–S9 (ESI).†
In complex 1, three signals for the aromatic protons of the 3-methoxysalicylaldehyde moiety were observed as a doublet at 6.90 ppm (J = 7.5 Hz), a doublet of doublets at 6.79 ppm (J = 1.5 Hz), and another doublet of doublets at 6.43 ppm (J = 7.5 Hz). The singlet at 8.03 ppm corresponded to the signals of benzylic protons (Ph–CHN). The peaks at the range of 3.85–3.75 ppm may be assigned to the protons of the methoxy group of 3-methoxysalicylaldehyde and methylene protons attached to imine nitrogen atoms of the Schiff base units. The peaks at the range of 3.18–1.24 ppm may be assigned to the hydrogen atoms of the methylene protons of 3-amino-N,N-dimethylpropan-1-aminium units and methyl protons of +NHMe2 groups. In this complex, the methyl protons of +NHMe2 groups are not in the same environment due to an anisotropy effect of the benzene rings. The 1H NMR spectrum of complex 1 is shown in Fig. S10.†
In complex 2, two signals for the aromatic protons of the 3,5-dichlorosalicylaldehyde moiety were observed as singlets at 7.47 ppm (J = 1.5 Hz) and at 7.40 ppm (J = 1.5 Hz). The singlet at 8.50 ppm corresponded to the signals of benzylic protons (Ph–CHN). Again, the peak at 7.40 ppm corresponded to the signals of protons attached to the N of the +NHMe2 group. The methylene protons of the 3-amino-N,N-dimethylpropan-1-aminium unit were observed at 3.18 ppm (J = 7.1 Hz). The methylene protons of N,N-dimethyl-1,2-diaminoethane and methyl protons of the +NHMe2 group were observed in the ranges of 2.91–2.05 ppm (J = 7.1 Hz). The other methyl protons of the NMe2 group were observed in the ranges of 1.57–1.24 ppm (J = 7.1 Hz). The 1H NMR spectrum of complex 2 is shown in Fig. S11.†
Concluding remarks
In this manuscript, we report the synthesis and X-ray characterization of three new cobalt complexes, with tridentate N2O-donor Schiff base ligands. Complex 1 contains a distorted octahedral cobalt(III), being bonded to two imine nitrogen atoms and two phenolate oxygen atoms of the zwitterionic forms of two Schiff base moieties and two nitrogen atoms of two terminal azides, along with a non-coordinate perchlorate. The structure of complex 2 consists of a distorted octahedral cobalt(III), being bonded to one amine nitrogen atom, one imine nitrogen atom, and one phenolate oxygen atom of a deprotonated Schiff base in a meridional fashion, one imine nitrogen atom and one phenolate oxygen atom of the zwitterionic Schiff base, and an azide nitrogen atom, along with a non-coordinate perchlorate and lattice water molecules. The asymmetric unit of complex 3 contains two crystallographically independent cationic units containing cobalt(III) and one anionic unit containing cobalt(II). The cobalt(III) centers are more or less octahedral in both cationic units, being bonded by one amine nitrogen atom, one imine nitrogen atom, and one phenolate oxygen atom of the deprotonated Schiff base in a meridional fashion, one imine nitrogen atom and one phenolate oxygen atom of the zwitterionic Schiff base, and one thiocyanate nitrogen atom. The anionic part of the complex consists of a tetra-coordinated distorted tetrahedral cobalt(II), being coordinated by two thiocyanate nitrogen atoms and two chloride ions. Examination of the X-ray packing of complexes 1–3 and their HS analyses revealed the structure-directing role of the CH⋯X (X = N, S, Cl) interactions. These were investigated using QTAIM and NCI plot analysis. Their combination is useful to characterize NCIs in real space. Although the individual interaction energies associated with these contacts are small, they are additive and can lead to a moderately strong stabilization of supramolecular synthons.
Finally, the advancements in structural diversity, intermolecular interaction studies, and the potential for tailored materials design showcase the significance of these complexes in advancing both fundamental knowledge and practical applications within the realm of coordination chemistry and beyond. Their flexible nature and unique features offer promising directions for future research and development, like catalysis, molecular design, and crystal engineering.
Conflicts of interest
The authors declare no conflict of interest.
Acknowledgements
Susovan Bera and Sudip Bhunia thank UGC, India for providing Junior Research Fellowship (JRF). A. F. is grateful to the Alexander von Humboldt Foundation for the J. C. Mutis Award. A. F. and R. G. M. acknowledge the financial support by the MICIU/AEI of Spain (project PID2020-115637GB-I00, FEDER funds).
References
- B. De Clercq and F. Verpoort, Macromolecules, 2002, 35, 8943–8947 CrossRef CAS.
- T. Opstal and F. Verpoort, Angew. Chem., Int. Ed., 2003, 42, 2876–2879 CrossRef CAS PubMed.
- B. De Clercq, F. Lefebvre and F. Verpoort, Appl. Catal., A, 2003, 247, 345–364 CrossRef CAS.
- S. L. Lambert, C. L. Spiro, R. R. Gagne and D. N. Hendriekson, Inorg. Chem., 1982, 21, 68–72 CrossRef CAS.
- S. Brooker, Coord. Chem. Rev., 2001, 222, 33–56 CrossRef CAS.
- V. Amendola, L. Fabbrizzi, C. Mangano, P. Pallavicini, A. Poggi and A. Taglietti, Coord. Chem. Rev., 2001, 219, 821–837 CrossRef.
- P. A. Vigato and S. Tamburini, Coord. Chem. Rev., 2004, 248, 1717–2128 CrossRef CAS.
- K. Ghosh, S. Pattanayak and A. Chakravorty, Organometallics, 1998, 17, 1956–1960 CrossRef CAS.
- S. Chattopadhyay, M. G. B. Drew, C. Diaz and A. Ghosh, Dalton Trans., 2007, 2492–2494 RSC.
- S. Roy, A. Dey, P. P. Ray, J. Ortega-Castro, A. Frontera and S. Chattopadhyay, Chem. Commun., 2015, 51, 12974–12976 RSC.
- S. Jana, B. K. Shaw, P. Bhowmik, K. Harms, M. G. B. Drew, S. Chattopadhyay and S. K. Saha, Inorg. Chem., 2014, 53, 8723–8734 CrossRef CAS PubMed.
- R. Biswas, C. Diaz, A. Bauzá, M. Barceló-Oliver, A. Frontera and A. Ghosh, Dalton Trans., 2014, 43, 6455–6467 RSC.
- R. Biswas, Y. Ida, M. L. Baker, S. Biswas, P. Kar, H. Nojiri, T. Ishida and A. Ghosh, Chem. - Eur. J., 2013, 19, 3943–3953 CrossRef CAS PubMed.
- A. Bhattacharyya, P. K. Bhaumik, P. P. Jana and S. Chattopadhyay, Polyhedron, 2014, 78, 40–45 CrossRef CAS.
- A. Bhattacharyya, B. N. Ghosh, S. Herrero, K. Rissanen, R. Jiménez-Aparicio and S. Chattopadhyay, Dalton Trans., 2015, 44, 493–497 RSC.
- R. Biswas, S. Giri, S. K. Saha and A. Ghosh, Eur. J. Inorg. Chem., 2012, 2916–2927 CrossRef CAS.
- R. Biswas, C. Diaz and A. Ghosh, Polyhedron, 2013, 56, 172–179 CrossRef CAS.
- P. Ghorai, A. Chakraborty, A. Panja, T. K. Mondal and A. Saha, RSC Adv., 2016, 6, 36020–36030 RSC.
- S. Mukherjee and P. S. Mukherjee, Dalton Trans., 2013, 42, 4019–4030 RSC.
- S. C. Manna, S. Mistri, A. Patra, M. K. Mahish, D. Saren, R. K. Manne, M. K. Santra, E. Zangrand and H. Puschmann, Polyhedron, 2019, 171, 77–85 CrossRef CAS.
- A. Bhunia, V. Bertolasi and S. C. Manna, Appl. Organomet. Chem., 2020, 34, e5424 CrossRef CAS.
- S. Chattopadhyay, P. Chakraborty, M. G. B. Drew and A. Ghosh, Inorg. Chim. Acta, 2009, 362, 502–508 CrossRef CAS.
- S. Chattopadhyay, M. G. B. Drew and A. Ghosh, Polyhedron, 2007, 26, 3513–3522 CrossRef CAS.
- M. Das, B. N. Ghosh, A. Valkonen, K. Rissanen and S. Chattopadhy, Polyhedron, 2013, 60, 68–77 CrossRef CAS.
- S. Chattopadhyay, G. Bocelli, A. Cantoni and A. Ghosh, Inorg. Chim. Acta, 2006, 359, 4441–4446 CrossRef CAS.
- K. Ghosh, M. G. B. Drew and S. Chattopadhyay, Inorg. Chim. Acta, 2018, 482, 23–33 CrossRef CAS.
- K. Ghosh and S. Chattopadhyay, Polyhedron, 2019, 170, 495–507 CrossRef CAS.
- K. Ghosh and K. Harms, ChemistrySelect, 2017, 2, 8207–8220 CrossRef CAS.
- N. C. Jana, M. Patra, P. Brandão and A. Panja, Inorg. Chim. Acta, 2019, 490, 163–172 CrossRef CAS.
- S. K. Dey and A. Mukherje, Coord. Chem. Rev., 2016, 310, 80–115 CrossRef CAS.
- K. Ghosh, K. Harms, A. Franconetti, A. Frontera and S. Chattopadhyay, J. Organomet. Chem., 2019, 883, 52–64 CrossRef CAS.
- S. Roy, S. Halder, K. Harms, P. P. Ray and S. Chattopadhyay, New J. Chem., 2020, 44, 1285–1293 RSC.
- J. L. Dempsey, B. S. Brunschwig, J. R. Winkler and H. B. Gray, Acc. Chem. Res., 2009, 42, 1995–2004 CrossRef CAS PubMed.
- T. M. McCormick, B. D. Calitree, A. Orchard, N. D. Kraut, F. V. Bright, M. R. Detty and R. Eisenberg, J. Am. Chem. Soc., 2010, 132, 15480–15483 CrossRef CAS PubMed.
- W. R. McNamara, Z. Han, C. Yin, W. W. Brennessel, P. L. Holland and R. Eisenberg, Natl. Acad. Sci. U.S.A., 2012, 109, 15594–15599 CrossRef CAS PubMed.
- S. Kal, A. S. Filatov and P. H. Dinolfo, Inorg. Chem., 2014, 53, 7137–7145 CrossRef CAS PubMed.
- W. R. McNamara, Z. Han, P. J. Alperin, W. W. Brennessel, P. L. Holland and R. Eisenberg, J. Am. Chem. Soc., 2011, 133, 15368–15371 CrossRef CAS PubMed.
- V. Artero, M. Chavarot-Kerlidou and M. Fontecave, Angew. Chem., Int. Ed., 2011, 50, 7238–7266 CrossRef CAS PubMed.
- X. Li, W. Ge, S. Guo, J. Bai and W. hong, Angew. Chem., Int. Ed., 2023, 62, e202216819 CrossRef CAS PubMed.
- M. H.-Y. Chan and V. W.-W. Yam, J. Am. Chem. Soc., 2022, 144, 22805–22825 CrossRef CAS PubMed.
- A. S. Novikov, Crystals, 2022, 12, 246 CrossRef CAS.
- W. L. Ping, J. S. Bo, Z. W. Feng, L. Y. Qi and Y. Gui, Chin. Sci. Bull., 2013, 58, 2733–2740 CrossRef.
- D. Sadhukhan, A. Ray, G. Pilet, C. Rizzoli, G. M. Rosair, C. J. Gomez-García, S. Signorella, S. Bell and S. Mitra, Inorg. Chem., 2011, 50, 8326–8339 CrossRef CAS PubMed.
- C.-L. Hu and J.-G. Mao, Coord. Chem. Rev., 2015, 288, 1–17 CrossRef CAS.
- G. N. D. Francesco, A. Gaillard, I. Ghiviriga, K. A. Abboud and L. J. Murray, Inorg. Chem., 2014, 53, 4647–4654 CrossRef PubMed.
- K. Kalyanasundaram and M. Gratzel, Coord. Chem. Rev., 1998, 77, 347–414 CrossRef.
- S. Roy, M. G. B. Drew, A. Bauzá, A. Frontera and S. Chattopadhyay, Dalton Trans., 2017, 46, 5384–5397 RSC.
- S. Mirdya, A. Frontera and S. Chattopadhyay, CrystEngComm, 2019, 21, 6859–6868 RSC.
- Y. Shen, N. Ma, L. Wu and H.-H. Song, Inorg. Chim. Acta, 2015, 429, 51–60 CrossRef CAS.
- Y.-S. Yang, Y.-P. Yang, M. Liu, Q.-M. Qiu, Q.-H. Jin, J.-J. Sun, H. Chen, Y.-C. Dai and Q.-X. Meng, Polyhedron, 2015, 85, 912–917 CrossRef CAS.
- S. Carboni, C. Gennari, L. Pignataro and U. Piarulli, Dalton Trans., 2011, 40, 4355–4373 RSC.
- L. Wang, B. Song, S. Khalife, Y. Li, L.-J. Ming, S. Bai, Y. Xu, H. Yu, M. Wang, H. Wang and X. Li, J. Am. Chem. Soc., 2020, 142, 1811–1821 CrossRef CAS PubMed.
- G. Mahmoudi, A. Bauzá and A. Frontera, Dalton Trans., 2016, 45, 4965–4969 RSC.
- C. A. Hunter and J. K. M. Sanders, J. Am. Chem. Soc., 1990, 112, 5525–5534 CrossRef CAS.
- S. K. Burley and G. A. Petsko, Science, 1985, 229, 23–28 CrossRef CAS PubMed.
- K. S. Kim, P. Tarakeshwar and J. Y. Lee, Chem. Rev., 2000, 100, 4145–4185 CrossRef CAS PubMed.
- J. C. Ma and D. A. Dougherty, Chem. Rev., 1997, 97, 1303–1324 CrossRef CAS PubMed.
- K. S. Kim, J. Y. Lee, S. J. Lee, T.-K. Ha and D. H. Kim, J. Am. Chem. Soc., 1994, 116, 7399–7400 CrossRef CAS.
- P. Middya, M. Karmakar, R. M Gomila, M. G. B. Drew, A. Frontera and S. Chattopadhyay, J. Chem., 2023, 47, 9346–9363 CAS.
- D. Quiñonero, C. Garau, C. Rotger, A. Frontera, P. Ballester, A. Costa and P. M. Deyà, Angew. Chem., Int. Ed., 2002, 41, 3389–3392 CrossRef.
- M. Egli and S. Sarkhel, Acc. Chem. Res., 2007, 40, 197–205 CrossRef CAS PubMed.
- T. J. Mooibroek, P. Gamez and J. Reedijk, CrystEngComm, 2008, 10, 1501–1515 RSC.
- J. Ran and P. Hobza, J. Chem. Theory Comput., 2009, 5, 1180–1185 CrossRef CAS PubMed.
- M. Barceló-Oliver, C. Estarellas, A. García-Raso, A. Terrón, A. Frontera, D. Quiñonero, E. Molins and P. M. Deyà, CrystEngComm, 2010, 12, 362–365 RSC.
- M. Nishio, CrystEngComm, 2004, 6, 130–156 RSC.
- J. Xiao, P. Broz, A. W. Puri, E. Deu, M. Morell, D. M. Monack and M. Bogyo, J. Am. Chem. Soc., 2013, 135, 9130–9138 CrossRef CAS PubMed.
- S. Naskar, D. Mishra, R. J. Butcher and S. K. Chattopadhyay, Polyhedron, 2007, 26, 3703–3714 CrossRef CAS.
- A. Bauzá, D. Quiñonero, P. M. Deyà and A. Frontera, Chem. - Asian J., 2013, 8, 2708–2713 CrossRef PubMed.
- L. M. Salonen, M. Ellermann and F. Diederich, Angew. Chem., Int. Ed., 2011, 50, 4808–4842 CrossRef CAS PubMed.
- S. Jana, K. Harms, A. Bauzá, A. Frontera and S. Chattopadhyay, Cryst. Growth Des., 2015, 15, 257–267 CrossRef CAS.
- S. Cañellas, A. Bauzá, A. Lancho, A. Garcia-Raso, J. J. Fiol, E. Molins, P. Ballesterc and A. Frontera, CrystEngComm, 2015, 17, 5987–5997 RSC.
- G. M. Sheldrick, Acta Crystallogr., 2015, 71A, 3–8 CrossRef PubMed.
- G. M. Sheldrick, SADABS, V2014/5, Software for Empirical Absorption Correction, University of Göttingen, Institute fur Anorganische Chemieder Universitat, Gottingen, Germany, pp. 1999–2003 Search PubMed.
- M. A. Spackman and D. Jayatilaka, CrystEngComm, 2009, 11, 19–32 RSC.
- F. L. Hirshfeld, Theor. Chim. Acta, 1977, 44, 129–138 CrossRef CAS.
- H. F. Clausen, M. S. Chevallier, M. A. Spackman and B. B. Iversen, New J. Chem., 2010, 34, 193–199 RSC.
- A. L. Rohl, M. Moret, W. Kaminsky, K. Claborn, J. J. McKinnon and B. Kahr, Cryst. Growth Des., 2008, 8, 4517–4525 CrossRef CAS.
- A. Parkin, G. Barr, W. Dong, C. J. Gilmore, D. Jayatilaka, J. J. McKinnon, M. A. Spackman and C. C. Wilson, CrystEngComm, 2007, 9, 648–652 RSC.
- M. A. Spackman and J. J. McKinnon, CrystEngComm, 2002, 4(66), 378–392 RSC.
- S. K. Wolff, D. J. Grimwood, J. J. McKinnon, D. Jayatilaka and M. A. Spackman, Crystal Explorer 2.0, University of Western Australia, Perth, Australia, 2007, http://hirshfeldsurfacenet.blogspot.com Search PubMed.
- F. H. Allen, O. Kennard, D. G. Watson, L. Brammer, A. G. Orpen and R. J. Taylor, J. Chem. Soc., Perkin Trans., 1987, 2, S1–S19 RSC.
- J. J. Kinnon, M. A. Spackman and A. S. Mitchell, Acta Crystallogr., 2004, B60, 627–668 Search PubMed.
- R. Ahlrichs, M. Bar, M. Hacer, H. Horn and C. Komel, Chem. Phys. Lett., 1989, 162, 165–169 CrossRef CAS.
- S. Grimme, J. Antony, S. Ehrlich and H. Krieg, J. Chem. Phys., 2010, 132, 154104–154122 CrossRef PubMed.
- F. Weigend, Phys. Chem. Chem. Phys., 2006, 8, 1057–1065 RSC.
- J. Contreras-Garcia, E. R. Johnson, S. Keinan, R. Chaudret, J. P. Piquemal, D. N. Beratan and W. Yang, J. Chem. Theory Comput., 2011, 7, 625–632 CrossRef CAS PubMed.
- R. F. W. Bader, Chem. Rev., 1991, 91, 893–928 CrossRef CAS.
- T. Lu and F. Chen, J. Comput. Chem., 2012, 33, 580–592 CrossRef CAS PubMed.
- J. W. Humphrey, A. Dalke and K. Schulten, J. Mol. Graphics, 1996, 14, 33–38 CrossRef PubMed.
- K. Ghosh, M. G. B. Drew and S. Chattopadhyay, Inorg. Chim. Acta, 2018, 482, 23–33 CrossRef CAS.
- S. Roy, P. K. Bhaumik, K. Harms and S. Chattopadhyay, Polyhedron, 2014, 75, 57–63 CrossRef CAS.
- M. Das, B. N. Ghosh, A. Bauzá, K. Rissanen, A. Frontera and S. Chattopadhyay, RSC Adv., 2015, 5, 73028–73039 RSC.
- S. Bhunia, R. M. Gomila, A. Frontera and S. Chattopadhyay, Polyhedron, 2022, 223, 115910 CrossRef CAS.
- S. Bhattacharya, S. Roy, K. Harms, A. Bauzá, A. Frontera and S. Chattopadhyay, Inorg. Chim. Acta, 2016, 442, 16–23 CrossRef CAS.
- K. Ghosh, K. Harms and S. Chattopadhyay, Polyhedron, 2017, 123, 162–175 CrossRef CAS.
- J. Müller, C. Würtele, O. Walter and S. Schindler, Angew. Chem., Int. Ed., 2007, 46, 7775–7777 CrossRef PubMed.
- D. Bandyopadhyay, M. Layek, M. Fleck, R. Saha and C. Rizzoli, Inorg. Chim. Acta, 2017, 461, 174–182 CrossRef CAS.
- D. Cremer and J. A. Pople, J. Am. Chem. Soc., 1975, 97, 1354–1358 CrossRef CAS.
- K. Ghosh, T. Dutta, M. G. B. Drew, A. Frontera and S. Chattopadhyay, Polyhedron, 2020, 182, 114432 CrossRef CAS.
- S. Sen, P. Talukder, S. K. Dey, S. Mitra, G. Rosair, D. L. Hughes, G. P. A. Yap, G. Pilet, V. Gramlich and T. Matsushita, Dalton Trans., 2006, 1758–1767 RSC.
- K. Kitaura and K. Morokuma, Int. J. Quantum Chem., 1976, 10, 325–340 CrossRef CAS.
- S. Jana, B. K. Shaw, P. Bhowmik, K. Harms, M. G. B. Drew, S. Chattopadhyay and S. K. Saha, Inorg. Chem., 2014, 53, 8723–8734 CrossRef CAS PubMed.
- M. Das, S. Chatterjee, K. Harms, T. K. Mondal and S. Chattopadhyay, Dalton Trans., 2014, 43, 2936–2947 RSC.
- S. Khan, S. Jana, M. G. B. Drew, A. Bauzá, A. Frontera and S. Chattopadhyay, RSC Adv., 2016, 6, 61214–61220 RSC.
- S. Chattopadhyay, M. S. Ray, S. Chaudhuri, G. Mukhopadhyay, G. Bocelli, A. Cantoni and A. Ghosh, Inorg. Chim. Acta, 2006, 359, 1367–1375 CrossRef CAS.
- S. Khan, A. A. Masum, M. M. Islam, M. G. B. Drew, A. Bauzá, A. Frontera and S. Chattopadhyay, Polyhedron, 2017, 123, 334–343 CrossRef CAS.
- A. Bhattacharyya, S. Sen, K. Harms and S. Chattopadhyay, Polyhedron, 2015, 88, 156–163 CrossRef CAS.
- K. Ghosh, K. Harms, A. Bauzá, A. Frontera and S. Chattopadhyay, CrystEngComm, 2018, 20, 7281–7292 RSC.
- P. Bhowmik, A. Bhattacharyya, K. Harms, S. Sproules and S. Chattopadhyay, Polyhedron, 2015, 85, 221–231 CrossRef CAS.
- N. Sarkar, M. G. B. Drew, K. Harms, A. Bauzá, A. Frontera and S. Chattopadhyay, CrystEngComm, 2018, 20, 1077–1086 RSC.
- A. Das, K. Bhattacharya, S. Giri and A. Ghosh, Polyhedron, 2017, 134, 295–301 CrossRef CAS.
- A. Ray, G. M. Rosair, G. Pilet, B. Dede, C. J. Gómez- García, S. Signorella, S. Bellú and S. Mitra, Inorg. Chim. Acta, 2011, 375, 20–30 CrossRef CAS.
- H. A. R. Pramanik, P. C. Paul, P. Mondal and C. R. Bhattacharjee, J. Mol. Struct., 2015, 1100, 496–505 CrossRef CAS.
- A. Saha, P. Majumdar and S. Goswami, J. Chem. Soc., Dalton Trans., 2000, 1703–1708 RSC.
- S. Thakurta, R. J. Butcher, G. Pilet and S. Mitra, J. Mol. Struct., 2009, 929, 112–119 CrossRef CAS.
- A. H. Kianfar, W. A. K. Mahmood, M. Dinari, M. H. Azarian and F. Z. Khafri, Spectrochim. Acta, Part A, 2014, 127, 422–428 CrossRef CAS PubMed.
- L. Mandal and S. Mohanta, Dalton Trans., 2014, 43, 15737–15751 RSC.
- A. K. Ghosh, M. Mitra, A. Fathima, H. Yadav, A. R. Choudhury, B. U. Nair and R. Ghosh, Polyhedron, 2016, 107, 1–8 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Figures from S1 to S9 and details of Hirshfeld surface analysis. CCDC 2259434, 2259435 and 2259436 contain the supplementary crystallographic data for complexes 1, 2, and 3, respectively. CCDC 2259434–2259436. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d3ra03828a |
|
| This journal is © The Royal Society of Chemistry 2023 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence b,
Michael G. B. Drewc,
Antonio Frontera
b,
Michael G. B. Drewc,
Antonio Frontera
















