
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceConstruction of the lactate-sensing fibremats by confining sensor fluorescent protein of lactate inside nanofibers of the poly(HPMA/DAMA)/ADH-nylon 6 core–shell fibremat†
Yuna Katoa,
Shuichi Iwataa,
Yusuke Nasub,
Akiko Obataa,
Kenji Nagataa,
Robert E. Campbellb and
Toshihisa Mizuno *ac
*ac
aDepartment of Life Science and Applied Chemistry, Graduate School of Engineering, Nagoya Institute of Technology, Gokiso-cho, Showa-ku, Nagoya, Aichi 466-8555, Japan. E-mail: toshitcm@nitech.ac.jp
bDepartment of Chemistry, School of Science, The University of Tokyo, Bunkyo-ku, Tokyo 113-0033, Japan
cDepartment of Nanopharmaceutical Sciences, Graduate School of Engineering, Nagoya Institute of Technology, Gokiso-cho, Showa-ku, Nagoya, Aichi 466-8555, Japan
First published on 10th October 2023
Abstract
The development of a new materials platform capable of sustaining the functionality of proteinous sensor molecules over an extended period without being affected by biological contaminants in living systems, such as proteases, is highly demanded. In this study, our primary focus was on fabricating new core–shell fibremats using unique polymer materials, capable of functionalizing encapsulated sensor proteins while resisting the effects of proteases. The core-fibre parts of core–shell fibremats were made using a newly developed post-crosslinkable water-soluble copolymer, poly(2-hydroxypropyl methacrylamide)-co-poly(diacetone methacrylamide), and the bifunctional crosslinking agent, adipic dihydrazide, while the shell layer of the nanofibers was made of nylon 6. Upon encapsulating the lactate-sensor protein eLACCO1.1 at the core-fibre part, the fibremat exhibited a distinct concentration-dependent fluorescence response, with a dynamic range of fluorescence alteration exceeding 1000% over the lactate concentration range of 0 to 100 mM. The estimated dissociation constant from the titration data was comparable to that estimated in a buffer solution. The response remained stable even after 5 cycles and in the presence of proteases. These results indicates that our core–shell fibremat platform could serve as effective immobilizing substrates for various sensor proteins, facilitating continuous and quantitative monitoring of various low-molecular-weight metabolites and catabolites in a variety of biological samples.
1. Introduction
Lactate, being a major catabolite crucial for adenosine triphosphate (ATP) synthesis via glycolysis, has drawn significant interest as a biomarker in blood, sweat, and urine samples.1,2 Its analysis offers valuable insights into physiological and cellular-level states, such as physical condition,3,4 diseases prevalence,5,6 anaplastic, or differentiated of cell state,7 etc. Recently, lactate monitoring has also gained attention as an index for food control.8 Various sensor molecules have been developed to quantify lactate concentration electrochemically, colorimetrically, or through fluorescence detection.9–11 Hansen et al. developed the BODIPY boronic acid derivatives and achieved successful fluorescence detection of lactate using them, with a Kd range of 30–54 mM.10 Zaryanov et al. developed lactate sensor, prepared by electropolymerization of 3-aminophenylboronic acid onto the working electrode and succeeded in electrochemical detection of lactate using this modified electrode.11 However, their simple mechanisms, such as cyclic complex formation with boronic acid derivatives,10,11 pose disadvantages for accurately quantifying lactate in biological samples containing diverse contaminants. To address this issue, the methods involving enzymatic reactions selective for lactate12,13 or the use of fluorescent protein mutants fused with lactate receptor proteins14–17 were developed. Nevertheless, concerns remain due to the presence of various proteases in biological samples, impacting the continuous monitoring of lactate using lactate-specific enzymes and sensor proteins with the aforementioned techniques. To overcome these challenges, combined use of the methods able to use proteinous sensor molecules irrespective of protease digestions as well as other contaminants has been recognized necessary and encapsulation of sensor proteins in the ‘(low-molecular weight (MW)) ligands-permeable materials’ matrix was found effective and examined by several researchers.18,19 However, methods that involve protein denaturation during the immobilization process of sensor proteins in materials' matrix20 or by which sensor proteins can detach from the materials' matrix during repeated usage21 are undesirable, especially for the use of fluorescence sensor proteins. Therefore, it is desirable for approaches to address these issues while meeting the required criteria.22Meanwhile, recently, we have been studying new methods that allow us to confine enzyme molecules within the nanofibers of water-insoluble fibremats without protein denaturation.23–26 Even upon encapsulating in nanofibers, the enzyme molecules can exhibit efficient enzymatic reactions, as it were in solution, for the substrate molecules that are capable of permeating inside the nanofibers. Fibermats obtained through the electrolytic spinning method consist of fiber assemblies with diameters ranging from approximately 50 nm to a few μm. One of their distinctive characteristics is their excellent permeability to solvents and various molecules included therein. In our approach, we have taken it a step further by introducing a molecular permeable feature within these fibres, preserving MW-dependent permeability. This method allows for efficient enzyme reactions with low-MW substrates that can penetrate the fibres while ensuring the immobilized enzymes remain firmly bound within the fiber matrix, preventing detachment.
To construct such enzyme-encapsulated fibremats, two elements are necessary to be achieved simultaneously – confining protein molecules inside water-insoluble nanofibers without denaturation and increasing the permeability of substrate molecules inside nanofibers. By employing post-crosslinkable hydrophilic polymers whose crosslinking reactions with crosslinkers can be accelerated by dehydration process and dose not react with proteins27 as a base material, we successfully satisfied them. According to electrospinning of the precursor aqueous solution, including the post-crosslinkable polymer, enzyme proteins, and crosslinker, the nanofiber formation of fibremats and confinement of enzyme molecules inside the spun nanofibers were simultaneously progressed, giving the protein-encapsulated fibremat in a single step operation. We named this electrospinning method as “in situ crosslinking during electrospinning (SCES) method”.25 To date, we have verified that SCES can be applied to poly(γ-glutamic acid)/3-(glycidyloxy)propyl trimethoxysilane23,28 and poly (acrylamide)-co-poly(diacetone acrylamide) [poly(AM/DAAM)]/adipic acid dihydrazide (ADH).24 Because these network polymers solely form hydrogels in aqueous buffers, the mechanical strengths of the fibremats prepared from them were too weak to use for practical applications.25 However, wrapping the fibres with a thin layer of hydrophobic poly(ε-caprolactone) (PCL)25 or nylon 6 (ref. 26) satisfied the required mechanical strength without sacrificing the efficient enzymatic activity against low-MW substrates. Such double-layered fibremats are called ‘core–shell fibremats’, and the PCL or nylon 6 layer is called a ‘shell’. We came up with the idea that the above core–shell fibremats with unique molecular permeability dependent on MW could be a suitable ‘lactate-permeable materials’ matrix for enabling proteinous lactate sensors to function without being affected by proteases and other contaminants.
In this study, therefore we constructed core–shell fibremats that incorporate the lactate-sensor fluorescent protein, eLACCO1.1,16 within their constituent nanofibers and characterized their abilities as lactate sensors (Fig. 1). eLACCO1.1 is a well-established lactate-sensor fluorescent protein designed by fusing the amino acid sequences of green fluorescent protein (GFP) and the lactate sensor protein (LSP) by Nasu and Campbell et al.16 Before binding to lactate, the tertiary structure of GFP-derived domain is partially disrupted. While after binding to lactate in the LSP-derived domain, the structure of the GFP-derived moiety is restored, thereby restoring its fluorescent properties. This enables a more than 10 times increase in its fluorescence intensity with an increase in lactate concentration. The core-fiber parts of the constituent double-layered nanofibers were composed of hydrogels prepared from the newly developed post-crosslinkable hydrophilic copolymer, poly(2-hydroxypropyl methacrylamide)-co-poly(diacetone methacrylamide) (poly(HPMA/DAMA)), and the bifunctional crosslinking agent, adipic dihydrazide (ADH). The core fibres were covered with a thin shell layer of the hydrophobic polymer, nylon 6. By adding eLACCO1.1 to the precursor solution of the core-fiber part, which included poly(HPMA/DAMA) and ADH, we successfully constructed the eLACCO1.1-encapsulated core–shell fibremats. We characterized the fibremats' ability as a sensor material for lactate based on its fluorescence response to lactate. Additionally, we discussed the effectiveness of the poly(HPMA/DAMA)/ADH-nylon 6 core–shell fibremats as a suitable ‘(low-MW) ligand-permeable materials’ matrix for enabling protein-based sensors to function without being affected by proteases and other contaminants.
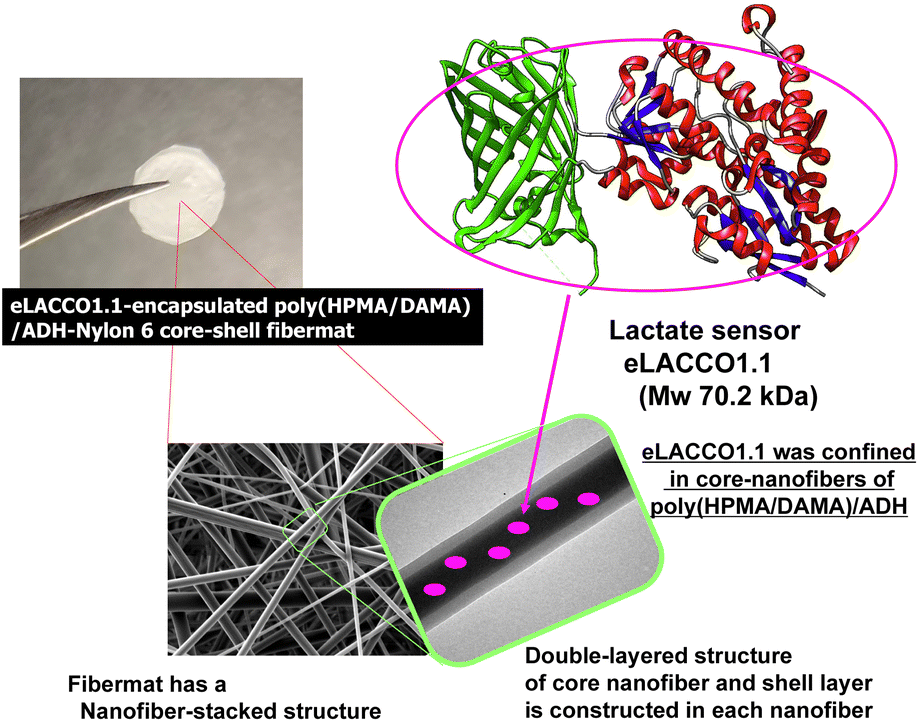 | ||
| Fig. 1 The eLACOO1.1-encapsulated poly(HPMA/DAMA)/ADH-nylon 6 core–shell fibremat, where eLACCO1.1 molecules are included in the core-fiber part of constitutive nanofibers of poly(HPMA/DAMA)/ADH. Nylon 6 was wrapped onto the core-fiber parts as a shell layer, giving the double-layered nanofibers of core–shell fibremat. Poly(HPMA/DAMA), poly(2-hydroxypropyl methacrylamide)-co-poly(diacetone methacrylamide); ADH, adipic dihydrazide; poly(HPMA/DAMA)/ADH, network polymers prepared by crosslinking of poly(HPMA/DAMA) with ADH. | ||
2. Results and discussion
2.1 Synthesis of poly(HPMA/DAMA) and characterization of its gel formation properties
Synthesis of the copolymer of 2-hydroxypropyl methacrylamide (HPMA)29 and diacetone methacrylamide (DAMA),30 poly(HPMA/DAMA), was performed through free radical polymerization of HPMA and DAMA with the molar ratio of 8![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2 (Fig. 2). DAMA has ketone group at the side chain as similar to DAAM, which is expected to serve as a crosslinking point when using ADH as a bifunctional crosslinker (Fig. 3(a)). While, the synthesis of each monomer was carried out according to previous reports.29,30 The molar ratio of monomer units in the synthesised copolymer was checked by 1H-NMR (Fig. S1†), and as a result, it was estimated to be 8:2, as reflecting the initial monomer ratio of HPMA and DAMA for polymer synthesis. While Mn and the polydispersity index (PDI) were analysed by a gel permeation chromatography and estimated to be 153 kDa and 1.51, respectively (Fig. S2†).
:2 (Fig. 2). DAMA has ketone group at the side chain as similar to DAAM, which is expected to serve as a crosslinking point when using ADH as a bifunctional crosslinker (Fig. 3(a)). While, the synthesis of each monomer was carried out according to previous reports.29,30 The molar ratio of monomer units in the synthesised copolymer was checked by 1H-NMR (Fig. S1†), and as a result, it was estimated to be 8:2, as reflecting the initial monomer ratio of HPMA and DAMA for polymer synthesis. While Mn and the polydispersity index (PDI) were analysed by a gel permeation chromatography and estimated to be 153 kDa and 1.51, respectively (Fig. S2†).
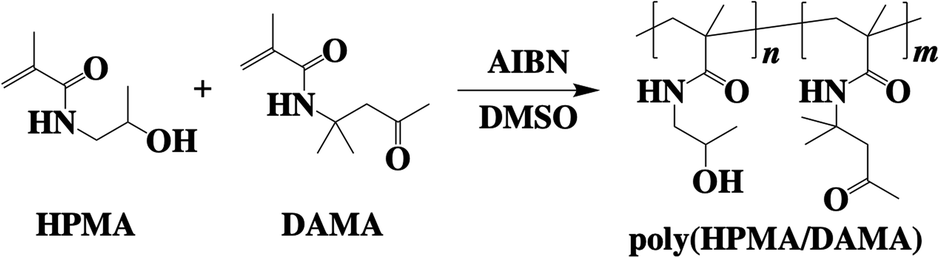 | ||
| Fig. 2 Synthesis of the copolymer of 2-hydroxypropyl methacrylamide (HPMA) and diacetone methacrylamide (DAMA), poly(HPMA/DAMA). | ||
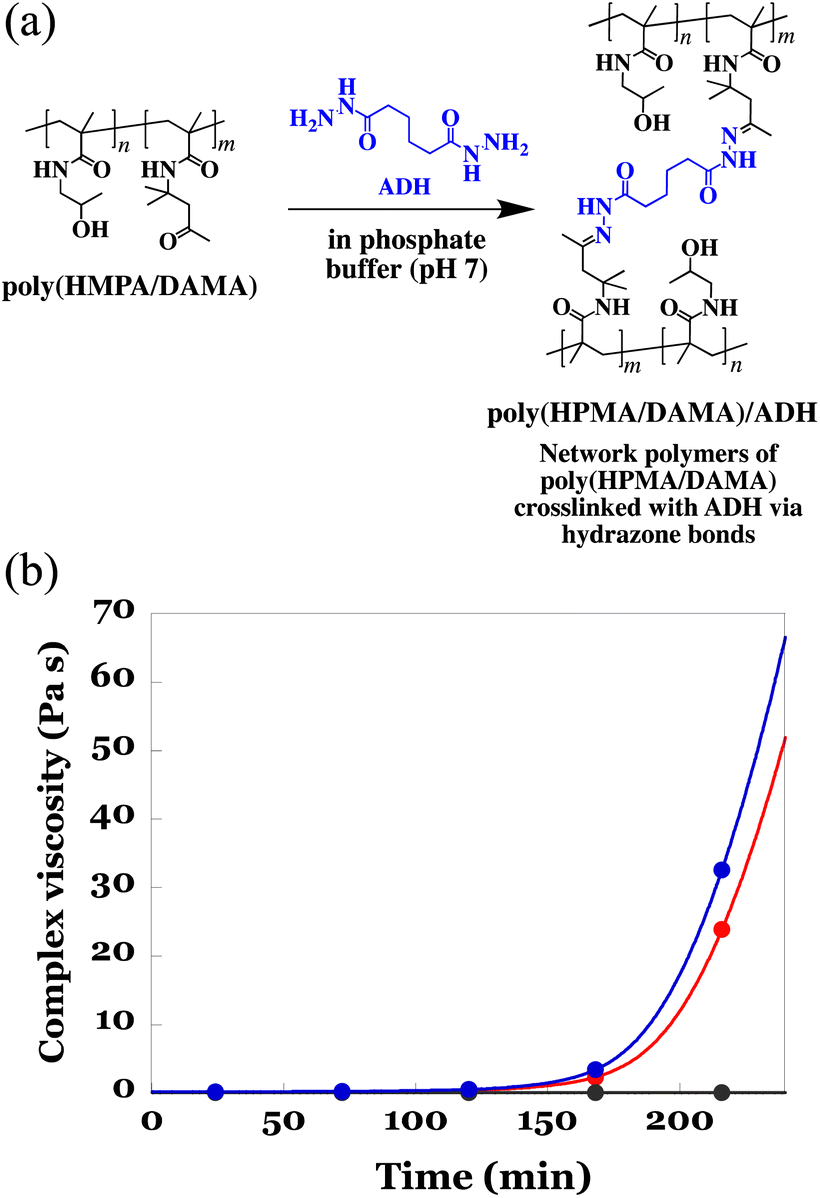 | ||
| Fig. 3 (a) Schematic illustration of crosslinking of poly(HPMA/DAMA) with ADH in a phosphate buffer (pH 7) via formation of hydrazone bonds between the ketone groups in poly(HPMA/DAMA) and the hydrazide groups in ADH, giving the network polymers, poly(HPMA/DAMA)/ADH. (b) The difference in the time course of complex viscosity (Pa s) was observed for a 25 wt% solution of poly(HPMA/DAMA) in the presence (blue line) and absence (red line) of lactase (5 wt%) in 100 mM phosphate buffer (pH 7) after the addition of ADH (0.5 molar ratio for DAMA unit). Additionally, as a reference, the same polymer solution without ADH addition was measured (black line). | ||
Prior to electrospinning of the poly(HPMA/DAMA) aqueous solution while crosslinking by ADH according to the SCES method, we need to check whether there is enough spinnable time for electro-spinning until the crosslinking was fully progressed to form an un-spinnable hard hydrogels. In our previous studies on fabrication of the poly(AM/DAAM)/ADH fibremats using poly(AM/DAAM) as a post-crosslinkable copolymer and ADH as a bifunctional crosslinker, until 80 min from the ADH addition the viscosity of 15 wt% poly(AM/DAAM) solution was less than 5 Pa s, evaluated by a rotational viscometer (RE80H, TOKI SANGYO CO., LTD, Japan), and could be electrospun in this time period (0–80 min, i.e., spinnable time).24 Therefore, to estimate the time period less than 5 Pa s could be used as an effective index to predict the spinnable time of poly(HPMA/DAMA) solution upon ADH addition. Further, it was known that inclusion of proteins to be encapsulated in poly(AM/DAAM) solution has a tendency to shorten spinnable time by accelerating crosslinking rate by ADH. Therefore, choosing lactase as a representative of proteins to be added, time-course of the complex viscosity (Pa s) of poly(HPMA/DAMA) solution in the presence of proteins (5 wt% of lactase with respect to the mass of poly(HPMA/DAMA)) after addition of ADH was also evaluated using a rheometer. According to Cox-Merz rule,31,32 it is known that there is a high correlation between the viscosity (Pa s) estimated by a rotational viscometer, and the complex viscosity (Pa s), estimated from a rheometer with rotational measurement. Therefore, in this study, we conducted the evaluation using complex viscosity.
To a 25 wt% solution of poly(HPMA/DAMA) in 100 mM phosphate buffer (pH 7) with or without lactase (5 wt% with respect to the mass of poly(HPMA/DAMA)), ADH was added at 0.5 equivalents per DAMA unit. Then the viscoelasticity measurement using a rotational rheometer at 25 °C was initiated. As a reference, the same measurement without addition of ADH was also performed. The results are summarized in Fig. 3(b). A gradual increase in complex viscosity of the solution was observed for both in the presence and absence of lactase after 105 min of ADH addition, and at 175 min or 183 min (in the presence or absence of lactase, respectively) its value reached more than 5 Pa s. These results meant that, as expected, via formation of hydrazone bonds between the ketone groups in poly(HPMA/DAMA) and the hydrazide groups in ADH, the network polymers of poly(HPMA/DAMA), named poly(HPMA/DAMA)/ADH, could be formed, irrespective of the presence or absence of protein and its solution finally became a hard hydrogel. Further, electrospinning of 25 wt% poly(HPMA/DAMA) solution with ADH according to the SCES method seemed to be applicable within 0–175 min time period after ADH addition as a spinnable time.
2.2 Construction of the core–shell fibremats, consisting of core-fibre part of poly(HPMA/DAMA)/ADH and shell layer of nylon 6
First, we fabricated the core–shell fibremats without encapsulating protein molecules inside the constitutive nanofibers. As a precursor solution for the core-fiber parts of constitutive nanofibers, poly(HPMA/DAMA) was dissolved in a 100 mM phosphate buffer (pH 7) (final concentration was set by 25 wt%), and then ADH was added at 1/2 molar equivalent per DAMA unit in poly(HPMA/DAMA). While, for the shell part 10 wt% TFE solution of nylon 6 was prepared. Both solutions were transferred to a separate syringe and subjected to electro-spinning through a co-axial spinneret, applying a high voltage of 18 kV and constant extrusion of the core and shell precursor solutions by 0.3 and 1.2 mL h−1, respectively (Fig. 4(a)). After coating the surfaces of fibremat with osmium vapor, SEM measurements were applied (Fig. 4(b)). The results showed a typical fibres-stacked nanostructure of electrospun fibremats with a uniform fiber diameter of 457 ± 54 nm. Furthermore, from TEM observation of single nanofibers uniform double-layered structure, with core fibres of 219 ± 14 nm diameter covered with a shell layer of 107 ± 5 nm was also ensured (Fig. 4(c)). These results meant successful construction of the core–shell fibremats using the combination of poly(HPMA/DAMA)/ADH for core-fiber part and nylon 6 for shell layer.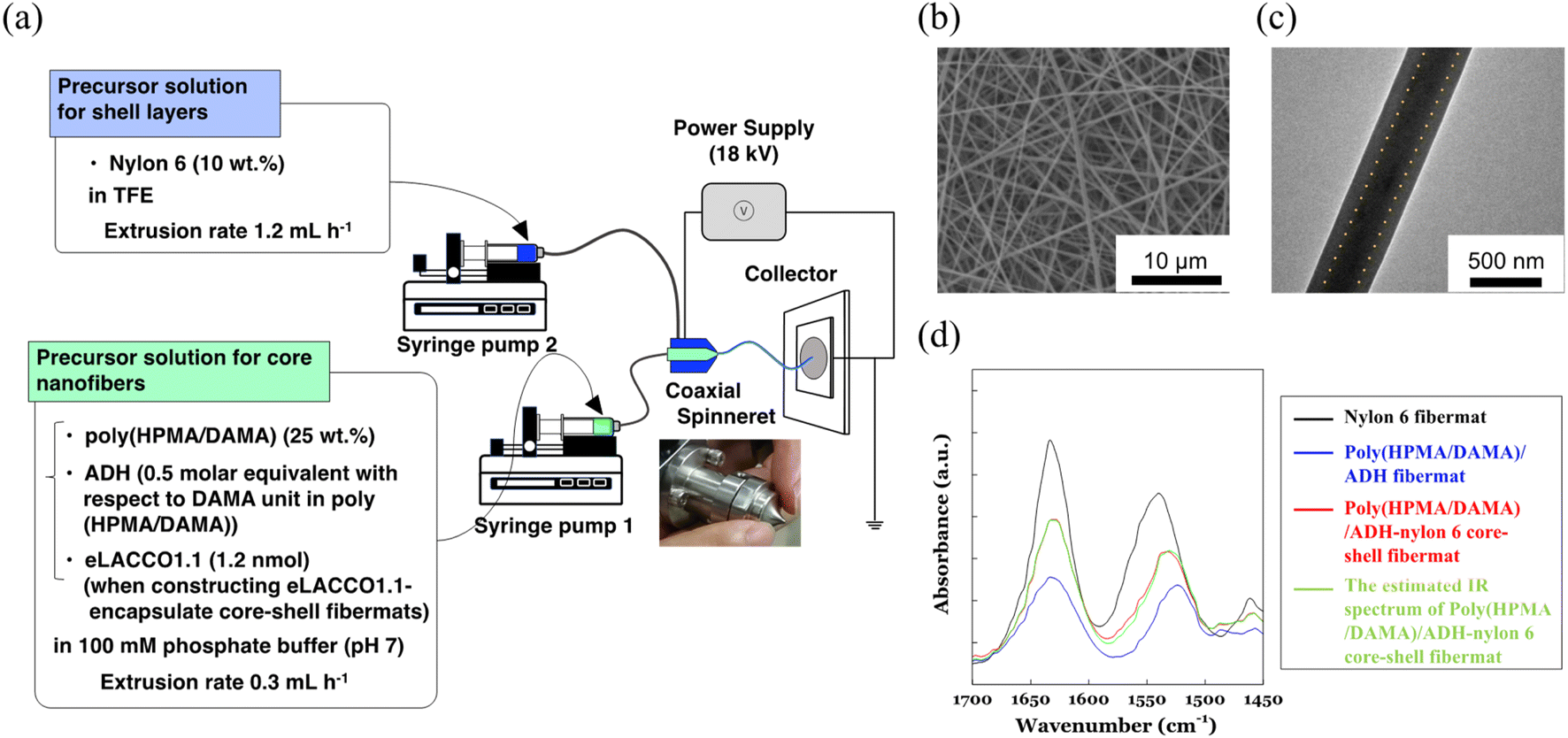 | ||
| Fig. 4 (a) Schematic illustration of the co-axial electrospinning set-up to fabricate the poly(HPMA/DAMA)/ADH-nylon 6 core–shell fibremats, encapsulating eLACCO1.1 or not at the core-fiber part of constitutive nanofibers. (b) SEM image of (c) TEM images of the core–shell fibremat, consisting of the poly(HPMA/DAMA)/ADH for core-fiber part and nylon 6 for shell part. To get contrast between core-fiber part and shell part in TEM image, phosphotungstate salt was beforehand added to the precursor solution of core-fiber part for electrospinning. (d) FT-IR spectra of the poly(HPMA/DAMA)/ADH-nylon 6 core–shell fibremat (red), poly(HPMA/DAMA)/ADH fibremat (blue), and nylon 6 fibremat (black), and the estimated IR spectra of poly(HPMA/DAMA)/ADH-nylon 6 core–shell fibremat (green). | ||
Using ATR-IR measurements, we further checked concomitance of both polymer materials in the obtained core-shell fibremat. The IR spectra of poly(HPMA/DAMA)/ADH-nylon 6 core–shell fibremats and the fibremats only of poly(HPMA/DAMA)/ADH or nylon 6 were observed and compared in Fig. 4(d). A linear additivity in the IR spectrum of poly(HPMA/DAMA)/ADH-nylon 6 core–shell fibremats with those of poly(HPMA/DAMA)/ADH fibremat and nylon 6 fibremat could be estimated (Fig. 6, red line). This data also supported successful construction of the core–shell fibremats, consisting of poly(HPMA/DAMA)/ADH and nylon 6.
2.3 Molecular permeability of the encapsulated molecules inside the core-fiber part in the core–shell fibremats
To utilize the core–shell fibremats as an immobilizing substrate for eLACCO1.1, where eLACCO1.1 molecules were entangled within the network structure of poly(HPMA/DAMA)/ADH in the core-fiber part of fibremat nanofibers, these nanofibers are necessary to possess the following compatible features; one is sufficient molecular permeability for low molecular weight (MW) molecules such as lactate, and the other one is effective retention of high MW proteins such as eLACCO1.1 inside nanofibers without leakage. Prior to constructing the eLACCO1.1-encapsulated fibremats, therefore we checked molecular permeability of the beforehand encapsulated molecules from the inside to outside of nanofibers of the poly(HPMA/DAMA)/ADH-nylon 6 core–shell fibremats. As a representative of low MW and high MW molecules to be encapsulated, we used fluorescein (MW, 332) and the superfolder GFP (MW, 24 kDa), respectively in this experiment. By addition of either molecule in the precursor solution for core-fiber part, the poly(HPMA/DAMA)/ADH-nylon 6 core–shell fibremat, encapsulating fluorescein or GFP at the core-fiber part was prepared.An assessment of leakage behaviours of the encapsulated molecules to an immersing buffer was performed by immersing the square pieces (0.5 cm × 0.5 cm, 1 mg) of fibremats in a 100 mM phosphate buffer (pH 7, 1 mL) for the defined time (0–170 h) and from differences in fluorescence intensities of the supernatant the leaked amounts of molecules at each time point were quantified. The results are summarized in Fig. 5. As similar to the previous report on the poly(AM/DAAM)/ADH-nylon 6 core–shell fibremats,22 quantitative release (>97%) of low-MW fluorescein to an immersing buffer within 5 min (Fig. 5(a)),as well as no leakage of the high MW protein even after 7 days' immersion (Fig. 5(b)) were both observed. This MW-dependent permeation features indicated that the poly(HPMA/DAMA)/ADH-nylon 6 core–shell fibremats should be a good immobilization platform to functionalize eLACCO1.1 for detection of lactate by immersing in the sample solutions.
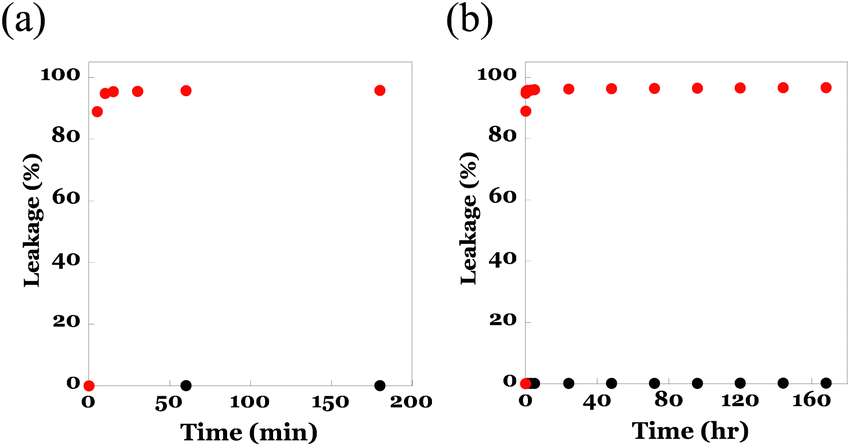 | ||
| Fig. 5 Leakage behaviours of fluorescein (red circles) and GFP (black circles) in (a) short time range (0–160 min) or (b) long time range (0–180 h) from the poly(HPMA/DAMA)/ADH-nylon 6 core–shell fibremat, encapsulating them inside the core-fiber part of constitutive nanofibers to an immersing phosphate buffer (100 mM, pH 7). | ||
2.4 Preparation of eLacco1.1-encapsulated core–shell fibremats and fluorescent response according to lactate concentration
Finally, we prepared the poly(HPMA/DAMA)/ADH-nylon 6 core–shell fibremat, encapsulating the lactate sensor protein, eLACCO1.1 inside the core-fiber part. eLACCO1.1 was produced using the E. coli DH10B strain and purified by His-tag affinity chromatography, according to the previous report (Fig. S3†).15 eLACCO1.1 (∼1.2 nmol) was added to the precursor solution (2 mL) for core-fiber part of 25wt% poly(HPMA/DAMA) solution, along with ADH (1/2 molar equivalent of DAMA unit in poly(HPMA/DAMA)). Then we conducted co-axial electrospinning, simultaneously using the precursor solution of shell layer of nylon 6 (10 wt% in TFE) as similar to that without encapsulating eLACCO1.1. The SEM and TEM images of the eLACCO1.1-encapsulted core–shell fibremat were summarized in Fig. 6. No significant impact on nanostructure (whole fiber morphology and average diameter, as well as the double-layered structure inside nanofibers) was observed, meaning successful construction of eLACCO1.1-encapsulated core–shell fibremats. | ||
| Fig. 6 SEM (a) and TEM (b) images of the eLACCO1.1-encapsulated core–shell fibremat, consisting of the poly(HPMA/DAMA)/ADH for core-fiber part and nylon 6 for shell part. To get contrast between core-fiber part and shell part in TEM image, phosphotungstate salt was beforehand added to the precursor solution of core-fiber part for electrospinning. | ||
To analyse fluorescence response of the eLACCO1.1-encapsulated fibremat to lactate, we first check time-course of fluorescence intensities after addition of 10 mM lactate by immersing the fibremat in 150 mM MOPS buffer (pH 7), supplemented with 10 mM lactate, 100 mM KCl and 1 mM CaCl2. eLACCO1.1 is a sensor protein based on GFP and is known to exhibit pH-dependent fluorescence properties.16 However, in this study, to demonstrate its general application for detecting lactate in biological samples, we evaluated its responsiveness to lactate at pH 7. The fibremat was cut in a circle shape (6.4 mmφ) and set at the bottom of 96 well black plate. Using a fluorescence plate reader, alteration in fluorescence intensities (λex 470 nm, λem 507 nm) from the fibremats was evaluated. Because structural alteration of eLACCO1.1 from off-state (less fluorescent) to on-state (fluorescent) according to lactate binding takes several min,16 gradual increase in fluorescence intensity after addition of lactate (final concentration was 10 mM) and its saturation over 10 min were both observed (Fig. 7(a) upper). Reflecting this original property, the eLACCO1.1-encapsulated core–shell fibremat also exhibited similar gradual increase in fluorescence intensity and the subsequent saturation by lactate addition (Fig. 7(b) upper). While, upon exchanging the immersing buffer to that without including lactate, decrease in fluorescence intensity was observed. Interestingly, the time order of decrease rate in fluorescence was similar to that of eLACCO1.1 in solution, which first added 10 mM lactate to increase fluorescence intensity (on-state) and then EGTA was subsequently added to return to the off-state (Fig. 7(a) upper). These consistence in time-dependent fluorescence response between in its solution and in the fibremat meant that the eLACCO1.1 in the core–shell fibremat maintained the original biological activities without protein denaturation and no significant interruption in structural alteration of eLACCO1.1 according to lactate binding irrespective of entangling in the network structure of poly(HPMA/DAMA)/ADH of nanofibers in the core–shell fibremats. eLACCO1.1 has been examined for its detection selectivity towards lactate at in vitro experiments,16 and this selectivity should be maintained in the current lactate sensor fibermat created by immobilizing eLACCO1.1.
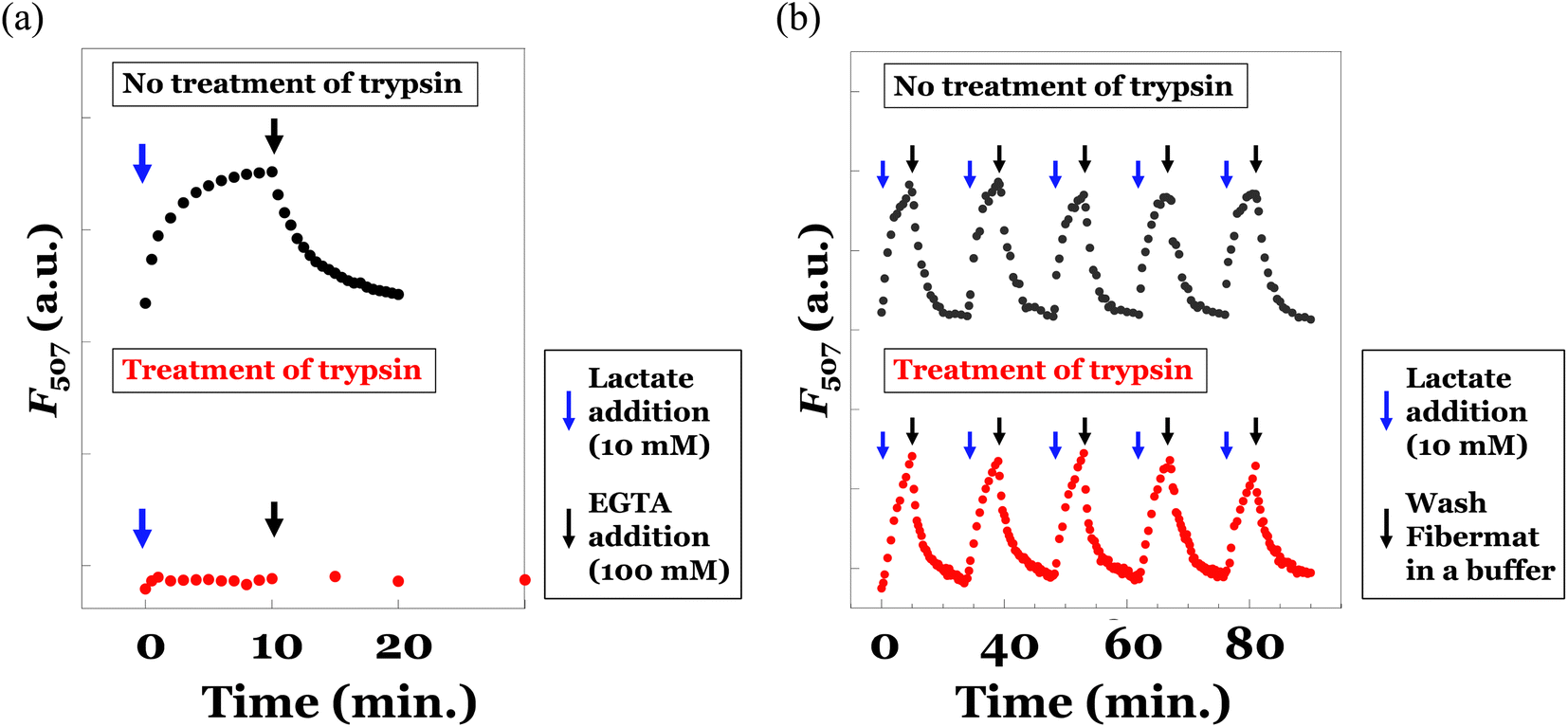 | ||
| Fig. 7 (a) Time-course of F507 of eLACCO1.1 (0.45 μM) in 100 mM MOPS buffer (pH 7), supplemented with 100 mM KCl and 1 mM CaCl2, after addition of lactate (10 mM, blue arrow) and after further adding EGTA (100 mM, black arrow). As a reference, the same experiment was performed after 2 h treatment of trypsin at 37 °C. Excitation wavelength was set by 470 nm. (b) Time-course of fluorescence intensities at 507 nm (F507) of the eLACCO1.1-encapsulated fibremat immersing in 100 mM MOPS buffer (pH 7), supplemented with 100 mM KCl and 1 mM CaCl2, after addition (10 mM) (blue arrow) or removement of lactate (i.e., washing fibremat with a buffer, black arrow), which were repeated in five times (black dots). To clarify the protection effects by encapsulating in the fibremat nanofibers from protease, the same experiment was performed after 2 h treatment of trypsin at 37 °C (red dots). Excitation wavelength was set by 470 nm. | ||
As an advantage to encapsulating in the nanofibers of fibremats, protection from the protease digestion of the encapsulating eLACCO1.1 was expected. In our previous study, when immobilizing enzyme proteins within the core-fiber part of the constitutive nanofibers in the poly(AM/DAAM)/ADH-PCL core–shell fibremat, we were able to prevent the penetration of an enzyme inhibition peptide (specifically, trypsin-chymotrypsin inhibitor from glycine max, soybean, with a MW of 7.9 kDa) into the core-fiber interior.25 This was achieved due to the characteristic molecular permeability properties that depend on MW. Because of similar no permeation property of high MW molecules through the nanofibers of poly(HPMA/DAMA)/ADH-nylon 6 core–shell fibremats, proteases could not approach to the encapsulated eLACCO1.1. To confirm whether this effect could function or not, we also did the same experiments after 2 h treatment of trypsin at 37 °C for both the solution sample of eLACCO1.1 and the fibremat sample of eLACCO1.1. The obtained results were summarized in the upper graphs of Fig. 7(a) and (b), respectively. For the solution sample of eLACCO1.1, the fluorescence response of eLACCO1.1 was perfectly disappeared by trypsin, probably due to trypsin digestion of eLACCO1.1. Whereas, for the fibremat sample of eLACCO1.1, no difference in fluorescence response according to lactate addition even after 5 times repeat was successfully observed. These results implied that encapsulation in the nanofibers of poly(HPMA/DAMA)/ADH-nylon 6 core–shell fibremat should have great advantage to functionalize eLACCO1.1 for biological samples, including various contaminants such as protease, for quantification of lactate concentration.
Next, we evaluated the titration curve of fluorescence intensity of eLACCO1.1 vs. lactate concentration. The results of the solution sample of eLACCO1.1 (as a reference) and the fibremat sample of eLACCO1.1 were summarized in Fig. 8(a) and (b). According to increase in lactate concentration, fluorescence intensity of eLACCO1.1 solution was increased and upon more than 100 mM concentration range its value became more than 14 times and saturated. Due to the lack of transparency in the current fibremats, there was a certain reduction (approximately 25%) in the apparent fluorescence emission from the fibremat. However, similarly, the fibremat sample of eLACCO1.1 also showed a significant increase in fluorescence intensity in response to lactate concentration, exceeding 10 times its initial value, and eventually reaching saturation in the 100 mM range of lactate concentration. The significant increase (more than 10 times) in fluorescence signal intensity observed in response to changes in lactate concentration with eLACCO1.1 effectively mitigated issues such as measurement errors and false positives in lactate concentration quantification.33 By applying the Benesi–Hildebrand method for the titration data, we could estimate the dissociation constants (Kd, mM) in solution and in fibremat and were 2.74 and 2.97 mM, respectively. Comparable Kds in solution and in fibremat also support that the encapsulated eLACCO1.1 maintained the original biological activity without protein denaturation and the poly(HPMA/DAMA)/ADH-nylon 6 fibremat did not hamper molecular interaction between eLACCO1.1 and lactate.
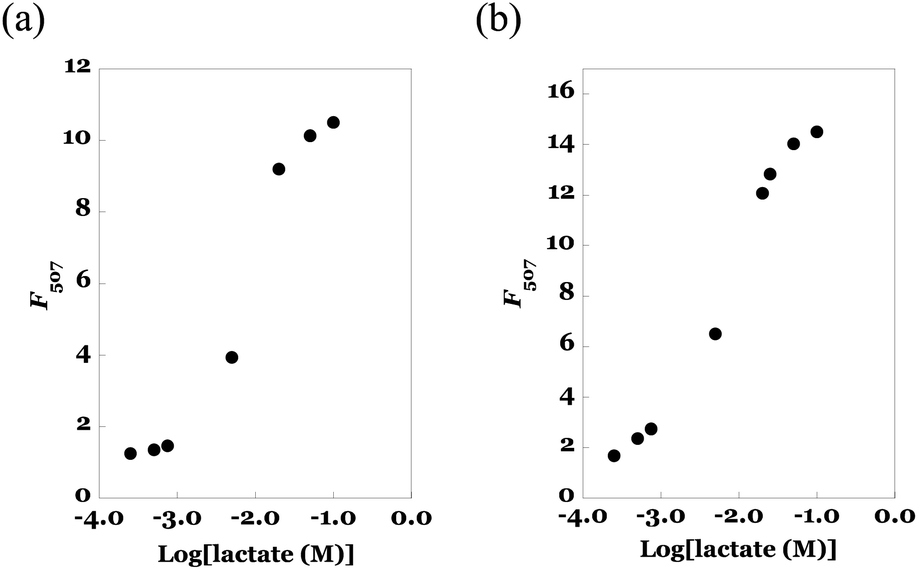 | ||
| Fig. 8 Alteration in fluorescence intensities at 507 nm (F507) of (a) the eLACCO1.1-encapsulated fibremat and (b) the eLACCO1.1 (0.45 μM) in a buffer solution in respond to addition of different concentration lactate (0–100 mM) at 25 °C. Excitation wavelength was 470 nm. | ||
Actual difference in fluorescence emission from the eLACCO1.1-encapsulated fibremats was evaluated using a confocal laser scanning microscope (CLSM). As shown in Fig. 9, fluorescence emission from each nanofiber, harbouring eLACCO1.1, was increased according to an increase in lactate encapsulation of eLACCO1.1 within the fibremat nanofibers and also support the successful encapsulation of eLACCO1.1 without protein denaturation. Furthermore, because the fluorescent emissions were only from the isolated nanofibers with sizes less than 1 μm, it suggests that the eLACCO1.1-encapsulated core–shell fibremat could be utilized to assess alterations in lactate concentration in the local area of approximately ∼10 μm square.
 | ||
| Fig. 9 Confocal microscopic images of the eLACCO1.1-encapsulated fibremats in respond to addition of different concentration of lactate (0–100 mM). | ||
3. Conclusions
As a summary, in this study we developed core–shell fibremats with eLACCO1.1 encapsulated in the core-fiber part and evaluated its fluorescence response to lactate. The poly (HPMA/DAMA)/ADH-nylon 6 core–shell fibremat exhibited a characteristic molecular permeability, allowing good permeability of low MW molecules like lactate while retaining high MW sensor-proteins like eLACCO1.1 without leakage. The lactate sensor protein eLACCO1.1 is retained by physically intertwining within the network structure of the hydrated polymer gels of poly(HPMA/DAMA)/ADH, forming the core-fiber part. This immobilization method does not rely on covalent bonds or adsorption onto a 2D material surface, thus preventing functional deactivation due to structural perturbations in protein tertiary structures. These features of core–shell fibermat is expected to enable immobilization with minimized denaturation for various sensor proteins. As a result, the encapsulated eLACCO1.1 showed a similar fluorescence response (dissociation constant and response rate) to lactate as in solution.Moreover, due to the filtering effect of the fibremat nanofibers, the fluorescence response was maintained even after treatment of protease. Even in cases where measurement samples collected from the body retain residual metabolite enzymes, the current lactate sensor core–shell fibermat would demonstrate the ability to mitigate the influence of metabolic enzymes, for the sake of unique molecular permeability of fibermat nanofibers. These findings suggest that eLACCO1.1-encapsulated poly(HPMA/DAMA)/ADH-nylon 6 core–shell fibremats can be used for continuous lactate monitoring in various biological samples with practical applications, such as wearable sensors for personal health monitoring.34 To the best of our knowledge, this is the first example of immobilizing sensor proteins inside nanofibers of fibremats and indicating its effectiveness as a matrix for functioning sensor proteins, irrespective of other contaminants, especially proteases. Not limited to eLACCO1.1, for the continuous quantification of specific metabolites or biological target molecules in biological samples, the combined use of methods that can utilize proteinous sensors of them irrespective of protease digestion and other contaminants has been recognized as necessary, and the encapsulation of sensor proteins in the ligand-permeable materials' matrix was found to be effective. Our fibremat technology should contribute as a new method in this category. Our research group is currently working on constructing core–shell fibremats encapsulating other sensor proteins, too and applying these sensor-protein-encapsulated core–shell fibremats as sensor materials for cultured cells.
4. Experimental section
4.1 Materials
Unless stated otherwise, all the chemicals and reagents were commercially obtained and used without further purification. Methacryloyl chloride, (±)-1-amino-2-propanol, 2,2,2-trifluoroethanol (TFE), methacrylonitrile, 2,2′-azobis(isobutyronitrile) (AIBN), disodium phosphate (Na2HPO4), sodium dihydrogen phosphate (NaH2PO4), and fluorescein were purchased from Wako Pure Chemical Industries, Ltd. (Japan). Diacetone alcohol and adipic acid dihydrazide (ADH) were purchased from Tokyo Chemical Industry Co., Ltd. (Japan). Nylon 6 (Cat. No., 181110) were purchased from Sigma–Aldrich (USA). Synthesis of the 2-hydroxypropyl methacrylamide (HPMA) was conducted according to the previous report.25 Green fluorescent protein was produced by E. coli BL21(DE3) strain using the pET-3b vector, harbouring the superfolder GFP gene. eLACCO1.1 was produced by E. coli DH10B strain (Thermo Fisher Scientific) using pBAD-eLACCO1.1.154.2 Synthesis of diacetone methacrylamide (DAMA)26
Methacrylonitrile (7.5 mL, 89.4 mmol) was added dropwise to diacetone alcohol (11.2 mL, 89.4 mmol) in con-H2SO4 (10 mL) with keeping the temperature below 10 °C using ice bath. After further 1 h stirring at 4 °C, the resulting solution was extracted with dichloromethane. The obtained organic layer was dried over Na2SO4 and solvent was removed in vacuo. The oily residue was subjected to silica gel chromatography (eluent; CH2Cl2 to CH2Cl2/MeOH = 10/1) and gave the target product as colourless oil. Yield 8.1 g (49%). 1H-NMR (400 MHz, CDCl3) 1.41 (s, 6H, C![[H with combining low line]](https://www.rsc.org/images/entities/char_0048_0332.gif) 3–), 1.89 (s, 3H, C3–C(
3–), 1.89 (s, 3H, C3–C(![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) CH2)–C(O)–), 2.11 (s, 3H, C3–C(O)–), 2.91 (s, 2H, –C2–C(O)), 5.72, 5.79 (s, 1H each, CH3–C(C2)–), 6.30 (brs, 1H, –C(O)N–).
CH2)–C(O)–), 2.11 (s, 3H, C3–C(O)–), 2.91 (s, 2H, –C2–C(O)), 5.72, 5.79 (s, 1H each, CH3–C(C2)–), 6.30 (brs, 1H, –C(O)N–).
4.3 Synthesis of the copolymer of HPMA and DAMA, poly(HPMA/DAMA)
DAMA (1.29 g, 7.0 mmol), HPMA (4.00 g, 27.9 mmol), and AIBN (8.70 mg, 53.0 μmol) were dissolved in DMSO (5 mL) and by freeze-pump threw the dissolved oxygen was removed. Then this mixture was heated at 90 °C for 3 h under N2 atmosphere to progress polymerization. To precipitate the target copolymer from the resulting reaction mixture, it was added to acetonitrile. By successive reprecipitation using acetonitrile as a poor solvent the purification of the target copolymer was performed. Yield 4.1 g (78%). 1H-NMR (400 MHz, CDCl3) 0.5–2.1 (m, 33H, –[CH2–C(C3)–C(O)]n–, –NH–C(C3)2–CH2– in DAMA unit, and C3–C(OH)– in HPMA unit), 2.10 (s, 3H,C3–C(O)–CH2– in DAMA), 2.52–3.1 (brs, 10H, –NH–C2–CH(OH)– in HPMA and –C(CH3)2–C2–C(O)– in DAMA), 3.70 (s, 4H, CH3–C(OH)–CH2–), 4.70 (s, 4H, CH3–CH(O)–CH2–), 6.52 (brs, 1H, –C(O)N– in the DAMA unit), 7.18 (brs, 4H, –C(O)N– in the HPMA unit). From the proton ratio at 7.18 ppm (for amide NH in HPMA) and 6.52 ppm (for amide NH in DAMA), we could estimate the molar ratio of HPMA to DAMA in the obtained copolymer to be 8:2. The Mn and polydispersity index (PDI) were determined by gel permeation chromatography by referring the poly(ethylene glycol) (PEG) standards and were 153 kDa and 1.51, respectively.
4.4 Complex viscosity measurement by a stress-controlled rheometer
The time-course of complex viscosity (Pa s) of a 25 wt% solution of poly(HPMA/DAMA) in 100 mM phosphate buffer (pH 7) was measured over 240 min after the addition of ADH (0.5 mol equivalent to DAMA unit) or without ADH using an Anton Paar 302 Rheometer (MCR 302/WESP, Anton Paar GmbH, Austria) with a parallel plate geometry (PP25) at 25 °C. The gap size was set to 1 mm. Rotational measurements were conducted with a shear strain of 50% and an angular frequency of 5 rad s−1. Additionally, to assess the impact of protein coexistence in this polymer solution on complex viscosity (Pa s), the same experiment was performed using a 25 wt% solution of poly(HPMA/DAMA) with 5 wt% lactase in 100 mM phosphate buffer (pH 7) over 240 min after the addition of ADH (0.5 mol equivalent to DAMA unit).4.5 Construction of the core–shell fibremats, consisting of the core nanofibers of poly(HPMA/DAMA)/ADH and the shell layer of nylon 6
ADH [0.5 molar equivalent with respect to the DAMA unit in poly(HPMA/DAMA)] was added to a poly(HPMA/DAMA) solution [0.5 g dissolved in 2.5 mL of phosphate buffer (100 mM; pH 7)] and transferred to a Luer-Lok syringe (5 mL). To encapsulate proteins such as GFP or eLACCO1.1, each protein (12.5 nmol of GFP or 1.2 nmol of eLACCO1.1) was added to this solution. To encapsulate fluorescent low MW molecule such as fluorescein, 0.142 nmol of fluorescein was added to this solution. Concomitantly, 10 wt% nylon 6 solution dissolved in TFE was prepared and transferred to another Luer-Lok syringe (5 mL). Both syringes were then placed in different syringe pumps fitted with a linear actuator (KDS-100, KD Scientific, USA) and connected to a co-axial spinneret (MECC Co., Ltd., Japan) equipped with 27 G needle (Terumo Corp.) for passing the poly(HPMA/DAAM)/ADH core solution through a polytetrafluoroethylene (PTFE) tube. The poly(HPMA/DAAM)/ADH and nylon 6 solutions were then electrospun (SD-02, MECC Co., Ltd., Japan) at linear extrusion velocities of 0.3, 1.2 mL h−1, respectively, at a high voltage (20 kV). The obtained fibremat core and shell components were poly(HPMA/DAMA)/ADH and nylon 6, respectively. The distance between the spinneret tip and electrically grounded collector (aluminium plate; 150 × 200 mm) was set to 180 mm for constructing the poly(HPMA/DAMA)/ADH-nylon 6 fibremats.4.6 Scanning electron microscopy (SEM)
The samples were coated with amorphous osmium by plasma chemical vapor deposition using an OPC60A vacuum evaporator (Filgen, Inc., Japan). The sample morphologies were examined using field-emission SEM (JSM-7800F, JEOL, Japan) operating at 3 kV acceleration. The fibremat nanofibre mean diameters and corresponding standard deviations were evaluated using the SEM images of 30 nanofibres and ImageJ software.4.7 Transmission electron microscopy (TEM)
The TEM samples were prepared by directly collecting the electrospun core–shell nanofibres from the co-axial spinneret on a TEM grid comprising a formvar carbon film on a 100-mesh copper wire grid (50) [Okenshoji Co., Ltd., Japan]. To obtain adequate image contrast between the core and shell, sodium phosphotungstate was mixed at a final concentration of 0.0001% (w/v) with the core precursor solution comprising poly(HPMA/DAMA) and ADH. The TEM images were acquired using a JEM-1400Plus instrument (JEOL, Japan) operating at 100 kV acceleration.4.8 Attenuated total-reflectance Fourier-transform infrared (ATR–FTIR) spectroscopy
The ATR–FTIR spectra were acquired using an FT-IR-4000 spectrometer (JASCO, Japan) equipped with an ATR PRO450-S unit (JASCO, Japan). The free-induction-decay (FID) spectra were scanned 200 times at 25 °C, accumulated, and Fourier-transformed to obtain the FTIR spectra at a resolution of 4 cm−1.Leakage/release of encapsulated fluorescein or GFP upon fibremat immersion in a buffer.
The loaded fibremats (1 mg) were cut in round pieces (5 × 5 mm; 1 mg) and placed in a 24-well plate and immersed in 1.0 mL of phosphate buffer (pH 7; 100 mM) for defined periods (0–170 h) at 25 °C and gently orbitally shaken at 100 rpm. The fibremats were then removed from the buffer, and the F520 values of the remaining solutions were measured using a plate reader (SH-9000Lab, Hitachi, Japan). By comparison to the standard curve, the fluorophore release degree was determined during the assay period.
4.9 Fluorescence response of eLACCO1.1 in fibremat or in solution
The eLACCO1.1-encapsulated fibremat was cut into a round shape (6.4 mm in diameter) using a steel punch and placed at the bottom of a 96-well black plate. The fibremat was initially pre-immersed in 200 μL of 150 mM MOPS buffer (pH 7) and then alternately immersed in 200 μL of MOPS buffer (150 mM, pH 7) containing 1 mM CaCl2, 100 mM KCl, and various concentrations of lactate (0–100 mM) for 10 min. The fluorescence emission at 507 nm (F507) from the fibremat (excited at 470 nm) was measured using a fluorescence microplate reader (SH-9000Lab, Hitachi, Japan). For the time-course measurements of F507 for the eLACCO1.1-encapsulated fibremat after immersion in a 10 mM lactate solution, fluorescence observations were performed every minute for the first 10 min following immersion in 200 μL of MOPS buffer (150 mM, pH 7), containing 1 mM CaCl2, 100 mM KCl, and 10 mM lactate. Subsequently, the solution was replaced with MOPS buffer (150 mM, pH 7) without lactate, and the decrease in F507 was recorded every minute for the next 10 min. This process was repeated five times. In a separate experiment involving trypsin treatment, the eLACCO1.1-encapsulated fibremat in a 96-well plate was initially immersed in 100 μL of 150 mM MOPS buffer (pH 7) containing trypsin (0.5 mg mL−1) for 2 hours at 37 °C. Then, successive additions of 100 μL of 150 mM MOPS buffer (pH 7), containing 2 mM CaCl2 (resulting in a final concentration of 1 mM), 200 mM KCl (resulting in a final concentration of 100 mM), and various concentrations of lactate (resulting in final concentrations of 0–100 mM), were made, and the time-course observation of the initial increase in F507 was conducted. The subsequent processes were performed similarly to the samples without trypsin treatment. To observe the changes in fluorescence emission of eLACCO1.1 in solution by lactate, 0.9 μM of eLACCO1.1 in 100 μL of 150 mM MOPS buffer (pH 7) was mixed with 100 μL of 150 mM MOPS buffer (pH 7), containing 2 mM CaCl2 (resulting in a final concentration of 1 mM), 200 mM KCl (resulting in a final concentration of 100 mM), and various concentrations of lactate (resulting in a final concentration of 0–100 mM). After 10 min of incubation, fluorescence emissions upon addition of different lactate concentrations were measured using a fluorescence microplate reader (SH-9000Lab, Hitachi, Japan). For the time-course alterations of F507 of eLACCO1.1 in solution with 10 mM lactate, fluorescence observations were performed every minute following successive additions of 100 μL of MOPS buffer (150 mM, pH 7), containing 2 mM CaCl2 (resulting in a final concentration of 1 mM), 200 mM KCl (resulting in a final concentration of 100 mM), and 20 mM lactate (resulting in a final concentration of 10 mM) to eLACCO1.1 in 100 μL of MOPS buffer (150 mM, pH 7). To examine fluorescence decrease due to lactate dissociation from eLACCO1.1, EGTA (100 mM) was added to this sample. Similarly, in the experiment with trypsin treatment, eLACCO1.1 (0.9 μM) in 100 μL of 150 mM MOPS buffer (pH 7) was treated with trypsin (final concentration of 0.5 mg mL−1) for 2 hours at 37 °C. Then, successive additions of 100 μL of 150 mM MOPS buffer, containing 2 mM CaCl2 (resulting in a final concentration of 1 mM), 200 mM KCl (resulting in a final concentration of 100 mM), and various concentrations of lactate (resulting in a final concentration of 0–100 mM), were made, and the time-course observation of F507 was performed.4.10 Confocal laser scanning microscope observation of the eLACCO1.1-encapsulated fibremat after addition of lactate
The eLACCO1.1-encapsulated fibremat was cut in a square (10 mm × 10 mm) and put onto a coverslip (24 mm × 60 mm, micro cover glass neo, Matsunami, Japan). After pre-immersion in 200 μL of 150 mM MOPS buffer (pH 7), the fibremat was immersed in the MOPS buffer (150 mM, pH 7), containing 1 mM CaCl2, 100 mM KCl, and the different concentration lactate (0–100 mM) for 10 min. By placing another coverslip on top, the fibremat sample was sandwiched between two coverslips and subjected to observation of fluorescence images from the fibremats using a confocal laser scanning microscope (LSM880 microscope, Zeiss, German). Samples were imaged using a 63× objective lens. Excitation was carried out using the 488 nm laser. To detect emission fiuorescence from eLACCO1.1, we used the band-pass filters of 493–634 nm.Author contributions
T. Mizuno conceived the project. Y. Kato and T. Mizuno designed the experiments. Y. Kato performed the experiments and data analysis. Y. Nasu and R. E. Campbell supplied eLACCO1.1. S. Iwata measured dynamic viscoelastic properties of polymer solutions using a rheometer. A. Obata supported fibremat construction. K. Nagata supported microscopic observations of fibremats. T. Mizuno wrote the manuscript. All the authors edited and approved the finalized manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We appreciate Dr Shinichi Matsuoka (Graduate School of Engineering, Nagoya Institute of Technology) helping GPC analysis of poly(HPMA/DAMA). This work was supported by the Toshiaki Ogasawara Memorial Foundation.Notes and references
- L. Rassaei, W. Olthuis, S. Tsujimura, E. J. Sudholter and A. van den Berg, Anal. Bioanal. Chem., 2014, 406, 123–137 CrossRef CAS PubMed.
- J. J. Garcia-Guzman, A. Sierra-Padilla, J. M. Palacios-Santander, J. J. Fernandez-Alba, C. G. Macias and L. Cubillana-Aguilera, Biosensors, 2022, 12, 919 CrossRef CAS PubMed.
- L. J. Currano, F. C. Sage, M. Hagedon, L. Hamilton, J. Patrone and K. Gerasopoulos, Sci. Rep., 2018, 8, 15890 CrossRef PubMed.
- D. Nguyen, M. M. Lawrence, H. Berg, M. A. Lyons, S. Shreim, M. T. Keating, J. Weidling and E. L. Botvinick, ACS Sens., 2022, 7, 441–452 CrossRef CAS PubMed.
- J. W. Kim and C. V. Dang, Cancer Res., 2006, 66, 8927–8930 CrossRef CAS PubMed.
- M. Solis-Maldonado, M. P. Miro, A. I. Acuna, A. Covarrubias-Pinto, A. Loaiza, G. Mayorga, F. A. Beltran, C. Cepeda, M. S. Levine, I. I. Concha, L. F. Batiz, M. A. Carrasco and M. A. Castro, CNS Neurosci. Ther., 2018, 24, 343–352 CrossRef CAS PubMed.
- W. Zhou, M. Choi, D. Margineantu, L. Margaretha, J. Hesson, C. Cavanaugh, C. A. Blau, M. S. Horwitz, D. Hockenbery, C. Ware and H. Ruohola-Baker, EMBO J., 2012, 31, 2103–2116 CrossRef CAS PubMed.
- R. Monosik, M. Stredansky, G. Greif and E. Sturdik, Food Control, 2012, 23, 238–244 CrossRef CAS.
- F. K. Sartain, X. P. Yang and C. R. Lowe, Chem.–Eur. J., 2008, 14, 4060–4067 CrossRef CAS PubMed.
- J. S. Hansen, T. Hoeg-Jensen and J. B. Christensen, Tetrahedron, 2017, 73, 3010–3013 CrossRef CAS.
- N. V. Zaryanov, V. N. Nikitina, E. V. Karpova, E. E. Karyakina and A. A. Karyakin, Anal. Chem., 2017, 89, 11198–11202 CrossRef CAS PubMed.
- D. B. Groegel, M. Link, A. Duerkop and O. S. Wolfbeis, Chembiochem, 2011, 12, 2779–2785 CrossRef CAS PubMed.
- N. Kumar, Y. J. Lin, Y. C. Huang, Y. T. Liao and S. P. Lin, Talanta, 2023, 265, 124888 CrossRef CAS PubMed.
- A. San Martin, S. Ceballo, I. Ruminot, R. Lerchundi, W. B. Frommer and L. F. Barros, PLoS ONE, 2013, 8, e57712 CrossRef CAS PubMed.
- A. Galaz, F. Cortes-Molina, R. Arce-Molina, I. Romero-Gomez, G. A. Mardones, L. Felipe Barros and A. San Martin, Anal. Chem., 2020, 92, 10643–10650 CrossRef CAS PubMed.
- Y. Nasu, C. Murphy-Royal, Y. Wen, J. N. Haidey, R. S. Molina, A. Aggarwal, S. Zhang, Y. Kamijo, M. E. Paquet, K. Podgorski, M. Drobizhev, J. S. Bains, M. J. Lemieux, G. R. Gordon and R. E. Campbell, Nat. Commun., 2021, 12, 7058 CrossRef CAS PubMed.
- D. Koveal, P. C. Rosen, D. J. Meyer, C. M. Diaz-Garcia, Y. Wang, L. H. Cai, P. J. Chou, D. A. Weitz and G. Yellen, Nat. Commun., 2022, 13, 2919 CrossRef CAS PubMed.
- C. I. Li, Y. H. Lin, C. L. Shih, J. P. Tsaur and L. K. Chau, Biosens. Bioelectron., 2002, 17, 323–330 CrossRef CAS PubMed.
- Y. Zhang, L. Xu and J. Ge, Nano Lett., 2022, 22, 5029–5036 CrossRef CAS PubMed.
- X. Xuan, C. Perez-Rafols, C. Chen, M. Cuartero and G. A. Crespo, ACS Sens., 2021, 6, 2763–2771 CrossRef CAS PubMed.
- M. E. Payne, A. Zamarayeva, V. I. Pister, N. A. D. Yamamoto and A. C. Arias, Sci. Rep., 2019, 9, 13720 CrossRef PubMed.
- J. J. Garcia-Guzman, A. Sierra-Padilla, J. M. Palacios-Santander, J. J. Fernandez-Alba, C. G. Macias and L. Cubillana-Aguilera, Biosensors, 2022, 12, 919 CrossRef CAS PubMed.
- S. Koeda, K. Ichiki, N. Iwanaga, K. Mizuno, M. Shibata, A. Obata, T. Kasuga and T. Mizuno, Langmuir, 2016, 32, 221–229 CrossRef CAS PubMed.
- Y. Ido, A. L. B. Macon, M. Iguchi, Y. Ozeki, S. Koeda, A. Obata, T. Kasuga and T. Mizuno, Polymer, 2017, 132, 342–352 CrossRef CAS.
- Y. Tanikawa, Y. Ido, R. Ando, A. Obata, K. Nagata, T. Kasuga and T. Mizuno, Bull. Chem. Soc. Jpn., 2020, 93, 1155–1163 CrossRef CAS.
- T. Ishiguro, A. Obata, K. Nagata, T. Kasuga and T. Mizuno, RSC Adv., 2022, 12, 34931–34940 RSC.
- R. E. Bird, S. A. Lemmel, X. Yu and Q. A. Zhou, Bioconjug Chem., 2021, 32, 2457–2479 CrossRef CAS PubMed.
- K. Mizuno, S. H. Koeda, A. Obata, J. Sumaoka, T. Kasuga, J. R. Jones and T. Mizuno, Langmuir, 2017, 33, 4028–4035 CrossRef CAS PubMed.
- M. F. Ebbesen, D. H. Schaffert, M. L. Crowley, D. Oupicky and K. A. Howard, J. Polym. Sci. Pol. Chem., 2013, 51, 5091–5099 CrossRef CAS.
- D. I. Hoke, J. Polym. Sci., Part A-1: Polym. Chem., 1971, 9, 2753–3081 CrossRef.
- R. B. Bird, Dynamics of polymeric liquids, Wiley, New York, 2nd edn, 1987 Search PubMed.
- C. W. Macosko and G. Strbac, Ebscohost and MyiLibrary, Rheology: Principles, Measurements, and Applications, John Wiley & Sons VCH, Chichester, West Sussex, England; Hoboken, NJ, New York, 2004 Search PubMed.
- V. Naresh and N. Lee, Sensors, 2021, 21, 1109 CrossRef CAS PubMed.
- X. Xuan, C. Perez-Rafols, C. Chen, M. Cuartero and G. A. Crespo, ACS Sens, 2021, 6, 2763–2771 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3ra06108f |
| This journal is © The Royal Society of Chemistry 2023 |