DOI:
10.1039/D3RA03957A
(Paper)
RSC Adv., 2023,
13, 29959-29974
Tailoring the intrinsic magneto-electronic, mechanical, thermo-physical and thermoelectric response of cobalt-based Heusler alloys: an ab initio insight
Received
13th June 2023
, Accepted 2nd October 2023
First published on 13th October 2023
Abstract
We conducted a comprehensive analysis of the fundamental properties of CoHfSi and CoHfGe half-Heusler alloys using density functional theory simulations implemented in Wien2k. To begin, structural optimization revealed that both alloys effectively adopt a cubic C1b structure, with Y1 as the dominant ferromagnetic phase. Electronic properties were computed using various approximation schemes, including the Generalized Gradient Approximation and the modified Becke–Johnson potential. The examination of electronic band structures and their accompanying density of states using the modified Becke–Johnson functional approach unveiled their half-metallic nature. In this context, the spin-up channel exhibited semiconductor behaviour, while the spin-down channel displayed metallic characteristics. Additionally, the spin-splitting observed in their resulting band structures contributed to a net magnetism within their lattice structure, making them promising candidates for spintronic applications. We also scrutinized Seebeck coefficients, electrical conductivity, thermal conductivity, and power factor to gain a better understanding of their thermoelectric properties.
1. Introduction
The rapid advancement in Heusler alloys is the subject of interest due to which they display high spin polarisation, great electronics, thermoelectric properties, etc. They came into the limelight for industrialists and researchers. They have been significantly used for various applications such as shape memory devices, spin filters, and so on.1 The applicability in numerous technologies, spintronic materials commonly used are single spin electron sources and spin injection. Spin-polarized charge carriers are a source for spintronics, and Heusler alloys are the best choice.2,3 A little bit of overview on Heusler alloys is one based on the arrangement of the atomic locations in a unit cell, such as (i) full Heusler (FH), (ii) half Heusler, (iii) inverse, and (iv) quaternary.4 The full Heusler alloys can be used to derive the structure of the HH alloys. The structural formula of FH alloys is X2YZ, where X and Y are transition metals (TM) or alkaline earth metals, respectively, and Z is the primary group element.5 FH alloys have an FCC (L21) crystal structure with four interpenetrating layers. The crystal structure of HH alloys is C1b, with the structural composition formula XYZ.6 Compared to FH alloys, HH alloys have three interpenetrating FCC lattices and one vacant position. A family of quaternary Heusler alloys is characterized by the structural formula XX′YZ where X, X′, and Y are all TM, and Z is the sp-3 element.7
Half-metallic ferromagnets (HMFs) are an excellent choice since the minority band is semiconducting with a gap at the Fermi level EF, resulting in 100% spin polarisation at EF, while the majority band acts like a metal.8–10 Thermoelectric, superconductors, topological insulators, spintronic devices, thin-film solar cells, and shape memory behaviour are just a few fields where Heusler alloys are used.
Among the most frequently used materials for practical spintronic applications, where magnetic sensors and spin injectors are employed, half-metal ferromagnets (HMFs) are commonly used. Two different ways of spin characteristics describe the half-metallic ferromagnets. One is a spin-up with a metallic nature, and another spin-down state has a semiconductor nature.
NiMnSb half-Heusler is the first half-metal ferromagnetic material discovered.11 There have since been many experimental and theoretical studies carried out on Heusler materials, full-Heusler alloys such Mn2IrAl, X2VSi (X = Ti, Co), M2IrSi (M = Ti, Cr, and Mn), Fe2−xCoxTiGe, and M2IrSi (M = Ti, Cr, and Mn), Fe2−xCoxTiGe (x = 0, 0.5, 1, 1.5, 2), Ir2MnSi, Fe2MnP,12–17 and half-Heusler alloys such as CrFeZ (Z = Si, Sn and Ge), MnZrX (X = In, C, Si, Ge)18–20 and quaternary Heusler's alloys such as CoRuFeSi and NiCoMnSb.21,22
Our choice of CoHfSi and CoHfGe half-Heusler alloys is motivated by their unique properties like electronic and thermoelectric properties. They exhibit features such as half-metallicity, high thermoelectric efficiency, and good mechanical stability. Thermoelectric materials like CoHfSi and CoHfGe have gained considerable attention due to their potential for converting waste heat into electricity, making them relevant in the context of sustainable energy solutions. The study of half-Heusler alloys and their properties is a relatively new and emerging research area in materials science. These materials offer opportunities to explore novel physical phenomena and develop innovative technologies, making them an attractive focus for research CoHfSi and CoHfGe alloys have demonstrated versatility in various applications, including thermoelectric generators and spintronic devices.
An investigation of the structural, mechanical, electronic, magnetic, thermodynamic and thermoelectric properties of half-Heusler compounds CoHfSi and CoHfGe are presented. Despite their half-metallic nature, these two compounds have not been previously investigated either experimentally or theoretically. In this study, the main goal is to analyse these alloys in detail and explore some of their new properties.
2. Computational method
At the atomic scale, computational methods have gained substantial significance for analyzing systems, be they molecules or solids. These methods serve as a crucial foundation for interpreting experimental results. In our study, we employed the Wien2k simulation package23 to compute various physical properties of CoHfSi and CoHfGe. This involved solving Kohn–Sham equations using density functional theory (DFT).24 During the DFT calculations, we utilized different exchange–correlation functionals, namely the generalized gradient approximation (GGA) and Tran and Blaha modified version of the Becke–Johnson (TB-mBJ) potential.25,26 In the GGA approach, the exchange–correlation (EXC) is determined by the local charge density and its associated gradient. However, GGA can encounter limitations when dealing with systems containing d/f electrons due to issues like self-interaction effects and inadequate potential representation for highly localized states. Consequently, to achieve more accurate descriptions of such systems, the GGA method often needs to be supplemented with modified approximations. Therefore, we adopted the mBJ method, which offers a reasonably straightforward and ab initio approach, to provide precise treatments for highly correlated d/f electron systems. Our computational approach, known as the FP-LAPW (Full-Potential Linear Augmented Plane Wave) method, divides the crystal space into non-overlapping muffin tin spheres, with RMT representing the radius of these muffin tin (MT) spheres. The regions beyond these muffin tin spheres, known as interstitial regions, are treated with a plane-wave basis set to achieve eigenvalue convergence. To ensure both charge and energy convergence, we employed a linearized augmented plane-wave basis set with lmax = 10 and RMTKmax = 7 (where Kmax denotes the highest possible K-value). To explore the thermoelectric response, we applied semi-classical Boltzmann theory, as implemented in the BoltzTraP code.27 To assess the mechanical properties of these alloys, we conducted elastic constant calculations using Charpin's Cubic-elastic package.28 Furthermore, we evaluated the thermal properties by utilizing the Gibbs2 package in conjunction with the Wien2k code.29
3. Results and discussions
A comprehensive analysis of CoHfSi and CoHfGe alloys is presented, covering various aspects, including their structural, electronic, magnetic, mechanical, thermoelectric, and thermodynamic properties, discussed separately under different sections.
3.1 Structural details
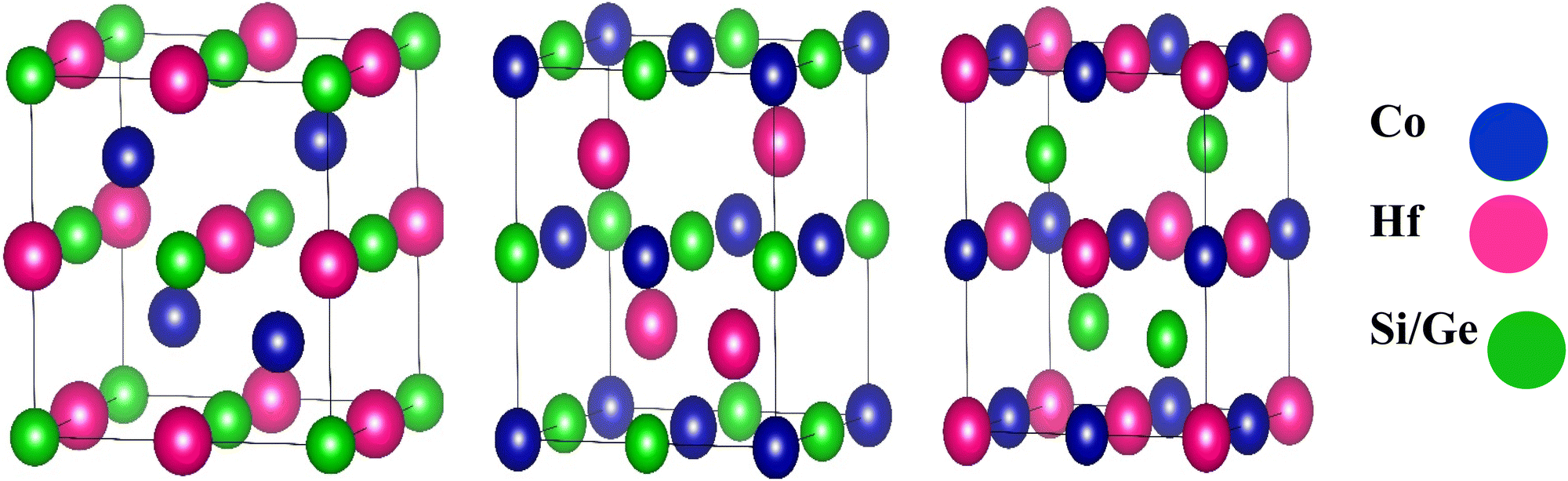
The half-Heusler compounds are ternary compounds with an elemental arrangement of 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1:1. Its chemical formula is XYZ, where X and Y represent the transition metals and Z describes the element in the III-VIA group.30 The half Heusler's alloy of C1b component XYZ consists of three interpenetrating FCC subunits, each occupied by X, Y, and Z atoms, as depicted in Table 1. The Wyckoff positions occupied are 4a (0, 0, 0), 4b (1/2, 1/2, 1/2), and 4c (1/4, 1/4, 1/4).31 The structural representation of half Heusler's alloys CoHfSi and CoHfGe is shown in Fig. 1.
:1:1. Its chemical formula is XYZ, where X and Y represent the transition metals and Z describes the element in the III-VIA group.30 The half Heusler's alloy of C1b component XYZ consists of three interpenetrating FCC subunits, each occupied by X, Y, and Z atoms, as depicted in Table 1. The Wyckoff positions occupied are 4a (0, 0, 0), 4b (1/2, 1/2, 1/2), and 4c (1/4, 1/4, 1/4).31 The structural representation of half Heusler's alloys CoHfSi and CoHfGe is shown in Fig. 1.
Table 1 Summarizes three possible equivalent atomic arrangements for CoHfSi and CoHfGe alloys
| Alloys |
Phase |
X |
Y |
Z |
| CoHfSi and CoHfGe |
Y1 |
(0.25, 0.25, 0.25) |
(0.50, 0.50, 0.50) |
(0, 0, 0) |
| Y2 |
(0, 0, 0) |
(0.25, 0.25, 0.25) |
(0.50, 0.50, 0.50) |
| Y3 |
(0.50, 0.50, 0.50) |
(0, 0, 0) |
(0.25, 0.25, 0.25) |
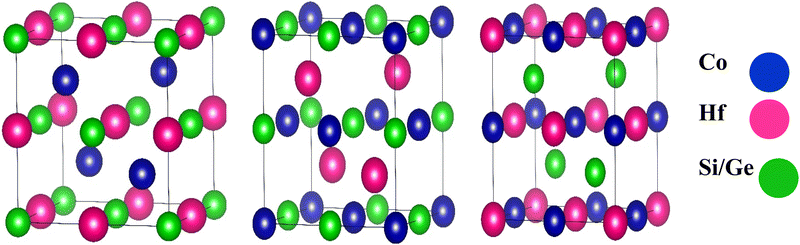 |
| | Fig. 1 Crystal structure of half Heusler alloys CoHfX (X = Si, Ge). | |
So, this completely defines the absolute geometry of the resulting half Heusler alloys CoHfSi and CoHfGe where the atoms are suitably allocated at their corresponding specified positions, thus creating an opportunity to study the structural stability in terms of their total ground state energies. For that we fit the total energy versus unit cell volume curve by the expedition of Murnaghan equation of state (EOS) to optimize the structural arrangement of atoms within three selected configurations i.e.; ferromagnetic (FM), antiferromagnetic (AFM) and non-magnetic (NM) phases. The analysis of the total ground state energy during structural optimization reveals that the ferromagnetic (FM) phase possesses the lowest energy when contrasted with the antiferromagnetic (AFM) and non-magnetic (NM) phases, hence, is energetically more stable as depicted in Fig. 2. Alongside this, it is worth mentioning that the other thermodynamic ground state parameters like volume V0 (nm3), bulk modulus B0 (GPa), its pressure derivative 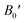 and the minimum energy E0 (eV) have been fetched as well as collected in Table 2.
and the minimum energy E0 (eV) have been fetched as well as collected in Table 2.
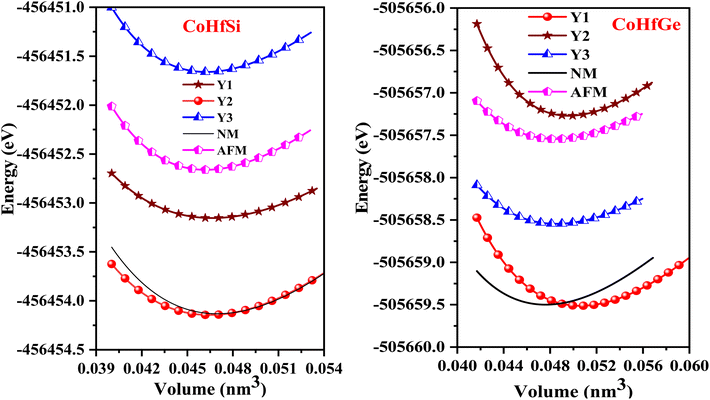 |
| | Fig. 2 Optimization curve in Y1, Y2, Y3, antiferromagnetic and non-magnetic phases for CoHfSi and CoHfGe. | |
Table 2 The computed values of lattice constant (a0 in 10−2 nm), unit cell volume (V0 in nm3), bulk modulus (B0 in GPa), derivative bulk modulus (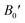 ) and minimum energy (E0 in eV) and cohesive energy (Ecoh) for CoHfSi and CoHfGe alloys
) and minimum energy (E0 in eV) and cohesive energy (Ecoh) for CoHfSi and CoHfGe alloys
| Alloys |
Phase |
a0 |
a0 |
V0 |
B0 |
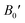
|
E0 |
Ecoh |
| CoHfSi |
FM |
58.73 |
59.95 (ref. 32) |
0.050 |
131.07 |
4.67 |
−456454.12 |
2.99 |
| NM |
58.70 |
|
0.048 |
154.71 |
4.23 |
−456453.08 |
|
| AFM |
58.67 |
|
0.046 |
145.65 |
4.56 |
−456452.65 |
|
| CoHfGe |
FM |
58.75 |
58.92 (ref. 33) |
0.051 |
145.02 |
4.71 |
−505659.56 |
4.22 |
| NM |
58.71 |
|
0.050 |
146.03 |
4.90 |
−505659.42 |
|
| AFM |
58.68 |
|
0.048 |
151.26 |
4.65 |
−505657.59 |
|
| CoHfSb34 |
FM |
59.91 |
|
… |
154.86 |
4.39 |
… |
|
| CoZrSb34 |
FM |
60.31 |
|
… |
148.63 |
4.39 |
… |
|
Cohesive energy (Ecoh) is employed to assess the bonding of strength, a value that corresponds to the numerical difference between the sum of energies of individual atoms and the overall energy of the alloy. For half Heusler alloys cohesive energy can be expressed as:
| |
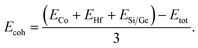 | (1) |
The computed value of Ecoh for both the alloys is recorded in Table 2.
By utilizing the formula, one can determine the enthalpy of formation energy (ΔE) to evaluate the stability of the compounds.
| | |
ΔE = Etotal − aEA − bEB − dEX
| (2) |
where,
Etotal represents the overall energy of CoHfX (X = Si, Ge) with
EA,
EB,
EX representing the energies of Co, Hf, Si and Ge respectively. The computed energy value for CoHfSi and CoHfGe are −2.65 and −3.89 eV respectively. These values demonstrate that the alloys possess negative enthalpy of formation energies, thereby substantiating their stability.
We have investigated the structural, mechanical, electronic, and thermoelectric properties of the CoHfSi and CoHfGe alloys using the GGA and mBJ methods. It is observed that the ferromagnetic phase is stable by the GGA method, while the mBJ method shows a half-metallic ferromagnetic (i.e., half-metallic) phase and can be used in spintronic applications.
3.2 Elastic properties
The important properties of any material are its mechanical properties, which indicate how the material behaves under pressure. Calculating the elastic constants of a material is necessary for investigating its mechanical properties. In technical applications, elastic constants represent the bonding of nature, mechanical strength, dynamic stability, hardness, and thermodynamic parameters of a system.35 There are three independent elastic constants of the structure under investigation based on its cubic symmetry: C11, C12, and C44.36 The C11 constants can be used to demonstrate longitudinal deformation in materials. Similarly, C12 can be used to measure the transverse expansion in materials. The C44 constant can be used to restrict the transverse deformation. Using these constants, we can define the material's stability and stiffness. The independent elastic constants must satisfy the Born-stability criteria,37 to ensure that these materials are mechanically stable.| | |
(C11 > 0; C44 > 0; C11 − C12 > 0; C11 + 2C12 > 0)
| (3) |
To determine the stiffness and compressibility of materials, two important parameters are often measured, namely the bulk modulus and the shear modulus,
| |
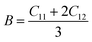 | (4) |
| |
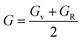 | (5) |
while,
| |
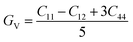 | (6) |
and
where,
GR and
GV are Voigt shear moduli and Reuss shear moduli respectively.
38–40
The magnitude of Young's modulus is the ratio of linear stress to strain, and its values are given in Table 3.
| |
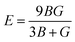 | (7) |
Table 3 Calculated values of elastic parameters (C11, C12, C44, CP, E, B, G in GPa), anisotropy factor (A), Poisson's ratio (σ), Pugh's ratio (B/G), and Debye temperature (θD in K)
| Parameters |
CoHfSi |
CoHfGe |
| C11 |
249.93 |
302.96 |
| C12 |
73.01 |
69.64 |
| C44 |
79.89 |
99.41 |
| B |
131.99 |
147.41 |
| GV |
83.31 |
106.31 |
| GR |
83.10 |
105.66 |
| G |
83.10 |
105.98 |
| E |
206.28 |
256.49 |
| B/G |
1.58 |
1.39 |
| σ |
0.23 |
0.21 |
| A |
0.90 |
0.85 |
| Vl (m s−1) |
3.63 |
3.73 |
| Vt (m s−1) |
6.20 |
6.15 |
| Vm (m s−1) |
4.02 |
4.12 |
| θD |
424.58 |
434.39 |
The Poisson's ratio is the ratio of the transverse strain to the axial strain.41
| |
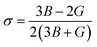 | (8) |
If σ is less than 0.5, the material is compressible, but if it is greater than 0.5, the material becomes incompressible. Based on the evaluated value of σ, it can be concluded that CoHfBi alloy is somewhat compressible. According to Pugh's ratio (B/G), a material is considered ductile if its value is 1.75 or larger; otherwise, it is brittle.42 The computed values of B/G, as reported in Table 3, show that both compounds are ductile. Another way to analyse the nature of the material to be ductile is if the value of Cauchy's pressure (CP = C11 − C12) is positive else it is brittle. The values of CP also signify the ductile character of the CoHfSi and CoHfGe alloy.
The Debye temperature (θD) is a property of solid materials that represents the temperature at which the vibrational energy of the atoms within the material is equivalent to the thermal energy of its surroundings. The Debye temperature is computed using the average sound velocity (νm) one can determine the temperature of a maximum mode of vibration.43
| |
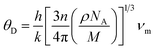 | (9) |
where
h is Plank's constant,
k is Boltzmann's constant,
NA is Avogadro's number,
n is the number of atoms per molecule or the number of atoms per formula unit,
M is the molar mass,
ρ is the density of the unit cell and
νmis the average sound velocity.
| |
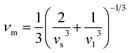 | (10) |
By applying Navier's equation,41 we have computed the longitudinal velocity νl and shear velocity νs by using the values of bulk and shear moduli as:
| |
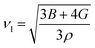 | (11) |
and
The calculated value of longitudinal velocity νl, shear velocity νs and average sound velocity are reported in Table 3. The calculated value of Debye temperature is 424.58 K and 434.39 K for CoHfSi and CoHfGe alloys respectively. When the Debye temperature of a material is greater than the room temperature, it indicates that the vibrational energy of the atoms inside the material is greater than the thermal energy of the surrounding. We determined the melting temperature of these materials with the use of an elastic constant C11 and C33 (ref. 44)
| |
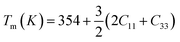 | (12) |
For CoZrSi and CoZrGe, the calculated values of melting temperature (Tm) are 1935.39 K and 1688.04 K, respectively. Since CoZrSi & CoZrGe has a high subscript value it is suggested that the material can maintain its ground state structure over a wide temperature range.
The anisotropic factor (A) is also an important physical property. It provides the nature of the bonding in different crystallographic directions and is described by the equation:
| |
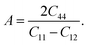 | (13) |
If A = 1, the crystal is considered totally isotropic; if the values are less than one, the crystal is elastically anisotropic. As a result of the computed data in the Table 3, both the half Heusler's is anisotropic.
3.3 Electronic properties
The significance of a material's electronic characteristics lies in their capacity to enable us to examine and comprehend the type of connections that have developed among the various elements within the material.45 In electronic band structure, the presence of energy gaps in materials has amazing applications in spintronic, memory devices, thermoelectric etc. This feature of electronic structure can significantly be used to alter the physical properties required for present-day technological needs. The essential data concerning band structure and density of states are fundamental tools for analysing the electronic characteristics of the alloys under study.
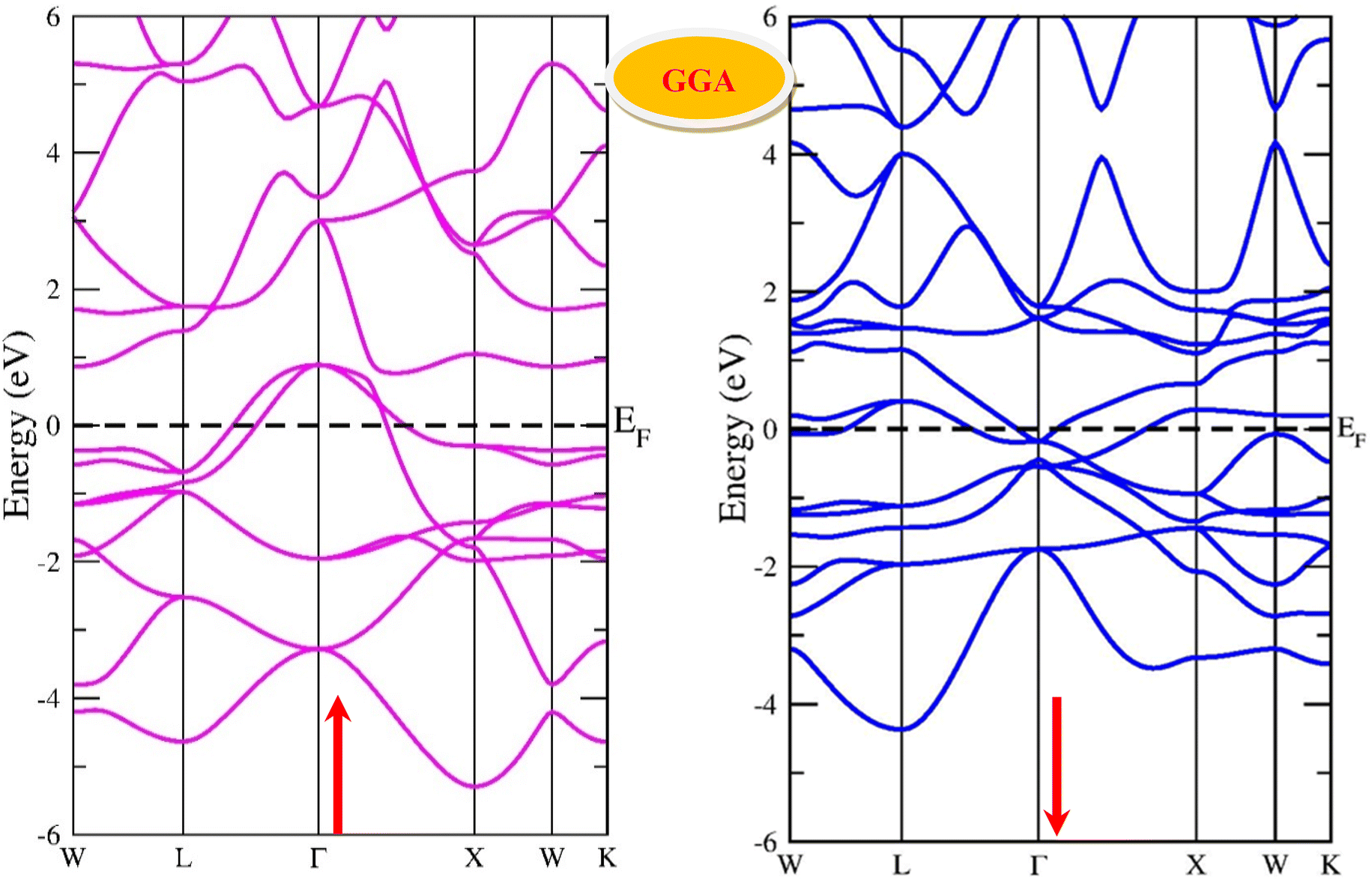
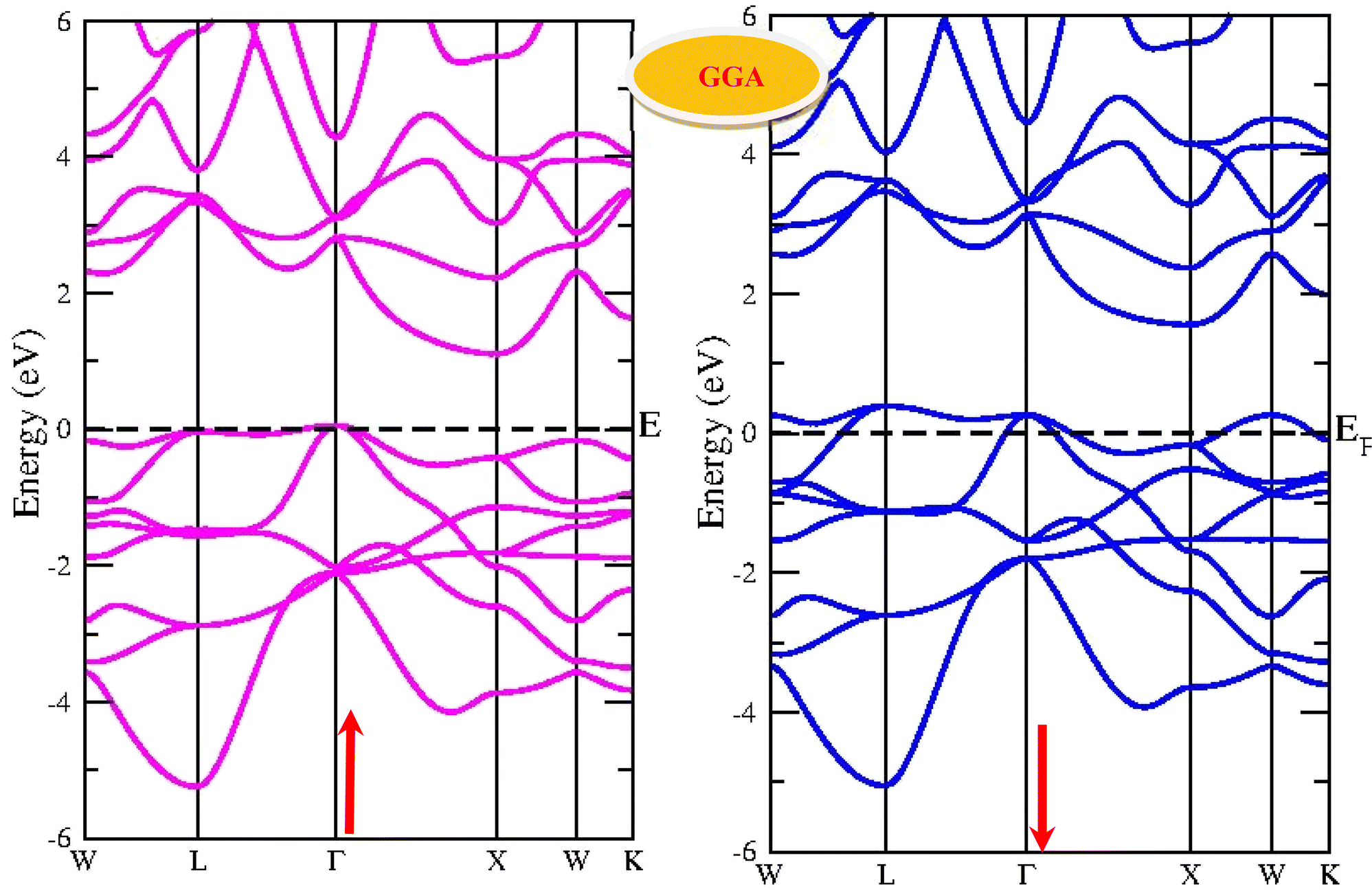
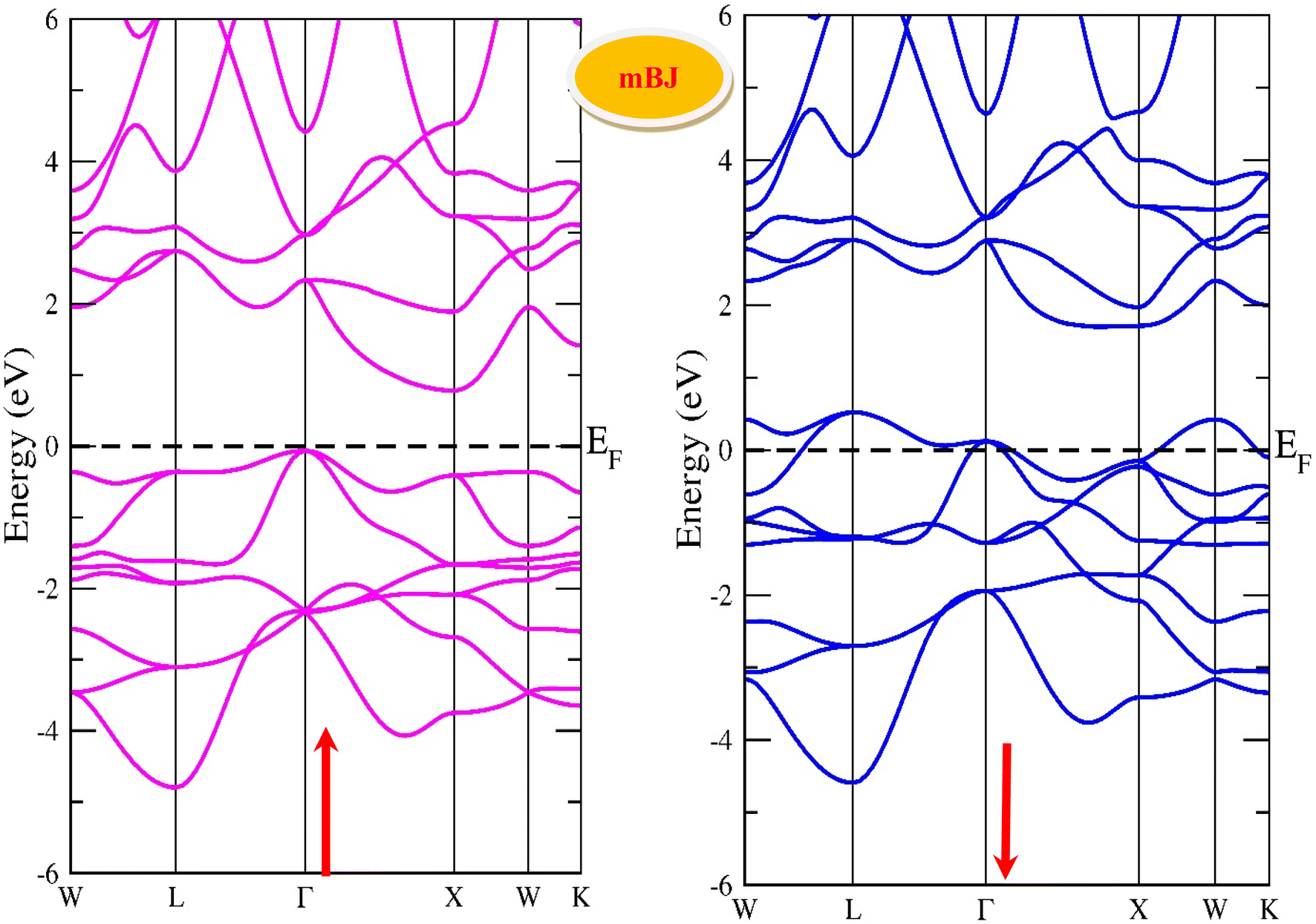
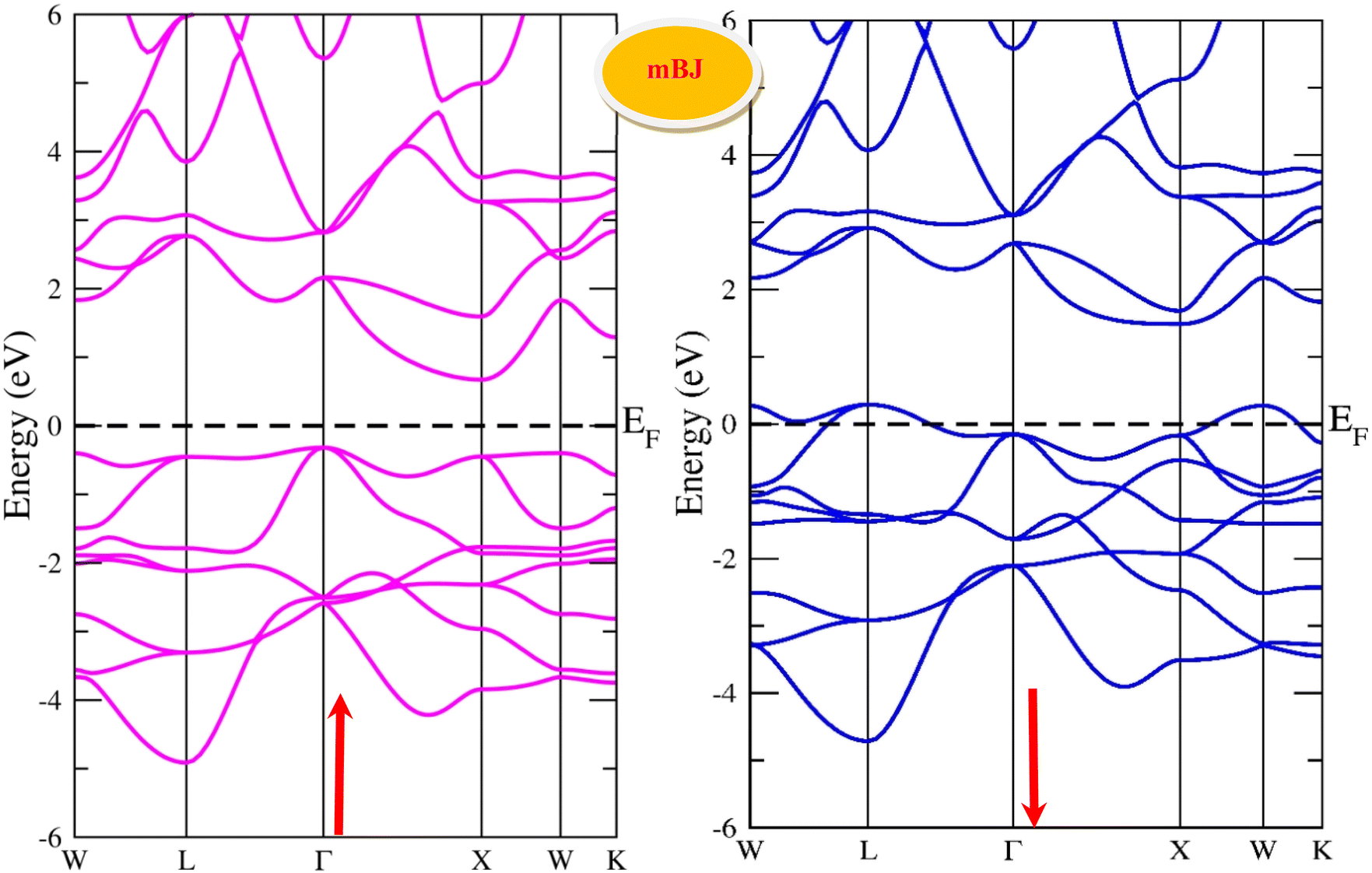
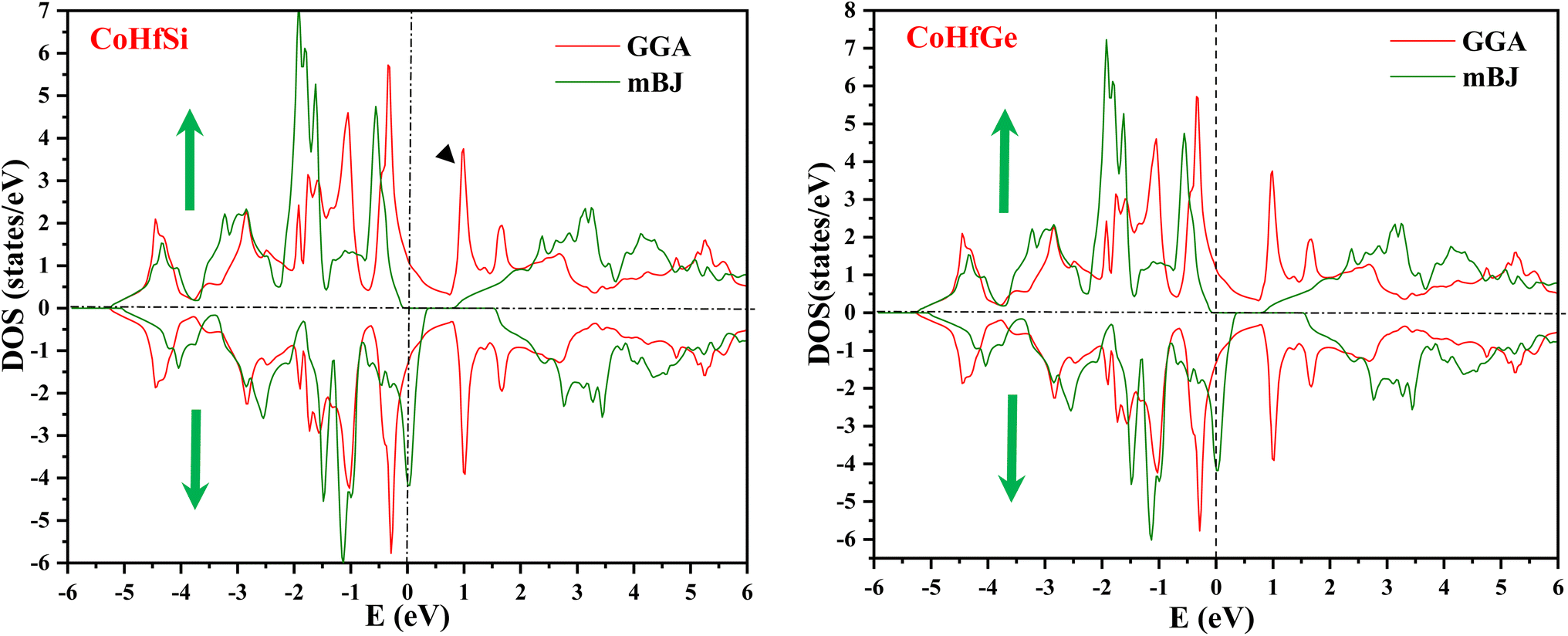
By using the GGA approximation we estimate the electronic band structure along with the Brillion zone. The obtained results are labelled in Fig. 3 and 4, which demonstrate that the alloys have a metallic character in the up-spin channel and down-spin channel of CoHfSi and CoHfGe in GGA approximation. To overcome this ambiguity, we have employed a more precise band structure based on the mBJ scheme, as well-known approximations GGA underestimate the overall electronic structure of these alloys. The band structures obtained from the mBJ method are profiled in Fig. 5 and 6. By adding mBJ potential to GGA, the alloys exhibit semiconducting behaviour, with an indirect band gap in the up-spin channel, whereas the opposite channel has a metallic character. The band gap in the spin-up channel increases and comes out to be 0.89 eV and 0.97 eV for CoHfSi and CoHfGe along Γ–X symmetric point, giving rise to the overall half-metallic nature. Additionally, the half-metallic nature of present alloys can also be revealed by analysing the calculated total density of states (TDOS) and partial density of states (pDOS), displayed in Fig. 7–9.
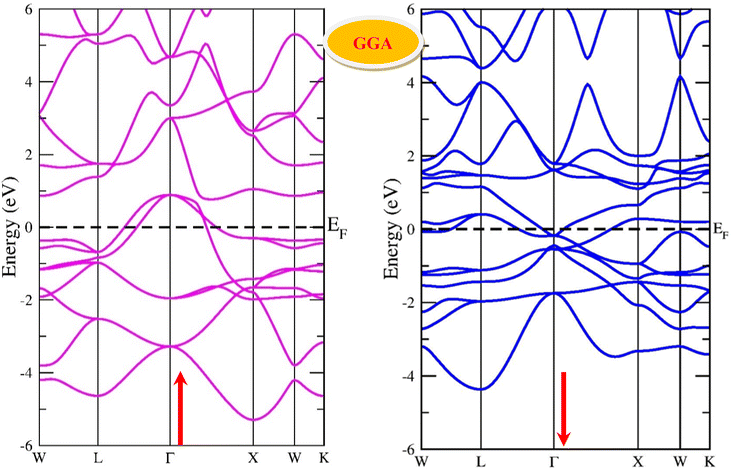 |
| | Fig. 3 Spin polarised band structure of CoHfSi alloy via GGA method. | |
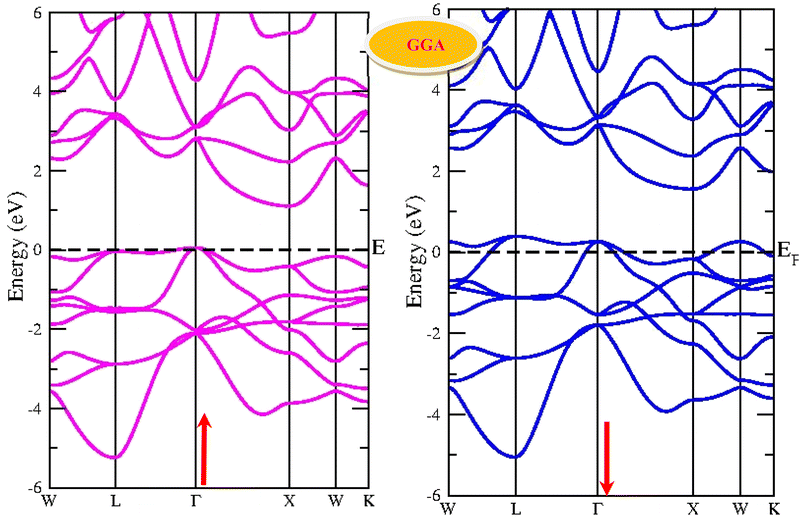 |
| | Fig. 4 Spin polarised band structure of CoHfGe alloy via GGA method. | |
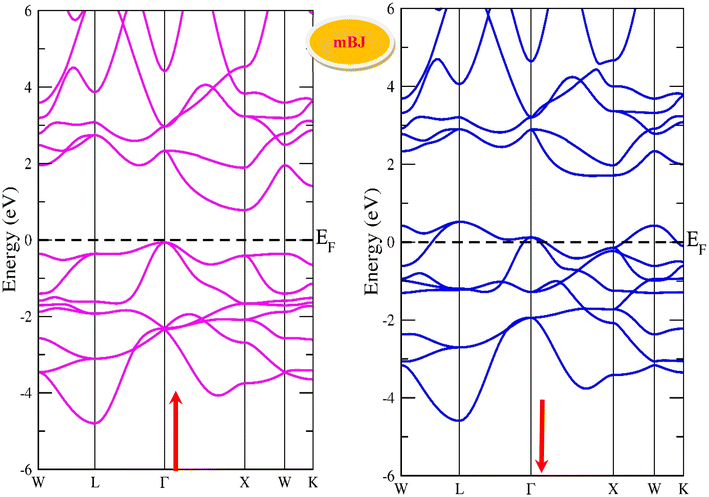 |
| | Fig. 5 Spin polarised band structure of CoHfSi alloy via mBJ method. | |
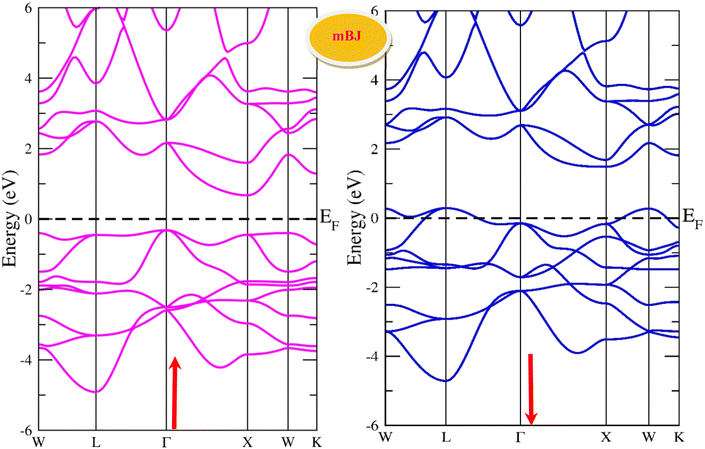 |
| | Fig. 6 Spin polarised band structure of CoHfGe alloy via mBJ method. | |
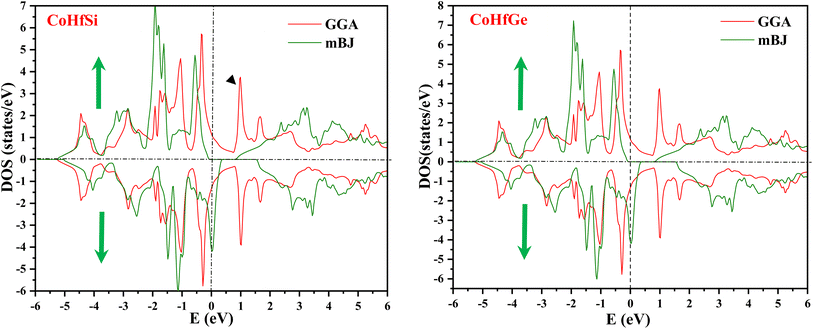 |
| | Fig. 7 Calculated total density of states for CoHfSi and CoHfGe by GGA and mBJ. | |
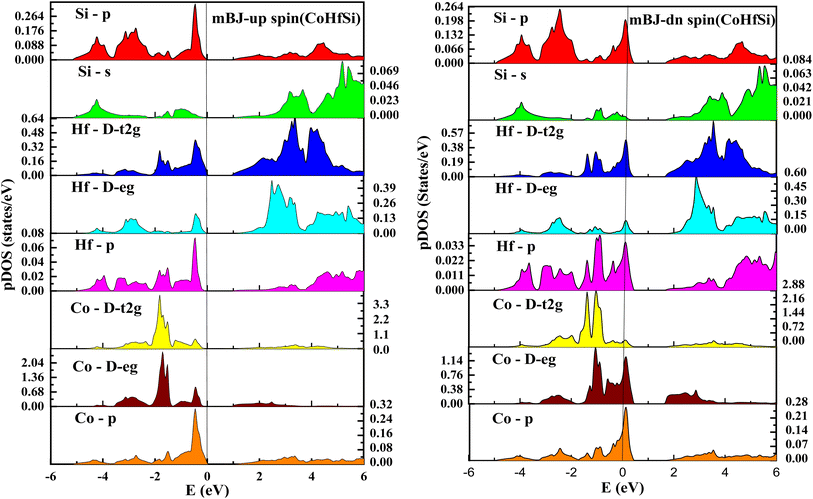 |
| | Fig. 8 pDOS for CoHfSi half Heusler's alloy by mBJ calculation in both spins. | |
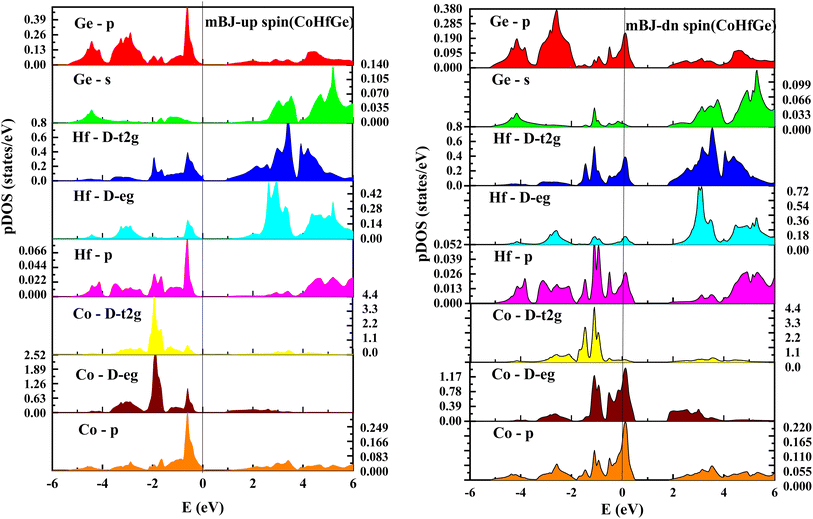 |
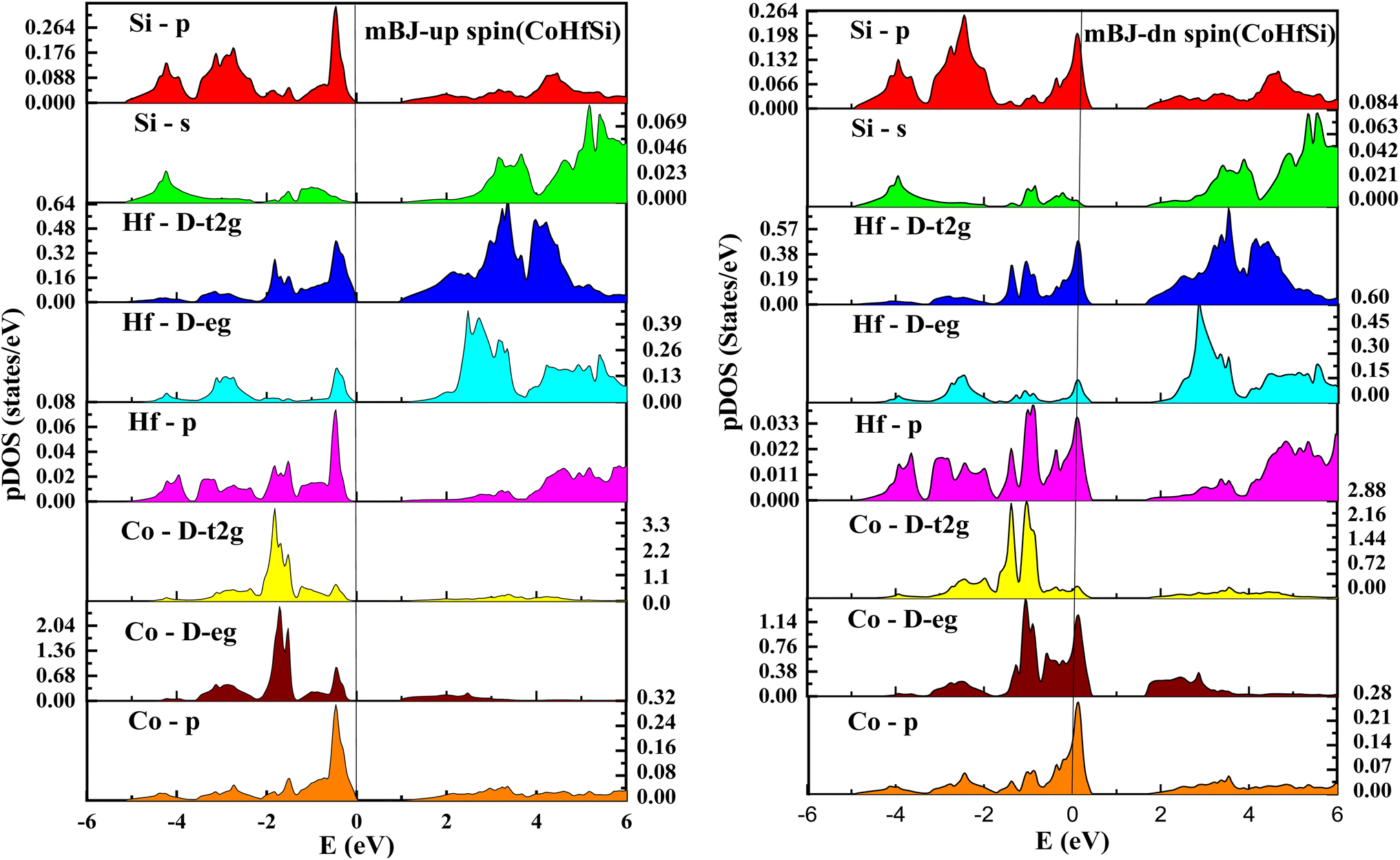
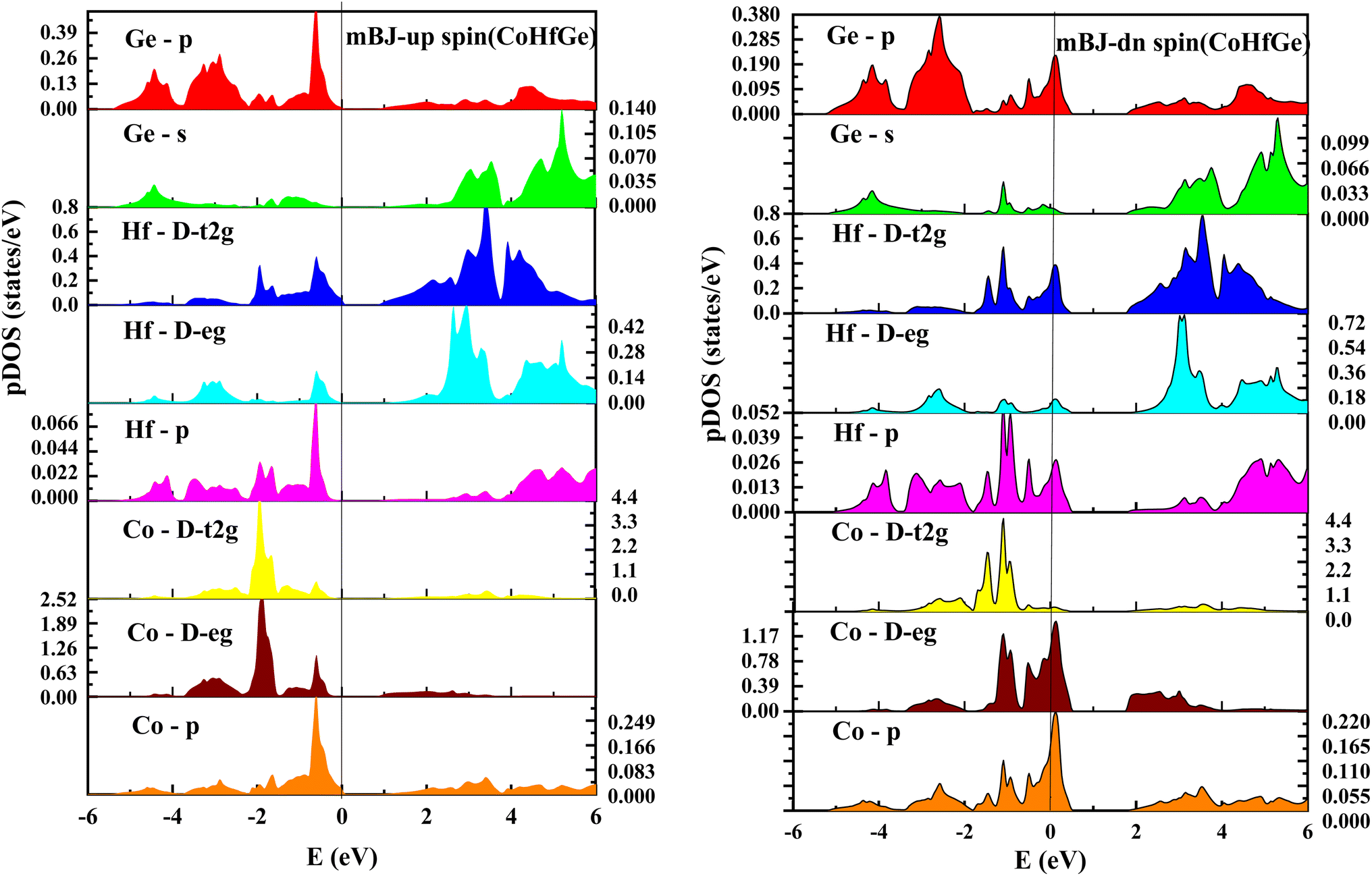
| | Fig. 9 pDOS for CoHfGe half Heusler's alloy by mBJ calculation in both spin. | |
From the total DOS, in the spin-down state, few energy states are occupied at the reference level, which is a clear indication of metallic nature in the respective state, but in the opposite spin state, there is a splitting of energy states, which creates a gap, can be seen in Fig. 7. There is no DOS at the Fermi level, indicating semiconducting nature in mBJ approximation. From Fig. 8, it can be seen that in the spin-up channel, the energy states are away from the Fermi level, exhibiting a gap near the Fermi level and revealing its semiconducting nature. In the spin-down channel, the energy states are occupying the Fermi level revealing its metallic character. In spin down channel all the atoms have their contribution to DOS (cobalt and hafnium have a majority contribution), and half-metallicity is also confirmed from the total DOS. This can be understood further from the partial density of states (pDOS) as shown in Fig. 8 and 9.
3.4 Magnetic properties
The Slater Pauling's rule (SP) is one of the best methods used to determine the magnetic moment of Heusler alloys. The total magnetic moment is expressed as Mtot, which is calculated by using the following equation:where (Mtot) is the spin magnetic moment and (Ztot) is the total number of valence electrons represented in this equation. This method can be used by subtracting 24 from the total valence electrons in full Heusler alloys and 18 from the total valence electrons in half Heusler alloys.46 The electronic configurations of the elements Co, Hf, Si and Ge are as follows:
| Co: [Ar] 3d74s2, Hf: [Kr] 4d25s2, Si: [Ne] 3s23p2 and Ge: [Ar] 3d104s24p2. |
In the CoHfSi and CoHfGe alloys, the total number of valence electrons (Ztot) is 17. The total magnetic moment of the CoHfSi and CoHfGe alloys is equal to 1.00 μB, according to the SP rule (Mtot = Ztot − 18). Both the GGA and mBJ approximation produce 1.00 μB total magnetic moments. Our findings are matched with the SP rule. The transition metal Co contributes the most to the total magnetic moment. The total magnetic moment of both alloys is recorded in Table 4.
Table 4 Evaluated magnetic moment (μB) and energy band gaps (eV) for CoHfSi and CoHfGe alloys
| Alloys |
Methods |
Co |
Hf |
Si/Ge |
Int |
Total |
Band gap |
TC |
| CoHfSi |
GGA |
0.20 |
0.62 |
0.11 |
0.03 |
0.97 |
0.00 |
491 |
| mBJ |
0.64 |
0.08 |
0.04 |
0.22 |
1.00 |
0.87 |
|
| CoHfGe |
GGA |
0.14 |
0.65 |
0.05 |
0.03 |
0.88 |
0.00 |
462 |
| mBJ |
0.75 |
0.04 |
0.05 |
0.14 |
0.98 |
0.97 |
|
Both CoHfSi and CoHfGe HH alloys exhibit a half-metallic nature, supported by the band graphs, DOS, and total magnetization numerical values. From an application perspective, spin injectors require a high TC. As such, we determined the TC for these alloys using the mean field approximation (MFA). The MFA calculates TC based on the energy difference between the NM and FM states, which is expressed in the equation below.
| |
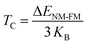 | (15) |
where
KB be the Boltzmann constant, is involved in the formula for energy difference between NM and FM phases, which is represented by Δ
ENM-FM =
ENM − E
FM. The
TC values are estimated and listed in
Table 4, with computed values greater than ambient temperature, indicating the suitability of these materials for spintronic applications.
3.5 Thermoelectric properties
The thermoelectric (TE) material can convert waste heat energy into beneficial electric energy and is employed as green energy.47 Thermoelectric properties are interconnected through an electronic band profile, which determines the electronic properties. The TE characteristics are obtained using the semi-classical Boltzmann theory under constant time relaxation. The TE performance is predicted by the dimensionless figure-of-merit mathematically illustrate as;| |
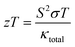 | (16) |
The quantity S2σ symbolizes the power factor, which comprises of Seebeck coefficient (S), electrical conductivity (σ), and κtotal total thermal conductivity. The total thermal conductivity is contributed by electronic (κe) and as well as lattice (kl) parts illustrated mathematically as κ = κe + κl.48–50
3.5.1. Seebeck coefficient. The ability of a substance to generate voltage as a function of temperature is explained by the vital phenomenon called as Seebeck coefficient (S). It serves as a useful parameter for characterizing a material's thermoelectric efficiency. The amount of voltage that a material develops in response to a temperature differential across it can be determined by the Seebeck coefficient by using the following equation.51| |
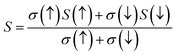 | (17) |
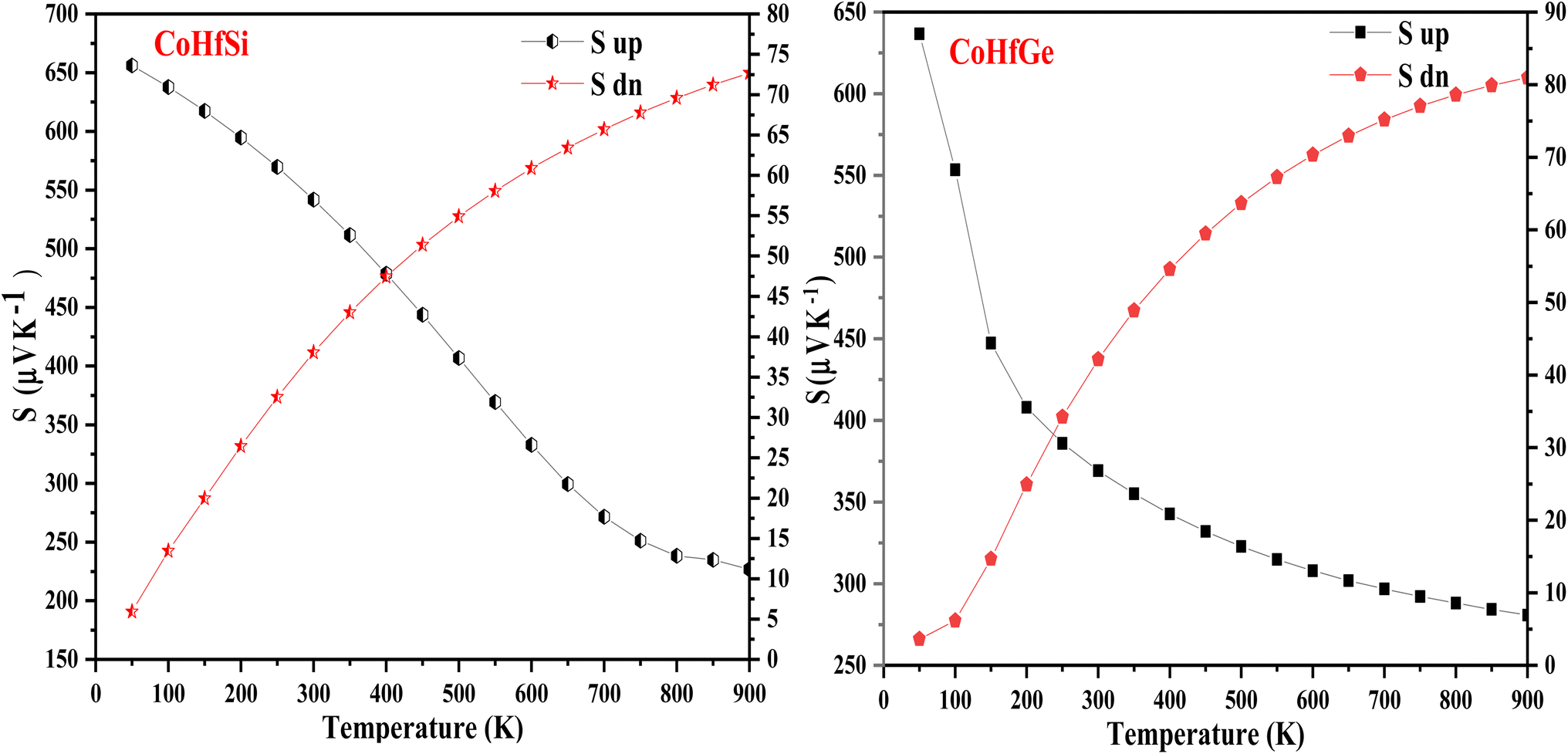
In this current analysis, we have examined CoHfSi & CoHfGe alloys to demonstrate the Seebeck throughout in a selected temperature range. The geometrical interpretation of the Seebeck coefficient (S) of the two half-Heusler alloys is exhibited in Fig. 10. The substantial reduction in S value dignifies the semiconducting performance of a material mainly because enhanced carrier transporters and therefore offers more and more electron–hole pair, cause scattering possessions and henceforth reduce the Seebeck coefficient value. The calculated value of Seeback coefficient for CoHfSi at 50 K and 900 K is 656.10 μVK−1 and 226.76 μVK−1 in spin-up channel, 5.91 μVK−1 and 72.68 μVK−1 in spin-dn channel respectively. For the CoHfGe the value is 636.60 μVK−1 and 280.76 μVK−1 in spin-up channel, 3.61 μVK−1 and 80.96 μVK−1 in spin-dn channel respectively. These both alloys which exhibit a good value of the Seebeck coefficient directly suggested that these materials may find a suitable root for the thermoelectric applicability.
 |
| | Fig. 10 Variation in Seeback coefficient (S) with temperature in both spin channels of CoHfSi and CoHfGe alloys. | |
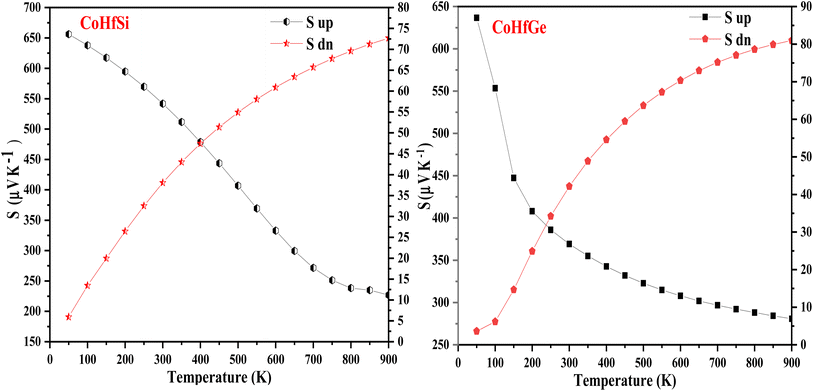
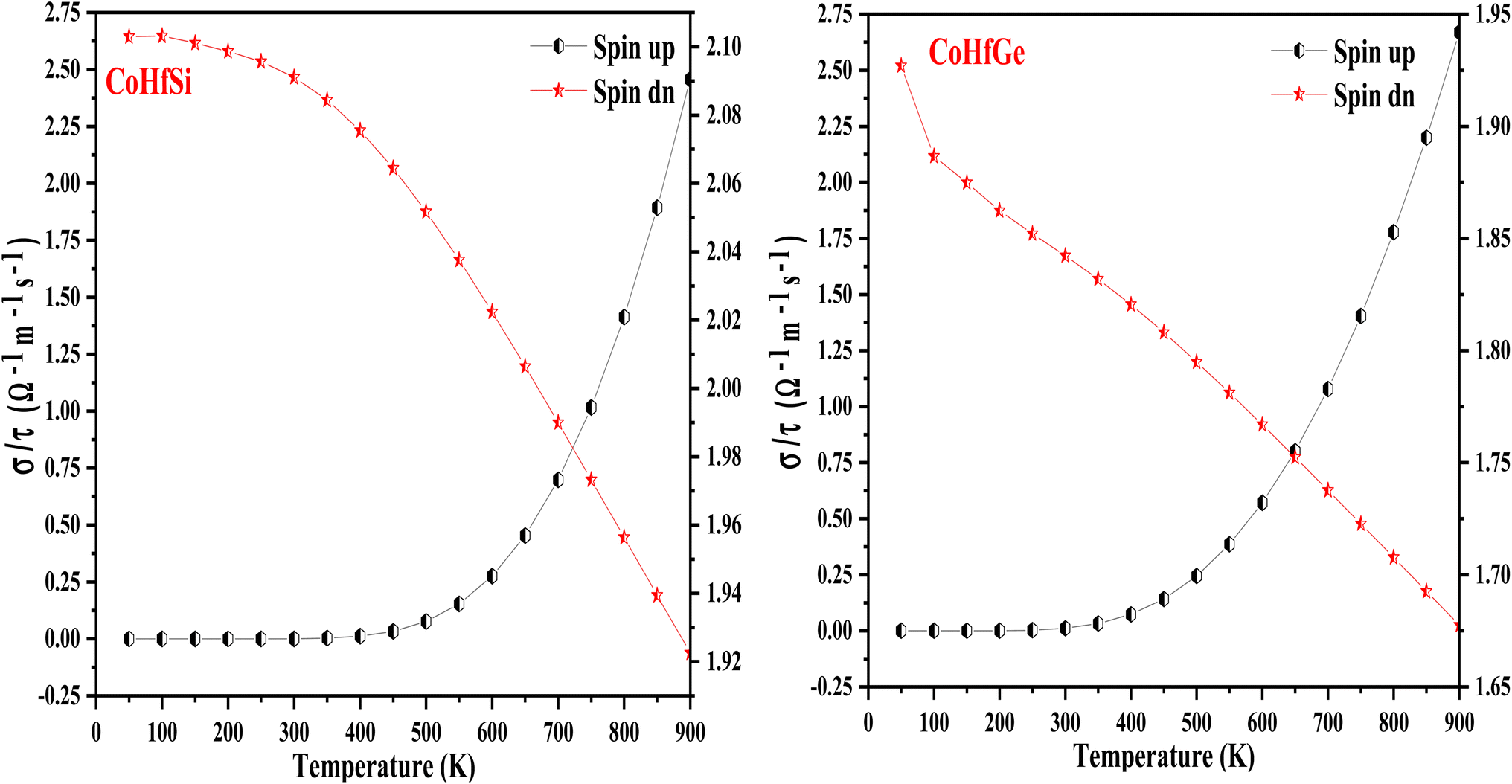
3.5.2. Electrical conductivity. Electrical conductivity is a crucial property in electronic devices, and materials can be categorized as metals, ceramics, semiconductors, etc. The electrical conductivity mechanism varies among these materials due to differences in their Fermi level width. Metals have free electrons and a narrow band gap, while semiconductors have a small band gap. External factors like temperature, electric field, and magnetic field influence the electrical conductivity of materials significantly. Among these factors, temperature variations play a vital role in affecting the electrical conductivity of the material. In Fig. 11 the electrical conductivity (σ/τ) of the Heusler materials is figured out in both spin configurations. In a spin-up channel graphical plot, (σ/τ) increases linearly as a function of temperature that display the semiconductor character, hence the charge carrier increase with temperature. In the spin-dn plot, (σ/τ) decreases with increasing temperature which shows the metallic character. However, the elevated temperature causes a rise in the charge carrier and hence defines the σ value. The electrical conductivity (σ), as well as the charge carrier, are interlinked through mathematical relations illustrated by σ = neμ, here e and μ symbolize the electric charge and mobility of the charge carrier. The calculated σ/τ values are 2.45 (Ω−1 m−1 s−1) in the up-spin channel and 1.92 (Ω−1 m−1 s−1) in the down-spin channel at a higher temperature (900 K) for CoHfSi alloy, while in, 2.66 (Ω−1 m−1 s−1) in up spin channel and 1.67 (Ω−1 m−1 s−1) in the down spin channel for CoHfGe Heusler alloys.
 |
| | Fig. 11 Variation in electrical conductivity (σ/τ) with temperature in both spin channels of CoHfSi and CoHfGe alloys. | |
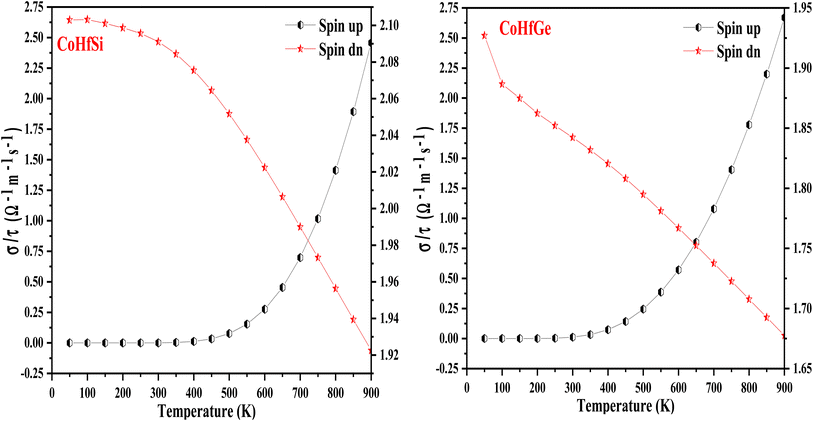
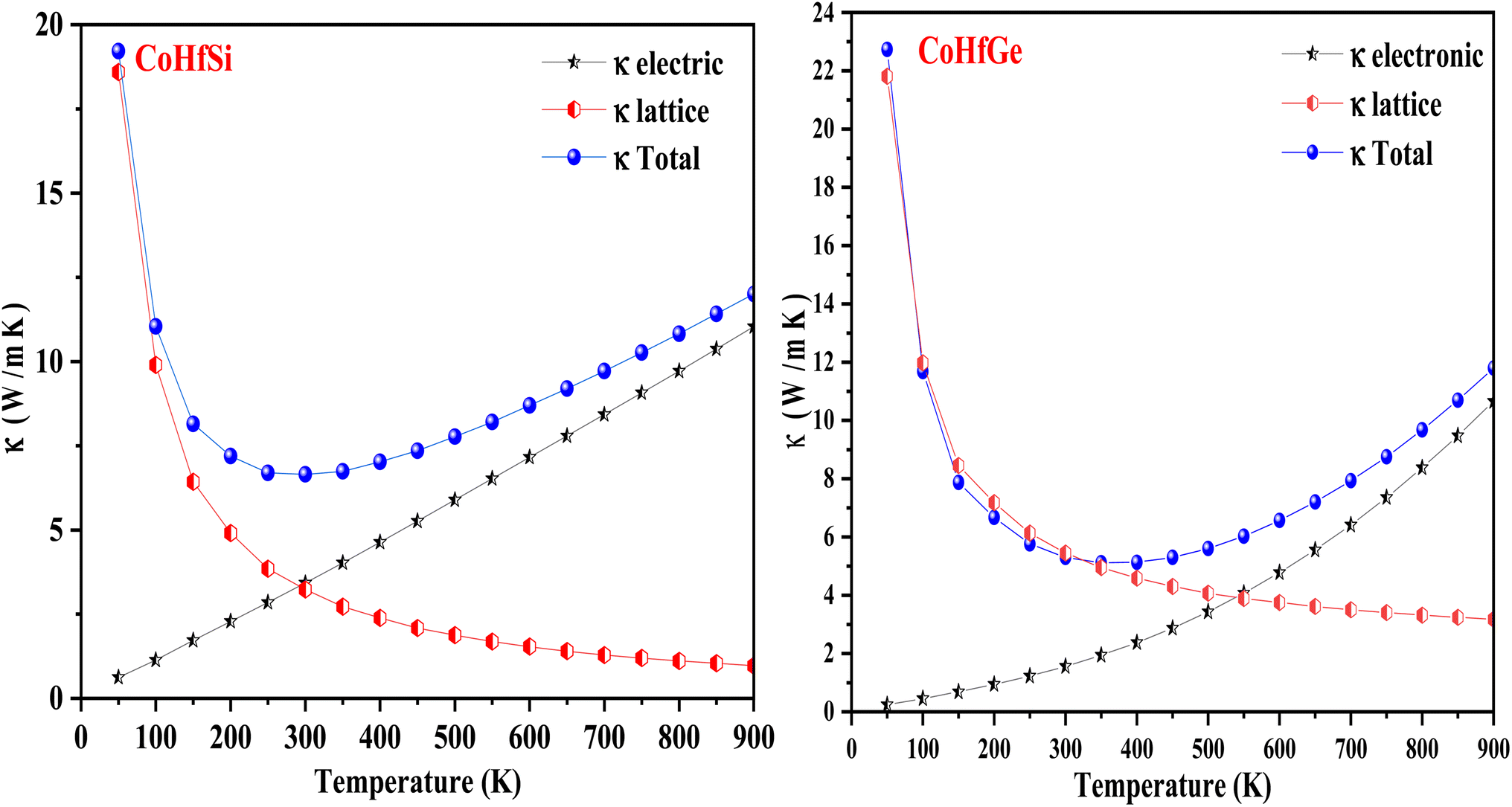
3.5.3. Thermal conductivity. Thermal conductivity (κ) pertains to the transmission of heat, primarily resulting from the continual motion of atoms within the material. This motion encompasses various forms, such as rotation, translation, and vibration. The atomic vibrations can produce heat or thermal energy in the material. As a result, two processes are defined, either through electrons or phonons or atoms vibrating at a single frequency to carry out the operation of transmitting heat inside a material. As a result, both the electronic and lattice vibrations of the total thermal conductivity κtotal = κelectronic + κlattice are taken into account for their overall contribution. Good conductors are those materials that possess a high value of electrical conductivity. The lattice thermal conductivity can be figured out through the Slacks equation.52,53 The geometrical plots depict the increasing trend in the electronic thermal conductivity (κe) of CoHfSi and CoHfGe from a low value of 3.42 W mK−1 and 1.55 W mK−1 at 300 K to their values of 11.03 W K−1 and 10.65 W mK−1 at 900 K respectively. Furthermore, the figured-out value for CoHfSi and CoHfGe alloys is 3.22 W mK−1 and 3.73 W mK−1 at 300 K and 0.97 W mK−1 to 1.14 W mK−1 at a higher temperature of 900 K, the lattice thermal conductivity in both of these half-Heusler exhibits an exponentially decreasing trend as a function of temperature as shown in Fig. 12.
 |
| | Fig. 12 Variation in thermal conductivity (κ) with a temperature of CoHfSi and CoHfGe alloys. | |
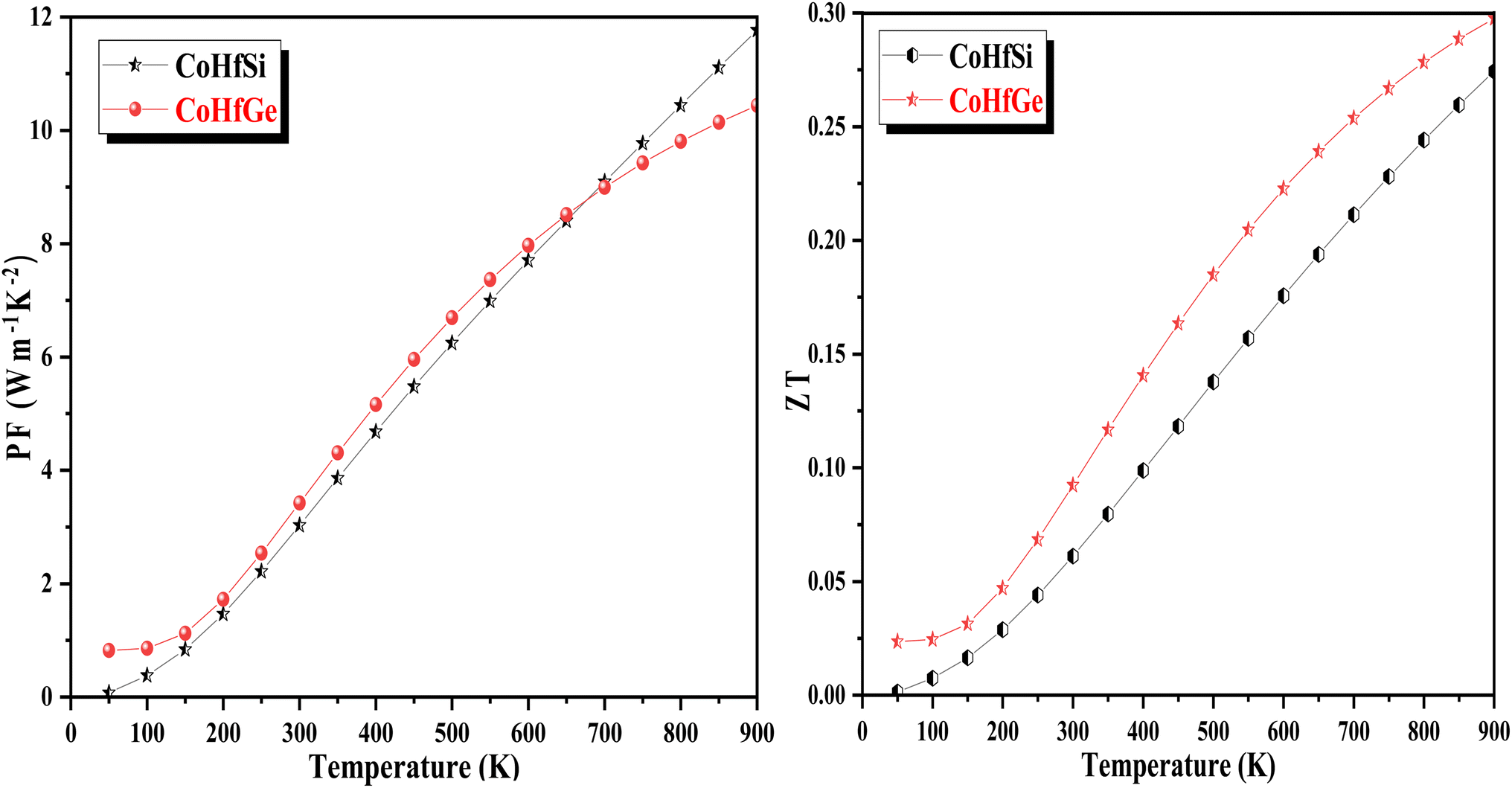
3.5.4. Power factor. The power factor (PF) is also an important parameter for determining the efficiency of the materials. The power factor is mathematically symbolized as PF = S2σ where S is the Seebeck coefficient and is the electrical conductivity, consequences in high output generation form low input from the compound. The power factor designates the interlinked electrons that execute a critical part in refining the TE response. Fig. 13 illustrates the variation of the PF as a function of temperature. The graphical plot of PF directly suggested a rising trend with temperature. The collected value of PF is 3.02 W mK−2 and 3.27 W mK−2 at room temperature for CoHfSi and CoHfGe alloys respectively.
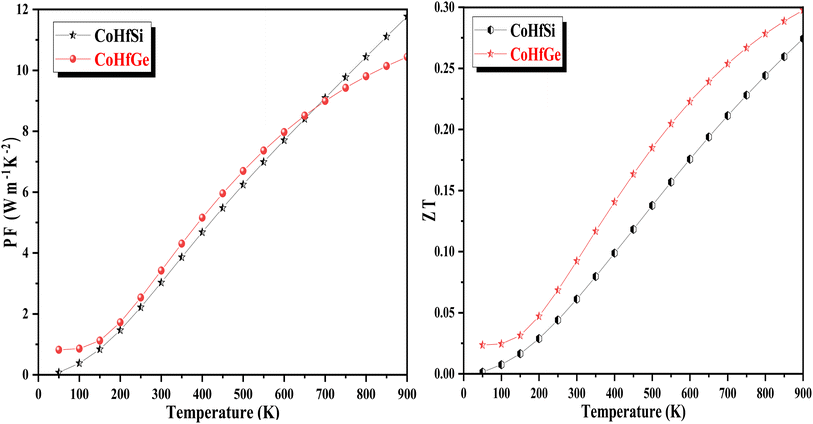 |
| | Fig. 13 Variation in power factor (PF) and figure of merit (ZT) with a temperature of CoHfSi and CoHfGe alloys. | |
3.5.5. The figure of merit (ZT). The typical way to assess the thermoelectric performance of a material is by utilizing a parameter known as the figure of merit (zT). The maximum reliability (η) of a thermoelectric device is defined as the ratio of the energy provided to the load (W) and the heat energy dissipated at the hot junction (Q). The figure of merit (zT) mathematically express as: 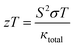 .
.The graphical plot of zT as a function of temperature is presented in Fig. 13. The observed value of zT is 0.27 and 0.29 for CoHfSi and CoHfGe alloys respectively at 900 K. This vital capability defines that both the materials find a suitable route towards thermoelectric applicability.
3.6 Thermodynamic properties
For reliable thermodynamic property calculations, we used the Gibbs2 package in conjunction with the Wien2k code. This comprehensive approach allowed us to obtain accurate data on the thermodynamic properties of the materials under investigation. Gibbs2 is a computational software package specifically designed for calculating various thermodynamic properties of materials. It's commonly used in conjunction with electronic structure calculation codes like Wien2k to provide a more complete characterization of materials. Gibbs2 takes this electronic structure information and performs thermodynamic calculations based on statistical mechanics principles. It can calculate properties such as heat capacity (CV), thermal expansion coefficient (α) and the Grüneisen parameter (γ) as a function of temperature and pressure. Through the use of a variety of thermodynamic potentials, we have attempted to describe the nature of the stability of these materials. The high temperature–pressure dependence of thermodynamic characteristics can be estimated via DFT calculations. The quasi-harmonic Debye model is used to calculate the approximate value of these thermodynamic potentials.54
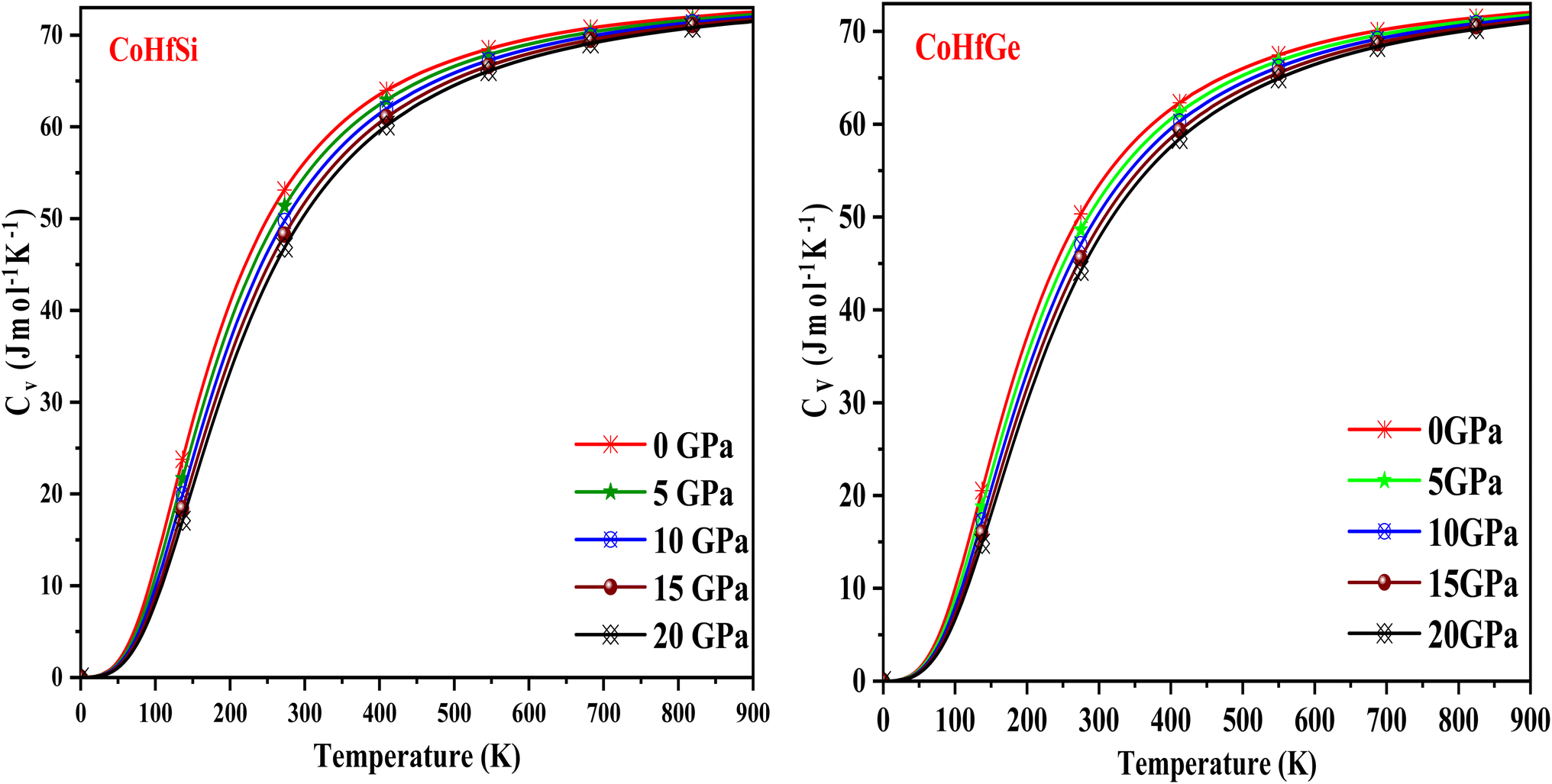
It is important to note that the specific heat at constant volume (CV) of any material is one of the main factors determining how it can transport waste heat. In addition, the study relates the cause and effect of the material's dynamics and microstructures. As seen in Fig. 14, the graphical variation in this material shows a greater ability to transport heat concerning temperature T3 at room temperature. This method of calculating the specific heat under a harmonic scheme indicates a robust relationship between the specific heat and the temperature. Due to the high-temperature limitation, the change becomes constant and follows the Dulong–Petit law.55
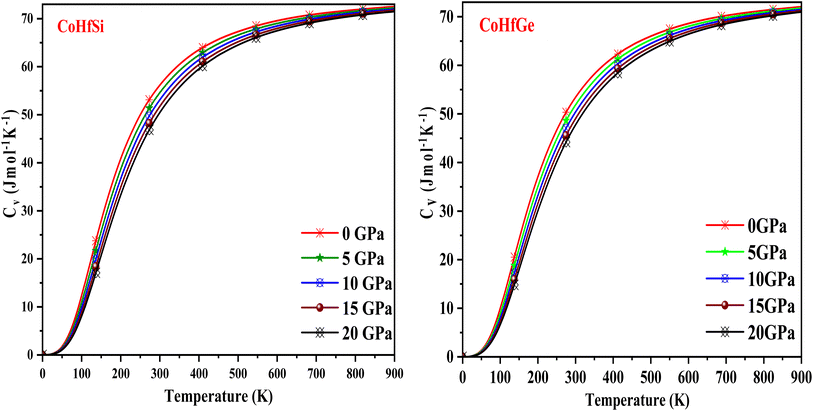 |
| | Fig. 14 The variation of specific heat with the temperature at different pressure of CoHfSi and CoHfGe half Heusler alloys. | |
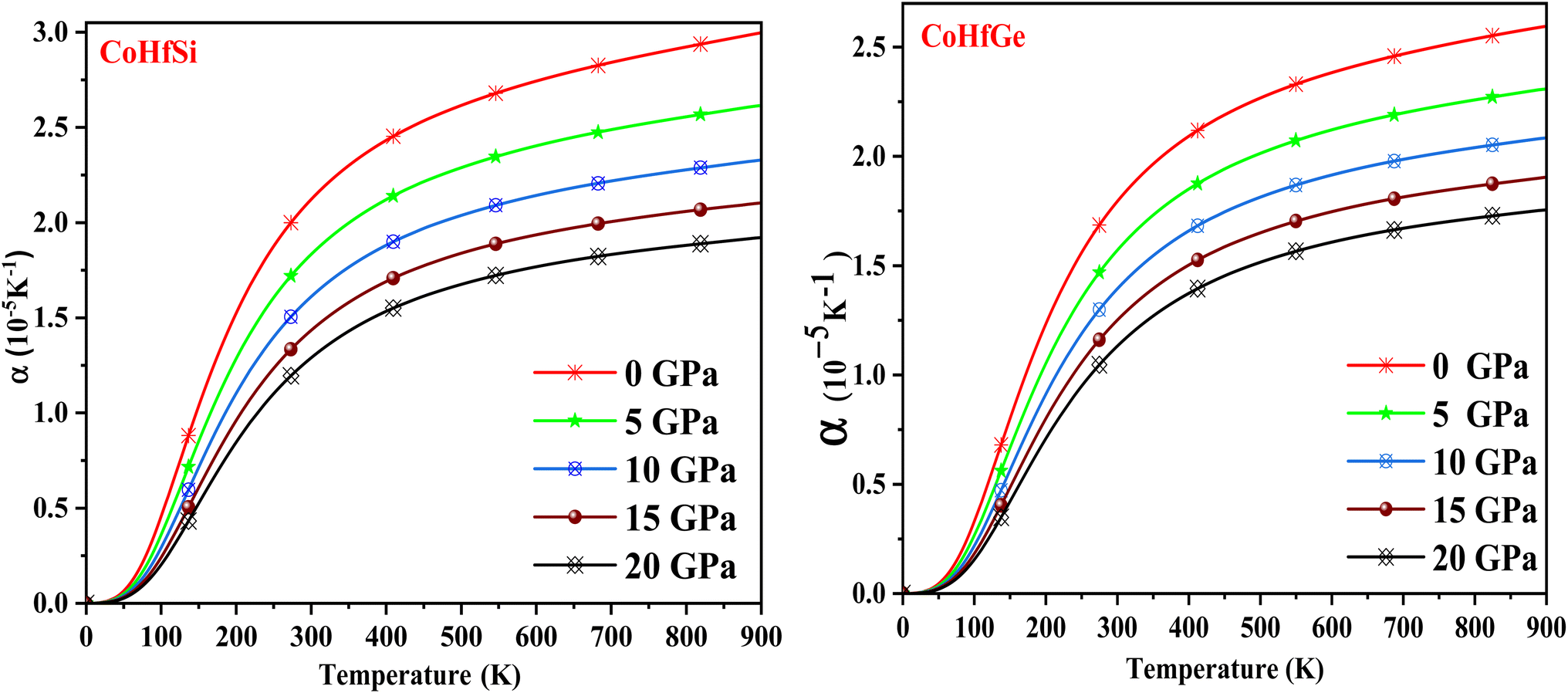
When the temperature of a material is increased, its arrangement (length, area and volume) changes. This phenomenon is known as thermal expansion (α). Fig. 15 depicts the evolution of the thermal expansion coefficient (α) vs. temperature, which explains the nature of bonding among the constituent atoms. These plots demonstrate that it is temperature-dependent and becomes constant with increasing temperature, while pressure-dependent and decreases with increasing temperature. The reason for this is that atoms and molecules conduct harmonic motion at their equilibrium positions when the temperature rises, they gain high kinetic energies, which causes them to vibrate and move, increasing their interatomic distance, which then causes the material to expand.
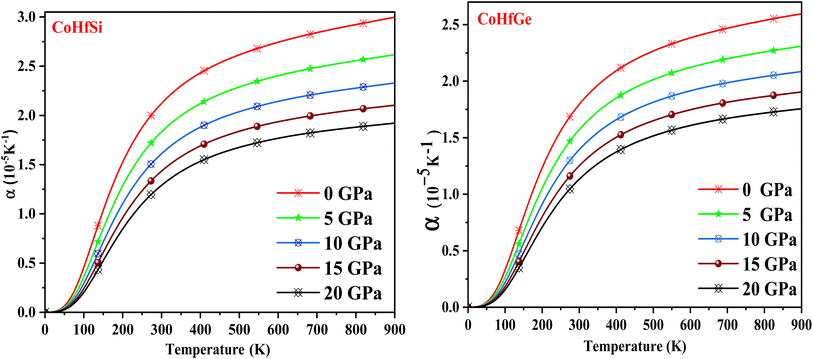 |
| | Fig. 15 The variation of thermal expansion coefficient with the temperature at different pressure of CoHfSi and CoHfGe half Heusler alloys. | |
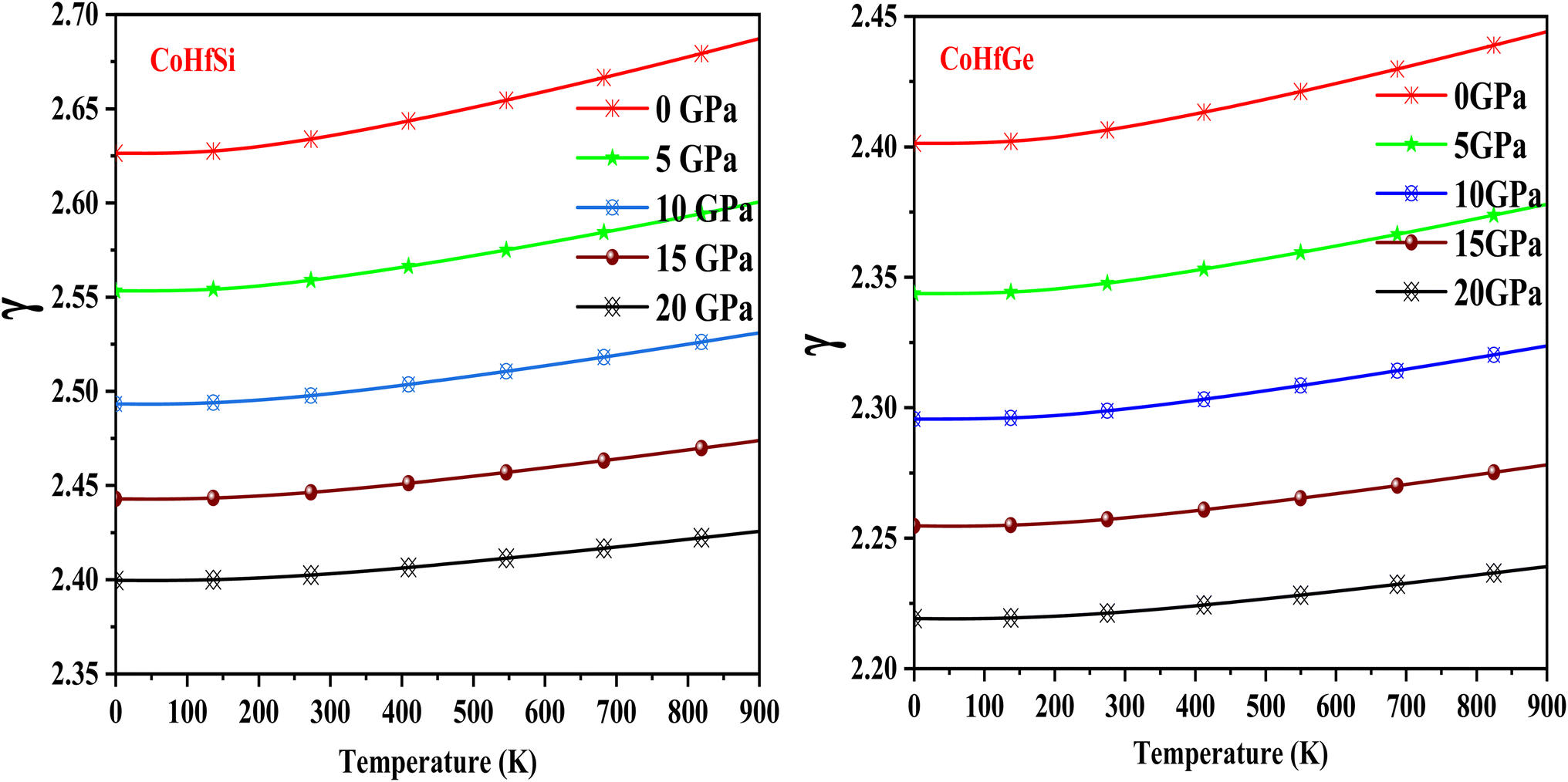
The Grüneisen parameter (γ) displays the relationship between phonon frequency and volume change, and it is essential parameter to determine thermo-mechanical properties of any materials. The Grüneisen parameter (γ) slowly grows with the rising temperature at constant pressure as seen in Fig. 16. A detailed description of the recorded room temperature is included in Table 5. Meanwhile, CoHfSi and CoHfGe half Heusler alloys give good results, indicating that these compounds have a lot of anharmonicities.
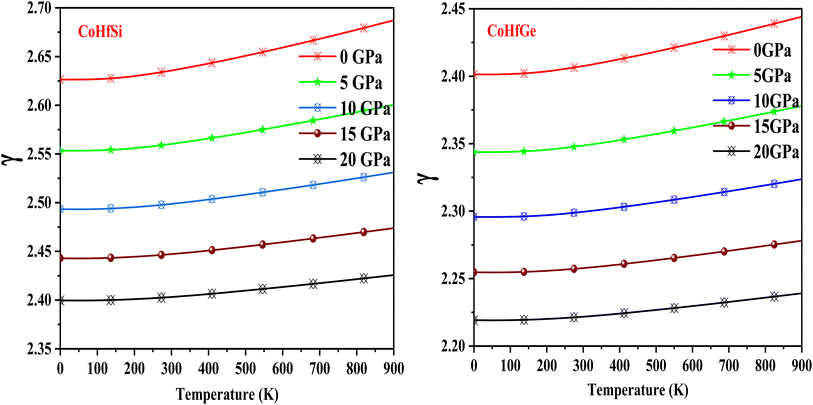 |
| | Fig. 16 The variation of the Grüneisen parameter with the temperature at different the pressure of CoHfSi and CoHfGe half Heusler alloys. | |
Table 5 Values of different thermodynamic parameters at room temperature and zero pressure of CoHfSi and CoHfGe alloy
| Parameters |
CoHfSi |
CoHfGe |
| Specific heat (CV) |
56.21 (J mol−1 K−1) |
53.69 (J mol−1 K−1) |
| Thermal expansion coefficient (α) |
2.12 (10−5 K−1) |
1.80 (10−5 K−1) |
| Grüneisen parameter (γ) |
2.63 |
2.40 |
4. Conclusion
By employing first-principle calculations, we conducted a comprehensive investigation into the structural, magneto-electronic, mechanical, thermoelectric, and thermophysical properties of CoHfSi and CoHfGe half-Heusler alloys. We assessed structural stability by calculating mechanical stability criteria. Both CoHfSi and CoHfGe alloys maintain their half-metallic characteristics when using GGA and GGA + mBJ approximations with exchange–correlation functions. According to the electronic band structures, CoHfSi exhibits an energy gap of 0.87 eV, while CoHfGe displays a gap of 0.97 eV. These gaps are both indirect and occur along the Γ–X symmetric point. The magnetic moment of these half-metallic alloys is precisely 1 μB, an integer value characteristic of half-metallic materials. Furthermore, both alloys exhibit ductile behavior, as indicated by their mechanical parameters such as Cauchy pressure and Pugh's ratio. Moreover, they exhibit favourable Seebeck coefficients and power factors at room temperature, making them suitable for various applications, including spintronics and thermoelectric technology. We have implemented a quasi-harmonic Debye model to describe their stability under high temperatures and pressures.
Author contributions
Bharti Gurunani: writing – review & editing, writing – original draft, methodology. Dinesh C. Gupta: supervision, software.
Conflicts of interest
There are no conflict to declare.
References
- S. A. Sofi and D. C. Gupta, Exploration of electronic structure, mechanical stability, magnetism, and thermophysical properties of L21 structured Co2XSb (X = Sc and Ti) ferromagnets, Int. J. Energy Res., 2020, 44(3), 2137–2149 CrossRef CAS.
- I. Choudhuri, P. Bhauriyal and B. Pathak, Recent advances in graphene-like 2D materials for spintronics applications, Chem. Mater., 2019, 31(20), 8260–8285 CrossRef CAS.
- X. Wang, Z. Cheng and G. Liu, Largest magnetic moments in the half-Heusler alloys XCrZ (X = Li, K, Rb, Cs; Z = S, Se, Te): a first-principles study, Materials, 2017, 10(9), 1078 CrossRef PubMed.
- D. P. Rai, Heusler compound: a novel material for optoelectronic, thermoelectric, and spintronic applications, in High-K Gate Dielectric Materials, 2020, pp. 201–237 Search PubMed.
- J. He, M. Amsler, Y. Xia, S. Naghavi, V. I. Hegde, S. Hao and C. Wolverton, Ultralow thermal conductivity in full Heusler semiconductors, Phys. Rev. Lett., 2016, 117(4), 046602 CrossRef PubMed.
- S. Singh, M. Zeeshan, J. V. D. Brink and H. C. Kandpal, {Ab initio} study of Bi-based half Heusler alloys as potential thermoelectric prospects, arXiv, 2019, preprint, arXiv:1904.02488, DOI:10.48550/arXiv.1904.02488.
- L. Zhang, Z. X. Cheng, X. T. Wang, R. Khenata and H. Rozale, First-principles investigation of equiatomic quaternary Heusler alloys NbVMnAl and NbFeCrAl and a discussion of the generalized electron-filling rule, J. Supercond. Novel Magn., 2018, 31, 189–196 CrossRef CAS.
- V. Baltz, A. Manchon, M. Tsoi, T. Moriyama, T. Ono and Y. Tserkovnyak, Antiferromagnetic spintronics, Rev. Mod. Phys., 2018, 90(1), 015005 CrossRef CAS.
- A. Bahnes, A. Boukortt, H. Abbassa, D. E. Aimouch, R. Hayn and A. Zaoui, Half-metallic ferromagnets behaviour of a new quaternary Heusler alloys CoFeCrZ (Z = P, As and Sb): ab initio study, J. Alloys Compd., 2018, 731, 1208–1213 CrossRef CAS.
- I. Galanakis, P. H. Dederichs and N. Papanikolaou, Slater–Pauling behaviour and origin of the half-metallicity of the full-Heusler alloys, Phys. Rev. B: Condens. Matter Mater. Phys., 2002, 66(17), 174429 CrossRef.
- R. A. De Groot, F. M. Mueller, P. V. van Engen and K. H. J. Buschow, New class of materials: half-metallic ferromagnets, Phys. Rev. Lett., 1983, 50(25), 2024 CrossRef CAS.
- E. G. Özdemir and Z. Merdan, First-principles predictions on structural, electronic, magnetic and elastic properties of Mn2IrAl Heusler alloy, Mater. Res. Express, 2018, 6(3), 036101 CrossRef.
- O. Cheref, F. Dahmane, S. Benalia, D. Rached, M. Mokhtari, L. Djoudi and N. Bettahar, First-principles study of half-metallic properties in X2VSi (X = Ti, Co) and their quaternary TiCoVSi and CoTiVSi compounds, Comput. Condens. Matter, 2019, 19, e00369 CrossRef.
- Z. Merdan and E. G. ÖZDEMİR, The electronic and magnetic properties of new full-Heusler compounds: M2IrSi (M = Ti, Cr and Mn), Gazi Univ. J. Sci., 2018, 31(3), 940–952 Search PubMed.
- A. Amudhavalli, M. Manikandan, R. Rajeswarapalanichamy and K. Iyakutti, Electronic and magnetic properties of Fe2−xCoxTiGe (x = 0, 0.5, 1, 1.5, 2) full Heusler alloys, Chin. J. Phys., 2019, 59, 166–174 CrossRef CAS.
- E. G. Özdemir and Z. Merdan, Theoretical calculations on half-metallic results properties of FeZrX (X = P, As, Sb and Bi) half-Heusler compounds: density functional theory, Mater. Res. Express, 2019, 6(8), 086102 CrossRef.
- R. Paudel and J. Zhu, Investigation of half-metallicity and magnetism of bulk and (111)-surfaces of Fe2MnP full Heusler alloy, Vacuum, 2019, 164, 336–342 CrossRef CAS.
- A. Beloufa, B. Bakhti, D. Bouguenna and M. R. Chellali, Computational investigation of CrFeZ [Z = Si, Sn and Ge] half-Heusler compounds ferromagnets, Phys. B, 2019, 563, 50–55 CrossRef CAS.
- E. G. Özdemir and Z. Merdan, First principle predictions on half-metallic results of MnZrX (X = In, Tl, C, Si, Ge, Sn, Pb, N, P, As, Sb, O, S, Se, Te) half-Heusler compounds, J. Magn. Magn. Mater., 2019, 491, 165567 CrossRef.
- D. Shrivastava and S. P. Sanyal, Theoretical study of structural, electronic, phonon and thermoelectric properties of KScX (X = Sn and Pb) and KYX (X = Si and Ge) half-Heusler compounds with 8 valence electrons count, J. Alloys Compd., 2019, 784, 319–329 CrossRef CAS.
- N. Ak, F. Şarlı, E. G. Özdemir, B. Saatçi and Z. Merdan, Key role of central antimony in magnetization of Ni0.5Co1.5MnSb quaternary Heusler alloy revealed by comparison between theory and experiment, Phys. B, 2019, 560, 46–50 CrossRef.
- K. Seema, The effect of pressure and disorder on half-metallicity of CoRuFeSi Quaternary Heusler alloy, Intermetallics, 2019, 110, 106478 CrossRef CAS.
- P. Blaha, K. Schwarz, G. K. H. Madsen and D. Kvasnicka, WIEN2k: An Augmented Plane Wave and Local Orbitals Program for Calculating Crystal Properties, Vienna University of Technology, Vienna, 2001, pp. 19–22 Search PubMed.
- L. J. Sham and W. Kohn, One-particle properties of an inhomogeneous interacting electron gas, Phys. Rev., 1966, 145(2), 561 CrossRef CAS.
- U. Von Barth and L. Hedin, A local exchange-correlation potential for the spin polarized case, J. Phys. C: Solid State Phys., 1972, 5(13), 1629 CrossRef CAS.
- J. P. Perdew, K. Burke and M. Ernzerhof, generalized gradient approximation made simple, Phys. Rev. Lett., 1996, 77(18), 3865 CrossRef CAS PubMed.
- B. G. Yalcin, Ground state properties and thermoelectric behavior of Ru2VZ (Z = Si, Ge, Sn) half-metallic ferromagnetic full-Heusler compounds, J. Magn. Magn. Mater., 2016, 408, 137–146 CrossRef CAS.
- M. Y. Sofi and D. C. Gupta, Scrutinized the inherent spin half-metallicity and thermoelectric response of f-electron-based RbMO3 (M = Np, Pu) perovskites: a computational assessment, Sci. Rep., 2022, 12(1), 19476 CrossRef CAS PubMed.
- S. A. Khandy and D. C. Gupta, Systematic understanding of f-electron-based semiconducting actinide perovskites Ba2MgMO6 (M = U, Np) from DFT ab initio calculations, Int. J. Energy Res., 2020, 44(4), 3066–3081 CrossRef CAS.
- E. G. Özdemir and Z. Merdan, Theoretical calculations on half-metallic results properties of FeZrX (X = P, As, Sb and Bi) half-Heusler compounds: density functional theory, Mater. Res. Express, 2019, 6(8), 086102 CrossRef.
- T. Graf, C. Felser and S. S. Parkin, Simple rules for the understanding of Heusler compounds, Prog. Solid State Chem., 2011, 39(1), 1–50 CrossRef CAS.
- H. Mokhtari, L. Boumia, M. Mokhtari, F. Dahmane, D. Mansour and R. Khenata, Mechanical Stability, Electronic, and Magnetic Properties of XZrAs (X = Cr, Mn, V) Half-Heusler Compounds, J. Supercond. Novel Magn., 2023, 36(4), 1217–1224 CrossRef CAS.
- Y. Rached, M. Caid, H. Rached, M. Merabet, S. Benalia, S. Al-Qaisi and D. Rached, Theoretical insight into the stability, magneto-electronic and thermoelectric properties of XCrSb (X: Fe, Ni) Half-Heusler alloys and their superlattices, J. Supercond. Novel Magn., 2022, 35(3), 875–887 CrossRef CAS.
- M. Zeeshan, K. Singh, J. van den Brink and H. C. Kandpa, Ab initio design of new cobalt-based half-Heusler materials for thermoelectric applications, Phys. Rev. Mater., 2017, 1(7), 075407 CrossRef.
- G. Surucu, M. Isik, A. Candan, X. Wang and H. H. Gullu, Investigation of structural, electronic, magnetic and lattice dynamical properties for XCoBi (X: Ti, Zr, Hf) Half-Heusler compounds, Phys. B, 2020, 587, 412146 CrossRef CAS.
- R. Hill, The elastic behaviour of a crystalline aggregate, Proc. Phys. Soc., London, Sect. A, 1952, 65(5), 349 CrossRef.
- F. Mouhat and F. X. Coudert, Necessary and sufficient elastic stability conditions in various crystal systems, Phys. Rev. B: Condens. Matter Mater. Phys., 2014, 90(22), 224104 CrossRef.
- S. F. Pugh, XCII. Relations between the elastic moduli and the plastic properties of polycrystalline pure metals, London, Edinburgh Dublin Philos. Mag. J. Sci., 1954, 45(367), 823–843 CrossRef CAS.
- S. A. Sofi, S. Yousuf and D. C. Gupta, Prediction of robustness of electronic, magnetic and thermoelectric properties under pressure and temperature variation in Co2MnAs alloy, Comput. Condens. Matter, 2019, 19, e00375 CrossRef.
- H. S. Yoon and R. E. Newnham, Elastic properties of fluorapatite, Am. Mineral.:J. Earth Planet. Mater., 1969, 54(7–8), 1193–1197 CAS.
- D. H. Wu, H. C. Wang, L. T. Wei, R. K. Pan and B. Y. Tang, First-principles study of structural stability and elastic properties of MgPd3 and its hydride, J. Magnesium Alloys, 2014, 2(2), 165–174 CrossRef CAS.
- S. A. Khandy and D. C. Gupta, Structural, elastic and magneto-electronic properties of half-metallic BaNpO3 perovskite, Mater. Chem. Phys., 2017, 198, 380–385 CrossRef CAS.
- O. L. Anderson, A simplified method for calculating the Debye temperature from elastic constants, J. Phys. Chem. Solids, 1963, 24(7), 909–917 CrossRef CAS.
- A. Belkacem, H. Rached, M. Caid, Y. Rached, D. Rached, T. Mahmoud and N. Benkhettou, The stability analysis and efficiency of the new MAX-phase compounds M3GaC2 (M: Ti or Zr): a first-principles assessment, Results Phys., 2022, 38, 105621 CrossRef.
- A. Elkoua and R. Masrour, Structural, thermodynamics, optical, electronic, magnetic and thermoelectric properties of Heusler Ni2MnGa: an ab initio calculation, Opt. Quantum Electron., 2022, 54(10), 667 CrossRef.
- S. A. Mir and D. C. Gupta, Understanding the origin of half-metallicity and thermophysical properties of ductile La2CuMnO6 double perovskite, Int. J. Energy Res., 2019, 43(9), 4783–4847 CrossRef CAS.
- H. Luo, G. Liu, F. Meng, L. Wang, E. Liu and C. Jiang, Slater–Pauling behaviour and half-metallicity in Heusler alloys Mn2CuZ (Z = Ge and Sb), Comput. Mater. Sci., 2011, 50(11), 3119–3122 CrossRef CAS.
- Y. Jia, Q. Jiang, H. Sun, P. Liu, D. Hu, Y. Pei and Y. Cao, Wearable thermoelectric materials and devices for self-powered electronic systems, Adv. Mater., 2021, 33(42), 2102990 CrossRef CAS PubMed.
- T. M. Bhat and D. C. Gupta, Robust thermoelectric performance and high spin polarisation in CoMnTiAl and FeMnTiAl compounds, RSC Adv., 2016, 6(83), 80302–80309 RSC.
- L. E. Bell, Cooling, heating, generating power, and recovering waste heat with thermoelectric systems, Science, 2008, 321(5895), 1457–1461 CrossRef CAS PubMed.
- E. Krimi and R. Masrour, Cobalt-based full Heusler compounds Co2FeZ (Z = Al, Si, and Ga): a comprehensive study of competition between XA and L21 atomic ordering with ab initio calculation, Mater. Sci. Eng., B, 2022, 284, 115906 CrossRef.
- S. A. Mir and D. C. Gupta, Analysis of cage structured halide double perovskites Cs2NaMCl6 (M = Ti, V) by spin polarized calculations, J. Alloys Compd., 2021, 854, 156000 CrossRef CAS.
- T. M. Bhat and D. C. Gupta, Analysis of electronic, thermal, and thermoelectric properties of the half-Heusler CrTiSi material using density functional theory, J. Phys. Chem. Solids, 2018, 119, 281–287 CrossRef CAS.
- S. A. Khandy and D. C. Gupta, Magneto-electronic, mechanical, thermoelectric and thermodynamic properties of ductile perovskite Ba2SmNbO6, Mater. Chem. Phys., 2020, 239, 121983 CrossRef CAS.
- F. Peng, H. Fu and X. Yang, Ab initio study of phase transition and thermodynamic properties of PtN, Phys. B, 2008, 403(17), 2851–2855 CrossRef CAS.
|
| This journal is © The Royal Society of Chemistry 2023 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence *
*
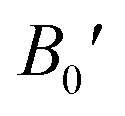 and the minimum energy E0 (eV) have been fetched as well as collected in Table 2.
and the minimum energy E0 (eV) have been fetched as well as collected in Table 2.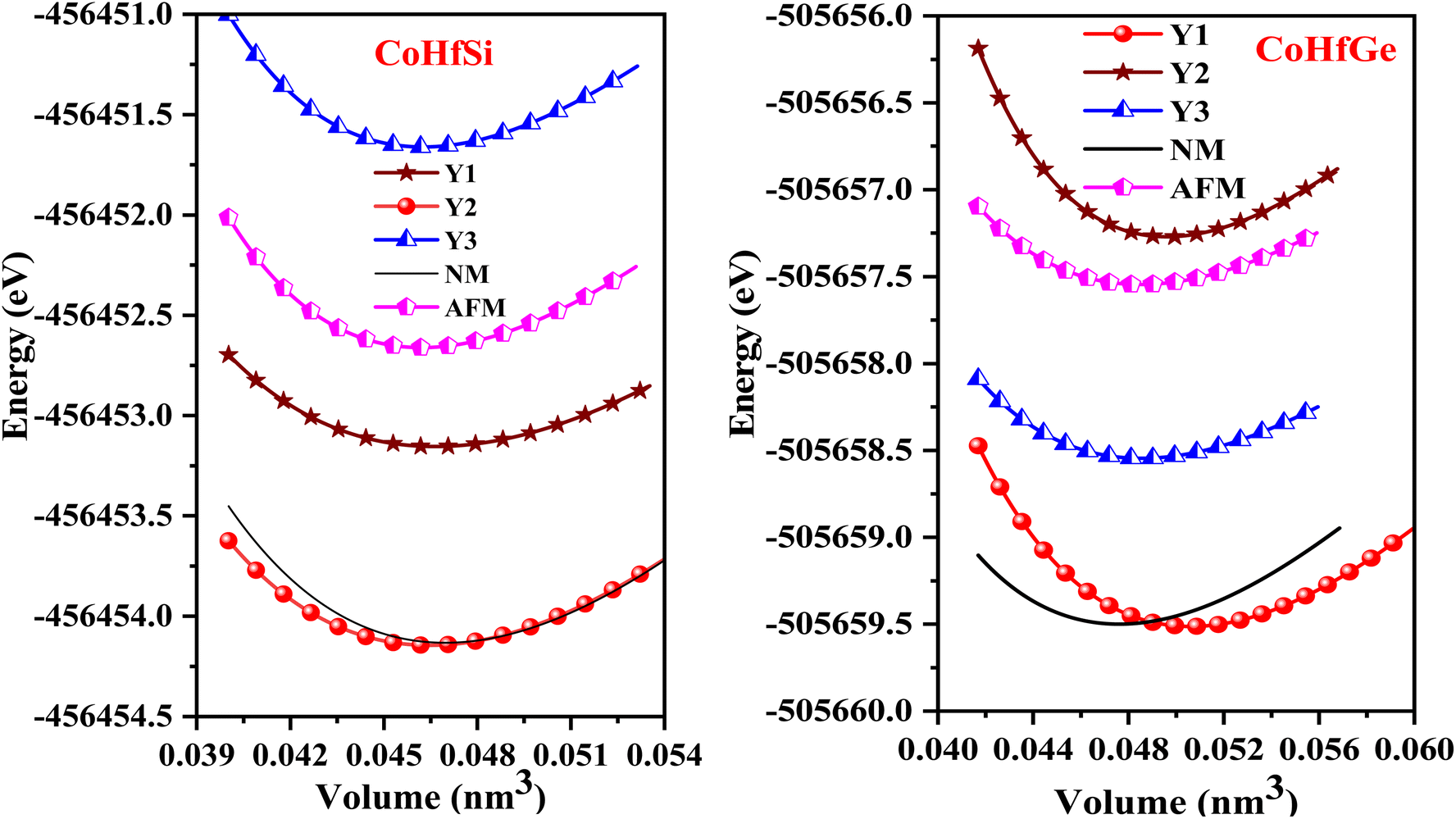
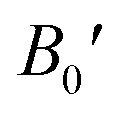 ) and minimum energy (E0 in eV) and cohesive energy (Ecoh) for CoHfSi and CoHfGe alloys
) and minimum energy (E0 in eV) and cohesive energy (Ecoh) for CoHfSi and CoHfGe alloys
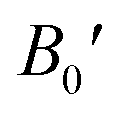
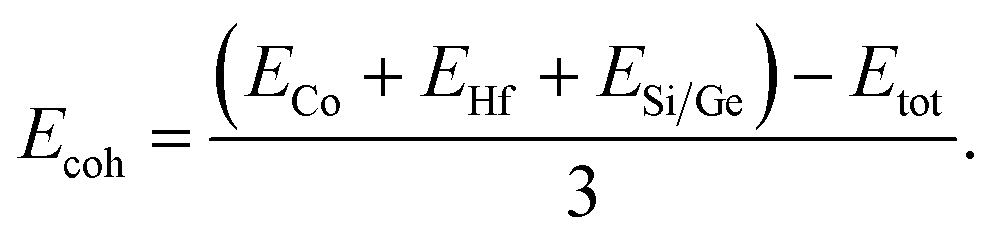
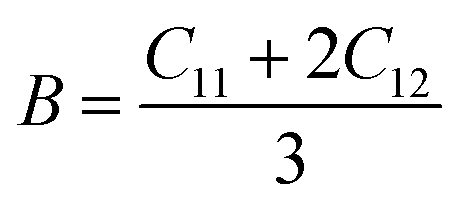
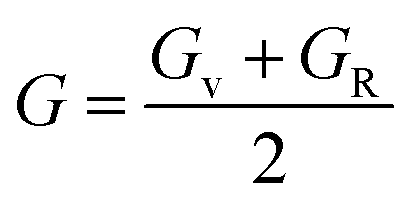
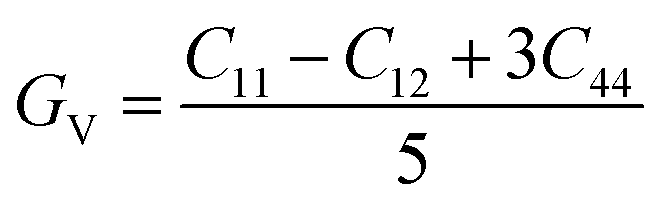
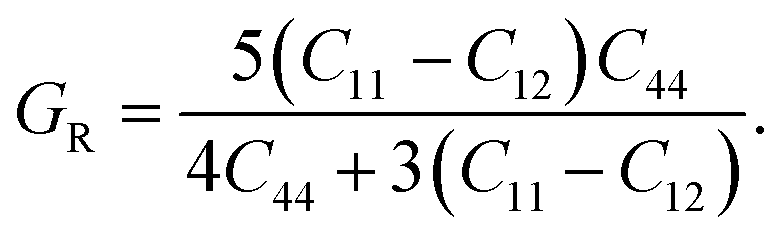
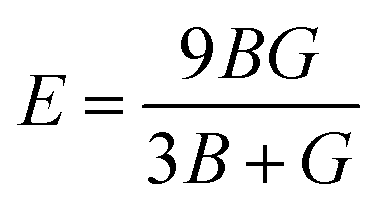
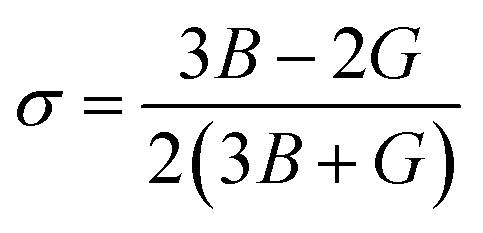
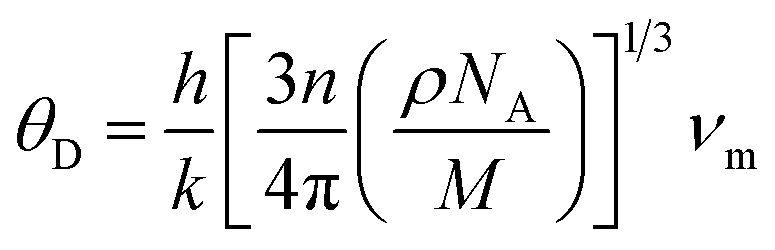
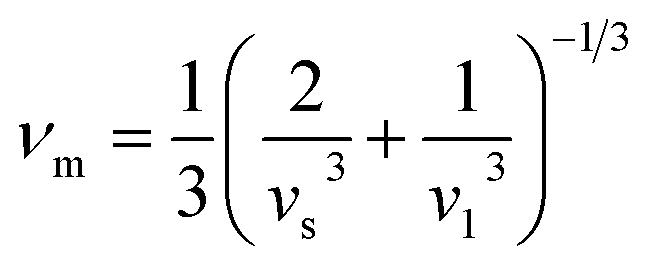
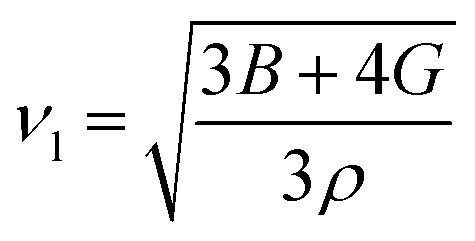
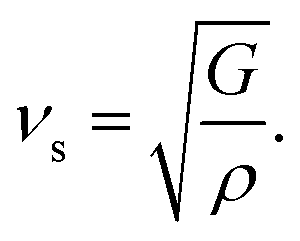
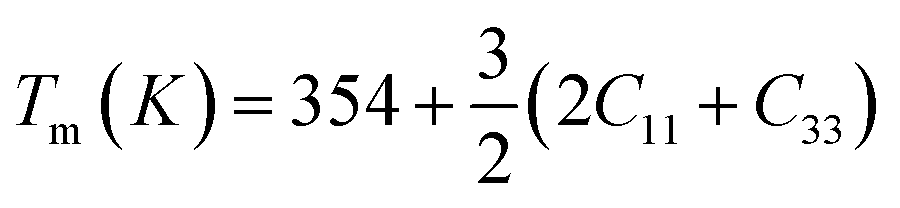
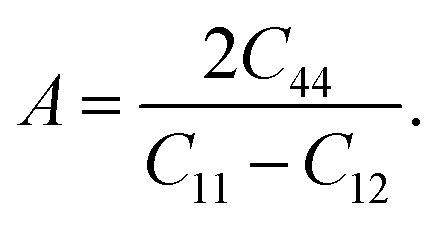
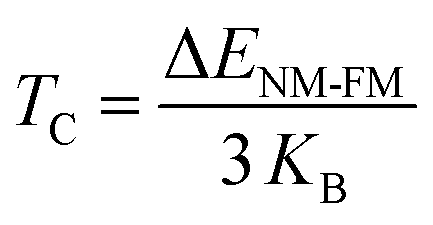
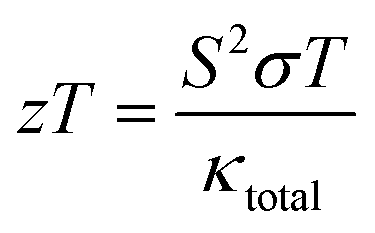
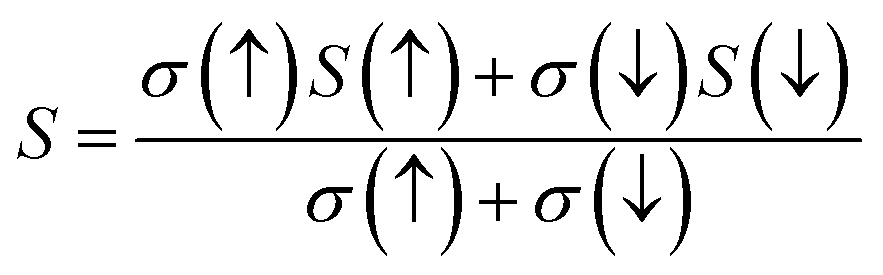
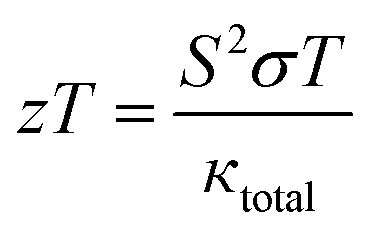 .
.