
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceInfluence of coordination structure of Fe-585DV/NxC4−x on the electrocatalytic performance of oxygen reduction reactions†
Ren Li‡
a,
Lei Zhang‡b,
Yi Wang a,
Jinbo Baic,
Xiaolin Li*d and
Chunmei Zhang*e
a,
Jinbo Baic,
Xiaolin Li*d and
Chunmei Zhang*e
aState Key Laboratory of Photoelectric Technology and Functional Materials, International Collaborative Center on Photoelectric Technology and Nano Functional Materials, Institute of Photonics & Photon-Technology, Northwest University, Xi'an, 710069, China
bState Energy Key Lab of Clean Coal Grading Conversion, Shaanxi Coal and Chemical Technology Institute Co., Ltd, Xi'an 710070, China
cUniversité Paris-Saclay, CentraleSupélec, ENS Paris-Saclay, CNRS, LMPS-Laboratoire de Mécanique Paris-Saclay, 8-10 Rue Joliot-Curie, Gif-sur-Yvette 91190, France
dInstitute of Intelligent Manufacturing Technology, Shenzhen Polytechnic University, Shenzhen 518055, China. E-mail: lixiaolin0427@szpt.edu.cn
eSchool of Physics, Northwest University, Xi'an, 710069, China. E-mail: chunmeizhang@nwu.edu.cn
First published on 19th September 2023
Abstract
Fe–N–C material, known for its high efficiency, cost-effectiveness, and environmental friendliness, is a promising electrocatalyst in the field of the oxygen reduction reaction (ORR). However, the influence of defects and coordination structures on the catalytic performance of Fe–N–C has not been completely elucidated. In our present investigation, based on density functional theory, we take an Fe adsorbed graphene structure containing a 5–8–5 divacancy (585DV) defect as a research model and investigate the influence of the coordination number of N atoms around Fe (Fe–NxC(4−x)) on the ORR electrocatalyst behavior in alkaline conditions. We find that the Fe–N4 structure exhibits superior ORR catalytic performance than other N coordination structures Fe–NxC4−x (x = 0–3). We explore the reasons for the improved catalytic performance through electronic structure analysis and find that as the N coordination number in the Fe–NxC(4−x) structure increases, the magnetic moment of the Fe single atom decreases. This reduction is conducive to the ORR catalytic performance, indicating that a lower magnetic moment is more favorable for the catalytic process of the ORR within the Fe–NxC(4−x) structure. This study is of great significance for a deeper understanding of the structure–performance relationship in catalysis, as well as for the development of efficient ORR catalysts.
1. Introduction
The oxygen reduction reaction (ORR), involving multiple electron transfers, is vital in a variety of modern devices for energy conversion and storage, such as fuel cells and metal–air batteries.1–4 However, challenges remain due to the slow kinetics and lack of catalyst stability.5,6 Commercial Pt-based materials exhibit superior ORR activity; however, their high cost and limited availability have ignited exploration into non-noble-metal catalysts.7 Over the past decade, there has been extensive research into identifying highly active and durable ORR electrocatalyst materials as viable alternatives to Pt-based catalysts.8–12 In 2011, Prof. Tao Zhang and his team pioneered the concept of single-atom catalysts (SACs), where individual atoms function as active sites.13 These catalysts have since demonstrated application potential due to their maximal usage of metal atoms, outstanding stability, and extraordinary activity.14–17 Various substrate materials have been reported for synthesizing SACs.15,18–20 Among various options, carbon-supported single-atom catalysts (CS-SACs) stand as highly potential and sustainable advanced hybrid nanocatalysts, owing to their impressive graphite structure, remarkable mechanical strength, and electrical conductivity. Carbon-based single-atoms are abundant in nature and can be synthesized from readily available and affordable precursors with ease. Prof. Xin-Bo Zhang's group synthesized the Fe–N-doped mesoporous carbon microspheres (Fe–NMCSs) catalyst, and found that it exhibited impressive catalytic performance, selectivity, and durability in alkaline conditions, with its activity matching that of Pt/C in acidic conditions.21 CS-SACs are sustainable and environmentally friendly, making them an attractive option for various applications. The sp2 hybridized graphene as a carbon-based nanomaterial has gained widespread usage as the standard theoretical model.22,23Graphene with vacancy defects has been proven to exhibit satisfactory ORR performance as a result of defect-induced charge transfer and charge redistribution.24,25 In particular, the 5–8–5 configuration of divacancy (585DV) in graphene is a common defect structure. Substituting N atoms for C atoms around these defects provides stability and has been shown to create effective anchoring points for single atoms.26–32 585DV unsaturated dangling bond in graphene trapping a single transition metal atom generates a single-atom electrocatalyst with excellent ORR catalytic activity.33 Especially, Fe–N–C, i.e., Fe single-atom anchored on N-doped carbon materials is promising to replace Pt electrocatalysts in proton exchange membrane fuel cells (PEMFCs).34 Numerous theoretical and experimental studies on Fe–N–C coordination structures have shown their excellent ORR activity, especially the Fe–N4 structure, which exhibits even more remarkable ORR performance.35–40 In addition, some experiments demonstrate that the catalytic performance of transition metal single atom can be influenced by the magnetic moment via the adsorption of intermediate products.41–43
Similarly, the catalytic performance of a single-atom catalyst (SAC) is not solely influenced by the nature of the metal single atom itself but is also affected by the local coordination environment surrounding it.44–48 Existing research indicates that the variation in hydrogen evolution reaction (HER) performance obtained by altering the coordination atoms around the SAC is comparable to the changes in performance achieved by only altering the type of transition metal atom while keeping the support material.49 This underscores the importance of the local coordination environment of the metal single atom. The influence of local coordination on metal single-atom catalysis is vital in both future catalyst design and the reconciliation of theoretical and experimental findings. This includes designing superior catalysts through controlled local coordination and investigating discrepancies between theoretical calculations and experiments, which may stem from subtle changes in the local environment due to complex experimental conditions. These subtleties can lead to significant shifts in catalytic behavior, making this field both important and challenging.
In this study, we employ density functional theory (DFT) to investigate the effect of coordination numbers of N on Fe atoms (FeNx) in 585DV/graphene on the binding strength of Fe single atom catalyst and the catalytic performance for ORR in alkaline solution. The introduction of nitrogen atoms can greatly enhance the binding ability of the substrate to the Fe single atom, where the Fe–N2C2(II) demonstrates the strongest binding capability. We observe that varying the coordination number and topological structure of N atoms can effectively modulate the binding energy of Fe single atom and regulate the performance of ORR. We explain the possible reasons for the variations in adsorption strength and find that different Fe–Nx coordination changes the electronic structure and magnetic moment of the Fe atom. These factors, in turn, alter the adsorption of intermediate products, and ultimately influence the catalytic performance.
2. Calculation methods
Spin-polarized DFT computations are carried out using the Vienna Ab initio Simulation Package (VASP).50,51 The exchange–correlation interaction is treated with generalized gradient approximation (GGA) of Perdew–Burke–Ernzerhof (PBE).52 The cutoff energy for the plane-wave is 500 eV. The convergence criteria were set with an energy threshold of 1 × 10−6 eV and a force threshold of 0.02 eV Å−1. The van der Waals (vdW) interaction was considered by the application of the DFT-D3 method.53 All the supercell of computational models in this study is constructed as the 5 × 5 × 1 of a unit cell of graphene. In order to prevent interaction between adjacent layers of graphene, a vacuum thickness of 15 Å is employed. The Brillouin zone is sampled with 3 × 3 × 1 and 4 × 4 × 1 gamma-point grids in all structural optimizations and electronic structure calculations, respectively. Using CI-NEB (climbing image-nudged elastic band), we obtained transition state geometries and activation energy calculations.54 Ab initio molecular dynamics simulations (AIMD) were conducted at 300 K for a duration of 10 ps to assess the stability of the material, utilizing the Nose–Hoover thermostat with a time step of 2 fs. The simulations were carried out on a 2 × 2 × 1 k-point grid.The 585DV/graphene structure of a single Fe atom loaded with various numbers of N dopants is expressed as Fe-585DV/NxC(4−x), where x represents the number of N atoms doped adjacent to the defect, and x varies from 1 to 4. We evaluate the stability of an adsorbed Fe single atom on Fe-585DV/NxC(4−x) (x = 0–4) by determining its binding energy when supported on 585DV/NxC4−x, in comparison to the cohesive energy of bulk Fe metal. The binding energy (Ebind) and cohesive (Ecohesive) energy are given by equations:
| Ebind = EFe/585DV − E585DV − EFe | (1) |
| Ecohesive = E(bulk/n) − EFe | (2) |
| O2* + 2H2O + 4e− → OOH* + OH− + H2O + 3e− | (3) |
| OOH* + OH− + H2O + 3e− → 2OH− + O* + H2O + 2e− | (4) |
| 2OH− + O* + H2O + 2e− → 3OH− + OH* + e− | (5) |
| 3OH− + OH* + e− → 4OH− | (6) |
| ΔG = ΔEads + ΔEZPE + TΔS + ΔGU + ΔGpH | (7) |
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 10 × pH kBT, where kB is the Boltzmann constant. In our calculations of the ORR for alkaline conditions, we assume a pH value of 14.56 We carry out Gibbs free energy correction through the VASPKIT software.59
10 × pH kBT, where kB is the Boltzmann constant. In our calculations of the ORR for alkaline conditions, we assume a pH value of 14.56 We carry out Gibbs free energy correction through the VASPKIT software.59
It is worth noting that although the PBE functional is commonly used in single-atom catalysis computational research, it may introduce errors due to an approximate treatment of the self-interaction between electrons. If the research goal is to accurately predict the most optimal catalysts, it would be advisable to use more precise methods, such as PBE+U or hybrid functionals like PBE0,60–62 to achieve more accurate results.63–65 However, since our study is not aimed at predicting the most exceptional catalysts but rather investigating the influence of local coordination structures on the ORR activity of Fe single atom, focusing on the variations brought by different coordination structures, the use of the more conventional density functional PBE for calculations is considered acceptable.
3. Results and discussion
3.1. Stability of Fe–N-585DV
The 585DV structure of a single Fe atom loaded with various numbers of N dopants is expressed as Fe-585DV/NxC(4−x), and the final optimized stable geometric structure is depicted in Fig. 1, where x represents the number of N atoms doped adjacent to the defect. Hereafter, the Fe-585DV/NxC(4−x) structure is called Fe–NxC4−x for simplicity. Stability is one of the most significant factors for catalyst synthesis. Owing to their high surface free energy, there's a propensity for single atoms to coalesce into clusters on the surface of the material. We check the stability of the Fe atom on 585DV/NxC4−x (x = 0–4) substrate by comparing the binding energies (Ebind) and cohesive energy of Fe atom. The binding energies for different N-coordination structures are shown in Table 1. The Fe–N2C2, Fe–N3C1, and Fe–N4 coordinated Fe single-atom structures are more stable. In contrast, the Fe–C4 without N doping has the lowest binding energy, higher than the cohesive energy of bulk Fe (−5.17 eV). This suggests that the Fe single atom loaded 585DV without N doping is easily aggregated to clusters. It is evident that the Fe single atom can be better stabilized by doping certain amount of N atoms around 585DV, and the stability is further enhanced when more than one N atom is doped.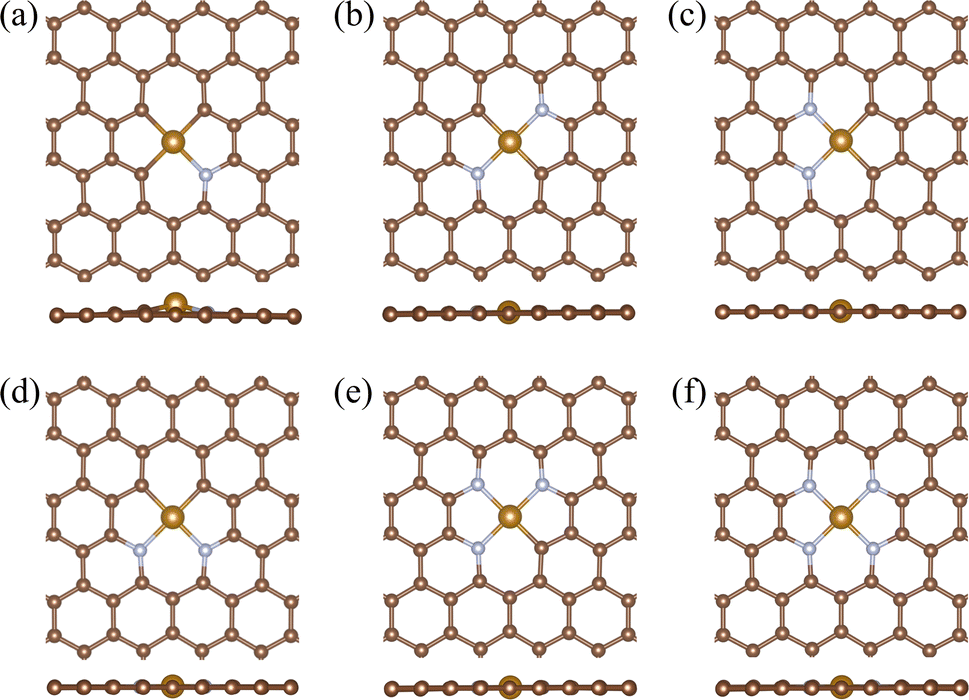 | ||
| Fig. 1 The geometry structures of (a) Fe–N1C3, (b) Fe–N2C2(I), (c) Fe–N2C2(II), (d) Fe–N2C2(III), (e) Fe–N3C1, (f) Fe–N4. The large yellow, small brown, and grey balls represent Fe, C, and N atoms, respectively. | ||
| Structure | EB/eV | Bond length/Å | ΔG1/eV | ΔG2/eV | ΔG3/eV | ΔG4/eV | GOH*/eV | η/eV | |
|---|---|---|---|---|---|---|---|---|---|
| a The ORR reaction in alkaline media at zero electrode potential (U = 0 V vs. NHE). | |||||||||
| Fe–N4 | −7.46 | Fe–N1 1.90 | Fe–N2 1.89 | −0.53 | −1.54 | 0.04 | 0.42 | −0.42 | 0.82 |
| Fe–N3 1.89 | Fe–N4 1.89 | ||||||||
| Fe–N1C3 | −6.63 | Fe–N1 1.99 | Fe–C1 1.94 | −0.78 | −1.90 | 0.34 | 0.73 | −0.73 | 1.13 |
| Fe–C2 1.93 | Fe–C3 1.93 | ||||||||
| Fe–N2C2(I) | −8.00 | Fe–N1 1.93 | Fe–N2 1.93 | −0.80 | −1.80 | 0.20 | 0.80 | −0.80 | 1.20 |
| Fe–C1 1.91 | Fe–C2 1.91 | ||||||||
| Fe–N2C2(II) | −8.31 | Fe–N1 1.94 | Fe–N2 1.94 | −0.74 | −1.56 | 0.01 | 0.68 | −0.68 | 1.08 |
| Fe–C1 1.90 | Fe–C2 1.90 | ||||||||
| Fe–N2C2(III) | −7.23 | Fe–N1 1.96 | Fe–N2 1.96 | −0.73 | −1.49 | −0.16 | 0.78 | −0.78 | 1.18 |
| Fe–C1 1.88 | Fe–C2 1.88 | ||||||||
| Fe–N3C1 | −7.83 | Fe–N1 1.93 | Fe–N2 1.92 | −0.75 | −1.42 | −0.14 | 0.70 | −0.70 | 1.10 |
| Fe–N3 1.90 | Fe–C1 1.88 | ||||||||
| Fe–C4 | −4.80 | Fe–C1 1.96 | Fe–C2 1.96 | −1.17 | −1.70 | 0.42 | 0.85 | −0.85 | 1.25 |
| Fe–C3 1.96 | Fe–C4 1.96 | ||||||||
After relaxation, the average bond lengths from the Fe atom to its neighbouring N and C atoms in the Fe–C4 and Fe–N1C3 structures are 1.96 Å and 1.95 Å, respectively. These bond lengths are larger than the average bond lengths of other coordination structures, with the Fe–C4 bond length being the largest. This result is consistent with the obtained binding energy, indicating an unstable structure of Fe–C4. The side view of Fig. 1 and S1† provide an intuitive observation that the Fe atom in both the Fe–C4 and Fe–N1C3 structures protrude from the plane. Table 1 provides specific calculation details.
To compare the bond strength between different coordination structures of Fe-585DV/NxC(4−x), we further calculate the crystal orbital Hamilton population (COHP) of Fe–N and Fe–C bonds of various Fe-585DV/NxC(4−x) structures. The traditional integrated COHP (ICOHP) of the Fe–N4 coordination structure (−10.174 eV) is more negative than the ICOHP of the Fe–C4 coordination structure (−9.530 eV), indicating stronger bonding. Fig. 2 demonstrates that the COHP of Fe–C4 has more antibonding molecular orbitals near the Fermi surface, which reduces the bonding strength of Fe–C, a conclusion aligned with the Ebind calculation. Fig. S2(a–g) of the ESI† shows that with increasing N coordination number, the bonding strength of Fe and the four adjacent atoms gradually increase. It is discovered that introducing N atoms not only enhances the bonding between Fe and N but also influences the electronic structure of the neighbouring C atom. This results in an increase in bonding strength, suggesting that the different Fe-585DV/NxC(4−x) coordination structures could regulate the electronic structure around the active Fe site. From the projected density of states (PDOS) for Fe and the neighbouring N and C atoms in various Fe-585DV/NxC(4−x) structures, we find that Fe and the coordinated N atoms overlap greatly, indicating a stronger orbital interaction. The details are in the ESI Fig. S3.†
 | ||
| Fig. 2 Fe–C4 coordination structure with Fe–C bond COHP and Fe–N4 coordination structure with Fe–N bond COHP. | ||
For the typical Fe–N4 structure, which demonstrates superior performance, AIMD simulations were conducted at 300 K to assess its stability. As illustrated in Fig. S4,† the structure oscillated around the equilibrium position in response to temperature changes. The trend is also consistent with the reports in the literature.66 Structures at 1 ps, 3 ps, and 5 ps were plotted, and no structural distortion was observed, further confirming its stability.
3.2. ORR reaction mechanism on Fe–N-585DV
To check the Fe–N coordination configurations of Fe-585DV/NxC(4−x) on the ORR catalytic activity, we further investigate the ORR catalytic activity. In this study, we initially focused on the traditional 4-electron oxygen reduction reaction (ORR) process, yielding intermediates such as OOH*, O*, and OH*.47,67,68 It is worth mentioning that recent findings by Gianfranco Pacchioni's group and other researchers have indicated the potential existence of other more stable intermediates, such as OH*OH* and OH*O*.69–73 Inspired by these findings, we later conducted a supplementary investigation into the effects of these two intermediates on the ORR reaction pathway under various N coordination structures.As illustrated in the method part, O2 eventually forms OH− through a continuous protonation process. The initial molecule O2 is first adsorbed on Fe active sites and forms *OOH by a proton–electron transfer. The *OOH transforms to *O and OH− through additional proton–electron transfer. O* can be further hydrogenated to form OH*. The final step is that the adsorbed OH* dissociates to release OH− ions. As can be seen from Fig. 3, the free energy diagrams for Fe single-atomic ORR reactions with different N coordination numbers in alkaline media at zero electrode potential (U = 0 V vs. NHE), the rate-determining step occurs at the final stage – the desorption of OH*. The Gibbs free energy of adsorption for OH* in different coordination structures is presented in Table 1. As the coordination number of N increases, it becomes more favorable for the desorption of OH*. The OH* adsorption free energy is the highest in the Fe–N4 structure, at −0.42 eV. It should be mentioned that in the Fe–N2C2 coordination structure, different topological structures have a significant impact on the desorption energy of OH*, with the OH* adsorption free energy being higher in the Fe–N2C2(II) topological structure. This demonstrates that N coordination affects the rate-determining step for 585DV graphene defects.
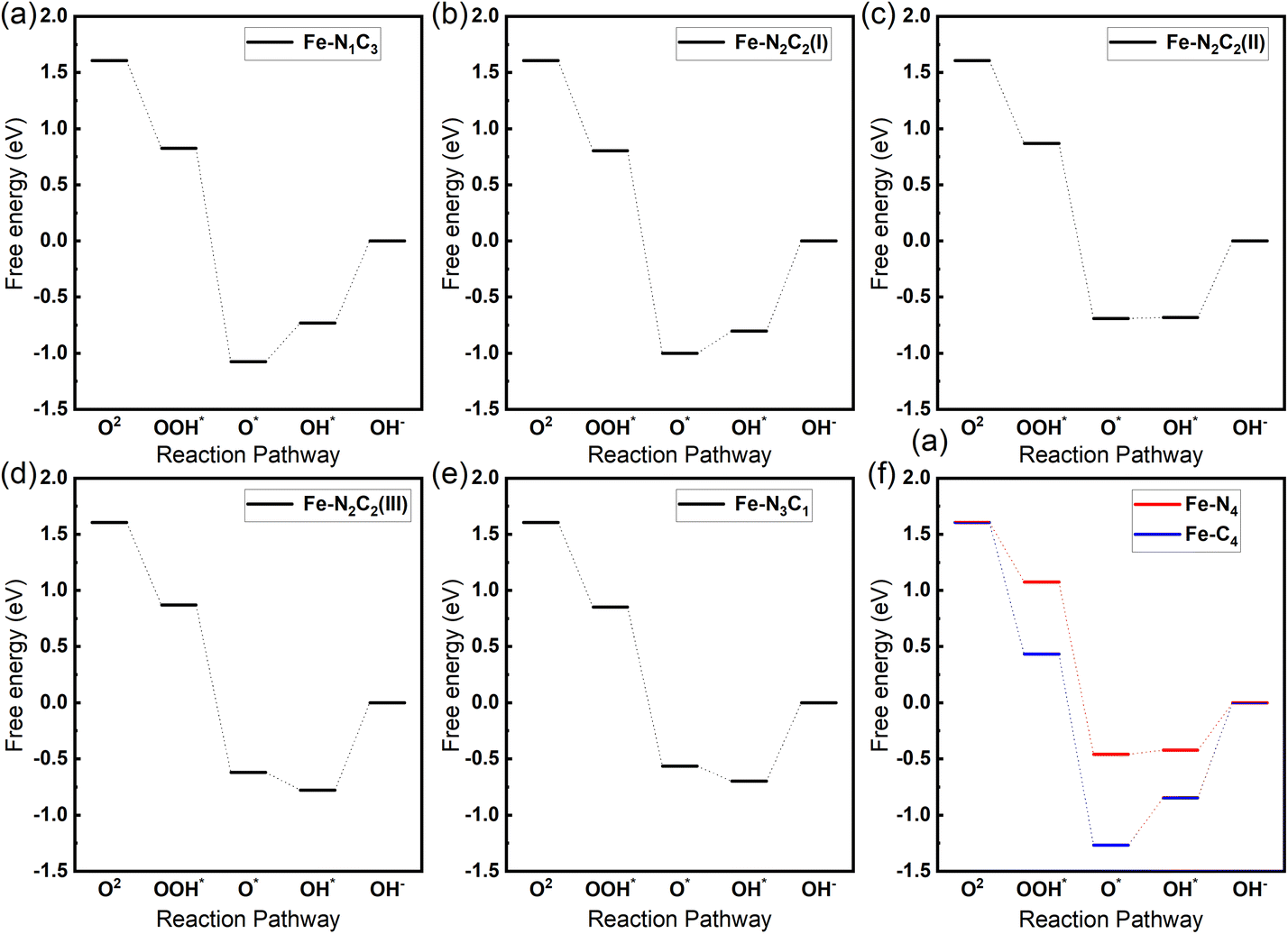 | ||
| Fig. 3 Gibbs energy profiles of ORR reaction in alkaline media at zero electrode potential (U = 0 V vs. NHE) with different Fe-585DV/NxC(4−x) structures: (a) Fe–N1C3, (b) Fe–N2C2(I), (c) Fe–N2C2(II), (d) Fe–N2C2(III), (e) Fe–N3C1, (f) Fe–N4. | ||
On the contrary, the overpotential of the Fe–C4 coordination structure is relatively large, providing lower ORR catalytic performance. The volcanic curve of Gibbs free energy adsorbing OH* and the negative overpotential relation of each coordination structure in Fig. 4(a) reveals that the Fe–N4 coordination structure is located near the peak of the volcanic curve. In contrast, the Fe–C4 coordination structure is positioned at the low point on the left, illustrating that the Fe–N4 coordination structure is more advantageous to the ORR catalytic performance of alkaline solutions. These coordination structures Fe–N2C2(I), Fe–N2C2(III), Fe–N1C3, Fe–N3C1, and Fe–N2C2(III) are relatively close in the position of the volcanic plot and are gradually shifting near the apex. Correspondingly, a gradual increase is observed in their catalytic performance for the alkaline oxygen reduction reaction (ORR). Further exploring the superior performance of the Fe–N4 coordination structure, we performed a kinetic behavior analysis, as shown in Fig. S5.† The final step of releasing OH* has the highest barrier of 1.81 eV among all steps. This is consistent with the thermodynamic result that the final step is the rate-determining step.
 | ||
| Fig. 4 (a) Overpotential as a function of *OH adsorption free energy (ΔGOH*). (b) OH* adsorption free energy (ΔG*OH) changes with magnetic moment (μB). | ||
We further explore whether the emergence of other intermediates such as OH*O* and OH*OH* during the ORR reaction process would impact the catalytic performance, as shown in Fig. S6.† Various N-coordination structures were chosen for examination, with a special note on the Fe–N2C2 coordination structure. We selected the higher-performing Fe–N2C2(II) as a representative. The specific catalytic process is depicted in eqn (1)–(4) in the ESI.† This remains a 4-electron process but with OH*O* substituting OOH*, where OH*O* represents the co-adsorption of OH* and O* on a single Fe atom. Similarly, the dissociation process of OOH* to OH− is replaced by the hydrogenation of OH*O*, and the dissociation of the first OH− from OH*OH* replaces the process of adding H to O*. The final step aligns with the traditional pathway, representing the dissociation of OH* and therelease of OH−. As observed in the computed Gibbs free energy process depicted in Fig. S7 and Tabel S1,† the intermediate OH*O* is more stable energetically than OOH* within N-coordinated structures, indicating that the intermediate OH*O* is more likely to form. Conversely, the energy of the intermediate OH*OH* is higher than that of the adsorbed O*, reflecting the ease of formation of the O* intermediate in the traditional ORR pathway. Interestingly, in the absence of N coordination in the Fe–C4 structure, both OH*O* and OH*OH* intermediates exhibit higher energy, making the traditional ORR pathway more likely to occur. This illustrates that the introduction of N atoms into the local coordination structure of single-atom Fe can affect the ORR reaction pathway. Furthermore, our findings revealed that regardless of the reaction pathway, the rate-determining step in the ORR reaction is the dissociation process of OH*, releasing OH−. The consideration of OH*O* and OH*OH* intermediates does not affect the overpotential of the Fe–Nx coordination structure.
From the electronic structure aspect, we illustrate how the Fe–Nx coordination structures impact catalytic performance. Calculate the Bader charge transfer of the Fe single atom after the adsorption of OH* on different coordination structures of Fe–Nx, as detailed in the Table 2. In structures containing N-doping, electrons are lost, with the Fe–N4 coordination structure losing the least amount, at 0.12e. Compared to other N coordination structures, this value strikes a moderate balance. Such a balanced charge transfer contributes to suitable adsorption energy (G), thus enhancing its performance as an ORR catalyst. This aligns with the Sabatier principle's proposition, emphasizing an effective catalyst's need for moderate interaction with the substrate – not too strong nor too weak in terms of bonding.74 On the contrary, the Fe–C4 coordination structure gains 0.01e, resulting in a higher OH* desorption energy, which hinders catalysis. Relative to the Fe–N4 structure, other coordination structures of Fe–Nx, due to varying N coordination numbers or topological differences, lead to the Fe single atom transferring either more or fewer charges after the adsorption of OH*. This results in catalytic performance that falls between that of Fe–N4 and Fe–C4. Further calculations of the density of states for Fe–NxC4−x indicate that within the energy range of −2 eV and 2 eV around the Fermi level, there is less hybridization in the Fe–N4 coordination structure between Fe's 3d orbital and the 2p orbital of O when compared to other coordination structures that contain N.
| Structure | Charge transfer/|e| | Magnetic moment/μB |
|---|---|---|
| Fe–N4 | −0.12 | 1.91 |
| Fe–N1C3 | −0.21 | 2.68 |
| Fe–N2C2(I) | −0.05 | 2.42 |
| Fe–N2C2(II) | −0.12 | 2.58 |
| Fe–N2C2(III) | −0.16 | 2.51 |
| Fe–N3C1 | −0.17 | 2.13 |
| Fe–C4 | 0.01 | 2.80 |
However, it shows more overlap compared to the Fe–C4 coordination structure, as shown in Fig. 5 and S4.† This suggests that the interaction between single-atom Fe and O atoms in the Fe–N4 coordination structure is more favourable for the ORR catalytic reaction.
 | ||
| Fig. 5 Projected density of states for Fe and O atoms after adsorption of OH* on different Fe-585DV/NxC(4−x) structures. (a) Fe–N1C3, (b) Fe–N2C2(I), (c) Fe–N2C2(II), (d) Fe–N2C2(III), (e) Fe–N3C1, (f) Fe–N4. | ||
The spin magnetic moment involves the unpaired electron count, which can lead to a certain degree of exchange interaction with the adsorbed intermediates. Calculating the magnetic moment of different coordination structures of Fe-585DV/NxC(4−x), as shown in Table 2 and Fig. 4(b), reveals that the magnetic moment is approximately proportional to the adsorption energy of OH* for different Fe–Nx configurations. The magnetic moment of the Fe–N4 coordination structure is the smallest, at 1.91 μB, which produces a more suitable adsorption energy for OH*, thereby enhancing the ORR reaction. In contrast, the Fe–C4 coordination structure, with a high magnetic moment of 2.80 μB, leads to stronger OH* adsorption. Interestingly, with an increase in the N coordination number within the Fe–NxC(4−x) structure, the magnetic moment of the Fe single atom progressively decreases. This decrease is accompanied by an enhancement in ORR catalytic performance. From this, we can deduce that the different N coordination structures within Fe–NxC(4−x) affect the central Fe single atom's magnetic moment, and a lower magnetic moment appears to be more favorable for the ORR catalytic performance of the Fe–NxC(4−x) structure.
4. Conclusions
We investigate the effect of various N coordination numbers on the binding strength of Fe single-atom and the ORR catalytic performance of Fe-585DV/NxC(4−x) in alkaline solutions. Through COHP and electronic structure analysis, we find that the Fe binding energy is greater in structures with 2N, 3N, and 4N coordination than in structures with 4C coordination. The latter has the weakest binding strength and could easily form clusters as its energy is lower than the Fe atomic cohesive energy. The COHP for Fe–C4 and Fe–N1C3 contain more antibonding orbitals near the Fermi level than other structures, which decreases the substrate's bonding ability with Fe. Additionally, AIMD simulations were conducted on the Fe–N4 coordination structure, further confirming the stability of this structure. We investigated the traditional ORR pathway under alkaline conditions, as well as the ORR catalytic performance involving the formation of OH*O* and OH*OH* intermediates. We concluded that irrespective of the pathway, the rate-determining step for different N-coordinated structures is the final dissociation of OH* into OH−, where Fe–N4 exhibits the best performance, while Fe–C4 shows the poorest. In the case of the Fe–N2C2 coordination structure, even with the same coordination number of N, different topological structures can influence the energy required for OH* dissociation. Furthermore, the introduction of the N atom also affects the formation of intermediates in the ORR reaction pathway. In N-containing coordination structures, the OH*O* intermediate has a relatively lower energy compared to OOH*, making it more likely to occur, whereas the OH*OH* intermediate has a higher relative energy. Conversely, in the 4C coordination structure, the relative energies of OH*O* and OH*OH* are higher, making the traditional pathway more favorable.Bader charge calculations demonstrate that Fe in the Fe–N4 coordination structure transfers a moderate amount of charges to the adsorbed OH*, resulting in adequate adsorption strength that is advantageous to ORR catalytic performance. We also analyzed the magnetic moment of the local coordination structure of the Fe single atom and discovered that as the number of N coordinations in the Fe–NxC(4−x) structure increases, the magnetic moment of the Fe single atom progressively decreases. This reduction is accompanied by an enhancement in ORR catalytic performance. Different N coordination structures within Fe–NxC(4−x) affect the central Fe single atom's magnetic moment, with a lower magnetic moment appearing to be more favorable for the ORR catalytic performance of the Fe–NxC(4−x) structure.
By adjusting the neighboring N coordination structure of the single-atom Fe located at a double vacancy on graphene, we have demonstrated that the catalytic performance of a single atom is influenced by the interactions with its surrounding local coordination environment. Even slight changes in the local coordination environment can affect the Fe single atom's electronic structure and spin magnetic moment, subsequently impacting the adsorption of intermediates and determining the catalytic effect. Therefore, whether in experimental catalyst design or in comparisons between theoretical calculations and experiments, the single-atom local coordination environment can easily be influenced by complex external experimental conditions. This may lead to changes in the local structure, and even minor alterations might bring about significant shifts in catalytic performance. This aspect is highly important and requires particular care and attention.
Conflicts of interest
The authors have no conflicts to disclose.Acknowledgements
We acknowledge the financial support from Guangdong Basic and Applied Basic Research Foundation (2021A1515011808), Shenzhen Science and Technology Program (RCBS20210609104444087), Research Funding of Post-doctoral Who Came to Shenzhen (4103-6021271018K1), High-level Talents Scientific Research Start-up Project of Shenzhen Polytechnic (4103-6023330010K), Natural Science Basic Research Program of Shaanxi (2022JM-018).References
- M. Shao, Q. Chang, J.-P. Dodelet and R. Chenitz, Chem. Rev., 2016, 116, 3594–3657 CrossRef CAS PubMed.
- Z.-L. Wang, D. Xu, J.-J. Xu and X.-B. Zhang, Chem. Soc. Rev., 2014, 43, 7746–7786 RSC.
- D. Banham and S. Ye, ACS Energy Lett., 2017, 2, 629–638 CrossRef CAS.
- Y. Liu, F. Chen, W. Ye, M. Zeng, N. Han, F. Zhao, X. Wang and Y. Li, Adv. Funct. Mater., 2017, 27, 1606034 CrossRef.
- M. Liu, Z. Zhao, X. Duan and Y. Huang, Adv. Mater., 2019, 31, 1802234 CrossRef PubMed.
- M. K. Debe, Nature, 2012, 486, 43–51 CrossRef CAS PubMed.
- X. Tian, X. F. Lu, B. Y. Xia and X. W. Lou, Joule, 2020, 4, 45–68 CrossRef CAS.
- A. Kulkarni, S. Siahrostami, A. Patel and J. K. Nørskov, Chem. Rev., 2018, 118, 2302–2312 CrossRef CAS PubMed.
- N. Kakati, J. Maiti, S. H. Lee, S. H. Jee, B. Viswanathan and Y. S. Yoon, Chem. Rev., 2014, 114, 12397–12429 CrossRef CAS PubMed.
- K. Ben Liew, W. R. W. Daud, M. Ghasemi, J. X. Leong, S. Su Lim and M. Ismail, Int. J. Hydrogen Energy, 2014, 39, 4870–4883 CrossRef CAS.
- Z. Xia, L. An, P. Chen and D. Xia, Adv. Energy Mater., 2016, 6, 1600458 CrossRef.
- D. K. Madheswaran and A. Jayakumar, Bull. Mater. Sci., 2021, 44, 287 CrossRef CAS.
- B. Qiao, A. Wang, X. Yang, L. F. Allard, Z. Jiang, Y. Cui, J. Liu, J. Li and T. Zhang, Nat. Chem., 2011, 3, 634–641 CrossRef CAS PubMed.
- X.-F. Yang, A. Wang, B. Qiao, J. Li, J. Liu and T. Zhang, Acc. Chem. Res., 2013, 46, 1740–1748 CrossRef CAS PubMed.
- A. Wang, J. Li and T. Zhang, Nat. Rev. Chem., 2018, 2, 65–81 CrossRef CAS.
- J. Yang, W. Li, D. Wang and Y. Li, Adv. Mater., 2020, 32, 2003300 CrossRef CAS PubMed.
- H. Zhang, G. Liu, L. Shi and J. Ye, Adv. Energy Mater., 2018, 8, 1701343 CrossRef.
- C. Zhu, S. Fu, Q. Shi, D. Du and Y. Lin, Angew. Chem., Int. Ed., 2017, 56, 13944–13960 CrossRef CAS PubMed.
- S. Swain, A. Altaee, M. Saxena and A. K. Samal, Coord. Chem. Rev., 2022, 470, 214710 CrossRef CAS.
- Y. Chen, S. Ji, C. Chen, Q. Peng, D. Wang and Y. Li, Joule, 2018, 2, 1242–1264 CrossRef CAS.
- F.-L. Meng, Z.-L. Wang, H.-X. Zhong, J. Wang, J.-M. Yan and X.-B. Zhang, Adv. Mater., 2016, 28, 7948–7955 CrossRef CAS PubMed.
- M. B. Gawande, P. Fornasiero and R. Zbořil, ACS Catal., 2020, 10, 2231–2259 CrossRef CAS.
- Y. Jiao, Y. Zheng, M. Jaroniec and S. Z. Qiao, J. Am. Chem. Soc., 2014, 136, 4394–4403 CrossRef CAS PubMed.
- X. Wang, Y. Jia, X. Mao, L. Zhang, D. Liu, L. Song, X. Yan, J. Chen, D. Yang, J. Zhou, K. Wang, A. Du and X. Yao, Chem, 2020, 6, 2009–2023 CAS.
- P. Cui, L. Zhao, Y. Long, L. Dai and C. Hu, Angew. Chem., Int. Ed., 2023, 62, e202218269 CrossRef CAS PubMed.
- Y. Fujimoto and S. Saito, Phys. Rev. B: Condens. Matter Mater. Phys., 2011, 84, 245446 CrossRef.
- Y. Jia, J. Chen and X. Yao, Mater. Chem. Front., 2018, 2, 1250–1268 RSC.
- J. Zhu and S. Mu, Adv. Funct. Mater., 2020, 30, 2001097 CrossRef CAS.
- Z. Hou, X. Wang, T. Ikeda, K. Terakura, M. Oshima, M. Kakimoto and S. Miyata, Phys. Rev. B: Condens. Matter Mater. Phys., 2012, 85, 165439 CrossRef.
- A. Zitolo, V. Goellner, V. Armel, M.-T. Sougrati, T. Mineva, L. Stievano, E. Fonda and F. Jaouen, Nat. Mater., 2015, 14, 937–942 CrossRef CAS PubMed.
- C. Qin, S. Ruan, K. Xu, C. He, Y. Shi, B. Feng and L. Zhang, Mol. Catal., 2023, 541, 113103 CrossRef CAS.
- J. Leverett, R. Daiyan, L. Gong, K. Iputera, Z. Tong, J. Qu, Z. Ma, Q. Zhang, S. Cheong, J. Cairney, R.-S. Liu, X. Lu, Z. Xia, L. Dai and R. Amal, ACS Nano, 2021, 15, 12006–12018 CrossRef CAS PubMed.
- F. Li, G. Han, Y. Bu, H. Noh, J. Jeon, T. J. Shin, S. Kim, Y. Wu, H. Y. Jeong, Z. Fu, Y. Lu and J. Baek, Angew. Chem., Int. Ed., 2020, 59, 23678–23683 CrossRef CAS PubMed.
- S. Kattel, P. Atanassov and B. Kiefer, Phys. Chem. Chem. Phys., 2014, 16, 13800 RSC.
- S. Kattel and G. Wang, J. Phys. Chem. Lett., 2014, 5, 452–456 CrossRef CAS PubMed.
- Y. Li, X. Liu, L. Zheng, J. Shang, X. Wan, R. Hu, X. Guo, S. Hong and J. Shui, J. Mater. Chem. A, 2019, 7, 26147–26153 RSC.
- D. Xia, X. Yang, L. Xie, Y. Wei, W. Jiang, M. Dou, X. Li, J. Li, L. Gan and F. Kang, Adv. Funct. Mater., 2019, 29, 1906174 CrossRef CAS.
- F. Liu, G. Zhu, D. Yang, D. Jia, F. Jin and W. Wang, RSC Adv., 2019, 9, 22656–22667 RSC.
- H. Xu, D. Wang, P. Yang, A. Liu, R. Li, Y. Li, L. Xiao, J. Zhang and M. An, Phys. Chem. Chem. Phys., 2020, 22, 28297–28303 RSC.
- Y. Qiao, P. Yuan, Y. Hu, J. Zhang, S. Mu, J. Zhou, H. Li, H. Xia, J. He and Q. Xu, Adv. Mater., 2018, 30, 1804504 CrossRef PubMed.
- G. Yang, J. Zhu, P. Yuan, Y. Hu, G. Qu, B.-A. Lu, X. Xue, H. Yin, W. Cheng, J. Cheng, W. Xu, J. Li, J. Hu, S. Mu and J.-N. Zhang, Nat. Commun., 2021, 12, 1734 CrossRef CAS PubMed.
- Z. Chen, H. Niu, J. Ding, H. Liu, P. Chen, Y. Lu, Y. Lu, W. Zuo, L. Han, Y. Guo, S. Hung and Y. Zhai, Angew. Chem., 2021, 133, 25608–25614 CrossRef.
- Y. Liu, X. Liu, Z. Lv, R. Liu, L. Li, J. Wang, W. Yang, X. Jiang, X. Feng and B. Wang, Angew. Chem., 2022, 134, e202117617 CrossRef.
- D. Jiao, Y. Liu, Q. Cai and J. Zhao, J. Mater. Chem. A, 2021, 9, 1240–1251 RSC.
- Y. Li, J. Chen, P. Cai and Z. Wen, J. Mater. Chem. A, 2018, 6, 4948–4954 RSC.
- F. Lin, F. Lv, Q. Zhang, H. Luo, K. Wang, J. Zhou, W. Zhang, W. Zhang, D. Wang, L. Gu and S. Guo, Adv. Mater., 2022, 34, 2202084 CrossRef CAS PubMed.
- J. Zhang, D. Cao, H. Liu, F. Wang, L. Liang, C. Liu, Q. Hao and Y. Li, ChemPhysChem, 2022, 23, e202100692 CrossRef CAS PubMed.
- X. Zhu, X. Tan, K. Wu, S. Haw, C. Pao, B. Su, J. Jiang, S. C. Smith, J. Chen, R. Amal and X. Lu, Angew. Chem., Int. Ed., 2021, 60, 21911–21917 CrossRef CAS PubMed.
- G. Di Liberto, L. A. Cipriano and G. Pacchioni, ChemCatChem, 2022, 14, e202200611 CrossRef CAS.
- G. Kresse and J. Furthmüller, Comput. Mater. Sci., 1996, 6, 15–50 CrossRef CAS.
- G. Kresse and J. Furthmüller, Phys. Rev. B: Condens. Matter Mater. Phys., 1996, 54, 11169–11186 CrossRef CAS PubMed.
- J. P. Perdew and W. Yue, Phys. Rev. B: Condens. Matter Mater. Phys., 1986, 33, 8800–8802 CrossRef PubMed.
- S. Grimme, J. Antony, S. Ehrlich and H. Krieg, J. Chem. Phys., 2010, 132, 154104 CrossRef PubMed.
- G. Henkelman and H. Jónsson, J. Chem. Phys., 2000, 113, 9978–9985 CrossRef CAS.
- S. Zhang, Y. Qin, S. Ding and Y. Su, ChemPhysChem, 2022, 23, e202200165 CrossRef CAS PubMed.
- J. K. Nørskov, J. Rossmeisl, A. Logadottir, L. Lindqvist, J. R. Kitchin, T. Bligaard and H. Jónsson, J. Phys. Chem. B, 2004, 108, 17886–17892 CrossRef.
- K. Mathew, R. Sundararaman, K. Letchworth-Weaver, T. A. Arias and R. G. Hennig, J. Chem. Phys., 2014, 140, 084106 CrossRef PubMed.
- K. Mathew, V. S. C. Kolluru, S. Mula, S. N. Steinmann and R. G. Hennig, J. Chem. Phys., 2019, 151, 234101 CrossRef PubMed.
- V. Wang, N. Xu, J.-C. Liu, G. Tang and W.-T. Geng, Comput. Phys. Commun., 2021, 267, 108033 CrossRef CAS.
- C. Adamo and V. Barone, J. Chem. Phys., 1999, 110, 6158–6170 CrossRef CAS.
- J. Hubbard, Proc. R. Soc. London, Ser. A, 1964, 277, 237–259 Search PubMed.
- S. L. Dudarev, G. A. Botton, S. Y. Savrasov, C. J. Humphreys and A. P. Sutton, Phys. Rev. B: Condens. Matter Mater. Phys., 1998, 57, 1505–1509 CrossRef CAS.
- A. M. Patel, S. Ringe, S. Siahrostami, M. Bajdich, J. K. Nørskov and A. R. Kulkarni, J. Phys. Chem. C, 2018, 122, 29307–29318 CrossRef CAS.
- I. Barlocco, L. A. Cipriano, G. Di Liberto and G. Pacchioni, Adv. Theory Simul., 2022, 2200513 CrossRef.
- G. Di Liberto, I. Barlocco, L. Giordano, S. Tosoni and G. Pacchioni, Curr. Opin. Electrochem., 2023, 40, 101343 CrossRef CAS.
- X. Zheng, Y. Yao, Y. Wang and Y. Liu, Nanoscale, 2020, 12, 9696–9707 RSC.
- F. Yu, J. Zhan, D. Chen, J. Guo, S. Zhang and L. Zhang, Adv. Funct. Mater., 2023, 2214425 CrossRef CAS.
- P. Zhu, P. Song, W. Feng, D. Zhao, T. Liu, J. Zhang and C. Chen, J. Mater. Chem. A, 2022, 10, 17948–17967 RSC.
- L. Zhong and S. Li, ACS Catal., 2020, 10, 4313–4318 CrossRef CAS.
- L. A. Cipriano, G. Di Liberto and G. Pacchioni, ACS Catal., 2022, 12, 11682–11691 CrossRef CAS.
- I. Barlocco, L. A. Cipriano, G. Di Liberto and G. Pacchioni, J. Catal., 2023, 417, 351–359 CrossRef CAS.
- I. Barlocco, G. Di Liberto and G. Pacchioni, Energy Adv., 2023, 2, 1022–1029 RSC.
- C. Saetta, G. Di Liberto and G. Pacchioni, Top. Catal., 2023 DOI:10.1007/s11244-023-01802-x.
- M. Che, Catal. Today, 2013, 218–219, 162–171 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3ra04270g |
| ‡ Ren Li and Lei Zhang contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2023 |