
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSynthesis of a 1-aza-2-phospha-acenaphthene complex profiting from coordination enabled chloromethane elimination†
David Biskup a,
Tom Bergmanna,
Gregor Schnakenburga,
Rosa M. Gomilab,
Antonio Fronterab and
Rainer Streubel*a
a,
Tom Bergmanna,
Gregor Schnakenburga,
Rosa M. Gomilab,
Antonio Fronterab and
Rainer Streubel*a
aInstitut für Anorganische Chemie, Rheinische Friedrich-Wilhelms-Universität Bonn, Gerhard-Domagk-Str. 1, 53121 Bonn, Germany. E-mail: r.streubel@uni-bonn.de
bDepartment of Chemistry, Universitat de les Illes Balears, crts de Valldemossa km 7.5, 07122 Palma de Mallorca, Baleares, Spain
First published on 14th July 2023
Abstract
Despite debate on intramolecular N→P interactions in peri-substituted naphthalene derivatives their coordination chemistry has not yet been reported. Herein, we describe bonding in and reactivity of dichloro(8-dimethylamino-1-naphthyl)phosphane towards pentacarbonyltungsten(0) reagents. A 1-aza-2-phospha-acenaphthene complex was obtained via the unexpected elimination of chloromethane enabled through P-coordination. Theoretical DFT calculations provide insights into P⋯N pnictogen bonding interaction as well as the reaction pathway of the elimination reaction.
Phosphanes are typically known and used as Lewis bases due to the availability of a lone electron pair, and are employed as tunable ligands in coordination chemistry.1–3 But they can also act as Lewis acids when electron withdrawing substituents are present.1 An early example was provided by Holmes and Bertaut in 1958 reporting on a thermally unstable adduct of trimethylphosphane and trichlorophosphane4 which was structurally confirmed by Müller and Winkler in 2001 in the case of PBr3.5 Since the 1960s, peri-substituted species play an important role as donor–acceptor complexes,6 e.g. in proton sponges,7–9 group 13,10 and group 15 and 16 element peri-substituted species.11 These compounds showed an increased stability towards redox decomposition pathways due to the geometrically enforced acid–base interaction.12 The coordination chemistry of group 13–15 element peri-substituted compounds (primarily of acenaphthene derivatives) are of great interest as illustrated in the review by Kilian in 2011,13 and many new developments have occurred since.14–21 Intramolecularly amino-stabilized phosphanes are of special interest due to a potentially increased electron density at the phosphorus. For example, the existence of an intramolecular N→P interaction in 8-amino-1-naphthylphosphane I was proposed early by Schiemenz and Papageorgiou (Fig. 1).22 This was further studied by Corriu observing a P–N distance of 2.706(6) Å being significantly shorter than the sum of the van der Waals radii (3.4 Å), and X-ray diffraction studies revealed an orientation of the lone electron pair at nitrogen towards the phosphorus centre.23
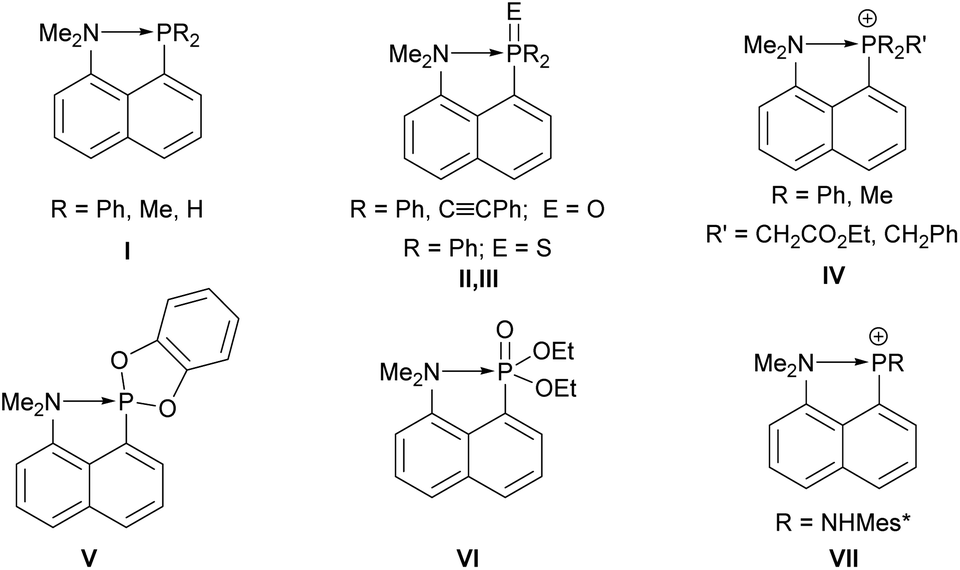 | ||
| Fig. 1 Examples of 8-amino-1-naphthyl-stabilized phosphorus species I–VII.22–28 | ||
In the same vein further studies were performed by Netzbandt.28 Additionally, Corriu and Chuit et al. reported on respective 8-amino-1-naphthyl-substituted phosphane oxides II, phosphane sulfide III, phosphonium IV, phosphonite V and phosphonate compounds VI (Fig. 1).25–27 Sanchez and Romanenko synthesized an 8-amino-1-naphthylphosphenium cation VII.24 The hypothesis of an intramolecular dative N→P bonding caused a controversial debate in the literature.29 Such dative interactions can be defined as intramolecular pnictogen bonds following the recent definition by the IUPAC.30 An especially interesting reactivity of some peri-substituted donor–acceptor complexes are coupling reactions to form a covalent bond of the donor and acceptor centres via elimination reactions. Kilian and co-workers reported on thermally induced P–P dehydro- and dealkacoupling reactions, and proposed a radical mechanism for the latter.31
We decided to focus on the dichloro(8-dimethylamino-1-naphthyl)phosphane 1 as starting material for the synthesis of an intramolecularly donor-stabilized phosphinidene complex; the synthesis was first described by Corriu, i.e., 8-amino-1-naphthyl lithium VIII was treated with 4 eq. of trichlorophosphane at low temperatures (−80 °C) yielding 62% of the product (Scheme 1).26 Surprisingly, 1 has been investigated only towards variation of the substitution pattern at phosphorus25,26 but coordination chemistry has not been reported, yet.
 | ||
| Scheme 1 Synthesis of dichloro(8-dimethylamino-1-naphthyl)phosphane 1.26 | ||
Herein, reactivity of compound 1 towards pentacarbonyltungsten(0) reagents is reported, including the elimination of chloromethane to form a 1-aza-2-phospha-acenaphthene-κP complex.
When compound 1 was prepared according to the protocol,26 an almost quantitative yield was obtained when the product was extracted from the residue using diethyl ether. The molecular structure of 1, determined by single crystal X-ray diffraction analysis (Fig. 2), revealed a very short P–N distance of 2.3280 Å. This is significantly shorter (Δ(dPN) = 0.38 Å) than the one of the aforementioned P–Ph2 substituted derivative (2.706(6) Å) and, thus, providing evidence for a stronger P–N interaction. This is further supported by two acute angles, i.e., the C2–C7–N angle of 114.5° and the C2–C1–P angle of 117.5°, due to the geometrically enforcing short N–P distance. As the N, P and Cl2 atoms are arranged in a linear fashion with an N–P–Cl2 angle of 178.4°, a donation of the lone electron pair at nitrogen to the σ*(P–Cltrans) orbital is assumed. This is corroborated by the significantly elongated P–Cl2 distance of 2.2046 Å (d(P–Cl1) = 2.1007 Å).
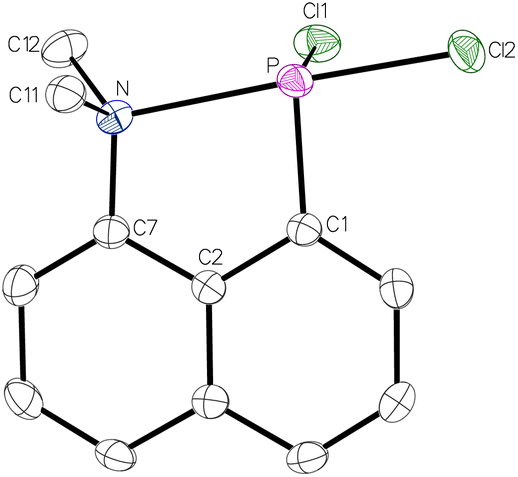 | ||
| Fig. 2 Molecular structure of 1. Thermal ellipsoids are set at 50% probability and hydrogen atoms are omitted for clarity. Selected bond lengths/Å and bond angles/°: P–N 2.32803(8), P–Cl1 2.1007(7), P–Cl2 2.2046(7), N–P–Cl2 173.38(2), Cl1–P–Cl2 96.8(5), C2–C1–P 117.51(13), C2–C7–N 114.48(16). | ||
To study the coordination chemistry of the dichlorophosphane 1 (Scheme 2), it was treated with different reagents such as freshly prepared pentacarbonyl(tetrahydrofuran)tungsten(0) (2a)32 or pentacarbonyl(dichloromethane)tungsten(0) (2b)33 but only the latter showed an almost clean conversion. The 31P{1H} NMR spectrum of the reaction mixture revealed the formation of two main products (ratio 71![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :29) with resonance signals at 181.5 ppm (1JW,P = 365.3 Hz) and 112.3 ppm (1JW,P = 332.4 Hz). The 31P NMR signal, downfield-shifted compared to 1, is tentatively assigned to the dichlorophosphane-κP complex 3. Unfortunately, all our attempts to isolate this compound failed. The yield could be significantly improved when 1 was treated with acetonitrile(pentacarbonyl)tungsten(0) (2c)34 in toluene and heated at 70 °C.
:29) with resonance signals at 181.5 ppm (1JW,P = 365.3 Hz) and 112.3 ppm (1JW,P = 332.4 Hz). The 31P NMR signal, downfield-shifted compared to 1, is tentatively assigned to the dichlorophosphane-κP complex 3. Unfortunately, all our attempts to isolate this compound failed. The yield could be significantly improved when 1 was treated with acetonitrile(pentacarbonyl)tungsten(0) (2c)34 in toluene and heated at 70 °C.
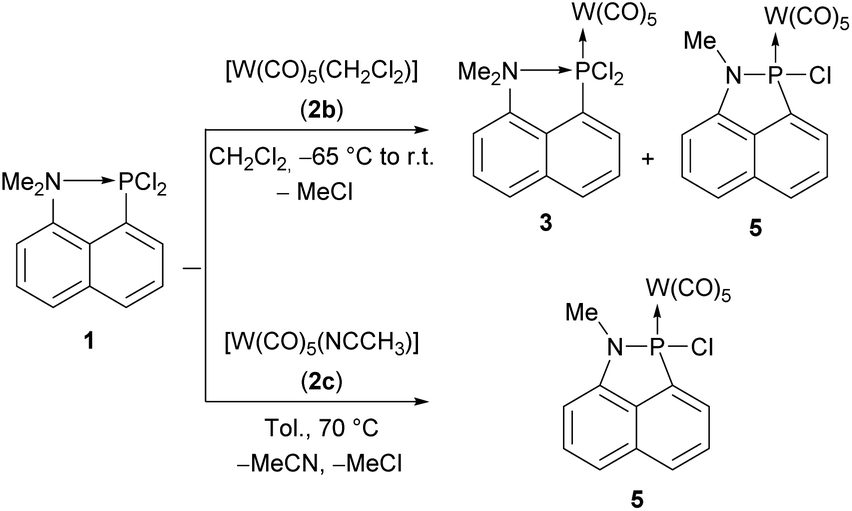 | ||
| Scheme 2 Complexation reaction of 1 with 2b and 2c. | ||
The product possessing the signal at 112.3 ppm was isolated via extraction of the residue with diethyl ether and identified as the 1-aza-2-phospha-acenaphthene-κP complex 5. This N–P coupling reaction is very reminiscent to the P–P dehydro- and dealkacoupling reactions of Kilian.31
The molecular structure was confirmed by single crystal X-ray diffraction analysis (Fig. 3). The P–N distance of 1.6970 Å is typical for a single bond as observed in similar systems, e.g., in a 1,2H-azaphosphole of Cain in 2018 (d(P–N) = 1.74 Å)35 or a respective complex from our earlier work (d(P–N) = 1.67 Å).36
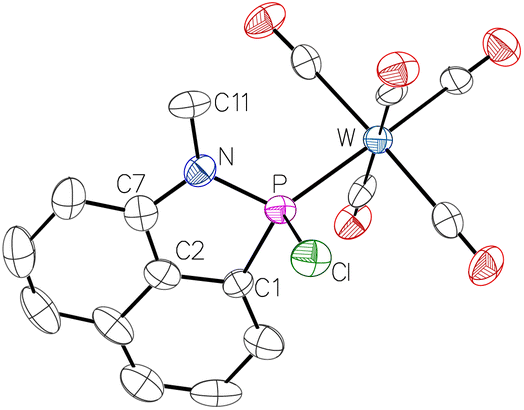 | ||
| Fig. 3 Molecular structure of 5. Thermal ellipsoids are set at 50% probability and hydrogen atoms are omitted for clarity. Selected bond lengths/Å and bond angles/°: P–N 1.697(5), P–Cl 2.099(3), P–W 2.4318(17), N–P–C1 91.9(3), N–P–Cl 104.4(2), N–P–W 120.6(2), C1–P–W 123.9(3), C1–P–Cl 101.0(2), Cl–P–W 111.48(8). | ||
At this point, we assumed that a further destabilization of the (elongated) P–Cl bond occurs due to the complexation at phosphorus, thus leading to a heterolytic P–Cl bond cleavage and, eventually, to the elimination of chloromethane to form 5 from 4, a transient phosphenium complex (see Fig. 5).
The proposed, rare elimination of a halomethane is somewhat similar to a reaction in which an azasilane was formed from an intramolecularly stabilized 8-dimethylamino-1-naphthylsilene treated with iodomethane.37
To examine if the non-ligated 1-aza-2-phospha-acenaphthene 1 is able to eliminate chloromethane a toluene solution was heated to 70 °C, but no reaction occurred. It should be also noted that related (acyclic) amino(chloro)phosphane complexes, being not constrained by an aromatic backbone, were investigated inter alia by Kuchen, and no elimination of chloromethane was reported either.38,39
In order to give theoretical support to the proposed mechanism, DFT calculations have been performed. Initially, compound 3 was fully optimized in dichloromethane. The resulting geometry exhibits a N⋯P distance of d(P–N) = 2.617 Å which is longer than the sum of the covalent radii (ΣRcov = 1.78 Å), and shorter than the sum of their van der Waals radii (ΣRvdW = 3.35 Å), thus suggesting a partial covalent character. The natural bond orbital (NBO) analysis has been performed for 3 in order to analyse orbital donor acceptor interactions (see the ESI,† theoretical methods) (Fig. 4).
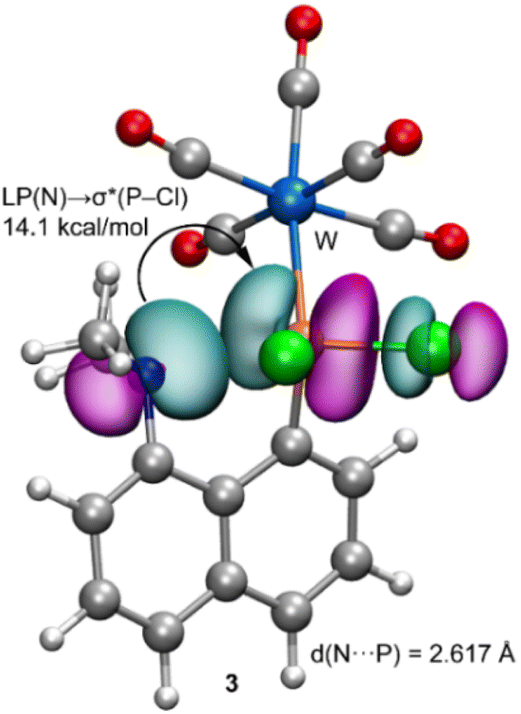 | ||
| Fig. 4 NBO involved in the LP(N) → σ*(P–Cl) transfer e transfer. | ||
We have found an important electron transfer from the lone pair (LP) orbital of N to the P–Cl antibonding orbital σ*(P–Cl), with a concomitant stabilisation energy of E(2) = 14.1 kcal mol−1. Such important donor–acceptor interaction is relevant, since the electron transfer to the antibonding σ*(P–Cl) orbital significantly weakens the P–Cl bond and, eventually, facilitates the subsequent nucleophilic attack.
The proposed mechanism is supported by DFT calculations as detailed in the energy profile shown in Fig. 5. That is, compound 5 is easily accessible from compound 3, via the TS1 that is only 6.5 kcal mol−1 higher in energy. Compound 4 is 2.1 kcal mol−1 higher in energy than compound 3, therefore the equilibrium is displaced toward the starting product. The N⋯P distance in the cationic intermediate 5 is 1.910 Å, significantly shorter than in 3 (2.617 Å). The final product 5 is obtained via a SN2 transition state (see TS2 in Fig. 5) where the chloride anion attacks one of the methyl groups with the concomitant formation of methylchloride. This is the rate determining step (barrier ΔG# = 18 kcal mol−1 from 4). The reaction is exergonic (−20.1 kcal mol−1) and irreversible since the reverse step barrier is very high (40.2 kcal mol−1). In the final product the N⋯P distance is 1.68 Å that is in excellent agreement with the experimental one (1.69 Å).
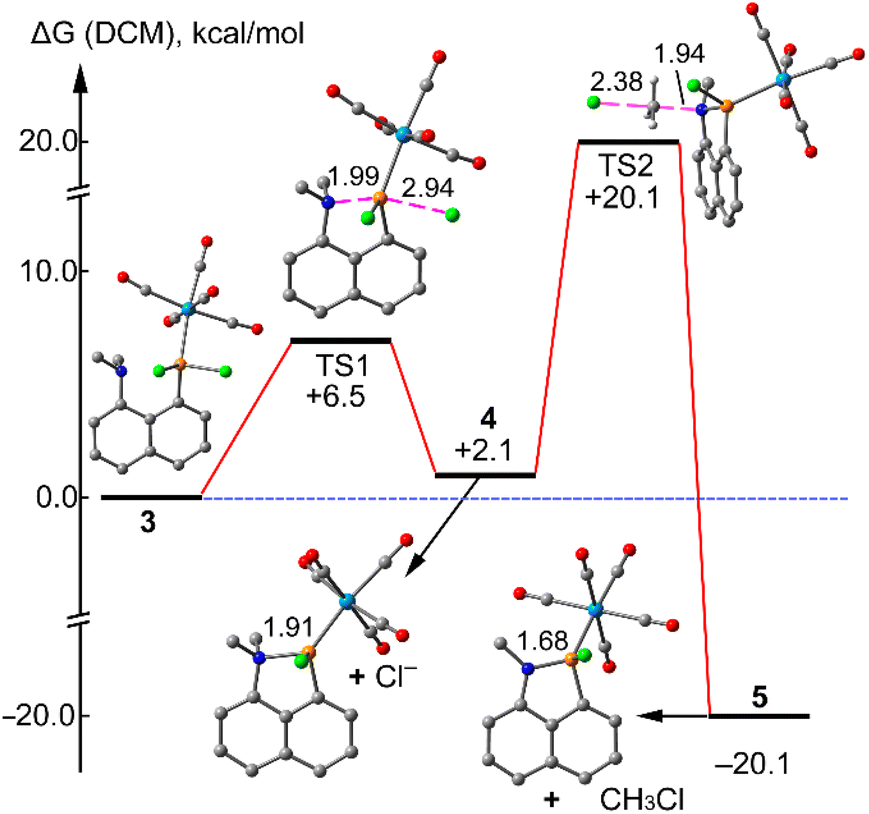 | ||
| Fig. 5 Energy profile for the transformation of compound 3 to 5 via the intermediate 4. | ||
The P–Cl bond in complex 5 is highly reactive and can be easily hydrolysed to form the respective P–OH substituted complex 6.
This became apparent in our attempts to further purify complex 5 via column chromatography (SiO2) at ambient temperature and without the use of inert gas atmosphere, which then led to a full conversion of 5 into complex 6 (Scheme 3).
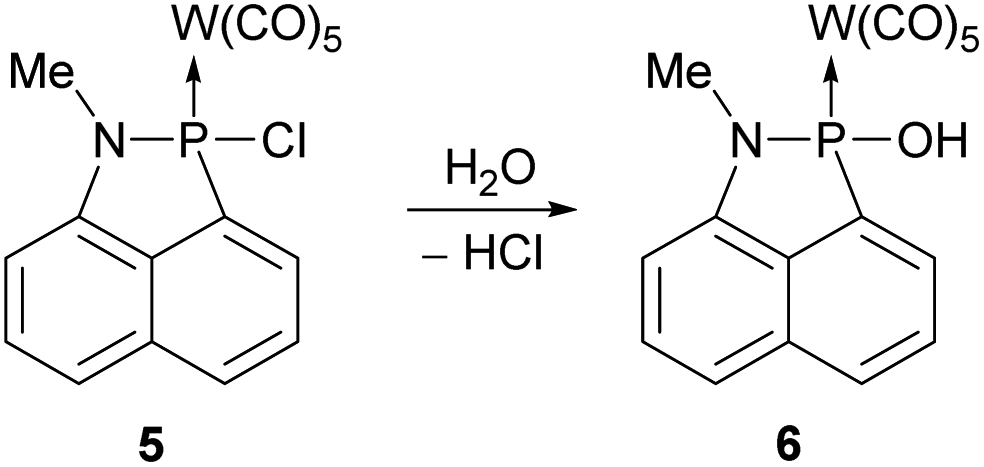 | ||
| Scheme 3 Hydrolysis of complex 5 to form the P–OH substituted complex 6. | ||
Complex 6 was fully characterised and showed, e.g., the broad signature vibrational band of the P-hydroxy substituent at 3473 cm−1 in the IR spectrum; the constitution was further confirmed by an X-ray diffraction study (see ESI Fig. S22†), but the data quality does not justify any further discussion.
In summary, the existence of a strong N→P interaction in a peri-substituted 8-dimethylamino-1-naphthyl)phosphane was clearly evidenced by X-ray diffraction data which was futher supported by DFT results. Complexation to pentacarbonyl-tungsten(0) led to a cleavage of a P–Cl bond and formation of a 1-aza-2-phospha-acenaphthene complex via N-dealkylation and a rare elimination of chloromethane. DFT calculations provide more insight into the reaction mechanism of the transformation 3 to 5 via the cationic intermediate 4 that is in equilibrium with 3. The subsequent elimination and irreversible transformation of 4 to 5 is the rate determining step.
Author contributions
DB and TB planned and carried out the experiments, collected and evaluated analytical data. GS measured and evaluated the crystallographic data. RGM and AF performed and evaluated the DFT computational study. RS had the supervision of this investigation. DB, RGM, AF and RS wrote this paper. The ESI† was prepared by DB, RGM, AF and RS.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We are grateful to Prof. Dr A. C. Filippou and Prof. Dr D. Menche for the use of X-ray facilities. A. F. is grateful to the Alexander von Humboldt Foundation for the J. C. Mutis Award.Notes and references
- R. H. Crabtree, The organometallic chemistry of the transition metals, Wiley, Hoboken, NJ, 5th edn, 2009 Search PubMed.
- M. Peruzzini and L. Gonsalvi, Phosphorus compounds. Advanced tools in catalysis and material sciences, Springer, Dordrecht, 2011 Search PubMed.
- N. N. Greenwood and A. Earnshaw, Chemistry of the elements, Elsevier-Butterworth-Heinemann, Amsterdam, 2nd edn, 1997 Search PubMed.
- R. R. Holmes and E. F. Bertaut, J. Am. Chem. Soc., 1958, 80, 2980–2983 CrossRef CAS.
- G. Müller, H.-J. Matheus and M. Winkler, Z. Naturforsch., B: J. Chem. Sci., 2001, 56, 1155–1162 CrossRef.
- V. Balasubramaniyan, Chem. Rev., 1966, 66, 567–641 CrossRef CAS.
- L. Sobczyk, J. Mol. Struct., 2010, 972, 59–63 CrossRef CAS.
- A. L. Llamas-Saiz, C. Foces-Foces and J. Elguero, J. Mol. Struct., 1994, 328, 297–323 CrossRef CAS.
- H. A. Staab and T. Saupe, Angew. Chem., 1988, 100, 895–909 (Angew. Chem. Int. Ed. Engl., 1988, 27, 865–879) CrossRef CAS.
- J. D. Hoefelmeyer, M. Schulte, M. Tschinkl and F. P. Gabbaı, Coord. Chem. Rev., 2002, 235, 93–103 CrossRef CAS.
- P. Kilian, F. R. Knight and J. D. Woollins, Chem. – Eur. J., 2011, 17, 2302–2328 CrossRef CAS PubMed.
- B. A. Chalmers, D. M. U. K. Somisara, B. A. Surgenor, K. S. Athukorala Arachchige, J. D. Woollins, M. Bühl, A. M. Z. Slawin and P. Kilian, Molecules, 2021, 26, 7222–7237 CrossRef CAS PubMed.
- P. Kilian, F. R. Knight and J. D. Woollins, Coord. Chem. Rev., 2011, 255, 1387–1413 CrossRef CAS.
- M. J. Ray, R. A. M. Randall, K. S. A. Arachchige, A. M. Z. Slawin, M. Bühl, T. Lebl and P. Kilian, Inorg. Chem., 2013, 52, 4346–4359 CrossRef CAS.
- M. J. Ray, M. Bühl, L. J. Taylor, K. S. Athukorala Arachchige, A. M. Z. Slawin and P. Kilian, Inorg. Chem., 2014, 53, 8538–8547 CrossRef CAS.
- B. A. Chalmers, P. S. Nejman, A. V. Llewellyn, A. M. Felaar, B. L. Griffiths, E. I. Portman, E.-J. L. Gordon, K. J. H. Fan, J. D. Woollins, M. Bühl, O. L. Malkina, D. B. Cordes, A. M. Z. Slawin and P. Kilian, Inorg. Chem., 2018, 57, 3387–3398 CrossRef CAS PubMed.
- S. Furan, E. Hupf, J. Boidol, J. Brünig, E. Lork, S. Mebs and J. Beckmann, Dalton Trans., 2019, 48, 4504–4513 RSC.
- M. Aman, L. Dostál, A. Růžička, J. Tydlitát, J. Beckmann, J. Turek and R. Jambor, Chem. Commun., 2021, 57, 12992–12995 RSC.
- S. Holsten, L. A. Malaspina, F. Kleemiss, S. Mebs, E. Hupf, S. Grabowsky and J. Beckmann, Organometallics, 2021, 40, 2027–2038 CrossRef CAS.
- S. Furan, M. Molkenthin, K. Winkels, E. Lork, S. Mebs, E. Hupf and J. Beckmann, Organometallics, 2021, 40, 3785–3796 CrossRef CAS.
- D. Duvinage, P. Puylaert, E. K. Wieduwilt, L. A. Malaspina, A. J. Edwards, E. Lork, S. Mebs, E. Hupf, S. Grabowsky and J. Beckmann, Inorg. Chem., 2022, 61, 8406–8418 CrossRef CAS.
- G. P. Schiemenz and E. Papageorgiou, Phosphorus Sulfur Relat. Elem., 1982, 13, 41–58 CrossRef CAS.
- C. Chuit, R. J. Corriu, P. Monforte, C. Reyé, J.-P. Declercq and A. Dubourg, J. Organomet. Chem., 1996, 511, 171–175 CrossRef CAS.
- M. Sanchez, F. Cosledan, J. M. Sotiropoulos, L. Lamandé, A. B. Drapailo, A. O. Gudima and V. D. Romanenko, Tetrahedron Lett., 1995, 36, 2085–2088 CrossRef CAS.
- S. B. Bushuk, F. H. Carré, D. M. Guy, W. E. Douglas, Y. A. Kalvinkovskya, L. G. Klapshina, A. N. Rubinov, A. P. Stupak and B. A. Bushuk, Polyhedron, 2004, 23, 2615–2623 CrossRef CAS.
- F. H. Carré, C. Chuit, R. J. P. Corriu, W. E. Douglas, D. M. H. Guy and C. Reyé, Eur. J. Inorg. Chem., 2000, 2000, 647–653 CrossRef.
- C. Chuit and C. Reyé, Eur. J. Inorg. Chem., 1998, 1998, 1847–1857 CrossRef.
- D. W. Netzbandt, PhD thesis, University of Bonn, 2008 Search PubMed.
- G. P. Schiemenz, Phosphorus, Sulfur Silicon Relat. Elem., 2009, 184, 2247–2262 CrossRef CAS.
- G. Resnati, Definition of the pnictogen bond, 2023, https://iupac.org/recommendation/definition-of-the-pnictogen-bond, (accessed 11 June 2023) Search PubMed.
- L. J. Taylor, M. Bühl, B. A. Chalmers, M. J. Ray, P. Wawrzyniak, J. C. Walton, D. B. Cordes, A. M. Z. Slawin, J. D. Woollins and P. Kilian, J. Am. Chem. Soc., 2017, 139, 18545–18551 CrossRef CAS PubMed.
- I. W. Stolz, H. Haas and R. K. Sheline, J. Am. Chem. Soc., 1965, 87, 716–718 CrossRef CAS.
- M. A. Zayed and H. Fischer, J. Therm. Anal. Calorim., 2000, 61, 897–908 CrossRef CAS.
- U. Koelle, J. Organomet. Chem., 1977, 133, 53–58 CrossRef CAS.
- J. Hyvl, W. Y. Yoshida, C. E. Moore, A. L. Rheingold and M. F. Cain, Polyhedron, 2018, 143, 99–104 CrossRef CAS.
- R. Streubel, H. Wilkens, U. Rohde, A. Ostrowski, J. Jeske, F. Ruthe and P. G. Jones, Eur. J. Inorg. Chem., 1999, 1999, 1567–1579 CrossRef.
- M. Mickoleit, K. Schmohl, M. Michalik and H. Oehme, Eur. J. Inorg. Chem., 2004, 2004, 1538–1544 CrossRef.
- K. Diemert, A. Hinz, W. Kuchen and D. Lorenzen, J. Organomet. Chem., 1990, 393, 379–387 CrossRef CAS.
- K. Diemert, W. Kuchen and D. Lorenzen, J. Organomet. Chem., 1989, 378, 17–31 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 2250016–2250018. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d3ra04352e |
| This journal is © The Royal Society of Chemistry 2023 |